Abstract
Cardiovascular disease (CVD) remains a leading cause of death globally, posing a major public health challenge. Due to the complexity of CVD's etiology, understanding its pathogenesis has been a significant challenge and research focus. In recent years, the communication between organelles has gained increasing attention, with mitochondria-associated endoplasmic reticulum (ER) membranes (MAMs) emerging as a key structural component that facilitates dialogue between the mitochondria and the ER. Numerous studies have highlighted that proteins located in MAMs may play a role in the development of CVD. Among these, mitofusin 2 (MFN2), a protein found on the outer mitochondrial and ER membranes, has garnered particular interest due to its widespread presence in MAMs. This review aims to sort out current research on MFN2, focusing on its potential involvement in myocardial protection through its mediation of MAMs. We discuss how MFN2-mediated MAMs may contribute to the protection against various CVDs, including myocardial ischemia/reperfusion injury, diabetic cardiomyopathy, dilated cardiomyopathy, pathological myocardial hypertrophy, cardiotoxicity, and heart failure. However, given the functional diversity of MFN2, the current body of research remains controversial, and further studies are urgently needed to clarify its precise mechanisms of action.
1 Introduction
Cardiovascular disease (CVD) is the leading cause of death from non-communicable diseases globally and represents a common endpoint for many chronic diseases (1). Effective programs for the prevention and treatment of CVD remain insufficient worldwide, primarily due to the diverse and multifactorial causes of the disease (2). The endoplasmic reticulum (ER) and mitochondria are two critical organelles involved in cellular protein production and energy metabolism, as well as in biological processes such as signal transduction, redox balance, calcium homeostasis, and apoptosis (3). It has been shown that some mitochondria and ER can be connected by tethering proteins to form mitochondria-associated ER membranes (MAMs) (4–6). MAMs are phospholipid bilayer structures composed of ER and mitochondria, including the outer mitochondrial membrane (OMM), the endoplasmic reticulum membrane, and the portion between them, and studies have shown that the distance of MAMs composed of smooth endoplasmic reticulum to mitochondria is 10–50 nm, and the distance of MAMs composed of rough endoplasmic reticulum to mitochondria is 50–80 nm (7). This structure is closely associated with the pathophysiology of CVD (8–10). Comparative analysis has shown that 1,347 proteins (approximately 96.56%) are highly conserved and expressed in human and mouse testis MAMs (11). Among these, some proteins are essential for the structural composition of MAMs, such as mitofusin 2/1 (MFN2/1), voltage-dependent anion channel (VDAC), inositol-1,4,5-triphosphate (IP3) receptor (IP3R), glucose-regulated protein 75 (GRP75), vesicle-associated membrane-protein-associated protein B, protein tyrosine phosphatase-interacting protein 51, B-cell receptor-associated protein 31, mitochondrial fission 1, and phosphofurin acidic cluster sorting protein 2. Other proteins serve regulatory functions, including calnexin, Sarco/ER calcium ion (Ca2+) ATPase, sigma-1 receptor, phosphatidylserine synthase, cyclophilin D, protein kinase B (AKT), mammalian TOR complex 2, long-chain fatty acid coenzyme A ligase 4, ER oxidoreductase-1α, and autophagy-related gene 14 (12). These proteins operate individually or in complexes, and our previous review highlighted the diverse functions of MAMs, which include, but are not limited to, the modulation of Ca2+ homeostasis, lipid homeostasis, mitochondrial dynamics, autophagy, mitophagy, apoptosis, and inflammation (3). Among the numerous proteins associated with MAMs, MFN2 stands out as the most specialized, being present not only on the OMM but also on the ER membrane (3, 13). Thus, MFN2 may play an important role in MAMs. In this review, we focus on the structure and function of MFN2 and its role in regulating MAMs to provide cardioprotection.
2 Structure of MFN2
MFN2 is a highly conserved GTPase composed of 757 amino acid residues. It has both an N-terminal and a C-terminal, each exposed in the cellular matrix, playing a central role in regulating mitochondrial fusion and cellular metabolism (14–17). The protein contains an N-terminal p21ras signature motif (amino acids 77–96), a GTPase-binding domain (amino acids 93–342), two coiled-coil regions (amino acids 391–434 and 695–738), and two transmembrane domains (amino acids 605–647) that span the OMM (Figure 1A) (14, 15, 17). MFN1 is structurally similar to MFN2, except that MFN1 contains 741 amino acid residues and the GTPase-binding domain is between amino acid residues 75–336 (18, 19). A recent comparative analysis and phylogenetic reconstruction study proposed a predictive model that contradicts current models. Bioinformatics analysis revealed that fungal Fuzzy onions 1 proteins possess two predicted transmembrane structural domains, while metazoan MFN2 contains only one. This newly predicted topology of MFN2 was biochemically confirmed, with the C-terminal oxidative reduction-sensitive cysteine residue located within the mitochondrial intermembrane space (IMS). Functional experiments further supported the notion that redox-mediated disulfide bond modification within this IMS structural domain is a key regulator, driving reversible MFN2/1 oligomerization necessary for fusion (Figure 1B) (20, 21). The discovery of this structural feature provides a mechanistic basis for the coordination between MFN2/1-dependent OMM fusion and optic atrophy factor 1 (OPA1)-dependent inner mitochondrial membrane (IMM) fusion (20, 22).
Figure 1
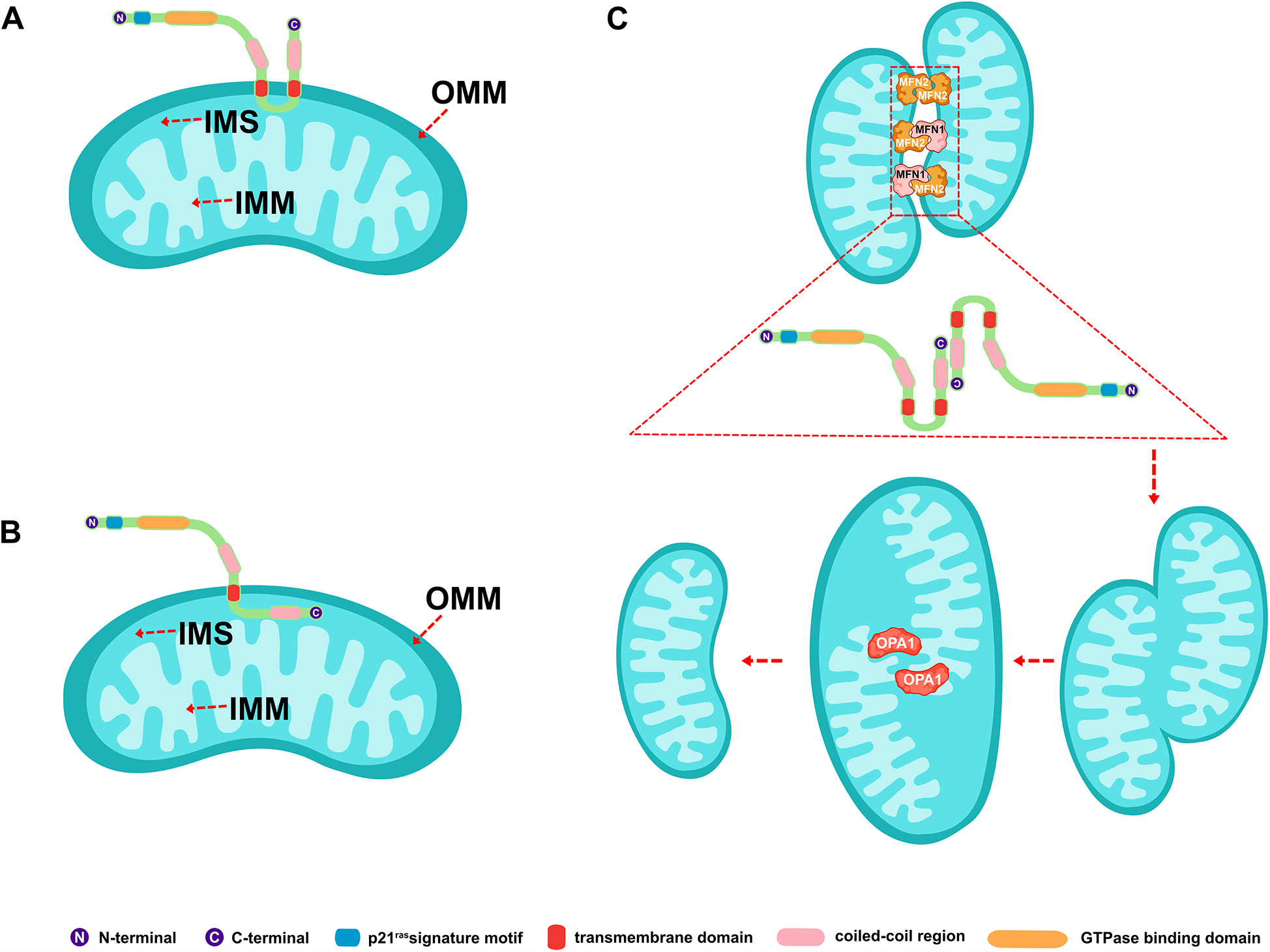
Schematic representation of the structure of mitofusin 2 and its involvement in mitochondrial fusion. (A) Classical MFN2 structural model. (B) Latest predicted MFN2 structural model. (C) MFN2-mediated mitochondrial fusion process. MFN2/1, mitofusin 2/1; IMM, inner mitochondrial membrane; OMM, outer mitochondrial membrane; IMS, mitochondrial intermembrane space; and OPA1, optic atrophy factor 1.
3 Function of MFN2
It is widely accepted that sequence determines structure, which in turn determines function (23, 24). The structure of MFN2 gives it a certain function, and we combed through the functions of MFN2 reported in existing studies.
