 P. Wambua
P. Wambua M. Wahinya2†
M. Wahinya2† Z. Khan
Z. Khan- 1Tigoni Level IV Hospital, Kiambu, Kenya
- 2Kenyatta University Teaching, Referral & Research Hospital, Nairobi, Kenya
- 3Cardiology Department, Bart’s Heart Centre, London, United Kingdom
Background: Calcific aortic valve stenosis (CAVS) is the most prevalent valvular heart disease and a growing global health concern. Aortic sclerosis (ASc) and aortic stenosis (AS) represent a continuum of progressive disease characterized by leaflet thickening, inflammation, lipid deposition, and calcification. Lipoprotein(a) [Lp(a)], with its pro-atherogenic, pro-inflammatory, and pro-calcific properties, has emerged as a key contributor to this process. While its role in atherosclerotic cardiovascular disease is well established, the relationship between Lp(a) and CAVS has been demonstrated in several key studies; however, the available evidence remains limited in volume, and important gaps persist in understanding mechanisms, risk stratification, and therapeutic implications.
Methods: A systematic literature search was conducted in PubMed, Cochrane Library, ScienceDirect, Medline, ResearchGate, Embase, and Google Scholar in accordance with PRISMA guidelines. Eligible studies included observational designs (cross-sectional, cohort, case-control) and randomized trials evaluating associations between Lp(a) levels, genetic variants, and CAVS. Study quality was assessed using the Newcastle–Ottawa Scale (NOS).
Results: Eighteen studies met the inclusion criteria, comprising six case-control, six cohort, and six cross-sectional studies with a total of 153,192 participants. No randomized controlled trials were identified. Elevated Lp(a) levels were consistently associated with an increased risk of AS and aortic valve calcification (AVC), with a dose-dependent effect. The risk was highest at levels ≥50 mg/dl, though some evidence supported risk at ≥30 mg/dl. Genetic analyses identified rs10455872 as a significant risk allele, while rs3798220 showed inconsistent associations. Multi-ethnic cohorts highlighted racial variability: Afro-Caribbean individuals had higher baseline Lp(a) levels but lower AVC prevalence than Caucasians.
Conclusion: Lp(a) is an independent risk factor for CAVS, influenced by both concentration and genetic variation. Early screening and emerging Lp(a)-lowering therapies, including antisense oligonucleotides, small interfering RNA, and PCSK9 inhibitors, may help mitigate disease progression. Further randomized trials are needed to determine whether Lp(a) reduction translates into cardiovascular and valvular benefit.
Systematic Review Registration: https://www.crd.york.ac.uk/PROSPERO/view/CRD42024533835, PROSPERO CRD42024533835.
Introduction
Calcific aortic valve stenosis (CAVS) is the most common valvular heart disease and is projected to impose a substantial health burden in the coming decades (1, 2). Aortic sclerosis (ASc) and aortic stenosis (AS) form a disease continuum, beginning with leaflet thickening and progressing to severe obstruction.
The pathophysiology of CAVS is multifactorial, but lipoprotein(a) [Lp(a)] has emerged as a key driver (4). Lp(a) consists of a low-density lipoprotein–like particle covalently bound to apolipoprotein(a) and carries pro-atherogenic, pro-inflammatory, and pro-thrombotic properties (3, 7, 8). Elevated Lp(a) is an established causal factor in coronary artery disease and myocardial infarction, and increasing evidence implicates it in aortic valve calcification and stenosis (6, 11, 13, 25).
Lp(a) was first described by Berg in 1963 (1), and subsequent advances—including cloning of the LPA gene—clarified its genetic basis (3, 6). Carriers of risk alleles such as rs10455872 and rs3798220 produce smaller apolipoprotein(a) isoforms, leading to higher plasma concentrations and increased risk of calcific valve disease (3, 6, 22). Early studies were hampered by inadequate assays, but modern techniques have confirmed the causal association between Lp(a), oxidized phospholipids, and fibrocalcific remodeling of the aortic valve (13, 25).
