Abstract
There exists a complex relationship between gut microbiota and cardiovascular diseases (CVD). On one hand, the plasma levels of various metabolites produced by gut microbiota, such as trimethylamine n-oxide (TMAO), short-chain fatty acid (SFCA), bile acid (BA), are closely related to the occurrence and development of CVD. On the other hand, CVD can affect gut microbiota, leading to gut microbiota dysbiosis or metabolic changes. Cardiovascular drugs are the cornerstone of treating CVD, especially oral medications that play an indispensable role in the long-term treatment of chronic CVD. Increasing research suggests that drugs entering the gastrointestinal environment interact with gut microbiota. Due to the individual differences in gut microbiota, the exploration of its mechanisms is insufficient. Therefore, the purpose of this review is to summarize the interactions between various common cardiovascular drugs and gut microbiota, and to highlight the impact of the gut microbiota on the therapeutical effects and side effects of cardiovascular drugs.
Introduction
There exists a complex bidirectional interaction between the gut microbiota and drugs, a mechanism that profoundly impacts the efficacy of drug therapies and host health. The gut microbiota can influence drug metabolism and absorption (1–3). The gut microbiota influences drug metabolism through several mechanisms, including the production of enzymes that degrade or activate drugs, altering the pH of the drug absorption environment, and performing biotransformation of drugs (such as demethylation, deamination, dehydroxylation, deacylation, decarboxylation, or oxidation) (4). These processes can significantly impact the efficacy, bioavailability, and pharmacokinetics of various medications. Drugs characterized by low solubility and/or permeability or sustained release are particularly susceptible to these effects, as they tend to have a longer residence time in the gastrointestinal tract. Additionally, the gut microbiota-bile acid axis appears to enhance the solubility of certain low-solubility drugs (5). The influence of the gut microbiota is not confined to the gastrointestinal tract, as certain metabolites derived from it may affect liver function. This occurs by mimicking and competing with intermediates generated during drug metabolism in liver or by influencing the expression of hepatic drug metabolism genes (6, 7).
Conversely, drugs can alter the composition and metabolism of the gut microbiota (8–10). The mechanisms involved include direct or indirect disruption of the gut microbiota, modulation of metabolite production, and changes in the gastrointestinal environment, among others. A few typical examples are worth mentioning. Antibiotics directly disrupt the gut microbiota through their bacteriostatic or bactericidal activities, altering the metabolic capabilities of the microbiota. For instance, ampicillin alters the characteristics of the gut microbiota, reducing its metabolic activity and enhancing the antithrombotic effect of aspirin (11). Aspirin affects the composition of the gut microbiota, reducing the production of mucosal protective metabolites in the intestine, thereby promoting aspirin-induced intestinal injury (12). Proton pump inhibitors (PPI) increase the pH of the gastrointestinal tract, leading to a significant reduction in the α-diversity of the intestinal microbiota (13). This promotes the abundance of opportunistic pathogens such as Enterococcaceae and Streptococcaceae, thereby increasing the risk of infections and inflammation. Additionally, PPIs enhance patients’ susceptibility to the pathogenic bacterium Clostridioides difficile (14), which is closely associated with numerous cardiovascular diseases (CVD) (15–18). Furthermore, drugs may induce modifications in genes or enzymes involved in their own metabolism or transport, accelerating their own transport and metabolism (19), a phenomenon known as autoinduction. This can have implications for other drugs that share the same metabolic or transport pathways. In summary,these interactions highlight the role of the gut microbiota in drug efficacy and side effects, while also offering potential targets for optimizing therapeutic strategies.
The gut microbiota plays a pivotal role in the pathogenesis and progression of CVD. Key metabolites derived from gut microbiota, such as trimethylamine n-oxide (TMAO), short-chain fatty acids (SCFA), and bile acids (BA), have been extensively studied for their roles in CVD. For instance, TMAO, a metabolite produced by gut bacteria from dietary choline and carnitine, is closely associated with adverse cardiovascular outcomes when its plasma levels are elevated (20). Similarly, SCFA, generated from the fermentation of dietary fibers, exhibit anti-inflammatory and vasoprotective effects (21), while BA are involved in regulating lipid metabolism and energy homeostasis through various signaling pathways (22, 23). Additionally, certain pathogenic bacteria in the gut microbiota, such as Shigella, can promote systemic inflammation, thereby increasing the risk of CVD (18, 24–27). Consequently, gut microbiota dysbiosis contributes to the development and exacerbation of CVD. Furthermore, CVD can induce changes in the composition and function of the gut microbiota (28–31), creating a vicious cycle that perpetuates disease progression.
Given the complex interactions between the gut microbiota and drugs, as well as the influence of the gut microbiota on CVD, the gut microbiota also plays a crucial role in shaping the therapeutic effects and side effects of cardiovascular drugs. The gut microbiota exhibits metabolic activity toward many cardiovascular drugs (2, 11, 32, 33) and can influence the transport and absorption of drugs such as aspirin (1), ultimately altering their bioavailability and pharmacokinetics and affecting their therapeutic efficacy. Conversely, cardiovascular drugs can modulate the composition and function of the gut microbiota, thereby influencing their own therapeutic effects and side effects. For example, aspirin alters the composition of the gut microbiota, reducing the production of mucosal protective metabolites and promoting aspirin-induced intestinal injury (12). Nifedipine, with its potential antibacterial activity, inhibits specific gut bacteria, thereby reducing the production of blood pressure-elevating metabolites by the gut microbiota (34). Additionally, different pathological conditions can influence the interactions between the gut microbiota and cardiovascular drugs. For instance, amlodipine increases the proportion of pro-inflammatory bacteria in the gut microbiota of healthy individuals, leading to intestinal inflammation and increased intestinal permeability (35), whereas in individuals with hypertension and non-alcoholic fatty liver disease (NAFLD), it promotes the restoration of intestinal integrity (36). Therefore, exploring the interactions between CVD, cardiovascular drugs and the gut microbiota holds significant implications for individualized drug therapy.
In this review, we will first summarize the associations between the gut microbiota and CVD, aiming to understand the role of gut microbiota and their metabolites in cardiovascular health. We will then explore the mechanisms underlying the interactions between common cardiovascular drugs and the gut microbiota, highlighting the impact of the microbiota on drug efficacy and adverse effects, propose potential therapeutic targets for microbiota-based interventions in cardiovascular medicine.
Cardiovascular diseases and gut microbiota
The gut microbiota refers to the complex microbial community colonizing the human gastrointestinal tract, comprising bacteria, fungi, viruses, and archaea, with bacteria being the predominant component. The metagenome (collective genetic material) of the gut microbiota far exceeds the human genome in size and establishes a symbiotic relationship with the host, participating in diverse physiological functions such as digestion, immune regulation, metabolic synthesis, and disease defense. Notably, it plays a critical role in maintaining cardiovascular health. Approximately 98% of human gut bacteria belong to the phyla Bacteroidetes, Firmicutes, Proteobacteria, and Actinobacteria. Among these, the ratio of Bacteroidetes to Firmicutes (F/B ratio) is relatively stable under normal conditions, and its dysregulation is associated with metabolic syndromes such as obesity and diabetes. The gut microbiota harbors numerous commensal bacteria, such as Lactobacillus species, which produce SCFA that confer cardiovascular benefits, including anti-inflammatory effects, prevention of atrial fibrillation, and reduction of insulin resistance (37–40). Blautia, Ruminococcaceae, and Akkermansia muciniphila are associated with lower triglyceride levels and exhibit cardioprotective properties (41). Bacteroides fragilis has been shown to mitigate high-salt diet-induced hypertension and prevent aging-related atrial fibrillation (42, 43). However, the gut microbiota also includes pathogenic bacteria, such as Clostridioides difficile, which significantly increases the risk of myocardial infarction, heart failure, and stroke in infected individuals (15–18). Pathogens like Shigella, Campylobacter, and Salmonella are linked to systemic inflammation and elevated CVD risk (18, 24–27). Certain bacteria exhibit dual roles in cardiovascular health. For example, Prevotella produces trimethylamine (TMA, a precursor of TMAO) and is associated with hypertension (44, 45). Conversely, Prevotella has also been shown to ameliorate diabetes-induced glucose dysmetabolism and produce SCFA (46–48), suggesting that dietary and other environmental factors may modulate its metabolic activity and impact on host health.
The metabolites of gut microbiota has a complex relationship with CVD. For instance, TMAO is a harmful metabolite associated with adverse cardiovascular outcomes. Meta-analyses have shown a positive dose-dependent association between plasma TMAO concentration and CVD risk (20). TMAO promotes the accumulation of cholesterol in macrophages, leading to their transformation into foam cells, one of the earliest cellular markers of atherosclerosis (49, 50). It also enhances the expression of the nuclear factor-kappa B (NF-κB) pathway and the production of reactive oxygen species (ROS) (51, 52), triggering inflammation and endothelial dysfunction, which contribute to the development of atherosclerosis. Elevated levels of TMAO further promote cardiac hypertrophy and fibrosis through the transforming growth factor-β-mothers against decapentaplegic homolog 2/3 (TGF-β-Smad2/3) signaling pathway, thereby inducing heart failure (53). Additionally, TMAO prolongs the activity of angiotensin II (AngII) by altering its structure, exacerbating hypertension. The gut microbiota also produces metabolites beneficial to cardiovascular health. SCFA, key metabolites derived from the microbial fermentation of dietary fibers, include acetate, propionate, and butyrate. These SCFA exert multiple cardioprotective effects. For instance, acetate and propionate bind to the G protein-coupled receptor 41(GPR41,specific SCFA receptors) on vascular endothelium, regulating vasodilation and reducing blood pressure (54). Acetate downregulates the expression of early growth response-1(Egr-1, a kind of transcription factor) in the heart and kidneys, a critical factor involved in cardiac hypertrophy, cardiorenal fibrosis, and inflammation (21). Butyrate suppresses cholesterol absorption by downregulating Niemann-Pick C1-Like 1 (NPC1L1, key protein for dietary cholesterol absorption) expression (55). Furthermore, BA in the gut are closely linked to metabolism, and their composition and levels are modulated by the gut microbiota (56). BA exert their effects by binding to BA receptors in various tissues. For example, activation of the farnesoid X receptor (FXR) in the liver reduces the expression of lipogenic genes such as sterol regulatory element-binding protein 1c (SREBP-1c), significantly lowering serum and hepatic triglyceride levels (22). Activation of intestinal FXR also reduces lipid levels by decreasing BA reabsorption (23). Takeda G protein-coupled receptor 5(TGR5), another BA receptor, improves insulin sensitivity, reduces inflammation, and ameliorates ventricular remodeling through multiple mechanisms, including modulating DHHC-type palmitoyltransferase 4 (DHHC4), the cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) pathway, and the cyclic adenosine monophosphate/protein kinase A (cAMP/PKA) pathway (57–59). Activation of TGR5 can also mitigate obesity by stimulating the sympathetic nervous system (60).
