Abstract
Copper (Cu) and iron (Fe) are essential trace elements that are involved in normal human metabolic processes. Disruption of their homeostasis contributes to disease pathogenesis through mechanisms such as cuproptosis and ferroptosis. Cuproptosis targets lipoylated proteins to disrupt mitochondrial respiration, whereas ferroptosis is driven by lipid peroxidation. These processes may independently or interactively exacerbate pulmonary hypertension (PH), a condition characterized by progressive pulmonary vascular remodeling, clinical manifestations of dyspnea, right-sided heart failure, and high mortality, via oxidative stress, metabolic reprogramming, and other mechanisms. This review systematically elucidates: (1) the updated molecular mechanisms of cuproptosis/ferroptosis, (2) research evidence for their roles in PH, and (3) synergistic crosstalk in different subtypes of PH progression. We propose that coordination and regulation of the crosstalk network between cuproptosis and ferroptosis may represent a novel therapeutic strategy for pulmonary vascular remodeling.
1 Introduction
Pulmonary hypertension (PH) is a cardiopulmonary disorder characterized by an elevated mean pulmonary arterial pressure (mPAP) of ≥20 mmHg at rest, accompanied by abnormal pulmonary vascular pressure. Hemodynamically, precapillary PH is defined by mPAP ≥20 mmHg, pulmonary vascular resistance (PVR) ≥3 Wood units, and pulmonary artery wedge pressure (PAWP) ≤15 mmHg (1), whereas postcapillary PH requires mPAP ≥20 mmHg with PAWP >15 mmHg (2). The World Health Organization classifies PH into five groups: pulmonary arterial hypertension (PAH), PH because of left heart disease (PH-LHD), PH associated with lung diseases/hypoxia (PH-LD), Chronic Thromboembolic Pulmonary Hypertension (CTEPH), and PH of unclear multifactorial mechanisms (2, 3). Recent epidemiological studies revealed an aging-related rise in PH incidence among industrialized nations, affecting approximately 1% of the global population—including ≥50% of heart failure patients, 10% of chronic obstructive pulmonary disease (COPD) cases, and 10% of adults aged >65 years (4, 5).
Although vasodilatory therapies (6) and initial combination strategies [e.g., dual targeting of the endothelin and nitric oxide pathways with the endothelin receptor antagonist macitentan and phosphodiesterase type 5 inhibitor (PDE5i) tadalafil] have demonstrated efficacy in arterial and thromboembolic PH, challenges persist. Notably, such combination therapy significantly reduces PVR compared to monotherapy but concurrently increases severe adverse events (7), underscoring the urgent need for novel pathway exploration to curb disease progression.
Recent studies have identified nonapoptotic cell death pathways as promising therapeutic targets (8). Ferroptosis, first proposed by Dixon in 2012 (9), correlates with PH severity (10) through iron (Fe) accumulation-driven Fenton reactions that generate oxygen radicals, induce lipid peroxidation, and disrupt membrane integrity. While physiologically involved in embryogenesis, ferroptosis also exhibits therapeutic potential in oncology, cardiovascular, and gastrointestinal disorders (11, 12). Recently, Tsvetkov et al. (13) identified cuproptosis, a ferredoxin 1(FDX1)-dependent process that disrupts mitochondrial respiration via the modulation of lipoylated proteins and iron-sulfur (Fe-S) proteins. This discovery has expanded research in the fields of cancer, neurology, and cardiovascular diseases (14, 15). Notably, cuproptosis and ferroptosis may synergistically exacerbate PH progression through mutual reinforcement, and their co-targeting strategies have shown preclinical promise in oncology and immunotherapy (16) (Table 1).
Table 1
| Abbreviation | Full name | Function | References |
|---|---|---|---|
| ACSL4 | Acyl-CoA synthetase long-chain family member 4 | Catalyzes the generation of CoA derivatives from polyunsaturated fatty acids, promotes lipid peroxidation, and drives ferroptosis | (17–19) |
| GPX4 | Glutathione peroxidase 4 | Core antioxidant enzymes. Inhibits ferroptosis by reducing lipid hydroperoxides and maintains redox homeostasis | (20, 21) |
| SLC7A11 | Solute carrier family 7 member 11 | Forms the Xc− system, mediating cystine uptake for glutathione synthesis to inhibit ferroptosis | (22, 23) |
| SLC7A5 | Solute carrier family 7 member 5 | Transports cystine, thereby promoting glutathione synthesis and reducing lipid peroxidation levels | (22–24) |
| FSP1 | Ferroptosis suppressor protein 1 | Reduces coenzyme Q10 and vitamin K to synergistically inhibit ferroptosis with GPX4 | (20) |
| TFR1 | Transferrin receptor 1 | Mediates iron uptake into cells via transferrin endocytosis, promoting iron accumulation | (25, 26) |
| 4-HNE | 4-Hydroxynonenal | Biomarkers of ferroptosis. Covalently binds to GPX4, inactivating it and thereby exacerbating lipid peroxidation | (27, 28) |
| CoQ10 | Coenzyme Q10 | Inhibits lipid peroxidation by reducing coenzyme Q10 through FSP1, thereby protecting cells from ferroptosis | (20, 29) |
| PRDX3 | Peroxiredoxin 6 | Inhibits cystine uptake to trigger ferroptosis; translocates to the plasma membrane after peroxidation | (30) |
| HMGB1 | High Mobility Group Box 1 | In ferroptosis, it is released to activate the TLR4/NLRP3 inflammasome pathway, thereby exacerbating inflammation | (31) |
Ferroptosis-related factors.
2 Ferroptosis
2.1 The concept of ferroptosis
Ferroptosis is a newly identified form of cell death that depends on iron ions and lipid peroxidation (20). Current research on its mechanisms relies on the detection of key biomarkers, such as the lipid peroxidation-related molecules Acyl-CoA Synthetase Long-Chain Family Member 4 (ACSL4) and Arachidonate 15-Lipoxygenase, as well as the dynamic changes in the lipid peroxidation products 4-Hydroxy-2-Nonenal (4-HNE) and malondialdehyde (17, 18). Lipid peroxidation involves two mechanisms: (1) the active site of lipoxygenase contains Fe3+, which catalyzes the oxidation of polyunsaturated fatty acids (PUFAs), and (2) polyunsaturated fatty acids undergo oxidation reactions with reactive oxygen species (ROS) and Fe2+. Both these processes can generate lipid hydroperoxides (LOOH), which ultimately participate in ferroptosis, The hallmark of ferroptosis is the inhibition of glutathione peroxidase 4 (GPX4), which can reduce LOOH to maintain cellular homeostasis (21). Additionally, ferroptosis suppressor protein 1 (FSP1) selectively inhibits ferroptosis by targeting the plasma membrane, thereby exerting a protective effect (20). These core regulatory mechanisms focus on the role of the lipid metabolism network in the maintenance of plasma membrane integrity. As the molecular mechanisms of ferroptosis are further elucidated, therapeutic strategies targeting this process may open new avenues for the treatment of related diseases.
2.2 Mechanisms of ferroptosis
2.2.1 The Xc− system
Ferroptosis is closely related to the Xc − system activity. This system is composed of serum solute carrier family 7 member 11 (SLC7A11) and solute carrier family 3 member 2 (SLC3A2) subunits, which mediate the exchange of cysteine and glutamate and provide cysteine for glutathione synthesis. Notably, impairment of the Xc− System leads to glutathione deficiency, which in turn results in decreased GPX4 activity, contributing to ferroptosis (22).
Solute carrier family 7 member 5 (SLC7A5), a component subunit of SLC3A2, can be induced by interleukin-3 (24) and plays a crucial role in the function of the Xc− system (32). Sulfasalazine and erastin (Er) promote ferroptosis by inhibiting the Xc− system: Er, as a small-molecule activator of ferroptosis, binds to SLC7A5, indirectly preventing cystine uptake, while Ras-selective lethal small molecule 3 (RSL3) covalently binds to GPX4. Both of these actions inhibit the function of the Xc− system, thereby regulating ferroptosis (23). Compared to GPX4, RSL3, and ML162, two ferroptosis inducers more directly inhibit selenoprotein thioredoxin reductase 1 (TXNRD1), which clears peroxides (33).
