 Shoshi Sphitzen1Mordechai Golomb2Mohammad Mowaswes2Refael Bitzur3Smadar Horowitz Cederboim1
Shoshi Sphitzen1Mordechai Golomb2Mohammad Mowaswes2Refael Bitzur3Smadar Horowitz Cederboim1 Ronen R. Leker4
Ronen R. Leker4 Marc Gotkine4Itai Chovers5Daniel Schurr1Eran Leitersdorf1
Marc Gotkine4Itai Chovers5Daniel Schurr1Eran Leitersdorf1 Ronen Durst1,2*
Ronen Durst1,2*
- 1Lipid Clinic and Center for Cardiovascular Precision Medicine, Hadassah Hebrew University Medical Center, Jerusalem, Israel
- 2Cardiology Department, Hadassah Hebrew University Medical Center, Jerusalem, Israel
- 3The Bert W. Strassburger Lipid Center, Sheba Medical Center, Ramat Gan, Israel
- 4Department of Neurology, Hadassah Hebrew University Medical Center, Jerusalem, Israel
- 5Ophthalmology Department, Hadassah Hebrew University Medical Center, Jerusalem, Israel
Introduction: High-Density Lipoprotein Cholesterol (HDL-C) plays a pivotal role in cardiovascular health, acting as a key component in lipid transport and atheroprotection. While low HDL-C levels in the general population are often the result of multifactorial causes, extremely low HDL-C levels (<20 mg/dl) are rare and may be attributed to underlying genetic defects. Mutations in genes such as LCAT, APOA1, and ABCA1—although exceedingly rare—have been linked to profound alterations in lipid metabolism, often resulting in significant morbidity and increased cardiovascular risk.
Methods: In this study, we used exome sequencing on patients with very low HDL-C.
Results: We identified three patients with pathogenic mutations associated with genetic low HDL-C syndrome, including ABCA1 [NM_005502.4(ABCA1):c.4175 + 1G > T, chr:9 91757308° C > A, rs375247413], LCAT [NM_000229.2(LCAT):c.349G > A p.Ala117Thr, rs28940886], and APOA1 [NM_000039.3(APOA1):c.388A > T, p.Lys130*].
Discussion: Each case presented a unique spectrum of clinical phenotypes, systemic complications, and biochemical abnormalities, illustrating the diverse impact of these genetic mutations. We provide a detailed analysis of the clinical and biochemical profiles of these patients, highlighting key aspects of disease manifestation and progression. This report underscores the importance of recognizing and characterizing rare genetic causes of low HDL-C, which may have profound implications for patient care and risk stratification.
Introduction
High-Density Lipoprotein (HDL-C) cholesterol plays a vital role in promoting cardiovascular health through its central function in lipoprotein metabolism. Severe HDL-C deficiency (<20 mg/dl) in the absence of secondary causes is extremely rare. Patients with such a marked deficiency are prone to early onset atherosclerotic disease and other systemic complications (1). HDL-C's primary function is to remove excess cholesterol from peripheral tissues through reverse cholesterol transport (RCT), delivering it to the liver, organs with high cholesterol needs, or exchanging it with apoB particles like low-density lipoprotein (LDL-C) for disposal. Cholesterol is transported to the liver and steroidogenic tissues via HDL-C's binding to the scavenger receptor B1 (SR-B1) and interacting with ATP-dependent transmembrane transporters, ATP-Binding Cassette Transporter A1 (ABCA1) and ATP-binding cassette sub-family G member 1 (ABCG1), which are highly expressed in tissues such as macrophages, adipose tissue, and the liver (2). HDL-C's core component, apolipoprotein A1 (apoA1), is synthesized in the liver and intestine, and through lipidation, nascent HDL-Cs mature into α-HDL-C, which undergoes constant remodelling. Genetic mutations in genes such as apoA1, LCAT, and ABCA1 can lead to severe HDL-C deficiency (3, 4). Partial HDL-C deficiency, or hypoalphalipoproteinemia, defined as plasma HDL-C levels below the 10th percentile, is a major risk factor for coronary heart disease and stroke.
