 Yuxuan Li
Yuxuan Li Ying Bai
Ying Bai Wujiao Wang
Wujiao Wang Zhaotian Ma3
Zhaotian Ma3 Dong Li
Dong Li Sinai Li
Sinai Li Jialin Jin
Jialin Jin- 1Department of Cardiology, Dongzhimen Hospital, Beijing University of Chinese Medicine, Beijing, China
- 2Department of Traditional Chinese Medicine, Peking Union Medical College Hospital, Beijing, China
- 3College of Pharmacy, Jining Medical University, Jining, China
- 4Department of Cardiology, Dongfang Hospital, Beijing University of Chinese Medicine, Beijing, China
- 5Beijing Hospital of Traditional Chinese Medicine, Capital Medical University, Beijing Institute of Traditional Chinese Medicine, Beijing, China
- 6China Science and Technology Development Center for Chinese Medicine, Beijing, China
Background: Patients with heart failure (HF) have a poor prognosis and continue to pose a global threat to human health. Consequently, it is crucial to employ bioinformatic approaches to analyze functional alterations within the transcriptome. This analysis should be conducted in conjunction with transcriptome sequencing data from a large sample of clinical myocardial tissue, in order to identify the core pathogenic mechanisms in heart failure myocardial tissue.
Method: Transcriptome data from HF patient myocardial biopsies underwent Robust Rank Aggregation (RRA) to identify differentially expressed genes (DEGs). These DEGs were intersected with key genes identified via Weighted Gene Co-expression Network Analysis (WGCNA) in HF. Functional enrichment analysis was performed on the DEGs. Selected key genes were experimentally validated using RT-qPCR in hypertrophic cardiomyocyte models. Single-cell data dimensionality reduction, clustering, and visualization were achieved using Principal component analysis (PCA) and uniform manifold approximation and projection (UMAP). Cell types were annotated with SingleR and CellMarker, and single-cell functional enrichment was performed using the “irGSEA” R package.
Results: RRA of transcriptome data from five studies identified 102 DEGs. Functional enrichment analyses (GO, KEGG, GSEA) revealed associated functional alterations. WGCNA highlighted a key module enriched for energy metabolism-related genes, with the mitochondrial matrix and inner membrane identified as their primary subcellular locations. Integrating RRA-derived DEGs with WGCNA key module genes yielded 14 crucial genes, validated experimentally in a hypertrophic cardiomyocyte model. Analysis of single-cell RNA-seq data identified cold shock domain containing C2 (CSDC2) and Single-pass membrane and coiled-coil domain-containing protein 4 (SMCO4) as cardiomyocyte-specific genes within this set. Subpopulations of cardiomyocytes with high or low expression of SMCO4 and CSDC2 showed strong associations with alterations in fatty acid metabolism, adipogenesis, and oxidative phosphorylation pathways.
Conclusion: Integrated transcriptomic analysis identified 12 key genes linked to HF, which were validated in a hypertrophy model. Single-cell data showed SMCO4 and CSDC2 are specifically expressed in cardiomyocytes and regulate fatty acid metabolism. This suggests SMCO4 and CSDC2 contribute to HF by altering fatty acid metabolism in heart cells, revealing new disease mechanisms.
1 Introduction
Heart failure(HF) poses a significant threat to human health on a global scale. Although important advances have been made in the treatment of HF in recent years, breakthroughs in pharmacotherapy have primarily been in diuretic and vasodilatory treatments that reduce systemic circulation load, thereby providing comprehensive benefits for patients. The search for new therapeutic targets to further reduce patient mortality remains a hot topic of research.
Basic research has revealed a significant correlation between mitochondrial function and pathological hypertrophy of cardiomyocytes (1, 2). In hypertrophied cardiomyocytes, increased mitochondrial fission and activated mitophagy are observed (3), with mice exhibiting severe cardiac dysfunction and structural alterations (4). Cardiomyocytes require mitochondria to generate substantial ATP for their enormous energy demands during contraction and relaxation. Fatty acids and glucose serve as the heart's primary substrates, each possessing distinct energy-yielding advantages. Fatty acids constitute the dominant cardiac fuel, with fatty acid oxidation accounting for 40%–60% of myocardial ATP production (5). While healthy hearts maintain flexible switching between fatty acid and glucose metabolism to sustain oxidative phosphorylation and ATP generation (6), HF triggers metabolic reprogramming that shifts substrate preference toward glycolysis. This transition from fatty acid to glucose metabolism proves detrimental since glycolysis yields far less ATP than oxidative phosphorylation, ultimately causing energy deficiency (7). Consequently, investigating HF pathogenesis through the lens of mitochondrial function and metabolic changes—particularly in fatty acid metabolism—holds significant clinical relevance for identifying therapeutic targets.
HF originates from pathological factors that alter multiple genes, involving gene mutations and complex pathophysiological processes (8). By employing bioinformatics techniques to analyze transcriptomic data, it is possible to perform massive computations to parse and predict gene changes, thereby revealing complex pathogenic mechanisms. This is of significant importance for exploring therapeutic targets for HF.
2 Materials and methods
2.1 Data source and processing
The sample sequencing datasets were selected according to the following criteria: Studies were retrieved from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) using “HF” as the MeSH term, with the organism filter set to Homo sapiens and the entry type restricted to series. The study types were limited to expression profiling by array and expression profiling by high-throughput sequencing. The tissue attribute was specified as left ventricular tissue. Through the screening process illustrated in the flow diagram (Supplementary Figure S1), six specific datasets (GSE57338, GSE42955, GSE52601, GSE21610, GSE76701, and GSE145154) were selected for inclusion in our study (Table 1) (9–14). For detailed clinical information of the GEO datasets, see Supplementary Table S1. Subsequently, the gene expression profiles and corresponding clinical information were downloaded for further analysis. Probe data from different platforms were preprocessed using the R packages tidyverse, affy, and annotationDbi for gene symbol conversion, removal of duplicate values, and handling of missing values. The limma package was utilized for normalization and batch effect correction. Additional batch effects were mitigated using the FactoMineR and factoextra packages. For single-cell transcriptome sequencing, the Seurat package in R was employed for data importation, quality control, filtering, normalization, selection of cell type-specific features, and extraction of cardiomyocyte expression profiles. Data quality control was based on gene counts per cell being >200 and <2,500, along with a mitochondrial gene percentage <20%. Subsequently, the “vst” method was applied in the FindVariableFeatures function to select the top 2,000 highly variable genes. Principal component analysis (PCA) and data clustering analysis were performed using uniform manifold approximation and projection (UMAP) via the RunPCA, FindClusters, and RunUMAP functions. Additionally, we utilized Single R V1.4.1 and CellMarker 2.0 for cell type annotation (15). Finally, single-cell functional enrichment analysis was conducted using the R package “irGSEA”. We obtained fatty acid metabolism-related genes from MSigDB (https://www.gsea-msigdb.org/gsea/msigdb/), which contains hallmark gene sets. A total of 158 genes were identified for further analysis.
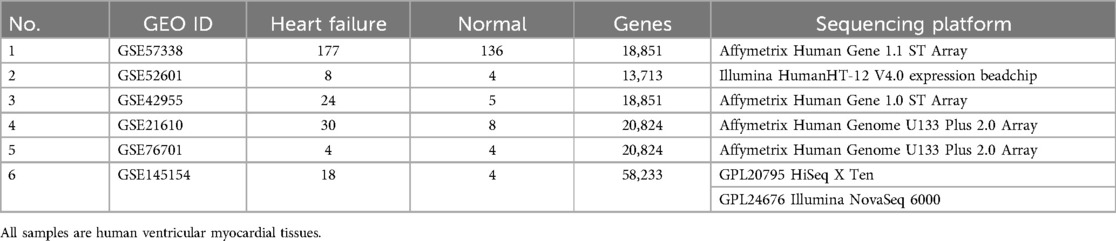
Table 1. GEO dataset basic information.
2.2 Identifying differential genes
After obtaining the gene expression matrix of the dataset in a common format, the Bioconductor package and the limma package were used to perform Bayesian multiple testing correction methods to calculate the differential changes between clinical grouping data; the threshold for significant differences was set at P value < 0.05 & |LogFC| > 0.3.
As a statistical algorithm for integrating prioritized gene lists from multiple datasets, Robust Rank Aggregation (RRA) identifies genes that are consistently ranked better than expected under the null hypothesis of random input lists. It assigns significance scores via order statistics to compare observed ranks with random expectations (16). Distinct from other algorithms, RRA is parameter-free, robust to noise/outliers, handles partial rankings, and efficiently computes statistically rigorous significance scores—unlike average rank (noise-sensitive) or the Stuart method (computationally expensive with non-P-value scores).
To minimize cross-study dependence, our study selected five GEO datasets derived from independent experiments and distinct cohorts. Each dataset underwent independent processing and normalization prior to gene list ranking, ensuring inter-dataset comparability.Subsequently, the “RobustRankAggreg” package was employed to integrate and analyze these ranked gene lists. This method internally validates ranking consistency using order statistics under the null hypothesis of random ordering, thereby ensuring statistical rigor.