Despite these advances, the relationship between Lp(a), race, genetic polymorphisms, and CAVS remains incompletely understood. This systematic review synthesizes the available evidence on the association between Lp(a) levels and the risk of AS and ASc, aiming to clarify its role in disease onset and progression, highlight gaps in knowledge, and inform therapeutic strategies.
Methods
Study design
This systematic review was conducted in accordance with PRISMA guidelines (9) and registered with PROSPERO (CRD42024533835).
Search strategy
A systematic search was performed across PubMed, Cochrane Library, ScienceDirect, Medline, ResearchGate, Embase, and Google Scholar between June 1st and July 1st, 2024. Keywords and MeSH terms included: lipoprotein(a) OR Lp(a) AND “calcific aortic valve disease,” “aortic valve sclerosis,” “aortic valve stenosis,” “aortic stenosis,” “aortic sclerosis,” and “aortic valve calcification.” The search and screening process is shown in Figure 1.
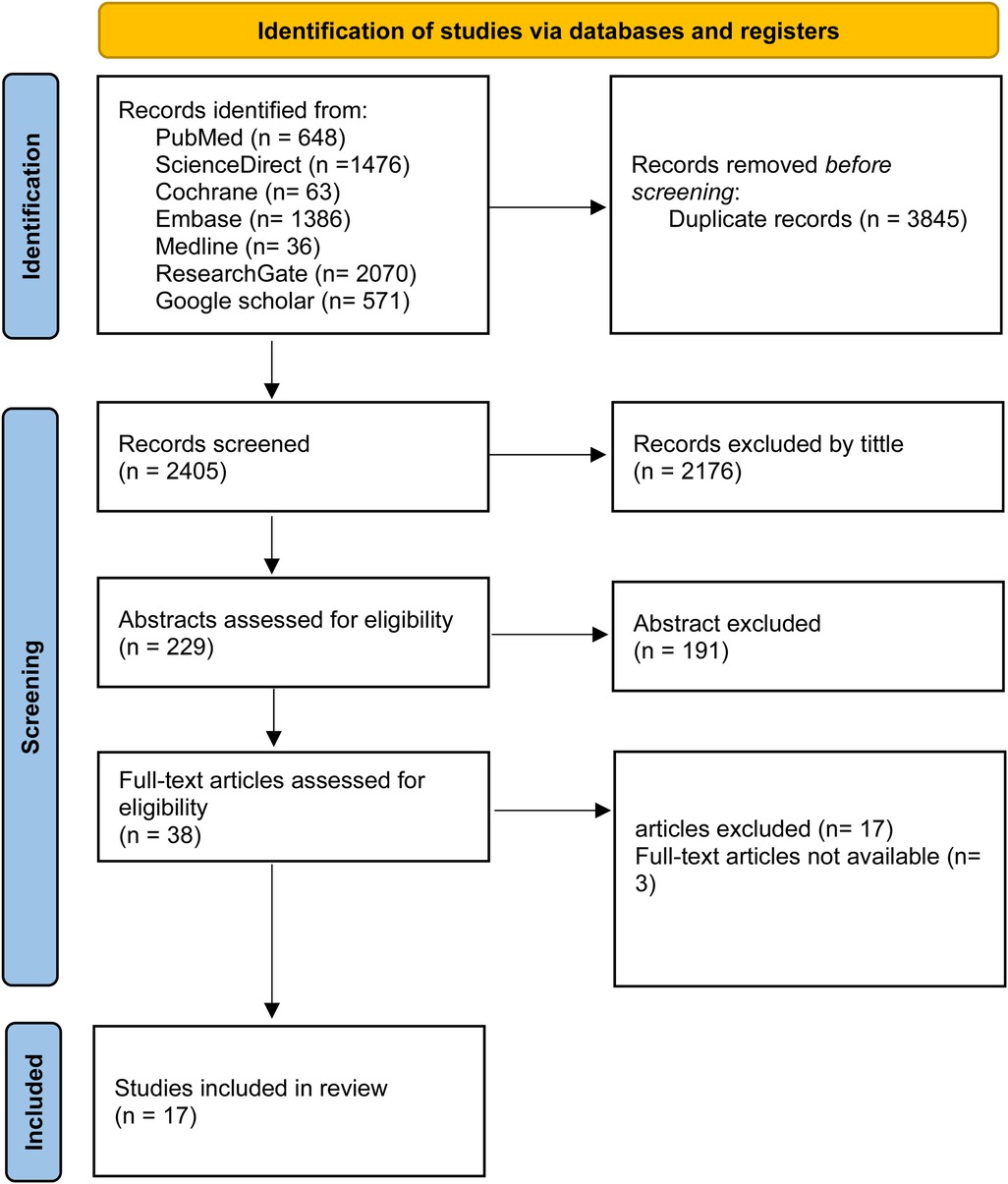
Figure 1. PRISMA flow diagram.