On the other hand, CVD can also affect the gut microbiota, leading to gut dysbiosis, which may, in turn, exacerbate cardiovascular conditions. For example, in patients with heart failure, reduced blood flow to the intestinal arteries increases the number of gut bacteria in the mucus layer near the apical surface of the colonic mucosa. This leads to increased permeability of the small and large intestines, allowing higher levels of inflammatory cytokines and endotoxins to enter the bloodstream (61). In addition, the composition of the gut microbiota in patients with heart failure undergoes significant alterations, characterized by a reduction in Coriobacteriaceae, Erysipelotrichaceae, and Ruminococcaceae (62, 34), and an increase in pathogenic bacteria including Shigella, Campylobacter, Salmonella, and Candida species (63). A national study in the United States also reported an increase in the number of pathogenic bacteria (such as Clostridium difficile) in fecal samples from patients with chronic heart failure, and this pathogenic bacterial infection was significantly associated with increased hospital mortality among heart failure patients (28). Furthermore, in patients with chronic heart failure, downregulation of microbial genes involved in the production of protective metabolites such as butyrate and significant upregulation of intestinal microbes metabolizing harmful metabolites like TMAO and lipopolysaccharide (LPS) have been observed (29). In patients with carotid atherosclerosis, there is an increase in infection-associated gut microbiota, such as Klebsiella and Streptococcus (30, 31). Patients with hypertension exhibit gene loss in their intestinal microbiome related to amino acid (particularly lysine, histidine, leucine, and serine) biosynthesis and transport, as well as a decrease in fatty acid utilization and carbohydrate transport modules, indicating impaired nutrient synthesis, absorption, and energy production capabilities. In contrast, LPS biosynthesis and export modules are enriched, and LPS has been shown to contribute to inflammation (45). It can be seen that there is an interaction between the gut microbiota and CVD, and this interaction forms a cycle that impacts the health of the organism.
Interaction between cardiovascular drugs and gut microbiota
Aspirin
Aspirin, also known as acetylsalicylic acid, is an antiplatelet agent that exerts its therapeutic effects by irreversibly acetylating cyclooxygenase-1 (COX-1), thereby inhibiting the synthesis of thromboxane A2 (TxA2) and suppressing platelet aggregation. This mechanism prevents thrombus formation (64). Compared to its primary metabolite, salicylic acid, the addition of the acetyl group enhances aspirin's water solubility and facilitates its rapid absorption into the bloodstream. Aspirin serves as a cornerstone medication for the treatment and prevention of atherosclerosis. As an antiplatelet agent, enteric-coated tablets are the main form of aspirin, which means it stays in the gut longer and interacts with the gut flora for a longer time.
Aspirin is closely associated with the characteristics of the gut microbiota. Studies have shown that aspirin and its metabolite, salicylate, possess antimicrobial activity. They can reduce cholesterol levels in the cell membranes of certain gut bacteria in a dose-dependent manner, thereby decreasing membrane fluidity. Additionally, aspirin inhibits the activity of dehydrogenase (DHA), a core energy metabolism enzyme that drives redox reactions and ATP production, as well as esterase (EA), a hydrolase involved in lipid metabolism, detoxification, and signal regulation (65). Furthermore, aspirin can induce the lysis of Helicobacter pylori (66). The effects of aspirin on gut bacteria are not uniform; they are more pronounced in suppressing the growth of pro-inflammatory and other harmful bacteria, thereby creating a favorable environment for the proliferation of beneficial bacteria. Research has demonstrated that individuals who take oral aspirin exhibit more prominent features of Prevotella, Bacteroides, Ruminococcaceae, and Barnesiella in their gut microbiota compared to control groups. These bacteria are closely linked to cardiovascular health (9). Animal studies have also revealed that aspirin can modulate the composition of the gut microbiota by balancing the ratio of Tregs to Th17 cells and enhancing the cluster of differentiation 39-cluster of differentiation 73 (CD39-CD73) adenosine signaling pathway, which is involved in purinergic signaling. This modulation leads to an increase in the levels of SCFA (67). A comprehensive analysis of the gut metagenomics, host clinical data, and metabolomics of 2,173 European residents found that aspirin improves cardiometabolic health by influencing the gut microbiota. This includes reductions in Ruminococcus, Clostridium citroniae, and Parvimonas micra, lower concentrations of plasma inflammatory markers such as C-reactive protein (CRP) and interleukin-6 (IL-6), and decreased levels of pyruvate (9). These studies collectively suggest that the cardiovascular therapeutic effects of aspirin may be partially mediated by the gut microbiota.
On the other hand, the gut microbiota can also influence the metabolism and absorption of aspirin. Kim et al. found that the gut microbiota metabolizes aspirin into salicylate through esterases produced by certain bacteria. Salicylate, compared to aspirin, is less readily absorbed, thereby reducing aspirin's plasma concentration and weakening its antiplatelet effects. When treated with ampicillin, the abundance of Enterococci, Enterobacteria, and Lactobacilli in the gut microbiota significantly decreased, enhancing the antiplatelet effects of aspirin. This suggests that these bacteria may possess or secrete esterases capable of metabolizing aspirin (11). Additionally, the gut microbiota can influence the absorption of aspirin in the small intestine. Multidrug Resistance Protein 4 (MRP4), an efflux transporter, pumps a variety of structurally diverse endogenous and exogenous organic anions out of cells, and aspirin is also a substrate of MRP4 (68). Studies have shown that the gut microbiota can regulate the expression of MRP4 in intestinal epithelial cells. Jeon et al. demonstrated that treating Caco-2 cells (which structurally and functionally resemble differentiated small intestinal epithelial cells) with coffee bean extract(CBE)-treated fecal microbiota suppressed MRP4 expression, whereas CBE alone did not have this effect. Furthermore, the CBE-treated gut microbiota showed an increase in Muribaculaceae and Lactobacillaceae and a decrease in Proteobacteria, Helicobacteriaceae, and Bacteroidaceae, indicating that these bacteria may play a role in regulating MRP4 expression in intestinal epithelial cells (1).
Gastrointestinal injury is a common side effect of aspirin, which is traditionally attributed to the inhibition of COX-1 and COX-2. However, recent studies suggest that the gut microbiota may also play a role in gastrointestinal injury caused by aspirin. WU et al. identified Parabacteroides goldsteinii, an intestinal microbe inhibited by aspirin. Supplementing with Parabacteroides goldsteinii or the BA metabolite 7-keto-lithocholic acid (7-keto-LCA) can promote intestinal epithelial repair by inhibiting FXR receptor signaling, alleviating aspirin-mediated intestinal microenvironment and intestinal barrier damage (12). Through the use of figures, we have provided a more intuitive summary of the interactions between aspirin and the gut microbiota, as detailed in Figure 1.
