 Francesco Tona1*†
Francesco Tona1*† Giovanni Civieri1,†
Giovanni Civieri1,† Marta Vadori2
Marta Vadori2 Giulia Masiero1
Giulia Masiero1 Laura Iop3
Laura Iop3 Martina Perazzolo Marra1
Martina Perazzolo Marra1 Annagrazia Cecere1Marika Martini1Donatella Tansella1
Annagrazia Cecere1Marika Martini1Donatella Tansella1 Giacomo Bernava3Benedetta Schiavon1Loira Leoni1
Giacomo Bernava3Benedetta Schiavon1Loira Leoni1 Emanuele Cozzi2,‡Sabino Iliceto1,4,‡
Emanuele Cozzi2,‡Sabino Iliceto1,4,‡
- 1Cardiology Division, Department of Cardiac, Thoracic, Vascular Sciences and Public Health, University of Padua, Padua, Italy
- 2Transplant Immunology Unit, Department of Cardiac, Thoracic, Vascular Sciences and Public Health, University of Padua, Padua, Italy
- 3Translational Biomedicine Group, Department of Cardiac, Thoracic, Vascular Sciences and Public Health, University of Padua, Padua, Italy
- 4School of Health, LUM-Libera Università Mediterranea “Giuseppe Degennaro”, Bari, Italy
Background: Functional autoantibodies against angiotensin II type 1 (AT1R-AAs) and endothelin-1 type A (ETAR-AAs) receptors are associated with microvascular obstruction and myocardial remodeling after ST-elevation myocardial infarction (STEMI). However, their role in the long-term prognosis after STEMI has not been investigated.
Methods: This is a prospective observational study enrolling STEMI patients undergoing early primary PCI. The incidence of major adverse cardiovascular events (MACE) was investigated during the follow-up. Autoantibody seropositivity was defined as a level >10 U/ml.
Results: 200 STEMI patients (89% male, median age 61 years) were enrolled. 110 (55%) were seronegative for both autoantibodies, 44 (22%) were seropositive for one autoantibody, and 46 (23%) were seropositive for both autoantibodies. Over a median follow-up of 1.2 years, the incidence of MACE was higher in patients with double (31%) and single (25%) seropositivity than in seronegative patients (13%, p = 0.02 among groups). Double seropositivity was independently associated with higher risk of MACE (HR 2.386, 95% CI 1.471–3.864, p < 0.001).
Conclusion: AT1R-AAs and ETAR-AAs are associated with an increased risk of MACE after STEMI. Assessment of autoantibody levels paves the way for future therapies targeting specific molecular pathways associated with poor prognosis after an acute coronary event.
1 Introduction
ST–elevation myocardial infarction (STEMI) is a prevalent cardiac emergency that results in significant morbidity and mortality worldwide. The occurrence of this condition is similar in both Europe and the United States (1, 2). Although interventional treatment with primary percutaneous coronary intervention (PPCI) can markedly reduce short-term morbidity and mortality rates, the prognosis of STEMI remains relatively poor. In the last decades, no changes in the severity of myocardial damage and long-term outcomes have been observed (3).
In addition to their effects on blood pressure (4, 5), vasoconstrictor peptides, such as angiotensin II (AngII) and endothelin−1 (ET1), appear to be important contributors to the progression of heart failure following STEMI via non-hemodynamic pathways that lead to adverse myocardial remodeling. There is increasing evidence that these vasoconstrictor peptides contribute to the development of myocardial inflammation and fibrosis within both infarct and non-infarct regions, with the latter contributing to reduced myocardial elasticity and contractile dysfunction (6, 7).
AngII and ET1 exert detrimental effects by binding to the angiotensin II receptor type 1 (AT1R) and endothelin−1 receptor type A (ETAR), respectively. AT1R and ETAR are G protein-coupled receptors (GPCRs) expressed in a wide variety of cell types. Recently, functional autoantibodies against AT1R (AT1R-AAs) and ETAR (ETAR-AAs) have been identified (8). AT1R-AAs and ETAR-AAs appear to bind the same AT1R and ETAR receptors, permanently causing their hyperactivation (8). This leads to the development of an abiding pro-inflammatory environment with the formation of pro-inflammatory cytokines that participate in microvascular inflammation (9, 10).
While these autoantibodies have been documented in various clinical contexts, research on their impact on cardiovascular conditions is limited (11). However, AT1R-AAs and ETAR-AAs may play a significant role in numerous cardiac disorders, with their vasoconstrictive and inflammatory properties potentially contributing to poor outcomes following STEMI revascularization. Our recent findings indicate that ETAR-AA seropositivity is linked to no-reflow in patients with STEMI, and both ETAR-AAs and AT1R-AAs elevate the risk of adverse left ventricular remodeling after myocardial infarction (12, 13).
Since both no-reflow and adverse left ventricular remodeling negatively influence clinical outcomes after myocardial infarction (14, 15), we undertook the present study to analyze the prognostic role of AT1R-AAs and ETAR-AAs in patients with STEMI treated with PPCI.
2 Methods
This prospective observational study was conducted between January 2022 and June 2023. We enrolled 226 adult (>18 years) STEMI patients who had undergone reperfusion therapy by PPCI within 12 h of symptom onset at the Padua University Medical Center, expanding our previously described cohort and using the same inclusion and exclusion criteria (13). STEMI was defined as chest pain with (1) typical characteristics; (2) duration of more than 30 min; (3) an ST-segment elevation on initial ECG of >0.1 mV in two contiguous leads; (4) increased levels of cardiac troponin (cTn), with at least one measurement exceeding the 99th percentile upper reference limit. Reperfusion time was calculated as the duration between the initial onset of symptoms and crossing of the wire. Seven patients with a previous myocardial infarction, six with significant valvular heart disease, three with chronic atrial fibrillation, and three with inadequate image quality on transthoracic echocardiography (TTE) were excluded. The remaining 207 patients underwent serum testing for AT1R-AAs and ETAR-AAs. Patients were then followed up for a maximum of two years with clinically indicated in-person visits or telephone interviews. The physicians who cared for the patients were blinded to the autoantibody levels. Seven patients were lost to follow-up, and the final cohort comprised 200 patients.
2.1 PPCI procedure
All patients received guideline-directed medical therapy (250 mg aspirin and heparin (60 U/kg body weight) intravenously before PPCI; prasugrel (60 mg) or ticagrelor (180 mg) were chosen according to patient's age and weight. Aspirin was given indefinitely at a dose of 100 mg/day, while prasugrel (10 mg/day) or ticagrelor (90 mg twice daily) were continued according to the current guidelines (16). The use of glycoprotein IIb/IIIa inhibitors, beta-blockers, angiotensin-converting enzyme inhibitors, and statins was determined by the treating physician according to current guidelines (16). The TIMI study group classification (17) was used to grade the coronary flow in the infarct-related artery before and after revascularization of the culprit vessel. The myocardial blush grade (MBG) (0–3) before and after PPCI was also evaluated (18). Angiographic assessments were performed in the angiographic core laboratory by two blinded experienced cardiologists (F.T., G.M).
2.2 Echocardiographic analysis
LV end-diastolic volume index (LVEDVi) and end-systolic volume index (LVESVi) were measured using Simpson's biplane method. LV ejection fraction (LVEF) and wall motion score index (WMSI) were calculated as recommended by current guidelines (19). The mitral inflow peak early velocity (E)/mitral annular peak early velocity (e′) (E/e′ ratio) was also evaluated. Right ventricular function was measured as previously described (19). Additional details are provided in the Supplementary Methods.