3.1 Involved in mitochondrial fusion
The mitochondrial fusion involves several proteins, including MFN2 and MFN1 on the OMM and OPA1 on the IMM. It was shown that mitochondrial phosphatase phosphoglycerate mutase 5 (PGAM5) regulates MFN2 phosphorylation, thereby protecting it from ubiquitination and degradation (25). While phosphorylation promotes mitochondrial fission and degradation, and dephosphorylation promotes mitochondrial fusion (25). MFN2 and MFN1 mediate the fusion of the OMM, while OPA1 mediates the fusion of the IMM (26, 27). The GTP hydrolyzed by these fusion proteins enables two neighboring mitochondria to fuse, allowing the sharing of mitochondrial DNA, proteins, and metabolites (27). MFN2 on one mitochondrion can form either a homodimer (i.e., MFN2 binding to another MFN2 molecule to form MFN2-MFN2 polymers) or a heterodimer (i.e., MFN2 binding to MFN1 to form MFN2-MFN1 polymers) with MFN2/1 on another mitochondrion. Studies have shown that combining MFN2 and MFN1 in a heterodimer is the most efficient mechanism for MFN2-mediated OMM fusion (28, 29). A schematic representation of mitochondrial fusion, based on the generally accepted double-transmembrane structure, is shown in Figure 1C. The mechanism of action involves the reverse parallel interaction of the coiled-coil region at the C-terminal end of MFN2 (or MFN2 on one mitochondrion and MFN1 on the other) in two mitochondria, facilitating docking. This is followed by GTP hydrolysis and, ultimately, OMM fusion (29–31) (Figure 2). The mechanism of OMM fusion based on the single-transmembrane structure of MFN2/1 remains under investigation.
Figure 2
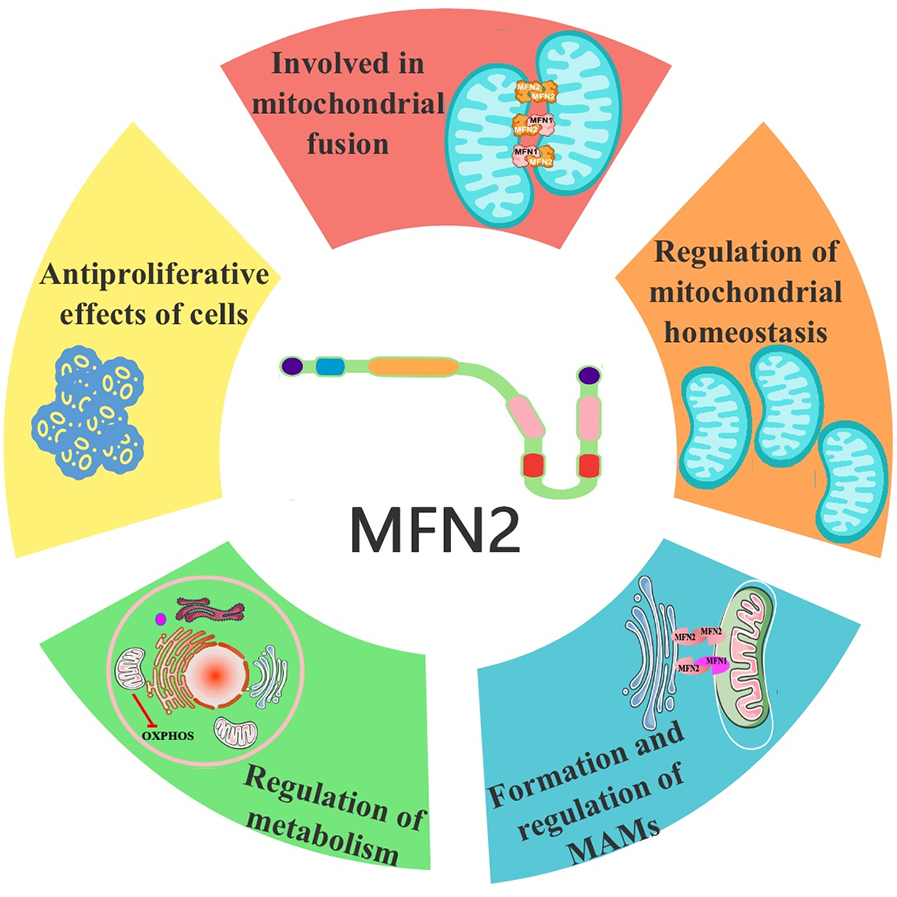
Functional schematic of MFN2. MFN2, mitofusin 2.
3.2 Regulation of mitochondrial homeostasis
MFN2 also plays a crucial role in regulating mitochondrial homeostasis. A study on Charcot-Marie-Tooth type 2A revealed that mutations in the Mfn2 gene lead to alterations in mitochondrial morphology and homeostasis (32). The activation of the MORN repeat-containing protein 4 (MORN4)-MFN2 axis regulates myocardial mitochondrial homeostasis. Mechanistically, MORN4 binds directly to MFN2 and promotes the phosphorylation of MFN2 at serine 442 via Rho-associated protein kinase 2, thereby mediating beneficial mitophagy induced by mitochondrial dynamics (33). A study in cultured neurons demonstrated that inhibition of USP30 promotes the ubiquitination of MFN2 by the E3 ubiquitin ligase (Parkin). This process separates damaged mitochondria from the healthy mitochondrial network and facilitates mitophagy, thereby removing damaged mitochondria from the cell (34). Modulation of mitochondrial morphology can likewise regulation of mitochondrial homeostasis. MFN2 also physically interacts with the protein kinase PERK. In cells ablated for Mfn2, sustained activation of PERK occurs under basal conditions. Silencing of PERK, however, reduces reactive oxygen species (ROS) production, normalizes mitochondrial Ca2+ levels, and improves mitochondrial morphology (35, 36). Similarly modulation of mitochondrial dynamics can regulation of mitochondrial homeostasis. In a vitro study in cardiomyocytes confirmed that overexpression of OPA1 and MFN2 inhibited hypoxia-induced mitochondrial division in H9c2 cardiomyocytes and reduced ROS production, thereby delaying the process of cardiomyocyte hypertrophy (37). A study in alveolar epithelial cells (A549) demonstrated that upregulation of MFN2 and OPA1 attenuates oxidative stress, mitochondrial damage, dysfunction, and mitophagy (38). Paeonol (chemically 2'-hydroxy-4'-methoxymethylphenol, a natural phenolic antioxidant extracted from the root bark of Paeonia lactiflora) stimulates mitochondrial fusion by activating the PKCε-Stat3-MFN2 pathway, protecting the heart from doxorubicin-induced injury (39). In contrast, resveratrol has been shown to restore mitochondrial quality control in myocardial ischemia/reperfusion injury via the Sirt1/Sirt3-MFN2-Parkin-PGC-1α pathway (40).
Mitophagy, the process of removing senescent or damaged mitochondria, is a key mechanism for maintaining mitochondrial homeostasis. Increased expression of MFN2 may upregulate mitophagy marker proteins, such as BNIP3l and Beclin1 (41). Both in vivo and in vitro studies have shown that activation of the MFN2/Pink1/Parkin mitophagy pathway reduces mitochondrial fragmentation in podocytes. Upregulation of MFN2 reduces puromycin aminonucleoside-induced podocyte injury, while downregulation of MFN2 limits the renal protective effects by modulating mitophagy (42). A study of atherosclerotic endothelial cells found that MFN2/Pink1/Parkin-mediated mitochondrial autophagy is critical for alleviating atherosclerosis (43). Metformin has been shown to cooperate with PINK1/MFN2 overexpression to inhibit ROS generation, reduce mitochondrial dysfunction, increase ATP generation, enhance mitochondrial membrane potential, and ultimately improve mitochondrial function, thus preventing cardiac injury (44). Research in astrocytes revealed that upregulation of MFN2 expression activates mitophagy through the PI3K/Akt/mTOR pathway, leading to the removal of damaged mitochondria in astrocytes (45) (Figure 2).
3.3 Formation and regulation of MAMs
In vitro assays, along with genetic and biochemical evidence, support the concept that MFN2 on the ER links the ER and mitochondria by forming homotypic and heterotypic complexes with MFN2 or MFN1 on the mitochondrial surface, effectively bridging the gap between the two organelles. Specifically, MFN2 on the ER interacts with MFN2 or MFN1 on the mitochondria to form either homotypic (MFN2-MFN2) or heterotypic (MFN2-MFN1) complexes, thereby linking the ER and mitochondria (20, 46). In contrast, the ablation or silencing of Mfn2 in mouse embryonic fibroblasts and HeLa cells disrupts ER morphology, weakens the interaction between the ER and mitochondria, and reduces the efficiency of mitochondrial Ca2+ uptake upon stimulation by IP3 production (17). Similarly, silencing Mfn2 in young mice reduces cardiomyocyte ER-mitochondrial contact (approximately 15 nm) by 30% and inhibits mitochondrial Ca2+ uptake from the neighboring ER (47). A recent study demonstrated that splicing of MFN2 generates two ER-specific variants, ERMIT2 and ERMIN2. ERMIN2 regulates ER morphology, while ERMIT2 localizes to the ER-mitochondrial interface, interacting with mitochondrial mitofusins to tether the ER and mitochondria. This interaction promotes mitochondrial Ca2+ uptake and phospholipid translocation. Notably, the expression of ERMIT2 ameliorated ER stress, inflammation, and fibrosis in liver-specific Mfn2 knockout (KO) mice (13). Furthermore, a separate study showed that the mitochondrial ubiquitin ligase MITOL regulates MAMs by activating K192 ubiquitination of the MFN2 GTPase structural domain (48). MITOL also prevents ER stress-induced apoptosis through IRE1α ubiquitination on MAMs (49). MAMs are disrupted during mitophagy and reduced ER-mitochondrial contact accelerates mitochondrial degradation. This process is driven by a rapid burst of Parkin/PINK1-catalyzed MFN2 phosphorylation, which triggers p97-dependent disassembly of the MFN2 complex from the OMM, thereby separating the mitochondria from the ER (50). Additionally, MAMs can contribute to MAMs for the formation of autophagic vesicles. A reduction in MFN2 significantly disrupts MAMs and attenuates autophagy, possibly through regulation by the AMP-activated protein kinase (AMPK)-MFN2 axis (51, 52). Knockdown of endogenous Mfn2 via stable small hairpin RNA (shRNA) expression targeting the 3′ untranslated region of Mfn2 increases the distance between the ER and the OMM. In contrast, tyrosine phosphorylation of MFN2 regulates the distance between the OMM and ER via c-Src, leading to elevated mitochondrial Ca2+ levels and oxidative stress (53).