2.3 Weighted gene co-expression network analysis
The WGCNA is an R package for constructing and analyzing weighted correlation networks, particularly focused on gene co-expression networks. It identifies clusters (modules) of highly correlated genes, summarizes modules using eigengenes or intramodular hub genes, relates modules to external sample traits (e.g., disease status), and quantifies gene-module relationships via fuzzy module membership measures (17). After excluding outlier samples, we clustered to obtain different gene modules. Subsequently, we used the pickSoftThreshold function in R to select a soft threshold (β). Based on the β value, we calculated the adjacency matrix and then used the TOMsimilarity function to obtain the topological overlap matrix. Dynamic tree cutting cluster analysis was applied to acquire gene modules with similar expression patterns. Functional enrichment was conducted on key modules, followed by intersection analysis with RRA differential genes. The intersecting genes are considered to be key differential genes in HF.
2.4 Gene set enrichment analysis
GSEA focuses on the collective analysis of genes, ranking all genes based on their differential expression fold changes. It then compares these rankings to existing gene function databases, aligning known gene sets to the entire gene ranking to determine the trend of changes in these gene sets under disease conditions. GSEA can be implemented using the clusterProfiler package functions in R. The specific process involves formatting the data, converting gene symbols to entrezid format, ranking genes by fold change, downloading the GSEA database GMT file, and analyzing and plotting the results.
2.5 Gene functional enrichment analysis
Gene functional enrichment can reveal the changes in biological functions underlying bioinformatics differences. To evaluate the biological processes (BP), molecular functions (MF), cellular components (CC), and signaling pathways associated with the target gene expression profiles obtained from various analysis methods, GO and KEGG functional enrichments are performed (18, 19). This can be achieved using the enrichGO and enrichKEGG functions from the clusterProfiler package, along with the org.Hs.eg.db and AnnotationDbi packages in R. The underlying mathematical principle involves hypergeometric distribution/Fisher's exact test. The false discovery rate (FDR) is controlled using the Benjamini-Hochberg procedure, with a selection criterion of FDR < 0.05. The relevant plotting is carried out using the ggplot2 package in R.
2.6 RT-qPCR
We employed the Trizol method to extract total RNA, and the collected RNA was placed on ice and then transferred to a spectrophotometer (Nanodrop 2,000) to assess concentration and quality. Subsequently, reverse transcription was performed to synthesize cDNA, and quantitative real-time PCR (qRT-PCR) was conducted using a fluorescence-based PCR instrument (Roche LightCycler480) to detect mRNA expression. The PCR reaction conditions were as follows: set at 95°C for 15 min for 1 cycle, and 95°C for 10 s, 60°C for 32 s for 40 cycles; to calculate the relative gene expression levels in each sample, we utilized the cycle threshold method (2−ΔΔCt). GAPDH was used as the internal control gene. Detailed information on the primer sequences can be found in the Table 2.
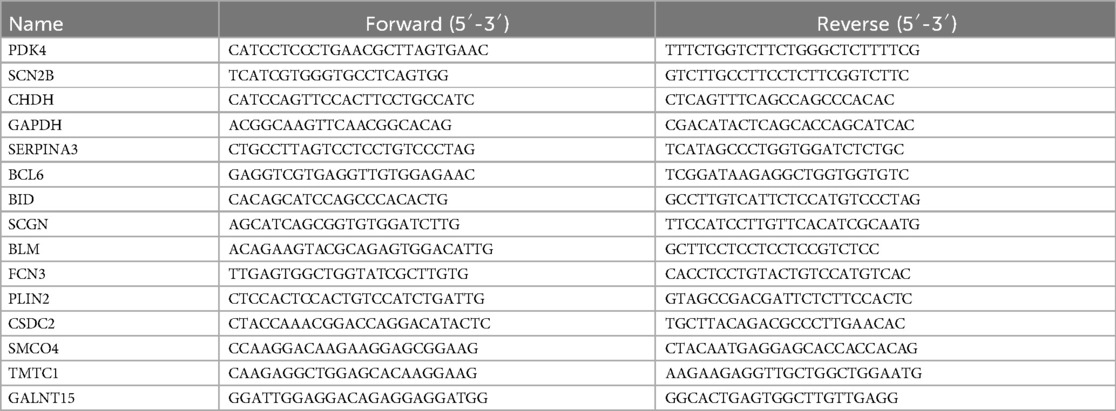
Table 2. Primer sequence table.
2.7 Neonatal rat primary cardiomyocyte hypertrophy model
Neonatal rat primary cardiomyocytes (NRCMs) were isolated from the hearts of 1- to 3-day-old male Sprague-Dawley rats. The rats were provided by Beijing Vital River Laboratory Animal Technology Co., Ltd. (SCXK Beijing 2021-0011). The entire research adhered to the principles of laboratory animal science and was approved by the Animal Ethics Committee of the Basic Research Institute, China Academy of Chinese Medical Sciences, with the ethical approval number (ERCCACMS 21-2406-02). The NRCMs were placed in a CO2 incubator to differentially adhere and enrich cardiomyocytes. After 1.5 h, the supernatant was collected to obtain a primary cardiomyocyte suspension, and the cell concentration was adjusted to 5 × 105/ml. The cells were then plated into culture plates or flasks as required, and incubated in the incubator under the same conditions for further culture. After 24 h of culture, the cells were used for experiments. A hypertrophy model of cardiomyocytes was prepared by stimulating the cells with angiotensin II (1 μM) (20).
2.8 Statistical analysis
All statistical analysis was performed using R (version4.2.3) and associated R packages. A significance level of P < 0.05 was used for all analyses to indicate statistical significance.
3 Results
3.1 Identification of key differential genes and functional enrichment analysis
Individual differential analyses were conducted on the GSE57338, GSE42955, GSE52601, GSE21610, and GSE76701 datasets (Figures 1A–E). Subsequently, using RRA with |LogFC| > 0.3 as the screening criterion, 913 differential genes were obtained. Since the joint analysis of multiple datasets increases its robustness, these genes should be included in subsequent analyses and are referred to as RRA trend genes. When using P value <0.05 & |LogFC| > 0.3 as the screening criteria, a total of 102 differential genes (DEGs) were obtained, consisting of 8 upregulated genes and 94 downregulated genes. These genes are referred to as RRA differential genes, and the heatmap displayed the genes with the top differential magnitudes (Figure 1F). The threshold of |LogFC| > 0.3 was selected to capture more subtle yet biologically meaningful transcriptional differences in myocardial tissue, particularly considering that mitochondrial or metabolic genes often exhibit moderate fold changes—a practice that is permissible in numerous studies (21–23). A more stringent threshold (e.g., |LogFC| > 1) would likely miss relevant genes with small but consistent changes across datasets. The p-value < 0.05 cutoff is standard and ensures statistical significance.
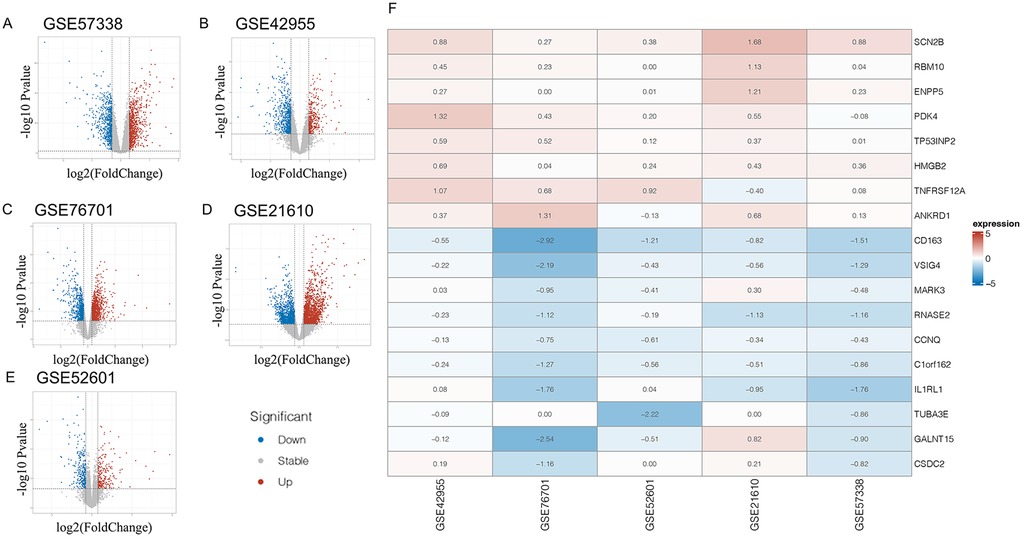
Figure 1. Panels (A–E) display volcano plots of differentially expressed genes (DEGs) across five independent heart failure transcriptomic datasets. Panel (F) shows a heatmap of the top 10 upregulated and downregulated DEGs identified through Robust Rank Aggregation (RRA) analysis, which integrates ranked gene lists from multiple studies.
To thoroughly understand the changes in biological functions associated with RRA differential genes, we conducted GO and KEGG enrichment analyses (Figures 2A,B). The GO functional enrichment analysis revealed that RRA differential genes were enriched in 2 genes involved in the biological processes of arachidonic acid metabolite production involved in inflammatory response and leukotriene production involved in inflammatory response.
