Abstract
Ventricular arrhythmia is the primary cause of sudden cardiac death in patients with myocardial infarction (MI). Myocardial inflammation and Na+/Ca2+ imbalance are the main triggering factors for life-threatening tachyarrhythmias after MI, which induce ion channel dysfunction, intracellular environment imbalance, tissue damage, and other alterations, subsequently resulting in modifications in cardiac conduction velocity and pathways. Subsequent adverse fibrotic remodeling provides a substrate for ventricular tachyarrhythmia (VT). Mitochondria, as the intersection site of these pathophysiological changes and the center of Na+/Ca2+ homeostasis and inflammatory crosstalk, may be key sites for the occurrence and development of ischemic arrhythmia. This review briefly outlines the roles of inflammation, Na+/Ca2+ homeostasis, and mitochondria in the damage, repair, and structural remodeling of infarcted hearts, in which these three are interconnected to provide a large number of substrates for VT.
1 Introduction
Ventricular arrhythmia (VA) is the main cause of sudden cardiac death (SCD) in patients with myocardial infarction (MI) (1). SCD accounts for approximately 50% of all cardiovascular deaths and is the primary manifestation of heart disease (2). Approximately 250,000–400,000 people die from SCD annually in the United States. In North America and Europe, the annual incidence of SCD in the general population ranges from 50 to 100 deaths per 100,000 individuals. In China, the incidence of SCD is approximately 41.84/100,000, which has significantly increased. While primary and secondary prevention has improved over recent years and the mortality rate from coronary heart disease has substantially decreased, the decline in SCD rate has been much smaller (3, 4). In addition to congenital cardiac structural abnormalities, cardiomyopathy, and primary cardiac ion channel diseases, slowing of conduction and increased dispersion of action potential repolarization caused by ischemia play important roles in SCD occurrence (5).
Myocardial inflammation and Na+/Ca2+ imbalance are the main triggering factors of life-threatening tachyarrhythmias after MI (6, 7). In the early stages, the aggregation of inflammatory factors mediates myocardial cell damage, leading to a change in ion channel function and a direct action on arrhythmia. In the subsequent healing phase of MI, inflammatory cells not only activate the repair of myofibroblasts and vascular cells but may also cause adverse fibrotic remodeling of the living segment. Cardiac hemodynamic and structural changes can cause left ventricular dilatation and dysfunction, providing a substrate for the ventricular tachyarrhythmia (VT) reentry circuit (8). With these structural modifications, alterations in intercellular coupling and ion channels further augment the susceptibility to VA. Therefore, the inhibition of myocardial inflammation, electrophysiological changes in the Na+/Ca2+ imbalance, and subsequent structural changes can significantly prevent the occurrence of VA and reduce SCD mortality in patients with MI.
Mitochondria are the main organelles of cardiomyocytes and are primarily responsible for adenosine triphosphate (ATP) production, metabolic regulation, oxidative stress, and inflammatory responses (9). In recent record, abnormal automaticity, triggered activity, and reentry are the three main mechanisms underlying cardiac arrhythmia (10). Mitochondrial dysfunction is closely associated with cardiac arrhythmia. For instance, triggered activity is caused by diastolic sarcoplasmic reticulum (SR) Ca2+ release. The mitochondrial Ca2+ content affects SR Ca2+ release by activating the ryanodine receptor 2 (RyR2) channels (11). Mitochondrial dysfunction causes abnormal ion channel function, Na+/Ca2+ imbalance, increased reactive oxygen species (ROS) production, changes in mitochondrial permeability, and activation of inflammatory factors, which in turn cause apoptosis and lead to fibrosis (12, 13). Fibrosis is one of the substrates for reentry (14). This review briefly outlines the roles of inflammation, Na+/Ca2+ homeostasis, and mitochondria in the damage, repair, and structural remodeling of the infarcted heart; describes how these three are interconnected in this dynamic process to provide a large number of substrates for VA; and discusses the difficulties and challenges faced by current related research and clinical practice.
2 Mechanism of ischemic arrhythmias
Electrophysiological changes in the ischemic region after acute MI are rapid, ranging from normal electrical activation to severe abnormal electrical activation, and repolarization occurs within 1 min. In the early stages of MI, the action potential duration (APD) is shortened, the amplitude is decreased, and the ascending velocity of the ascending branch is slowed, followed by a significant post-repolarization refractory period. The excitation threshold decreases at 1–3 min after coronary artery occlusion and then increases rapidly. At approximately 5 min after coronary artery occlusion, the threshold is 10 times higher than that before coronary artery occlusion, and excitability progressively increases as the tissue transitions from the normal area to the ischemic area. The absolute and relative refractory periods of the ischemic myocardium are shortened by 40–50 ms. At approximately 15 min after coronary occlusion, the cells completely lose their ability to respond, and their excitability gradually disappears. Coronary artery occlusion occurs at 20 min to 2 h after the occurrence of conduction disorders, along with prolonged mild simple conduction time and severe complete atrioventricular block, resulting in arrhythmia. Electrocardiography reveals ST segment elevation, delayed activation, QRS fragmentation, t-waves and QRS alternans, and conduction block (15).
2.1 Altered functions of multiple ion channels
Changes in ion channel function play an important role in the electrophysiological changes that occur during arrhythmias. The dysfunction of various cardiac ion channels, such as Na+, K+, and Ca2+ channels, increases the susceptibility to arrhythmia after MI (16, 17). K+ starts to change during the early stages of acute myocardial ischemia. Long-term ischemia leads to an increase in extracellular K+ concentration, which is the substrate and trigger for cardiac conduction velocity (CV) changes and arrhythmia (18). An increase in extracellular K+ concentration changes the resting membrane potential of cardiomyocytes, reduce the activity of voltage-gated Na+ channels, leads to a decrease in cell excitability and CV, and promotes unidirectional blockage and reentry (19, 20). CV and APD are important factors in the occurrence of arrhythmias, and changes in CV play important roles in the generation and maintenance of arrhythmias. Voltage-optical mapping studies of isolated hearts have shown that the induction of ventricular fibrillation (VF) at high activation frequencies is associated with decreased CV (21). It has also been suggested that arrhythmias may result from local CV heterogeneity (22). Many factors affect the CV, including coupling with non-muscle cells (23, 24), extracellular gaps, gap links, and extracellular ion concentrations (25–27). Among these, the upstroke velocity of the action potential is the key factor that affects CV, and the upstroke velocity mainly depends on the recovery of the Na+ channel. Therefore, the CV is closely related to Na+ channels (28).
The major subtype of voltage-gated Na+ channels, Nav1.5, encoded by the SCN5A gene, is mainly expressed in the intercalated discs of the heart. It is the key channel for maintaining a normal CV and determining the excitability and conductivity of the heart. It also interacts with cAMP-dependent protein kinase A (PKA) and calmodulin-dependent kinase II (CaMKII). It can bind to various proteins such as CaMKII and membrane-associated guanylate kinase (MAGUK) to form macromolecular compounds that regulate gene transcription, protein synthesis, trafficking, membrane incorporation, channel function, and ultimately degradation (29, 30). Nav1.5 remodeling is a key basis for the occurrence of VA reentry into the border zone of MI. Recent studies have shown that the autoimmune response against Nav1.5 can cause conduction defects (31) and complete inactivation of Nav1.5 due to a molecular dynamics disorder, can cause long QT syndrome type 3 (LQT3). Non-equilibrium gating leading to decreased availability of the Nav1.5 closed conformation can cause Brugada syndrome (BrS). Moreover, Nav1.5, dysfunction can lead to VA in pathophysiological conditions of heart disease, such as heart failure, dilated cardiomyopathy, and diabetic heart disease (32–34). Previous studies have determined that SCN5A mutations can induce a decrease in the amount and function of Nav1.5, using genetic, electrophysiological, and molecular methods, leading to a series of VA such as LQT3, BrS, torsades de pointes, and idiopathic ventricular fibrillation. This process can be affected by time, temperature, environmental factors, and genetic factors (35, 36).