2.2.2 Lipid oxidation
Fatty acids are critical components of cell membranes and signal transduction pathways. Peroxidation of PUFAs is central to ferroptosis. Lipid peroxidation typically requires a combination of peroxyl radicals (RO•) and hydroxyl radicals (HO•) with fatty acids (34). The redox cycling of Fe³+/Fe²+ in the Haber–Weiss reaction and the Fe²+-mediated Fenton reaction both produce large amounts of ROS. PUFAs, such as arachidonic acid (AA) and adrenic acid (AdA), are prone to lipid peroxidation by ROS because of their bis-allylic hydrogen atoms. This process is catalyzed by ACSL4, which converts PUFAs into Coenzyme A derivatives, which are esterified into phosphatidylethanolamines (PEs) by lysophosphatidylcholine acyltransferase 3. These PEs, rich in easily oxidizable AA/AdA, disrupt the lipid bilayer, leading to cell death and participating in pathological processes such as aortic endothelial cell atherosclerosis (19, 35). Ma et al. (9) showed that the binding of the 3′ untranslated region of ACSL4 with miR-424-5p can suppress ACSL4 expression, thereby alleviating ferroptosis induced by Erastin and RSL3.
2.2.3 Fe metabolism
Disorders in Fe metabolism are closely linked to ferroptosis. Dietary Fe exists in two forms: Fe²+ and Fe³+. Fe³+, which is poorly soluble in water, must bind to proteins for transport to participate in the synthesis of erythrocytes and other Fe-dependent cells. Once bound to transferrin (TF), Fe³+ is internalized via receptor-mediated endocytosis through transferrin receptor 1 and subsequently reduced to Fe²+ by metalloreductase STEAP3, Fe²+, which is highly water-soluble and reactive, exerts cytotoxic effects. It is then transported into the cytosolic labile Fe pool via divalent metal transporter 1 (DMT1) (25, 36). Ferritin, composed of ferritin light chain 1 (FTL1) and ferritin heavy chain 1 (FTH1), stores Fe³+ after FTH1 converts Fe²+. Ferritin can be released via exosomes to enable Fe efflux. When Fe²+ accumulates excessively, ferritin interacts with NCOA4, undergoes autophagy, and releases Fe²+ within the cell, promoting lipid peroxidation and triggering ferroptosis (26). Cytosolic Fe²+ is extruded through ferroportin 1 (FPN1) on the basolateral membrane, oxidized to Fe³+ by the membrane Fe oxidase hephaestin (HEPH), and then binds to TF to enter the circulation. Hepcidin binds to ferroportin to inhibit Fe²+ efflux, whereas DMT1 promotes Fe²+ uptake. These processes collectively maintain Fe homeostasis (37, 38). Experimental evidence (39, 40) shows that Fe-responsive element-binding protein 2 (IREB2) enhances its stability by binding to the deubiquitinase OTU Deubiquitinase 1, which promotes IREB2 deubiquitination. This increases the expression of downstream genes, Transferrin Receptor and DMT1, leading to Fe²+ accumulation and facilitating ferroptosis. In summary, Fe metabolism imbalance drives ferroptosis via multiple pathways.
3 Cuproptosis
3.1 Definition and discovery of cuproptosis
Maintaining Copper (Cu) ion homeostasis is crucial for human health. Cu can primarily exist in two oxidative states: Cu+, which accounts for 95% of the total intracellular Cu content, and Cu2+, which constitutes 5% of extracellular Cu. The reduction of Cu²+ in the gut to Cu+ by metal reductases is followed by Cu uptake in a Ctr1-dependent manner. Notably, the form of Cu(copper complex or copper ions) also affects the accumulation efficiency (41, 42). Following absorption, Cu is exported into the bloodstream via the P-type ATPases ATP7A and ATP7B (43). Cu+ in the blood is easily oxidized to Cu²+, which can bind with plasma proteins, among which Ceruloplasmin (CP) transports Cu²+ to various organs. However, Intracellular Cu+ is delivered to specific targets by chaperones such as antioxidant 1 copper chaperone (Atox1), cytochrome c oxidase copper chaperone (Cox17), and copper chaperone for superoxide dismutase (CCS), or it can be stored in metallothionein (44). Dysregulated Cu metabolism has been implicated in metabolic disorders, providing a rationale for targeting Cu homeostasis in therapies (14, 45). Even before the formal concept of cuproptosis was proposed, studies showed that Cu imbalance could suppress cytochrome c oxidase (COX) activity, impair mitochondrial membrane potential, reduce energy production, activate the AMP-activated protein kinase (AMPK) pathway, shift cellular metabolism toward glycolysis, and inhibit cell growth (46, 47). In 2022, researchers discovered that Cu ions, when delivered by carriers such as elesclomol, bind to lipoylated proteins in the tricarboxylic acid (TCA) cycle. The intracellular reductase FDX1 promotes the synthesis of lipoic acid synthase (LIAS), which regulates lipoylation. Under Cu overload conditions, reduced FDX1 specifically interacts with the Cu ionophore elesclomol or elesclomol-Cu(II), facilitating the conversion of excess Cu²+ into more toxic Cu+ (48, 49). The released Cu+ subsequently binds to lipoylated proteins, inducing proteotoxic stress and ultimately leading to cell death (50). This process, termed cuproptosis, is accompanied by the loss of Fe-S cluster proteins. This form of cell death is uniquely reversible by Cu chelators, distinguishing it from other regulated cell death pathways (Table 2).
Table 2
| Abbreviation | Full name | Function | References |
|---|---|---|---|
| FDX1 | Ferredoxin 1 | Cuproptosis core regulatory factor. mediating copper ion-dependent cell death by modulating Lipoylated proteins and iron-sulfur cluster proteins | (13, 45) |
| LIAS | Lipoic acid synthetase | The necessary condition for cuproptosis. Catalyzing the conjugation of lipoic acid with target proteins to generate Lipoylation | (51, 52) |
| DLAT | Dihydrolipoamide Acetyltransferase | Lipoylated target proteins. Inducing mitochondrial proteotoxic stress through oligomerization during copper overload | (13, 53) |
| DLST | Dihydrolipoamide S-succinyltransferase | The components of the key enzyme α-ketoglutarate dehydrogenase complex in the tricarboxylic acid cycle. Binding to copper leads to loss of its own function, thereby inhibiting energy metabolism | (13, 54) |
| GLS | Glutaminase | Anti-cuproptosis genes. Maintaining glutamine metabolism, alleviate copper-induced mitochondrial dysfunction. | (55) |
| MTF1 | Metal-regulatory transcription factor 1 | Anti-cuproptosis genes. Activating metallothioneins to chelate excess copper when cells encounter excess copper | (53) |
| SLC31A1 | Solute carrier family 31 member 1 | Mediating copper ion uptake. Promoting cuproptosis under copper overload conditions | (39, 56) |
| PDH | Pyruvate Dehydrogenase Complex | The key enzyme linking glycolysis and the tricarboxylic acid cycle (TCA cycle). Inhibition of activity during cuproptosis affects mitochondrial energy metabolism | (53, 57) |
| ATOX1 | Antioxidant 1 Copper Chaperone | Copper chaperone proteins. Regulates copper transport and SOD expression, It can alleviate oxidative stress | (59, 60) |
Cuproptosis-related factors.