However, conflicting reports also exist. Studies in HeLa cells using shRNA to silence Mfn2 indicate that such silencing increases the ER-mitochondrial linkage (54, 55). Additionally, the total number of ER-mitochondrial associations detected using a novel bifluorescence complementation method that labels a subset of ER-mitochondrial associations in fixed and living cells increased as MFN2 levels decreased (56). After the ablation or knockdown of Mfn2, there was a net increase in the number of narrow (∼10 nm) ER-mitochondrial contacts, while the number of wider (∼50 nm) contacts decreased (55, 56) (Figure 2).
3.4 Regulation of metabolism
An earlier study demonstrated that MFN2 is associated with mitochondrial energy metabolism. Specifically, loss of MFN2 function inhibits the oxidation of pyruvate, glucose, and fatty acids while decreasing mitochondrial membrane potential. Conversely, the gain of MFN2 function increases glucose oxidation and mitochondrial membrane potential. Mechanistically, in the Mfn2 cause Charcot-Marie-Tooth neuropathy type 2A, loss of MFN2 function inhibits the nuclear-encoded subunits of the oxidative phosphorylation (OXPHOS) complexes I, II, III, and V, whereas MFN2 overexpression induces the subunits of complexes I, IV, and V (57). Studies on liver-specific Mfn2 KO mice show that MFN2 connects mitochondrial and ER function with insulin signaling and is essential for normal glucose homeostasis (58). Furthermore, MFN2 influences the tricarboxylic acid cycle through sirtuin 3/isocitrate dehydrogenase 2 (NADP+) and mitochondrial activation, modulating glucose-stimulated insulin secretion in β-cells (59). In addition, MFN2 in pro-opiomelanocortin neurons regulates whole-body energy homeostasis by fine-tuning the function of the mitochondria-ER axis. In the brain, pro-opiomelanocortin neuron-specific deletion of Mfn2 results in binge eating and obesity, accompanied by reduced energy expenditure (60). A study showed that MFN2, highly expressed in brown adipose tissue, mediates mitochondria-lipid droplet interactions, influencing lipolytic processes and overall energy homeostasis. Mfn2 KO mice exhibit severe BAT dysfunction, impaired respiratory capacity, and a sluggish response to adrenergic stimuli (61). In contrast, both loss and gain of function of GRP75 or MFN2 significantly alter cholesterol metabolism, promoting triglyceride accumulation in hepatocytes (62). Furthermore, analysis of muscle Mfn2-KO mice revealed that aging-induced reductions in MFN2 underlie age-related changes in metabolic homeostasis and muscle loss (63) (Figure 2).
3.5 Antiproliferative effects of cells
MFN2 may also exert antiproliferative effects. Hypoxia-induced downregulation of MFN2 has been identified as a key determinant of smooth muscle cell (SMC) proliferation in pulmonary arteries (64). The mechanism may involve MFN2 counteracting cell proliferation by attenuating glycolysis. A study in cancer cells showed that MFN2 can interact with the M2 isoform of pyruvate kinase, one of the rate-limiting enzymes of glycolysis, to promote mitochondrial fusion, enhance OXPHOS, and reduce glycolysis (65). In vascular SMCs, mutations in the protein kinase A phosphorylation site on MFN2 inhibited its antiproliferative activity, an effect independent of its regulation of mitochondrial morphology (66). In cultured human vascular SMCs, GATA2 can bind to the Mfn2 promoter region and promote MFN2 expression. Interference with GATA2 reduces MFN2 expression and impedes the proliferation of human vascular SMCs (67). Further studies showed that MFN2 inhibits glycolysis in vascular SMCs by degrading phosphofructokinase-1 and prevents neointimal hyperplasia in vein grafts (68). Another study indicated that activation-induced degradation of MFN2 is a prerequisite for T-cell entry into the cell cycle. This supports the idea that MFN2 inhibits cell proliferation, with the PI3K-AKT-mTOR pathway playing an important role in the activation-induced downregulation of MFN2 and subsequent T-cell proliferation (15). In contrast, overexpression of MFN2 effectively attenuates astrocyte proliferation and halts the cell cycle. This is accompanied by downregulation of labeling and inhibition of wound healing, as MFN2 overexpression inhibits reactive astrocyte formation by blocking the Raf1-ERK1/2 and PI3K-AKT signaling pathways (69) (Figure 2).
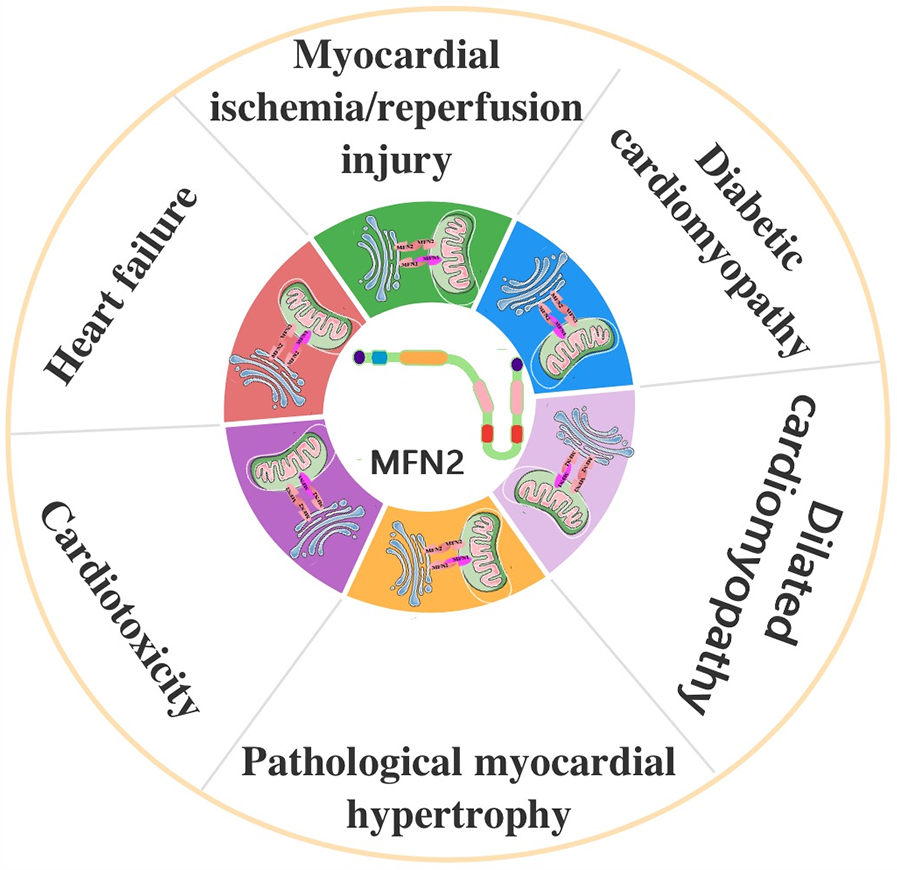
4 The role of MFN2-mediated MAMs in CVD
CVD has become the leading cause of death worldwide (70). Increasing evidence suggests that MAMs may be involved in the pathogenesis of CVD. As an essential tethering protein at MAMs, MFN2 plays a pivotal role in regulating MAM function and, consequently, influencing the disease processes associated with CVD. Furthermore, MFN2-associated strategies to mitigate CVD are being increasingly explored and reported (2, 3, 10, 71–78). In the following, we focused on the existing reports that MFN2 acts in CVD by regulating MAMs, with the aim of providing research ideas for further studies.
4.1 Myocardial ischemia/reperfusion (I/R) injury (MIRI)
MIRI is widely recognized as a major contributor to cardiac dysfunction and cardiomyocyte death in coronary artery disease (3, 36). A study revealed that MFN2 acts as a key tethering junction at MAMs. In hearts from Mfn2 and Mfn1 KO mice, the loss of MFN2 reduces mitochondrial-ER interactions, attenuates mitochondrial Ca2+ overload, and protects against acute I/R injury by reducing ROS production (79). In addition, Mfn2-KO adult cardiomyocytes were found to be protected against various stimuli that induce cell death, and Mfn2 KO hearts exhibited better recovery following reperfusion injury (80). Similarly, research using a mouse model of MIRI showed that downregulation of MFN2 expression promotes cardiomyocyte proliferation and inhibits apoptosis (81). However, other studies present contrasting findings. In a different application, ischemic preconditioning significantly increased MFN2 expression in skeletal muscle during total knee arthroplasty combined with tourniquet use. This increase partially protected postoperative quadriceps strength by enhancing mitochondrial fusion proteins and preventing tourniquet-induced I/R injury, suggesting that MFN2 upregulation may have protective effects against I/R injury (82). Additionally, the upregulation of MFN2 expression through MiR-93 demonstrated inhibition of myocardial apoptosis in rats with MIRI (83). In another study, the knockdown of Mfn2 using shRNA prevented the fusion of autophagosomes with lysosomes in neonatal cardiomyocytes. Re-expression of Mfn2 restored this fusion, whereas cardiac-specific Mfn2 KO mice, which exhibit abnormal cardiac mitochondria and cellular metabolism, were more susceptible to I/R challenges (84) (Figure 3). This may be related to the course of the disease, as evidenced by the fact that these effects may play an isotropic or diametrically opposed role in different courses of the same disease (85, 86). For example, it has been shown that increasing autophagy during ischemia relieves MIRI and increasing autophagy during reperfusion increases MIRI (87–89). It may also be related to the multiple roles of MFN2, as evidenced by differences in the degree of each role, which ultimately presents a combination of roles, i.e., a balanced result of the various roles. It was suggested, for instance, that physiologic autophagy may play a role in protecting the myocardium from MIRI, whereas excessive autophagy exacerbates MIRI (90–93). The above studies present conflicting results, highlighting ongoing controversy in this area. These discrepancies underscore the need for further research to elucidate the underlying mechanisms, especially in CVD that are comorbid with other diseases. For example, upregulation of AMPK/MFN2-dependent mitochondrial fusion has been shown to reduce MIRI in diabetic hearts (94).