The heart beats rhythmically to drive the blood through the body. Cardiac action potentials are generated by the simultaneous opening and closing of many transmembrane ion channels. Dynamic changes in Ca2+ concentration play a key role in this process (37). RyR2 is the main Ca2+ release channel during systole and sarcoendoplasmic reticulum Ca2+-ATPase (SERCA) is the main Ca2+ uptake channel during systole in the SR, which is involved in excitation-contraction coupling. The amount of Ca2+ released by the SR through RyR2 largely determines the Ca2+ transient state (38). Ca2+ flows into the cell and is released from the SR via RyR2 to trigger the contractile myocardium, after which Ca2+ is mostly taken up or loaded into the SR via SERCA to trigger the diastolic myocardium. Genetic and acquired defects in RyR2 or SERCA have been suggested to be associated with a range of heart diseases, including life-threatening arrhythmias and heart failure (39) (40). These defects typically manifest as an impaired ability of RyR2 to remain closed during the diastolic phase of the cardiac cycle, resulting in enhanced diastolic Ca2+ release (DCR, manifested as Ca2+ sparks and Ca2+ waves) (41). The diastolic release of Ca2+ from the SR leads to prolonged APD and increased arrhythmic risk (10). Increasing the RyR2 activity in the ventricle and alleviating its inhibitory effect on RyR2-mediated Ca2+ release have been reported to independently cause catecholaminergic polymorphic VT (42). Furthermore, Xie et al. discovered that type 2a SERCA (SERCA2a) knockdown mice had a reduced arrhythmic risk of ischemic cardiomyopathy due to decreased SR diastolic Ca2+ leak (40). Therefore, intracellular Ca2+ homeostasis imbalance is a key factor in ischemic arrhythmia (43).
CaMKII is a multifunctional serine/threonine protein kinase widely expressed in vivo. Its activity is mainly modified and regulated by changes in intracellular Ca2+ content, and regulates intracellular Ca2+ dynamics, contractility, metabolism, and gene expression by phosphorylating various downstream targets (44). CaMKII has been identified as an important modulator of excitation-contraction and excitation-transcription coupling, a key determinant of the response to pathological cardiac remodeling, and is activated upon MI. CaMKII exerts proarrhythmic signaling through a large number of ion channels and SR-related proteins. CaMKII is known to activate L-type calcium channels, various K+ channels, Nav1.5 and Nav1.8 (45, 46). Stimulation of these channels results in early and delayed depolarization and spatially dispersed increases in repolarization, which promote arrhythmias, such as atrial fibrillation, ventricular tachycardia, and VF. CaMKII can also phosphorylate RyR2 and promote Ca2+ release from the SR into the cytoplasm (47). This SR Ca2+ leakage can activate proarrhythmic Ca2+-sensitive conductance. Overexpression of CaMKII has also been shown to induce structural and electrical remodeling of the heart, leading to impaired contractility and an increased risk of SCD. Conversely, inhibition of CaMKII helps maintain intracellular Ca2+ homeostasis after pressure overload and ischemic stress to prevent adverse electrical remodeling after MI (48, 49). Previous studies have demonstrated that CaMKII co-immunoprecipitates with Nav1.5, and experiments have shown a stable physical interaction between phosphorylated CaMKIIδC and L1 of Nav1.5. Phosphorylation of CaMKII enhances the inhibition of the late depolarization current of Nav1.5, leading to the prolongation of the action potential, further disrupting Ca2+ homeostasis, and providing additional substrates for arrhythmia formation (50).
2.2 Inflammation
Inflammation is thought to trigger arrhythmia following MI. In an analysis of 478,524 individuals from the UK Biobank cohort, C-reactive protein (CRP) levels were found to be significantly and positively associated with the risk of developing atrial fibrillation. The heart rate for atrial fibrillation events increased significantly with increasing neutrophil count, monocyte count, and neutrophil-to-lymphocyte ratio (NLR), whereas the levels of systemic infection markers had an even stronger relationship with VA risk than the levels of systemic infection markers with atrial fibrillation risk. Restricted cubic spline analysis of the fully adjusted model showed that the risk of developing VA increased monotonically with increasing CRP levels and neutrophil counts; a similar association was observed between monocyte count, NLR, and VA occurrence (51). It adds precipitants and substrates to the VA in both the early acute injury phase and the subsequent chronic repair phase.
Repair of the infarcted heart depends on the timely suppression of the inflammatory response and the resolution of inflammatory infiltration after infarction. Damage-associated molecular pattern proteins released by necrotic cells after early MI trigger local and systemic inflammatory responses. Various inflammatory factors directly induce arrhythmias and recruit large numbers of neutrophils and monocytes. Under the action of inflammatory factors, recruited white blood cells change the function of ion channels and cause membrane potential changes (52). Simultaneously, many white blood cells infiltrate and exchange ions, nucleotides, metabolites, and electrical signals with the cardiomyocytes via connexins. The quantitative change and redistribution of connexins leads to gap junction remodeling, which is an important factor in inducing arrhythmia (53). Macrophages couple with cardiomyocytes through gap junctions containing connexin 43(Cx43), undergo synchronous depolarization, and participate in normal and abnormal cardiac conduction. Moreover, computer simulations have shown that an increased number of such junctions reduces the action potential upshoot and overshoot, leading to earlier repolarization and a shorter refractory period (54).
In addition, fever, increased heart rate, and increased oxygen consumption caused by a systemic inflammatory response can further promote arrhythmia and expansion of the lesion area. Subsequently, white blood cells remove dead cells and matrix debris through phagocytosis, thereby providing an environment for subsequent repair of the infarct area. An appropriate early inflammatory response can reduce the infarct area, promote scar formation, maintain the stability of the environment in the peri-infarct area, and contribute to recovery of the ischemic myocardium. Excessive and prolonged inflammatory responses can lead to apoptosis of myocardial cells, hypertrophy, and fibrosis of myocardial tissue in the non-infarcted area, leading to adverse remodeling of ischemia-related tissues and myocardial electrophysiological dysfunction. Monocyte and macrophage subsets secrete cytokines and growth factors that coordinate the repair, recruitment, and activation of mesenchymal cells, such as cardiac fibroblasts and vascular endothelial cells (55). Activated mesenchymal cells secrete a large number of extracellular matrix proteins (56), promote the formation of myocardial fibrosis scars, and thus provide a matrix for reentry, which is closely related to arrhythmia (57).
2.3 Increased fibrosis
The heart adaptively responds to pathological injury, leading to cardiac remodeling characterized by cardiomyocyte hypertrophy and fibrosis. This remodeling includes chronic remodeling under volume/pressure load and acute repair under ischemia-hypoxia injury. During MI, phagocytosis of necrotic cells and tissues activates anti-inflammatory pathways that inhibit cytokine and chemokine signaling (58). Activation of the renin-angiotensin-aldosterone system and release of transforming growth factor-β induce the transformation of fibroblasts into myofibroblasts, causing ventricular fibrosis, similar to the macrophages mentioned above. When the number of myofibrocytes is considerable, nonmyocardial cells in the heart become obstacles to the propagation of action potentials. The differentiation state of fibroblasts has been previously shown to be associated with a change in the expression profile of ion channels, and the transition from fibroblasts to myofibroblasts can increase Nav1.5; furthermore, they display rapid inward voltage-gated sodium currents that exhibit biophysical properties similar to the sodium currents found in cardiomyocytes (59). in vitro-cultured fibroblasts can electrically couple with cardiomyocytes to participate in excitation conduction through Cx43, and these intercellular connections allow myofibroblasts to influence the electrical activity of cardiomyocytes (60). Previous studies have generated mice that specifically express the optogenetic cation channel ChR2(H134R) in cardiac fibroblasts. After MI, fibroblasts are highly expressed in the injured area and close to cardiomyocytes in scar tissue, and light stimulation of the scar tissue can cause excitation of the whole heart and induce arrhythmia. Cx43 and other gap junction proteins, which are thought to mediate the coupling between cardiomyocytes and fibroblasts, are not required. Gap junctions and ephaptic coupling mediate the coupling between cardiomyocytes and fibroblasts in a cooperative but functionally redundant manner; however, this fibroblast-muscle coupling is not as strong as myofibroblast-myocyte coupling (61). These results suggest that electrical coupling between myofibroblasts and cardiomyocytes can destroy the original electrophysiological activity of the myocardium, induce electrophysiological abnormalities such as ectopic automaticity, posterior depolarization, and reentry, and promote the occurrence and development of arrhythmia.