Located in a tertiary referral center, our lipid clinic provides genetic counseling for lipid disorders for Israel. Of the cases referred to us for genetic evaluation we describe three cases of genetically determined HDL-C deficiency in the Israeli population and discuss the importance of identifying such patients. Our routine laboratory tests are done early morning after an overnight fast using the Roche COBASTM system. LDL-C values are calculated.
Case reports
Case 1: tangier disease in an Ashkenazi Jewish family
Tangier disease is an extremely rare genetic disorder caused by mutations in the ABCA1 gene. It is characterized by severe plasma deficiency or absence of HDL-C and apolipoprotein A-I (apoA-I), leading to the accumulation of cholesteryl esters in various tissues throughout the body. A classic finding on physical examination is the presence of orange-colored tonsils. The disease results in reduced cholesterol efflux from peripheral cells (5–7).
A 77-year-old woman of Ashkenazi Jewish ancestry with a history of consanguinity was diagnosed with Tangier disease in 1997. She presented with syringomyelia-like syndrome (8) and abnormally low levels of HDL-C, LDL-C, and total cholesterol.
Genetic testing through exome sequencing identified a homozygous splice site sequence variation in the ABCA1 gene [NM_005502.4(ABCA1):c.4175 + 1G > T, chr:9 91757308° C > A, rs375247413, minor allele frequency 0.0000134] Subsequently, a CDNA library was prepared from lymphoblasts of the propositus. PCR flanking the splice variation demonstrated the skipping of the exon downstream of the sequence variation, confirming its pathogenicity (Figure 1A). Her sister was homozygote for the same sequence variation (Figure 1A). Over the past 10 years, the patient's lipid profile has been consistently abnormal (Table 1).
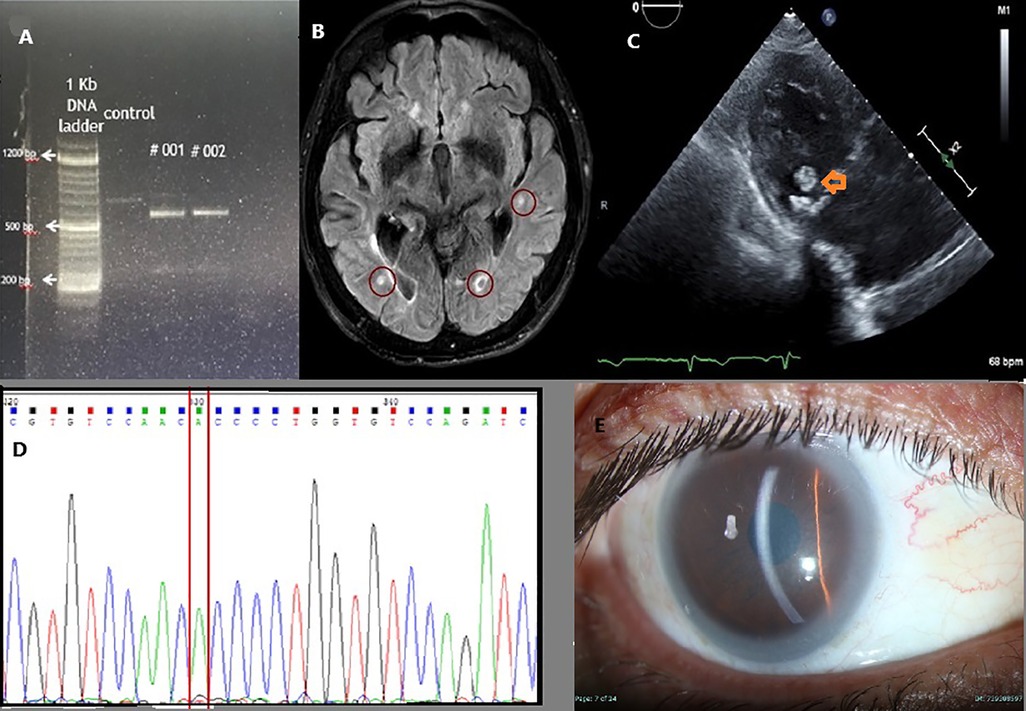
Figure 1. (A) Polymerase chain reaction (PCR) products of cDNA extracted from the patient lymphoblasts, spanning the adjacent exons of rs375247413 in the ABCA1 gene are presented. Lane 1 is control, and the two Tangier Diseased sisters in Lanes #1 & #2. Lower molecular weight fragments in Tangier Disease patients confirm a splice site at the ABCA1 gene, resulting in shorter fragments due to exon skipping in these. (B) MRI FLAIR imaging showing multiple bilateral emboli (red circles). (C) Echocardiography with a para-sternal long axis view of the heart. A mobile mass swinging on the posterior leaflet of the mitral valve (orange arrow) suggests a probable cholesterol vegetation. (D) Sequencing of the LCAT gene demonstrating homozygous splice site error at position 349 substituting G to A. (E) Slit lamp picture showing corneal opacities and lipemic arch in the patient with apoA1 deficiency.