2.3 Laboratory assays
Blood samples were drawn from a peripheral vein from all patients within 12 h of reperfusion. AT1R-AAs and ETAR-AAs were determined by enzyme-linked immunosorbent technique (ELISA) using a 96-well microtiter plate coated with AT1R and ETAR in their native configurations, according to the manufacturer's instructions (CellTrend, Luckenwalde, Germany). These kits are the most commonly used in studies on this topic (20–33). Seropositivity was defined using > 10.0 U/ml as the cutoff value for AT1R-AAs and ETAR-AAs, based on previous studies and following manufacturer recommendations (20, 22, 27, 29, 30, 33). Further details on the choice of the cutoff are reported in the Supplementary Methods. Patients were defined based on their serum autoantibody status as follows: (1) seronegative, when neither AT1R-AAs nor ETAR-AAs were above the cutoff value; (2) single seropositive, when either AT1R-AAs or ETAR-AAs (but not both) were positive; and (3) double seropositive, when both AT1R-AAs and ETAR-AAs were positive. Additional details in the Supplementary Methods.
2.4 Outcome definition
The study endpoint was the occurrence of major adverse cardiovascular events (MACE) during follow-up. MACE included cardiovascular mortality, nonfatal myocardial re-infarction, and hospitalization for heart failure. The time to the first event was considered for patients with more than one event during follow-up.
2.5 Follow-up and event adjudication
Patients were followed up for the occurrence of the study endpoint for up to 2 years (730 days) after enrollment. Structured follow-up was performed via telephone after inclusion. The endpoint was verified using medical records from the center, primary care clinicians, and other medical centers. A clinical endpoint committee consisting of two experienced cardiologists blinded to the autoantibody data reviewed and adjudicated all (possible) events.
2.6 Statistical analysis
The statistical packages R (version 4.3.1, R Foundation for Statistical Computing, Vienna, Austria) and IBM SPSS Statistics version 28 (Chicago, SPSS, Inc., Chicago, IL) were employed for statistical analysis. Continuous variables are presented as mean [standard deviation (SD)] if normally distributed or median [interquartile range (IQR)] if not normally distributed. Categorical variables are presented as numbers (percentages). Differences between two groups were analyzed using the Student t-test (for continuous variables with Gaussian distribution), the Mann–Whitney test (for continuous variables with non-parametric distributions), or Pearson χ2 test (for categorical variables). Differences between >2 groups were analyzed using one-way ANOVA for continuous variables with Gaussian distribution, the Kruskal–Wallis test for non-normally distributed continuous variables, and the Pearson χ2 test for categorical variables. Multiple comparisons of continuous variables were performed using the Bonferroni correction. After adjusting for related variables, multivariable logistic regression was used to analyze the association between continuous autoantibody serum levels and the risk of MACE. The resulting odds ratios are relative to a 1-unit increase in AT1R-AAs and ETAR-AAs levels. Survival curves were obtained using the R-package `survival’ (version 3.5.5) and compared using the log-rank test. The relationship between the continuous autoantibody levels and the risk of MACE was explored using Cox regression models by entering AT1R-AAs and ETAR-AAs as a restricted cubic spline, with three knots located at the 10th, 50th, and 90th percentiles (for AT1R-AAs: 5.09, 7.67, and 15.3 U/ml; for ETAR-AAs: 4.14, 8.11, and 21.42 U/ml). Restricted cubic spline regression was performed using the R-package ‘rms’ (version 6.7.1). A univariate Cox proportional hazard regression model was used to estimate the hazard ratios (HR). A multivariable Cox proportional hazards regression analysis, using backward stepwise elimination (if p > 0.10), was performed to assess the risk factors independently associated with MACE. Possible confounders with a significant p-value in the univariate analysis were included in the multivariable regression analysis (p < 0.05) after checking for collinearity. Validity of assumptions for regression models were confirmed as appropriate. Model discrimination was assessed using c-statistics. All tests were two-sided, and the α level was set at 0.05. The authors had full access to and took full responsibility for the integrity of the data. All authors have read and agreed to the manuscript.
2.7 Ethical approval
This study was approved by the Institutional Ethics Committee (Comitato Etico per la Sperimentazione Clinica della Provincia di Padova; code number CESC 5478/AO) and was conducted in accordance with the Declaration of Helsinki and Italian laws. Informed consent was obtained from all patients included in this study.
3 Results
3.1 Clinical characteristics
The clinical characteristics, both of the overall population and divided according to seropositivity status, are reported in Table 1. A total of 110 (55%) patients were seronegative for both autoantibodies, 44 (22%) patients were seropositive for one of the two autoantibodies (AT1R-AAs or ETAR-AAs), and 46 (23%) patients were seropositive for both autoantibodies (AT1R-AAs and ETAR-AAs). Diabetes mellitus and known coronary artery disease had a different prevalence between the groups (p = 0.029 and p = 0.024, respectively) and were more frequent in patients with double seropositivity. The peak levels of troponin and BNP at admission were higher in seropositive patients (difference between groups, p = 0.046 and p = 0.001, respectively).
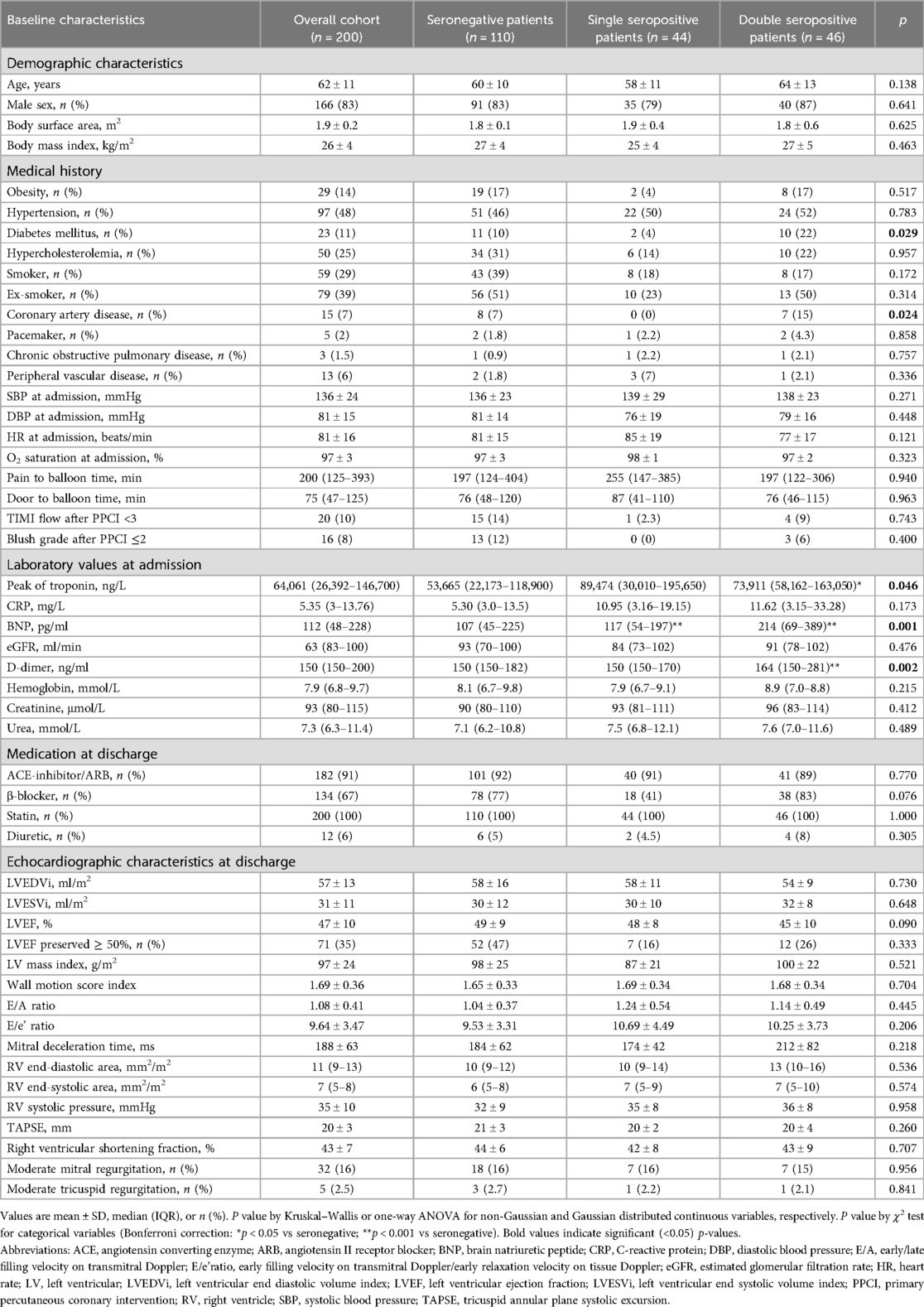
Table 1. Clinical and laboratory data for patients with STEMI (n = 200).