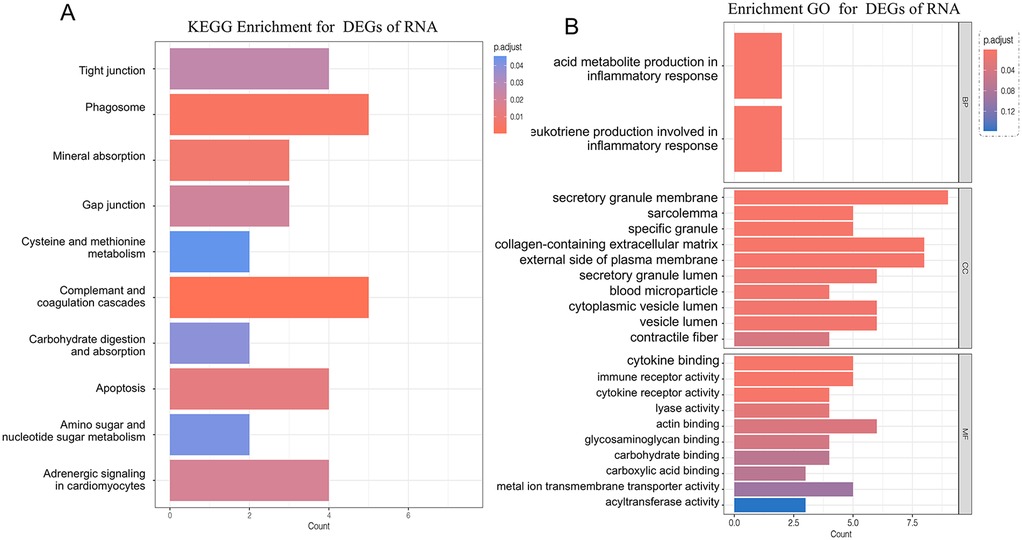
Figure 2. Panel (A) shows the KEGG pathway enrichment analysis of DEGs identified through RRA. Panel (B) displays the gene ontology (GO) functional enrichment analysis of RRA-derived DEGs, categorized into biological processes, molecular functions, and cellular components.
In terms of cellular components, enrichment was observed in secretory granule membrane, collagen-containing extracellular matrix, external side of the plasma membrane, cytoplasmic vesicle lumen, secretory granule lumen, vesicle lumen, sarcolemma, specific granule, blood microparticle, and contractile fiber locations.
In molecular function, RRA differential genes were primarily enriched in actin binding, cytokine binding, immune receptor activity, metal ion transmembrane transporter activity, carbohydrate binding, cytokine receptor activity, glycosaminoglycan binding, lyase activity, acyltransferase activity, and carboxylic acid binding.
KEGG analysis showed that genes were involved in the complement and coagulation cascades, phagosome pathway, mineral absorption pathway, apoptosis pathway, adrenergic signaling in cardiomyocytes pathway, gap junction pathway, tight junction pathway, carbohydrate digestion and absorption pathway, amino sugar and nucleotide sugar metabolism pathway, and cysteine and methionine metabolism pathway.
3.2 Gene set enrichment analysis
Since the functional enrichment analysis of differential genes did not show a significant trend and covered a wide range of cellular life activities, a GSEA was subsequently conducted to thoroughly examine the RRA trend genes. Among the RRA trend genes (913 genes), not all gene changes were statistically significant. However, considering that the joint analysis of the five datasets increased the robustness of these 913 genes, to reduce the loss of data due to statistical rules, GSEA analysis was used to reflect the genes with common regulatory trends and their functional changes across the five datasets.
Among the RRA trend genes, the downregulated biological functions included the Proteasome, P53 signaling pathway, Phagosome, Ferroptosis, Glycerophospholipid metabolism, Biosynthesis of nucleotide sugars, and Apoptosis—multiple species (Figures 3A–G). The upregulated biological functions included Th1 and Th2 cell differentiation, Renin secretion, Wnt signaling pathway, Regulation of lipolysis in adipocytes, Vascular smooth muscle contraction, cGMP−PKG signaling pathway, and Hippo signaling pathway (Figures 3H–O).
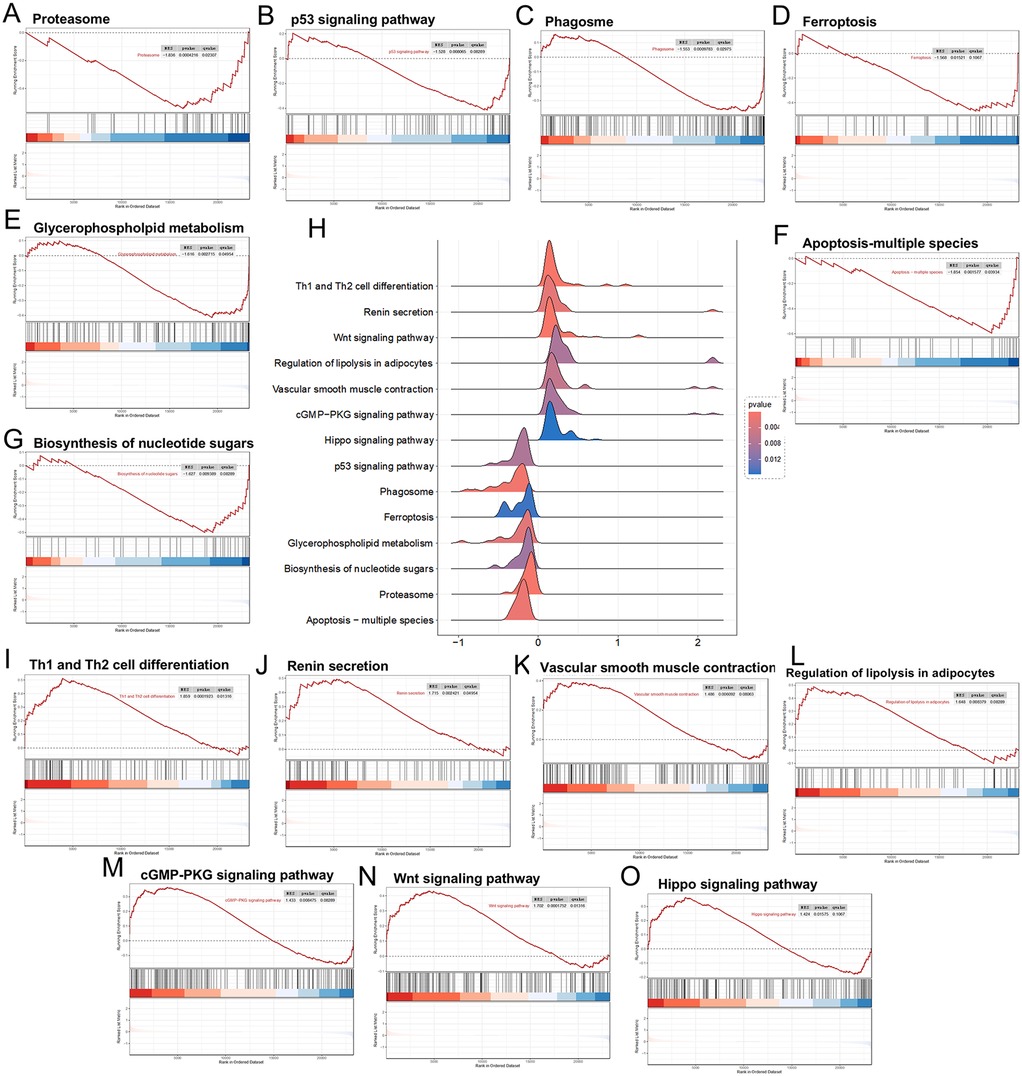
Figure 3. Panels (A–G), downregulated differential genes in the GSEA analysis; (H) landscape plot of GSEA results; (I–O) upregulated differential genes in the GSEA analysis.
In summary, the GSEA results still covered a wide range of cellular life activities, and the aspects involved were functionally consistent with the pathological changes in myocardial tissues of HF, but no obvious trend was observed.
3.3 Construction and identification of key modules in the WGCNA network for HF
When constructing a WGCNA network, it is necessary for the data to conform as closely as possible to a scale-free distribution. After performing power calculations on the five datasets, it was determined that the GSE76701 dataset had the distribution closest to a scale-free distribution (R2 = 0.84) (Figures 4A,B). Dynamic tree cutting cluster analysis was applied to obtain gene modules with similar expression patterns (Figure 4C), with different colors used to distinguish between modules. A total of 22 gene modules were obtained after dynamic cutting and merging of similar modules, and specific module information can be found in Table 3.
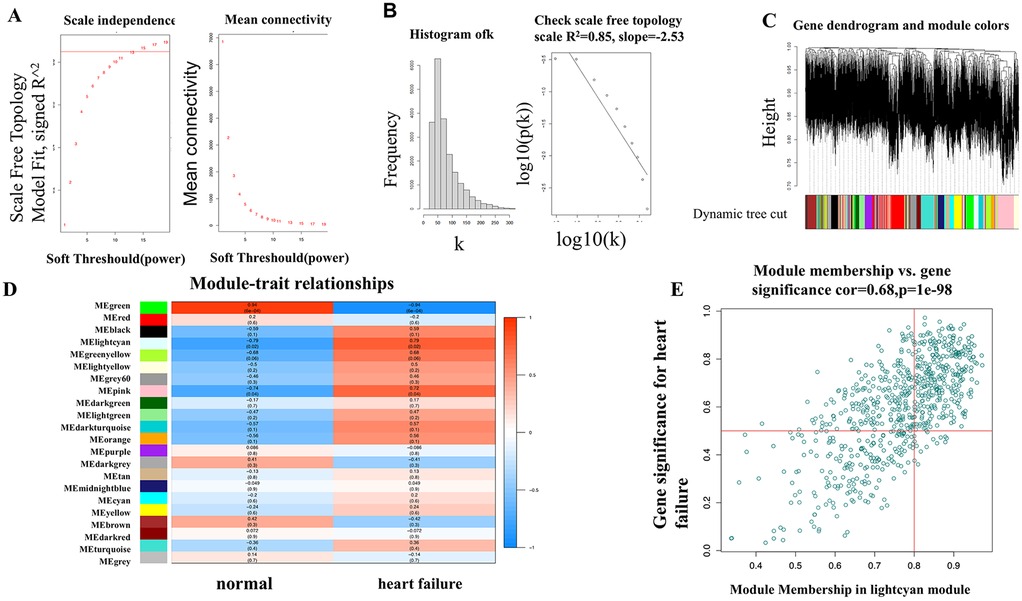
Figure 4. (A,B) Selection of the soft threshold in WGCNA analysis; (C) modular partitioning of the overall expression matrix for the GSE76701 dataset; (D) association analysis between gene modules and heart failure phenotypes for the GSE76701 dataset; (E) Scatter plot of the association between genes in the lightcyan module and heart failure phenotypes.