Table 1. Cholesterol panel of the three patients.
Notably, during her clinical follow-up, a dense, mobile mass measuring 11 × 9 mm was identified on the mitral valve, suggestive of cholesterol accumulation (Figure 1C). This finding correlated with recurrent strokes, emphasizing the cardiovascular implications of Tangier disease. Her sibling had a milder form of the disease. Although she experienced peripheral neuropathy, it was not severe enough to require wheelchair assistance. She had a cerebrovascular accident (CVA), but it was not debilitating. Worth mentioning that she underwent a tonsillectomy during childhood. Interestingly, during a colonoscopy, her mucosa appeared orange, resembling the description of orange tonsils seen in Tangier disease.
The risk of cerebrovascular disease in Tangier's patients was described previously (Figure 1B) (9). Of note, her sister had a CVA episode later on during follow up. A prior report of early onset valve disease as well as our report may suggest valve involvement with Tangier's disease (10).
Case 2: lecithin-cholesterol acyltransferase (LCAT) deficiency
LCAT deficiency, also known as Fish Eye Disease (FED), is a rare recessive genetic disorder with only a few cases reported worldwide (11–14). The condition arises due to mutations in the LCAT gene, resulting in a lack of lecithin-cholesterol acyltransferase, an enzyme critical for HDL-C formation (11). Clinical manifestations include corneal opacifications (“boiled fish eye”) and dyslipidemia characterized by low HDL-C levels.
A 64-year-old male with a known history of dyslipidemia presented with new-onset visual impairment. Clinical examination revealed corneal opacifications, which raised the suspicion of FED. Genetic testing identified a missense variation [NM_000229.2(LCAT):c.349G > A, p.Ala117Thr, rs28940886, minor allele frequency 0.0000057] (Figure 1D) in the LCAT gene. This change, causing alanine to threonine amino acid substitution in exon 3, was previously documented as pathogenic (15) due to its association with reduced enzyme activity. A detailed lipid profile of this patient is presented in Table 1.
Case 3: apoA1 deficiency
apoA1 deficiency is a rare disorder of lipoprotein metabolism characterized by the complete absence of apolipoprotein A1, leading to extremely low levels of plasma HDL-C cholesterol. This condition can manifest with significant clinical features, including corneal opacities, xanthomas, and premature coronary heart disease (CHD).
We report a case involving a 30-year-old male proband of Muslim ancestry, who is the only child of first-cousin parents, though he has healthy half-siblings from both sides. He has three healthy daughters aged 17 to 2. The patient experienced a myocardial infarction at the age of 33, which prompted further investigation revealing corneal lipid deposits compatible with systemic hyperlipidemia (as shown in Figure 1E) and undetectable plasma HDL-C cholesterol levels. Genetic analysis confirmed that he was homozygous for the novel apoA1 [NM_000039.3(APOA1):c.388A > T, p.Lys130*, novel mutation] single nucleotide change, resulting in the premature termination of apolipoprotein A1 translation on exon 4. This change is novel, not reported before. It is likely pathogenic because of the early termination codon. Over a 10-year follow-up period, he underwent five coronary revascularization procedures, underscoring the severe cardiovascular consequences associated with apoA1 deficiency and highlighting the urgent need for awareness and management strategies for affected individuals.