Study population and eligibility (inclusion/exclusion)
Eligibility Criteria: The criteria were structured using the PECO framework (10):
○ Population: Adults from the general population.
○ Exposure: Elevated plasma Lp(a) levels or presence of LPA genetic variants.
○ Comparator: Individuals with normal Lp(a) levels or non-risk variants.
○ Outcomes: Development or progression of AS or ASc, or presence of AVC.
Inclusion criteria: observational studies (cohort, case-control, cross-sectional) and RCTs evaluating Lp(a) and calcific aortic valve disease; studies published in English since 2010.
Exclusion criteria: studies of non-calcific valvular disease (e.g., rheumatic), those not reporting Lp(a) in mg/dl, or non-English publications.
Quality assessment
Methodological quality was assessed using the Newcastle–Ottawa Scale (NOS). Scores of 0–3 indicated high risk, 4–6 moderate, and 7–9 low risk of bias. Two reviewers independently assessed studies, resolving disagreements by consensus with a third reviewer. Sixteen studies were high quality, and two were of moderate quality (Table 1).
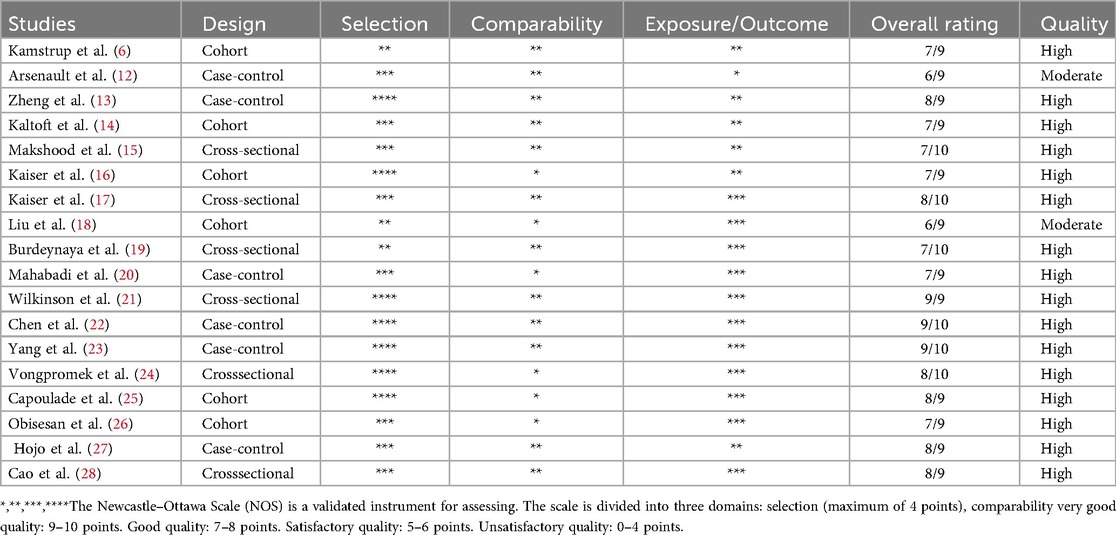
Table 1. Newcastle-Ottawa quality assessment scale of nonrandomised studies.
Figure 2 shows summary plot for risk of bias assessment.
Figure 2. Risk of bias assessment of included studies using the ROBINS.
