 Adil Ijaz
Adil Ijaz Bhavya Yarlagadda
Bhavya Yarlagadda Marco Orecchioni
Marco Orecchioni- 1Immunology Center of Georgia, Augusta University, Augusta, GA, United States
- 2Department of Pharmacology and Toxicology, Augusta University, Augusta, GA, United States
Atherosclerosis is a complex immuno-metabolic disease characterized by lipid accumulation and chronic inflammation within arterial walls, leading to cardiovascular events such as stroke and myocardial infarction. Central to the disease are arterial plaques initiated by modified low-density lipoproteins (LDL), particularly oxidized LDL, deposited in the arterial intima. This deposition activates tissue-resident macrophages (TRMs), inducing a lipid-loaded “foamy” phenotype. Additionally, endothelial dysfunction promotes monocyte recruitment, differentiation into macrophages, and further foam cell formation. Foamy macrophages were initially identified as anti-inflammatory but have recently shown dual functionality, possibly depending on the disease stage and phenotype. Recent mouse and human studies also identified subsets of “foamy” macrophages with both pro and anti-inflammatory features. This review examines “foamy” macrophage complex roles and phenotypic diversity in atherosclerosis, emphasizing their potential as therapeutic targets to reduce inflammation and slow disease progression.
1 Introduction
Atherosclerosis is a lipid-driven chronic inflammatory condition of the arteries that leads to stenosis of the blood vessels, hence causing cardiovascular complications such as ischemia, stroke, and heart attack, which is a leading cause of death in the western countries (1). Evidence suggests that damage to the integrity of endothelial cells lining the arterial intima due to multiple insults, such as oxidative stress, leads to the infiltration and accumulation of low-density lipoproteins (LDL) in vascular intimal space (2, 3). Excessive reactive oxygen species (ROS) in arterial intima due to high-fat diet feeding and oxidative stress causes oxidation of LDL to transform into oxidized LDL (ox-LDL), a primary driver of atherogenesis (4). The accumulation of ox-LDL and other modified LDLs, such as acetylated LDL, triggers tissue-resident macrophages (TRM), such as aortic intima resident macrophages (MacAIR) to acquire a lipid-loaded phenotype (defined here as “foamy” throughout the manuscript) to clear the lipid-rich microenvironment and restore homeostasis (5). Concurrently, ox-LDL, as well as endothelial dysfunction, drives monocyte chemotaxis into the intimal space, where they further differentiate into macrophages, leading to the formation of additional foam cells in the process of getting rid of excessive cholesterol from the microenvironment (6, 7). Macrophage-derived foam cells play a significant role in shaping subsequent immune responses and atherosclerotic disease progression (8). Thus, it is known that the number of foam cells increases in the lesion as atherosclerosis progresses. Normal blood vessels have indeed few tissue-resident macrophages, most of which reside in the adventitial space (9).
Macrophages are key innate immune cells that respond to pathogens, tissue-derived signals, metabolites, and dying cells using a wide range of sensors (10). They originate early in development (11–15), populating most tissues as tissue-resident macrophages (TRMs), a population conserved from Drosophila to Humans (16–19). TRMs typically persist within tissues, closely interacting with local cells (20). These embryonically derived macrophages differ developmentally and functionally from monocyte-derived macrophages (MDMs), which arise from bone marrow monocytes sharing a common progenitor with dendritic cells, known as the monocyte dendritic cell precursor (MDP) (11, 19, 21–25). Distinguishing TRMs can be challenging due to tissue-specific marker expression. For example, F4/80 labels resident macrophages in the spleen (26) and liver (27), MerTK is found in the liver (28) and lungs (29) macrophages, CD64 along with F4/80 in the aorta (9), whereas TREM2 is specific for macrophages residing in the brain (30) in mice. Similarly, to identify monocyte-derived macrophages, Ccr2, Cd11bhi, and Ly6c are commonly used markers (31). Vascular TRM derives from CX3CR1+ embryonic precursors with a postnatal contribution from BM-derived monocytes that colonize the arterial adventitia immediately after birth (11, 32). These vascular macrophages persist into adulthood through local proliferation (33). In atherosclerosis, TRM as well as MDMs can acquire a foamy phenotype characterized by the expression of TREM2. These foam cells were initially viewed as anti-inflammatory and pro-resolving (34). However, recent findings show that myeloid-specific deletion of TREM2 cells markedly attenuates plaque progression (35). On the other hand, TREM2 has also been shown to enhance plaque stability and fibrous cap formation in established atherosclerosis (36). These evidences underscore the need to clarify the inflammatory mechanisms driving foamy macrophage behavior.
This review explores the complex and sometimes controversial processes underlying foamy macrophage formation, functions, and phenotypes in atherosclerosis. By dissecting these aspects, we aim to pinpoint possible therapeutic targets in foamy macrophages that can help reduce inflammation and slow atherosclerosis progression.
2 Endothelial dysfunction and oxidized LDL
The vascular endothelium plays a crucial role in maintaining blood vessel homeostasis, acting as a gatekeeper that regulates the movement of macromolecules and fluids between the vascular lumen and the surrounding stroma (37). Cholesterol is essential for proper cell function, as it is an integral component of the cell membrane. LDL acts as a chief carrier of cholesterol to cells (38). Under normal physiological conditions, excessive free cholesterol is transported out of the cells to the liver, mediated by high-density lipoprotein (HDL) (39).
The vascular endothelium is critical for maintaining vascular homeostasis by regulating vasodilation, vasoconstriction, and vascular permeability through tightly controlled mechanisms involving endothelin-1, angiotensin II, prostacyclin, and nitric oxide (40, 41). Impaired vasodilation in response to stimuli (e.g., bradykinin) often signals the onset of endothelial dysfunction. Under oxidative stress, reduced nitric oxide production and excessive ROS disrupt normal vasodilation (42). Simultaneously, increased ROS quenches nitric oxide to form peroxynitrite, which further exacerbates endothelial dysfunction (43). This oxidative environment also increases the expression of adhesion molecules such as VCAM-1 and ICAM-1, promoting monocyte adhesion and inflammation (44, 45). Endothelial dysfunction is widely recognized as an early event in atherosclerosis (46, 47).
Following the initial stage, the progression of endothelial dysfunction is characterized by the retention of LDL in endothelial cells, leading to its modification to oxidized LDL (ox-LDL) through the activity of enzymes like NADPH oxidases (NOX), lipoxygenases, xanthine oxidase (XO), myeloperoxidase (MPO), mitochondria reactive oxygen species (ROS) and uncoupled endothelial nitric oxide synthase (eNOS) (48, 49). Elevated levels of ox-LDL further activate the endothelial cells, leading to the expression of surface adhesion molecules that trigger circulating monocyte transmigration into sub-endothelial layers, where they differentiate into macrophages (50, 51). To reduce the elevated levels of ox-LDL at the lesion site, both TRM and MDMs recognize this modified LDL and engulf it via the scavenger receptors such as CD36, lectin-like ox-LDL receptor-1 (LOX-1) and scavenger receptor A1(SR-A1) leading to the formation of foamy macrophages during the initial stage of plaque formation (52, 53). It has been shown that up to 90% of modified LDL uptake by macrophages is mediated by CD36 and SR-A1 (54).
3 Macrophage profiling in atherosclerotic plaque
In the atherosclerotic neointima, macrophages play diverse and critical roles, including clearing cholesterol, promoting an anti-inflammatory phenotype that promotes tissue repair and plaque stabilization, as well as driving a proinflammatory microenvironment that favors the progression of atherosclerosis and leads to unstable plaques (8, 55). Because of these divergent roles, macrophages represent an attractive therapeutic target for atherosclerosis, including (but not limited to) inhibiting monocyte recruitment to the lesion site, reducing their proinflammatory phenotype, and increasing their anti-inflammatory responses (8).