3.2 Prognostic impact of AT1R-AAs and ETAR-AAs
During a median follow-up of 429 days (IQR 235–730 days), MACE occurred in 39 (19.5%) patients (22 cardiovascular-related deaths, 2 nonfatal myocardial re-infarctions, and 15 hospitalizations for heart failure).
The incidence of MACE was 13% in seronegative patients, 25% in single-seropositive patients, and 31% in double-seropositive patients (p = 0.021) (Figure 1). The incidence of the outcome was higher in patients with double seropositivity than in seronegative patients (31% vs. 13%, OR 3.0, 95% CI 1.29–6.96, p = 0.009) and was also higher also in patients with single seropositivity than in seronegative ones (25% vs. 13%, OR 2.28, 95% CI 0.94–5.52, p = 0.06). The characteristics of the patients according to the occurrence of the endpoint are shown in Supplementary Table S1.
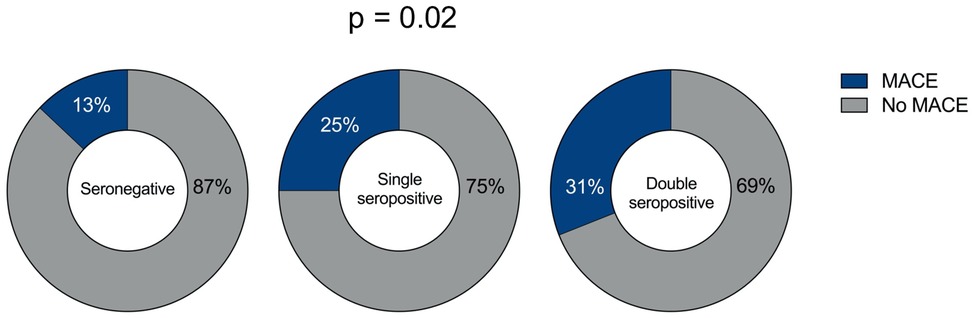
Figure 1. Frequency of MACE in STEMI patients based on serum autoantibody status. MACE in seronegative STEMI patients (left pie chart); MACE in single seropositive (AT1R-AAs or ETAR-AAs seropositive) STEMI patients (middle pie chart); and MACE in double seropositive (AT1R-AAs and ETAR-AAs seropositive) STEMI patients (right pie chart). The P-value for the difference between the groups is reported.
To further clarify the association between serum levels of AT1R-AAs/ETAR-AAs and the risk of MACE, multivariable logistic regression analysis was performed. After adjusting for confounding factors, the risk of MACE was related to both AT1R-AAs (OR 1.07, 95% CI 1.02–1.16, p = 0.03) and ETAR-AAs (OR 1.08, 95% CI 1.01–1.15, p = 0.01) serum levels (Supplementary Table S2).
Survival analysis using Kaplan–Meier curves showed a significantly higher incidence of MACE in patients with single or double autoantibody seropositivity than in seronegative patients (Figure 2).
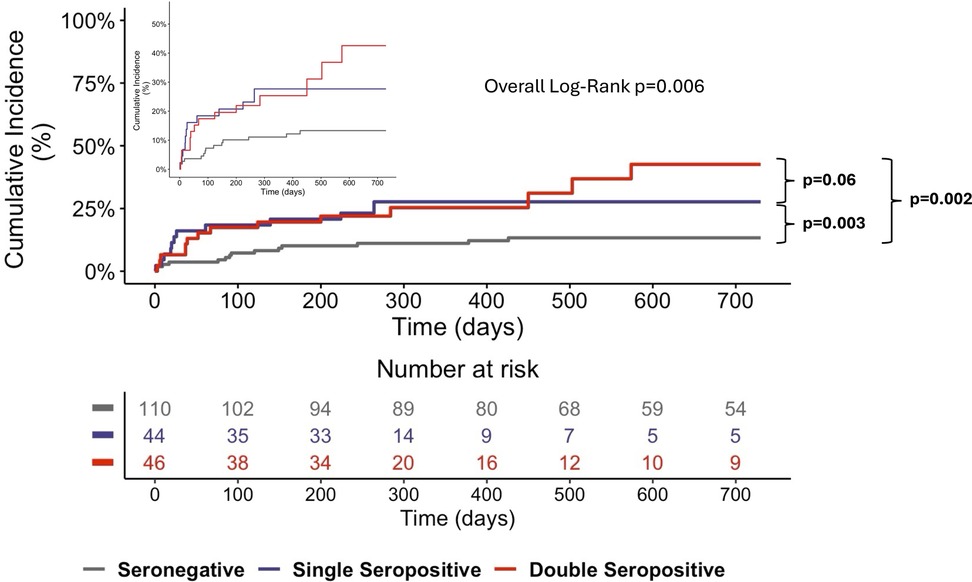
Figure 2. Kaplan–meier curves for MACE at the 4-year follow-up. The Kaplan–Meier curves show significantly higher MACE rates for single- and double-seropositive patients than for seronegative patients. The inset graph shows the data on an expanded y axis.
Univariable Cox regression analysis showed that autoantibody seropositivity (p = 0.009) was associated with an increased risk of MACE (Table 2). At multivariable Cox regression analysis, double seropositivity was independently associated with an increased risk of MACE (HR 2.386, 95% CI 1.471–3.864, p < 0.001) (Table 2). The c-statistic for the Cox multivariable model was 0.907 for MACE (SE 0.034, 95% CI 0.840–0.973; p < 0.0001), indicating a fair to good discriminatory ability.