In addition to directly affecting the electrical activity by coupling with cardiomyocytes, fibroblasts, which are the main cells producing extracellular matrix, are activated in large numbers after MI, promoting the deposition of extracellular matrix proteins (62). Various extracellular matrix proteins bind to cytokines, growth factors, and cell surface receptors to regulate the cell phenotype, thereby indirectly affecting arrhythmia. Structural remodeling of atrial fibrillation has been reported to involve the accumulation of cross-linked collagen in atrial fibroblasts. In a previous report, calcitonin receptor-knockout mice exhibited atrial fibrosis and increased susceptibility to atrial fibrillation due to collagen accumulation (63). A large amount of collagen and extracellular matrix can mechanically separate cardiomyocytes, destroy the continuity of myocardial bundles, interfere with the gap junction of cardiomyocytes, destroy the electrical coupling between cardiomyocytes, and cause discontinuous or “zigzag” conduction, leading to slow CV, unidirectional conduction block, and prolongation of the conduction path between cardiomyocytes, thus inducing arrhythmia. Myocardial fibrosis is an essential substrate for arrhythmias (64).
After MI, three distinct structural regions emerge in the left ventricle: the infarct, transition boundary, and remote zones (65). Magnetic resonance imaging shows that a large peri-infarct transition border zone is the single factor in the inducibility of monomorphic VT, providing mechanistic support for the association between peri-infarct size and mortality. Tissue inhomogeneity in the infarct border may provide a substrate for underlying reentrant arrhythmias, leading to SCD (66). In a study of 686 patients with apparent idiopathic nonsustained VA, left ventricular scars with annular patterns were associated with malignant arrhythmic events on cardiac magnetic resonance imaging. All patients with annular scars showed VA with a right bundle branch block, and multifocal VA was observed in 46% of patients. The prevalence of multifocal VA is much higher in patients with annular scars than in those with non-annular scars, suggesting that the influence of a specific infarct shape on VA should not be ignored (67). In addition, other studies have shown that the effect of fibrotic areas <20% and >80% on arrhythmia is relatively benign and that the arrhythmogenic effect is usually maximal at 30%–50% of the fibrotic area (64). Thus, inhibiting the progression of fibrosis without affecting MI healing may reduce the risk of arrhythmias.
2.4 Crosstalk among Na+/Ca2+ homeostasis, inflammation, and fibrosis
In most pathological conditions, arrhythmia is often accompanied by inflammation and structural and electrical remodeling, with crosstalk. Upon initial ischemic injury, monocytes and macrophages are recruited and polarized to a proinflammatory phenotype, secreting inflammatory factors (such as IL-6, IL-1, and TNF-α), triggering a cytokine cascade. Inflammation rapidly induces the effect of cytokines on the expression of ion channels, which directly prolongs the QTc interval, and changes in these ion channels are negatively correlated with changes in CRP and IL-1 in patients. Although these changes are transient, they may significantly increase the risk of developing life-threatening VA in these patients (68). In addition, cardiac fibroblasts respond to IL-1 by acquiring a proinflammatory and matrix-degrading phenotype, delaying myofibroblast transformation and preventing premature acquisition of a matrix-synthetic phenotype until the infarct clears dead cells and matrix debris (69). In addition, some members of the chemokine family may also affect non-hematopoietic cells, such as cardiomyocytes, fibroblasts, and vascular cells, which mediate the transformation of fibroblasts into myofibroblasts or the recruitment of monocytes and neutrophils with fibroblast characteristics to promote the development of fibrosis (70, 71), which increases the axial resistance of the sarcoplasm. This enhances the coupling between fibroblasts and cardiomyocytes, both of which reduce the CV and increase the CV dispersion. As the activation of pro-inflammatory signals leads to cardiomyocyte death, mitochondrial membrane permeability transition pores open, perturb the intracellular Ca2+ balance, and increase ROS, triggering arrhythmic events. When intracellular Ca2+ increases in cardiomyocytes, CaMKII phosphorylation increases and activates IκB kinase, nuclear factor kappa B (NF-κB) is deinhibited, macrophage infiltration in the ischemic area increases, fibrosis scars become larger, and cardiac function is weakened (72). In the absence of endogenous CaMKII inhibitor 1 (CaMK2n1), the increased activation of CaMKIIδ-p38/JNK-NLRP3 inflammasome pathway leads to aggravated cardiomyocyte inflammation, aggravated ventricular remodeling and malignant VA (73). Single-cell sequencing of the infarcted and non-infarcted regions of ischemic cardiomyopathy revealed a large amount of leukocyte infiltration in the fibrotic myocardium, especially of proinflammatory CD4+ T cells (74). The presence of these inflammatory cells suggests that myofibroblast apoptosis occurs during the transition from the proliferative to mature phase of healing in the infarcted area, which may be regulated by inflammation (75). The increase in fibrosis can cause partial uncoupling of muscle fibers, a zigzag path of wave conduction, and slow or blocked conduction, which eventually leads to the occurrence of arrhythmia (76). Therefore, ischemic arrhythmia results from the crosstalk between the ion channel-fibrosis-inflammatory response and other factors.
3 The role of mitochondrial function as a central cross-linking point in ischemic arrhythmia
Mitochondria are widely distributed in cardiomyocytes, accounting for 30% of the total volume of adult cardiomyocytes (77). They produce ATP and regulate metabolism, oxidative stress, and inflammatory responses, which are common pathological changes in ischemic arrhythmia (9). Mitochondria also sense intracellular Ca2+ signals, mediate energy production and cell death (78), and play important roles in Ca2+ homeostasis in cardiomyocytes (79). Under normal conditions, fatty acids are the preferred energy substrates for ATP production in the myocardium. Fatty acids undergo β-oxidation in the mitochondria to produce acetyl-CoA, which enters the tricarboxylic acid cycle to produce ATP for energy (80).
3.1 Reduced mitochondrial function can cause mitochondrial Ca2+ overload in ischemic state
When the body undergoes hypoxia-ischemia, the mitochondrial metabolism changes from oxidative phosphorylation to glycolysis, which reduces oxygen consumption and ensures ATP output (81). This metabolic transition leads to an increase in lactic acid, a decrease in intracellular pH, and an increase in the concentration of H+ in both the intercellular space and within the cell (82). H+ is exchanged with Na+ through the Na+/H+ exchanger, resulting in an increase in intracellular Na+ concentration. Simultaneously, owing to the decrease in Na+/ K + -ATPase activity, the extracellular transport of Na+ is reduced, leading to its accumulation in cells. In addition, the influx of Na+ ions into cells through nonselective cation channels activated by membrane stretching also leads to an increase in intracellular Na+ concentration. This increase in Na+ further activates the sodium-calcium exchanger (NCX) to operate in a “reverse mode,” eventually leading to intracellular Ca2+ overload (83) and causing early afterdepolarization or delayed afterdepolarization. When multiple depolarization events reach the threshold for sodium channel activation, a series of tachyarrhythmias can be induced. If this is extremely insufficient to induce an action potential, it may exacerbate regional differences in repolarization, leading to alternating or unidirectional conduction blocks and reentry (84). When Ca2+ overload occurs, a large amount of Ca2+ enters mitochondria through the mitochondrial Ca2+ uniporter (MCU) complex (85). Ultimately, this results in mitochondrial Ca2+ overload.