Table 3. The number of genes in each module of the GSE76701 dataset.
We visualized the correlation between each gene module and HF phenotypes (Figure 4D). The results showed that the gene module with the highest correlation with the HF disease phenotype was the lightcyan module (correlation coefficient 0.79, P = 0.02), which contains 722 genes. We presented the correlation between these genes and the HF phenotype using a scatter plot. It can be observed that as the gene expression in the lightcyan module increases, the correlation between the gene dots and the HF phenotype significantly increases in a positively correlated linear relationship (Figure 4E).
The lightcyan module genes are the most highly associated gene modules with HF in the WGCNA analysis. Functional analysis of this module can uncover key pathogenic mechanisms of HF. Following GO and KEGG analyses, the results indicate a certain propensity towards energy metabolism. The GO analysis is primarily enriched in biological functions such as energy derivation by oxidation of organic compounds, fatty acid metabolic process, mitochondrial gene expression, long-chain fatty acid transport, and energy reserve metabolic process, and in terms of cellular components, it highlights the mitochondrial matrix and mitochondrial intermembrane space as the main locations where key genes are primarily localized (Figure 5A). The KEGG analysis also enriches in functional pathways related to energy cycle utilization, such as the mTOR signaling pathway, phagosome signaling pathway, and lysosome signaling pathway (Figure 5B).
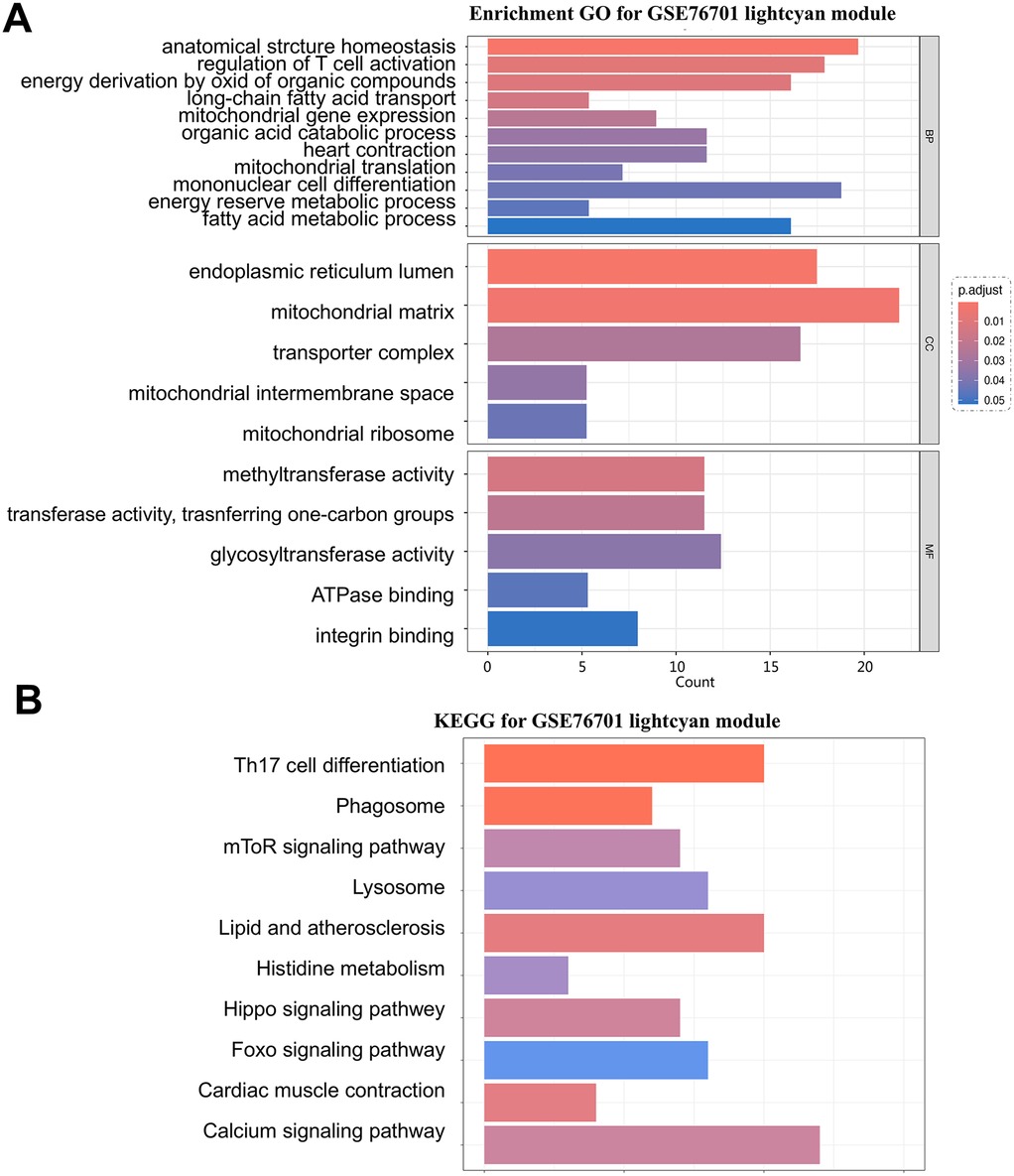
Figure 5. (A) GO analysis of the lightcyan module in the GSE76701 dataset; (B) representative results of KEGG analysis for the key module in the GSE76701 dataset.
3.4 Intersection analysis of RRA and WGCNA differential genes and validation by RT-qPCR
The intersection analysis of RRA differential genes and WGCNA module analysis uncovered pathogenic genes highly associated with HF from two perspectives. The intersection of RRA differential genes with key module genes from WGCNA, followed by validation of the expression of the intersecting genes using the AngII-induced primary rat cardiomyocyte hypertrophy model, is of significant importance. A total of 14 genes were obtained after the intersection (Figure 6A) (Table 4).
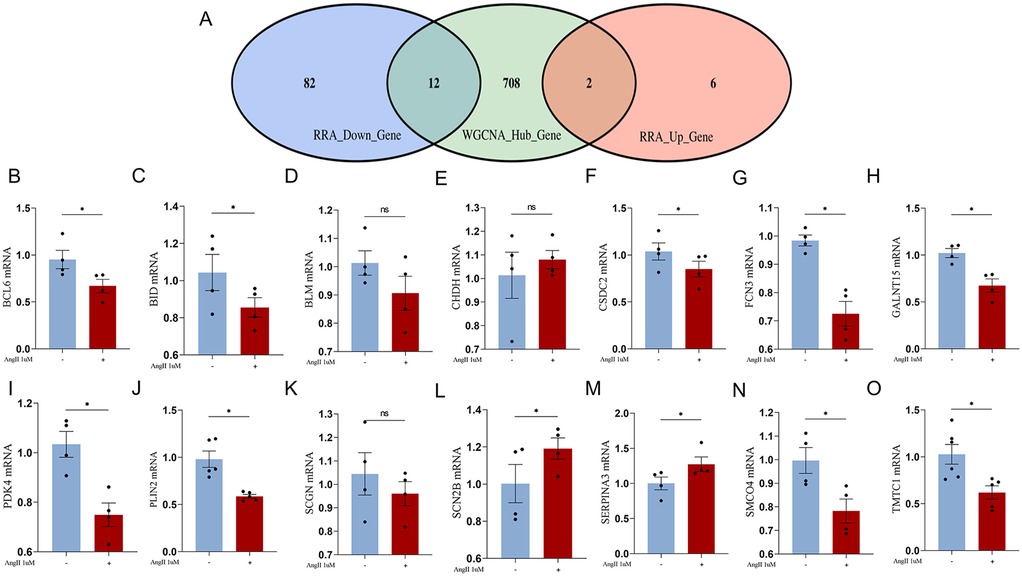
Figure 6. (A) Venn diagram of RRA differential genes intersecting with WGCNA lightcyan module genes; (B–O), RT-qPCR validation of the intersection genes between RRA differential genes and WGCNA lightcyan module genes.