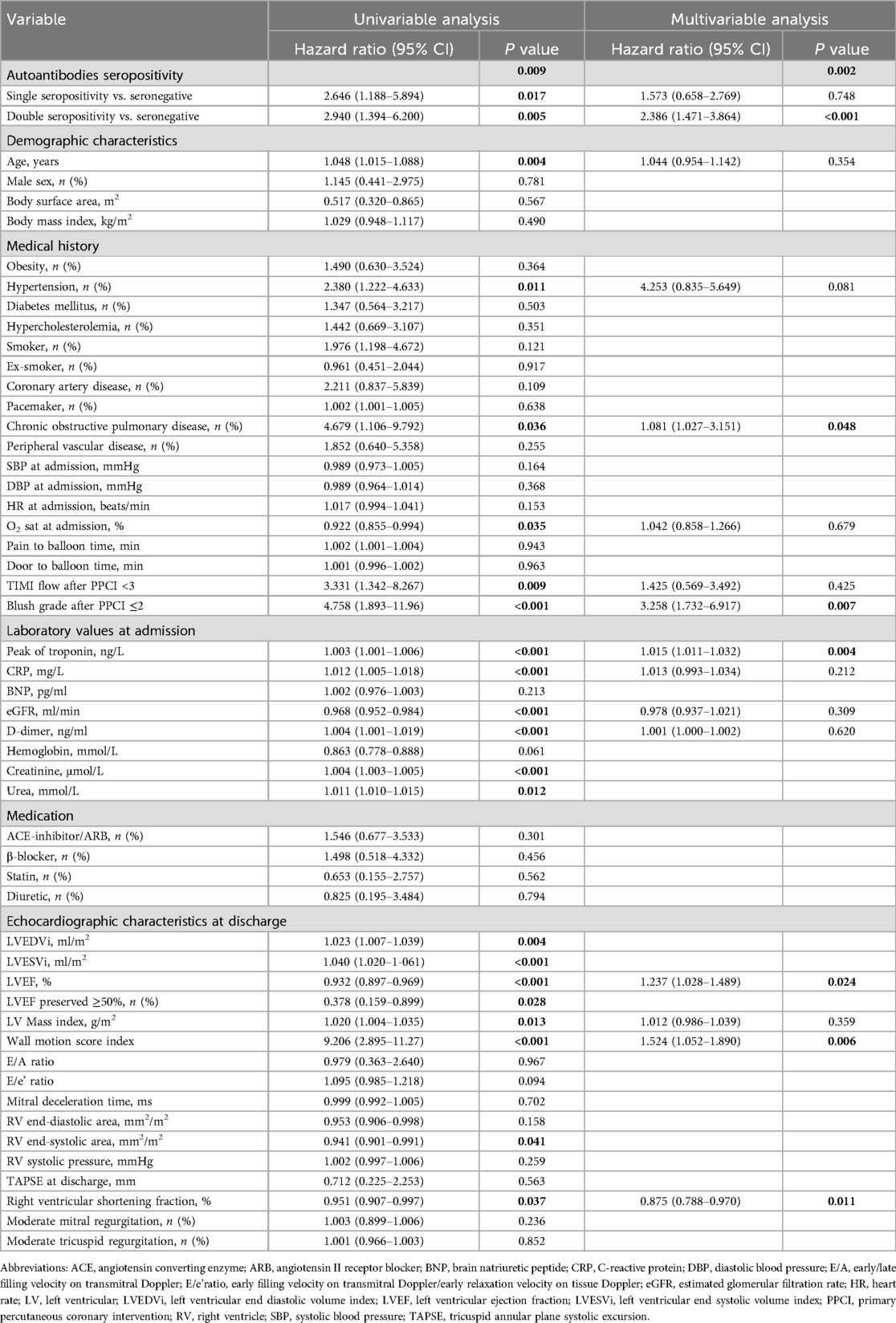
Table 2. Univariable and multivariable Cox proportional hazard models for MACE (n = 200).
Levels of AT1R-AAs and ETAR-AAs, when analyzed as continuous variables, were shown to correlate with a higher risk of experiencing MACE (Figure 3).
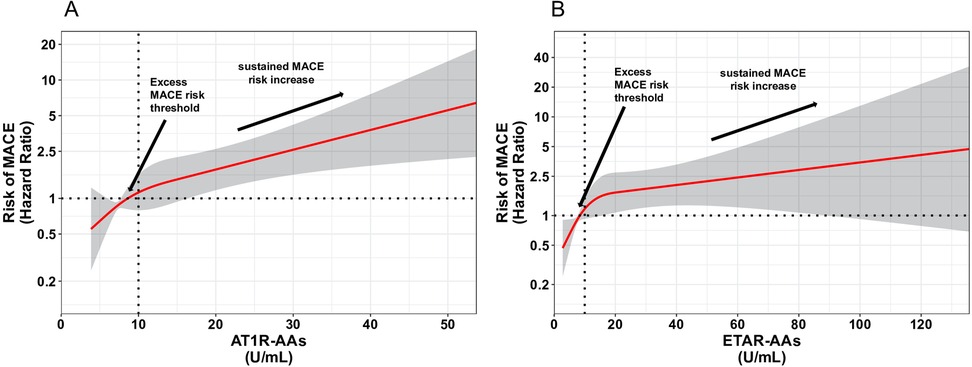
Figure 3. Spline curves for AT1R-AAs and ETAR-AAs versus MACE. Changes in the hazard ratio (HR) across the AT1R-AAs (A) and ETAR-AAs (B) are demonstrated in spline curves on a hazard scale with overlaid 95% confidence intervals (gray shaded area). This scale shows the relationship between AT1R-AAs, ETAR-AAs, and MACE.
4 Discussion
In this study, we report, for the first time, the prognostic role of AT1R-AAs and ETAR-AAs in patients with STEMI. Ninety (45%) patients were seropositive for at least one autoantibody. Specifically, 44 (22%) patients were seropositive for AT1R-AAs or ETAR-AAs, and 46 (23%) patients were seropositive for both AT1R-AAs and ETAR-AAs. The incidence of MACE in AT1R-AAs- and ETAR-AA-seropositive patients was significantly higher than that in seronegative patients. During follow-up, the MACE risk was significantly higher in both single- and double-seropositive patients than in seronegative patients.
STEMI continues to be a global health issue, despite notable progress in its diagnosis and treatment. Furthermore, patients with STEMI undergoing PPCI have shown no improvement in myocardial infarction severity over the last few decades (3). Post-STEMI complications, including heart failure and recurrent infarction, can substantially affect patient prognosis. Timely recognition and treatment of these complications are essential for improving patient outcomes. In addition to comorbidities and myocardial infarction extension, the role of inflammation and fibrosis in STEMI outcomes has also received increasing attention (34, 35).
AT1R and ETAR are GPCRs expressed in different cell types, including vascular myocytes, immune cells, endothelial cells, and fibroblasts (36). Notably, AT1R and ETAR can be activated not only by their natural ligands but also by specific autoantibodies (AT1R-AAs and ETAR-AAs, respectively), which can bind AT1R and ETAR and regulate their function. AT1R-AAs and ETAR-AAs, which mirror (and amplify) the effects of the natural ligands AngII and ET1, induce vasoconstriction and stimulate pro-inflammatory and pro-fibrotic pathways (37, 38). Compared with AngII and ET1, receptor desensitization secondary to binding of AT1R-AAs and ETAR-AAs to their target receptors is more difficult, so that the autoantibodies can impose a sustained pathological stimulatory effect on receptors (8, 9, 11). Furthermore, the interaction between autoantibodies and receptors, combined with increased sensitivity to natural binding molecules, may contribute to an additional aspect of irregular vascular function (39). The concurrent presence of AT1R-AAs and ETAR-AAs also suggests the significance of receptor pairing, which could modify the impact of agonists on signal transmission (40).