3.2 Crosstalk between mitochondrial ROS and Na+/Ca2+ homeostasis
When mitochondrial calcium levels increase, the activity of the electron transport chain is stimulated, leading to a higher ROS release. Overproduction of mitochondrial-derived ROS may lead to the oxidation of RyR2 and further leakage of endoplasmic reticulum Ca2+, forming a vicious cycle (86). ROS also increases CaMKII phosphorylation. In ischemic heart disease, ROS production is increased in the infarct border region, which overactivates CaMKII phosphorylation and reduces INa density, thereby slowing the recovery rate after Nav1.5 inactivation. In addition, the delay in repolarization and prolongation of the effective refractory period lead to a decrease in cardiac CV and even a conduction block (87). Mitochondria are the central hub for immune system activation, and their dysfunction leads to many inflammatory diseases. On the one hand, it relies on ROS to trigger an inflammatory response. Impaired mitochondrial function leads to activation of the tricarboxylic acid cycle and an increase in nitric oxide synthase, which eventually causes an increase in ROS. However, this does not depend on ROS for triggering inflammatory responses.
3.3 Crosstalk among mitochondrial permeability transition pore and Na+/Ca2+ homeostasis and mROS
Mitochondrial Ca2+ binds to oxidized cardiolipin and triggers the release of the membrane gap protein cytochrome c into the cytoplasm. At the same time, the increase in Ca2+ and ROS levels can further open the mitochondrial permeability transition pore, leading to the immediate collapse of the mitochondrial membrane potential, release of cytochrome c, and activation of caspase protease, resulting in cardiomyocyte apoptosis (88). In the process, myocardial cells produce inflammatory factors such as TNF-α, IL-1β, and IL-6, leading to inflammatory response or dysfunction when myocardial injury occurs (89).
In recent years, studies have shown that, apart from the above effects through metabolism, inflammation, and oxidative stress, mitochondria are directly related to cell membrane Nav1.5 in structure and function, and this association is different in various mitochondrial subsets. Subsarcolemmal mitochondria are more closely related to Nav1.5 than to interfibrillar mitochondria and perinuclear subdomain mitochondria. This link may be established through the NCX, and its functional crosstalk includes sodium currents, Ca2+ dynamics, transcriptomics, and oxidative stress. Interestingly, the transcriptomics results showed that the negative correlation between SCN5A and SCL8B1 may be a compensatory result, indicating that the functions of the mitochondria and sodium channels are complementary. However, the mechanism by which Na+ channel expression or activity affects mitochondrial function or integrity requires further studied (90).
4 Limitations
Mitochondrial research currently faces several challenges and limitations in the study of arrhythmias. In different regions of the heart and even within individual myocardial cells, there is significant functional heterogeneity in mitochondria. Traditional batch detection methods (such as western blotting and PCR) mask this heterogeneity. Single-cell sequencing and spatial transcriptomics have partially addressed this issue; however, they have limitations in mitochondria-specific analysis. Most existing studies focus on the overall mitochondrial function changes in the entire myocardial cell but neglect the precise spatial localization of mitochondria within the cells and the interactions between organelles. Bidirectional communication between the mitochondria and the T tubules, SR, and endoplasmic reticulum is accomplished through mitochondria-associated endoplasmic reticulum membranes, forming a complex structure. Disruption of this specific spatial relationship not only leads to calcium regulation imbalance but also causes dysregulation of multicellular organelles, becoming a triggering factor for arrhythmia. Nevertheless, the details of this spatially specific regulatory mechanism have not yet been fully elucidated (91, 92). In addition, there is real-time coupling between the cardiomyocyte energy status and ion channel function, and this metabolic-electrical coupling occurs rapidly and precisely (93). However, owing to the lack of dynamic detection technology, there is a lack of research on the simultaneous monitoring of metabolic changes and electrical activity at the millisecond level. This has led to a fundamental gap in our understanding of how metabolic alterations translate into electrical instability, with most conclusions remaining at the correlation level.
5 Future directions
Despite these difficulties, the potential use of mitochondria in the treatment of arrhythmias remains promising. The therapeutic implications of targeting mitochondria are anticipated. For example, the coenzyme Q10(antioxidants protect mitochondria and transfer electrons to facilitate energy metabolism) that is currently widely used in clinical practice has demonstrated the potential for anti-arrhythmic effects. Studies have shown that it can reduce the incidence of atrial fibrillation. Supplementation of mitochondria-targeted antioxidants such as mitoquinone can reduce mitochondrial membrane damage, maintain Na+/K+-ATPase activity, and reduce NCX-mediated Ca²+ influx. It is particularly applicable to arrhythmias related to mitochondrial oxidative stress (such as ischemia-reperfusion injury and metabolic cardiomyopathy), and has achieved good clinical evidence. Targeted MCU inhibitors, such as Ru360, reduce mitochondrial calcium overload, whereas the activation of NCX promoted Ca²+ efflux and restores intracellular calcium homeostasis in cardiomyocytes. The activation of the TLR4/NF-κB pathway can be suppressed by improving mitochondrial membrane permeability, such as using cyclosporine A to inhibit the opening of the mitochondrial permeability transition pore and reducing the release of mitochondrial DNA and ROS, thereby alleviating the inflammatory response. Peptide inhibitors have been used to selectively block the pathological mitochondrial fission protein Drp1 to prevent excessive mitochondrial fission in the early stages of mitochondrial injury and to reduce cardiomyocyte apoptosis (94).
For arrhythmias caused by specific mitochondrial DNA mutations or nuclear DNA mutations, gene therapy is at the forefront of exploration. Normal genes are introduced into the lesion model by using adeno-associated virus for gene replacement therapy. Another strategy is heterologous expression, where mitochondrial genes are re-encoded and then introduced into the cell nucleus, allowing them to be synthesized in the cytoplasm and targeted into mitochondria to compensate for the function of the mutated genes. Although gene therapy is not yet mature, it represents an important direction for the future.
In addition, Recent research has indicated that injecting healthy donor mitochondria into the induced pluripotent stem cells of patients with Barth syndrome can promote mitochondrial autophagy and biogenesis, improve mitochondrial respiratory function, and reduce the APD and frequency of arrhythmia (95). Although the existence of mitochondrial transplantation is relatively short-lived, this exciting discovery provides new directions for future research. In the future, we can differentiate high-purity cardiomyocytes from the induced pluripotent stem cells of patients themselves or immunologically matched donors, extract healthy mitochondria, and achieve efficient delivery through microinjection, nanotube-mediated delivery, or mitochondrial-targeting vectors (such as MITO-Porter). Thus, we can overcome the limitations of traditional drug thinking and develop mitochondrial replacement therapies. Although there are many studies on the role of mitochondria in arrhythmias, those that are actually used in clinical practice are relatively few. How to achieve clinical transformation in the future is also a major issue, and more clinical trials are needed for exploration.
6 Conclusion
In summary, the mitochondria, as a central site that regulates various cellular functions, are an important factor contributing to ischemic arrhythmia, which is a key point in the crosstalk among inflammation, Na+/Ca2+ homeostasis, and mitochondria and is also a potential therapeutic target. The specific mechanism is illustrated in Figure 1. Targeting the mitochondria to improve or restore function has been a popular topic in the treatment of ischemic and metabolic cardiomyopathies (96). However, because different etiologies of the disease produce different responses (97), preclinical data and clinical studies on such therapies remain insufficient. Herein, we reviewed and analyzed the role of mitochondria in the development of arrhythmia. If the mechanism between mitochondria and arrhythmia can be further clarified in the future, it may provide a new direction different from that of traditional arrhythmia treatment.