Results
Search results and study characteristics
From 6,250 articles screened, 18 studies met inclusion criteria, including six cohorts, six case–controls, and six cross-sectional studies from Europe, the USA, and Asia, with a total of 153,192 participants. Ten studies evaluated Lp(a) and AS, six focused on AVC, and four assessed genetic variants. Participants' mean or median age ranged from 46 to 80 years, with varied sex distribution.
Lp(a) and aortic valve stenosis
Most studies demonstrated that elevated Lp(a) was associated with higher risk of AS, with thresholds ≥50 mg/dl consistently linked to incident disease (12, 13, 15, 19). A dose–response effect was reported by Kamstrup et al. (6), where risk increased at >20 mg/dl (HR: 1.6, 95% CI 1.1–2.4) and peaked above 90 mg/dl (HR: 2.9, 95% CI 1.8–4.9). In contrast, Mahabadi et al. (20) did not find a significant association.
Lp(a) and aortic valve calcification
All six studies confirmed an association between elevated Lp(a) and AVC. Higher concentrations correlated with greater calcification severity. Multi-ethnic cohorts highlighted variability: in MESA (28), Afro-Caribbean participants had higher baseline Lp(a) (35 mg/dl) than Hispanics (13 mg/dl), Caucasians (13 mg/dl), and Chinese (12.9 mg/dl). Although Caucasians exhibited lower median levels, they had a higher baseline prevalence of AVC. After adjustment, the association between Lp(a) and AVC persisted in Caucasians but not in other groups. ARIC and Makshood et al. reported similar findings, with associations in Afro-Caribbean and Caucasian populations but not in South Asians (15, 16).
Association between Lp(a) genetic variants and aortic valve disease
Two Lp(a) genotypes were evaluated. The rs10455872 allele was consistently associated with increased risk of aortic valve stenosis or sclerosis across four studies (6). In contrast, the rs3798220 variant showed no significant association with AVS (6). Chen et al. (22) reported that carriers of two risk alleles (heterozygous, homozygous, or compound heterozygous) had a two-fold higher risk of AVS compared to those with one allele.
Table 2 below shows the characteristics and findings of the included studies.
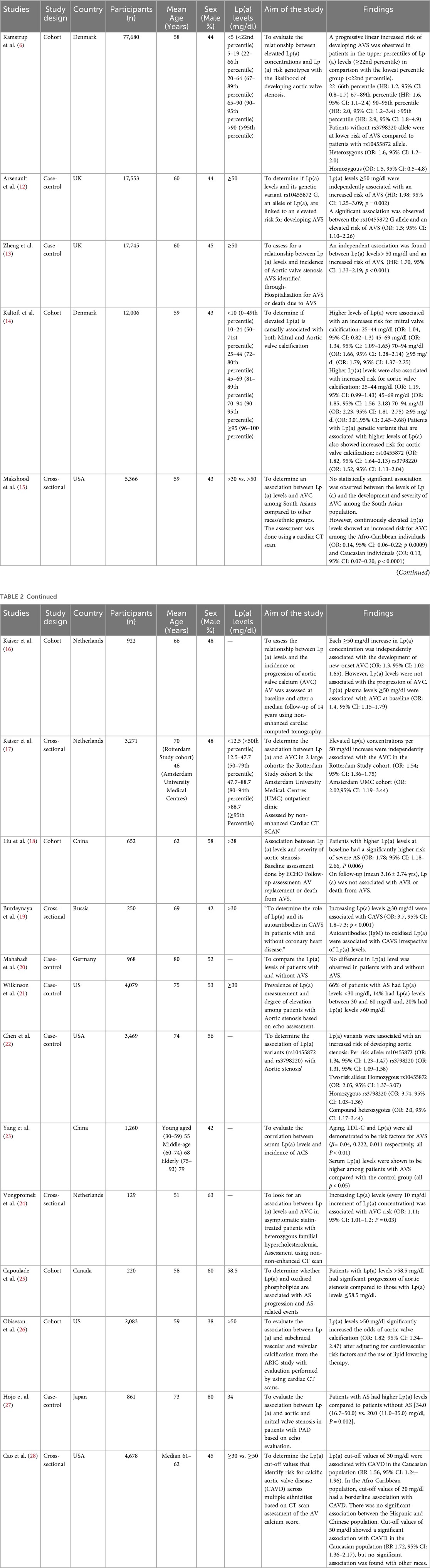
Table 2. Summary of key observational studies evaluating the association.