Figure 1
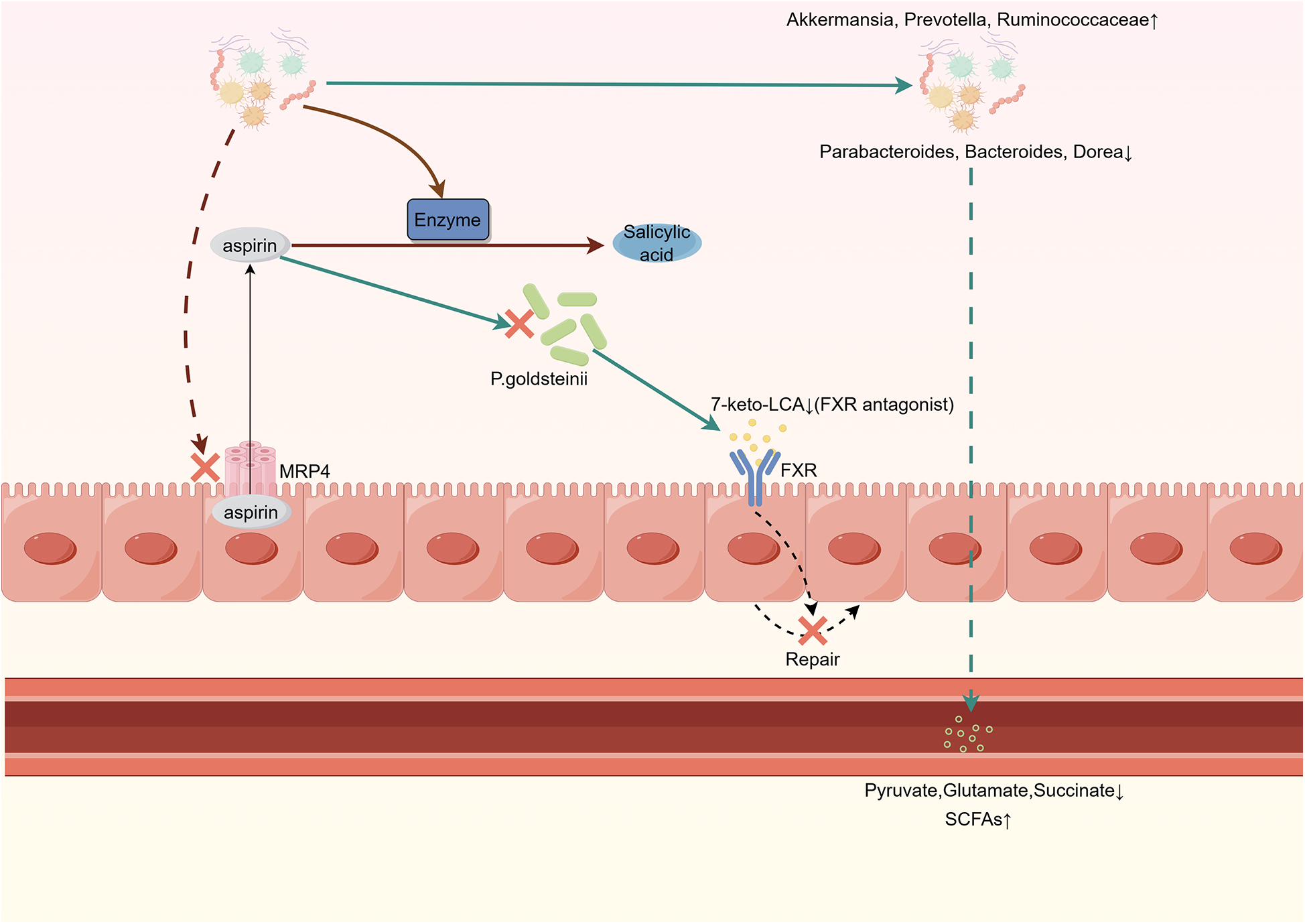
Interaction between gut microbiota and aspirin.Gut microbiota inhibits MRP4 expression, reducing the efflux of aspirin from intestinal cells. Aspirin inhibits P. goldsteinii, leading to decreased production of 7-keto-LCA. This weakens its inhibitory effect on FXR, thereby impairing the self-repair ability of intestinal cells. Aspirin increases the abundance of anti-inflammatory bacteria while reducing the levels of pro-inflammatory bacteria in the gut. Gut microbiota metabolic enzymes convert aspirin into salicylic acid, which is less readily absorbed compared to aspirin. MRP4, multidrug resistance protein 4; P. goldsteinii, Parabacteroides goldsteinii; 7-keto-LCA, 7-keto-lithocholic acid; FXR, farnesoid X receptor.
Statins
Statins are the most commonly used lipid-lowering drugs in clinical practice, achieving cardiovascular protection by reducing blood lipid levels. Along with aspirin, they are cornerstone medications for the treatment and prevention of atherosclerosis. It is traditionally believed that the antilipidemic effect of statins is mainly achieved by inhibiting 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase. Additionally, statins are associated with an increased risk of type 2 diabetes mellitus (T2DM), although the mechanism underlying this side effect remains unclear. Recent studies have revealed that the gut microbiota plays a role in both the lipid-lowering effects and the blood glucose-elevating effects of statins. Kari's cross-sectional analysis of cohort studies revealed that the gut microbiota has an additive effect on the risk of statin-associated new-onset type 2 diabetes mellitus (T2DM). Specifically, the abundance of Ruminococcus torques, Blautia obeum, and Blautia sp. KLE 1732 was positively correlated with the risk of statin-associated new-onset diabetes (69). Jose et al. discovered that statins activate the liver pregnane x receptor (PXR), which suppresses the expression of cytochrome P450 family 7 subfamily A member 1(CyP7A1, a key rate-limiting enzyme in BA synthesis). This expands the BA pool and alters the proportion of BA, ultimately leading to a deficiency in butyrate-producing bacterial communities and increasing the risk of T2DM (10). Additionally, BA play a significant role in the relationship between statins and the gut microbiota. She et al. found that statins reduce the abundance of Clostridium in the gut microbiota, leading to a decrease in the proportion of ursodeoxycholic acid (UDCA) in total BA. UDCA is a ligand for the TGR5 receptor, which stimulates glucagon-like peptide-1(GLP-1) secretion and enhances insulin sensitivity (70). This partially explains the mechanism behind statin-induced elevated blood glucose. In the same study, an increase in the proportion of chenodeoxycholic acid (CDCA) was also observed. CDCA is an endogenous ligand for FXR, and its activation can reduce lipid absorption in intestinal cells and control lipid synthesis in liver cells (23). However, upregulation of liver FXR may attenuate the lipid-lowering effects of statins. He et al. found that FXR upregulation can reverse the inhibition of CyP7A1 induced by a high-fat diet, reducing BA synthesis. Since cholesterol is the precursor for BA synthesis, this may counteract the lipid-lowering effects of statins (3). Furthermore, the gut microbiota metabolizes many drugs, and statins are no exception. in vitro studies have shown that simvastatin undergoes bioaccumulation in gut bacteria and is biotransformed by bacterial enzymes (e.g., from Lactobacillus and Bifidobacterium), leading to delayed absorption and reduced concentration of simvastatin (2). This may involve the off-target effects of statins on the gut microbiota (71), ultimately weakening the lipid-lowering efficacy of simvastatin. Therefore, the regulation of statins’ lipid-lowering effects by the gut microbiota is comprehensive. The individual variability of the gut microbiota may influence its modulation of statins’ effects, which aligns with the observed individual differences in statins’ lipid-lowering efficacy. Future research is needed to further explore the relationship between statins and the gut microbiota, providing more comprehensive strategies for the personalized treatment of statins.
With further research into statins, it has been discovered that statins may protect the cardiovascular system through mechanisms independent of lipid-lowering, referred to as pleiotropic effects (72). The pleiotropic effects of statins on the cardiovascular system may be related to the gut microbiota. Several studies support the potential of statins to improve gut microbial dysbiosis caused by hyperlipidemia. Bacteroides2 (Bact2) enterotype is a gut microbiota structure associated with systemic inflammation, and its prevalence is related to body mass index. However, statins can reduce its prevalence (8). Administration of atorvastatin increases the abundance of anti-inflammatory bacteria (Faecalibacterium prausnitzii, Akkermansia muciniphila, and Oscillospira) in the gut microbiota of hyperlipidemia patients, while reducing the abundance of proinflammatory species Desulfovibrio sp. and bile-related species (Bifidobacterium bifidum) (73). Statins significantly increase the abundance of Bifidobacteria, which are generally considered beneficial, and reduce the abundance of bacteria related to cardiovascular outcomes, such as Ruminococcus and Parabacteroides (74). Atorvastatin and rosuvastatin significantly increase the abundance of Bacteroides, Butyricimonas, and Mucispirillum species, whose abundances are associated with inflammation (75). Statins are also associated with reduced plasma levels of TMAO (76). Through the use of figures and tables, we have provided a more intuitive summary of the interactions between statins and the gut microbiota, as detailed in Figure 2.
Figure 2
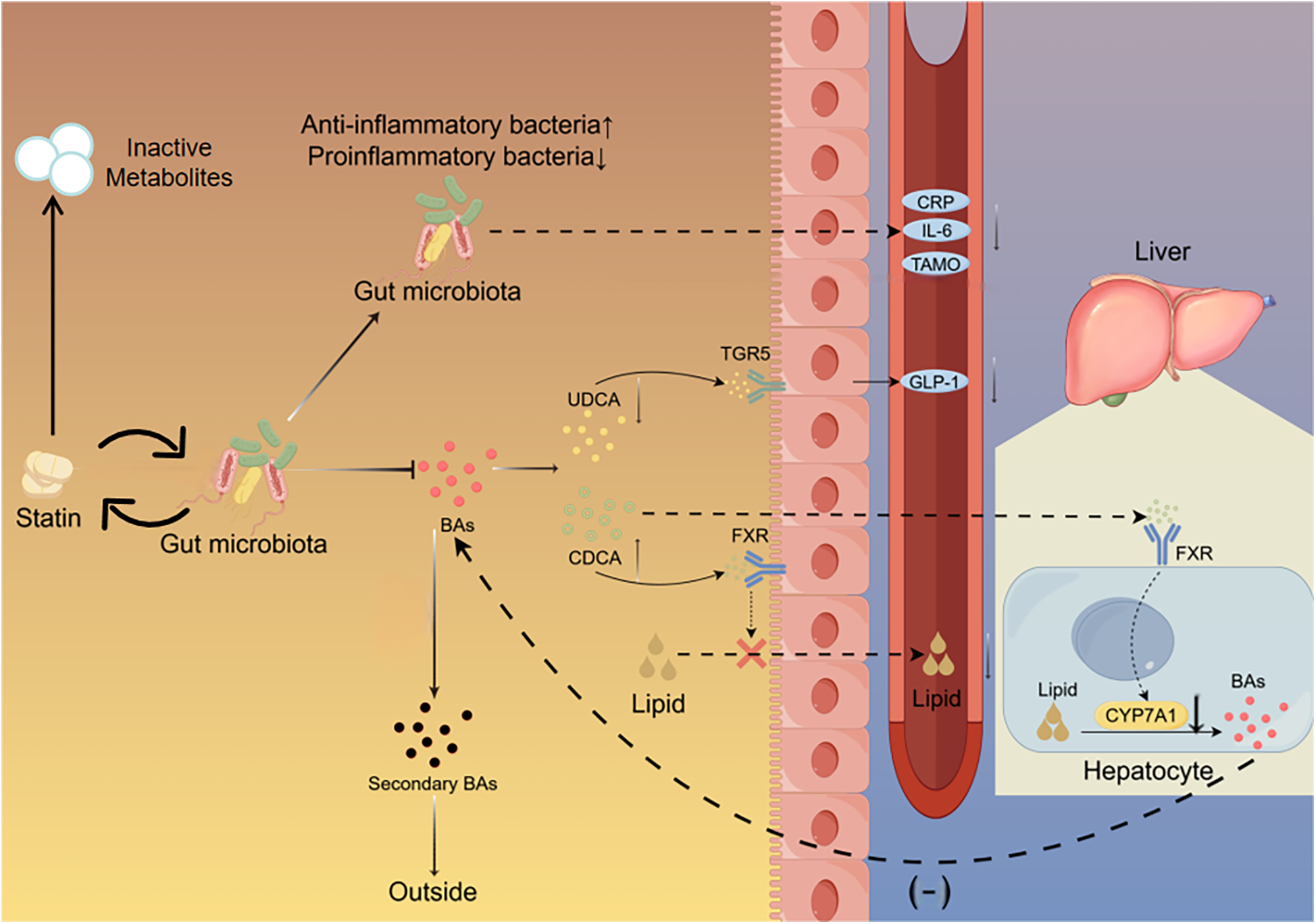
Interaction between gut microbiota and statin. Statins reduce the proportion of pro-inflammatory bacteria in the gut while increasing the proportion of anti-inflammatory bacteria. Statins decrease the proportion of UDCA which binds to the TGR5 on intestinal cells to stimulate the secretion of GLP-1. Statins increase the proportion of CDCA which binds to the FXR in the intestine to inhibit lipid absorption and the FXR in the liver to suppress the expression of Cyp7A1.The gut microbiota metabolizes statins, leading to a decrease in their bioavailability. UDCA, ursodeoxycholic acid; CDCA, chenodeoxycholic acid; TGR5, takeda G protein-coupled receptor 5; FXR, farnesoid X receptor; CyP7A1, cholesterol 7α-Hydroxylase; Bas, bile acids.