Figure 1
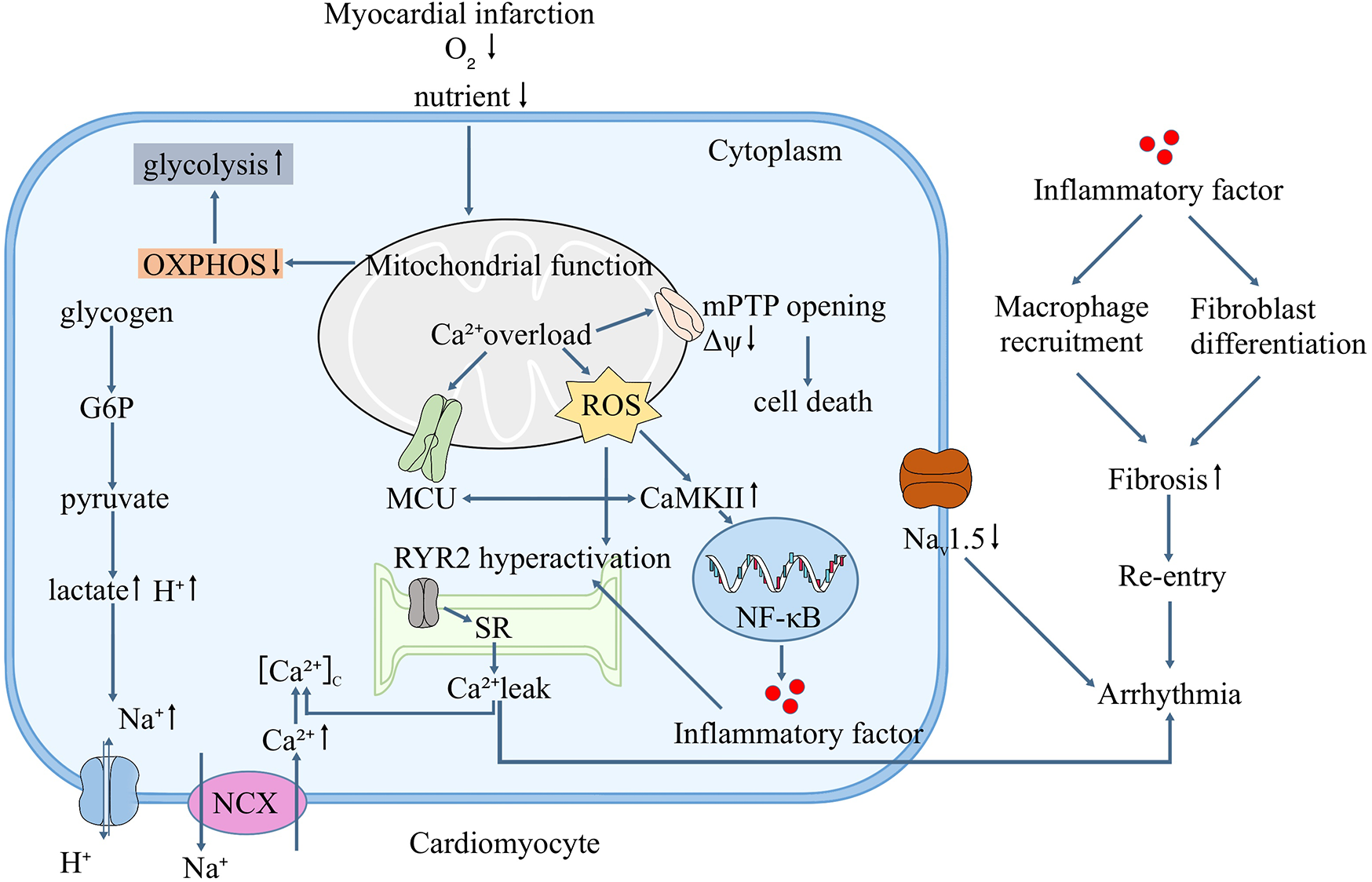
Mitochondrial function and the mechanism of ischemic arrhythmia.
Statements
Author contributions
SS: Writing – original draft, Writing – review & editing. ZZ: Writing – review & editing. YL: Writing – original draft, Writing – review & editing. HZ: Writing – review & editing. HG: Writing – review & editing. GC: Writing – review & editing. PW: Writing – review & editing. FL: Project administration, Supervision, Writing – review & editing. GZ: Funding acquisition, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Research Projects of Higher Education Institutions in Henan Province (No: 25B360015).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
Waks JW Buxton AE . Risk stratification for sudden cardiac death after myocardial infarction. Annu Rev Med. (2018) 69:147–64. 10.1146/annurev-med-041316-090046
2.
Tfelt-Hansen J Winkel BG de Riva M Zeppenfeld K . The ‘10 Commandments’ for the 2022 esc guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J. (2023) 44(3):176–7. 10.1093/eurheartj/ehac699
3.
Lehnart SE Ackerman MJ Benson DW Brugada R Clancy CE Donahue JK et al Inherited arrhythmias: a national heart, lung, and blood institute and office of rare diseases workshop consensus report about the diagnosis, phenotyping, molecular mechanisms, and therapeutic approaches for primary cardiomyopathies of gene mutations affecting Ion channel function. Circulation. (2007) 116(20):2325–45. 10.1161/circulationaha.107.711689
4.
Go AS Mozaffarian D Roger VL Benjamin EJ Berry JD Blaha MJ et al Heart disease and stroke statistics–2014 update: a report from the American Heart Association. Circulation. (2014) 129(3):e28–e292. 10.1161/01.cir.0000441139.02102.80
5.
Liang C Li Q Wang K Du Y Wang W Zhang H . Mechanisms of ventricular arrhythmias elicited by coexistence of multiple electrophysiological remodeling in ischemia: a simulation study. PLoS Comput Biol. (2022) 18(4):e1009388. 10.1371/journal.pcbi.1009388
6.
McKenna WJ Caforio ALP . Myocardial inflammation and sudden death in the inherited cardiomyopathies. Can J Cardiol. (2022) 38(4):427–38. 10.1016/j.cjca.2022.01.004
7.
Wagner S Maier LS Bers DM . Role of sodium and calcium dysregulation in tachyarrhythmias in sudden cardiac death. Circ Res. (2015) 116(12):1956–70. 10.1161/circresaha.116.304678
8.
Stamatis KV Kontonika M Daskalopoulos EP Kolettis TM . Electrophysiologic effects of growth hormone post-myocardial infarction. Int J Mol Sci. (2020) 21(3):918. 10.3390/ijms21030918
9.
Li Y Meng W Hou Y Li D Wang X Wu K et al Dual role of mitophagy in cardiovascular diseases. J Cardiovasc Pharmacol. (2021) 78(1):e30–e9. 10.1097/fjc.0000000000001046
10.
Xie A Kang GJ Kim EJ Liu H Feng F Dudley SC . C-Src is responsible for mitochondria-mediated arrhythmic risk in ischemic cardiomyopathy. Circ Arrhythm Electrophysiol. (2024) 17(10):e013054. 10.1161/circep.124.013054
11.
Hamilton S Terentyeva R Kim TY Bronk P Clements RT OU J et al Pharmacological modulation of mitochondrial Ca2+ content regulates sarcoplasmic reticulum Ca2+ release via oxidation of the ryanodine receptor by mitochondria-derived reactive oxygen Species. Front Physiol. (2018) 9:1831. 10.3389/fphys.2018.01831
12.
Takeuchi A Matsuoka S . Physiological and pathophysiological roles of mitochondrial na+-Ca2+ exchanger, nclx, in hearts. Biomolecules. (2021) 11(12):1876. 10.3390/biom11121876
13.
Andrieux P Chevillard C Cunha-Neto E Nunes JPS . Mitochondria as a cellular hub in infection and inflammation. Int J Mol Sci. (2021) 22(21):11338. 10.3390/ijms222111338
14.