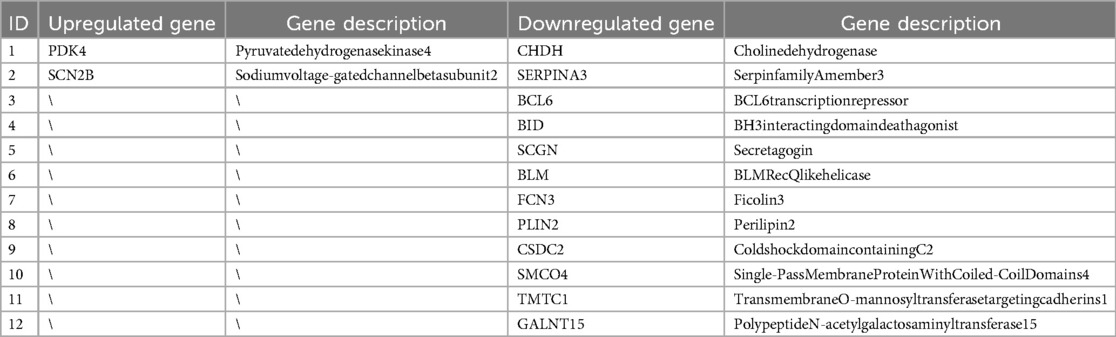
Table 4. The intersection genes of RRA differential genes and key module genes from WGCNA.
We constructed an AngII-induced primary rat cardiomyocyte hypertrophy model to simulate the pathological changes in HF myocardial tissue. The expression of the aforementioned intersecting genes were verified by RT-qPCR. Among the upregulated genes, the expression of SCN2B gene increased, which is consistent with the trend predicted by bioinformatics analysis (Figure 6L). The expression trend of PDK4 gene was opposite to the prediction (Figure 6I). Among the downregulated genes, the expression of BCL6, BID, CSDC2, FCN3, GALNT15, PLIN2, SMCO4, and TMTC1 genes decreased, which is consistent with the predicted trend. The expression trend of SERPINA3 gene was opposite to the prediction. The changes in BLM, CHDH, and SCGN genes were not statistically significant, but the trends were consistent with the predictions (Figures 6B–O).
3.5 CSDC2 and SMCO4 may influence the progression of HF by modulating fatty acid metabolism in cardiomyocytes
The single-cell sequencing data in the GSE145154 dataset were obtained from ventricular tissues. Through PCA and UMAP, similar cells were clustered into the same subgroup. Both normal myocardial tissue and the myocardial tissue in the HF group showed that the cell populations were divided into 10 types: Monocytes, Natural Killer (NK) cells, Fibroblasts, M2 macrophages, Cardiomyocytes, B cells, Endothelial cells (ECs), Cluster of Differentiation 8 positive T cells (CD8+ T cells), Pericytes, and Smooth muscle cells (SMCs) (Figures 7A,B).
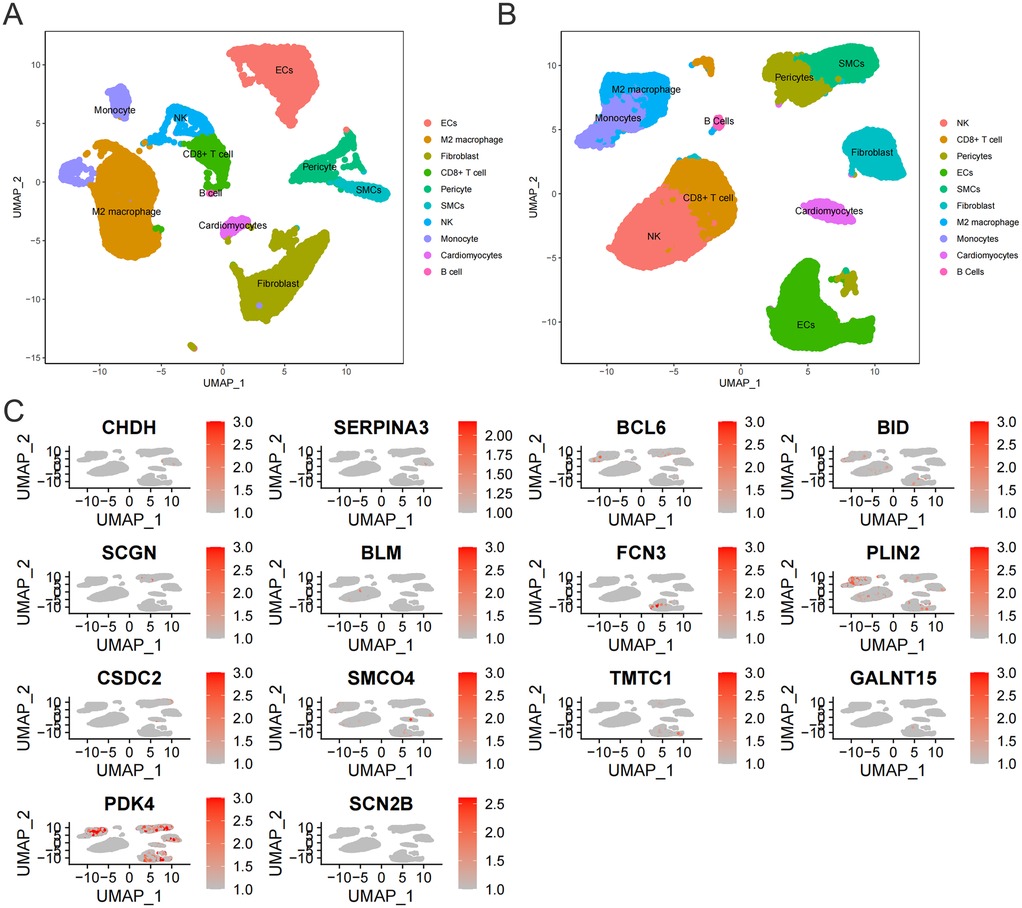
Figure 7. (A) Cell subtypes in normal myocardial tissue. (B) Cell subtypes in myocardial tissue of HF patients. (C) Expression of 14 core genes in myocardial tissue of HF patients.
To experimentally validate the association between the 14 key genes and cardiomyocytes in a real-world context, we selected 11 genes exhibiting inter-group differential expression for further investigation. The expression levels of these 11 genes were subsequently measured in cardiomyocytes isolated from the HF group. Our findings revealed that only three genes (CSDC2, PDK4, SMCO4) were detectable in HF cardiomyocytes (Figure 7C). Among these, PDK4 has established functional links to mitochondrial energy metabolism in cardiomyocytes. Given that bulk RNA-seq analyses consistently identified disrupted fatty acid metabolism as a mitochondrial energy metabolism subtype associated with HF, and considering that the roles of CSDC2 and SMCO4 in cardiomyocyte lipid metabolism remain uncharacterized, we further investigated the functional contributions of CSDC2 and SMCO4 to fatty acid metabolism within cardiomyocytes of HF patients.
The analysis of cell proportion revealed that there were differences in the expression of CSDC2 and SMCO4 in cardiomyocytes between the normal group and the HF group (Figures 8A,B). The differentially expressed genes between the cardiomyocytesCSDC2 high subset and the cardiomyocytesCSDC2 low subset were mainly concentrated in processes related to myocardial remodeling, such as Cytoskeleton in muscle cells, Mitophagy—animal, Apelin signaling pathway, and Diabetic cardiomyopathy (Figure 8C). Meanwhile, the differentially expressed genes between the cardiomyocytesSMCO4 high subset and the cardiomyocytesSMCO4 low subset were mainly concentrated in processes related to lipid and energy metabolism, such as Insulin signaling pathway, Focal adhesion, Pyrimidine metabolism, and Arachidonic acid metabolism (Figure 8D).