3.1 Macrophage subsets defined in mouse atherosclerotic plaque
Due to the initial TRM and the infiltration of MDMs, combined with distinct transcriptional signatures shaped by the microenvironment (including MacAIR), a diverse range of populations of macrophages arises in the plaque. Recent advances in single-cell technologies, such as single-cell RNA sequencing and CyTOF, have greatly improved our ability to identify different macrophage subsets in atherosclerotic plaques. At least 9 phenotypically distinct macrophage subsets with possible distinct functions have been observed in mice (56, 57). These subsets were primarily defined as resident macrophages (MacAIR, and aorta adventitia resident), TREM2 + foamy, interferon-inducible, inflammatory, and smooth muscle-derived macrophages (58). To this extent, multiple independent studies have described a huge heterogeneity in macrophages present in the atherosclerotic plaques and identified various markers to distinguish resident macrophages (5, 7, 9, 59–64), TREM2 (35, 63–67), Interferon inducible macrophages (5, 62, 66), Inflammatory macrophages (63, 65–67), and smooth muscle-derived macrophages (68, 69), all of which may have the ability to transform into foamy macrophages in mouse models of atherosclerosis (Figure 1). Recently, a metanalysis of 12 single-cell RNA sequencing datasets of healthy and atherosclerotic mouse aortas revealed distinct subpopulations with unique transcriptomic signatures within established macrophage groups, such as inflammatory (IL-1β), aortic resident (LYVE1) and foamy (TREM2hi) macrophages (56). The authors also observed 2 resident subsets based on CD209low or high expression and 2 foamy (TREM2hi) macrophage subsets differentiated based on the expression of Gpnmb and Slamf9, speculating about possible differences in pro or anti-inflammatory features among them (56). These single-cell transcriptomic analyses of mouse aortas have demonstrated more significant heterogeneity in plaque macrophages than traditional immunophenotyping (6). However, currently, most newly identified macrophage subsets are defined solely by gene expression profiles, lacking comprehensive validation through protein marker characterization and functional analyses.
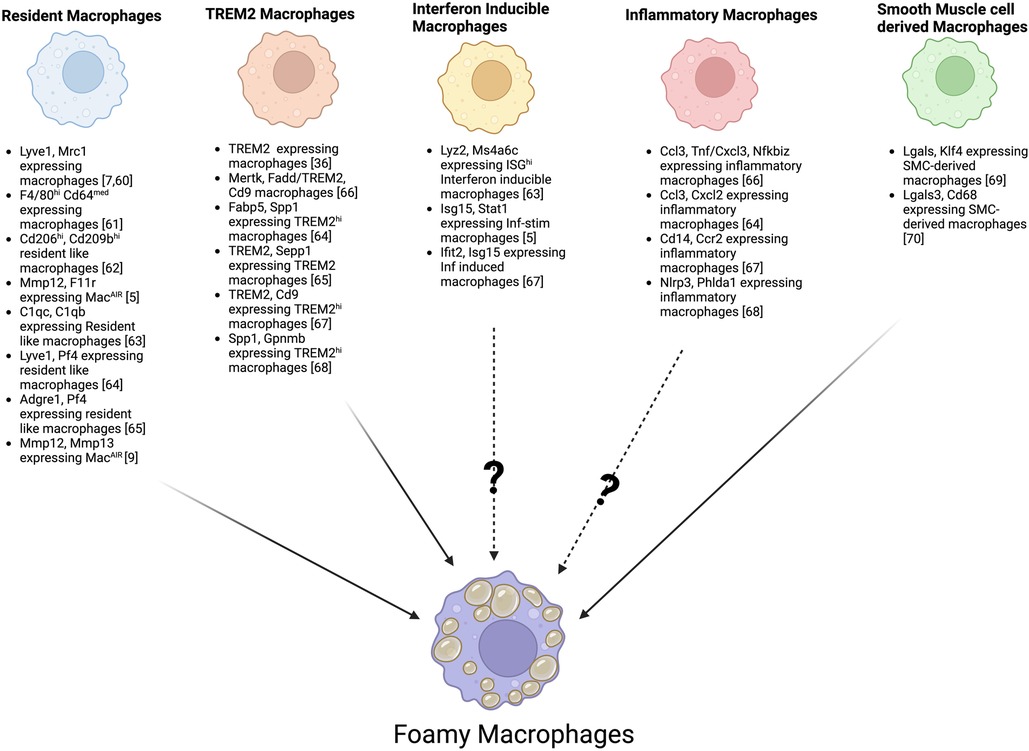
Figure 1. Foamy macrophages possible origin from distinct aortic macrophage subsets based on specific marker genes. Schematic overview illustrating how multiple macrophage subsets, including resident macrophages, TREM2-expressing macrophages, interferon-inducible macrophages, inflammatory macrophages, and smooth muscle cells–derived macrophages, may each give rise to foamy macrophages. Key publications defining various subsets of macrophages based on specific marker genes are shown, highlighting the known and potential transitions (indicated by?) that may lead to lipid accumulation and the “foamy” phenotype.
3.2 Macrophage subsets defined in human atherosclerotic plaque
Transcriptional analysis of the macrophage landscape in human atherosclerotic plaques uncovered five distinct macrophage clusters, each exhibiting different functional profiles. Most clusters showed a pro-inflammatory and macrophage activation gene signature, while one cluster demonstrated a foamy transcriptional signature (70). This foamy cluster expressed Apoe, Apoc1, and Plin2—genes involved in lipid uptake, metabolism, and accumulation (70). A recent meta-analysis identified macrophage populations in humans with gene expression patterns similar to those found in mice; notably, key transcripts from the foamy/TREM2hi signature (TREM2, Spp1, Gpnmb, Cd9) defined a distinct macrophage population in human lesions (56).
3.3 Foamy macrophages and macrophage-like cells
As discussed, macrophages present in the atherosclerotic plaque may have a foamy or non-foamy phenotype (57). Those that internalize modified LDL or aggregated LDLs via specific receptors become foamy macrophages, whereas those that do not are classified as non-foamy macrophages. Among foamy macrophage phenotypes, MacAIR are the earliest macrophages to transform into foamy macrophages, even prior to the recruitment of monocytes to the lesion site (5). The origin of foam cells in atherosclerotic plaque has been an interesting topic of discussion. Growing evidence suggests that foam cells are majorly macrophages. However, several studies have shown that also smooth muscle cells and endothelial cells transition into foam cells (71). Thus, multiple studies suggest that almost half of the foam cells may originate from smooth muscle cells (72–74). More importantly, it has been shown that upon taking up modified LDL, smooth muscle cells can even lose their contractile phenotype and express macrophage markers such as CD68, acquiring foamy macrophage-like features (75). Recently, Pan et al. demonstrated by lineage tracing studies that smooth muscle cells can express macrophage-like features and acquire a foamy phenotype in atherosclerotic plaques (76). Li et al. also suggested that smooth muscle cells derived macrophage-like cells may be transient cells as they lose macrophage markers in late atherosclerosis stages (77). This shows that smooth cells-derived macrophages can significantly influence plaque progression and stability. Similarly, macrophages and endothelial cells may also express smooth muscle cell markers (78–80).
The functionality of foamy macrophages may vary according to the macrophage subtype that undergoes this transformation and depending on the disease stage. It is overall accepted that excessive accumulation of foam cells in atherosclerotic lesions leads to the release of matrix-degrading enzymes, tissue factors, and proinflammatory cytokines ultimately creating a necrotic core with a weakened fibrous cap (81). Such unstable plaques are prone to rupture and are a major cause of myocardial infarction and stroke.
4 Oxidized LDL uptake and export in foamy macrophages
Macrophages in the arterial intima take up modified LDLs like ox-LDL as part of a lipid homeostasis mechanism, ultimately transforming into foamy macrophages (82). Once internalized, ox-LDL is delivered to lysosomes, where esterified cholesterol is converted into free (unesterified) cholesterol. This free cholesterol is then transported to the endoplasmic reticulum, re-esterified, and stored in lipid droplets (83). The formation of these foam cells is a pivotal event in atherosclerosis, influencing both lesion development and late-stage clinical outcomes such as stroke and myocardial infarction (71, 84). Hence foam cells have emerged as an attractive target to prevent atherosclerosis (85).