The role of AT1R-AAs and ETAR-AAs in cardiovascular diseases is well established (9); however, their specific impact on outcomes after myocardial infarction has not been previously investigated. Two distinct processes, potentially initiated by AT1R-AAs and ETAR-AAs, and working together to promote myocardial fibrosis (41), play crucial roles in determining poorer outcomes following myocardial infarction: microvascular dysfunction (14) and myocardial inflammation (34). In terms of the inflammatory response, AT1R-AAs and ETAR-AAs are recognized for their ability to activate specific intracellular inflammatory cascades (8), which may contribute to the inflammatory process within the myocardium. Like AngII and ET1, AT1R-AAs and ETAR-AAs can indeed activate signal transduction pathways in non-immune and immune cells (8, 11, 42). Although the signaling molecules activated by AT1R-AAs and ETAR-AAs in non-immune cells are well characterized (42), the signaling pathways modulated by these autoantibodies in immune cells remain to be elucidated. Regarding microvascular dysfunction, the potential association with AT1R-AAs and ETAR-AAs is even more intriguing. Although the overall vasoconstrictive effects of these autoantibodies have long been recognized (8, 38), their particular impact on heart disease and coronary microcirculation has only recently become a focus of research (9, 12, 13). Notably, ETAR-AAs have been linked to the no-reflow phenomenon following acute myocardial infarction, a condition in which the coronary microcirculation becomes completely dysfunctional, hindering myocardial reperfusion (12). We have also recently demonstrated the association of AT1R-AAs and ETAR-AAs with post-infarction left ventricular remodeling (13). After myocardial infarction, both no-reflow and adverse left ventricular remodeling negatively influence clinical outcomes (14, 15). The association between AT1R-AAs and ETAR-AAs and these important determinants of myocardial infarction outcomes suggests that these autoantibodies may be involved in the prognosis of myocardial infarction and may explain the results of the present study. However, further investigation using preclinical and animal studies is required to confirm these hypotheses.
In this study, we used a unique approach to assess not only the levels of each autoantibody but also the overall degree of seropositivity. Thus, we did not only examine the effect of a single autoantibody but rather investigated the overall burden of anti-AT1R/ETAR autoimmunity, hypothesizing that the simultaneous presence of both autoantibodies exerts a larger effect by activating similar complementary intracellular pathways, which amplify their effects in a sort of “double hit” phenomenon. We decided to take this approach given (1) a high similarity of effects between the two autoantibodies (8, 38); (2) autoantibody-mediated receptor cross-talk (32, 43, 44); and (3) previous literature on the topic. For example, in preeclampsia, all patients have high levels of AT1R-AAs, but only those with severe disease are seropositive for ETAR-AAs (45). In pediatric kidney disease, the presence of both antibodies is significantly associated with arteritis, elevated inflammatory markers, and a decline in renal function, suggesting possible interaction effects (46). Notably, a combined effect has been observed with other autoantibodies, such as those targeting heat-shock protein 60 (anti-Hsp60). These antibodies enhance the likelihood of arterial vascular events only when antiphospholipid antibodies are also present (47). Similarly, in idiopathic nephrotic syndrome, circulating podocyte autoantibodies exert their full effect only once antibodies to glomerular endothelial cells have damaged the endothelial cells (48). Based on these previous findings, we believe that AT1R-AAs and ETAR-AAs have a cumulative effect, and that the simultaneous presence of both autoantibodies is associated with a higher probability of recurrent cardiovascular adverse events due to the amplification of the effects elicited by each single autoantibody.
AT1R-AAs and ETAR-AAs are both IgG autoantibodies. The autoantibodies detected during the acute stage of myocardial infarction are therefore likely to be pre-existing rather than induced by myocardial ischemia. AT1R-AAs and ETAR-AAs are compelling components of our immune system (9, 37, 38, 49). Pivotal to the scope of understanding no-reflow and adverse remodeling after myocardial infarction, as we recently described (12, 13), autoantibodies anti-GPCRs have recently been described as powerful markers of chronic GPCR expression, reflecting chronic individual exposure to different environmental factors (“Exposome”). This antibody network (“Antibodiom”) is thus shaped by both environmental exposure and an individual's immune system (49). Although speculative, we hypothesize that each patient possesses unique levels of AT1R-AAs and ETAR-AAs, which serve as a signature of their “exposome” and influence their reaction to an acute ischemic event and their susceptibility to no-reflow and left ventricular remodeling, regardless of infarct size (12, 13). This hypothesis aligns with emerging evidence suggesting the immune system's role in homeostasis beyond host defence (50). Additionally, specific metabolic conditions, such as those induced by ischemia and reperfusion, may be necessary for the full activity of AT1R-AAs and ETAR-AAs (51).
Following myocardial infarction, the endothelium produces AngII and ET1, which are not merely passive observers but indicators of infarct size and potential enhancers of AT1R-AAs and ETAR-AAs effects (52). This situation is further complicated by factors beyond natural ligands and autoantibodies. Studies on rat models have shown that ischemia and reperfusion lead to an increase in AngII and ET1 binding sites on cardiac membranes (53). Consequently, both ligands (elevated AngII and ET1 production and higher AT1R-AAs and ETAR-AAs levels) and receptors (increased AT1R and ETAR expression) can affect infarct extension and no-reflow.
As regards the potential clinical implications of our findings, the first and most straightforward one is that of having a biomarker that allows stratification of the risk of adverse events after STEMI. Seropositivity for AT1R-AAs/ETAR-AAs (and most of all double seropositivity) allows the identification, even in the first phases after the acute event, of STEMI patients with a risk of poor prognosis who could benefit from closer monitoring and follow-up. However, we believe that the potential implications of AT1R-AAs/ETAR-AAs in ischemic heart disease go beyond those of simple risk biomarkers. Our findings open the first window on the interconnection between autoimmunity and acute coronary syndromes, potentially leading to major changes in the treatment of patients with acute myocardial infarction. Currently, no available therapy specifically inhibits AT1R-AAs and ETAR-AAs and/or regulates the expression of AT1R and ETAR. Nevertheless, laboratory studies have shown that angiotensin receptor blockers (ARBs) and ETAR antagonists can counteract the stimulatory effects of AT1R-AAs and ETAR-AAs (54). Clinical research has also demonstrated that combining plasmapheresis with ARBs reduces the risk of acute rejection in transplant patients (55), and recent findings indicate that ARBs are linked to lower overall mortality and fewer heart failure hospitalizations than ACE inhibitors in myocardial infarction patients (56). Notably, a recent clinical trial also showed that sparsentan, a non-immunosuppressive, dual endothelin and angiotensin receptor antagonist with high selectivity for ETAR and AT1,R is highly effective in IgA nephropathy (57). This evidence supports the hypothesis that blocking the effects of AT1R-AAs/ETAR-AAs with already approved and well-tolerated drugs might be beneficial in patients with STEMI. Due to the observational design and limited sample size, the therapeutic implications remain speculative and warrant further investigation. However, advancing our understanding of the cell signaling pathways and mechanisms underlying autoantibody-induced pathology triggered by AT1R-AAs and ETAR-AAs could lead to the development of novel therapies. These treatments can specifically target autoantibody effects while preserving the normal physiological functions of the RAAS and endothelin systems.