Discussion
HDL-C cholesterol facilitates reverse cholesterol transport from peripheral tissues to the liver, playing a crucial role in cardiovascular health. Genetic factors significantly influence HDL-C cholesterol levels, affecting the body's ability to maintain this essential biological function. Cholesterol ester transfer protein (CETP), LCAT, and ABCA1 are critical genes involved in regulating HDL-C levels (2). Several genetic mutations have been identified that contribute to low HDL-C cholesterol levels, each associated with distinct clinical phenotypes and consequences. Alves et al. (16) presented cases of rare dyslipidemias and in cases with reduced HDL-C levels, they defined mutation in either the ABCA1, apoA1, and LCAT genes.
Similarly, we present three cases of genetically determined HDL-C deficiency associated with pathogenic DNA sequence variation in the ABCA1, LCAT, and apoA1 genes. The diverse mutations, along with their varying vascular and extravascular phenotypes, highlight the importance of modern genetic testing in determining the underlying genetic cause. Early-onset atherogenic disease was a common feature across all three cases, while the extravascular manifestations differed depending on the specific mutated gene. Although the phenotype may provide clues to the genetic cause of the disease, genotyping is crucial in most cases -especially those with predominantly vascular phenotypes -for accurately identifying the cause of low HDL-C and predicting potential future organ system involvement. In Tangier disease, clinical manifestations may include a highly atypical neuropathy, mimicking syringomyelia or leprosy (8), and orange discoloration of lymphatic tissues (6). Notably, we describe the first novel splice site error in the ABCA1 gene within an Ashkenazi Jewish population, where a tendency toward consanguinity suggests that other undiagnosed cases of Tangier disease may exist (17). Additionally, the presence of cholesterol deposits on the mitral valve and their association with stroke further delineate the systemic consequences of cholesterol dysregulation. The cases of LCAT and apoA1 deficiencies demonstrate the diverse phenotypic spectrum of HDL-C-related metabolic disorders, highlighting how early genetic screening can guide clinical management and follow-up strategies. A significant manifestation in these patients is corneal opacification, which serves as a critical diagnostic feature (13). The CETP gene encodes a protein that transfers cholesteryl esters and triglycerides between lipoproteins. LCAT is responsible for the esterification of free cholesterol on HDL-C particles, a key step in HDL-C maturation. ABCA1 mediates the efflux of cholesterol and phospholipids to apolipoprotein AI, forming nascent HDL-C particles. The role of ABCA1 actually extends beyond lipid efflux, including plasma membrane remodeling and apoAI binding (18, 19).
Clinical management of individuals with genetically low HDL-C cholesterol levels requires a nuanced approach. Lifestyle modifications remain fundamental but have negligible effects on HDL-C levels. Similarly, reducing traditional risk factors, such as lowering LDL cholesterol and physical activity, has a very limited impact on HDL-C metabolism. Unfortunately, pharmacological interventions designed to raise HDL-C levels have yielded negative results in large clinical trials with CETP inhibitors and niacin (20, 21). Trials with HDL-C mimetics in patients with genetically determined very low HDL-C have also failed to show clinical benefit, despite significant increases in cholesterol efflux among treated patients (22). Thus, the current state of the art is early diagnosis of HDL-C deficiency syndrome patients and targeting modifiable risk factors. Recent advances in genetic research focus on gene therapy and CRISPR technologies. For example, researchers have used AAV vector to deliver the human ApoA-I gene in mice subjected to cardiac pressure overload. Several beneficial cardiovascular outcomes were demonstrated including reduced septal wall thickness, improved myocardial capillary density, and reduced interstitial cardiac fibrosis (23). Such technologies may, in the future, strengthen our ability to treat genetic HDL-C deficiency; however, these disorders currently do not have targeted treatment.
Summary
This report shares our experience in a tertiary referral centre in identifying and treating genetically determined very low HDL-C syndrome. We described three patients with mutations in CETP, LCAT, and ABCA1, demonstrating the clinical presentation and risk for cardiovascular disease. There remains an unmet need for effective treatments for this condition, as targeted therapies have thus far failed to show clinical benefit.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/snp/, rs375247413.