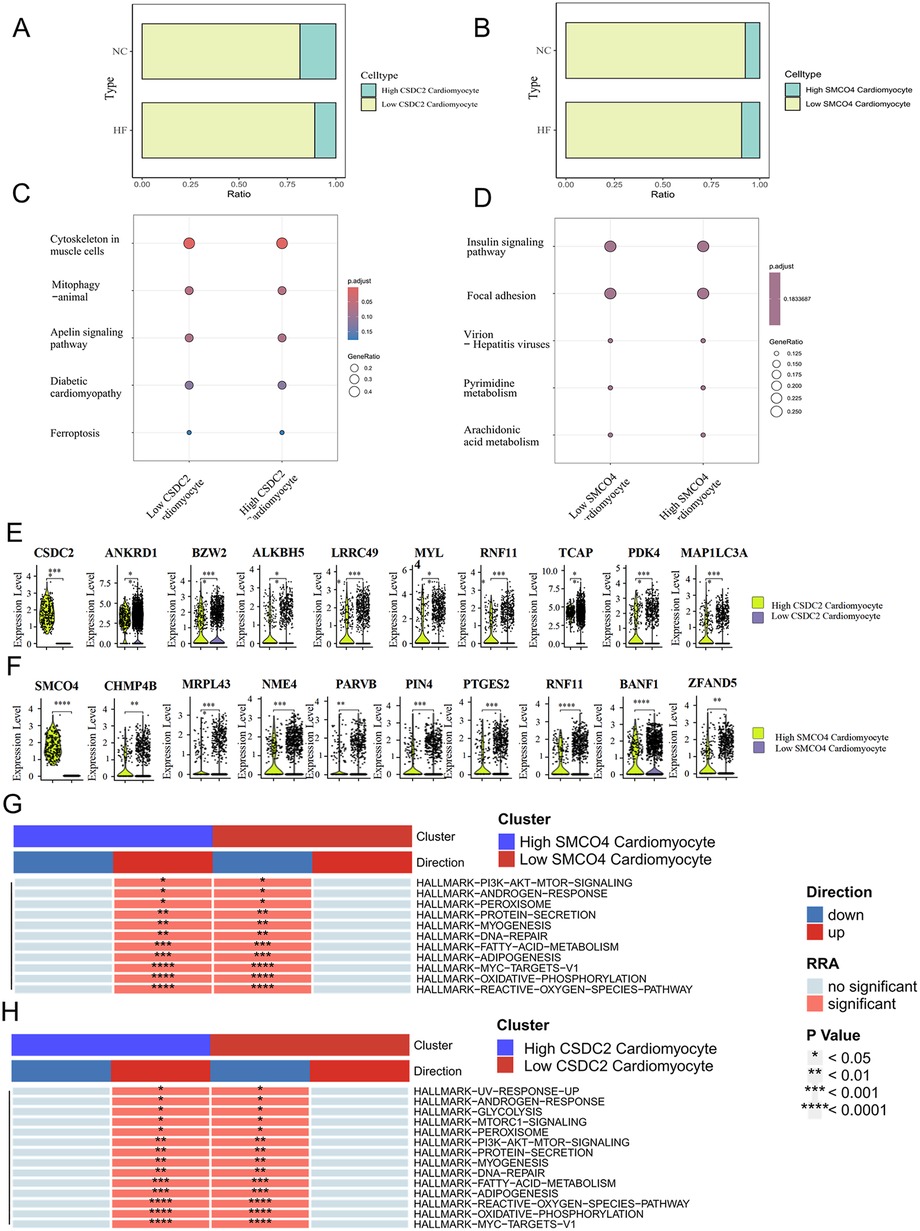
Figure 8. (A) Proportional differences between the CardiomyocytesCSDC2 high subset and the CardiomyocytesCSDC2 low subset in normal and HF patients' myocardial tissues. (B) Proportional differences between the CardiomyocytesSMCO4 high subset and the CardiomyocytesSMCO4 low subset in normal and HF patients' myocardial tissues. (C) KEGG analysis of differentially expressed genes between the CardiomyocytesCSDC2 high subset and the CardiomyocytesCSDC2 low subset. (D) KEGG analysis of differentially expressed genes between the CardiomyocytesSMCO4 high subset and the CardiomyocytesSMCO4 low subset. (E) Lipid metabolism-related genes differentially expressed between the CardiomyocytesCSDC2 high subset and the CardiomyocytesCSDC2 low subset. (F) Lipid metabolism-related genes differentially expressed between the CardiomyocytesSMCO4 high subset and the CardiomyocytesSMCO4 low subset. (G) Changes in the enrichment pathways of marker gene sets of differentially expressed genes between the CardiomyocytesSMCO4 high subset and the CardiomyocytesSMCO4 low subset. (H) Changes in the enrichment pathways of marker gene sets of differentially expressed genes between the CardiomyocytesCSDC2 high subset and the CardiomyocytesCSDC2 low subset.
To further clarify the correlation between CSDC2, SMCO4 and fatty acid metabolism, we examined the expression differences of fatty acid metabolism-related genes in the cardiomyocytesCSDC2 high subset, cardiomyocytesCSDC2 low subset, cardiomyocytesSMCO4 high subset, and cardiomyocytesSMCO4 low subset. The results revealed that ANKRD1, BZW2, ALKBH5, LRRC49, MYL, RNF11, TCAP, PDK4, and MAP1LC3A exhibited statistically significant differences (P < 0.05) between the cardiomyocytesCSDC2 high subset and cardiomyocytesCSDC2 low subset (Figure 8E). Additionally, CHMP4B, MRPL43, NME4, PARVB, PIN4, PTGES2, RNF11, BANF1, and ZFAND5 displayed statistically significant differences (P < 0.05) between the cardiomyocytesSMCO4 high subset and cardiomyocytesSMCO4 low subset (Figure 8F). Further pathway enrichment analysis corroborated these findings: the FATTY-ACID-METABOLISM, ADIPOGENESIS, and OXIDATIVE-PHOSPHORYLATION pathways were significantly upregulated in the cardiomyocytesSMCO4 high subset, while the same pathways (FATTY-ACID-METABOLISM, ADIPOGENESIS, and OXIDATIVE-PHOSPHORYLATION) were also significantly upregulated in the cardiomyocytesCSDC2 high subset (Figures 8G,H). These results suggest that CSDC2 and SMCO4 may influence the progression of HF by modulating lipid metabolism in cardiomyocytes.
4 Discussion
HF is a set of systemic syndromes caused by the decompensation of the heart's pumping function, leading to congestion in the pulmonary and systemic circulation. It represents the final stage of many cardiac diseases and continues to pose a challenge to healthcare systems due to its high incidence and mortality rates (24).
In recent years, the treatment of HF has primarily focused on reducing the hemodynamic burden on the heart (25), yet the pathological mechanisms of HF remain unclear. Cardiomyocytes are the main working cell type in the heart and are the most severely affected cells. Therefore, research on the cellular mechanisms and therapeutic strategies targeting cardiomyocytes continues to be the greatest unmet need in cardiology. Cardiomyocytes rely on mitochondrial ATP production to meet high energy demands during contraction/relaxation. Fatty acids serve as the heart's primary fuel, generating 40%–60% of cardiac ATP through oxidation (5). HF initiates pathological metabolic reprogramming characterized by a substrate shift from fatty acid oxidation to glycolysis. This transition reduces ATP yield as glycolysis produces far less energy than oxidative phosphorylation (7), creating an energy deficit that directly contributes to HF progression. Consequently, dysregulation of fatty acid metabolism represents a critical therapeutic target in HF pathophysiology.With the advancement of bioinformatics and multi-omics technologies, leveraging bioinformatics techniques to uncover complex pathogenic mechanisms is of significant importance for the treatment of HF (26).
In this study, we conducted differential gene screening and functional enrichment analysis on the transcriptome sequencing results of myocardial tissue from HF patients in six datasets, including GSE57338, GSE42955, GSE52601, GSE21610, GSE76701, and GSE145154 (which includes one single-cell sequencing transcriptome dataset), using RRA and WGCNA. In this study, RRA's strengths include amplifying biological signals in noisy/heterogeneous data (e.g., integrating microarray results across platforms or incomplete metadata), enabling meta-analysis by consolidating diverse experiments, and retaining meaningful information even with top-ranked elements only, thus enhancing reliability in high-throughput genomic data integration (16). In applications such as HF research, WGCNA's advantages include its ability to identify disease-relevant modules (e.g., gene clusters associated with HF subtypes, clinical traits, or pathological pathways), validate modules via functional enrichment analysis, and link module eigengenes to HF phenotypes (e.g., left ventricular dysfunction or disease progression). This enhances the discovery of potential biomarkers or therapeutic targets by leveraging systems-level gene interactions in complex cardiac diseases (17).
The results indicated that the functional enrichment of RRA differential genes lacked directional characteristics, covering a broad range of cellular life activities. However, the analysis of the key module in the GSE76701 WGCNA dataset showed a certain tendency towards energy metabolism. The GO analysis was primarily enriched in biological functions such as energy production through the oxidation of organic compounds, fatty acid metabolism, mitochondrial genes, long-chain fatty acid transport, and energy reserve metabolism, and in terms of cellular components, it highlighted that the mitochondrial matrix and mitochondrial intermembrane space are the main locations where key genes are primarily localized. The KEGG analysis also enriched in functional pathways related to energy cycle utilization, such as the mTOR signaling pathway, phagosome signaling pathway, and lysosome signaling pathway. After taking the intersection of the key gene modules from the RRA analysis and the WGCNA analysis, 14 key genes were identified.
Research has found that when HF occurs, there is a dramatic change in myocardial energy metabolism, with a loss of metabolic flexibility (27, 28) and insufficient ATP production (29, 30). A normal heart requires a large amount of ATP, 95% of which is derived from oxidative phosphorylation in mitochondria, with the remaining 5% coming from glycolysis. After the onset of HF, cardiac metabolism is reprogrammed to favor glycolysis as the energy substrate, shifting from fatty acid metabolism to glucose metabolism. The energy output efficiency of glycolysis is much lower than that of oxidative phosphorylation, resulting in an energy deficiency. Currently, there is no consensus on how substrate metabolism is transformed after HF and the exact issues with each metabolic pathway (7). However, it is generally believed that the energy deficiency is attributed to impaired mitochondrial oxidative phosphorylation and changes in ATP substrate sources.
Fatty acids constitute the primary fuel for the heart, with fatty acid oxidation accounting for 40%–60% of cardiac ATP production. Fatty acids are supplied to the myocardium through three principal pathways: (1) albumin-bound free fatty acids (FFAs) from systemic circulation, (2) triglyceride (TAG) delivery via chylomicrons and very-low-density lipoproteins (VLDL) hydrolyzed by lipoprotein lipase (31–33), and (3) mobilization from intracellular TAG stores within cardiomyocytes (34). FFAs enter cardiomyocytes through facilitated diffusion or via fatty acid transport proteins and fatty acid translocase (35).
Within the cytosol, approximately 90% of fatty acids undergo mitochondrial shuttling for oxidation, while the remaining 10% are esterified into TAG for cellular storage as reserve substrates (36). Mitochondrial fatty acid oxidation requires sequential activation and transport steps (37): Cytosolic FFAs are first esterified to fatty acyl-CoA, which is then converted to long-chain acylcarnitine by carnitine palmitoyltransferase-1 (CPT-1) at the outer mitochondrial membrane (37). After traversing the inner membrane via carnitine-acylcarnitine translocase, CPT2 reconverts acylcarnitine to fatty acyl-CoA in the matrix. Subsequent β-oxidation generates acetyl-CoA, which enters the TCA cycle to produce NADH and FADH2 (38). These electron donors fuel the electron transport chain to drive oxidative phosphorylation—the primary ATP-generating pathway in cells. Consequently, targeting fatty acid metabolism to sustain oxidative phosphorylation capacity represents a critical therapeutic strategy in HF.