This study had some limitations, including a small sample size, single center, single ethnicity, and short follow-up time. Moreover, although this is a common issue in studies investigating acute coronary events (58, 59), most patients enrolled in the present study were men. Although the current literature does not report sex-based differences in the effects of AT1R-AAs and ETAR-AAs, the autoimmune profile is significantly different between men and women (60); thus, our findings cannot be confidently extended to female patients. The relatively small number of events did not allow for the investigation of the association between autoantibody levels and the individual components of the primary outcome. Additionally, the observational nature of the study implies that residual confounding might be present; although we adjusted our multivariate survival analysis for variables associated with worse outcomes, unmeasured comorbidities/factors might still be present. Dynamic changes in antibody titers were not investigated, and we do not know whether the levels of autoantibodies changed during follow-up. We also investigated only medical therapy at discharge and did not know which medical therapy patients were prescribed during follow-up. However, given that all patients were followed up at the same center, we can hypothesize a homogeneous approach. Additionally, although the cutoff of 10 U/ml to define seropositivity has been widely used in previous studies, its accuracy in patients with cardiovascular disease should be tested in larger cohorts. Finally, the observational study design and correlations observed in this study do not allow for the inference of causality and should therefore be considered hypothesis-generating. To validate our hypothesis, ELISA tests for antibody detection are not sufficient. Additional preclinical studies are necessary to gain a deeper understanding of the molecular pathways through which AT1R-AAs and ETAR-AAs exert their effects. Future multicenter clinical studies with longer follow-up periods and larger cohorts of patients are warranted to validate our findings in independent patient cohorts and to capture events occurring beyond the early years following STEMI.
In conclusion, our research provides the first evidence suggesting that functional autoantibody-mediated vascular receptor activation plays a role in revascularized STEMI. Additionally, it suggests that AT1R-AAs and ETAR-AAs may serve as potential prognostic markers for acute myocardial infarction. These findings may lead to the development of innovative treatments to improve outcomes for STEMI patients.
Data availability statement
The datasets presented in this article are available from the corresponding author on reasonable request. Requests to access the datasets should be directed to Francesco Tona,ZnJhbmNlc2NvLnRvbmFAdW5pcGQuaXQ=.
Ethics statement
The studies involving humans were approved by institutional ethics committee (Comitato Etico per la Sperimentazione Clinica della Provincia di Padova; code number CESC 5478/AO). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
FT: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. GC: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. MV: Data curation, Formal analysis, Investigation, Writing – review & editing. GM: Data curation, Formal analysis, Investigation, Writing – review & editing. LI: Conceptualization, Data curation, Formal analysis, Investigation, Writing – review & editing. MPM: Data curation, Formal analysis, Investigation, Writing – review & editing. AC: Data curation, Formal analysis, Investigation, Methodology, Writing – review & editing. MM: Data curation, Formal analysis, Investigation, Methodology, Writing – review & editing. DT: Data curation, Formal analysis, Investigation, Writing – review & editing. GB: Data curation, Formal analysis, Investigation, Methodology, Writing – review & editing. BS: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – review & editing. LL: Conceptualization, Supervision, Writing – review & editing. EC: Conceptualization, Supervision, Writing – review & editing. SI: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by a grant from DSCTV prot. BIRD202539 – project code U-Gov_BIRD2020_01.
Acknowledgments
This study would not have been possible without the tireless support of the catheter laboratory staff of Padua University Hospital. Special mention is also deserved by the nursing staff of the Cardiac Intensive Care Unit and Cardiology ward of Padua University Hospital, whose help in obtaining patients' blood samples has been invaluable. We are grateful to the patients who participated in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2025.1515693/full#supplementary-material
References
1. Widimsky P, Wijns W, Fajadet J, De Belder M, Knot J, Aaberge L, et al. Reperfusion therapy for ST elevation acute myocardial infarction in Europe: description of the current situation in 30 countries. Eur Heart J. (2010) 31:943–57. doi: 10.1093/eurheartj/ehp492
2. Mensah GA, Fuster V, Murray CJL, Roth GA, Mensah GA, Abate YH, et al. Global burden of cardiovascular diseases and risks, 1990–2022. J Am Coll Cardiol. (2023) 82:2350–473. doi: 10.1016/j.jacc.2023.11.007
3. Lechner I, Reindl M, Tiller C, Holzknecht M, Fink P, Troger F, et al. Temporal trends in infarct severity outcomes in ST-segment–elevation myocardial infarction: a cardiac magnetic resonance imaging study. JAHA. (2023) 12(15):e028932. doi: 10.1161/JAHA.122.028932
4. Dhaun N, Webb DJ. Endothelins in cardiovascular biology and therapeutics. Nat Rev Cardiol. (2019) 16(8):491–502. doi: 10.1038/s41569-019-0176-3
5. Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. (2007) 292:C82–97. doi: 10.1152/ajpcell.00287.2006
6. Kvakan H, Kleinewietfeld M, Qadri F, Park J-K, Fischer R, Schwarz I, et al. Regulatory T cells ameliorate angiotensin II–induced cardiac damage. Circulation. (2009) 119:2904–12. doi: 10.1161/CIRCULATIONAHA.108.832782
7. Yeoh SE, Docherty KF, Campbell RT, Jhund PS, Hammarstedt A, Heerspink HJL, et al. Endothelin−1, outcomes in patients with heart failure and reduced ejection fraction, and effects of dapagliflozin: findings from DAPA-HF. Circulation. (2023) 147:1670–83. doi: 10.1161/CIRCULATIONAHA.122.063327
8. Philogene MC, Johnson T, Vaught AJ, Zakaria S, Fedarko N. Antibodies against angiotensin II type 1 and endothelin A receptors: relevance and pathogenicity. Hum Immunol. (2019) 80:561–7. doi: 10.1016/j.humimm.2019.04.012
9. Civieri G, Iop L, Tona F. Antibodies against angiotensin II type 1 and endothelin 1 type A receptors in cardiovascular pathologies. IJMS. (2022) 23:927. doi: 10.3390/ijms23020927
10. Günther J, Kill A, Becker M, Heidecke H, Rademacher J, Siegert E, et al. Angiotensin receptor type 1 and endothelin receptor type A on immune cells mediate migration and the expression of IL-8 and CCL18 when stimulated by autoantibodies from systemic sclerosis patients. Arthritis Res Ther. (2014) 16:R65. doi: 10.1186/ar4503