3.2 Mechanisms of cuproptosis
3.2.1 Mitochondrial mechanism: targeting the TCA cycle
Cu-induced cell death has been extensively studied over the past decade (61). Tsvetkov et al. first demonstrated its effect on mitochondrial respiration (13). Mitochondria serve as key hubs for energy metabolism and apoptotic signaling and harbor a Cu reservoir. Excessive Cu accumulation disrupts mitochondrial enzyme activity and impairs respiration (62). Intracellular Cu+, a cofactor for COX and SOD1, can have its deficiency restored by Cu ionophores to regain enzyme activity. This genetic disorder is caused by pathogenic mutations in the Cu transporter ATP7A, leading to systemic Cu deficiency. Cu ionophores have also shown potential for therapeutic applications (63), among which SOD1 has antioxidant functions. Cu+ binds to SOD1 and promotes the formation of its disulfide (-S-S-) bonds through the mediation of the CCS (64). In addition, the synthesis of cytochrome c oxidase (SCO) proteins significantly influences the maintenance of Cu ion homeostasis within the mitochondria. For instance, the mitochondrial cytochrome C oxidase Cu chaperone and SCO2 mediate Cu transport into the mitochondria and its allocation to COX (65, 66). Oxidized cysteine residues in mitochondrial SCO1 bind Cu+, generating redox signals transmitted via cytochrome c oxidase assembly factor 19 to cytosolic ATP7A, promoting Cu efflux (67). Furthermore, Cu forms complexes with ligands in the mitochondrial matrix, interfering with FDX2-dependent Fe-S cluster protein maturation and exacerbating mitochondrial dysfunction (54–52). Thus, Cu dyshomeostasis poses a severe threat to mitochondrial integrity.
Several enzymes involved in mitochondrial metabolism are associated with apoptosis. Cu-induced cell death depends on protein lipoylation, which is a post-translational modification of lysine residues. First described in the 1960s, lipoylation occurs in only five proteins: dihydrolipoamide acetyltransferase (DLAT), pyruvate dehydrogenase complex componentX(PDHX), dihydrolipoamide succinyltransferase (DLST), dihydrolipoamide branched-chain transacylase, and glycine cleavage system protein H (58). FDX1 directly binds to LIAS and provides electrons to enable LIAS-catalyzed reactions. Therefore, upon LIAS knockdown, the level of lipoylation decreases (53–69). Consequently, in Cu overload conditions, Cu+ binds to acylated DLAT and DLST, inducing their oligomerization and depleting Fe-S cluster proteins, leading to cell death in an FDX1-dependent manner (13). Recent studies have identified seven pro-cuproptosis genes [FDX1, LIAS, lipoyltransferase 1 (LIPT1), DLD, DLAT, PDHA1, and PDHB] and three anti-cuproptosis genes [MTF1, Glutaminase(GLS), and CDKN2A], Notably, the pyruvate dehydrogenase (PDH) complex plays a central role in these pathways (70), highlighting its critical role in the TCA cycle during cuproptosis.
3.2.2 Cu-induced oxidative stress
Cu-induced oxidative stress plays a pivotal role in disease progression. The TCA cycle, a primary site for ROS generation, is targeted by Cu ions, leading to cuproptosis, suggesting that oxidative stress is an inevitable consequence of cuproptosis. Moreover, erythroid 2-related factor 2 (Nrf2), a central regulator of oxidative stress, is activated by Cu overload to initiate autophagy and antioxidant responses (71). This process can be rescued by Cu chelators, such as tetrathiomolybdate (TTM), potentially through the AMPK/mTOR/ULK1 pathway-mediated degradation of Kelch-like ECH-associated protein 1(KEAP1), a negative regulator of Nrf2 (72). This indicates that Cu overload can exert oxidative stress via Nrf2, and that Cu-induced oxidative stress damage may involve both apoptosis and autophagy (59). Autophagy, a process of cellular self-digestion and degradation (60), has been shown to ameliorate the progression of pulmonary arterial hypertension (73), suggesting that an imbalance in Cu homeostasis may exacerbate disease progression through the Nrf2-autophagy-oxidative stress axis. In addition, within the vasculature, the generation of ROS can occur not only through the reduction of Cu2+ to Cu+, but also via the ATOX1-TRAF axis. This process can disrupt Fe-S clusters and induce damage to vascular endothelial and smooth muscle cells, which has been demonstrated in cardiovascular pathologies (74). Antioxidant 1, a chaperone protein for Cu+, maintains the redox balance by regulating Cu transport and superoxide dismutase [SOD])expression. A reduction in ATOX1 can trigger oxidative stress (75) and chelating Cu ions can mitigate this oxidative stress (30, 31). These intricate interactions not only highlight that dysregulation of Cu metabolism induces oxidative stress through multiple pathways, but also underscore the potential of targeting these pathways as novel therapeutic strategies for cardiovascular diseases. Therefore, the molecular mechanisms underlying these processes require further investigation.
4 Therapeutic potential of cuproptosis and ferroptosis research
Pulmonary arterial remodeling, which leads to increased vascular pressure, often results in secondary myocardial dysfunction, which progressively develops into heart failure, ultimately resulting in mortality. Both cuproptosis and ferroptosis have significant potential in this context.
4.1 Ferroptosis as a potential therapeutic target in PH
In PH, ferroptosis often exacerbates disease progression, primarily through a lipid peroxidation imbalance, which disrupts the redox equilibrium in pulmonary vascular endothelial and smooth muscle cells. In the vascular endothelium, promotion of the binding of SLC3A2 to ubiquitin induces ferroptosis, thereby causing endothelial injury. Similarly, PAH endothelial cells often exhibit pro-ferroptotic phenotypes. For instance, the overexpression of ACSL4, which is involved in lipid peroxidation, in PAECs induces ferroptosis, leading to an inflammatory pulmonary vascular burden, triggering PAH and subsequently inducing right ventricular dysfunction (10, 76). Additionally, peroxiredoxin 3 (PRDX3) serves as a crucial antioxidant protein. After peroxidation, it translocates from the mitochondria to the plasma membrane, where it triggers ferroptosis by inhibiting cystine uptake (77). Based on this, Liao et al. (78) found that peroxiredoxin 6, which belongs to the same family as that of PRDX3, can prevent the release of high-mobility group box 1 (HMGB1) induced by ferroptosis in PAECs and inhibit the TLR4/NLRP3 inflammasome pathway, thereby alleviating inflammatory responses and reversing PH. Similarly, Xie et al. (79) demonstrated that Fer-1 can also alleviate ferroptosis, which inhibits the release of HMGB1 and the subsequent inflammatory response, thus exerting a protective effect in PH. The ROS generated by ferroptosis can regulate the expression of endothelial to mesenchymal transition (EndMT)-related genes via the ROS-TGF-β axis, thereby endowing endothelial cells with the characteristics of mesenchymal cell migration and invasion capabilities, and thus promoting vascular remodeling (80).
Moreover, maintaining an appropriate level of Coenzyme Q10 (CoQ10) is crucial for protecting pulmonary cells. FSP1 synergizes with GPX4 to inhibit ferroptosis (20). Chen et al. (81) showed in aortic vessels that blocking the FSP1-CoQ10 pathway reduces cell viability and increases lipid peroxidation levels associated with ferroptosis in smooth muscle cells, exacerbating inflammatory damage, suggesting that the FSP1-CoQ10 pathway's role in counteracting ferroptosis may be significant in pulmonary vascular pathology. Rutin, a naturally occurring flavonoid, protects PAH rats from ferroptosis damage by directly interacting with protein kinase C (PKC)α and altering its structure and activity (82), highlighting the therapeutic potential of small and large molecule interactions. These findings not only provide new insights into PH pathogenesis but also offer potential interventions and therapeutic targets (Figure 1).