Amoni M Vermoortele D Ekhteraei-Tousi S Doñate Puertas R Gilbert G Youness M et al Heterogeneity of repolarization and cell-cell variability of cardiomyocyte remodeling within the myocardial infarction border zone contribute to arrhythmia susceptibility. Circ Arrhythm Electrophysiol. (2023) 16(5):e011677. 10.1161/circep.122.011677
15.
Orini M Taggart P Hayward M Lambiase PD . Spatiotemporal characterization of the transition from Sinus rhythm to ventricular fibrillation during an acute ischemic event in the intact human heart by whole-heart sock-mapping. HeartRhythm Case Rep. (2017) 3(5):259–63. 10.1016/j.hrcr.2017.01.002
16.
Varró A Tomek J Nagy N Virág L Passini E Rodriguez B et al Cardiac transmembrane Ion channels and action potentials: cellular physiology and arrhythmogenic behavior. Physiol Rev. (2021) 101(3):1083–176. 10.1152/physrev.00024.2019
17.
Kang GJ Xie A Kim E Dudley SC . Mir-448 regulates potassium voltage-gated channel subfamily a member 4 (Kcna4) in ischemia and heart failure. Heart Rhythm. (2023) 20(5):730–6. 10.1016/j.hrthm.2023.01.021
18.
Ferrero JM Gonzalez-Ascaso A Matas JFR . The mechanisms of potassium loss in acute myocardial ischemia: new insights from computational simulations. Front Physiol. (2023) 14:1074160. 10.3389/fphys.2023.1074160
19.
Han B Trew ML Zgierski-Johnston CM . Cardiac conduction velocity, remodeling and arrhythmogenesis. Cells. (2021) 10(11):2923. 10.3390/cells10112923
20.
Weiss JN Qu Z Shivkumar K . Electrophysiology of hypokalemia and hyperkalemia. Circ Arrhythm Electrophysiol. (2017) 10(3):e004667. 10.1161/circep.116.004667
21.
Hernández-Romero I Guillem MS Figuera C Atienza F Fernández-Avilés F Climent AM . Optical imaging of voltage and calcium in isolated hearts: linking spatiotemporal heterogeneities and ventricular fibrillation initiation. PLoS One. (2019) 14(5):e0215951. 10.1371/journal.pone.0215951
22.
Roney CH Whitaker J Sim I O'Neill L Mukherjee RK Razeghi O et al A technique for measuring anisotropy in atrial conduction to estimate conduction velocity and atrial fibre direction. Comput Biol Med. (2019) 104:278–90. 10.1016/j.compbiomed.2018.10.019
23.
Quinn TA Camelliti P Rog-Zielinska EA Siedlecka U Poggioli T 'Toole O et al Electrotonic coupling of excitable and nonexcitable cells in the heart revealed by optogenetics. Proc Natl Acad Sci U S A. (2016) 113(51):14852–7. 10.1073/pnas.1611184114
24.
Rohr S . Arrhythmogenic implications of fibroblast-myocyte interactions. Circ Arrhythm Electrophysiol. (2012) 5(2):442–52. 10.1161/circep.110.957647
25.
George SA Bonakdar M Zeitz M Davalos RV Smyth JW Poelzing S . Extracellular sodium dependence of the conduction velocity-calcium relationship: evidence of ephaptic self-attenuation. Am J Physiol Heart Circ Physiol. (2016) 310(9):H1129–39. 10.1152/ajpheart.00857.2015
26.
Dhillon PS Gray R Kojodjojo P Jabr R Chowdhury R Fry CH et al Relationship between gap-junctional conductance and conduction velocity in mammalian myocardium. Circ Arrhythm Electrophysiol. (2013) 6(6):1208–14. 10.1161/circep.113.000848
27.
Trew ML Engelman ZJ Caldwell BJ Lever NA LeGrice IJ Smaill BH . Cardiac intramural electrical mapping reveals focal delays but No conduction velocity slowing in the peri-infarct region. Am J Physiol Heart Circ Physiol. (2019) 317(4):H743–h53. 10.1152/ajpheart.00154.2019
28.
Qu Z Karagueuzian HS Garfinkel A Weiss JN . Effects of na(+) channel and cell coupling abnormalities on vulnerability to reentry: a simulation study. Am J Physiol Heart Circ Physiol. (2004) 286(4):H1310–21. 10.1152/ajpheart.00561.2003
29.
Iqbal SM Lemmens-Gruber R . Phosphorylation of cardiac voltage-gated sodium channel: potential players with multiple dimensions. Acta Physiol (Oxf). (2019) 225(3):e13210. 10.1111/apha.13210
30.
Eichel CA Beuriot A Chevalier MY Rougier JS Louault F Dilanian G et al Lateral membrane-specific maguk cask down-regulates Nav1.5 channel in cardiac myocytes. Circ Res. (2016) 119(4):544–56. 10.1161/circresaha.116.309254
31.
Korkmaz S Zitron E Bangert A Seyler C Li S Hegedüs P et al Provocation of an autoimmune response to cardiac voltage-gated sodium channel Nav1.5 induces cardiac conduction defects in rats. J Am Coll Cardiol. (2013) 62(4):340–9. 10.1016/j.jacc.2013.04.041
32.
Yu P Hu L Xie J Chen S Huang L Xu Z et al O-Glcnacylation of cardiac Nav1.5 contributes to the development of arrhythmias in diabetic hearts. Int J Cardiol. (2018) 260:74–81. 10.1016/j.ijcard.2018.02.099
33.
Hu CC Wei X Liu JM Han LL Xia CK Wu J et al Cardiac-targeted piasy gene silencing mediates desumoylation of caveolin-3 and prevents ischemia/reperfusion-induced Nav1.5 downregulation and ventricular arrhythmias. Mil Med Res. (2022) 9(1):58. 10.1186/s40779-022-00415-x
34.
Moreau A Gosselin-Badaroudine P Mercier A Burger B Keller DI Chahine M . A leaky voltage sensor domain of cardiac sodium channels causes arrhythmias associated with dilated cardiomyopathy. Sci Rep. (2018) 8(1):13804. 10.1038/s41598-018-31772-0
35.
Li W Yin L Shen C Hu K Ge J Sun A . Scn5a variants: association with cardiac disorders. Front Physiol. (2018) 9:1372. 10.3389/fphys.2018.01372
36.
Abdelsayed M Ruprai M Ruben PC . The efficacy of ranolazine on E1784k is altered by temperature and calcium. Sci Rep. (2018) 8(1):3643. 10.1038/s41598-018-22033-1
37.
Kohajda Z Farkas-Morvay N Jost N Nagy N Geramipour A Horváth A et al The effect of a novel highly selective inhibitor of the sodium/calcium exchanger (ncx) on cardiac arrhythmias in in vitro and in vivo experiments. PLoS One. (2016) 11(11):e0166041. 10.1371/journal.pone.0166041
38.
Dobrev D Wehrens XH . Role of Ryr2 phosphorylation in heart failure and arrhythmias: controversies around ryanodine receptor phosphorylation in cardiac disease. Circ Res. (2014) 114(8):1311–9. 10.1161/circresaha.114.300568
39.
Zhang Y Jiao L Sun L Li Y Gao Y Xu C et al Lncrna Zfas1 as a Serca2a inhibitor to cause intracellular Ca2+ overload and Contractile dysfunction in a mouse model of myocardial infarction. Circ Res. (2018) 122(10):1354–68. 10.1161/circresaha.117.312117
40.
Xie A Liu H Kang GJ Feng F Dudley SC . Reduced sarcoplasmic reticulum Ca2+ pump activity is antiarrhythmic in ischemic cardiomyopathy. Heart Rhythm. (2022) 19(12):2107–14. 10.1016/j.hrthm.2022.08.022
41.