We have summarized clinical trials examining the interactions between aspirin or statins and the gut microbiota (Tables 1, 2), as these two drugs are the most widely used in the treatment of CVD and have been the subject of extensive research.
Table 1
| Drugs | Number of patients | People characteristics | Changes of gut microbiota | Results |
|---|---|---|---|---|
| Aspirin (109) | 18 | Healthy people | ↓:TMAO in blood | |
| Aspirin (110) | 50 | Healthy people | ↑:Akkermansia, Prevotella, Ruminococcaceae. ↓:Parabacteroides, Bacteroides,Dorea. |
|
| Aspirin (111) | 2,173 | Healthy people and cardiovascular disease Patients | ↓:Neococcus, Clostridium glycyrrhizinilyticum, Micromonas. | |
| Statin (8) | 888 | Obese patients | ↑:Faecalis. ↓: Bacteroides. |
|
| Atorvastatin (112) | 20 | Colon cancer patients | ↑:Lactobacillus reuteri. | ↑:Tryptophan metabolic derivatives. |
| Rosuvastatin (113) | 66 | Healthy people | ↑:Betaine, gamma-butylbetaine in blood. | |
| Statin (69) | 5,755 | Healthy people | ↑:Clostridium salmonellosus. ↓: Ysobacterium cellulorum. |
|
| Statin (74) | 143 | Acute coronary syndrome patients | ↑:Actinomycete, blautia, Bifidobacterium, anaerobic bacteria. ↓:Parabacteroides. |
↓:Some metabolites in serum that are associated with disease in blood. |
Clinical trials of cardiovascular drugs affecting gut microbiota.
Table 2
| Measure of change the gut microbiota | Drugs | The number of patients | People characteristics | Effect on the action of drugs |
|---|---|---|---|---|
| Probiotics (Bifidobacteriumbreve Bif195) (114) | Aspirin | 66 | Healthy people | Reduce the risk of small bowel injury. |
| Probiotics (Lactobacillus gasseri) (115) | Aspirin | 64 | People treated with aspirin | Gastrointestinal symptoms were reduced, and the small intestinal mucosal rupture and turning red lesions were significantly reduced. |
| PPI (116) | Aspirin | 32 | People treated with aspirin | Serum gastrin levels were increased |
| Vancomycin (117) | Simvastatin | 6 | Healthy people | No change. |
| Probiotics (Lactobacillus casei Zhang, Bifidobactetium animalis subsp. lactis V9, and Lactobacillus plantarum P-8) (118) | Atorvastatin | 33 | Patients with hyperlipidemia | The lipid-lowering effect did not change significantly compared with the control group. |
| Probiotics (Clostridium butyricum) (119) | Rosuvastatin | 96 | Patients with nonalcoholic fatty liver disease | The lipid-lowering and anti-inflammatory effect was enhanced, Reduces the risk of elevated liver enzymes. |
Clinical trials of gut microbiota affecting cardiovascular drugs.
Ezetimibe
Ezetimibe is a cholesterol absorption inhibitor that reduces blood cholesterol levels by inhibiting the absorption of cholesterol in the small intestine. It has been established that the molecular target of ezetimibe is the sterol carrier NPC1L1, a cholesterol transport protein primarily expressed in the epithelial cells of the small intestine. The gut microbiota is closely linked to NPC1L1, as studies have shown that NPC1L1 gene knockout mice exhibit a decrease in Proteobacteria and an increase in Bacteroides when fed a high-fat diet (77). Ezetimibe can similarly induce these changes, along with a reduction in Desulfovibrio (78). Notably, an increase in Proteobacteria and a decrease in Bacteroides are associated with obesity (79), while a higher abundance of Desulfovibrio has been observed in individuals with diabetes (80). Therefore, ezetimibe may regulate the composition of the gut microbiota by inhibiting NPC1L1, ultimately influencing glucose and lipid metabolism. On the other hand, metabolites produced by gut bacteria are also related to NPC1L1 expression. For instance, propionate can increase the number of regulatory T cells and the level of interleukin-10 (IL-10) in the intestinal microenvironment, thereby suppressing NPC1L1 expression in an immune-dependent manner (81). Similarly, inhibiting the production of TMAO can also suppress NPC1L1 expression (82). Thus, dietary interventions, supplementation with SCFA, or the use of TMAO production inhibitors may synergistically enhance the inhibitory effect of ezetimibe on NPC1L1, further strengthening its ability to inhibit cholesterol absorption. However, more research is needed to validate these findings.
Calcium channel blockers
Calcium channel blockers (CCB) are commonly used antihypertensive drugs in clinical practice, known for their rapid onset of action. They achieve vasodilation by inhibiting calcium channels, thereby reducing the harm of hypertension to the cardiovascular system. The gut microbiota plays a role in the metabolism of CCB. Yoo et al. incubated amlodipine with human and rat feces and observed a gradual decrease in residual amlodipine with increasing incubation time (83). Zimmermann et al. found that diltiazem can be metabolized by Bacteroides thetaiotaomicron (32). Zhou et al. found that Bacteroides dorei in the gut microbiota of spontaneously hypertensive rat (SHR) was negatively correlated with the maximum concentration and elimination half-life of nifedipine, suggesting that Bacteroides dorei may possess enzyme activity capable of directly metabolizing nifedipine. Additionally, serum glycoursodeoxycholic acid (GUDCA) was significantly elevated in SHR, which can upregulate the expression of PXR, leading to a significant increase in the expression of target genes cytochrome P450 family 3 subfamily A member 1 (CyP3A1) and multidrug resistance gene 1a (Mdr1a). CyP3A1 is a key enzyme in drug metabolism, and the P-glycoprotein (P-gp) encoded by Mdr1a limits drug absorption (7). Therefore, the gut microbiota may indirectly reduce the bioavailability of nifedipine by either directly metabolizing it or upregulating hepatic drug-metabolizing enzymes. The question arises: Could improving gut microbiota dysbiosis induced by hypertension be beneficial for increasing the bioavailability of CCB? The answer is likely yes.Studies have shown that under high-altitude hypoxia conditions, the metabolic activity of the gut microbiota weakens, significantly reducing the metabolic rate of nifedipine (84). Direct supplementation with probiotics (85) or antibiotic treatment (83) to alter the composition of the gut microbiota can also increase the bioavailability of CCB.
CCB, in turn, affect the gut microbiota. Many representative drugs of CCB such as amlodipine (86–88), lacidipine (89, 90), felodipine (91), and verapamil (92), have been demonstrated have potential antibacterial activity due to their synergistic or additive effects with many antibiotics.Their chemical structures often contain two benzene rings, and many compounds with biphenyl structures exhibit significant antimicrobial activity, such as quinolone antibiotics. The hydrophobicity of the benzene ring helps the compound penetrate bacterial cell membranes (93), the planarity of the biphenyl structure can promote interaction with bacterial targets (such as enzymes or DNA), inhibiting bacterial DNA replication (94). Studies have shown that amlodipine besylate and amlodipine aspartate can increase the abundance of Akkermansia, Bacteroides, and Lactobacillus in mice with NAFLD and hypertension (36). Akkermansia muciniphila is considered a paradigm for the next generation of probiotics, capable of improving insulin resistance, reducing blood lipids, and exerting anti-inflammatory effects (95). Bacteroides is generally considered beneficial for host metabolism and immunity in the gut (96). Nifedipine significantly increased the abundance of Eubacterium and induced changes in metabolites related to hypertension, such as reduced corticosterone. Eubacterium rectale may increase γ-aminobutyric acid (GABA) production by regulating amino acid metabolic pathways (97), and GABA can exert antihypertensive effects through central and peripheral mechanisms, including inhibiting sympathetic activity, dilating blood vessels, and promoting sodium excretion (98, 99). Amlodipine can reverse gut microbiota dysbiosis in SHR, restoring the proportion of bacteria that produce lactic acid and acetic acid (100).