Ethics statement
The studies involving humans were approved by Hadassah Medical Center IRB, Jerusalem Israel. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
SS: Formal analysis, Writing – original draft, Writing – review & editing. MG: Data curation, Formal analysis, Writing – review & editing. MM: Investigation, Writing – review & editing. RB: Writing – review & editing. SH: Investigation, Writing – review & editing. RL: Investigation, Writing – original draft. MG: Investigation, Writing – review & editing. IC: Investigation, Supervision, Visualization, Writing – review & editing. DS: Investigation, Writing – review & editing. EL: Supervision, Writing – review & editing. RD: Formal analysis, Funding acquisition, Project administration, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. RD is supported by the BSF 2017265 grant.
Acknowledgments
This research paper is dedicated to the memory of the late Dr. Rafael Bitzur, a cherished friend, esteemed physician, and accomplished researcher, who unfortunately passed away during the preparation of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Schaefer EJ, Anthanont P, Diffenderfer MR, Polisecki E, Asztalos BF. Diagnosis and treatment of high density lipoprotein deficiency. Prog Cardiovasc Dis. (2016) 59(2):97–106. doi: 10.1016/j.pcad.2016.08.006
2. von Eckardstein A, Nordestgaard BG, Remaley AT, Catapano AL. High-density lipoprotein revisited: biological functions and clinical relevance. Eur Heart J. (2023) 44(16):1394–407. doi: 10.1093/eurheartj/ehac605
3. Jomard A, Osto E. High density lipoproteins: metabolism, function, and therapeutic potential. Front Cardiovasc Med. (2020) 7:39. doi: 10.3389/fcvm.2020.00039
4. Kiss RS, Kavaslar N, Okuhira K, Freeman MW, Walter S, Milne RW, et al. Genetic etiology of isolated low HDL syndrome: incidence and heterogeneity of efflux defects. Arterioscler Thromb Vasc Biol. (2007) 27(5):1139–45. doi: 10.1161/ATVBAHA.106.137646
5. Puntoni M, Sbrana F, Bigazzi F, Sampietro T. Tangier disease: epidemiology, pathophysiology, and management. Am J Cardiovasc Drugs. (2012) 12(5):303–11. doi: 10.1007/BF03261839
6. Hooper AJ, Hegele RA, Burnett JR. Tangier disease: update for 2020. Curr Opin Lipidol. (2020) 31(2):80–4. doi: 10.1097/MOL.0000000000000669
7. Muratsu J, Koseki M, Masuda D, Yasuga Y, Tomoyama S, Ataka K, et al. Accelerated atherogenicity in tangier disease. J Atheroscler Thromb. (2018) 25(10):1076–85. doi: 10.5551/jat.43257
8. Gibbels E, Schaefer HE, Runne U, Schröder JM, Haupt WF, Assmann G. Severe polyneuropathy in tangier disease mimicking syringomyelia or leprosy. Clinical, biochemical, electrophysiological, and morphological evaluation, including electron microscopy of nerve, muscle, and skin biopsies. J Neurol. (1985) 232(5):283–94. doi: 10.1007/BF00313867
9. Liang Z, Li W, Yang S, Liu Z, Sun X, Gao X, et al. Tangier disease may cause early onset of atherosclerotic cerebral infarction: a case report. Medicine (Baltimore). (2018) 97(39):e12472. doi: 10.1097/MD.0000000000012472
10. Serfaty-Lacrosniere C, Civeira F, Lanzberg A, Isaia P, Berg J, Janus ED, et al. Homozygous tangier disease and cardiovascular disease. Atherosclerosis. (1994) 107(1):85–98. doi: 10.1016/0021-9150(94)90144-9
11. Pavanello C, Calabresi L. Genetic, biochemical, and clinical features of LCAT deficiency: update for 2020. Curr Opin Lipidol. (2020) 31(4):232–7. doi: 10.1097/MOL.0000000000000697
12. Calabresi L, Pisciotta L, Costantin A, Frigerio I, Eberini I, Alessandrini P, et al. The molecular basis of lecithin: cholesterol acyltransferase deficiency syndromes: a comprehensive study of molecular and biochemical findings in 13 unrelated Italian families. Arterioscler Thromb Vasc Biol. (2005) 25(9):1972–8. doi: 10.1161/01.ATV.0000175751.30616.13