Surface receptors primarily involved in the uptake of modified LDL (e.g., ox-LDL) by macrophages include scavenger receptors such as CD36 and LOX1 (86). In addition, foamy macrophages upregulate TREM2, which can govern ox-LDL uptake alongside these scavenger receptors (35). Counterbalancing this internalization process, macrophages can export cholesterol via specialized efflux pathways. ATP-binding cassette (ABC) transporters, particularly ABCA1 and ABCG1, mediate the transfer of cholesterol and phospholipids onto lipid acceptors like apolipoprotein A-I (ApoA-I) and high-density lipoprotein (HDL) (87). The expression of these transporters is tightly regulated by nuclear receptors such as the liver X receptor (LXR), which responds to elevated intracellular oxysterol levels (88) and peroxisome proliferator-activated receptors (PPAR)-gamma (89). Conversely, aberrant downregulation or functional impairment of LXR and ABC transporters can tilt the balance toward pathological lipid accumulation and inflammation (90, 91). Several studies have demonstrated that cholesterol transporters like ABCA1 are crucial for preventing foam cell formation (92). However, findings in this area are not entirely consistent. For instance, mice lacking ABCA1 and SR-B1 exhibit hypocholesterolemia and foam cell accumulation but do not develop atherosclerosis (93). Meanwhile, upregulation of ABCA1 in LDLR knockout mice has been shown to exacerbate atherosclerosis (94). Thus, the precise role of these transporters in atherosclerosis remains somewhat controversial. Overall, it's clear that when cholesterol transporters are downregulated, intracellular ox-LDL accumulates, sustaining the foamy phenotype of macrophages.
5 Role of foamy macrophages in atherosclerosis
The foamy transformation not only alters the macrophage phenotype but also reprograms their functionality. Internalization of ox-LDL via the scavenger receptors CD36, SR-A1, and LOX1 (95) and possibly its signaling through other receptors like TREM2 and Olfr2 (96), ultimately activates downstream pathways that can either promote an anti-inflammatory, pro-resolving response by enhancing cholesterol clearance and efflux or trigger a pro-inflammatory response through NF-κB and inflammasome activation, thereby inducing IL-6 and IL-1β production and exacerbating inflammation and disease progression (97). IL-1β is a crucial cytokine involved in the progression of atherosclerosis, as highlighted by findings from the CANTOS trial (98). Produced through inflammasome activation, IL-1β is released during pyroptosis and primarily signals through the receptor IL-1R1, activating downstream NF-κB pathways that amplify inflammation (99). Alternatively, IL-1β can bind IL-1R2, a structurally similar decoy receptor expressed by anti-inflammatory foamy macrophages, thereby limiting inflammatory responses (100). Impaired efferocytosis and the activation of multiple cell death pathways, including apoptosis (mediated by activation of caspases 3, 7, and 9, resulting in DNA fragmentation) (101), dysfunctional autophagy (which disrupts lipid homeostasis and promotes cell death) (100), and pyroptosis (mediated by caspases 1 and 11) (102), can further contribute to the proinflammatory foamy phenotype ultimately increasing necrotic core size and plaque instability (103). Figure 2 illustrates how ox-LDL interaction with some macrophage surface receptors may lead to its export or intracellular storage, ultimately influencing the inflammatory phenotype and disease progression.
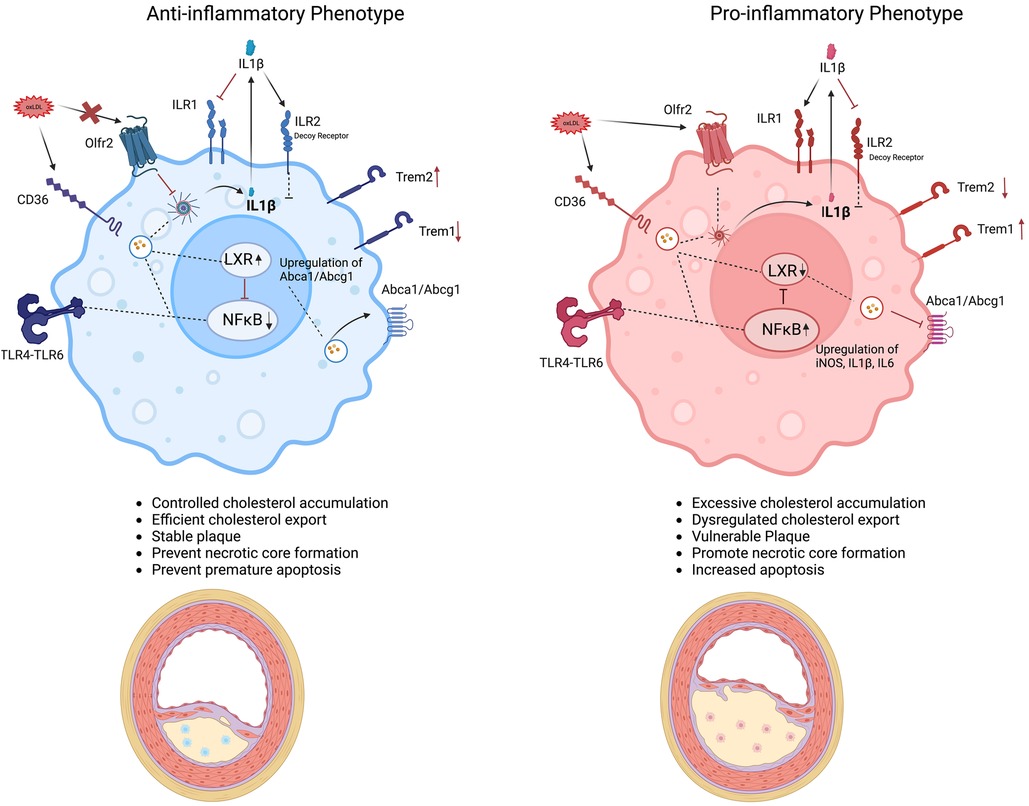
Figure 2. Drivers of foamy macrophage function and phenotype in atherosclerosis. Schematic representation of distinct macrophage foamy phenotypes within the atherosclerotic plaque environment. The anti-inflammatory foamy phenotype (left, blue) maintains controlled cholesterol accumulation, efficient cholesterol export, and reduced inflammatory signaling particularly IL-1β through increasing the expression of IL-1R2 (a decoy receptor) as well as having a dampening effect on NF-κB pathway, and maintaining higher anti-inflammatory gene signature especially IL10 contributing to plaque stability. The anti-inflammatory phenotype is also proposed to have a higher TREM2 expression and lower TREM1 expression contributing to the anti-inflammatory gene signature. In contrast, the pro-inflammatory foamy phenotype (right, pink) is marked by excessive cholesterol accumulation, dysregulated cholesterol export, and heightened production of inflammatory mediators like IL-1β and other inflammatory mediators like iNOS due to activation of NF-κB pathway and reduced expression of TREM2, promoting plaque progression and necrotic core formation. Key molecular pathways (e.g., NF-κB, LXR, TREM2, Olfr2) are highlighted, illustrating how foamy macrophage polarization and the interplay between these pathways can influence atherosclerotic lesion outcomes.
5.1 TREM2 expressing foamy macrophages
Foam cells, especially foamy macrophages, express several surface receptors that can modulate ox-LDL processing. One such receptor is TREM2, a transmembrane receptor belonging to the immunoglobulin family that is expressed on myeloid cells (104, 105). TREM2 is associated with multiple disease conditions and functions, including diabetes, apoptosis, central nervous system dysfunction, and inflammation (106–109). Previous studies using scRNA-seq analysis have demonstrated the expression of TREM2 in CD45 + cells isolated from aortic plaques of atherosclerotic-prone mice (both Ldlr−/− and Apoe−/−) (7, 59). TREM2 is particularly expressed in macrophages in the plaques and it is associated with foamy macrophage formation and differentiation (35). It has been shown that the deficiency of TREM2 in macrophages increases the phosphorylation of PPARγ, leading to its decreased transcriptional activity, which subsequently lowers transcription and surface expression of CD36, ultimately decreasing ox-LDL uptake and foam cell formation (110). Similarly, conditional deletion of TREM2 in macrophages reduces atherosclerotic plaque size and en face lesions in mice, negatively impacting foamy macrophage survival and proliferation through increased ER stress and impaired LXR signaling showing the pathogenic role of TREM2 in promoting atherosclerosis (35). However, another independent manuscript described that TREM2 function can stabilize the plaque in late stage of the disease by limiting necrotic core formation, promoting ox-LDL uptake, and enhancing foam cell survival through increased efferocytosis, suggesting the protective role of TREM2 in plaque progression (36). This dual function of TREM2 + foam macrophages could be explained by the recently described different subsets of TREM2 + macrophages that may play diverse functions during atherogenesis (56, 57). These TREM2-expressing aortic foamy macrophages were defined as TREM2hiSlamf9 and TREM2hi Gpnmb in atherosclerotic Ldlr−/− mice (36, 56). By discussing differential gene expression profiles as well as proportions, the authors proposed that TREM2hiSlamf9 macrophages show a pro-inflammatory profile characterized by elevated expression of CD72, Ch25h, Tnf, and Il1b. On the other hand, TREM2hi Gpnmb macrophages possess a specialized gene signature, including Gpnmb and Fabp5, suggesting an osteoclast-like differentiation and possible macrophage fusion (56). Interestingly, TREM2 may control these phenotypes as TREM2 deficient foamy macrophages have a reduced expression of scavenger receptors (CD36, Msr1), foam cell markers (Gpnmb, Spp1, Cd5L), and antioxidant heme oxygenase (Hmox1), and efferocytosis compared to TREM2+ foamy macrophages suggesting a role of TREM2 in lipid uptake, and foaminess in foamy macrophages (36). Overall, these data show that foamy macrophage populations in atherosclerosis have conserved yet functionally specialized transcriptional states. However, their distinct roles in disease progression still need to be established.