Discussion
This systematic review confirms a robust, dose-dependent association between elevated Lp(a) and CAVD, despite heterogeneity across study designs. Concentrations ≥50 mg/dl were reliably associated with disease risk (12, 13, 15, 19), while thresholds around ≥30 mg/dl yielded mixed findings (12, 15, 20). Elevated Lp(a) has also been implicated in disease progression, accelerating hemodynamic deterioration and adverse outcomes (13, 25), although Kaiser et al. (16) observed associations only with incident, not progressive, AVC.
Genetic determinants provide strong causal evidence. Variants such as rs10455872 consistently predict elevated Lp(a) and higher risk of CAVD (3, 6, 22). Carriage of multiple risk alleles more than doubled AS risk, with stronger effects in younger and male populations (12, 22). Instrumental variable analyses suggest a relative genetic risk of 1.6 for AS with a tenfold increase in Lp(a) (5, 6). These findings align with the biological mechanism whereby fewer kringle IV type 2 (KIV-2) repeats produce smaller apo(a) isoforms that are synthesized at higher rates, leading to elevated plasma concentrations.
Ethnic variability was also evident. Afro-Caribbean and Caucasian individuals demonstrated the strongest associations (15, 28), whereas South Asian, Hispanic, and Chinese populations showed weaker or inconsistent links (15, 28). Interestingly, despite lower median levels in Caucasians compared to Afro-Caribbeans, subclinical CAVD was more prevalent in the former (15), suggesting complex gene–environment interactions.
Mechanistically, Lp(a) is increasingly recognized as a multifactorial driver of CAVD. Circulating Lp(a) carries oxidized phospholipids (OxPLs), which promote endothelial activation, inflammatory cell infiltration, and osteogenic signaling (e.g., bone morphogenetic proteins), thereby accelerating valve fibrosis and calcification in a manner paralleling atherosclerosis (26, 27). Oxidized Lp(a) also impairs fibrinolysis and potentiates thrombosis, further contributing to valvular injury. A conceptual framework can thus be summarized:
Therapeutically, conventional lipid-lowering therapies have limited impact on Lp(a). Statins are ineffective and may modestly increase levels (29, 30), whereas PCSK9 inhibitors provide modest reductions (∼20%–25%) and have demonstrated cardiovascular benefit in outcomes trials, partly attributable to Lp(a) lowering (31, 32). More potent agents are in development: siRNAs (Olpasiran) reduce Lp(a) by up to 90% (33) and are under evaluation in the Phase 3 OCEAN[a]-Outcomes trial (NCT05581303); ASOs (Pelacarsen) reduce Lp(a) by ∼80% (34), with the large HORIZON trial underway. ANGPTL3 inhibitors also show potential in lowering Lp(a) alongside other lipids (35). These developments highlight a paradigm shift toward precision therapeutics for genetically mediated risk factors such as Lp(a).
Overall, the evidence indicates that Lp(a) is not only a biomarker but also a causal mediator of CAVD, supported by consistent epidemiological, genetic, and mechanistic data. These insights reinforce the rationale for incorporating Lp(a) into risk stratification models and prioritizing Lp(a)-specific therapies to prevent both cardiovascular and valvular events.
Future directions
Future research should focus on integrating Lp(a) measurement into routine cardiovascular and valvular risk assessment, especially for individuals with a family history of premature atherosclerotic disease or CAVD. Large-scale registries and multi-ethnic cohorts are required to clarify ancestry-specific risks, as evidence suggests important variability across populations. Mechanistic studies should further elucidate the roles of oxidized phospholipids, inflammation, and valve interstitial cell activation in disease progression, which may uncover new therapeutic targets. Most importantly, the outcomes of ongoing Phase 3 trials (HORIZON, OCEAN[a]-Outcomes) will determine whether targeted Lp(a) lowering can alter the natural history of CAVD. If successful, these therapies could establish a new standard of care, shifting management from late-stage intervention to early, precision-based prevention.