Figure 3
The role of MFN2-mediated MAMs in cardiovascular disease. MFN2, mitofusin 2; MAMs, mitochondria-associated endoplasmic reticulum (ER) membranes.
4.2 Diabetic cardiomyopathy (DCM)
DCM is a distinct subtype of CVD observed in patients with type 1 or type 2 diabetes mellitus. It is characterized by alterations in myocardial structure and function (36). In cardiomyocytes from mice with DCM, increased connectivity between the sarcoplasmic reticulum and mitochondria has been observed, representing an enhanced formation of MAMs (95). A study involving obese diabetic (db/db) mice and lean control (db/+) mice showed excessive mitochondrial fragmentation and significantly reduced MFN2 expression in the hearts of 12-week-old diabetic (db/db) mice (96). Notably, reconstitution of Mfn2 in diabetic hearts inhibited mitochondrial fission and prevented the development of DCM (96). Further research has demonstrated that MFN2 is crucial in high glucose-induced ER stress (ERS) in atrial cardiomyocytes. High glucose levels upregulate ERS, mitochondrial oxidative stress, and the MAMs-enriched proteins GRP75 and MFN2 in atrial myocytes vitro. Silencing Mfn2 prevents mitochondrial dysfunction caused by mitochondrial Ca2+ overload, thereby reducing ERS-induced cardiomyocyte death (97). In HL-1 cells under ERS conditions, silencing Mfn2 reduced Ca2+ transfer from the ER to mitochondria, and electron microscopy confirmed that Mfn2 siRNA significantly disrupted ER-mitochondrial tethering in ER-injured HL-1 cells (97). Additionally, Paeonol was found to stimulate mitochondrial fusion through the PKCε-Stat3-Mfn2 pathway, protecting the heart from doxorubicin-induced injury (39). The pathogenesis of DCM is also believed to be linked to AMPK inactivation. Under energy stress, AMPK activation triggers mitochondrial fission and autophagy, increasing the number of MAMs. During mitochondrial fission, a large proportion of AMPK translocates from the cytoplasm to the MAMs, where it interacts with MFN2. This interaction initiates mitochondrial autophagy, contributing to cardiac protection (36, 52). In contrast, although metformin is widely recommended as a first-line treatment for type 2 diabetes, and its ability to reduce the risk of CVD in these patients remains under investigation, evidence suggests that it may reduce the incidence of myocardial infarction in patients with diabetes. This effect is thought to be mediated through AMPK activation (98) (Figure 3). Since patients with CVD may have other diseases in combination, related comprehensive studies can be added to future research to clarify the prevention and treatment of related diseases.
4.3 Dilated cardiomyopathy
Dilated cardiomyopathy is a CVD in which the walls of one of the heart's chambers become dilated and thin, resulting in the heart's inability to pump blood properly. MFN2 has been implicated in various CVDs, including I/R injury, heart failure, and dilated cardiomyopathy (99, 100). Genetic analyses have linked MFN2 dysfunction to dilated cardiomyopathy (101). Mitochondrial fusion is essential for maintaining normal mitochondrial morphology and the proper respiratory and contractile function of the heart. Conditional ablation of Mfn1 and Mfn2 in the adult heart induces mitochondrial fragmentation, cardiomyocyte dysfunction, and impaired mitochondrial respiration, ultimately leading to rapidly progressive and lethal dilated cardiomyopathy (102). Further studies have shown that the accumulation of morphologically and functionally abnormal mitochondria induces respiratory dysfunction, which leads to dilated cardiomyopathy in Mfn2-KO mouse embryonic fibroblasts and cardiomyocytes, as well as in Parkin-KO Drosophila heart tubes (103) (Figure 3). While these findings indicate a clear association between MFN2 and dilated cardiomyopathy, further research is needed to elucidate the underlying mechanisms fully.
4.4 Pathological myocardial hypertrophy
MAMs may play a critical role in pathological myocardial hypertrophy. A significant reduction in ER-mitochondrial contact has been observed in norepinephrine-induced cardiac hypertrophic cells. This reduction leads to decreased Ca2+ uptake, AKT activation, glucose uptake, and mitochondrial oxygen consumption in response to insulin (104). Earlier studies have shown that Mfn2 expression is downregulated in hypertrophied hearts, with its expression level being linked to both the etiology and time course of hypertrophy (105). Consistent with this, Mfn2-KO mice exhibited moderate cardiac hypertrophy and mild functional deterioration, suggesting mild mitochondrial dysfunction (80). Furthermore, upregulation of MFN2 has been shown to inhibit angiotensin II-induced cardiac hypertrophy, with the inhibition of AKT activation playing a significant role in this process. These findings suggest that MFN2 is a key protein regulating cardiomyocyte hypertrophy (106). Additionally, hypertrophied myocardium exhibits a distinct microRNA (miRNA) expression profile compared to normal myocardium, with miRNA-20 promoting cardiomyocyte hypertrophy by reducing Mfn2 expression (107). In contrast, upregulation of Mfn2 expression ameliorated cardiac hypertrophy induced by angiotensin II (107). The luciferase reporter system has confirmed that Mfn2 is a target gene of miR-195-5p, which negatively regulates Mfn2 expression in H9c2 cells and promotes cardiac hypertrophy (108). Mechanistically, miR-17-5p may also contribute to cardiac hypertrophy by inhibiting Mfn2 expression, activating the PI3K/AKT/mTOR pathway, and inhibiting autophagy (109). Analogously, an opposing study indicated that lncRNA ZNF593-AS inhibits cardiac hypertrophy and myocardial remodeling by upregulating MFN2 expression (110). Similarly, ubiquitin-specific peptidase 2 overexpression mediates deubiquitination to upregulate MFN2, attenuating Ca2+ overload-induced mitochondrial dysfunction and cardiac hypertrophy (111). In contrast, 8 weeks of aerobic exercise and choline intervention inhibited myocardial MFN2 expression, attenuated cardiac hypertrophy, and improved cardiac function in damaged cardiac tissue after aortic constriction (112). However, it has also been shown that moderate exercise cannot cause changes in MFN2 expression in adult spontaneously hypertensive rats (113). Additionally, transient receptor potential vanilloid 1 (TRPV1), a nonselective cation channel, may also be involved in pathological cardiac hypertrophy. One study suggested that TRPV1 activation exacerbates hypoxia/reoxygenation-induced apoptosis in H9c2 cells via Ca2+ overload and mitochondrial dysfunction (114). In contrast, in a phenylephrine-treated model of cardiomyocyte hypertrophy, TRPV1 activation reduced mitochondrial ROS, decreased cardiomyocyte size, and improved mitochondrial function by promoting the formation of MAMs. Notably, interfering with Mfn2 via single-stranded RNA interference or silencing Mfn2 blocks the function of TRPV1 (115). Similarly, disrupting MAM formation via siMfn2 abrogates the protective effects mediated by TRPV1 (115) (Figure 3). These findings suggest that MFN2 may function through its regulated MAMs. Because of the multiple roles of MFN2, further studies are needed to elucidate its specific mechanisms and to determine whether MFN2 acts through the regulation of MAMs or whether the disruption of the structure of MAMs restricts TRPV1 from acting accordingly. This will help to clarify the therapeutic targets for this disease.
4.5 Cardiotoxicity
Sorafenib, an antitumor agent, induces cardiotoxicity, resulting in cardiomyocyte necrosis. The mechanistic basis of this effect may involve sorafenib-induced inactivation of mTOR and the activation of transcription factor EB, which translocates to the nucleus and promotes mitophagy. This process leads to the degradation of MFN2. Further studies have shown that both global and cardiac-specific overexpression of MFN2 can suppress cardiac dysfunction and inhibit cardiomyocyte necrosis. This protective effect occurs through inhibiting the MAM-calmodulin-dependent protein kinase II delta-RIP3/mixed-lineage kinase domain-like protein pathway, which is implicated in sorafenib-induced cardiomyocyte necrosis (116). Targeting MFN2-mediated mitochondrial fusion may offer dual therapeutic benefits in doxorubicin-based chemotherapy by protecting against cardiotoxicity and enhancing its antitumor efficacy through metabolic shifts (117). A similar study revealed that total flavonoids of Selaginella tamariscina (P.Beauv.) Spring (a perennial herb that belongs to the genus Selaginella in the family Selaginellaceae, contains abundant flavonoid bioactive substances) ameliorated doxorubicin-induced cardiotoxicity by attenuating mitochondrial dysfunction and ERS by activating the MFN2/PERK pathway (118). Additionally, MFN2 may play a role in heavy metal-induced cardiotoxicity by modulating MAMs (4). A study involving sheep hearts revealed that exposure to heavy metals such as molybdenum and/or cadmium led to myocardial morphological damage, impaired oxidative function, and a significant decrease in mitochondrial Ca2+ levels. An increase in the distance between MAMs and disruption of the MAM structure accompanied this. Furthermore, lower MAM-related genes (including IP3R, FUNDC1, MFN2, and VDAC1) were observed. The findings suggest that molybdenum and/or cadmium induce cardiotoxicity via the MFN2 pathway, leading to the disorganization of MAMs (119). In contrast, the sirtuin1 agonist resveratrol inhibits cardiomyocyte apoptosis by upregulating MFN2, antagonizing oxidative stress, and alleviating doxorubicin-induced cardiotoxicity (120). Similarly, trophoblast stem cell-derived exosomes have been shown to attenuate doxorubicin-induced cardiotoxicity through anti-apoptotic effects and improve mitochondrial fusion by increasing MFN2 expression (121) (Figure 3). Therefore, Mfn2 may be a key target for the treatment of cardiotoxicity, and methods such as promoting the expression of Mfn2 or targeting the administration of certain Mfn2 protein preparations may be developed in the future to alleviate cardiotoxicity. Thus, Mfn2 is a key target for the treatment of cardiotoxicity, which can be alleviated by promoting MFN2 expression.