Figure 1
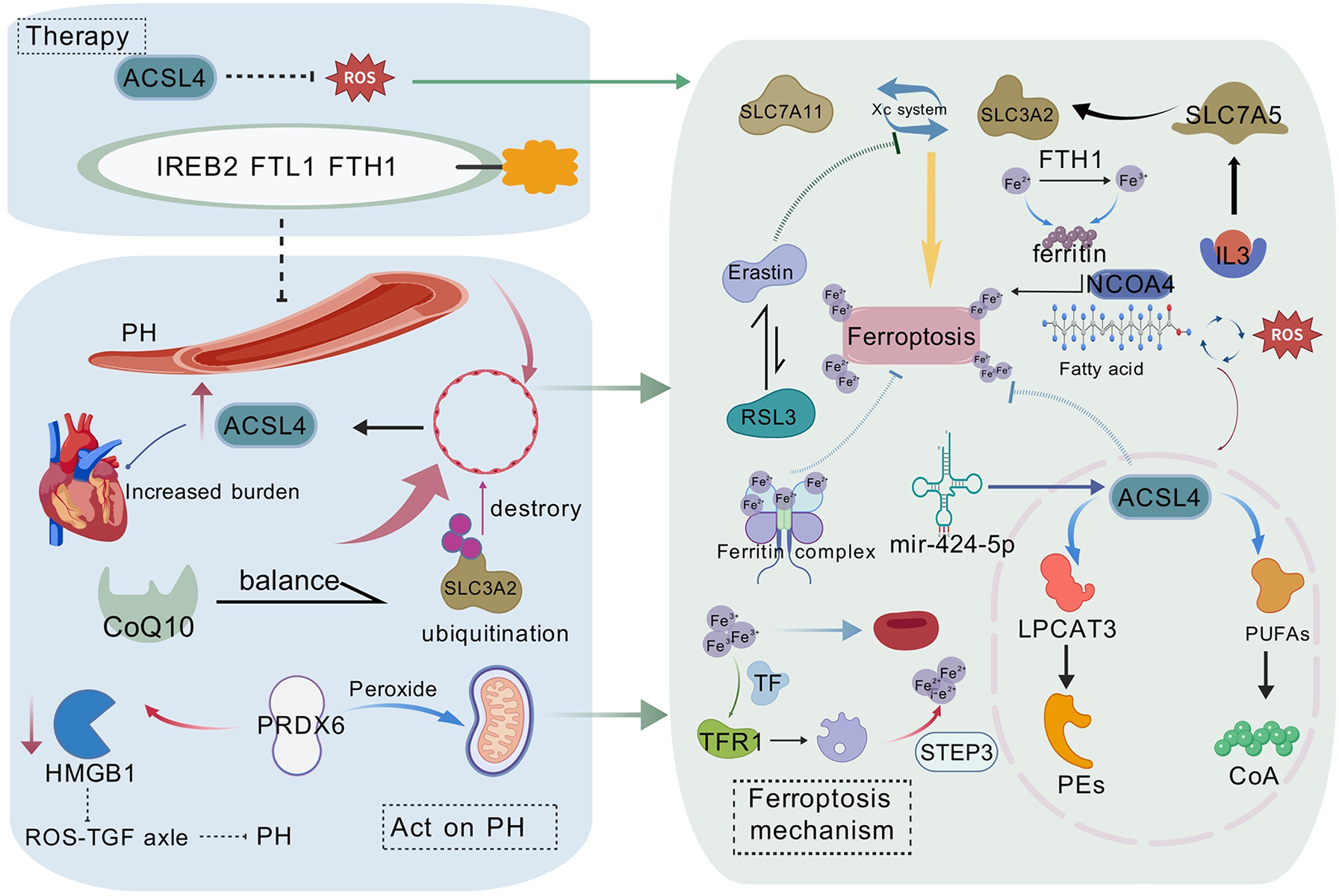
The molecular mechanisms of ferroptosis in pulmonary hypertension (PH) and potential therapeutic strategies are illustrated. PH affects endothelial cells, leading to lipid peroxidation and ferroptosis. ACSL4 plays a central role in lipid peroxidation, whereas iron metabolism disorders promote ferroptosis by affecting ferritin (FTL1/FTH1) and ferroportin (FPN1). The figure also presents potential therapeutic strategies targeting the Xc− system (including SLC7A11 and SLC3A2), ACSL4, and ferroptosis suppressor protein 1 (FSP1), offering new perspectives and therapeutic avenues for PH treatment. This figure is based on Bio Gdp.
4.2 Cuproptosis as a potential therapeutic target in PH
Cu, an essential trace element, may exacerbate PAH progression, with metabolic factors likely playing a dominant role. Cu affects vascular endothelium and smooth muscle, is associated with vascular aging, and participates in energy metabolism, signal transduction, and biomolecule synthesis (83, 84). Endothelial cell-derived nitric oxide (NO) diffuses to pulmonary smooth muscle cells (PSMCs) to induce vasodilation and oppose vascular remodeling. Cu contributes to NO synthesis and release, and restoring NO levels can prevent EndMT in PAECs, thereby counteracting the endothelial-to-mesenchymal transition that promotes a proliferative phenotype (85). Thus, inhaled nitric oxide (iNO) therapy effectively reduces PVR but is less effective in advanced stages, which are often accompanied by right heart failure (86). Moreover, a recent report showed that in children with PH who experienced in-hospital cardiac arrest, iNO treatment was associated with a lower uncorrected return of spontaneous circulation rate than that in controls (56), highlighting the need for further investigation of PH treatment.
Animal studies have shown that Cu overload induces mitochondrial abnormalities and inhibits angiogenesis, ultimately leading to Cuproptosis. Mo et al. (87) demonstrated that chelator-mediated reduction of Cu²+ in rat cerebral vessels inhibited cuproptosis, improved blood perfusion, and alleviated mitochondrial damage. Additionally, data from Phase II clinical trials indicated that the Cu ion chelator triethanolamine reduces Cu²+ toxicity on mitochondrial function and energy metabolism, significantly improving left ventricular systolic function (88). This also implies the potential involvement of cuproptosis in the pulmonary vasculature. However, direct studies linking cuproptosis to PH are lacking, although evidence suggests that PH severity correlates with elevated Cu ion levels in pulmonary vascular cells, possibly because of hypoxia or other factors upregulating Cu transporters (89). Excessive Cu accumulation may exacerbate PH progression by promoting cell death. Cu chelators have been shown to inhibit PAEC proliferation in patients with idiopathic pulmonary arterial hypertension (IPAH) by activating Apoptosis-Inducing Factor protein and triggering a non-caspase-dependent apoptotic pathway (90). Moreover, a diet deficient in Cu will not cause further damage to the right atrium (90, 91). These findings suggest that the role of Cuproptosis in PH and the activation or inhibition of associated signaling pathways in pulmonary vascular cells represent crucial directions for future research (Figure 2).