Additionally, the side effects of CCB may be related to gut microbiota. Recent studies have proposed that s-amlodipine can cause liver inflammation and dysfunction in rats by affecting gut microbiota rather than liver cells. Through in vitro experiments, it was demonstrated that gut microbiota treated by s-amlodipine happened to a proliferation of Escherichia coli and a reduction in Mucispirillum and Bacillus uniformis in the rat intestine, resulting in intestinal inflammation, increased intestinal permeability, and increased production of LPS by intestinal bacteria, which led to an increase in blood LPS levels, causing final liver inflammation and dysfunction (35). However, another study showed that amlodipine has the potential to restore intestinal integrity in NAFLD mice with hypertension, alleviating liver injury and steatosis caused by nonalcoholic fatty liver disease (36). The differences between these two studies may be due to the different models. Therefore, it is necessary to explore the effects of the gut microbiota on CCB under different physiological or pathological conditions.
Angiotensin-converting enzyme inhibitor/angiotensin II receptor blocker
Angiotensin-converting enzyme inhibitors (ACEI) are compounds that inhibit the activity of angiotensin-converting enzyme (ACE). ACE catalyzes the conversion of Ang I to Ang II, the latter being a potent vasoconstrictor and activator of aldosterone release from the adrenal cortex, significantly elevating blood pressure. Angiotensin II receptor blockers (ARB) selectively block the angiotensin II receptor (AT1 type), thereby inhibiting the effects of Ang II, such as vasoconstriction, increased blood pressure, aldosterone secretion, sodium and water retention, and sympathetic nervous system activation, producing pharmacological effects similar to those of ACEI. Ang II is a key component of the renin-angiotensin-aldosterone system (RAAS) and a major contributor to hypertension and myocardial fibrosis. Recent studies have shown that gut microbiota is involved in the pathophysiological mechanism of AngII in hypertension. AngII affects the α and β diversity of gut microbiota in mice, leading to gut dysbiosis, characterized by an increase in bacteria producing TMAO (101) and a decrease in bacteria producing SCFA (102), both of which have significant impacts on the cardiovascular system. AngII can induce Th17 cells to produce interleukin-17A (IL-17A) which is a key mediator of AngII-induced hypertension and vascular dysfunction (103). Gut microbiota can promote this process because Ang II-induced IL-17A production is attenuated in mice without gut microbiota (104). Can ACEI/ARB reverse the above changes? The answer is possibly yes. Long-term candesartan treatment increases the SCFA level in intestine of SHR (105). SCFA, particularly propionate, can directly act on Th17 cells located in the cecum, reducing IL-17A secretion by inhibiting histone deacetylase (106). Enalapril can improve intestinal permeability and reduce TMAO absorption in SHR. The abundance of Coprococcus which is considered a butyrate producer was increased in SHR fed with Enalapril, which is beneficial for lowering blood pressure (107). However, Yang et al. found that Coprococcus has esterase activity, which can decompose ester-type ACEI (33), impairing the antihypertensive effect of ACEI. Lactobacillus can produce GABA through L-glutamate metabolism. High-salt diet is an important factor for hypertension, which can deplete Lactobacillus in mice (108), while candesartan can counteract the decrease of Lactobacillus caused by hypertension, and candesartan treatment also increases the intestinal expression of genes encoding tight junction proteins (such as zonula occludens, occludin, and claudin-1), improving increased intestinal permeability caused by hypertension and preventing gut microbiota translocation (105). These findings indicate that ACEI and ARB exert part of their therapeutic effects through the gut microbiota.Future research is needed to elucidate the molecular mechanisms of the interaction between ACEI/ARB and the gut microbiota.
Conclusion
There are complex interactions between gut microbiota and cardiovascular drugs. We discussed the main interactions between the two. On the one hand, cardiovascular drugs cause changes in the composition and metabolism of gut microbiota, affecting the production and absorption of metabolites related to cardiovascular health such as TMAO, SCFA and BA. On the other hand, many cardiovascular drugs will be metabolized by gut microbial enzymes due to the ability of gut microbiota to metabolize exogenous substances and the self-induction of drugs. Cardiovascular drugs will not only affect specific intestinal bacteria, but also change the overall intestinal microbial characteristics of people with CVD, making them closer to the intestinal microbial characteristics of healthy people. It has potential impact on the overall health level of the body. Personalized treatment of CVD is promising based on individual differences in the gut microbiota. By evaluating the characteristics of a patient's gut microbiota, more suitable cardiovascular drugs and dosages can be selected for their specific needs, thereby improving treatment effectiveness and reducing side effects. Further research on the mechanism of the interaction between the two is needed to provide new perspectives and new strategies for the treatment of CVD.
Statements
Author contributions
TW: Methodology, Software, Writing – original draft. QL: Data curation, Investigation, Methodology, Validation, Visualization, Writing – review & editing. HD: Formal analysis, Resources, Validation, Writing – review & editing. WH: Conceptualization, Formal analysis, Supervision, Writing – review & editing. RZ: Data curation, Resources, Visualization, Writing – review & editing. JZ: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Natural Science Foundation of China (82270331) and Qingdao Key Clinical Specialty Elite Discipline (QDZDZK-2022008).
Acknowledgments
We thank to the team that has focused on the study of gut microbiota and cardiovascular drugs, as their research findings have contributed to the formation of this review.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
Kim JK Choi MS Yoo HH Kim DH . The intake of coffee increases the absorption of aspirin in mice by modifying gut microbiome. Pharmaceutics. (2022) 14(4):746. 10.3390/pharmaceutics14040746
2.
Đanić M Pavlović N Lazarević S Stanimirov B Vukmirović S Al-Salami H et al Bioaccumulation and biotransformation of simvastatin in probiotic bacteria: a step towards better understanding of drug-bile acids-microbiome interactions. Front Pharmacol. (2023) 14:1111115. 10.3389/fphar.2023.1111115
3.
He X Zheng N He J Liu C Feng J Jia W et al Gut microbiota modulation attenuated the hypolipidemic effect of simvastatin in high-fat/cholesterol-diet fed mice. J Proteome Res. (2017) 16(5):1900–10. 10.1021/acs.jproteome.6b00984
4.
Pant A Maiti TK Mahajan D Das B . Human gut microbiota and drug metabolism. Microb Ecol. (2023) 86(1):97–111. 10.1007/s00248-022-02081-x
5.
Enright EF Joyce SA Gahan CG Griffin BT . Impact of gut microbiota-mediated bile acid metabolism on the solubilization capacity of bile salt micelles and drug solubility. Mol Pharm. (2017) 14(4):1251–63. 10.1021/acs.molpharmaceut.6b01155
6.
Clayton TA Baker D Lindon JC Everett JR Nicholson JK . Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism. Proc Natl Acad Sci U S A. (2009) 106(34):14728–33. 10.1073/pnas.0904489106
7.
Zhou R Yang H Zhu P Liu Y Zhang Y Zhang W et al Effect of gut microbiota on the pharmacokinetics of nifedipine in spontaneously hypertensive rats. Pharmaceutics. (2023) 15(8):2085. 10.3390/pharmaceutics15082085
8.
Vieira-Silva S Falony G Belda E Nielsen T Aron-Wisnewsky J Chakaroun R et al Statin therapy is associated with lower prevalence of gut microbiota dysbiosis. Nature. (2020) 581(7808):310–5. 10.1038/s41586-020-2269-x
9.
Rogers MAM Aronoff DM . The influence of non-steroidal anti-inflammatory drugs on the gut microbiome. Clin Microbiol Infect. (2016) 22(2):178.e1–.e9. 10.1016/j.cmi.2015.10.003
10.
Caparrós-Martín JA Lareu RR Ramsay JP Peplies J Reen FJ Headlam HA et al Statin therapy causes gut dysbiosis in mice through a PXR-dependent mechanism. Microbiome. (2017) 5(1):95. 10.1186/s40168-017-0312-4
11.
Kim IS Yoo DH Jung IH Lim S Jeong JJ Kim KA et al Reduced metabolic activity of gut microbiota by antibiotics can potentiate the antithrombotic effect of aspirin. Biochem Pharmacol. (2016) 122:72–9. 10.1016/j.bcp.2016.09.023
12.
Li T Ding N Guo H Hua R Lin Z Tian H et al A gut microbiota-bile acid axis promotes intestinal homeostasis upon aspirin-mediated damage. Cell Host Microbe. (2024) 32(2):191–208.e9. 10.1016/j.chom.2023.12.015
13.
Imhann F Bonder MJ Vich Vila A Fu J Mujagic Z Vork L et al Proton pump inhibitors affect the gut microbiome. Gut. (2016) 65(5):740–8. 10.1136/gutjnl-2015-310376
14.
Freedberg DE Toussaint NC Chen SP Ratner AJ Whittier S Wang TC et al Proton pump inhibitors alter specific taxa in the human gastrointestinal microbiome: a crossover trial. Gastroenterology. (2015) 149(4):883–5.e9. 10.1053/j.gastro.2015.06.043
15.
Huang YJ Ferrari MW Lin S Wang ZH . Recent advances on the role of gut microbiota in the development of heart failure by mediating immune metabolism. Curr Probl Cardiol. (2024) 49(3):102128. 10.1016/j.cpcardiol.2023.102128
16.
Hediyal TA Vichitra C Anand N Bhaskaran M Essa SM Kumar P et al Protective effects of fecal microbiota transplantation against ischemic stroke and other neurological disorders: an update. Front Immunol. (2024) 15:1324018. 10.3389/fimmu.2024.1324018
17.