5.2 Strategies to lower lipid uptake and foam cell formation
Several surface or intracellular receptors or proteins have been described as either reducing or increasing foam cell formation, thereby mitigating or exacerbating the impact on atherosclerosis. Deleting these receptors or intracellular proteins primarily affects scavenger receptor expression or function, ultimately influencing lipid uptake. Li et al. have shown that ablation of the pyrimidinergic receptor P2Y6 in macrophages limits foamy macrophage formation by lowering SR-A expression and ox-LDL uptake in a mouse model (111). In another study, the macrophage-specific deletion of NFATc3 (nuclear factor of activated T cells cytoplasmic 3) promoted foam cell formation by enhancing ox-LDL uptake via CD36 and SR-A1 in mice (112). Moreover, genetic or pharmacological inhibition of Gsα, G protein stimulatory subunit α, has been shown to decrease atherosclerosis progression in mice by downregulating CD36 and SR-A1 expression (113). A recent study has described the role of a ubiquitin enzyme, USP9X, in suppressing lipid intake in macrophages both in rodents as well as humans (114). USP9X deficient macrophages exhibited increased lipid uptake and deposition as well as increased infiltration into the lesion site, which resulted in an enlarged necrotic core compared to control Apoe−/− mice (114). USP9X is a factor that suppresses lipid uptake in macrophages by targeting SR-A1 for degradation upon ox-LDL contact, thereby reducing SR-A1 internalization and foam cell formation. Conversely, USP9X genetic ablation or pharmacological inhibition promotes SR-A1 internalization and foam cell development (114).
As expected, cholesterol transporters, ABCA1 and ABCG1, are also important targets through which foamy macrophages influence atherosclerosis progression (115). A recent study has shown that YXTMD, a traditional Chinese decoction, effectively increases cholesterol efflux by activating PPARγ-LXR-ABCA1/ABCG1 pathway in foamy macrophages and attenuates atherosclerosis in Apoe−/− mice (116). Similarly, disruption of LXR signaling in Mφ can intensify both lipid accumulation and inflammatory activation, dramatically increasing atherosclerosis and plaque inflammation (117).
5.3 Foamy macrophages in human plaques
In humans, plaque macrophages, are not a homogenous cell type but rather comprise multiple subsets with diverse functional roles ranging from inflammatory mediators to plaque-stabilizing cells. To this extent, immunohistochemical analyses of human carotid atherosclerotic plaques (n = 27) also confirmed the presence of both M1-like and M2-like macrophages (118). Thus, carotid artery plaques from symptomatic coronary artery disease (CAD) patients were enriched with pro-inflammatory M1-like macrophages, whereas plaques from asymptomatic patients contained more anti-inflammatory M2-like macrophages (119). In a 2023 study, Patterson et al. analyzed symptomatic and asymptomatic carotid endarterectomy samples (70), identifying 19 distinct myeloid cell populations. Monocyte and macrophage clustering revealed four subsets expressing the myeloid markers CD14 and PTPRC, each with unique gene signatures ranging from inflammatory genes (IL-1β, NLRP3) to lipid-processing genes (FABP5, LGALS3). TREM2 expression was exclusive to foamy macrophages in human plaques (35), whereas foamy macrophage–associated genes (CD9, LPL, FABP4) were predominantly observed in asymptomatic plaques, suggesting that these foam cells may play a stabilizing role (7).
6 Inflammatory phenotype of foamy macrophages and possible therapeutic potential
The inflammatory profile of foamy macrophages remains under active investigation, and different receptors may determine this process. Recent studies have presented contrasting findings on whether foamy macrophages predominantly exhibit pro- or anti-inflammatory characteristics. For example, myeloid-specific deletion of TREM2 markedly attenuates plaque progression (35). The same authors have, however, also demonstrated that using a TREM2-specific agonist (AL002a) increases lipid uptake and cholesterol efflux, driving an anti-inflammatory phenotype that leads to plaque stability (120). This finding is in agreement with another recent manuscript, which suggests that TREM2 activation decreased the necrotic core and improved macrophage efferocytosis in late atherosclerosis (36). Transcriptomic analysis by Kim et al. showed that, in murine atherosclerotic models, non-foamy macrophages are more pro-inflammatory than foamy macrophages and substantially contribute to disease progression (7). Moreover, non-foamy macrophages not only have higher expression of genes involved in pro-inflammatory responses such as IL-1β, Tnf, Ccl2, Cx3cr1, Nlrp3, and TREM1 (7, 64) but also atherosclerosis-associated genes such as Egr1, Nlrp3, Cebpb (56, 121, 122). In contrast, foamy macrophages upregulate genes involved in lipid metabolism rather than genes involved in inflammation (103, 123). In addition to differences in the inflammatory signature of foamy and non-foamy macrophages, they are also distributed differently in the atherosclerotic plaques. Non-foamy macrophages expressing inflammatory phenotype are predominantly present in aortic adventitia, while foamy macrophages are homed in aortic intima in atherosclerotic plaques and mainly express lipid metabolism gene signature (7, 124). On the other hand, another study has demonstrated upregulation of TREM2 expression in foamy macrophages in atherosclerotic plaques in Apoe−/− mice fed on high-fat diet (110). Using a TREM2/Apoe double-KO model, others demonstrated that atherosclerotic lesions were significantly smaller, with fewer foamy cells and lower lipid load compared to Apoe−/− mice alone (110). A better characterization of foamy macrophage function may help define their role in atherosclerosis progression and identify effective therapeutic targets. Interestingly, Dib et al. identified a transition in lipid-associated macrophages from a TREM2-expressing, more reparative phenotype to a TREM1-expressing, pro-inflammatory state in human atherosclerotic plaques. While TREM2 promotes lipid handling and limits inflammation, TREM1 amplifies inflammatory signaling, driving plaque progression and instability (125).
To this extent, our group and others also recently described another receptor that regulates macrophage function and atherosclerosis progression: Olfactory Receptor 2 (Olfr2) (96). Our group has previously demonstrated that macrophages can express Olfr2 and its activation through octanal, a known ligand of Olfr2, induces cyclic adenosine monophosphate (cAMP), Calcium flux, ROS production, and Nlpr3 inflammasome activation to produce IL-1β in BMDMs. Thus, Olfr2 depletion has been shown to reduce atherosclerosis in mice (126). However, the actual physiological mechanism of Olfr2 in vivo has not yet been comprehensively described and it is an active area of investigation. Recently by using mass cytometry, we identified that Olfr2-expressing macrophages account for at least 30% of aortic macrophages and are characterized by the high expression of CD64, CCR2, and CD11c (127). Furthermore, Gene Set Enrichment Analysis (GSEA) using defined gene signatures revealed that these Olfr2 + macrophages were enriched with MDMs, aortic resident macrophages (MacAIR), and a TREM2 + subsets (127). Approximately half of the Olfr2 + macrophages are foamy, as identified by the fluorescent lipid probe BODIPY, and produce elevated levels of IL-6 and TNF compared to BODIPY + foamy macrophages lacking Olfr2. These findings suggest that Olfr2 + macrophages can be both foamy and pro-inflammatory, offering a potential means to distinguish different foamy macrophage phenotypes in atherosclerosis (127).