4.6 Heart failure
Heart failure has become an increasingly significant public health issue due to the aging global population. Despite substantial progress in understanding the disease, the development of effective therapies for heart failure continues to face several challenges (122). MAMs may provide viable targets for intervention in heart failure treatment (6). Dysfunction of MFN2, a key protein involved in forming and regulating MAMs, has long been associated with CVDs, including heart failure (99, 100). In the failing heart, an accumulation of MFN2 and MFN1 typically occurs in response to proteasome inactivation. Compared to sham-treated animals, cardiac Mfn1, Mfn2, and Dnm1l mRNA levels were significantly lower (123). This suggests that the elevated levels of MFN1 and MFN2 observed in the failing heart may result from impaired proteasome activity, consistent with findings from animal models of heart failure and human cardiac failure (124, 125). Furthermore, Mfn1/Mfn2 double-KO mice develop heart failure during embryonic life, leading to death (102) (Figure 3). While these findings highlight the role of MFN2 in heart failure, relevant reports remain scarce, indicating the need for further research to uncover the specific mechanisms involved. Future studies are especially needed to clarify whether it is the action of MFN2 or MFN1 alone or the result of the combined action of MFN2 and MFN1.
5 Conclusion
MFN2 is a highly conserved GTPase comprising 757 amino acid residues, widely distributed in the outer mitochondrial and ER membranes. MFN2 plays a crucial role in mitochondrial fusion, regulation of mitochondrial homeostasis, formation, modulation of MAMs, modulation of cellular metabolism, and the antiproliferative effects on cells. We have found that MFN2-mediated MAMs may play significant roles in a variety of CVDs, including MIRI, DCM, pathological myocardial hypertrophy, cardiotoxicity, and heart failure. However, due to the diverse range of functions of MFN2, its role in these diseases is not always consistent. Some studies report opposing findings, suggesting that understanding its precise role in these conditions remains unclear. This highlights the need for further research to elucidate the mechanisms by which MFN2 contributes to these diseases.
Statements
Author contributions
YL: Formal analysis, Funding acquisition, Methodology, Project administration, Writing – original draft, Writing – review & editing. LiyC: Formal analysis, Writing – original draft. LL: Writing – review & editing, Formal analysis. ZL: Writing – review & editing, Funding acquisition, Project administration. ZF: Funding acquisition, Project administration, Writing – review & editing. LinC: Writing – review & editing, Conceptualization, Formal analysis, Methodology, Writing – original draft. FP: Conceptualization, Formal analysis, Methodology, Writing – original draft, Writing – review & editing, Funding acquisition, Project administration, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the 2024 Annual “Challenge Board” Research Project at Guangdong University of Education (2024JBGS004), the “National Natural Science Foundation of China” (32360218), the “Guangdong Province Education Science Planning Project” (2024GXJK642), the “Sports Performance Analysis and Training Scientific Innovation Team” (2024WCXTD030), the “Research Initiation Program for Doctoral Talents of Guangdong University of Education” (202304670451), and the “Guangdong University of Education 2024 Research Capacity Enhancement Program for Key Construction Disciplines in Guangdong Province” (2024ZDJS049). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
Dhaun N Webb DJ . Endothelins in cardiovascular biology and therapeutics. Nat Rev Cardiol. (2019) 16:491–502. 10.1038/s41569-019-0176-3
2.
Lv Y Yu Z Zhang P Zhang X Li H Liang T et al The structure and function of FUN14 domain-containing protein 1 and its contribution to cardioprotection by mediating mitophagy. Front Pharmacol. (2024) 15:1389953. 10.3389/fphar.2024.1389953
3.
Lv YH Cheng L Peng FL . Compositions and functions of mitochondria-associated endoplasmic Reticulum membranes and their contribution to cardioprotection by exercise preconditioning. Front Physiol. (2022) 13:910452. 10.3389/fphys.2022.910452
4.
Lu B Chen X Ma Y Gui M Yao L Li J et al So close, yet so far away: the relationship between MAM and cardiac disease. Front Cardiovasc Med. (2024) 11:1353533. 10.3389/fcvm.2024.1353533
5.
Liu Y Huo J-L Ren K Pan S Liu H Zheng Y et al Mitochondria-associated endoplasmic reticulum membrane (MAM): a dark horse for diabetic cardiomyopathy treatment. Cell Death Discov. (2024) 10:148. 10.1038/s41420-024-01918-3
6.
Zhang Y Yao J Zhang M Wang Y Shi X . Mitochondria-associated endoplasmic reticulum membranes (MAMs): possible therapeutic targets in heart failure. Front Cardiovasc Med. (2023) 10:1083935. 10.3389/fcvm.2023.1083935
7.
Sukhorukov VS Voronkova AS Baranich TI Gofman AA Brydun AV Knyazeva LA et al Molecular mechanisms of interactions between mitochondria and the endoplasmic reticulum: a new look at how important cell functions are supported. Mol Biol. (2022) 56:59–71. 10.1134/s0026893322010071
8.
Merle A Jollet M Britto FA Goustard B Bendridi N Rieusset J et al Endurance exercise decreases protein synthesis and ER-mitochondria contacts in mouse skeletal muscle. J Appl Physiol. (2019) 127:1297–306. 10.1152/japplphysiol.00196.2019
9.
Montesinos J Area-Gomez E . Chapter 2 – Isolation of mitochondria-associated ER membranes. In: PonLASchonEA, editors. Methods in Cell Biology. vol. 155. Cambridge, MA: Academic Press (2020). p. 33–44.
10.
Luan Y Luan Y Yuan R-X Feng Q Chen X Yang Y . Structure and function of mitochondria-associated endoplasmic Reticulum membranes (MAMs) and their role in cardiovascular diseases. Oxid Med Cell Longev. (2021) 2021:4578809. 10.1155/2021/4578809
11.
Wang X Wen Y Dong J Cao C Yuan S . Systematic in-depth proteomic analysis of mitochondria-associated endoplasmic reticulum membranes in mouse and human testes. Proteomics. (2018) 18:e1700478. 10.1002/pmic.201700478
12.
Sun Y Ding S . ER-mitochondria contacts and insulin resistance modulation through exercise intervention. Int J Mol Sci. (2020) 21:9587. 10.3390/ijms21249587
13.
Naon D Hernandez-Alvarez MI Shinjo S Wieczor M Ivanova S Martins de Brito O et al Splice variants of mitofusin 2 shape the endoplasmic reticulum and tether it to mitochondria. Science. (2023) 380:1237+. 10.1126/science.adh9351
14.
Abati E Manini A Velardo D Del Bo R Napoli L Rizzo F et al Clinical and genetic features of a cohort of patients with MFN2-related neuropathy. Sci Rep. (2022) 12:6181. 10.1038/s41598-022-10220-0
15.
Dasgupta A Chen K-H Munk RB Sasaki CY Curtis J Longo DL et al Mechanism of activation-induced downregulation of mitofusin 2 in human peripheral blood T cells. J Immunol. (2015) 195:5780–6. 10.4049/jimmunol.1501023
16.
Li Y-J Cao Y-L Feng J-X Qi Y Meng S Yang J-F et al Structural insights of human mitofusin-2 into mitochondrial fusion and CMT2A onset. Nat Commun. (2019) 10:4914. 10.1038/s41467-019-12912-0
17.
de Brito OM Scorrano L . Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. (2008) 456:605–10. 10.1038/nature07534
18.
Cao Y-L Meng S Chen Y Feng J-X Gu D-D Yu B et al MFN1 structures reveal nucleotide-triggered dimerization critical for mitochondrial fusion. Nature. (2017) 542:372+. 10.1038/nature21077
19.
Sloat SR Whitley BN Engelhart EA Hoppins S . Identification of a mitofusin specificity region that confers unique activities to Mfn1 and Mfn2. Mol Biol Cell. (2019) 30:2309–19. 10.1091/mbc.E19-05-0291
20.
Yu F Xu T Wang M Chang W Li P Wang J . Function and regulation of mitofusin 2 in cardiovascular physiology and pathology. Eur J Cell Biol. (2018) 97:474–82. 10.1016/j.ejcb.2018.07.003
21.
Mottie S Riemer J Wideman JG McBride HM . A new mitofusin topology places the redox-regulated C terminus in the mitochondrial intermembrane space. J Cell Biol. (2018) 217:507–15. 10.1083/jcb.201611194
22.
Giacomello M Scorrano L . The INs and OUTs of mitofusins. J Cell Biol. (2018) 217:439–40. 10.1083/jcb.201801042
23.
Heulitt MJ . Structure determines function. J Pediatr Intensive Care. (2013) 2:1–3. 10.3233/pic-13041
24.