In this study, after validation via qPCR, it was confirmed that 12 key differentially expressed genes indeed showed differential expression in Ang II-induced hypertrophic cardiomyocytes. To experimentally validate the association between the 14 key genes and cardiomyocytes, we selected 11 intergroup differentially expressed genes for further investigation. Leveraging the GSE145154 single-cell sequencing dataset derived from ventricular tissue, our findings revealed that only three genes (CSDC2, PDK4, SMCO4) were detectable in HF cardiomyocytes. Given that extensive RNA-seq analyses have consistently identified dysregulated fatty acid metabolism as a mitochondrial energy metabolic subtype associated with HF, and considering the undetermined roles of CSDC2 and SMCO4 in lipid metabolism within cardiomyocytes, we further explored their functional contributions to fatty acid metabolism in cardiomyocytes of HF patients.
CSDC2, a cardiac-enriched RNA binding protein downregulated in failing hearts, is identified as a critical regulator of cardio-metabolic stress. Consistent with our findings, Jared M McLendon et al. also identified through bioinformatic analyses that CSDC2 directly participates in regulating fatty acid metabolism pathways, as well as ion channels and sarcomere gene networks. Validation using Csdc2-knockout (KO) mice revealed that, after 10 weeks of high-fat diet feeding, Csdc2-KO mice exhibited a 20% greater weight gain than wildtype littermates (p = 0.04, n = 13–18) and developed insulin resistance (p < 0.01). Furthermore, Csdc2-KO mice displayed larger infarcts, ventricular remodeling, and systolic dysfunction following microsurgery-induced MI (39).
SMCO4 is a single-pass transmembrane protein with a coiled-coil domain. Studies by Nicholas A Wachowski et al. demonstrated that knockdown of SMCO4 expression in pancreatic β-cells significantly enhances insulin secretion (40). Insulin plays a crucial role in cardiac fatty acid metabolism and energy metabolism, influencing the utilization of fatty acids and glucose by regulating the metabolic flexibility of cardiomyocytes (41). Under conditions of elevated insulin levels, the heart tends to increase glucose oxidation while inhibiting fatty acid oxidation, thereby affecting cardiac mechanical efficiency and function (42). These findings are consistent with our study, where we observed that the CardiomyocytesSMCO4 low subset exhibit inhibition of fatty acid metabolic pathways. This suggests that despite the current paucity of similar studies, SMCO4 still demonstrates potential as a therapeutic target for HF. Future investigations into its underlying mechanisms are warranted to facilitate the prevention and treatment of HF.
This study has certain limitations. First, it remains imperative to objectively evaluate its alignment with and distinctions from prior large-scale transcriptomic meta-analyses. For example, Ramirez Flores et al. consolidated 16 studies (n = 916) to validate consensus transcriptional signatures in end-stage HF and developed the open platform ReHeat for biomarker discovery and therapeutic target identification (43). By contrast, although we included fewer datasets, these datasets were not entirely overlapping. We utilized multiple algorithms to analyze and identify key hub genes associated with HF, introduced RT-qPCR and single-cell sequencing datasets to validate the results, and ultimately identified SMCO4 and CSDC2 as key genes regulating fatty acid metabolism and influencing HF progression. Nonetheless, constrained by the limited sample size (n = 422), the statistical sensitivity of this study may be suboptimal, necessitating future expansion of cohorts to validate the generalizability of the metabolic modules. Further validation of the roles of CSDC2 and SMCO4 in fatty acid metabolism still requires basic experimental verification.
Additionally, this study used a primary neonatal rat cardiomyocyte hypertrophy model to validate the results. Although such cell models are classic and commonly used in HF research (20, 44), the transcriptomic characteristics of primary neonatal rat cardiomyocytes are also difficult to be fully consistent with those of adult human cardiomyocytes. Although single-cell sequencing datasets were introduced to address this issue, it still indicates that further validation is required in future studies.
Another limitation of our study is the absence of formal sensitivity analyses, such as leave-one-out or bootstrap-based testing, to further assess the robustness of RRA-derived results. Although the inclusion of multiple independent datasets enhances the credibility of consistently identified DEGs, future work will incorporate systematic sensitivity analyses to quantitatively evaluate the impact of individual datasets and strengthen the reliability of integrative meta-analysis approaches.
5 Conclusion
Integrated transcriptomic analysis identified 12 key genes linked to HF, which were validated in a hypertrophy model. Single-cell data showed SMCO4 and CSDC2 are specifically expressed in cardiomyocytes and regulate fatty acid metabolism. This suggests SMCO4 and CSDC2 contribute to HF by altering fatty acid metabolism in heart cells, revealing new disease mechanisms.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics statement
The entire research adhered to the principles of laboratory animal science and was approved by the Animal Ethics Committee of the Basic Research Institute, China Academy of Chinese Medical Sciences, with the ethical approval number (ERCCACMS21-179 2406-02).
Author contributions
YL: Conceptualization, Methodology, Validation, Writing – original draft. YB: Conceptualization, Software, Validation, Writing – original draft. WW: Formal analysis, Validation, Writing – original draft. ZM: Formal analysis, Writing – review & editing. PL: Investigation, Writing – original draft. DL: Resources, Writing – original draft. SL: Data curation, Writing – original draft. JJ: Conceptualization, Visualization, Writing – original draft, Writing – review & editing. QL: Project administration, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
Acknowledgements to the authors who provided the original dataset.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2025.1559429/full#supplementary-material
Abbreviations
AngII, angiotensin II; ATP, adenosine triphosphate; BP, biological processes; CC, cellular components; CPT-1, carnitine palmitoyltransferase-1; CPT2, carnitine palmitoyltransferase-2; DEGs, differentially expressed genes; FFAs, free fatty acids; FADH₂, flavin adenine dinucleotide; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GEO, gene expression omnibus; GMT, gene matrix transposed; GO, gene ontology; GSEA, gene set enrichment analysis; KEGG, kyoto encyclopedia of genes and genomes; KO, knockout; MI, myocardial infarction; MF, molecular functions; NADH, nicotinamide adenine dinucleotide; NRCMs, neonatal rat primary cardiomyocytes; PCA, principal component analysis; RRA, robust rank aggregation; RT-qPCR, reverse transcription quantitative polymerase chain reaction; TAG, triglyceride; TCA, tricarboxylic acid cycle; UMAP, uniform manifold approximation and projection; VLDL, very-low-density lipoproteins; WGCNA, weighted gene co-expression network analysis.
References
1. Noone J, O'Gorman DJ, Kenny HC. OPA1 Regulation of mitochondrial dynamics in skeletal and cardiac muscle. Trends Endocrinol Metab. (2022) 33(10):710–21. doi: 10.1016/j.tem.2022.07.003
2. Liu BH, Xu CZ, Liu Y, Lu ZL, Fu TL, Li GR, et al. Mitochondrial quality control in human health and disease. Mil Med Res. (2024) 11(1):32. doi: 10.1186/s40779-024-00536-5
3. Wang Y, Dai X, Li H, Jiang H, Zhou J, Zhang S, et al. The role of mitochondrial dynamics in disease. MedComm. (2023) 4(6):e462. doi: 10.1002/mco2.462
4. Song M, Franco A, Fleischer JA, Zhang L, Dorn GW 2nd. Abrogating mitochondrial dynamics in mouse hearts accelerates mitochondrial senescence. Cell Metab. (2017) 26(6):872–83.e5. doi: 10.1016/j.cmet.2017.09.023
5. Brown DA, Perry JB, Allen ME, Sabbah HN, Stauffer BL, Shaikh SR, et al. Expert consensus document: mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol. (2017) 14(4):238–50. doi: 10.1038/nrcardio.2016.203
6. Lopaschuk GD, Karwi QG, Tian R, Wende AR, Abel ED. Cardiac energy metabolism in heart failure. Circ Res. (2021) 128(10):1487–513. doi: 10.1161/CIRCRESAHA.121.318241
7. Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. (2005) 85(3):1093–129. doi: 10.1152/physrev.00006.2004
8. Shah S, Henry A, Roselli C, Lin H, Sveinbjörnsson G, Fatemifar G, et al. Genome-wide association and Mendelian randomisation analysis provide insights into the pathogenesis of heart failure. Nat Commun. (2020) 11(1):163. doi: 10.1038/s41467-019-13690-5