Dawwas GK Brensinger CM Vajravelu RK Wu Q Kelly CR Laine L et al Long-term outcomes following multiply recurrent clostridioides difficile infection and fecal microbiota transplantation. Clin Gastroenterol Hepatol. (2022) 20(4):806–16.e6. 10.1016/j.cgh.2020.12.004
18.
Kichloo A El-Amir Z Dahiya DS Singh J Solanki D Wani F et al Impact of coexisting pneumonia in the patients admitted with Clostridium difficile infection: a retrospective study from a national inpatient database. J Investig Med. (2021) 69(5):976–82. 10.1136/jim-2021-001820
19.
Maurice CF Haiser HJ Turnbaugh PJ . Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell. (2013) 152(1–2):39–50. 10.1016/j.cell.2012.10.052
20.
Schiattarella GG Sannino A Toscano E Giugliano G Gargiulo G Franzone A et al Gut microbe-generated metabolite trimethylamine-N-oxide as cardiovascular risk biomarker: a systematic review and dose-response meta-analysis. Eur Heart J. (2017) 38(39):2948–56. 10.1093/eurheartj/ehx342
21.
Marques FZ Nelson E Chu PY Horlock D Fiedler A Ziemann M et al High-fiber diet and acetate supplementation change the gut microbiota and prevent the development of hypertension and heart failure in hypertensive mice. Circulation. (2017) 135(10):964–77. 10.1161/CIRCULATIONAHA.116.024545
22.
Watanabe M Houten SM Wang L Moschetta A Mangelsdorf DJ Heyman RA et al Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest. (2004) 113(10):1408–18. 10.1172/JCI21025
23.
Clifford BL Sedgeman LR Williams KJ Morand P Cheng A Jarrett KE et al FXR activation protects against NAFLD via bile-acid-dependent reductions in lipid absorption. Cell Metab. (2021) 33(8):1671–84.e4. 10.1016/j.cmet.2021.06.012
24.
Luo Q Hu Y Chen X Luo Y Chen J Wang H . Effects of gut microbiota and metabolites on heart failure and its risk factors: a two-sample Mendelian randomization study. Front Nutr. (2022) 9:899746. 10.3389/fnut.2022.899746
25.
Belfeki N Zayet S Yassin M Alloujami M Lefoulon A Pezel T et al Cardiovascular magnetic resonance imaging pattern in Campylobacter jejuni-related myocarditis. Microorganisms. (2022) 10(2):208. 10.3390/microorganisms10020208
26.
Hibbert B Costiniuk C Hibbert R Joseph P Alanazi H Simard T et al Cardiovascular complications of Salmonella enteritidis infection. Can J Cardiol. (2010) 26(8):323–5. 10.1016/S0828-282X(10)70444-X
27.
Pandeya A Zhang Y Cui J Yang L Li J Zhang G et al Inflammasome activation and pyroptosis mediate coagulopathy and inflammation in Salmonella systemic infection. Microbiol Res. (2023) 275:127460. 10.1016/j.micres.2023.127460
28.
Mamic P Heidenreich PA Hedlin H Tennakoon L Staudenmayer KL . Hospitalized patients with heart failure and common bacterial infections: a nationwide analysis of concomitant Clostridium Difficile infection rates and in-hospital mortality. J Card Fail. (2016) 22(11):891–900. 10.1016/j.cardfail.2016.06.005
29.
Cui X Ye L Li J Jin L Wang W Li S et al Metagenomic and metabolomic analyses unveil dysbiosis of gut microbiota in chronic heart failure patients. Sci Rep. (2018) 8(1):635. 10.1038/s41598-017-18756-2
30.
Chen J Qin Q Yan S Yang Y Yan H Li T et al Gut microbiome alterations in patients with carotid atherosclerosis. Front Cardiovasc Med. (2021) 8:739093. 10.3389/fcvm.2021.739093
31.
Lv H Zhang Z Fu B Li Z Yin T Liu C et al Characteristics of the gut microbiota of patients with symptomatic carotid atherosclerotic plaques positive for bacterial genetic material. Front Cell Infect Microbiol. (2023) 13:1296554. 10.3389/fcimb.2023.1296554
32.
Zimmermann M Zimmermann-Kogadeeva M Wegmann R Goodman AL . Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature. (2019) 570(7762):462–7. 10.1038/s41586-019-1291-3
33.
Yang T Mei X Tackie-Yarboi E Akere MT Kyoung J Mell B et al Identification of a gut commensal that compromises the blood pressure-lowering effect of ester angiotensin-converting enzyme inhibitors. Hypertension. (2022) 79(8):1591–601. 10.1161/HYPERTENSIONAHA.121.18711
34.
Yan X . The role and mechanism of intestinal flora in high salt-induced hypertension and Nifedipine Therapy (dissertation). Shandong University, Shandong, China (2020).
35.
Liu X Fang H Pan L Zhang P Lin H Gao H et al S-amlodipine induces liver inflammation and dysfunction through the alteration of intestinal microbiome in a rat model. Gut Microbes. (2024) 16(1):2316923. 10.1080/19490976.2024.2316923
36.
Li Y Zhao D Qian M Liu J Pan C Zhang X et al Amlodipine, an anti-hypertensive drug, alleviates non-alcoholic fatty liver disease by modulating gut microbiota. Br J Pharmacol. (2022) 179(9):2054–77. 10.1111/bph.15768
37.
Shi L Duan Y Fang N Zhang N Yan S Wang K et al Lactobacillus gasseri prevents ibrutinib-associated atrial fibrillation through butyrate. Europace. (2025) 27(2):euaf018. 10.1093/europace/euaf018
38.
Wen Y Yang L Wang Z Liu X Gao M Zhang Y et al Blocked conversion of Lactobacillus johnsonii derived acetate to butyrate mediates copper-induced epithelial barrier damage in a pig model. Microbiome. (2023) 11(1):218. 10.1186/s40168-023-01655-2
39.
Deng M Wu X Duan X Xu J Yang X Sheng X et al Lactobacillus paracasei L9 improves colitis by expanding butyrate-producing bacteria that inhibit the IL-6/STAT3 signaling pathway. Food Funct. (2021) 12(21):10700–13. 10.1039/D1FO02077C
40.
Gao Z Yin J Zhang J Ward RE Martin RJ Lefevre M et al Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes. (2009) 58(7):1509–17. 10.2337/db08-1637
41.
Warmbrunn MV Boulund U Aron-Wisnewsky J de Goffau MC Abeka RE Davids M et al Networks of gut bacteria relate to cardiovascular disease in a multi-ethnic population: the HELIUS study. Cardiovasc Res. (2024) 120(4):372–84. 10.1093/cvr/cvae018
42.
Zhang Y Sun D Zhao X Luo Y Yu H Zhou Y et al Bacteroides fragilis prevents aging-related atrial fibrillation in rats via regulatory T cells-mediated regulation of inflammation. Pharmacol Res. (2022) 177:106141. 10.1016/j.phrs.2022.106141
43.
Yan X Jin J Su X Yin X Gao J Wang X et al Intestinal flora modulates blood pressure by regulating the synthesis of intestinal-derived corticosterone in high salt-induced hypertension. Circ Res. (2020) 126(7):839–53. 10.1161/CIRCRESAHA.119.316394
44.
Li Z-H Weng J Yan J Zeng Y-H Hao Q-Y Sheng H-F et al Puerarin alleviates atherosclerosis via the inhibition of Prevotella copri and its trimethylamine production. Gut. (2024) 73(12):1934–43. 10.1136/gutjnl-2024-331880
45.
Li J Zhao F Wang Y Chen J Tao J Tian G et al Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome. (2017) 5(1):14. 10.1186/s40168-016-0222-x
46.
Yang C Lan R Zhao L Pu J Hu D Yang J et al Prevotella copri alleviates hyperglycemia and regulates gut microbiota and metabolic profiles in mice. mSystems. (2024) 9(7):e0053224. 10.1128/msystems.00532-24
47.
Kovatcheva-Datchary P Nilsson A Akrami R Lee YS De Vadder F Arora T et al Dietary fiber-induced improvement in glucose metabolism is associated with increased abundance of prevotella. Cell Metab. (2015) 22(6):971–82. 10.1016/j.cmet.2015.10.001
48.
Li YJ Chen X Kwan TK Loh YW Singer J Liu Y et al Dietary fiber protects against diabetic nephropathy through short-chain fatty acid-mediated activation of G protein-coupled receptors GPR43 and GPR109A. J Am Soc Nephrol. (2020) 31(6):1267–81. 10.1681/ASN.2019101029
49.
Wang Z Klipfell E Bennett BJ Koeth R Levison BS Dugar B et al Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. (2011) 472(7341):57–63. 10.1038/nature09922
50.
Koeth RA Wang Z Levison BS Buffa JA Org E Sheehy BT et al Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. (2013) 19(5):576–85. 10.1038/nm.3145
51.
Zhang X Gérard P . Diet-gut microbiota interactions on cardiovascular disease. Comput Struct Biotechnol J. (2022) 20:1528–40. 10.1016/j.csbj.2022.03.028
52.
Zhou S Xue J Shan J Hong Y Zhu W Nie Z et al Gut-flora-dependent metabolite trimethylamine-N-oxide promotes atherosclerosis-associated inflammation responses by indirect ROS stimulation and signaling involving AMPK and SIRT1. Nutrients. (2022) 14(16):3338. 10.3390/nu14163338
53.
Khalil H Kanisicak O Prasad V Correll RN Fu X Schips T et al Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest. (2017) 127(10):3770–83. 10.1172/JCI94753
54.