Wang K Samudrala R . FSSA: a novel method for identifying functional signatures from structural alignments. Bioinformatics. (2005) 21:2969–77. 10.1093/bioinformatics/bti471
25.
Nag S Szederkenyi K Gorbenko O Tyrrell H Yip CM McQuibban GA . PGAM5 is an MFN2 phosphatase that plays an essential role in the regulation of mitochondrial dynamics. Cell Rep. (2023) 42:112895. 10.1016/j.celrep.2023.112895
26.
Escobar-Henriques M Joaquim M . Mitofusins: disease gatekeepers and hubs in mitochondrial quality control by E3 ligases. Front Physiol. (2019) 10:517. 10.3389/fphys.2019.00517
27.
Hall AR Burke N Dongworth RK Hausenloy DJ . Mitochondrial fusion and fission proteins: novel therapeutic targets for combating cardiovascular disease. Brit J Pharmacol. (2014) 171:1890–906. 10.1111/bph.12516
28.
Hoppins S Edlich F Cleland MM Banerjee S McCaffery JM Youle RJ et al The soluble form of bax regulates mitochondrial fusion via MFN2 homotypic complexes. Mol Cell. (2011) 41:150–60. 10.1016/j.molcel.2010.11.030
29.
Koshiba T Detmer SA Kaiser JT Chen H McCaffery JM Chan DC . Structural basis of mitochondrial tethering by mitofusin complexes. Science. (2004) 305:858–62. 10.1126/science.1099793
30.
Escobar-Henriques M Anton F . Mechanistic perspective of mitochondrial fusion: tubulation vs. Fragmentation. Bba-Mol Cell Res. (2013) 1833:162–75. 10.1016/j.bbamcr.2012.07.016
31.
Colpman P Dasgupta A Archer SL . The role of mitochondrial dynamics and mitotic fission in regulating the cell cycle in cancer and pulmonary arterial hypertension: implications for dynamin-related protein 1 and Mitofusin2 in hyperproliferative diseases. Cells-Basel. (2023) 12:1897. 10.3390/cells12141897
32.
Das R Maity S Das P Kamal IM Chakrabarti S Chakrabarti O . CMT2A-linked MFN2 mutation, T206I promotes mitochondrial hyperfusion and predisposes cells towards mitophagy. Mitochondrion. (2024) 74:101825. 10.1016/j.mito.2023.101825
33.
Zhou J Liu H Zhang T Wang Z Zhang J Lu Y et al MORN4 protects cardiomyocytes against ischemic injury via MFN2-mediated mitochondrial dynamics and mitophagy. Free Radical Bio Med. (2023) 196:156–70. 10.1016/j.freeradbiomed.2023.01.016
34.
Liu Y Yao C Sheng B Zhi S Chen X Ding P et al Inhibition of USP30 promotes mitophagy by regulating ubiquitination of MFN2 by parkin to attenuate early brain injury after SAH. Transl Stroke Res. (2023) 16:448–66. 10.1007/s12975-023-01228-3
35.
Pablo Munoz J Ivanova S Sanchez-Wandelmer J Martinez-Cristobal P Noguera E Sancho A et al Mfn2 modulates the UPR and mitochondrial function via repression of PERK. Embo J. (2013) 32:2348–61. 10.1038/emboj.2013.168
36.
Zhao W-b Sheng R . The correlation between mitochondria-associated endoplasmic reticulum membranes (MAMs) and Ca2+ transport in the pathogenesis of diseases. Acta Pharmacol Sin. (2024):1–21. 10.1038/s41401-024-01359-9
37.
Luo F Fu M Wang T Qi Y Zhong X Li D et al Down-regulation of the mitochondrial fusion protein Opa1/Mfn2 promotes cardiomyocyte hypertrophy in Su5416/hypoxia-induced pulmonary hypertension rats. Arch Biochem Biophys. (2023) 747:109743. 10.1016/j.abb.2023.109743
38.
Li C Liu Q Chang Q Xie M Weng J Wang X et al Role of mitochondrial fusion proteins MFN2 and OPA1 on lung cellular senescence in chronic obstructive pulmonary disease. Resp Res. (2023) 24:319. 10.1186/s12931-023-02634-9
39.
Ding M Shi R Fu F Li M De D Du Y et al Paeonol protects against doxorubicin-induced cardiotoxicity by promoting Mfn2-mediated mitochondrial fusion through activating the PKCε-Stat3 pathway. J Adv Res. (2023) 47:151–62. 10.1016/j.jare.2022.07.002
40.
Zheng M Bai Y Sun X Fu R Liu L Liu M et al Resveratrol reestablishes mitochondrial quality control in myocardial ischemia/reperfusion injury through Sirt1/Sirt3-Mfn2-parkin-PGC-1α pathway. Molecules. (2022) 27:5545. 10.3390/molecules27175545
41.
Wu B Qi B Duan L Chen J . Lidamycin induces mitophagy in pancreatic cancer cells by regulating the expression of Mfn2. Sci Rep. (2024) 14:20713. 10.1038/s41598-024-71377-4
42.
Yuan Y Wu Y He M Jiang X . Astragaloside IV protects against podocyte injury by upregulating mitophagy via Mfn2/Pink1/parkin axis. Curr Mol Med. (2024). 10.2174/0115665240310818240531080353
43.
Kong DZ Sun P Lu Y Yang Y Min DY Zheng SC et al Yi Mai granule improve energy supply of endothelial cells in atherosclerosis via miRNA-125a-5p regulating mitochondrial autophagy through Pink1-Mfn2-parkin pathway. J Ethnopharmacol. (2024) 319:117114. 10.1016/j.jep.2023.117114
44.
Ma Z Liu Z Li X Zhang H Han D Xiong W et al Metformin collaborates with PINK1/Mfn2 overexpression to prevent cardiac injury by improving mitochondrial function. Biology-Basel. (2023) 12:582. 10.3390/biology12040582
45.
Yang L Ao Y Li Y Dai B Li J Duan W et al Morinda officinalis oligosaccharides mitigate depression-like behaviors in hypertension rats by regulating Mfn2-mediated mitophagy. J Neuroinflamm. (2023) 20:31. 10.1186/s12974-023-02715-y
46.
Huang S Zeng Z Chu Y Zhang S Zhou J Hu Z et al Mitigation of lipopolysaccharide-induced intestinal injury in rats by Chimonanthus nitens Oliv. Essential oil via suppression of mitochondrial fusion protein mitofusin 2 (MFN2)-mediated mitochondrial-associated endoplasmic reticulum membranes (MAMs) formation. J Ethnopharmacol. (2025) 337:118856. 10.1016/j.jep.2024.118856
47.
Chen Y Csordas G Jowdy C Schneider TG Csordas N Wang W et al Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca2+ crosstalk. Circ Res. (2012) 111:863+. 10.1161/circresaha.112.266585
48.
Sugiura A Nagashima S Tokuyama T Amo T Matsuki Y Ishido S et al MITOL regulates endoplasmic reticulum-mitochondria contacts via Mitofusin2. Mol Cell. (2013) 51:20–34. 10.1016/j.molcel.2013.04.023
49.
Takeda K Nagashima S Shiiba I Uda A Tokuyama T Ito N et al MITOL prevents ER stress-induced apoptosis by IRE1α ubiquitylation at ER-mitochondria contact sites. Embo J. (2019) 38:e100999. 10.15252/embj.2018100999
50.
McLelland G-L Goiran T Yi W Dorval G Chen CX Lauinger ND et al Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. Elife. (2018) 7:e32866. 10.7554/eLife.32866
51.
Manganelli V Matarrese P Antonioli M Gambardella L Vescovo T Gretzmeier C et al Raft-like lipid microdomains drive autophagy initiation via AMBRA1-ERLIN1 molecular association within MAMs. Autophagy. (2021) 17:2528–48. 10.1080/15548627.2020.1834207
52.
Hu Y Chen H Zhang L Lin X Li X Zhuang H et al The AMPK-MFN2 axis regulates MAM dynamics and autophagy induced by energy stresses. Autophagy. (2021) 17:1142–56. 10.1080/15548627.2020.1749490
53.
Chaput I Kelly M Landherr M Polina I Nieto B Cypress M et al Role of tyrosine phosphorylation of Mfn2 in endoplasmic reticulum-mitochondria coupling. Physiology. (2023) 38. 10.1152/physiol.2023.38.S1.5733327
54.
Chen L Liu B Qin Y Li A Gao M Liu H et al Mitochondrial fusion protein Mfn2 and its role in heart failure. Front Mol Biosci. (2021) 8:681237. 10.3389/fmolb.2021.681237
55.
Cieri D Vicario M Giacomello M Vallese F Filadi R Wagner T et al SPLICS: a split green fluorescent protein-based contact site sensor for narrow and wide heterotypic organelle juxtaposition. Cell Death Differ. (2018) 25:1131–45. 10.1038/s41418-017-0033-z
56.
Harmon M Larkman P Hardingham G Jackson M Skehel P . A bi-fluorescence complementation system to detect associations between the endoplasmic reticulum and mitochondria. Sci Rep. (2017) 7:17467. 10.1038/s41598-017-17278-1
57.
Pich S Bach D Briones P Liesa M Camps M Testar X et al The charcot-marie-tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum Mol Genet. (2005) 14:1405–15. 10.1093/hmg/ddi149
58.
Sebastian D Isabel Hernandez-Alvarez M Segales J Sorianello E Pablo Munoz J Sala D et al Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. P Natl Acad Sci U S A. (2012) 109:5523–8. 10.1073/pnas.1108220109
59.
Li L Li Z Cui N Huang M Hu X Hong D et al Mitofusin2 promotes β cell maturation from mouse embryonic stem cells via Sirt3/Idh2 activation. Stem Cells Int. (2022) 2022:1172795. 10.1155/2022/1172795
60.