Limitations
This review has several limitations. First, it included only observational studies, and no randomized controlled trials (RCTs) are yet available to establish causality between elevated Lp(a) and CAVD. Second, the included studies were conducted predominantly in high-income countries (Europe, the United States, China, and Japan), with limited data from developing regions and Sub-Saharan Africa, restricting global generalizability. Third, although the overall risk of bias was low, there was significant heterogeneity in study design, population characteristics, and Lp(a) thresholds, which may influence interpretation.
Conclusion
In summary, elevated Lp(a) is consistently associated with an increased risk of atherosclerotic cardiovascular disease, aortic valve sclerosis, and stenosis, supported by genetic and mechanistic evidence. While RCTs confirming that lowering Lp(a) reduces CAVD risk are lacking, emerging therapies such as siRNAs and antisense oligonucleotides offer great promise. Large, multi-ethnic RCTs are urgently needed to determine whether targeted Lp(a) reduction can modify the natural history of CAVD and should specifically include underrepresented populations such as Sub-Saharan Africa. Establishing effective Lp(a)-directed interventions could transform management paradigms, moving from symptomatic treatment of advanced valve disease to early, precision-based prevention.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
PW: Validation, Writing – original draft, Methodology, Conceptualization, Data curation, Investigation, Software, Funding acquisition, Resources, Visualization, Writing – review & editing, Formal analysis, Project administration. MW: Validation, Methodology, Formal analysis, Data curation, Project administration, Conceptualization, Software, Investigation, Writing – original draft, Resources, Writing – review & editing. ZK: Software, Data curation, Investigation, Visualization, Writing – original draft, Resources, Conceptualization, Methodology, Validation, Project administration, Funding acquisition, Writing – review & editing, Supervision, Formal analysis.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Berg K. A new serum type system in man—the Lp system. Acta Pathol Microbiol Scand. (1963) 59:369–82. Available online at: https://doi.org/10.1111/j.1699-0463.1963.tb01808.x14064818
2. Stewart BF, Siscovick D, Lind BK, Gardin JM, Gottdiener JS, Smith VE, et al. Clinical factors associated with calcific aortic valve disease. J Am Coll Cardiol. (1997) 29(3):630–4. doi: 10.1016/S0735-1097(96)00563-3
3. Thanassoulis G, Campbell CY, Owens DS, Smith JG, Smith AV, Peloso GM, et al. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. (2013) 368(6):503–12. doi: 10.1056/NEJMoa1109034
4. Rajamannan NM, Evans FJ, Aikawa E, Grande-Allen KJ, Demer LL, Heistad DD, et al. Calcific aortic valve disease: not simply a degenerative process. Circulation. (2011) 124(16):1783–91. doi: 10.1161/CIRCULATIONAHA.110.006767
5. Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol. (2014) 63(5):470–7. doi: 10.1016/j.jacc.2013.09.038
6. Kamstrup PR, Neely RDG, Nissen S, Landmesser U, Haghikia A, Costa-Scharplatz M, et al. Lipoprotein(a) and cardiovascular disease: sifting the evidence to guide future research. Eur J Prev Cardiol. (2024) 31(7):903–14. doi: 10.1093/eurjpc/zwae032
7. Nishimura RA, Otto CM, Bonow RO, Carabello BA, Erwin JP, Guyton RA, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease: executive summary. Circulation. (2014) 129(23):2440–92. doi: 10.1161/CIR.0000000000000029