9. Liu Y, Morley M, Brandimarto J, Hannenhalli S, Hu Y, Ashley EA, et al. RNA-Seq identifies novel myocardial gene expression signatures of heart failure. Genomics. (2015) 105(2):83–9. doi: 10.1016/j.ygeno.2014.12.002
10. Akat KM, Moore-McGriff D, Morozov P, Brown M, Gogakos T, Correa Da Rosa J, et al. Comparative RNA-Sequencing analysis of myocardial and circulating small RNAs in human heart failure and their utility as biomarkers. Proc Natl Acad Sci U S A. (2014) 111(30):11151–6. doi: 10.1073/pnas.1401724111
11. Molina-Navarro MM, Roselló-Lletí E, Ortega A, Tarazón E, Otero M, Martínez-Dolz L, et al. Differential gene expression of cardiac ion channels in human dilated cardiomyopathy. PLoS One. (2013) 8(12):e79792. doi: 10.1371/journal.pone.0079792
12. Schwientek P, Ellinghaus P, Steppan S, D'Urso D, Seewald M, Kassner A, et al. Global gene expression analysis in nonfailing and failing myocardium pre- and postpulsatile and nonpulsatile ventricular assist device support. Physiol Genomics. (2010) 42(3):397–405. doi: 10.1152/physiolgenomics.00030.2010
13. Kim EH, Galchev VI, Kim JY, Misek SA, Stevenson TK, Campbell MD, et al. Differential protein expression and basal lamina remodeling in human heart failure. Proteom Clin Appl. (2016) 10(5):585–96. doi: 10.1002/prca.201500099
14. Rao M, Wang X, Guo G, Wang L, Chen S, Yin P, et al. Resolving the intertwining of inflammation and fibrosis in human heart failure at single-cell level. Basic Res Cardiol. (2021) 116(1):55. doi: 10.1007/s00395-021-00897-1
15. Hu C, Li T, Xu Y, Zhang X, Li F, Bai J, et al. Cellmarker 2.0: an updated database of manually curated cell markers in human/mouse and web tools based on scRNA-Seq data. Nucleic Acids Res. (2023) 51(D1):D870–d6. doi: 10.1093/nar/gkac947
16. Kolde R, Laur S, Adler P, Vilo J. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics (Oxford, England). (2012) 28(4):573–80. doi: 10.1093/bioinformatics/btr709
17. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC bioinformatics. (2008) 9:559. doi: 10.1186/1471-2105-9-559
18. Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, et al. Clusterprofiler 4.0: a universal enrichment tool for interpreting omics data. Innovation (Cambridge (Mass)). (2021) 2(3):100141. doi: 10.1016/j.xinn.2021.100141
19. Lin K, Wang T, Tang Q, Chen T, Lin M, Jin J, et al. IL18R1-related molecules as biomarkers for asthma severity and prognostic markers for idiopathic pulmonary fibrosis. J Proteome Res. (2023) 22(10):3320–31. doi: 10.1021/acs.jproteome.3c00389
20. An D, Zeng Q, Zhang P, Ma Z, Zhang H, Liu Z, et al. Alpha-ketoglutarate ameliorates pressure overload-induced chronic cardiac dysfunction in mice. Redox Biol. (2021) 46:102088. doi: 10.1016/j.redox.2021.102088
21. Wang X, Tan Y, Liu F, Wang J, Liu F, Zhang Q, et al. Pharmacological network analysis of the functions and mechanism of kaempferol from Du Zhong in intervertebral disc degeneration (IDD). J Orthop Translat. (2023) 39:135–46. doi: 10.1016/j.jot.2023.01.002
22. Izzo A, Manco R, Bonfiglio F, Calì G, De Cristofaro T, Patergnani S, et al. NRIP1/RIP140 siRNA-mediated attenuation counteracts mitochondrial dysfunction in down syndrome. Hum Mol Genet. (2014) 23(16):4406–19. doi: 10.1093/hmg/ddu157
23. Wang H, Jin H, Liu Z, Tan C, Wei L, Fu M, et al. Screening and identification of key chromatin regulator biomarkers for ankylosing spondylitis and drug prediction: evidence from bioinformatics analysis. BMC Musculoskelet Disord. (2023) 24(1):389. doi: 10.1186/s12891-023-06490-y
24. Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, et al. Heart disease and stroke statistics-2020 update: a report from the American Heart Association. Circulation. (2020) 141(9):e139–596. doi: 10.1161/CIR.0000000000000757
25. Timmis A, Townsend N, Gale CP, Torbica A, Lettino M, Petersen SE, et al. European society of cardiology: cardiovascular disease statistics 2019. Eur Heart J. (2020) 41(1):12–85. doi: 10.1093/eurheartj/ehz859
26. DeGroat W, Abdelhalim H, Peker E, Sheth N, Narayanan R, Zeeshan S, et al. Multimodal AI/ML for discovering novel biomarkers and predicting disease using multi-omics profiles of patients with cardiovascular diseases. Sci Rep. (2024) 14(1):26503. doi: 10.1038/s41598-024-78553-6
27. Karwi QG, Uddin GM, Ho KL, Lopaschuk GD. Loss of metabolic flexibility in the failing heart. Front Cardiovasc Med. (2018) 5:68. doi: 10.3389/fcvm.2018.00068
28. Neubauer S. The failing heart–an engine out of fuel. N Engl J Med. (2007) 356(11):1140–51. doi: 10.1056/NEJMra063052
29. Bottomley PA, Panjrath GS, Lai S, Hirsch GA, Wu K, Najjar SS, et al. Metabolic rates of ATP transfer through creatine kinase (CK flux) predict clinical heart failure events and death. Sci Transl Med. (2013) 5(215):215re3. doi: 10.1126/scitranslmed.3007328
30. Ingwall JS, Weiss RG. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ Res. (2004) 95(2):135–45. doi: 10.1161/01.RES.0000137170.41939.d9
31. Augustus AS, Kako Y, Yagyu H, Goldberg IJ. Routes of FA delivery to cardiac muscle: modulation of lipoprotein lipolysis alters uptake of TG-derived FA. Am J Physiol Endocrinol Metab. (2003) 284(2):E331–9. doi: 10.1152/ajpendo.00298.2002
32. Panov AV. The structure of the cardiac mitochondria respirasome is adapted for the β-oxidation of fatty acids. Int J Mol Sci. (2024) 25(4):2410. doi: 10.3390/ijms25042410
33. Niu YG, Hauton D, Evans RD. Utilization of triacylglycerol-rich lipoproteins by the working rat heart: routes of uptake and metabolic fates. J Physiol (Lond). (2004) 558(Pt 1):225–37. doi: 10.1113/jphysiol.2004.061473
34. Saddik M, Gamble J, Witters LA, Lopaschuk GD. Acetyl-CoA carboxylase regulation of fatty acid oxidation in the heart. J Biol Chem. (1993) 268(34):25836–45. doi: 10.1016/S0021-9258(19)74465-2
35. Da Dalt L, Cabodevilla AG, Goldberg IJ, Norata GD. Cardiac lipid metabolism, mitochondrial function, and heart failure. Cardiovasc Res. (2023) 119(10):1905–14. doi: 10.1093/cvr/cvad100
36. Sun Q, Karwi QG, Wong N, Lopaschuk GD. Advances in myocardial energy metabolism: metabolic remodelling in heart failure and beyond. Cardiovasc Res. (2024) 120(16):1996–2016. doi: 10.1093/cvr/cvae231
37. Gibb AA, Hill BG. Metabolic coordination of physiological and pathological cardiac remodeling. Circ Res. (2018) 123(1):107–28. doi: 10.1161/CIRCRESAHA.118.312017
38. Altamimi TR, Thomas PD, Darwesh AM, Fillmore N, Mahmoud MU, Zhang L, et al. Cytosolic carnitine acetyltransferase as a source of cytosolic acetyl-CoA: a possible mechanism for regulation of cardiac energy metabolism. Biochem J. (2018) 475(5):959–76. doi: 10.1042/BCJ20170823
39. McLendon JM, Zhang X, Boudreau RL. Abstract P3052: RNA binding protein Csdc2 is required for metabolic and cardiac stress responses. Circ Res. (2023) 133(Suppl_1):AP3052–AP. doi: 10.1161/res.133.suppl_1.P3052
40. Wachowski NA, Pippin JA, Boehm K, Lu S, Leonard ME, Manduchi E, et al. Implicating type 2 diabetes effector genes in relevant metabolic cellular models using promoter-focused capture-C. Diabetologia. (2024) 67(12):2740–53. doi: 10.1007/s00125-024-06261-x
41. Guo CA, Guo S. Insulin receptor substrate signaling controls cardiac energy metabolism and heart failure. J Endocrinol. (2017) 233(3):R131–r43. doi: 10.1530/JOE-16-0679
42. Elnwasany A, Ewida HA, Menendez-Montes I, Mizerska M, Fu X, Kim CW, et al. Reciprocal regulation of cardiac β-oxidation and pyruvate dehydrogenase by insulin. J Biol Chem. (2024) 300(7):107412. doi: 10.1016/j.jbc.2024.107412
43. Ramirez Flores RO, Lanzer JD, Holland CH, Leuschner F, Most P, Schultz JH, et al. Consensus transcriptional landscape of human End-stage heart failure. J Am Heart Assoc. (2021) 10(7):e019667. doi: 10.1161/JAHA.120.019667
Keywords: heart failure, myocardial tissue, transcriptome, weighted correlation network analysis, bioinformatics analysis, gene set enrichment analysis, single-cell transcriptome
Citation: Li Y, Bai Y, Wang W, Ma Z, Li P, Li D, Li S, Jin J and Lin Q (2025) Analysis and validation of characteristic genes in RNA sequencing datasets from heart failure patients based on multiple algorithms. Front. Cardiovasc. Med. 12:1559429. doi: 10.3389/fcvm.2025.1559429
Received: 12 January 2025; Accepted: 7 August 2025;
Published: 26 August 2025.
Edited by:
Martina Semenzato, University of Padua, ItalyReviewed by:
Akshay Shekhar, Regeneron Pharmaceuticals, Inc., United StatesVarun Sharma, NMC Healthcare (NMC Genetics), India
Copyright: © 2025 Li, Bai, Wang, Ma, Li, Li, Li, Jin and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jialin Jin, amlhbGlucG9zdEBmb3htYWlsLmNvbQ==; Qian Lin, bGlucWlhbjYyQDEyNi5jb20=
†These authors have contributed equally to this work and share first authorship