Natarajan N Hori D Flavahan S Steppan J Flavahan NA Berkowitz DE et al Microbial short chain fatty acid metabolites lower blood pressure via endothelial G protein-coupled receptor 41. Physiol Genomics. (2016) 48(11):826–34. 10.1152/physiolgenomics.00089.2016
55.
Chen Y Xu C Huang R Song J Li D Xia M . Butyrate from pectin fermentation inhibits intestinal cholesterol absorption and attenuates atherosclerosis in apolipoprotein E-deficient mice. J Nutr Biochem. (2018) 56:175–82. 10.1016/j.jnutbio.2018.02.011
56.
Jiang Z Zhuo LB He Y Fu Y Shen L Xu F et al The gut microbiota-bile acid axis links the positive association between chronic insomnia and cardiometabolic diseases. Nat Commun. (2022) 13(1):3002. 10.1038/s41467-022-30712-x
57.
Wang H Wang J Cui H Fan C Xue Y Liu H et al Inhibition of fatty acid uptake by TGR5 prevents diabetic cardiomyopathy. Nat Metab. (2024) 6(6):1161–77. 10.1038/s42255-024-01036-5
58.
Hu X Yan J Huang L Araujo C Peng J Gao L et al INT-777 attenuates NLRP3-ASC inflammasome-mediated neuroinflammation via TGR5/cAMP/PKA signaling pathway after subarachnoid hemorrhage in rats. Brain Behav Immun. (2021) 91:587–600. 10.1016/j.bbi.2020.09.016
59.
Huang S Ma S Ning M Yang W Ye Y Zhang L et al TGR5 agonist ameliorates insulin resistance in the skeletal muscles and improves glucose homeostasis in diabetic mice. Metab Clin Exp. (2019) 99:45–56. 10.1016/j.metabol.2019.07.003
60.
Castellanos-Jankiewicz A Guzmán-Quevedo O Fénelon VS Zizzari P Quarta C Bellocchio L et al Hypothalamic bile acid-TGR5 signaling protects from obesity. Cell Metab. (2021) 33(7):1483–92.e10. 10.1016/j.cmet.2021.04.009
61.
Sandek A Swidsinski A Schroedl W Watson A Valentova M Herrmann R et al Intestinal blood flow in patients with chronic heart failure: a link with bacterial growth, gastrointestinal symptoms, and cachexia. J Am Coll Cardiol. (2014) 64(11):1092–102. 10.1016/j.jacc.2014.06.1179
62.
Pasini E Aquilani R Testa C Baiardi P Angioletti S Boschi F et al Pathogenic gut flora in patients with chronic heart failure. JACC Heart Fail. (2016) 4(3):220–7. 10.1016/j.jchf.2015.10.009
63.
Luedde M Winkler T Heinsen FA Rühlemann MC Spehlmann ME Bajrovic A et al Heart failure is associated with depletion of core intestinal microbiota. ESC Heart Fail. (2017) 4(3):282–90. 10.1002/ehf2.12155
64.
Eikelboom JW Hirsh J Spencer FA Baglin TP Weitz JI . Antiplatelet drugs: antithrombotic therapy and prevention of thrombosis, 9th ed: American college of chest physicians evidence-based clinical practice guidelines. Chest. (2012) 141(2 Suppl):e89S–e119S. 10.1378/chest.11-2293
65.
Upreti RK Kannan A Pant AB . Experimental impact of aspirin exposure on rat intestinal bacteria, epithelial cells and cell line. Hum Exp Toxicol. (2010) 29(10):833–43. 10.1177/0960327110363333
66.
Wang WH Wong WM Dailidiene D Berg DE Gu Q Lai KC et al Aspirin inhibits the growth of Helicobacter pylori and enhances its susceptibility to antimicrobial agents. Gut. (2003) 52(4):490–5. 10.1136/gut.52.4.490
67.
Bai Z Liu Y Zhao Y Yan R Yang L Ma H et al Aspirin ameliorates atherosclerotic immuno-inflammation through regulating the treg/Th17 axis and CD39-CD73 adenosine signaling via remodeling the gut microbiota in ApoE(-/-) mice. Int Immunopharmacol. (2023) 120:110296. 10.1016/j.intimp.2023.110296
68.
Mattiello T Guerriero R Lotti LV Trifirò E Felli MP Barbarulo A et al Aspirin extrusion from human platelets through multidrug resistance protein-4-mediated transport: evidence of a reduced drug action in patients after coronary artery bypass grafting. J Am Coll Cardiol. (2011) 58(7):752–61. 10.1016/j.jacc.2011.03.049
69.
Koponen K Kambur O Joseph B Ruuskanen MO Jousilahti P Salido R et al Role of gut microbiota in statin-associated new-onset diabetes-A cross-sectional and prospective analysis of the FINRISK 2002 cohort. Arterioscler Thromb Vasc Biol. (2024) 44(2):477–87. 10.1161/ATVBAHA.123.319458
70.
She J Tuerhongjiang G Guo M Liu J Hao X Guo L et al Statins aggravate insulin resistance through reduced blood glucagon-like peptide-1 levels in a microbiota-dependent manner. Cell Metab. (2024) 36(2):408–21.e5. 10.1016/j.cmet.2023.12.027
71.
Escalante V Nayak RR Noecker C Babdor J Spitzer M Deutschbauer AM et al Simvastatin induces human gut bacterial cell surface genes. Mol Microbiol. (2024) 122(3):372–86. 10.1111/mmi.15151
72.
Oesterle A Laufs U Liao JK . Pleiotropic effects of statins on the cardiovascular system. Circ Res. (2017) 120(1):229–43. 10.1161/CIRCRESAHA.116.308537
73.
Khan TJ Ahmed YM Zamzami MA Siddiqui AM Khan I Baothman OAS et al Atorvastatin treatment modulates the gut Microbiota of the hypercholesterolemic patients. Omics. (2018) 22(2):154–63. 10.1089/omi.2017.0130
74.
Hu X Li H Zhao X Zhou R Liu H Sun Y et al Multi-omics study reveals that statin therapy is associated with restoration of gut microbiota homeostasis and improvement in outcomes in patients with acute coronary syndrome. Theranostics. (2021) 11(12):5778–93. 10.7150/thno.55946
75.
Kim J Lee H An J Song Y Lee CK Kim K et al Alterations in gut microbiota by statin therapy and possible intermediate effects on hyperglycemia and hyperlipidemia. Front Microbiol. (2019) 10:1947. 10.3389/fmicb.2019.01947
76.
Li DY Wang Z Li XS Hazen SL Tang WHW . Relationship between statin use and trimethylamine N-oxide in cardiovascular risk assessment. J Am Coll Cardiol. (2018) 71(11 Supplement):A115. 10.1016/S0735-1097(18)30656-9
77.
Zhong CY Sun WW Ma Y Zhu H Yang P Wei H et al Microbiota prevents cholesterol loss from the body by regulating host gene expression in mice. Sci Rep. (2015) 5:10512. 10.1038/srep10512
78.
Jin J Wang J Cheng R Ren Y Miao Z Luo Y et al Orlistat and ezetimibe could differently alleviate the high-fat diet-induced obesity phenotype by modulating the gut microbiota. Front Microbiol. (2022) 13:908327. 10.3389/fmicb.2022.908327
79.
Everard A Lazarevic V Gaïa N Johansson M Ståhlman M Backhed F et al Microbiome of prebiotic-treated mice reveals novel targets involved in host response during obesity. ISME J. (2014) 8(10):2116–30. 10.1038/ismej.2014.45
80.
Murphy R Tsai P Jüllig M Liu A Plank L Booth M . Differential changes in gut microbiota after gastric bypass and sleeve gastrectomy bariatric surgery vary according to diabetes remission. Obes Surg. (2017) 27(4):917–25. 10.1007/s11695-016-2399-2
81.
Haghikia A Zimmermann F Schumann P Jasina A Roessler J Schmidt D et al Propionate attenuates atherosclerosis by immune-dependent regulation of intestinal cholesterol metabolism. Eur Heart J. (2022) 43(6):518–33. 10.1093/eurheartj/ehab644
82.
Pathak P Helsley RN Brown AL Buffa JA Choucair I Nemet I et al Small molecule inhibition of gut microbial choline trimethylamine lyase activity alters host cholesterol and bile acid metabolism. Am J Physiol Heart Circ Physiol. (2020) 318(6):H1474–h86. 10.1152/ajpheart.00584.2019
83.
Yoo HH Kim IS Yoo DH Kim DH . Effects of orally administered antibiotics on the bioavailability of amlodipine: gut microbiota-mediated drug interaction. J Hypertens. (2016) 34(1):156–62. 10.1097/HJH.0000000000000773
84.
Zhang J Chen Y Sun Y Wang R Zhang J Jia Z . Plateau hypoxia attenuates the metabolic activity of intestinal flora to enhance the bioavailability of nifedipine. Drug Deliv. (2018) 25(1):1175–81. 10.1080/10717544.2018.1469687
85.
Kato R Yuasa H Inoue K Iwao T Tanaka K Ooi K et al Effect of Lactobacillus casei on the absorption of nifedipine from rat small intestine. Drug Metab Pharmacokinet. (2007) 22(2):96–102. 10.2133/dmpk.22.96
86.
Ugurel E Turgut-Balik D . Synergistic combination of carvedilol, amlodipine, amitriptyline, and antibiotics as an alternative treatment approach for the susceptible and multidrug-resistant A. Baumannii infections via drug repurposing. Eur J Clin Microbiol Infect Dis. (2023) 42(9):1063–72. 10.1007/s10096-023-04634-5
87.
Kumar KA Ganguly K Mazumdar K Dutta NK Dastidar SG Chakrabarty AN . Amlodipine: a cardiovascular drug with powerful antimicrobial property. Acta Microbiol Pol. (2003) 52(3):285–92.