8. Tsimikas S. A test in context: lipoprotein(a). J Am Coll Cardiol. (2017) 69(6):692–711. doi: 10.1016/j.jacc.2016.11.042
9. Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. Br Med J. (2021) 372:n71. doi: 10.1136/bmj.n71
10. Aslam S, Emmanuel P. Formulating a researchable question: a critical step for facilitating good clinical research. Indian J Sex Transm Dis AIDS. (2010) 31(1):47–50. doi: 10.4103/0253-7184.69003
11. Stang A. Critical evaluation of the Newcastle-Ottawa scale for the assessment of the quality of nonrandomised studies in meta-analyses. Eur J Epidemiol. (2010) 25(9):603–5. doi: 10.1007/s10654-010-9491-z
12. Arsenault BJ, Boekholdt SM, Dubé MP, Rhéaume E, Wareham NJ, Khaw KT, et al. Lipoprotein(a) levels, genotype, and incident aortic valve stenosis: a prospective Mendelian randomisation study and replication in a case-control cohort. Circ Cardiovasc Genet. (2014) 7(3):304–10. doi: 10.1161/CIRCGENETICS.113.000400
13. Zheng KH, Tsimikas S, Pawade T, Kroon J, Jenkins WS, Doris MK, et al. Lipoprotein(a) and oxidised phospholipids promote valve calcification in patients with aortic stenosis. J Am Coll Cardiol. (2019) 73(17):2150–62. doi: 10.1016/j.jacc.2019.01.070
14. Kaltoft M, Sigvardsen PE, Afzal S, Langsted A, Fuchs A, Kühl JT, et al. Elevated lipoprotein(a) in mitral and aortic valve calcification and disease: the Copenhagen general population study. Atherosclerosis. (2022) 349:166–74. doi: 10.1016/j.atherosclerosis.2021.11.029
15. Makshood M, Joshi PH, Kanaya AM, Ayers C, Budoff M, Tsai MY, et al. Lipoprotein(a) and aortic valve calcium in South Asians compared to other race/ethnic groups. Atherosclerosis. (2020) 313:14–9. doi: 10.1016/j.atherosclerosis.2020.10.007
16. Kaiser Y, van der Toorn JE, Singh SS, Zheng KH, Kavousi M, Sijbrands EJG, et al. Lipoprotein(a) is associated with the onset but not the progression of aortic valve calcification. Eur Heart J. (2022) 43(39):3960–7. doi: 10.1093/eurheartj/ehac377
17. Kaiser Y, Singh SS, Zheng KH, Verbeek R, Kavousi M, Pinto SJ, et al. Lipoprotein(a) is robustly associated with aortic valve calcium. Heart. (2021) 107(17):1422–8. doi: 10.1136/heartjnl-2021-319044
18. Liu L, Rozi R, Shi W, Gao Y, Guo L, Tang D, et al. Association of serum lipoprotein(a) level with the severity and prognosis of calcific aortic valve stenosis: a Chinese cohort study. J Geriatr Cardiol. (2020) 17(3):133–40. doi: 10.11909/j.issn.1671-5411.2020.03.009
19. Burdeynaya AL, Afanasieva OI, Ezhov MV, Klesareva EA, Saidova MA, Pokrovsky SN. Lipoprotein(a) and its autoantibodies in association with calcific aortic valve stenosis. Diseases. (2023) 11(1):43. doi: 10.3390/diseases11010043
20. Mahabadi AA, Kahlert P, Kahlert HA, Dykun I, Balcer B, Forsting M, et al. Comparison of lipoprotein(a) levels in patients ≥70 years of age with versus without aortic valve stenosis. Am J Cardiol. (2018) 122(4):645–9. doi: 10.1016/j.amjcard.2018.04.046
21. Wilkinson MJ, Ma GS, Yeang C, Ang L, Strachan M, DeMaria AN, et al. The prevalence of lipoprotein(a) measurement and degree of elevation among 2710 patients with calcific aortic valve stenosis in an academic echocardiography laboratory. Angiology. (2017) 68(9):795–8. doi: 10.1177/0003319716688415
22. Chen HY, Dufresne L, Burr H, Ambikkumar A, Yasui N, Luk K, et al. Association of LP(A) variants with aortic stenosis: a large-scale study using diagnostic and procedural codes from electronic health records. JAMA Cardiol. (2018) 3(1):18–23. doi: 10.1001/jamacardio.2017.4266