Targeting these pathways with inhibitors of Olfr2 or agonists such as AL002a for TREM2 could be valuable in modulating foam macrophage phenotypes, promoting plaque stability, ultimately improving patient outcomes, and reducing complications. However, further understanding of the physiological mechanisms controlling these pathways, and possibly others, is essential to achieve possible clinical applications. Moreover, to determine the translational impact of the TREM2/TREM1, and Olfr2 therapies, the development of induced pluripotent stem cells derived in-vitro vascular organoids may be beneficial to screen drugs that are more effective in lowering atherosclerosis disease burden in humans.
7 Conclusions and outlook
In conclusion, foamy macrophages emerge as key players in atherosclerosis, exhibiting both protective and pathological roles according to their origin, stage of disease progression, and receptor expression profiles. While these foam cells can initially help clear modified lipids and maintain arterial homeostasis, their prolonged accumulation and phenotypic transitions driven by differential regulation of receptors such as TREM2, TREM1, and possibly Olfr2, together with compromised cholesterol uptake and export pathways (e.g., LXR, ABCA1, ABCG1), ultimately fuel chronic inflammation and promote plaque instability. Recent single-cell analyses highlight the functional heterogeneity of foamy macrophages, revealing subsets that either mitigate or exacerbate disease. Targeting specific pathways that govern modified-LDL uptake, cholesterol efflux, and pro-inflammatory signaling, potentially through modulation of TREM2, TREM1 and possibly others, may offer a promising avenue to rebalance macrophage function and improve plaque stability. Future work to pinpoint these regulatory mechanisms and to harness them for safe, effective interventions could advance the therapeutic landscape for atherosclerotic cardiovascular disease prevention and cure.
Author contributions
AI: Data curation, Writing – original draft, Writing – review & editing. BY: Data curation, Writing – original draft, Writing – review & editing. MO: Conceptualization, Writing – original draft, Writing – review & editing, Data curation, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by grant to MO from the American Heart Association (CDA 941152).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Poznyak AV, Nikiforov NG, Markin AM, Kashirskikh DA, Myasoedova VA, Gerasimova EV, et al. Overview of OxLDL and its impact on cardiovascular health: focus on atherosclerosis. Front Pharmacol. (2021) 11:613780. doi: 10.3389/fphar.2020.613780
2. Svindland A. The localization of sudanophilic and fibrous plaques in the main left coronary bifurcation. Atherosclerosis. (1983) 48:139–45. doi: 10.1016/0021-9150(83)90100-4
3. Jiang H, Zhou Y, Nabavi SM, Sahebkar A, Little PJ, Xu S, et al. Mechanisms of oxidized LDL-mediated endothelial dysfunction and its consequences for the development of atherosclerosis. Front Cardiovasc Med. (2022) 9:925923. doi: 10.3389/fcvm.2022.925923
4. Björkegren JL, Lusis AJ. Atherosclerosis: recent developments. Cell. (2022) 185:1630–45. doi: 10.1016/j.cell.2022.04.004
5. Williams JW, Zaitsev K, Kim K-W, Ivanov S, Saunders BT, Schrank PR, et al. Limited proliferation capacity of aortic intima resident macrophages requires monocyte recruitment for atherosclerotic plaque progression. Nat Immunol. (2020) 21:1194–204. doi: 10.1038/s41590-020-0768-4
6. Roy P, Orecchioni M, Ley K. How the immune system shapes atherosclerosis: roles of innate and adaptive immunity. Nat Rev Immunol. (2022) 22:251–65. doi: 10.1038/s41577-021-00584-1
7. Kim K, Shim D, Lee JS, Zaitsev K, Williams JW, Kim K-W, et al. Transcriptome analysis reveals nonfoamy rather than foamy plaque macrophages are proinflammatory in atherosclerotic murine models. Circ Res. (2018) 123:1127–42. doi: 10.1161/CIRCRESAHA.118.312804
8. Bobryshev YV, Ivanova EA, Chistiakov DA, Nikiforov NG, Orekhov AN. Macrophages and their role in atherosclerosis: pathophysiology and transcriptome analysis. Biomed Res Int. (2016) 2016:9582430. doi: 10.1155/2016/9582430
9. Hernandez GE, Ma F, Martinez G, Firozabadi NB, Salvador J, Juang LJ, et al. Aortic intimal resident macrophages are essential for maintenance of the non-thrombogenic intravascular state. Nat Cardiovasc Res. (2022) 1:67–84. doi: 10.1038/s44161-021-00006-4
10. Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity. (2016) 44:439–49. doi: 10.1016/j.immuni.2016.02.024
11. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. (2015) 518:547–51. doi: 10.1038/nature13989
12. Palis J, Robertson S, Kennedy M, Wall C, Keller G. Development of erythroid and myeloid progenitors in the yolk sac and embryo proper of the mouse. Development. (1999) 126:5073–84. doi: 10.1242/dev.126.22.5073
13. Palis J, Chan R, Koniski A, Patel R, Starr M, Yoder M. Spatial and temporal emergence of high proliferative potential hematopoietic precursors during murine embryogenesis. Proc Natl Acad Sci U S A. (2001) 98:4528–33. doi: 10.1073/pnas.071002398
14. Li Z, Liu S, Xu J, Zhang X, Han D, Liu J, et al. Adult connective tissue-resident mast cells originate from late erythro-myeloid progenitors. Immunity. (2018) 49:640–53.e5. doi: 10.1016/j.immuni.2018.09.023
15. Gentek R, Ghigo C, Hoeffel G, Bulle MJ, Msallam R, Gautier G, et al. Hemogenic endothelial fate mapping reveals dual developmental origin of mast cells. Immunity. (2018) 48:1160–71.e5. doi: 10.1016/j.immuni.2018.04.025
16. Alliot F, Godin I, Pessac B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Dev Brain Res. (1999) 117:145–52. doi: 10.1016/S0165-3806(99)00113-3
17. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. (2010) 330:841–5. doi: 10.1126/science.1194637
18. Mase A, Augsburger J, Brückner K. Macrophages and their organ locations shape each other in development and homeostasis–a Drosophila perspective. Front Cell Dev Biol. (2021) 9:630272. doi: 10.3389/fcell.2021.630272
19. Yona S, Kim K-W, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. (2013) 38:79–91. doi: 10.1016/j.immuni.2012.12.001
20. Perdiguero EG, Geissmann F. Myb-independent macrophages: a family of cells that develops with their tissue of residence and is involved in its homeostasis. Cold Spring Harb Symp Quant Biol. (2013) 78:91–100. doi: 10.1101/sqb.2013.78.020032
21. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. (2013) 38:792–804. doi: 10.1016/j.immuni.2013.04.004
22. Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. (2007) 10:1538–43. doi: 10.1038/nn2014
23. Merad M, Manz MG, Karsunky H, Wagers A, Peters W, Charo I, et al. Langerhans cells renew in the skin throughout life under steady-state conditions. Nat Immunol. (2002) 3:1135–41. doi: 10.1038/ni852
24. Perdiguero EG, Geissmann F. The development and maintenance of resident macrophages. Nat Immunol. (2016) 17:2–8. doi: 10.1038/ni.3341
25. Fogg DK, Sibon C, Miled C, Jung S, Aucouturier P, Littman DR, et al. A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science. (2006) 311:83–7. doi: 10.1126/science.1117729