Figure 2
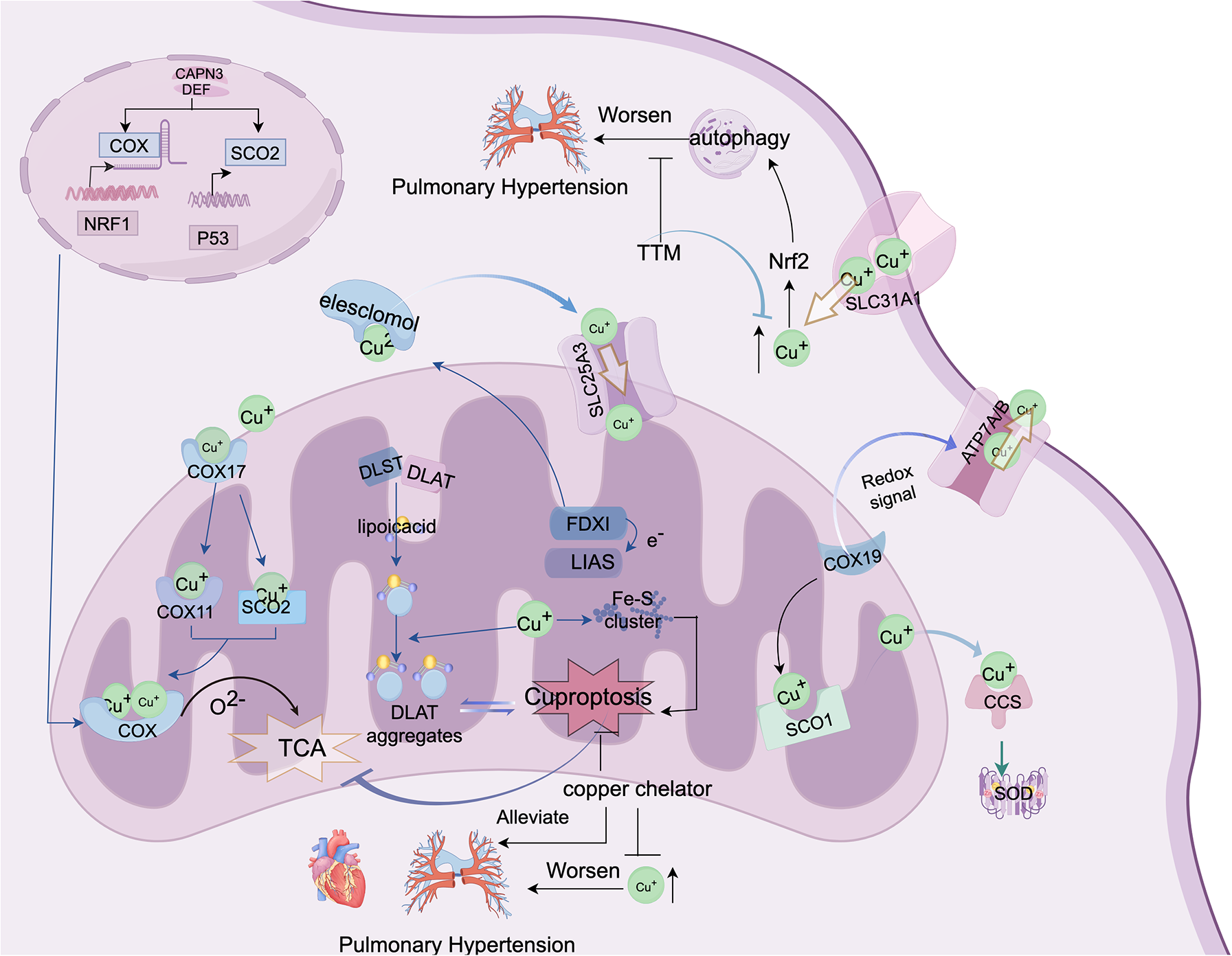
This figure depicts the molecular mechanisms of cuproptosis and potential therapeutic strategies for PH. This shows how abnormal copper metabolism and cuproptosis may contribute to PH progression. Cytochrome c oxidase (COX) and lipoic acid-containing proteins are the central components of copper metabolism, both localized in the mitochondria. The figure also demonstrates that copper can directly bind to lipoic acid components to induce cuproptosis. Additionally, elevated copper levels may exacerbate pulmonary vascular remodeling by activating autophagy/cuproptosis, thus offering new insights and potential therapeutic avenues for PH treatment. The figure was created using FigDraw. ID: AYOYOFCAAB.
4.3 Therapeutic potential of synergistic cuproptosis and ferroptosis in clinical and animal studies
Aberrant Cu and Fe metabolism has been implicated in the pathogenesis of cardiovascular diseases (92). In the context of pulmonary vascular remodeling, exploring the interplay between these two forms of cell death may offer a more effective strategy for mitigating PH progression. However, current research on the synergistic effects of ferroptosis and cuproptosis remains largely limited to preclinical studies, with nanotechnology showing promising results. For instance, the MitCuOHA nanozyme has been shown to deplete cysteine (Cys), release Cu ions, and induce lipoylated protein aggregation, thereby simultaneously triggering cuproptosis and ferroptosis both in vitro and in vivo, resulting in enhanced tumor growth suppression (93). Similarly, Erastin and Cu—conjugated nanoparticles deplete glutathione (GSH), increase lipid peroxidation, inhibit the TCA cycle, and promote T-cell infiltration (16, 94). A nanocarrier system responsive to GSH via disulfide bonds (-S-S-) was also found to inhibit GPX4 and induce DLAT oligomerization, ultimately activating both cuproptosis and ferroptosis in tumors (95). Moreover, in treating myelodysplastic syndromes, combining the ferroptosis inducer imidazole ketone erastin (IKE) with Elesclomol-Cu (ES-Cu) induces severe mitochondrial damage, elevates ROS levels, and enhances lipoylation-dependent DLAT oligomerization, resulting in more pronounced cell death and greater inhibition of proliferation compared to monotherapy (96). Likewise, compared to the toxic effects of Cu chelation therapy, the traditional Chinese medicine curcumin has demonstrated marked efficacy in Wilson's disease by lowering intracellular Cu levels, inhibiting ferroptosis, and promoting the expression of oxidative stress-related markers such as GPX4, heme oxygenase 1 (HO-1), and Nrf2 (97). These findings underscore the therapeutic potential of co-targeting ferroptosis and cuproptosis in disease treatment. Nevertheless, clinical reports on the combined induction of these two forms of cell death remain scarce.
Although research on the interplay between cuproptosis and ferroptosis has primarily focused on cancer, a notable gap remains in cardiovascular research. Cancer cells often exhibit a Warburg (glycolytic) phenotype to meet excessive growth demands. In mouse models of early-stage PH, reduced activity of electron transport chain complexes in pulmonary endothelial cells elevates mitochondrial ROS, which upregulates hypoxia-inducible factor-1 alpha (HIF-1α) and enhances glycolysis. This results in a metabolic profile resembling that of cancer cells—albeit without invasion or metastasis—ultimately contributing to vascular remodeling (98, 99). These observations suggest that insights from cancer research on the interaction between cuproptosis and ferroptosis may also be relevant to pulmonary vascular remodeling.
5 Crosstalk between cuproptosis and ferroptosis in different types of PH
As more forms of regulated cell death are being discovered, a strong correlation between ferroptosis and cuproptosis has become increasingly evident (100). Similarly, the dysregulation of Fe and Cu homeostasis plays a significant role in the pathogenesis of PH (27, 28, 70) (Figure 3). In PH, the diminished antioxidant capacity of pulmonary vascular cells exacerbates the insufficient GPX4 supply, which in turn promotes oxidative stress and elevated levels of lipid peroxidation (101). Recent studies have shown that Cu can directly bind to GPX4, inducing its oligomerization, which is subsequently degraded by the autophagy receptor Tax1 Binding Protein 1 during autophagy (102), thereby driving ferroptosis. 4-HNE, as a marker of ferroptosis, can also covalently bind to and inactivate the GPX4 protein (103) and inhibit antioxidant systems such as SOD and Glutathione Peroxidase(GPX), leading to cellular oxidative damage and activation of stress signaling pathways (104–106). GSH serves as a cofactor for GPX. It can not only inhibit lipid peroxidation to block ferroptosis but has also been found in pancreatic cancer tissues, where it can act as a ferroptosis inhibitor. This may involve the increased transport of GSH into the mitochondria to exert its effects via solute carrier family 25 member 39 (SLC25A39) (107), whereas SLC25A3 facilitates the transport of Cu into the mitochondrial matrix for enzymatic use (108). These findings highlight the pivotal role of GSH in the regulatory networks of cuproptosis and ferroptosis in PH.