13. Carlson LA. Fish eye disease: a new familial condition with massive corneal opacities and dyslipoproteinaemia. Eur J Clin Invest. (1982) 12(1):41–53. doi: 10.1111/j.1365-2362.1982.tb00938.x
14. Savel J, Lafitte M, Pucheu Y, Pradeau V, Tabarin A, Couffinhal T. Very low levels of HDL cholesterol and atherosclerosis, a variable relationship–a review of LCAT deficiency. Vasc Health Risk Manag. (2012) 8:357–61. doi: 10.2147/VHRM.S29985
15. Funke H, von Eckardstein A, Pritchard PH, Hornby AE, Wiebusch H, Motti C, et al. Genetic and phenotypic heterogeneity in familial lecithin: cholesterol acyltransferase (LCAT) deficiency. Six newly identified defective alleles further contribute to the structural heterogeneity in this disease. J Clin Invest. (1993) 91(2):677–83. doi: 10.1172/JCI116248
16. Alves AC, Miranda B, Moldovan O, Santo RE, Gouveia Silva R, Soares Cardoso S, et al. Rare primary dyslipidaemias associated with low LDL and HDL cholesterol values in Portugal. Front Genet. (2023) 13:1088040. doi: 10.3389/fgene.2022.1088040
17. Durst R, Colombo R, Shpitzen S, Avi LB, Friedlander Y, Wexler R, et al. Recent origin and spread of a common Lithuanian mutation, G197del LDLR, causing familial hypercholesterolemia: positive selection is not always necessary to account for disease incidence among Ashkenazi Jews. Am J Hum Genet. (2001) 68(5):1172–88. doi: 10.1086/320123
18. Qian H, Zhao X, Cao P, Lei J, Yan N, Gong X. Structure of the human lipid exporter ABCA1. Cell. (2017) 169(7):1228–39.e10. doi: 10.1016/j.cell.2017.05.020
19. Wang S, Smith JD. ABCA1 and nascent HDL biogenesis. Biofactors. (2014) 40(6):547–54. doi: 10.1002/biof.1187
20. Taheri H, Filion KB, Windle SB, Reynier P, Eisenberg MJ. Cholesteryl ester transfer protein inhibitors and cardiovascular outcomes: a systematic review and meta-analysis of randomized controlled trials. Cardiology. (2020) 145(4):236–50. doi: 10.1159/000505365
21. Rashid S. Lower LDL is better—can this be achieved with CETP inhibition therapy? Expert Rev Cardiovasc Ther. (2020) 18(1):1–5. doi: 10.1080/14779072.2020.1715797
22. Zheng KH, Kaiser Y, van Olden CC, Santos RD, Dasseux JL, Genest J, et al. No benefit of HDL mimetic CER-001 on carotid atherosclerosis in patients with genetically determined very low HDL levels. Atherosclerosis. (2020) 311:13–9. doi: 10.1016/j.atherosclerosis.2020.08.004
23. Amin R, Muthuramu I, Aboumsallem JP, Mishra M, Jacobs F, De Geest B. Selective HDL-raising human apo A-I gene therapy counteracts cardiac hypertrophy, reduces myocardial fibrosis, and improves cardiac function in mice with chronic pressure overload. Int J Mol Sci. (2017) 18(9):2012. doi: 10.3390/ijms18092012
Keywords: HDL-C, tangies, LCAT, ABCA1, apoA1
Citation: Sphitzen S, Golomb M, Mowaswes M, Bitzur R, Horowitz Cederboim S, Leker RR, Gotkine M, Chovers I, Schurr D, Leitersdorf E and Durst R (2025) Missing in action: the genetic mysteries of extremely low HDL cholesterol. Front. Cardiovasc. Med. 12:1553259. doi: 10.3389/fcvm.2025.1553259
Received: 30 December 2024; Accepted: 2 May 2025;
Published: 22 May 2025.
Edited by:
Yanqiao Zhang, University of Arizona, United StatesReviewed by:
Xiaoyue Pan, New York University, United StatesAna Catarina Alves, National Health Institute Doutor Ricardo Jorge (INSA), Portugal
Copyright: © 2025 Sphitzen, Golomb, Mowaswes, Bitzur, Horowitz Cederboim, Leker, Gotkine, Chovers, Schurr, Leitersdorf and Durst. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ronen Durst, ZHVyc3RAaGFkYXNzYWgub3JnLmls