26. Dos Anjos Cassado A. F4/80 as a major macrophage marker: the case of the peritoneum and spleen. Results Probl Cell Differ. (2017) 62:161–79. doi: 10.1007/978-3-319-54090-0_7
27. Morris L, Graham CF, Gordon S. Macrophages in haemopoietic and other tissues of the developing mouse detected by the monoclonal antibody F4/80. Development. (1991) 112:517–26. doi: 10.1242/dev.112.2.517
28. Triantafyllou E, Pop OT, Possamai LA, Wilhelm A, Liaskou E, Singanayagam A, et al. MerTK expressing hepatic macrophages promote the resolution of inflammation in acute liver failure. Gut. (2018) 67:333–47. doi: 10.1136/gutjnl-2016-313615
29. Morse C, Tabib T, Sembrat J, Buschur KL, Bittar HT, Valenzi E, et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur Respir J. (2019) 54. doi: 10.1183/13993003.02441-2018
30. Fahrenhold M, Rakic S, Classey J, Brayne C, Ince PG, Nicoll JA, et al. TREM2 expression in the human brain: a marker of monocyte recruitment? Brain Pathol. (2018) 28:595–602. doi: 10.1111/bpa.12564
31. Yang J, Zhang L, Yu C, Yang X-F, Wang H. Monocyte and macrophage differentiation: circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark Res. (2014) 2:1–9. doi: 10.1186/2050-7771-2-1
32. Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol. (2014) 14:392–404. doi: 10.1038/nri3671
33. Ensan S, Li A, Besla R, Degousee N, Cosme J, Roufaiel M, et al. Self-renewing resident arterial macrophages arise from embryonic CX3CR1(+) precursors and circulating monocytes immediately after birth. Nat Immunol. (2016) 17:159–68. doi: 10.1038/ni.3343
34. Spann NJ, Garmire LX, McDonald JG, Myers DS, Milne SB, Shibata N, et al. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell. (2012) 151:138–52. doi: 10.1016/j.cell.2012.06.054
35. Patterson MT, Firulyova MM, Xu Y, Hillman H, Bishop C, Zhu A, et al. Trem2 promotes foamy macrophage lipid uptake and survival in atherosclerosis. Nat Cardiovasc Res. (2023) 2:1015–31. doi: 10.1038/s44161-023-00354-3
36. Piollet M, Porsch F, Rizzo G, Kapser F, Schulz DJ, Kiss MG, et al. TREM2 protects from atherosclerosis by limiting necrotic core formation. Nat Cardiovasc Res. (2024) 3:269–82. doi: 10.1038/s44161-024-00429-9
37. Karnovsky MJ. The ultrastructural basis of capillary permeability studied with peroxidase as a tracer. J Cell Biol. (1967) 35:213–36. doi: 10.1083/jcb.35.1.213
38. Goldstein JL, Brown MS. A century of cholesterol and coronaries: from plaques to genes to statins. Cell. (2015) 161:161–72. doi: 10.1016/j.cell.2015.01.036
39. Duerrschmidt N, Stielow C, Muller G, Pagano PJ, Morawietz H. NO-mediated regulation of NAD (P) H oxidase by laminar shear stress in human endothelial cells. J Physiol. (2006) 576:557–67. doi: 10.1113/jphysiol.2006.111070
40. Ghisi GL, Durieux A, Pinho R, Benetti M. Physical exercise and endothelial dysfunction. Arq Bras Cardiol. (2010) 95:e130–7. doi: 10.1590/S0066-782X2010001500025
41. Hadi HA, Carr CS, Al Suwaidi J. Endothelial dysfunction: cardiovascular risk factors, therapy, and outcome. Vasc Health Risk Manag. (2005) 1:183–98. doi: 10.2147/vhrm.s12187331
42. Endemann DH, Schiffrin EL. Endothelial dysfunction. J Am Soc Nephrol. (2004) 15:1983–92. doi: 10.1097/01.ASN.0000132474.50966.DA
43. Koppenol W, Moreno J, Pryor WA, Ischiropoulos H, Beckman J. Peroxynitrite, a cloaked oxidant formed by nitric oxide and superoxide. Chem Res Toxicol. (1992) 5:834–42. doi: 10.1021/tx00030a017
44. Khan BV, Harrison DG, Olbrych MT, Alexander RW, Medford RM. Nitric oxide regulates vascular cell adhesion molecule 1 gene expression and redox-sensitive transcriptional events in human vascular endothelial cells. Proc Natl Acad Sci U S A. (1996) 93:9114–9. doi: 10.1073/pnas.93.17.9114
45. Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. (2002) 105:1135–43. doi: 10.1161/hc0902.104353
46. Celermajer DS, Sorensen KE, Gooch VM, Spiegelhalter D, Miller O, Sullivan I, et al. Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet. (1992) 340:1111–5. doi: 10.1016/0140-6736(92)93147-F
47. Suwaidi JA, Hamasaki S, Higano ST, Nishimura RA, Holmes DR Jr, Lerman A. Long-term follow-up of patients with mild coronary artery disease and endothelial dysfunction. Circulation. (2000) 101:948–54. doi: 10.1161/01.CIR.101.9.948
48. Steinberg D, Witztum JL. Oxidized low-density lipoprotein and atherosclerosis. Arterioscler Thromb Vasc Biol. (2010) 30:2311–6. doi: 10.1161/ATVBAHA.108.179697
49. Witztum JL, Steinberg D. Role of oxidized low density lipoprotein in atherogenesis. J Clin Invest. (1991) 88:1785–92. doi: 10.1172/JCI115499
50. Johnson JL, Newby AC. Macrophage heterogeneity in atherosclerotic plaques. Curr Opin Lipidol. (2009) 20:370–8. doi: 10.1097/MOL.0b013e3283309848
51. Paulson KE, Zhu S-N, Chen M, Nurmohamed S, Jongstra-Bilen J, Cybulsky MI. Resident intimal dendritic cells accumulate lipid and contribute to the initiation of atherosclerosis. Circ Res. (2010) 106:383–90. doi: 10.1161/CIRCRESAHA.109.210781
52. Haberland ME, Mottino G, Le M, Frank JS. Sequestration of aggregated LDL by macrophages studied with freeze-etch electron microscopy. J Lipid Res. (2001) 42:605–19. doi: 10.1016/S0022-2275(20)31170-6
53. Palinski W, Rosenfeld ME, Ylä-Herttuala S, Gurtner GC, Socher SS, Butler SW, et al. Low density lipoprotein undergoes oxidative modification in vivo. Proc Natl Acad Sci U S A. (1989) 86:1372–6. doi: 10.1073/pnas.86.4.1372
54. Kunjathoor VV, Febbraio M, Podrez EA, Moore KJ, Andersson L, Koehn S, et al. Scavenger receptors class AI/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J Biol Chem. (2002) 277:49982–8. doi: 10.1074/jbc.M209649200
55. Libby P, Clinton SK. The role of macrophages in atherogenesis. Curr Opin Lipidol. (1993) 4:355–63. doi: 10.1097/00041433-199310000-00003
56. Zernecke A, Erhard F, Weinberger T, Schulz C, Ley K, Saliba AE, et al. Integrated single-cell analysis-based classification of vascular mononuclear phagocytes in mouse and human atherosclerosis. Cardiovasc Res. (2023) 119:1676–89. doi: 10.1093/cvr/cvac161
57. Willemsen L, de Winther MP. Macrophage subsets in atherosclerosis as defined by single-cell technologies. J Pathol. (2020) 250:705–14. doi: 10.1002/path.5392
58. Zernecke A, Winkels H, Cochain C, Williams JW, Wolf D, Soehnlein O, et al. Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ Res. (2020) 127:402–26. doi: 10.1161/CIRCRESAHA.120.316903
59. Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, et al. Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ Res. (2018) 122:1661–74. doi: 10.1161/CIRCRESAHA.117.312509
60. Winkels H, Ehinger E, Vassallo M, Buscher K, Dinh HQ, Kobiyama K, et al. Atlas of the immune cell repertoire in mouse atherosclerosis defined by single-cell RNA-Sequencing and mass cytometry. Circ Res. (2018) 122:1675–88. doi: 10.1161/CIRCRESAHA.117.312513
61. Cole JE, Park I, Ahern DJ, Kassiteridi C, Danso Abeam D, Goddard ME, et al. Immune cell census in murine atherosclerosis: cytometry by time of flight illuminates vascular myeloid cell diversity. Cardiovasc Res. (2018) 114:1360–71. doi: 10.1093/cvr/cvy109