Figure 3
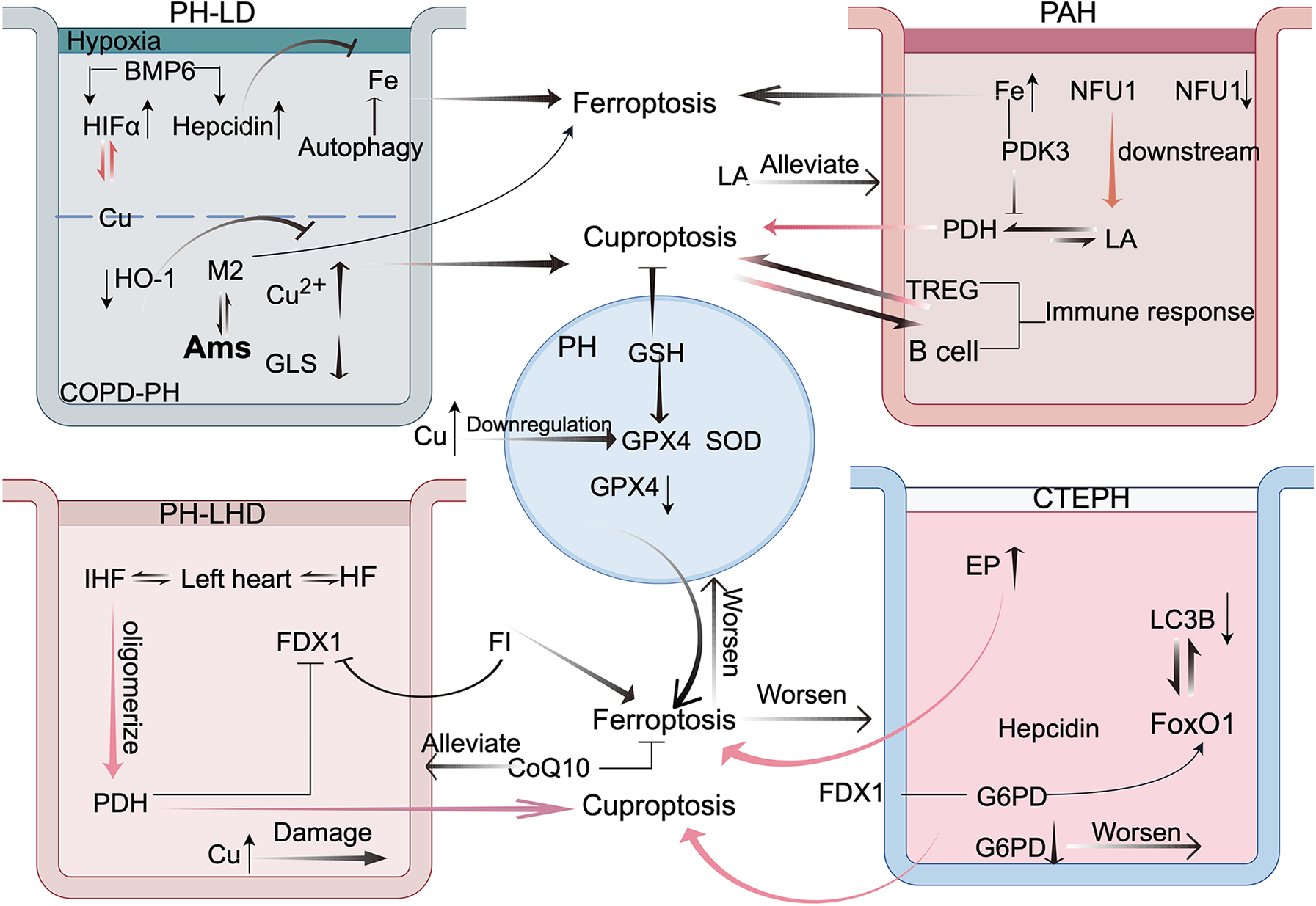
This figure illustrates the crosstalk between cuproptosis and ferroptosis in the four types of pulmonary hypertension (PH). This highlights the mechanisms of interaction between these two forms of cell death in pulmonary disease-associated PH, emphasizing the roles of hypoxia-inducible factor-1 alpha (HIF-1α) and alveolar macrophages (AMs). In arterial PH, immune responses and pyruvate dehydrogenase (PDH) are central to the crosstalk. In PH because of left heart disease, frataxin (FDX1) is the key mediator. In chronic thromboembolic pulmonary hypertension, glucose-6-phosphate dehydrogenase (G6PD) and forkhead box protein O1 (FoxO1) play pivotal roles. These findings offer new perspectives and potential therapeutic avenues for PH treatment. FI: ferroptosis inducer. This figure was created based on FigDraw. ID: OIPSOe5796.
5.1 PH-LD
PH associated with chronic pulmonary disease often exhibits unique pathological characteristics, primarily involving the loss of small pulmonary vessels, leading to vascular remodeling and increased mortality. However, the efficacy of conventional pharmacological treatments for PH remains limited (109), highlighting the urgent need for further research.
5.1.1 High-altitude pulmonary hypertension (HPH)
In the development of HPH, it is proposed that under hypoxic conditions, the hypoxia-induced mitochondrial autophagy receptor FUN14 domain containing 1 (FUNDC1) modulates HIF1α activity, thereby stimulating pulmonary artery smooth muscle cells (PASMCs) proliferation and triggering PH (110). In hypoxic PH rats, both hepcidin and HIF1α levels were elevated via the mediation of bone morphogenetic protein 6 (BMP6). BMP6 regulates systemic Fe levels, whereas hepcidin reduces intracellular Fe release (111). Consistent with this, Hu et al. observed that during early hypoxia, ferroptosis and ferroptosis resistance coexist, with PASMCs proliferation outweighing cell death, This may result from low pulmonary artery Fe levels and ROS accumulation, enhancing cellular defenses (112). Similarly, clinical reports indicate that patients with Fe deficiency exhibit higher rates of cardiovascular mortality and heart failure (113). In this context, ferroptosis inhibition paradoxically exacerbates PASMCs proliferation. However, when ferroptosis is induced via autophagy, such as through the induction of the long non-coding RNA MIR210HG, it can aggravate HPH by promoting a synthetic phenotype in PASMCs (114). Therefore, maintaining a balance between ferroptosis and proliferation is crucial in hypoxic PH. Additionally, in tumor models, MURR1 domain upregulation in Cu metabolism reduces Cu content, promotes ubiquitin-mediated HIF1α degradation, and downregulates CP and SLC7A11 transcription, thereby enhancing ferroptosis (115). Moreover, during cuproptosis induction, SLC7A11 degradation also occurs, triggering severe oxidative stress and lipid peroxidation (116), thereby providing further evidence of a potential link between these two forms of cell death. Thus, the interaction between ferroptosis and cuproptosis can be mediated through HIF1α, although research on the role of Cu in hypoxic pulmonary vasculature remains limited.
5.1.2 COPD-PH
COPD is one of the most common diseases worldwide and is characterized primarily by chronic inflammation and destruction of the lung tissue. Pulmonary vascular remodeling can be an early pathological change that is highly likely to induce irreversible emphysema. As the disease progresses, the accumulation of inflammation may trigger cardiovascular diseases, such as heart failure and PH (117). Smoking is the primary etiological factor. Recently, it was discovered that in patients with COPD, overexpression of fibroblast growth factor (FGF) 10 not only reversed cigarette smoke-induced emphysema but also exerted a protective effect on the pulmonary vasculature, preventing the development of PH (118). In pulmonary tissues, FGF 10 exerts its effects via the Nrf2-mediated antioxidant pathway, which activates the downstream SLC7A11/GPX4 axis. This process reduces free Fe and lipid peroxidation, thereby alleviating ferroptosis and ROS (119). These findings suggest that ferroptosis is involved in COPD-associated PH. However, excessive activation of Nrf2 or exposure to cigarette smoke can over-activate heme oxygenase 1 (HO-1), which catalyzes heme to release large amounts of Fe²+. This can increase Fe-dependent lipid peroxidation and potentially cause ferroptosis-related lung tissue damage (120). However, the biliverdin generated in this process exhibits antioxidant properties. Recent studies have suggested that HO-1 can exert antioxidant effects in alveolar cells, which are regulated by the glycoprotein CEACAM6, a member of the carcinoembryonic antigen family. When high levels of CEACAM6 inhibit HO-1, it fails to resist the oxidative damage caused by cigarette smoke, thereby potentially promoting COPD development (55). These findings highlighted the dual role of HO-1.