Tow BD Deb A Neupane S Patel SM Reed M Loper AB et al Sr-Mitochondria crosstalk shapes ca signalling to impact pathophenotype in disease models marked by dysregulated intracellular ca release. Cardiovasc Res. (2022) 118(13):2819–32. 10.1093/cvr/cvab324
42.
Sun Z Wang L Han L Wang Y Zhou Y Li Q et al Functional calsequestrin-1 is expressed in the heart and its deficiency is causally related to malignant hyperthermia-like arrhythmia. Circulation. (2021) 144(10):788–804. 10.1161/circulationaha.121.053255
43.
Reyes Gaido OE Nkashama LJ Schole KL Wang Q Umapathi P Mesubi OO et al Camkii as a therapeutic target in cardiovascular disease. Annu Rev Pharmacol Toxicol. (2023) 63:249–72. 10.1146/annurev-pharmtox-051421-111814
44.
Ashpole NM Herren AW Ginsburg KS Brogan JD Johnson DE Cummins TR et al Ca2+/calmodulin-dependent protein kinase ii (camkii) regulates cardiac sodium channel Nav1.5 gating by multiple phosphorylation sites. J Biol Chem. (2012) 287(24):19856–69. 10.1074/jbc.M111.322537
45.
Ai X Curran JW Shannon TR Bers DM Pogwizd SM . Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. (2005) 97(12):1314–22. 10.1161/01.Res.0000194329.41863.89
46.
Wagner S Dybkova N Rasenack EC Jacobshagen C Fabritz L Kirchhof P et al Ca2+/calmodulin-dependent protein kinase ii regulates cardiac na+ channels. J Clin Invest. (2006) 116(12):3127–38. 10.1172/jci26620
47.
Meng Y Ding P Wang H Yang X Wang Z Nie D et al Ca2+/calmodulin-dependent protein kinase ii inhibition reduces myocardial fatty acid uptake and oxidation after myocardial infarction. Biochim Biophys Acta Mol Cell Biol Lipids. (2022) 1867(6):159120. 10.1016/j.bbalip.2022.159120
48.
Nassal D Gratz D Hund TJ . Challenges and opportunities for therapeutic targeting of calmodulin kinase ii in heart. Front Pharmacol. (2020) 11:35. 10.3389/fphar.2020.00035
49.
Sun N Finkel T . Cardiac mitochondria: a surprise about size. J Mol Cell Cardiol. (2015) 82:213–5. 10.1016/j.yjmcc.2015.01.009
50.
Yao L Fan P Jiang Z Viatchenko-Karpinski S Wu Y Kornyeyev D et al Nav1.5-dependent persistent na+ influx activates camkii in rat ventricular myocytes and N1325s mice. Am J Physiol Cell Physiol. (2011) 301(3):C577–86. 10.1152/ajpcell.00125.2011
51.
Yang X Zhao S Wang S Cao X Xu Y Yan M et al Systemic inflammation indicators and risk of incident arrhythmias in 478,524 individuals: evidence from the UK biobank cohort. BMC Med. (2023) 21(1):76. 10.1186/s12916-023-02770-5
52.
Vicente R Escalada A Villalonga N Texidó L Roura-Ferrer M Martín-Satué M et al Association of Kv1.5 and Kv1.3 contributes to the Major voltage-dependent K+ channel in macrophages. J Biol Chem. (2006) 281(49):37675–85. 10.1074/jbc.M605617200
53.
Jongsma HJ Wilders R . Gap junctions in cardiovascular disease. Circ Res. (2000) 86(12):1193–7. 10.1161/01.res.86.12.1193
54.
Hulsmans M Clauss S Xiao L Aguirre AD King KR Hanley A et al Macrophages facilitate electrical conduction in the heart. Cell. (2017) 169(3):510–22.e20. 10.1016/j.cell.2017.03.050
55.
Arslan F de Kleijn DP Pasterkamp G . Innate immune signaling in cardiac ischemia. Nat Rev Cardiol. (2011) 8(5):292–300. 10.1038/nrcardio.2011.38
56.
Frangogiannis NG . The extracellular matrix in myocardial injury, repair, and remodeling. J Clin Invest. (2017) 127(5):1600–12. 10.1172/jci87491
57.
Frangogiannis NG . Regulation of the inflammatory response in cardiac repair. Circ Res. (2012) 110(1):159–73. 10.1161/circresaha.111.243162
58.
Wan E Yeap XY Dehn S Terry R Novak M Zhang S et al Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ Res. (2013) 113(8):1004–12. 10.1161/circresaha.113.301198
59.
Chatelier A Mercier A Tremblier B Thériault O Moubarak M Benamer N et al A distinct de novo expression of Nav1.5 sodium channels in human atrial fibroblasts differentiated into myofibroblasts. J Physiol. (2012) 590(17):4307–19. 10.1113/jphysiol.2012.233593
60.
Chilton L Giles WR Smith GL . Evidence of intercellular coupling between co-cultured adult rabbit ventricular myocytes and myofibroblasts. J Physiol. (2007) 583(Pt 1):225–36. 10.1113/jphysiol.2007.135038
61.
Wang Y Li Q Tao B Angelini M Ramadoss S Sun B et al Fibroblasts in heart scar tissue directly regulate cardiac excitability and arrhythmogenesis. Science. (2023) 381(6665):1480–7. 10.1126/science.adh9925
62.
Frangogiannis NG . Pathophysiology of myocardial infarction. Compr Physiol. (2015) 5(4):1841–75. 10.1002/cphy.c150006
63.
Moreira LM Takawale A Hulsurkar M Menassa DA Antanaviciute A Lahiri SK et al Paracrine signalling by cardiac calcitonin controls atrial fibrogenesis and arrhythmia. Nature. (2020) 587(7834):460–5. 10.1038/s41586-020-2890-8
64.
Zhou Y Suo W Zhang X Lv J Liu Z Liu R . Roles and mechanisms of quercetin on cardiac arrhythmia: a review. Biomed Pharmacother. (2022) 153:113447. 10.1016/j.biopha.2022.113447
65.
Amoni M Dries E Ingelaere S Vermoortele D Roderick HL Claus P et al Ventricular arrhythmias in ischemic cardiomyopathy-new avenues for mechanism-guided treatment. Cells. (2021) 10(10):2629. 10.3390/cells10102629
66.
Schmidt A Azevedo CF Cheng A Gupta SN Bluemke DA Foo TK et al Infarct tissue heterogeneity by magnetic resonance imaging identifies enhanced cardiac arrhythmia susceptibility in patients with left ventricular dysfunction. Circulation. (2007) 115(15):2006–14. 10.1161/circulationaha.106.653568
67.
Muser D Nucifora G Muser D Nucifora G Pieroni M Castro SA et al Prognostic value of nonischemic ringlike left ventricular scar in patients with apparently idiopathic nonsustained ventricular arrhythmias. Circulation. (2021) 143(14):1359–73. 10.1161/circulationaha.120.047640
68.
Lazzerini PE Acampa M Laghi-Pasini F Bertolozzi I Finizola F Vanni F et al Cardiac arrest risk during acute infections: systemic inflammation directly prolongs qtc interval via cytokine-mediated effects on potassium channel expression. Circ Arrhythm Electrophysiol. (2020) 13(8):e008627. 10.1161/circep.120.008627
69.
Saxena A Chen W Su Y Rai V Uche OU Li N et al Il-1 induces proinflammatory leukocyte infiltration and regulates fibroblast phenotype in the infarcted myocardium. J Immunol. (2013) 191(9):4838–48. 10.4049/jimmunol.1300725
70.
Frangogiannis NG . Cardiac fibrosis. Cardiovasc Res. (2021) 117(6):1450–88. 10.1093/cvr/cvaa324
71.
Rusciano MR Sommariva E Douin-Echinard V Ciccarelli M Poggio P Maione AS . Camkii activity in the inflammatory response of cardiac diseases. Int J Mol Sci. (2019) 20(18):4374. 10.3390/ijms20184374
72.