62. Cai J, Deng J, Gu W, Ni Z, Liu Y, Kamra Y, et al. Impact of local alloimmunity and recipient cells in transplant arteriosclerosis. Circ Res. (2020) 127:974–93. doi: 10.1161/CIRCRESAHA.119.316470
63. Fidler TP, Xue C, Yalcinkaya M, Hardaway B, Abramowicz S, Xiao T, et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature. (2021) 592:296–301. doi: 10.1038/s41586-021-03341-5
64. Zhang X, Sun J, Canfrán-Duque A, Aryal B, Tellides G, Chang YJ, et al. Deficiency of histone lysine methyltransferase SETDB2 in hematopoietic cells promotes vascular inflammation and accelerates atherosclerosis. JCI Insight. (2021) 6:e147984. doi: 10.1172/jci.insight.147984
65. McArdle S, Buscher K, Ghosheh Y, Pramod AB, Miller J, Winkels H, et al. Migratory and dancing macrophage subsets in atherosclerotic lesions. Circ Res. (2019) 125:1038–51. doi: 10.1161/CIRCRESAHA.119.315175
66. Park I, Goddard ME, Cole JE, Zanin N, Lyytikäinen L-P, Lehtimäki T, et al. C-type lectin receptor CLEC4A2 promotes tissue adaptation of macrophages and protects against atherosclerosis. Nat Commun. (2022) 13:215. doi: 10.1038/s41467-021-27862-9
67. van Kuijk K, Demandt JA, Perales-Patón J, Theelen TL, Kuppe C, Marsch E, et al. Deficiency of myeloid PHD proteins aggravates atherogenesis via macrophage apoptosis and paracrine fibrotic signalling. Cardiovasc Res. (2022) 118:1232–46. doi: 10.1093/cvr/cvab152
68. Chattopadhyay A, Kwartler CS, Kaw K, Li Y, Kaw A, Chen J, et al. Cholesterol-induced phenotypic modulation of smooth muscle cells to macrophage/fibroblast–like cells is driven by an unfolded protein response. Arterioscler Thromb Vasc Biol. (2021) 41:302–16. doi: 10.1161/ATVBAHA.120.315164
69. Depuydt MA, Prange KH, Slenders L, Örd T, Elbersen D, Boltjes A, et al. Microanatomy of the human atherosclerotic plaque by single-cell transcriptomics. Circ Res. (2020) 127:1437–55. doi: 10.1161/CIRCRESAHA.120.316770
70. Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir ED, Amadori L, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. (2019) 25:1576–88. doi: 10.1038/s41591-019-0590-4
71. Owsiany KM, Alencar GF, Owens GK. Revealing the origins of foam cells in atherosclerotic lesions. Am Heart Assoc. (2019) 39:836–8. doi: 10.1161/ATVBAHA.119.312557
72. Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res. (2016) 118:653–67. doi: 10.1161/CIRCRESAHA.115.306256
73. Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation. (2014) 129:1551–9. doi: 10.1161/CIRCULATIONAHA.113.005015
74. Feil S, Fehrenbacher B, Lukowski R, Essmann F, Schulze-Osthoff K, Schaller M, et al. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ Res. (2014) 115:662–7. doi: 10.1161/CIRCRESAHA.115.304634
75. Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. (2015) 21:628–37. doi: 10.1038/nm.3866
76. Pan H, Xue C, Auerbach BJ, Fan J, Bashore AC, Cui J, et al. Single-cell genomics reveals a novel cell state during smooth muscle cell phenotypic switching and potential therapeutic targets for atherosclerosis in mouse and human. Circulation. (2020) 142:2060–75. doi: 10.1161/CIRCULATIONAHA.120.048378
77. Li Y, Zhu H, Zhang Q, Han X, Zhang Z, Shen L, et al. Smooth muscle-derived macrophage-like cells contribute to multiple cell lineages in the atherosclerotic plaque. Cell Discov. (2021) 7:111. doi: 10.1038/s41421-021-00328-4
78. Chen P-Y, Qin L, Baeyens N, Li G, Afolabi T, Budatha M, et al. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J Clin Invest. (2015) 125:4514–28. doi: 10.1172/JCI82719
79. Wang Y-Y, Jiang H, Pan J, Huang X-R, Wang Y-C, Huang H-F, et al. Macrophage-to-myofibroblast transition contributes to interstitial fibrosis in chronic renal allograft injury. J Am Soc Nephrol. (2017) 28:2053–67. doi: 10.1681/ASN.2016050573
80. Souilhol C, Harmsen MC, Evans PC, Krenning G. Endothelial–mesenchymal transition in atherosclerosis. Cardiovasc Res. (2018) 114:565–77. doi: 10.1093/cvr/cvx253
81. Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and rupture. Circ Res. (2014) 114:1852–66. doi: 10.1161/CIRCRESAHA.114.302721
82. Wang T, Palucci D, Law K, Yanagawa B, Yam J, Butany J. Atherosclerosis: pathogenesis and pathology. Diagn Histopathol. (2012) 18:461–7. doi: 10.1016/j.mpdhp.2012.09.004
83. Rader DJ, Puré E. Lipoproteins, macrophage function, and atherosclerosis: beyond the foam cell? Cell Metab. (2005) 1:223–30. doi: 10.1016/j.cmet.2005.03.005
84. Poznyak AV, Nikiforov NG, Starodubova AV, Popkova TV, Orekhov AN. Macrophages and foam cells: brief overview of their role, linkage, and targeting potential in atherosclerosis. Biomedicines. (2021) 9:1221. doi: 10.3390/biomedicines9091221
85. Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, Van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. (2016) 354:472–7. doi: 10.1126/science.aaf6659
86. Kzhyshkowska J, Neyen C, Gordon S. Role of macrophage scavenger receptors in atherosclerosis. Immunobiology. (2012) 217:492–502. doi: 10.1016/j.imbio.2012.02.015
87. Rhoads JP, Major AS. How oxidized low-density lipoprotein activates inflammatory responses. Crit Rev Immunol. (2018) 38:333–42. doi: 10.1615/CritRevImmunol.2018026483
88. Gelissen IC, Harris M, Rye KA, Quinn C, Brown AJ, Kockx M, et al. ABCA1 and ABCG1 synergize to mediate cholesterol export to apoA-I. Arterioscler Thromb Vasc Biol. (2006) 26:534–40. doi: 10.1161/01.ATV.0000200082.58536.e1
89. Chinetti G, Lestavel S, Bocher V, Remaley AT, Neve B, Torra IP, et al. PPAR-α and PPAR-γ activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat Med. (2001) 7:53–8. doi: 10.1038/83348
90. Fayad ZA, Swirski FK, Calcagno C, Robbins CS, Mulder W, Kovacic JC. Monocyte and macrophage dynamics in the cardiovascular system: JACC macrophage in CVD series (part 3). J Am Coll Cardiol. (2018) 72:2198–212. doi: 10.1016/j.jacc.2018.08.2150
91. Pownall HJ, Rosales C, Gillard BK, Gotto AM Jr. High-density lipoproteins, reverse cholesterol transport and atherogenesis. Nat Rev Cardiol. (2021) 18:712–23. doi: 10.1038/s41569-021-00538-z
92. Bochem AE, van Wijk DF, Holleboom AG, Duivenvoorden R, Motazacker MM, Dallinga-Thie GM, et al. ABCA1 mutation carriers with low high-density lipoprotein cholesterol are characterized by a larger atherosclerotic burden. Eur Heart J. (2013) 34:286–91. doi: 10.1093/eurheartj/ehs376
93. Zhao Y, Pennings M, Vrins CL, Calpe-Berdiel L, Hoekstra M, Kruijt JK, et al. Hypocholesterolemia, foam cell accumulation, but no atherosclerosis in mice lacking ABC-transporter A1 and scavenger receptor BI. Atherosclerosis. (2011) 218:314–22. doi: 10.1016/j.atherosclerosis.2011.07.096
94. Joyce CW, Wagner EM, Basso F, Amar MJ, Freeman LA, Shamburek RD, et al. ABCA1 overexpression in the liver of LDLr-KO mice leads to accumulation of pro-atherogenic lipoproteins and enhanced atherosclerosis. J Biol Chem. (2006) 281:33053–65. doi: 10.1074/jbc.M604526200
95. Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, et al. CD36 Coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol. (2013) 14:812–20. doi: 10.1038/ni.2639