88.
Dutta NK Mazumdar K DasGupta A Dastidar SG . In vitro and in vivo efficacies of amlodipine against listeria monocytogenes. Eur J Clin Microbiol Infect Dis. (2009) 28(7):849–53. 10.1007/s10096-009-0703-y
89.
Dasgupta A Chaki S Mukherjee S Lourduraja J Mazumdar K Dutta NK et al Experimental analyses of synergistic combinations of antibiotics with a recently recognised antibacterial agent, lacidipine. Eur J Clin Microbiol Infect Dis. (2010) 29(2):239–43. 10.1007/s10096-009-0845-y
90.
Dasgupta A Jeyaseeli L Dutta NK Mazumdar K Karak P Dastidar SG et al Studies on the antimicrobial potential of the cardiovascular drug lacidipine. In Vivo. (2007) 21(5):847–50.
91.
Truong M Monahan LG Carter DA Charles IG . Repurposing drugs to fast-track therapeutic agents for the treatment of cryptococcosis. PeerJ. (2018) 6:e4761. 10.7717/peerj.4761
92.
Viljoen A Raynaud C Johansen MD Roquet-Banères F Herrmann JL Daher W et al Verapamil improves the activity of bedaquiline against Mycobacterium abscessus in vitro and in macrophages. Antimicrob Agents Chemother. (2019) 63(9):e00705–19. 10.1128/AAC.00705-19
93.
Lee JK Park SC Hahm KS Park Y . Antimicrobial HPA3NT3 peptide analogs: placement of aromatic rings and positive charges are key determinants for cell selectivity and mechanism of action. Biochim Biophys Acta. (2013) 1828(2):443–54. 10.1016/j.bbamem.2012.09.005
94.
Pereira JA Pessoa AM Cordeiro MN Fernandes R Prudêncio C Noronha JP et al Quinoxaline, its derivatives and applications: a state of the art review. Eur J Med Chem. (2015) 97:664–72. 10.1016/j.ejmech.2014.06.058
95.
Everard A Belzer C Geurts L Ouwerkerk JP Druart C Bindels LB et al Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci U S A. (2013) 110(22):9066–71. 10.1073/pnas.1219451110
96.
Zafar H Saier MH Jr . Gut bacteroides species in health and disease. Gut Microbes. (2021) 13(1):1–20. 10.1080/19490976.2020.1848158
97.
Hao Z Meng C Li L Feng S Zhu Y Yang J et al Positive mood-related gut microbiota in a long-term closed environment: a multiomics study based on the “lunar palace 365” experiment. Microbiome. (2023) 11(1):88. 10.1186/s40168-023-01506-0
98.
Gajić Bojić M Aranđelović J Škrbić R Savić MM . Peripheral GABA(A) receptors—physiological relevance and therapeutic implications. Pharmacol Ther. (2025) 266:108759. 10.1016/j.pharmthera.2024.108759
99.
Dupont AG Légat L . GABA is a mediator of brain AT(1) and AT(2) receptor-mediated blood pressure responses. Hypertens Res. (2020) 43(10):995–1005. 10.1038/s41440-020-0470-9
100.
González-Correa C Moleón J Miñano S Robles-Vera I Toral M Barranco AM et al Differing contributions of the gut microbiota to the blood pressure lowering effects induced by first-line antihypertensive drugs. Br J Pharmacol. (2024) 181(18):3420–44. 10.1111/bph.16410
101.
Jiang S Shui Y Cui Y Tang C Wang X Qiu X et al Gut microbiota dependent trimethylamine N-oxide aggravates angiotensin II-induced hypertension. Redox Biol. (2021) 46:102115. 10.1016/j.redox.2021.102115
102.
Muralitharan RR Nakai ME Snelson M Zheng T Dinakis E Xie L et al Influence of angiotensin II on the gut microbiome: modest effects in comparison to experimental factors. Cardiovasc Res. (2024) 120(10):1155–63. 10.1093/cvr/cvae062
103.
Madhur MS Lob HE McCann LA Iwakura Y Blinder Y Guzik TJ et al Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension. (2010) 55(2):500–7. 10.1161/HYPERTENSIONAHA.109.145094
104.
Karbach SH Schönfelder T Brandão I Wilms E Hörmann N Jäckel S et al Gut microbiota promote angiotensin II-induced arterial hypertension and vascular dysfunction. J Am Heart Assoc. (2016) 5(9):e003698. 10.1161/JAHA.116.003698
105.
Wu D Tang X Ding L Cui J Wang P Du X et al Candesartan attenuates hypertension-associated pathophysiological alterations in the gut. Biomed Pharmacother. (2019) 116:109040. 10.1016/j.biopha.2019.109040
106.
Dupraz L Magniez A Rolhion N Richard ML Da Costa G Touch S et al Gut microbiota-derived short-chain fatty acids regulate IL-17 production by mouse and human intestinal γδ T cells. Cell Rep. (2021) 36(1):109332. 10.1016/j.celrep.2021.109332
107.
Li HB Yang T Richards EM Pepine CJ Raizada MK . Maternal treatment with captopril persistently alters gut-brain communication and attenuates hypertension of male offspring. Hypertension. (2020) 75(5):1315–24. 10.1161/HYPERTENSIONAHA.120.14736
108.
Wilck N Matus MG Kearney SM Olesen SW Forslund K Bartolomaeus H et al Salt-responsive gut commensal modulates TH17 axis and disease. Nature. (2017) 551(7682):585–9. 10.1038/nature24628
109.
Zhu W Wang Z Tang WHW Hazen SL . Gut microbe-generated trimethylamine N-oxide from dietary choline is prothrombotic in subjects. Circulation. (2017) 135(17):1671–3. 10.1161/CIRCULATIONAHA.116.025338
110.
Prizment AE Staley C Onyeaghala GC Vivek S Thyagarajan B Straka RJ et al Randomised clinical study: oral aspirin 325 mg daily vs placebo alters gut microbial composition and bacterial taxa associated with colorectal cancer risk. Aliment Pharmacol Ther. (2020) 52(6):976–87. 10.1111/apt.16013
111.
Forslund SK Chakaroun R Zimmermann-Kogadeeva M Markó L Aron-Wisnewsky J Nielsen T et al Combinatorial, additive and dose-dependent drug–microbiome associations. Nature. (2021) 600(7889):500–5. 10.1038/s41586-021-04177-9
112.
Han JX Tao ZH Wang JL Zhang L Yu CY Kang ZR et al Microbiota-derived tryptophan catabolites mediate the chemopreventive effects of statins on colorectal cancer. Nat Microbiol. (2023) 8(5):919–33. 10.1038/s41564-023-01363-5
113.
Kummen M Solberg OG Storm-Larsen C Holm K Ragnarsson A Trøseid M et al Rosuvastatin alters the genetic composition of the human gut microbiome. Sci Rep. (2020) 10(1):5397. 10.1038/s41598-020-62261-y
114.
Mortensen B Murphy C O'Grady J Lucey M Elsafi G Barry L et al Bifidobacteriumbreve Bif195 protects against small-intestinal damage caused by acetylsalicylic acid in healthy volunteers. Gastroenterology. (2019) 157(3):637–46.e4. 10.1053/j.gastro.2019.05.008
115.
Suzuki T Masui A Nakamura J Shiozawa H Aoki J Nakae H et al Yogurt containing Lactobacillus gasseri mitigates aspirin-induced small bowel injuries: a prospective, randomized, double-blind, placebo-controlled trial. Digestion. (2017) 95(1):49–54. 10.1159/000452361
116.
Tsujimoto H Hirata Y Ueda Y Kinoshita N Tawa H Tanaka Y et al Effect of a proton-pump inhibitor on intestinal microbiota in patients taking low-dose aspirin. Eur J Clin Pharmacol. (2021) 77(11):1639–48. 10.1007/s00228-021-03167-0
117.
Sunwoo J Ji SC Kim AH Yu KS Cho JY Jang IJ et al Impact of vancomycin-induced changes in the intestinal Microbiota on the pharmacokinetics of simvastatin. Clin Transl Sci. (2020) 13(4):752–60. 10.1111/cts.12761
118.
Tian Y Wu G Zhao X Zhang H Ren M Song X et al Probiotics combined with atorvastatin administration in the treatment of hyperlipidemia: a randomized, double-blind, placebo-controlled clinical trial. Medicine (Baltimore). (2024) 103(21):e37883. 10.1097/MD.0000000000037883
119.
Zhu W Yan M Cao H Zhou J Xu Z . Effects of Clostridium butyricum capsules combined with rosuvastatin on intestinal Flora, lipid metabolism, liver function and inflammation in NAFLD patients. Cell Mol Biol (Noisy-le-grand). (2022) 68(2):64–9. 10.14715/cmb/2021.67.5.9
Summary
Keywords
gut microbiota, cardiovascular diseases, drugs, aspirin, statin
Citation
Wang T, Lan Q, Deng H, Han W, Zhang R and Zhong J (2025) Interactions between gut microbiota and cardiovascular drugs: effects on drug therapeutic effect and side effect. Front. Cardiovasc. Med. 12:1570008. doi: 10.3389/fcvm.2025.1570008
Received
02 February 2025
Accepted
27 June 2025
Published
10 July 2025
Volume
12 - 2025
Edited by
Mateusz Szudzik, Medical University of Warsaw, Poland
Reviewed by
Katongo Hope Mutengo, University of Zambia, Zambia
Ayoola Awosika, University of Illinois at Chicago, United States
Updates
Copyright
© 2025 Wang, Lan, Deng, Han, Zhang and Zhong.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
* Correspondence: Jingquan Zhong 198762000778@email.sdu.edu.cn
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.