In COPD, alveolar macrophages (AMs) are the predominant immune cells and are classified into two phenotypes: M1 and M2. The proinflammatory M1 phenotype promotes lipid peroxidation and ferroptosis in alveolar cells via the paracrine secretion of LTB4, thereby facilitating COPD progression (121). The anti-inflammatory M2 phenotype is susceptible to ferroptosis and consequently loses its anti-inflammatory function, exacerbating the inflammatory response and lung injury. Thus, HO-1 inhibition can reverse the inflammatory damage caused by ferroptosis in AMs (122). Nevertheless, M2 macrophages, often implicated in abnormal tissue repair, not only secrete fibrotic factors that promote airway remodeling in patients (123), but also stimulate the proliferation of PASMCs via secretion of the chemokine CX3CL1 (124). However, recent findings indicate that, in the context of smoke exposure alone, the absence of inducible nitric oxide synthase—commonly expressed in macrophages—can prevent the pro-proliferative effects of M2 macrophages on PASMCs. This may involve the extracellular signal-regulated kinase (ERK) signaling pathway and contribute to pulmonary vascular remodeling in patients with COPD (125). Additionally, in a COPD rat model, AMs showed decreased cuproptosis-related gene glutaminase (GLS) and increased Cu²+. Because GLS converts glutamine to glutamate during the TCA cycle, its reduction may cause energy deficits in AMs, inducing the release of inflammatory factors and further lung damage (126). Notably, plastic particles entering AMs can also increase SLC31A1 expression, promoting cuproptosis and tumor necrosis factor-α secretion, which activates alveolar epithelial inflammation and destroys lung tissue (127). Moreover, exposure to cigarette smoke can activate a large number of macrophage infiltrations within blood vessels, triggering oxidative stress and endothelial dysfunction, which, in turn, exacerbates PH (128). Cigarette smoke extract can elevate the levels of superoxide anions (O₂−) in PASMCs (129). This leads to a paradoxical decrease in the levels of nitric oxide (NO) generated by the catalytic action of nitric oxide synthase, thereby impeding the vasodilatory effect of NO on PASMCs (130). However, in lung tissue, excessive NO can react with O₂− to generate the potent oxidant peroxynitrite (ONOO−), further exacerbating COPD-associated damage. In conclusion, in the context of AMs, the concurrent occurrence of cuproptosis and ferroptosis has been implicated in the worsening of COPD progression. Nevertheless, the role of cuproptosis/ferroptosis in macrophages, particularly M2 macrophages, in pulmonary vascular remodeling requires further investigation.
Furthermore, experimental validation has shown that circSAV1 promotes the translation of IREB2 mRNA through m6A modification, leading to Fe overload and ferroptosis, thereby inducing COPD. IREB2 binds to Fe-responsive elements (IREs) to maintain Fe homeostasis (131). When Fe levels are low, Fe regulatory protein 1 loses its Fe-S cluster and binds to IREs in target mRNAs to promote Fe uptake and reduce Fe storage and utilization (132). Upregulation of the Fe-S cluster assembly protein IscU2 can inhibit this process because IscU2 is a scaffold protein that stabilizes Fe-S cluster-containing proteins. In pancreatic cancer tissues, IscU2 enhances the expression of the cuproptosis-related factor DLST during the TCA cycle (133). Additionally, researchers have discovered that the human Fe-S cluster assembly enzyme (ISCU) protein, owing to its strong Cu-binding activity, can hinder the assembly of Fe-S clusters. In Menkes disease, as Cu accumulates, the activities of alpha-ketoglutarate dehydrogenase (KGDH) and PDH decrease, both of which require Fe-S clusters (134). Collectively, these findings suggest that Fe-S clusters may serve as regulatory switches in the interactions between Cu and Fe metabolism.
Moreover, clinical investigations have revealed that the degree of pulmonary fibrosis is associated with the risk of mortality in PH (135). Furthermore, fibroblasts and PASMCs exhibit excessive resistance to cell death and hyperproliferation (136). Both Cu and Fe overload result in the accumulation of reduced ferrous and cuprous ions, initiating the Fenton reaction and generating large amounts of ROS (137). ROS induce TGF-β1, promoting fibroblast activation and extracellular matrix deposition, thereby exacerbating pulmonary fibrosis. Simultaneously, TGF-β1 increases Fe accumulation and induces ferroptosis (138). In summary, in pulmonary diseases, HIFα, AMs, and Fe-S clusters serve as hubs linking cuproptosis and ferroptosis, potentially emerging as new research hotspots in the future.
5.2 PAH
PAH is a vascular dysfunction associated with inflammatory infiltration and is closely related to mitochondrial dysfunction (139). For instance, studies have shown that serum ceruloplasmin (CP) levels are significantly elevated in patients with systemic sclerosis-associated PAH and PAH mouse models (140). CP is a vital protein responsible for binding Cu and transporting it to target organs. It relies on ferroxidase (FOX) activity to oxidize Fe2 + to Fe3+, thereby facilitating Fe transport. Excess Cu in the liver promotes FOX synthesis, potentially enhancing Fe absorption (141). Elevated Cu levels in the body can also induce oxidative stress and vascular dysfunction, both of which are detrimental to cardiovascular health (142).
When superoxide exacerbates endothelial injury, it contributes to the pathogenesis of PAH, as evidenced by the decreased levels of the antioxidant enzyme superoxide dismutase (SOD) in idiopathic PAH (143). Additionally, cohort studies have suggested that this disease is closely linked to autoimmunity, characterized by abnormal immune cell infiltration (144), including increased regulatory T-cell (Treg) concentrations and B cell frequencies. In hereditary PAH (HPAH) patients, the severity of pulmonary vascular lesions is associated with TIM-3-positive T cells (92). Although immune inflammation is a common feature of this disease, the precise relationship between immunity and IPAH remains unclear. Cuproptosis induces immunogenic cell death, thereby activating numerous immune cells (145). Similarly, Fe accumulation activates immune cells (146), both of which contribute to inflammatory responses. Cheng et al. (57) reported that lung cancer exhibits pulmonary vascular remodeling features of PH, possibly because of tumor cell-induced inflammation affecting the pulmonary vessels. Therefore, the immune effects of cuproptosis and ferroptosis may provide new perspectives for treating PH.
Cuproptosis, ferroptosis, and metabolism are closely linked to mitochondrial damage and cellular respiration (147). NFU1, a mitochondrial Fe-S scaffold protein, assembles and transfers Fe-S clusters to target proteins such as complex II and LIAS. Mutations in NFU1 impair these electron transport proteins, weaken mitochondrial respiration, and promote pulmonary artery smooth muscle cell proliferation (148). Accordingly, humanized NFU1 mutations leading to mitochondrial dysfunction are closely associated with pulmonary vascular remodeling, with a marked NFU1 deficiency observed in patients with IPAH. Supplementation of the downstream target LA in PAECs from patients with PAH has been shown to improve mitochondrial function (149). High Cu concentrations can also compromise the mitochondrial membrane (150). LA is a component of the PDH complex, and lipoyltransferase 1 (LIPT1), a recently identified cuproptosis-related gene, transfers LA from glycine cleavage system protein H to the E2 subunit of lipoylated substrates, thereby influencing PDH activity, as observed in cancer and metabolic studies (151–153). Further investigation revealed that elevated Fe levels may bind to pyruvate dehydrogenase kinase (PDK) 3, inhibiting PDH complex activity (154). Clinically, a 4-month trial demonstrated that administration of the PDK inhibitor dichloroacetate to patients with IPAH activated PDH, reduced PVR, and improved lung function (155). Conversely, another study found that PDK4-mediated inhibition of PDH could reduce pyruvate oxidation, thereby decreasing ROS generation in the TCA cycle, limiting lipid peroxidation, and mitigating ferroptosis (29). Thus, targeting mitochondrial ROS reduction may alleviate PDH-mediated ferroptotic damage and curb abnormal PASMC proliferation. Additionally, LIPT1 participates in the lipoylation of the mitochondrial enzymes PDH and KGDH. Studies on fibroblasts from patients with LIPT1 mutations have shown markedly reduced activity of these enzymes and intracellular Fe accumulation. In this setting, α-LA, an Fe chelator, inhibits ferroptosis and modulates PDH activity (85, 156). Consequently, suppressed PDH by iron metabolism may contribute to both cuproptosis and the inflammatory response, leading to adverse pathological outcomes.