11. Akbarzadeh R, Müller A, Humrich JY, Riemekasten G. When natural antibodies become pathogenic: autoantibodies targeted against G protein-coupled receptors in the pathogenesis of systemic sclerosis. Front Immunol. (2023) 14:1213804. doi: 10.3389/fimmu.2023.1213804
12. Tona F, Vadori M, Civieri G, Masiero G, Iop L, Antonelli G, et al. Association of autoantibodies targeting endothelin type-A receptors with no-reflow in ST-elevation myocardial infarction. Atherosclerosis. (2023) 378:117179. doi: 10.1016/j.atherosclerosis.2023.06.970
13. Tona F, Civieri G, Vadori M, Masiero G, Iop L, Marra MP, et al. Association of angiotensin II receptor type 1 and endothelin-1 receptor type A agonistic autoantibodies with adverse remodeling and cardiovascular events after acute myocardial infarction. J Am Heart Assoc. (2024) 13(4):e032672. doi: 10.1161/JAHA.123.032672
14. Galli M, Niccoli G, De Maria G, Brugaletta S, Montone RA, Vergallo R, et al. Coronary microvascular obstruction and dysfunction in patients with acute myocardial infarction. Nat Rev Cardiol. (2023) 21(5):283–98. doi: 10.1038/s41569-023-00953-4
15. Heusch G, Libby P, Gersh B, Yellon D, Böhm M, Lopaschuk G, et al. Cardiovascular remodelling in coronary artery disease and heart failure. Lancet. (2014) 383:1933–43. doi: 10.1016/S0140-6736(14)60107-0
16. Byrne RA, Rossello X, Coughlan JJ, Barbato E, Berry C, Chieffo A, et al. 2023 ESC guidelines for the management of acute coronary syndromes. Eur Heart J. (2023) 44:3720–826. doi: 10.1093/eurheartj/ehad191
17. TIMI Study Group. The thrombolysis in myocardial infarction (TIMI) trial. Phase I findings. N Engl J Med. (1985) 312:932–6. doi: 10.1056/NEJM198504043121437
18. Henriques JPS, Zijlstra F, Van ‘T Hof AWJ, De Boer M-J, Dambrink J-HE, Gosselink M, et al. Angiographic assessment of reperfusion in acute myocardial infarction by myocardial blush grade. Circulation. (2003) 107:2115–9. doi: 10.1161/01.CIR.0000065221.06430.ED
19. Lang RM, Badano LP, Mor-Avi V, Afilalo J, Armstrong A, Ernande L, et al. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European association of cardiovascular imaging. Eur Heart J Cardiovasc Imaging. (2015) 16:233–71. doi: 10.1093/ehjci/jev014
20. Miedema J, Schreurs M, van der Sar-van der Brugge S, Paats M, Baart S, Bakker M, et al. Antibodies against angiotensin II receptor type 1 and endothelin A receptor are associated with an unfavorable COVID19 disease course. Front Immunol. (2021) 12:684142. doi: 10.3389/fimmu.2021.684142
21. Budding K, van de Graaf EA, Hoefnagel T, Kwakkel-van Erp JM, van Kessel DA, Dragun D, et al. Anti-ETAR and anti-AT1R autoantibodies are elevated in patients with endstage cystic fibrosis. J Cyst Fibros. (2015) 14:42–5. doi: 10.1016/j.jcf.2014.07.007
22. Vythoulkas D, Lazana I, Kroupis C, Gavriilaki E, Konstantellos I, Bousiou Z, et al. Endothelial injury syndromes after allogeneic hematopoietic stem cell transplantation: angiopetin-2 as a novel predictor of the outcome and the role of functional autoantibodies against angiotensin II type 1 and endothelin A receptor. IJMS. (2023) 24:6960. doi: 10.3390/ijms24086960
23. Catar RA, Wischnewski O, Chen L, Heidecke H, Rutz C, Schülein R, et al. Non-HLA antibodies targeting angiotensin II type 1 receptors and endothelin-1 type A receptors impair endothelial repair via a β2-arrestin link to the mTOR pathway. Kidney Int. (2021) 101(3):498–509. doi: 10.1016/j.kint.2021.09.029
24. Hiemann NE, Meyer R, Wellnhofer E, Schoenemann C, Heidecke H, Lachmann N, et al. Non-HLA antibodies targeting vascular receptors enhance alloimmune response and microvasculopathy after heart transplantation. Transplant J. (2012) 94:919–24. doi: 10.1097/TP.0b013e3182692ad2
25. Banasik M, Boratyńska M, Kościelska-Kasprzak K, Kamińska D, Zmonarski S, Mazanowska O, et al. Non-HLA antibodies: angiotensin II type 1 receptor (anti-AT1R) and endothelin-1 type A receptor (anti-ETAR) are associated with renal allograft injury and graft loss. Transplant Proc. (2014) 46:2618–21. doi: 10.1016/j.transproceed.2014.09.029
26. Riemekasten G, Philippe A, Näther M, Slowinski T, Müller DN, Heidecke H, et al. Involvement of functional autoantibodies against vascular receptors in systemic sclerosis. Ann Rheum Dis. (2011) 70:530–6. doi: 10.1136/ard.2010.135772
27. Hall J, Bourne KM, Vernino S, Hamrefors V, Kharraziha I, Nilsson J, et al. Detection of G protein–coupled receptor autoantibodies in postural orthostatic tachycardia syndrome using standard methodology. Circulation. (2022) 146:613–22. doi: 10.1161/CIRCULATIONAHA.122.059971
28. Cabral-Marques O, Marques A, Giil LM, De Vito R, Rademacher J, Günther J, et al. GPCR-specific autoantibody signatures are associated with physiological and pathological immune homeostasis. Nat Commun. (2018) 9:5224. doi: 10.1038/s41467-018-07598-9
29. Ohe H, Uchida Y, Yoshizawa A, Hirao H, Taniguchi M, Maruya E, et al. Association of anti-human leukocyte antigen and anti-angiotensin II type 1 receptor antibodies with liver allograft fibrosis after immunosuppression withdrawal. Transplantation. (2014) 98:1105–11. doi: 10.1097/TP.0000000000000185
30. Giral M, Foucher Y, Dufay A, Van Huyen JPD, Renaudin K, Moreau A, et al. Pretransplant sensitization against angiotensin II type 1 receptor is a risk factor for acute rejection and graft loss. Am J Transplant. (2013) 13:2567–76. doi: 10.1111/ajt.12397
31. Piazza M, Seccia TM, Caroccia B, Rossitto G, Scarpa R, Persichitti P, et al. AT1AA (angiotensin II type-1 receptor autoantibodies): cause or consequence of human primary aldosteronism? Hypertension. (2019) 74:793–9. doi: 10.1161/HYPERTENSIONAHA.119.13388
32. Becker MO, Kill A, Kutsche M, Guenther J, Rose A, Tabeling C, et al. Vascular receptor autoantibodies in pulmonary arterial hypertension associated with systemic sclerosis. Am J Respir Crit Care Med. (2014) 190:808–17. doi: 10.1164/rccm.201403-0442OC
33. O’Leary JG, Philippe A, Freeman R, Heidecke H, Jennings LW, Catar R, et al. Non-HLA autoantibodies at 1 year negatively affect 5-year native renal function in liver transplant recipients. Transplant Proc. (2021) 53:1019–24. doi: 10.1016/j.transproceed.2021.01.013
34. Matter MA, Paneni F, Libby P, Frantz S, Stähli BE, Templin C, et al. Inflammation in acute myocardial infarction: the good, the bad and the ugly. Eur Heart J. (2024) 45:89–103. doi: 10.1093/eurheartj/ehad486
35. Scalise RFM, De Sarro R, Caracciolo A, Lauro R, Squadrito F, Carerj S, et al. Fibrosis after myocardial infarction: an overview on cellular processes, molecular pathways, clinical evaluation and prognostic value. Med Sci (Basel). (2021) 9:16. doi: 10.3390/medsci9010016
36. Horinouchi T, Terada K, Higashi T, Miwa S. Endothelin receptor signaling: new insight into its regulatory mechanisms. J Pharmacol Sci. (2013) 123:85–101. doi: 10.1254/jphs.13R02CR