Schneeberger M Dietrich MO Sebastian D Imbernon M Castano C Garcia A et al Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell. (2013) 155:172–87. 10.1016/j.cell.2013.09.003
61.
Boutant M Kulkarni SS Joffraud M Ratajczak J Valera-Alberni M Combe R et al Mfn2 is critical for brown adipose tissue thermogenic function. Embo J. (2017) 36:1543–58. 10.15252/embj.201694914
62.
Bassot A Prip-Buus C Alves A Berdeaux O Perrier J Lenoir V et al Loss and gain of function of Grp75 or mitofusin 2 distinctly alter cholesterol metabolism, but all promote triglyceride accumulation in hepatocytes. Bba-Mol Cell Biol L. (2021) 1866:159030. 10.1016/j.bbalip.2021.159030
63.
Sebastian D Sorianello E Segales J Irazoki A Ruiz-Bonilla V Sala D et al Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. Embo J. (2016) 35:1677–93. 10.15252/embj.201593084
64.
Ryan J Dasgupta A Huston J Chen K-H Archer SL . Mitochondrial dynamics in pulmonary arterial hypertension. J Mol Med. (2015) 93:229–42. 10.1007/s00109-015-1263-5
65.
Li T Han J Jia L Hu X Chen L Wang Y . PKM2 coordinates glycolysis with mitochondrial fusion and oxidative phosphorylation. Protein Cell. (2019) 10:583–94. 10.1007/s13238-019-0618-z
66.
Zhou W Chen K-H Cao W Zeng J Liao H Zhao L et al Mutation of the protein kinase A phosphorylation site influences the anti-proliferative activity of mitofusin 2. Atherosclerosis. (2010) 211:216–23. 10.1016/j.atherosclerosis.2010.02.012
67.
Wang Z Wang H Wu Q Chen Y Liu J Liu Y et al GATA2 promotes human vascular smooth muscle cell proliferation via mitofusin2-mediated ras/raf/MEK/ERK signaling pathway. Int J Cardiol. (2022) 346:62–70. 10.1016/j.ijcard.2021.11.012
68.
Tang Y Jia Y Fan L Liu H Zhou Y Wang M et al MFN2 prevents neointimal hyperplasia in vein grafts via destabilizing PFK1. Circ Res. (2022) 130:E26–43. 10.1161/circresaha.122.320846
69.
Shi Y Luo P Yi C Xie J Zhang Q . Effects of Mitofusin2 on astrocytes proliferation in vitro induced by scratch injury. Neurosci Lett. (2020) 729:134969. 10.1016/j.neulet.2020.134969
70.
Lynch S Boyett JE Smith MR Giordano-Mooga S . Sex hormone regulation of proteins modulating mitochondrial metabolism, dynamics and inter-organellar cross talk in cardiovascular disease. Front Cell Dev Biol. (2021) 8:610516. 10.3389/fcell.2020.610516
71.
Zhang Y Zhao X-Y Xie W-J Zhang Y . The role of mitochondria-associated endoplasmic reticulum membranes in age-related cardiovascular diseases. Shengli Xuebao. (2023) 75:799–816. 10.13294/j.aps.2023.0081
72.
Liu H Liu X Zhuang H Fan H Zhu D Xu Y et al Mitochondrial contact sites in inflammation-induced cardiovascular disease. Front Cell Dev Biol. (2020) 8:692. 10.3389/fcell.2020.00692
73.
Yu H-Y Guo Y-H Gao W . Mitochondrial fusion protein Mfn2 and cardiovascular diseases. Sheng Li Ke Xue Jin Zhan. (2010) 41:11–6.
74.
Ji L Han H Shan X Zhao P Chen H Zhang C et al Ginsenoside Rb1 ameliorates lipotoxicity-induced myocardial injury in diabetes mellitus by regulating Mfn2. Eur J Pharmacol. (2024) 974:176609. 10.1016/j.ejphar.2024.176609
75.
Yang A Guo L Zhang Y Qiao C Wang Y Li J et al MFN2-mediated mitochondrial fusion facilitates acute hypobaric hypoxia-induced cardiac dysfunction by increasing glucose catabolism and ROS production. Bba-Gen Subjects. (2023) 1867:130413. 10.1016/j.bbagen.2023.130413
76.
Xiong Y Leng Y Tian H Deng X Li W Li W et al Decreased MFN2 activates the cGAS-STING pathway in diabetic myocardial ischaemia-reperfusion by triggering the release of mitochondrial DNA. Cell Commun Signal. (2023) 21:192. 10.1186/s12964-023-01216-y
77.
Chen Y Li S Yin M Li Y Chen C Zhang J et al Isorhapontigenin attenuates cardiac microvascular injury in diabetes via the inhibition of mitochondria-associated ferroptosis through PRDX2-MFN2-ACSL4 pathways. Diabetes. (2023) 72:389–404. 10.2337/db22-0553
78.
Burtscher M Burtscher J . MFN2: shaping mitochondria and cardiac adaptations to hypoxia. Acta Physiol. (2023) 239:e14026. 10.1111/apha.14026
79.
Hall AR Burke N Dongworth RK Kalkhoran SB Dyson A Vicencio JM et al Hearts deficient in both Mfn1 and Mfn2 are protected against acute myocardial infarction. Cell Death Dis. (2016) 7:e2238. 10.1038/cddis.2016.139
80.
Papanicolaou KN Khairallah RJ Ngoh GA Chikando A Luptak I O’Shea KM et al Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol. (2011) 31:1309–28. 10.1128/mcb.00911-10
81.
Qin L Yang W Wang Y-X Wang Z-J Li C-C Li M et al MicroRNA-497 promotes proliferation and inhibits apoptosis of cardiomyocytes through the downregulation of Mfn2 in a mouse model of myocardial ischemia-reperfusion injury. Biomed Pharmacother. (2018) 105:103–14. 10.1016/j.biopha.2018.04.181
82.
Leurcharusmee P Sawaddiruk P Punjasawadwong Y Sugandhavesa N Klunklin K Tongprasert S et al Ischemic preconditioning upregulates Mitofusin2 and preserves muscle strength in tourniquet-induced ischemia/reperfusion. J Orthop Translat. (2022) 35:113–21. 10.1016/j.jot.2022.09.012
83.
Zhu ZD Chem YF Shi HM . MiR-93 inhibits myocardial apoptosis in rats with myocardial ischemia-reperfusion injury through regulating Mfn2 expression. J Biol Reg Homeos Ag. (2020) 34:909–16.
84.
Zhao T Huang X Han L Wang X Cheng H Zhao Y et al Central role of mitofusin 2 in autophagosome-lysosome fusion in cardiomyocytes. J Biol Chem. (2012) 287:23615–25. 10.1074/jbc.M112.379164
85.
Yang X Wu H Liu D Zhou G Zhang D Yang Q et al The link between ferroptosis and autophagy in myocardial ischemia/reperfusion injury: new directions for therapy. J Cardiovasc Transl. (2025) 18:408–23. 10.1007/s12265-025-10590-6
86.
Khan AA Sabartrasso S . Autophagy in myocardial ischemia and ischemia/reperfusion. Cardiovasc Pathol. (2025) 74:107691. 10.1016/j.carpath.2024.107691
87.
Kanamori H Takemura G Goto K Maruyama R Ono K Nagao K et al Autophagy limits acute myocardial infarction induced by permanent coronary artery occlusion. Am J Physiol-Heart C. (2011) 300:H2261–71. 10.1152/ajpheart.01056.2010
88.
Chen G Phan V Luo X Cao DJ . The mechanistic target of rapamycin complex 1 critically regulates the function of mononuclear phagocytes and promotes cardiac remodeling in acute ischemia. J Mol Cell Cardiol. (2021) 159:62–79. 10.1016/j.yjmcc.2021.06.004
89.
Nah J Zhai P Huang C-Y Fernandez AF Mareedu S Levine B et al Upregulation of rubicon promotes autosis during myocardial ischemia/reperfusion injury. J Clin Invest. (2020) 130:2978–91. 10.1172/jci132366
90.
Wen L Cheng X Fan Q Chen Z Luo Z Xu T et al TanshinoneIIA inhibits excessive autophagy and protects myocardium against ischemia/reperfusion injury via 14-3-3η/akt/Beclin1 pathway. Eur J Pharmacol. (2023) 954:175865. 10.1016/j.ejphar.2023.175865
91.
Wang L Wang J Cretoiu D Li G Xiao J . Exercise-mediated regulation of autophagy in the cardiovascular system. J Sport Health Sci. (2020) 9:203–10. 10.1016/j.jshs.2019.10.001
92.
Gao C Wang R Li B Guo Y Yin T Xia Y et al TXNIP/Redd1 signalling and excessive autophagy: a novel mechanism of myocardial ischaemia/reperfusion injury in mice. Cardiovasc Res. (2020) 116:645–57. 10.1093/cvr/cvz152
93.
Su F Shi M Zhang J Zheng Q Zhang D Zhang W et al Simvastatin protects heart from pressure overload injury by inhibiting excessive autophagy. Int J Med Sci. (2018) 15:1508–16. 10.7150/ijms.28106
94.
Yu H Hong X Liu L Wu Y Xie X Fang G et al Cordycepin decreases ischemia/reperfusion injury in diabetic hearts via upregulating AMPK/Mfn2-dependent mitochondrial fusion. Front Pharmacol. (2021) 12:754005. 10.3389/fphar.2021.754005
95.
Wu S Lu Q Ding Y Wu Y Qiu Y Wang P et al Hyperglycemia-driven inhibition of AMP-activated protein kinase α2 induces diabetic cardiomyopathy by promoting mitochondria-associated endoplasmic Reticulum membranes in vivo. Circulation. (2019) 139:1913–36. 10.1161/circulationaha.118.033552
96.