Beckendorf J van den Hoogenhof MMG Backs J . Physiological and unappreciated roles of camkii in the heart. Basic Res Cardiol. (2018) 113(4):29. 10.1007/s00395-018-0688-8
73.
Wei Z Fei Y Wang Q Hou J Cai X Yang Y et al Loss of Camk2n1 aggravates cardiac remodeling and malignant ventricular arrhythmia after myocardial infarction in mice via Nlrp3 inflammasome activation. Free Radic Biol Med. (2021) 167:243–57. 10.1016/j.freeradbiomed.2021.03.014
74.
Rao M Wang X Guo G Wang L Chen S Yin P et al Resolving the intertwining of inflammation and fibrosis in human heart failure at single-cell level. Basic Res Cardiol. (2021) 116(1):55. 10.1007/s00395-021-00897-1
75.
Ren G Michael LH Entman ML Frangogiannis NG . Morphological characteristics of the microvasculature in healing myocardial infarcts. J Histochem Cytochem. (2002) 50(1):71–9. 10.1177/002215540205000108
76.
Verheule S Schotten U . Electrophysiological consequences of cardiac fibrosis. Cells. (2021) 10(11). 10.3390/cells10113220
77.
De la Fuente S Sheu SS . Sr-mitochondria communication in adult cardiomyocytes: a close relationship where the Ca2+ has a lot to say. Arch Biochem Biophys. (2019) 663:259–68. 10.1016/j.abb.2019.01.026
78.
Hamilton S Terentyeva R Clements RT Belevych AE Terentyev D . Sarcoplasmic reticulum-mitochondria communication; implications for cardiac arrhythmia. J Mol Cell Cardiol. (2021) 156:105–13. 10.1016/j.yjmcc.2021.04.002
79.
Deng J Jiang Y Chen ZB Rhee JW Deng Y Wang ZV . Mitochondrial dysfunction in cardiac arrhythmias. Cells. (2023) 12(5). 10.3390/cells12050679
80.
Zhu H Toan S Mui D Zhou H . Mitochondrial quality surveillance as a therapeutic target in myocardial infarction. Acta Physiol (Oxf). (2021) 231(3):e13590. 10.1111/apha.13590
81.
Oknińska M Mączewski M Mackiewicz U . Ventricular arrhythmias in acute myocardial ischaemia-focus on the ageing and sex. Ageing Res Rev. (2022) 81:101722. 10.1016/j.arr.2022.101722
82.
Wongtanasarasin W Siri-Angkul N Wittayachamnankul B Chattipakorn SC Chattipakorn N . Mitochondrial dysfunction in fatal ventricular arrhythmias. Acta Physiol (Oxf). (2021) 231(4):e13624. 10.1111/apha.13624
83.
Alevriadou BR Patel A Noble M Ghosh S Gohil VM Stathopulos PB et al Molecular nature and physiological role of the mitochondrial calcium uniporter channel. Am J Physiol Cell Physiol. (2021) 320(4):C465–c82. 10.1152/ajpcell.00502.2020
84.
Tse G Wong ST Tse V Lee YT Lin HY Yeo JM . Cardiac dynamics: alternans and arrhythmogenesis. J Arrhythm. (2016) 32(5):411–7. 10.1016/j.joa.2016.02.009
85.
Di Lisa F Bernardi P . Mitochondrial function and myocardial aging. A critical analysis of the role of permeability transition. Cardiovasc Res. (2005) 66(2):222–32. 10.1016/j.cardiores.2005.02.009
86.
Takla M Huang CL Jeevaratnam K . The cardiac camkii-Nav1.5 relationship: from physiology to pathology. J Mol Cell Cardiol. (2020) 139:190–200. 10.1016/j.yjmcc.2019.12.014
87.
Zorov DB Juhaszova M Sollott SJ . Mitochondrial reactive oxygen Species (ros) and ros-induced ros release. Physiol Rev. (2014) 94(3):909–50. 10.1152/physrev.00026.2013
88.
Ponnalagu D Singh H . Insights into the role of mitochondrial Ion channels in inflammatory response. Front Physiol. (2020) 11:258. 10.3389/fphys.2020.00258
89.
Manolis AS Manolis AA Manolis TA Apostolaki NE Apostolopoulos EJ Melita H et al Mitochondrial dysfunction in cardiovascular disease: current status of translational research/clinical and therapeutic implications. Med Res Rev. (2021) 41(1):275–313. 10.1002/med.21732
90.
Pérez-Hernández M Leo-Macias A Keegan S Jouni M Kim JC Agullo-Pascual E et al Structural and functional characterization of a Nav1.5-mitochondrial couplon. Circ Res. (2021) 128(3):419–32. 10.1161/circresaha.120.318239
91.
Tse G Yan BP Chan YW Tian XY Huang Y . Reactive oxygen species, endoplasmic reticulum stress and mitochondrial dysfunction: the link with cardiac arrhythmogenesis. Front Physiol. (2016) 7:313. 10.3389/fphys.2016.00313
92.
Miragoli M Sanchez-Alonso JL Bhargava A Wright PT Sikkel M Schobesberger S et al Microtubule-dependent mitochondria alignment regulates calcium release in response to nanomechanical stimulus in heart myocytes. Cell Rep. (2016) 14(1):140–51. 10.1016/j.celrep.2015.12.014
93.
Reyat JS Sommerfeld LC O'Reilly M Roth Cardoso V Thiemann E Khan AO et al Pitx2 deficiency leads to atrial mitochondrial dysfunction. Cardiovasc Res. (2024) 120(15):1907–23. 10.1093/cvr/cvae169
94.
Wang J Zhou H . Mitochondrial quality control mechanisms as molecular targets in cardiac ischemia-reperfusion injury. Acta Pharm Sin B. (2020) 10(10):1866–79. 10.1016/j.apsb.2020.03.004
95.
Kim YS Yoo S Jung YJ Yoon JW Kwon YS Lee N et al Allogenic mitochondria transfer improves cardiac function in ips-cell-differentiated cardiomyocytes of a patient with barth syndrome. Exp Mol Med. (2025) 57(6):1260–71. 10.1038/s12276-025-01472-7
96.
Zhu L Chen Z Han K Zhao Y Li Y Li D et al Correlation between mitochondrial dysfunction, cardiovascular diseases, and traditional Chinese medicine. Evid Based Complement Alternat Med. (2020) 2020:2902136. 10.1155/2020/2902136
97.
Finck BN Kelly DP . Peroxisome proliferator-activated receptor gamma coactivator-1 (pgc-1) regulatory cascade in cardiac physiology and disease. Circulation. (2007) 115(19):2540–8. 10.1161/circulationaha.107.670588
Summary
Keywords
myocardial infarction, ventricular arrhythmia, mitochondrial, inflammation, Na+/Ca2+ homeostasis, fibrosis
Citation
Sun S, Zhang Z, Li Y, Zhang H, Guo H, Chen G, Wei P, Lin F and Zhao G (2025) Mitochondrial dysfunction as a central hub linking Na+/Ca2+ homeostasis and inflammation in ischemic arrhythmias: therapeutic implications. Front. Cardiovasc. Med. 12:1506501. doi: 10.3389/fcvm.2025.1506501
Received
05 October 2024
Accepted
29 July 2025
Published
12 August 2025
Volume
12 - 2025
Edited by
Tommaso Gori, Johannes Gutenberg University Mainz, Germany
Reviewed by
Gentaro Ikeda, Stanford University, United States
Richard M. Monaghan, The University of Manchester, United Kingdom
Updates
Copyright
© 2025 Sun, Zhang, Li, Zhang, Guo, Chen, Wei, Lin and Zhao.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
* Correspondence: Fei Lin linfeixixi@aliyun.com Guoan Zhao guoanzhao@xxmu.edu.cn
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.