23. Yang N, Zhang G, Li X, Zhou L. Correlation analysis between serum lipoprotein(a) and the incidence of aortic valve sclerosis. Int J Clin Exp Med. (2015) 8(10):19318–24. Available online at: https://pubmed.ncbi.nlm.nih.gov/26770570/26770570
24. Vongpromek R, Bos S, Kate T, Yahya GJ, Verhoeven R, J A, et al. Lipoprotein(a) levels are associated with aortic valve calcification in asymptomatic patients with familial hypercholesterolaemia. J Intern Med. (2015) 278(2):166–73. doi: 10.1111/joim.12335
25. Capoulade R, Chan KL, Yeang C, Mathieu P, Bossé Y, Dumesnil JG, et al. Oxidised phospholipids, lipoprotein(a), and progression of calcific aortic valve stenosis. J Am Coll Cardiol. (2015) 66(11):1236–46. doi: 10.1016/j.jacc.2015.07.020
26. Obisesan OH, Kou M, Wang FM, Boakye E, Honda Y, Uddin SMI, et al. Lipoprotein(a) and subclinical vascular and valvular calcification on cardiac computed tomography: the ARIC study. J Am Heart Assoc. (2022) 11(11):e024870. doi: 10.1161/JAHA.121.024870
27. Hojo Y, Kumakura H, Kanai H, Iwasaki T, Ichikawa S, Kurabayashi M. Lipoprotein(a) is a risk factor for aortic and mitral valvular stenosis in peripheral arterial disease. Eur Heart J Cardiovasc Imaging. (2016) 17(5):492–7. doi: 10.1093/ehjci/jev338
28. Cao J, Steffen BT, Budoff M, Post WS, Thanassoulis G, Kestenbaum B, et al. Lipoprotein(a) levels are associated with subclinical calcific aortic valve disease in white and black individuals: the multi-ethnic study of atherosclerosis. Arterioscler Thromb Vasc Biol. (2016) 36(5):1003–9. doi: 10.1161/ATVBAHA.115.306683
29. de Boer LM, Oorthuys AOJ, Wiegman A, Stroes ESG. The paradox of lipoprotein(a) lowering by statins: a meta-analysis. Atherosclerosis. (2022) 349:240–6. doi: 10.1016/j.atherosclerosis.2022.05.001
30. Tsimikas S, Fazio S, Ferdinand KC, Ginsberg HN, Koschinsky ML, Marcovina SM, et al. NHLBI Working group recommendations to reduce lipoprotein(a)–mediated risk of cardiovascular disease and aortic stenosis. J Am Coll Cardiol. (2018) 71(2):177–92. doi: 10.1016/j.jacc.2017.11.014
31. Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. (2017) 376(18):1713–22. doi: 10.1056/NEJMoa1615664
32. Schwartz GG, Steg PG, Szarek M, Bhatt DL, Bittner VA, Diaz R, et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. (2018) 379(22):2097–107. doi: 10.1056/NEJMoa1801174
33. Koren MJ, Moriarty PM, Massaro JM, Swerdlow DI, Scrimgeour AC, Rambaran C, et al. A phase 1 study of olpasiran, a small interfering RNA targeting lipoprotein(a). JAMA. (2022) 327(17):1680–9. doi: 10.1001/jama.2022.4709
34. Tsimikas S, Karwatowska-Prokopczuk E, Gouni-Berthold I, Tardif JC, Baum SJ, Steinhagen-Thiessen E, et al. Lipoprotein(a) reduction in persons with cardiovascular disease. N Engl J Med. (2020) 382(3):244–55. doi: 10.1056/NEJMoa1905239
Keywords: lipoprotein(a), aortic stenosis, aortic sclerosis, aortic valve calcification, genetic polymorphisms, cardiovascular disease, targeted therapies
Citation: Wambua P, Wahinya M and Khan Z (2025) The role of elevated lipoprotein(a) in aortic valve disease: a systematic review. Front. Cardiovasc. Med. 12:1610395. doi: 10.3389/fcvm.2025.1610395
Received: 11 April 2025; Accepted: 22 September 2025;
Published: 14 October 2025.
Edited by:
Xuchu Que, University of California, San Diego, United StatesReviewed by:
Shenglin Li, University of California, San Diego, United StatesTapan Ghose, Fortis Flt. Lt. Rajan Dhall Hospital, India
Copyright: © 2025 Wambua, Wahinya and Khan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Z. Khan, ZHJ6YWhpZDE5ODNAeWFob28uY29t
†These authors share first authorship