37. Cabral-Marques O, Moll G, Catar R, Preuß B, Bankamp L, Pecher A-C, et al. Autoantibodies targeting G protein-coupled receptors: an evolving history in autoimmunity. Report of the 4th international symposium. Autoimmun Rev. (2023) 22:103310. doi: 10.1016/j.autrev.2023.103310
38. Cabral-Marques O, Riemekasten G. Functional autoantibodies targeting G protein-coupled receptors in rheumatic diseases. Nat Rev Rheumatol. (2017) 13:648–56. doi: 10.1038/nrrheum.2017.134
39. Lukitsch I, Kehr J, Chaykovska L, Wallukat G, Nieminen-Kelhä M, Batuman V, et al. Renal ischemia and transplantation predispose to vascular constriction mediated by angiotensin II type 1 receptor-activating antibodies. Transplant J. (2012) 94:8–13. doi: 10.1097/TP.0b013e3182529bb7
40. Barnes PJ. Receptor heterodimerization: a new level of cross-talk. J Clin Invest. (2006) 116:1210–2. doi: 10.1172/JCI28535
41. Moysidou G-S, Dara A, Arvanitaki A, Skalkou A, Pagkopoulou E, Daoussis D, et al. Understanding and managing cardiac involvement in systemic sclerosis. Expert Rev Clin Immunol. (2023) 19:293–304. doi: 10.1080/1744666X.2023.2171988
42. Catar R, Herse-Naether M, Zhu N, Wagner P, Wischnewski O, Kusch A, et al. Autoantibodies targeting AT1- and ETA-receptors link endothelial proliferation and coagulation via ets-1 transcription factor. IJMS. (2021) 23:244. doi: 10.3390/ijms23010244
43. Hur E-M, Kim K-T. G protein-coupled receptor signalling and cross-talk. Cell Signal. (2002) 14:397–405. doi: 10.1016/S0898-6568(01)00258-3
44. Lin Y-J, Kwok C-F, Juan C-C, Hsu Y-P, Shih K-C, Chen C-C, et al. Angiotensin II enhances endothelin-1-induced vasoconstriction through upregulating endothelin type A receptor. Biochem Biophys Res Commun. (2014) 451:263–9. doi: 10.1016/j.bbrc.2014.07.119
45. Buttrup Larsen S, Wallukat G, Schimke I, Sandager A, Tvilum Christensen T, Uldbjerg N, et al. Functional autoantibodies against endothelin-1 receptor type A and angiotensin II receptor type 1 in patients with preeclampsia. Pregnancy Hypertens. (2018) 14:189–94. doi: 10.1016/j.preghy.2018.10.002
46. Pearl MH, Chen L, ElChaki R, Elashoff D, Gjertson DW, Rossetti M, et al. Endothelin type A receptor antibodies are associated with angiotensin II type 1 receptor antibodies, vascular inflammation, and decline in renal function in pediatric kidney transplantation. Kidney Int Rep. (2020) 5:1925–36. doi: 10.1016/j.ekir.2020.09.004
47. Dieudé M, Correa JA, Neville C, Pineau C, Levine JS, Subang R, et al. Association of autoantibodies to heat-shock protein 60 with arterial vascular events in patients with antiphospholipid antibodies. Arthritis Rheum. (2011) 63:2416–24. doi: 10.1002/art.30411
48. Ye Q, Wang D, Zhou C, Meng H, Liu H, Mao J. A spectrum of novel anti-vascular endothelial cells autoantibodies in idiopathic nephrotic syndrome patients. Clin Immunol. (2023) 249:109273. doi: 10.1016/j.clim.2023.109273
49. Riemekasten G, Petersen F, Heidecke H. What makes antibodies against G protein-coupled receptors so special? A novel concept to understand chronic diseases. Front Immunol. (2020) 11:564526. doi: 10.3389/fimmu.2020.564526
50. Matzinger P. The danger model: a renewed sense of self. Science. (2002) 296:301–5. doi: 10.1126/science.1071059
51. Silvis MJM, Kaffka genaamd Dengler SE, Odille CA, Mishra M, van der Kaaij NP, Doevendans PA, et al. Damage-associated molecular patterns in myocardial infarction and heart transplantation: the road to translational success. Front Immunol. (2020) 11:599511. doi: 10.3389/fimmu.2020.599511
52. Nio Y, Matsubara H, Murasawa S, Kanasaki M, Inada M. Regulation of gene transcription of angiotensin II receptor subtypes in myocardial infarction. J Clin Invest. (1995) 95:46–54. doi: 10.1172/JCI117675
53. Liu J, Chen R, Casley DJ, Nayler WG. Ischemia and reperfusion increase 125I-labeled endothelin-1 binding in rat cardiac membranes. Am J Physiol Heart Circ Physiol. (1990) 258:H829–35. doi: 10.1152/ajpheart.1990.258.3.H829
54. Kimm MA, Haas H, Stölting M, Kuhlmann M, Geyer C, Glasl S, et al. Targeting endothelin receptors in a murine model of myocardial infarction using a small molecular fluorescent probe. Mol Pharmaceutics. (2020) 17:109–17. doi: 10.1021/acs.molpharmaceut.9b00810
55. Carroll RP, Riceman M, Hope CM, Zeng A, Deayton S, Bennett GD, et al. Angiotensin II type-1 receptor antibody (AT1Rab) associated humoral rejection and the effect of peri operative plasma exchange and candesartan. Hum Immunol. (2016) 77:1154–8. doi: 10.1016/j.humimm.2016.08.009
56. Chen C-W, Chang C-W, Lin Y-C, Chen W-T, Chien L-N, Huang C-Y. Comparison of clinical outcomes of angiotensin receptor blockers with angiotensin-converting enzyme inhibitors in patients with acute myocardial infarction. PLoS One. (2023) 18:e0290251. doi: 10.1371/journal.pone.0290251
57. Rovin BH, Barratt J, Heerspink HJL, Alpers CE, Bieler S, Chae D-W, et al. Efficacy and safety of sparsentan versus irbesartan in patients with IgA nephropathy (PROTECT): 2-year results from a randomised, active-controlled, phase 3 trial. Lancet. (2023) 402:2077–90. doi: 10.1016/S0140-6736(23)02302-4
58. Mehran R, Steg PG, Pfeffer MA, Jering K, Claggett B, Lewis EF, et al. The effects of angiotensin receptor-neprilysin inhibition on major coronary events in patients with acute myocardial infarction: insights from the PARADISE-MI trial. Circulation. (2022) 146:1749–57. doi: 10.1161/CIRCULATIONAHA.122.060841
59. Mehta SR, Wang J, Wood DA, Spertus JA, Cohen DJ, Mehran R, et al. Complete revascularization vs culprit lesion-only percutaneous coronary intervention for angina-related quality of life in patients with ST-segment elevation myocardial infarction: results from the COMPLETE randomized clinical trial. JAMA Cardiol. (2022) 7:1091–9. doi: 10.1001/jamacardio.2022.3032
Keywords: autoimmunity, no-reflow, coronary microcirculation, G protein coupled receptors, acute coronary syndrome, adverse prognosis, autoantibodies
Citation: Tona F, Civieri G, Vadori M, Masiero G, Iop L, Perazzolo Marra M, Cecere A, Martini M, Tansella D, Bernava G, Schiavon B, Leoni L, Cozzi E and Iliceto S (2025) Prognostic role of angiotensin-II receptor type 1 and endothelin-1 receptor type A agonistic autoantibodies in patients with acute myocardial infarction. Front. Cardiovasc. Med. 12:1515693. doi: 10.3389/fcvm.2025.1515693
Received: 23 October 2024; Accepted: 16 July 2025;
Published: 12 August 2025.
Edited by:
Sabrina Pagano, University Hospitals of Geneva, SwitzerlandReviewed by:
Dmytro Dziuba, National University of Healthcare of Ukraine, UkraineStylianos Daios, Aristotle University of Thessaloniki, Greece
Copyright: © 2025 Tona, Civieri, Vadori, Masiero, Iop, Perazzolo Marra, Cecere, Martini, Tansella, Bernava, Schiavon, Leoni, Cozzi and Iliceto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesco Tona, ZnJhbmNlc2NvLnRvbmFAdW5pcGQuaXQ=
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share senior authorship