Hu L Ding M Tang D Gao E Li C Wang K et al Targeting mitochondrial dynamics by regulating Mfn2 for therapeutic intervention in diabetic cardiomyopathy. Theranostics. (2019) 9:3687–706. 10.7150/thno.33684
97.
Yuan M Gong M Zhang Z Meng L Tse G Zhao Y et al Hyperglycemia induces endoplasmic Reticulum stress in atrial cardiomyocytes, and mitofusin-2 downregulation prevents mitochondrial dysfunction and subsequent cell death. Oxid Med Cell Longev. (2020) 2020:6569728. 10.1155/2020/6569728
98.
Griffin SJ Leaver JK Irving GJ . Impact of metformin on cardiovascular disease: a meta-analysis of randomised trials among people with type 2 diabetes. Diabetologia. (2017) 60:1620–9. 10.1007/s00125-017-4337-9
99.
Yeh J-N Yue Y Chu Y-C Huang C-R Yang C-C Chiang JY et al Entresto protected the cardiomyocytes and preserved heart function in cardiorenal syndrome rat fed with high-protein diet through regulating the oxidative stress and Mfn2-mediated mitochondrial functional integrity. Biomed Pharmacother. (2021) 144:112244. 10.1016/j.biopha.2021.112244
100.
Zungu M Schisler J Willis MS . All the little pieces - regulation of mitochondrial fusion and fission by ubiquitin and small ubiquitin-like modifier and their potential relevance in the heart. Circ J. (2011) 75:2513–21. 10.1253/circj.CJ-11-0967
101.
Svagusa T Sikiric S Milavic M Sepac A Seiwerth S Milicic D et al Heart failure in patients is associated with downregulation of mitochondrial quality control genes. Eur J Clin Invest. (2023) 53:e14054. 10.1111/eci.14054
102.
Chen Y Liu Y Dorn GW II . Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res. (2011) 109:1327–U1336. 10.1161/circresaha.111.258723
103.
Chen Y Dorn GW II . PINK1-phosphorylated mitofusin 2 is a parkin receptor for culling damaged mitochondria. Science. (2013) 340:471–5. 10.1126/science.1231031
104.
Gutierrez T Parra V Troncoso R Pennanen C Contreras-Ferrat A Vasquez-Trincado C et al Alteration in mitochondrial Ca2+ uptake disrupts insulin signaling in hypertrophic cardiomyocytes. Cell Commun Signal. (2014) 12:68. 10.1186/s12964-014-0068-4
105.
Fang L Moore X-L Gao X-M Dart AM Lim YL Du X-J . Down-regulation of mitofusin-2 expression in cardiac hypertrophy in vitro and in vivo. Life Sci. (2007) 80:2154–60. 10.1016/j.lfs.2007.04.003
106.
Yu H Guo Y Mi L Wang X Li L Gao W . Mitofusin 2 inhibits angiotensin II-induced myocardial hypertrophy. J Cardiovasc Pharm T. (2011) 16:205–11. 10.1177/1074248410385683
107.
Sun D Li C Liu J Wang Z Liu Y Luo C et al Expression profile of microRNAs in hypertrophic cardiomyopathy and effects of microRNA-20 in inducing cardiomyocyte hypertrophy through regulating gene MFN2. DNA Cell Biol. (2019) 38:796–807. 10.1089/dna.2019.4731
108.
Wang L Qin D Shi H Zhang Y Li H Han Q . MiR-195-5p promotes cardiomyocyte hypertrophy by targeting MFN2 and FBXW7. Biomed Res Int. (2019) 2019:1580982. 10.1155/2019/1580982
109.
Xu X Su Y-l Shi J-y Lu Q Chen C . MicroRNA-17-5p promotes cardiac hypertrophy by targeting Mfn2 to inhibit autophagy. Cardiovasc Toxicol. (2021) 21:759–71. 10.1007/s12012-021-09667-w
110.
Nie X Fan J Wang Y Xie R Chen C Li H et al lncRNA ZNF593-AS inhibits cardiac hypertrophy and myocardial remodeling by upregulating Mfn2 expression. Front Med. (2024) 18:484–98. 10.1007/s11684-023-1036-4
111.
Fu D Luo J Wu Y Zhang L Li L Chen H et al Angiotensin II-induced calcium overload affects mitochondrial functions in cardiac hypertrophy by targeting the USP2/MFN2 axis. Mol Cell Endocrinol. (2023) 571:111938. 10.1016/j.mce.2023.111938
112.
Ma M Chen W Hua Y Jia H Song Y Wang Y . Aerobic exercise ameliorates cardiac hypertrophy by regulating mitochondrial quality control and endoplasmic reticulum stress through M2AChR. J Cell Physiol. (2021) 236:6581–96. 10.1002/jcp.30342
113.
Quiroga C Mancilla G Oyarzun I Tapia A Caballero M Gabrielli LA et al Moderate exercise in spontaneously hypertensive rats is unable to activate the expression of genes linked to mitochondrial dynamics and biogenesis in cardiomyocytes. Front Endocrinol. (2020) 11:546. 10.3389/fendo.2020.00546
114.
Sun Z Han J Zhao W Zhang Y Wang S Ye L et al TRPV1 activation exacerbates hypoxia/reoxygenation-induced apoptosis in H9C2 cells via calcium overload and mitochondrial dysfunction. Int J Mol Sci. (2014) 15:18362–80. 10.3390/ijms151018362
115.
Wang Y Li X Xu X Qu X Yang Y . Transient receptor potential vanilloid type 1 protects against pressure overload-induced cardiac hypertrophy by promoting mitochondria-associated endoplasmic Reticulum membranes. J Cardiovasc Pharm. (2022) 80:430–41. 10.1097/fjc.0000000000001301
116.
Song Z Song H Liu D Yan B Wang D Zhang Y et al Overexpression of MFN2 alleviates sorafenib-induced cardiomyocyte necroptosis via the MAM-CaMKIIδ pathway in vitro and in vivo. Theranostics. (2022) 12:1267–85. 10.7150/thno.65716
117.
Ding M Shi R Cheng S Li M De D Liu C et al Mfn2-mediated mitochondrial fusion alleviates doxorubicin-induced cardiotoxicity with enhancing its anticancer activity through metabolic switch. Redox Biol. (2022) 52:102311. 10.1016/j.redox.2022.102311
118.
Gao L Yuan P Wei Y Fu Y Hou Y Li P et al Total flavonoids of Selaginella tamariscina (P.beauv.) spring ameliorates doxorubicin-induced cardiotoxicity by modulating mitochondrial dysfunction and endoplasmic reticulum stress via activating MFN2/PERK. Phytomedicine. (2022) 100:154065. 10.1016/j.phymed.2022.154065
119.
Peng C Yang S Yang F Xiong Z Liu Q Liao S et al Crosstalk between Mfn2-mediated mitochondria associated membranes disorder and autophagy induced by molybdenum and cadmium in sheep heart. Food Chem Toxicol. (2023) 174:113660. 10.1016/j.fct.2023.113660
120.
Zhang Q Zhang Y Xie B Liu D Wang Y Zhou Z et al Resveratrol activation of SIRT1/MFN2 can improve mitochondria function, alleviating doxorubicin-induced myocardial injury. Cancer Innovation. (2023) 2:253–64. 10.1002/cai2.64
121.
Duan J Liu X Shen S Tan X Wang Y Wang L et al Trophoblast stem-cell-derived exosomes alleviate cardiotoxicity of doxorubicin via improving Mfn2-mediated mitochondrial fusion. Cardiovasc Toxicol. (2023) 23:23–31. 10.1007/s12012-022-09774-2
122.
Lou Q Janardhan A Efimov IR . Remodeling of calcium handling in human heart failure. In: IslamMS, editor. Calcium Signaling. vol. 740. Dordrecht: Springer (2012). p. 1145–74.
123.
Campos JC Queliconi BB Bozi LHM Bechara LRG Dourado PMM Andres AM et al Exercise reestablishes autophagic flux and mitochondrial quality control in heart failure. Autophagy. (2017) 13:1304–17. 10.1080/15548627.2017.1325062
124.
Campos JC Queliconi BB Dourado PMM Cunha TF Zambelli VO Bechara LRG et al Exercise training restores cardiac protein quality control in heart failure. PLoS One. (2012) 7:e52764. 10.1371/journal.pone.0052764
125.
Ferreira JCB Boer BN Grinberg M Brum PC Mochly-Rosen D . Protein quality control disruption by PKCβII in heart failure; rescue by the selective PKCβII inhibitor, βIIV5-3. PLoS One. (2012) 7:e33175. 10.1371/journal.pone.0033175
Summary
Keywords
mitochondria-associated endoplasmic reticulum membranes, mitofusin 2, cardioprotection, cardiovascular disease, mitochondria, endoplasmic reticulum
Citation
Lv Y, Chen L, Li L, Liao Z, Fang Z, Cheng L and Peng F (2025) The structure and function of mitofusin 2 and its role in cardiovascular disease through mediating mitochondria-associated endoplasmic reticulum membranes. Front. Cardiovasc. Med. 12:1535401. doi: 10.3389/fcvm.2025.1535401
Received
27 November 2024
Accepted
13 May 2025
Published
30 May 2025
Volume
12 - 2025
Edited by
Yi Zhao, First Affiliated Hospital of Zhengzhou University, China
Reviewed by
Satvik Mareedu, The State University of New Jersey, United States
Yang Yang, First Affiliated Hospital of Zhengzhou University, China
Updates
Copyright
© 2025 Lv, Chen, Li, Liao, Fang, Cheng and Peng.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
* Correspondence: Lin Cheng naxilin2022@stu.gxnu.edu.cn Fenglin Peng pengflin@mailbox.gxnu.edu.cn
† Present Address: Lin Cheng, College of Physical Education and Health, Guangdong Polytechnic Normal University, Guangzhou, China
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.