96. He Z, Wang DW. Olfactory receptor 2 activation in macrophages: novel mediator of atherosclerosis progression. Signal Transduct Target Ther. (2022) 7:247. doi: 10.1038/s41392-022-01115-7
97. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. (2010) 464:1357–61. doi: 10.1038/nature08938
98. Thompson PL, Nidorf SM. Anti-inflammatory therapy with canakinumab for atherosclerotic disease: lessons from the CANTOS trial. J Thorac Dis. (2018) 10:695. doi: 10.21037/jtd.2018.01.119
99. Weber A, Wasiliew P, Kracht M. Interleukin-1 (IL-1) pathway. Sci Signal. (2010) 3:cm1. doi: 10.1126/scisignal.3105cm1
100. Peters VA, Joesting JJ, Freund GG. IL-1 receptor 2 (IL-1R2) and its role in immune regulation. Brain Behav Immun. (2013) 32:1–8. doi: 10.1016/j.bbi.2012.11.006
101. Arai S, Shelton JM, Chen M, Bradley MN, Castrillo A, Bookout AL, et al. A role for the apoptosis inhibitory factor AIM/spα/Api6 in atherosclerosis development. Cell Metab. (2005) 1:201–13. doi: 10.1016/j.cmet.2005.02.002
102. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. (2014) 157:1013–22. doi: 10.1016/j.cell.2014.04.007
103. Gui Y, Zheng H, Cao RY. Foam cells in atherosclerosis: novel insights into its origins, consequences, and molecular mechanisms. Front Cardiovasc Med. (2022) 9:845942. doi: 10.3389/fcvm.2022.845942
104. Deczkowska A, Weiner A, Amit I. The physiology, pathology, and potential therapeutic applications of the TREM2 signaling pathway. Cell. (2020) 181:1207–17. doi: 10.1016/j.cell.2020.05.003
105. Shi Y, Holtzman DM. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat Rev Immunol. (2018) 18:759–72. doi: 10.1038/s41577-018-0051-1
106. Kober DL, Brett TJ. TREM2-ligand Interactions in health and disease. J Mol Biol. (2017) 429:1607–29. doi: 10.1016/j.jmb.2017.04.004
107. Zhang J, Liu Y, Zheng Y, Luo Y, Du Y, Zhao Y, et al. TREM-2-p38 MAPK signaling regulates neuroinflammation during chronic cerebral hypoperfusion combined with diabetes mellitus. J Neuroinflammation. (2020) 17:1–16. doi: 10.1186/s12974-019-1688-9
108. Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, et al. Lipid-associated macrophages control metabolic homeostasis in a Trem2-dependent manner. Cell. (2019) 178:686–98.e14. doi: 10.1016/j.cell.2019.05.054
109. Damisah EC, Rai A, Grutzendler J. TREM2: modulator of lipid metabolism in microglia. Neuron. (2020) 105:759–61. doi: 10.1016/j.neuron.2020.02.008
110. Guo X, Li B, Wen C, Zhang F, Xiang X, Nie L, et al. TREM2 promotes cholesterol uptake and foam cell formation in atherosclerosis. Cell Mol Life Sci. (2023) 80:137. doi: 10.1007/s00018-023-04786-9
111. Li Y, Zhou M, Li H, Dai C, Yin L, Liu C, et al. Macrophage P2Y6 receptor deletion attenuates atherosclerosis by limiting foam cell formation through phospholipase Cβ/store-operated calcium entry/calreticulin/scavenger receptor A pathways. Eur Heart J. (2024) 45:268–83. doi: 10.1093/eurheartj/ehad796
112. Liu X, Guo J-W, Lin X-C, Tuo Y-H, Peng W-L, He S-Y, et al. Macrophage NFATc3 prevents foam cell formation and atherosclerosis: evidence and mechanisms. Eur Heart J. (2021) 42:4847–61. doi: 10.1093/eurheartj/ehab660
113. Ma C, Li Y, Tian M, Deng Q, Qin X, Lu H, et al. Gsα regulates macrophage foam cell formation during atherosclerosis. Circ Res. (2024) 134:e34–51. doi: 10.1161/CIRCRESAHA.123.323156
114. Wang B, Tang X, Yao L, Wang Y, Chen Z, Li M, et al. Disruption of USP9X in macrophages promotes foam cell formation and atherosclerosis. J Clin Invest. (2022) 132. doi: 10.1172/JCI154217
115. Matsuo M. ABCA1 and ABCG1 as potential therapeutic targets for the prevention of atherosclerosis. J Pharmacol Sci. (2022) 148:197–203. doi: 10.1016/j.jphs.2021.11.005
116. Zheng S, Huang H, Li Y, Wang Y, Zheng Y, Liang J, et al. Yin-xing-tong-mai decoction attenuates atherosclerosis via activating PPARγ-LXRα-ABCA1/ABCG1 pathway. Pharmacol Res. (2021) 169:105639. doi: 10.1016/j.phrs.2021.105639
117. Endo-Umeda K, Kim E, Thomas DG, Liu W, Dou H, Yalcinkaya M, et al. Myeloid LXR (liver X receptor) deficiency induces inflammatory gene expression in foamy macrophages and accelerates atherosclerosis. Arterioscler Thromb Vasc Biol. (2022) 42:719–31. doi: 10.1161/ATVBAHA.122.317583
118. Bouhlel MA, Derudas B, Rigamonti E, Dièvart R, Brozek J, Haulon S, et al. PPARγ activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. (2007) 6:137–43. doi: 10.1016/j.cmet.2007.06.010
119. De Gaetano M, Crean D, Barry M, Belton O. M1-and M2-type macrophage responses are predictive of adverse outcomes in human atherosclerosis. Front Immunol. (2016) 7:275. doi: 10.3389/fimmu.2016.00275
120. Patterson MT, Xu Y, Hillman H, Osinski V, Schrank PR, Kennedy AE, et al. Trem2 agonist reprograms foamy macrophages to promote atherosclerotic plaque stability. Arterioscler Thromb Vasc Biol. (2024) 44:1646–57. doi: 10.1161/ATVBAHA.124.320797
121. Albrecht C, Preusch MR, Hofmann G, Morris-Rosenfeld S, Blessing E, Rosenfeld ME, et al. Egr-1 deficiency in bone marrow-derived cells reduces atherosclerotic lesion formation in a hyperlipidaemic mouse model. Cardiovasc Res. (2010) 86:321–9. doi: 10.1093/cvr/cvq032
122. Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT-H, et al. Fatty acid–induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. (2011) 12:408–15. doi: 10.1038/ni.2022
123. Kim K, Park SE, Park JS, Choi JH. Characteristics of plaque lipid-associated macrophages and their possible roles in the pathogenesis of atherosclerosis. Curr Opin Lipidol. (2022) 33:283–8. doi: 10.1097/MOL.0000000000000842
124. Li C, Qu L, Matz AJ, Murphy PA, Liu Y, Manichaikul AW, et al. Atherospectrum reveals novel macrophage foam cell gene signatures associated with atherosclerotic cardiovascular disease risk. Circulation. (2022) 145:206–18. doi: 10.1161/CIRCULATIONAHA.121.054285
125. Dib L, Koneva LA, Edsfeldt A, Zurke Y-X, Sun J, Nitulescu M, et al. Lipid-associated macrophages transition to an inflammatory state in human atherosclerosis, increasing the risk of cerebrovascular complications. Nat Cardiovasc Res. (2023) 2:656–72. doi: 10.1038/s44161-023-00295-x
126. Orecchioni M, Kobiyama K, Winkels H, Ghosheh Y, McArdle S, Mikulski Z, et al. Olfactory receptor 2 in vascular macrophages drives atherosclerosis by NLRP3-dependent IL-1 production. Science. (2022) 375:214–21. doi: 10.1126/science.abg3067
Keywords: atherosclerosis, inflammation, macrophages, foamy, TREM2, Olfr2
Citation: Ijaz A, Yarlagadda B and Orecchioni M (2025) Foamy macrophages in atherosclerosis: unraveling the balance between pro- and anti-inflammatory roles in disease progression. Front. Cardiovasc. Med. 12:1589629. doi: 10.3389/fcvm.2025.1589629
Received: 7 March 2025; Accepted: 16 April 2025;
Published: 2 May 2025.
Edited by:
Masanori Aikawa, Brigham and Women’s Hospital and Harvard Medical School, United StatesReviewed by:
Santosh Karnewar, University of Virginia, United StatesPopovics Petra, Eastern Virginia Medical School, United States
Copyright: © 2025 Ijaz, Yarlagadda and Orecchioni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marco Orecchioni, bW9yZWNjaGlvbmlAYXVndXN0YS5lZHU=