5.3 PH-LHD
In left heart disease, the loss of cardiac function leads to reduced myocardial contractility, ultimately resulting in heart failure (HF). This condition increases PVR and decreases pulmonary arterial compliance, thereby inducing postcapillary pulmonary hypertension (pcPH). HF is the primary cause of pcPH, with ischemic heart disease (IHF) being a major contributor to HF. IHF results from insufficient coronary blood supply, which initially damages the left ventricular cells (157, 158). Excessive Cu ion levels in patients with HF induce oxidative stress and severe cardiac injury (159). Furthermore, cuproptosis has been implicated in IHF and is characterized by the oligomerization of PDH complex proteins. In response, the body initiates a self-defense mechanism by downregulating FDX1 expression. Moreover, a strong positive correlation has been observed between cuproptosis-related genes and Treg cells, which can promote cardiac remodeling in IHF through inflammatory pathways (160). In hepatocellular carcinoma studies, ferroptosis inducers have been shown to inhibit FDX1 degradation while reducing the Cu chelator GSH, leading to the accumulation of lipoylated proteins (161) and, ultimately, cuproptosis. Ferroptosis is widely associated with various cardiac diseases (162). CoQ10, a core electron carrier in the mitochondrial respiratory chain, exerts its antioxidant effects by inhibiting lipid/DNA oxidation, thereby suppressing ferroptosis. Clinical trials have demonstrated that high-dose CoQ10 improves hepatic steatosis and enhances cardiovascular function (163). In a study by Sharp et al., patients with PAH treated with CoQ10 exhibited increased hemoglobin and mean corpuscular hemoglobin levels, along with improved right and left ventricular function (164). Therefore, ferroptosis regulated by CoQ10 may play a significant role in left-sided heart disease. Thus, FDX1 may serve as a hub for these two types of cell death. However, this requires further investigation.
5.4 CTEPH
CTEPH often develops in patients following pulmonary embolism. Despite at least 3 months of anticoagulation therapy, thrombi in the pulmonary artery become organized and resistant to conventional anticoagulation, resulting in vascular obstruction, impaired blood flow, and elevated pulmonary arterial pressure, typically presenting as dyspnea (165). Increased erythrophagocytosis in pulmonary microvascular endothelial cells enhances procoagulant activity by inducing ferroptosis, thereby contributing to thrombotic vascular remodeling (166). PAEC dysfunction caused by thrombi represents a key pathological feature of CTEPH. Experimental studies have demonstrated impaired autophagic activity in PAECs of CTEPH rats, as indicated by the reduced expression of microtubule-associated protein 1 light chain 3 (LC3B), a protein involved in autophagosome formation (167). Forkhead box O1 (FOXO1), a member of the FOXO family, interacts with LC3 to regulate autophagy. FOXO1, a glucose regulator, has been shown to activate hepatic hepcidin transcription (168), thereby reducing Fe release. Bioinformatic data from studies on FOXO signaling indicate that glucose-6-phosphate dehydrogenase (G6PD), a key enzyme in the pentose phosphate pathway, is central to cellular processes in lung cancer tissues (169). During PH progression, G6PD mutations drive metabolic reprogramming and inflammation, thereby promoting pulmonary arterial remodeling (170). Animal studies have revealed that under conditions of Cu overload, G6PD binds to FDX1, reducing its stability and downregulating NADPH and GSH levels, thereby enhancing cuproptosis (171). Cu chelators also reduce lipid peroxidation levels; therefore, FOXO1 and G6PD may mediate the interaction between Cu and ferroptosis in CTEPH, which requires further study using CTEPH models. In summary, future research on cuproptosis and ferroptosis may offer valuable insights into improving patient survival.
6 Future perspectives
With a deeper understanding of ferroptosis and its mechanisms, the targeted regulation of cell death offers a novel direction for the treatment of PH. Although significant progress has been made in PH classification and therapy, researchers continue to identify new biomarkers and targeted drugs to improve diagnostic precision and slow disease progression. For instance, sotatercept, a BMP/TGF-β inhibitor, improves patient outcomes but may elevate heme oxygenase-1 levels, increase free Fe, and raise inflammatory mediator interleukin levels (172, 173). Given the complexity of the disease, the chronic nature of treatment, and the substantial economic burden, PH remains a highly challenging condition. Therefore, the distinct mechanisms of ferroptosis and cuproptosis may provide promising therapeutic avenues for PH and other diseases, thereby offering a theoretical basis for the development of new drugs and treatment strategies. Future research should focus on (1) further elucidating the mechanisms and pathophysiological roles of ferroptosis and cuproptosis in PH, particularly regarding mitochondrial respiration and oxidative stress, to address the current gaps in clinical and experimental evidence; and (2) examining the interplay between ferroptosis and cuproptosis, as well as the associations among different forms of cell death in various PH subtypes. These investigations will not only enhance our understanding of PH pathophysiology but also yield new perspectives for its treatment.
Statements
Author contributions
YY: Visualization, Writing – original draft, Writing – review & editing, Conceptualization, Methodology. LL: Writing – original draft, Writing – review & editing, Visualization. WP: Conceptualization, Writing – review & editing. YS: Funding acquisition, Writing – review & editing, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Key Projects of the Hunan Provincial Department of Education, “Exploring the Role of TAK1-Mediated Pan-Apoptosis of PASMCs in High-Altitude Pulmonary Hypertension (HPH) and the Study of Herbal Intervention” (23A0296); Funder: Yinhui Sun. Exploring the Role of Cuproptosis in Pulmonary Hypertension via the AMPK-mTOR-ATG3 Pathway and the Regulatory Effects of Fei Xin Tang” (2024BKS094); Funder: Yinhui Sun.
Acknowledgments
We would like to thank Editage (https://www.editage.cn) for English language editing.
Conflict of interest
The authors declare that this study was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
Simonneau G Montani D Celermajer DS Denton CP Gatzoulis MA Krowka M et al Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. (2019) 53(1):1801913. 10.1183/13993003.01913-2018
2.
Humbert M Kovacs G Hoeper MM Badagliacca R Berger RMF Brida M et al 2022 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. (2023) 61(1):2200879. 10.1183/13993003.00879-2022
3.
Culley MK Chan SY . Endothelial senescence: a new age in pulmonary hypertension. Circ Res. (2022) 130(6):928–41. 10.1161/CIRCRESAHA.121.319815
4.
Collaborators USBoD, MokdadAHBallestrosKEchkoMGlennSOlsenHEet alThe state of US health, 1990–2016: burden of diseases, injuries, and risk factors among US states. JAMA. (2018) 319(14):1444–72. 10.1001/jama.2018.0158
5.
Hoeper MM Humbert M Souza R Idrees M Kawut SM Sliwa-Hahnle K et al A global view of pulmonary hypertension. Lancet Respir Med. (2016) 4(4):306–22. 10.1016/S2213-2600(15)00543-3
6.
Mocumbi A Humbert M Saxena A Jing ZC Sliwa K Thienemann F et al Pulmonary hypertension. Nat Rev Dis Primers. (2024) 10(1):1. 10.1038/s41572-023-00486-7
7.
Grunig E Jansa P Fan F Hauser JA Pannaux M Morganti A et al Randomized trial of macitentan/tadalafil single-tablet combination therapy for pulmonary arterial hypertension. J Am Coll Cardiol. (2024) 83(4):473–84. 10.1016/j.jacc.2023.10.045
8.
Hadian K Stockwell BR . The therapeutic potential of targeting regulated non-apoptotic cell death. Nat Rev Drug Discov. (2023) 22(9):723–42. 10.1038/s41573-023-00749-8
9.
Fearnhead HO Vandenabeele P Vanden Berghe T . How do we fit ferroptosis in the family of regulated cell death?Cell Death Differ. (2017) 24(12):1991–8. 10.1038/cdd.2017.149
10.
Kazmirczak F Vogel NT Prisco SZ Patterson MT Annis J Moon RT et al Ferroptosis-mediated inflammation promotes pulmonary hypertension. Circ Res. (2024) 135(11):1067–83. 10.1161/CIRCRESAHA.123.324138
11.
Yu H Guo P Xie X Wang Y Chen G . Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J Cell Mol Med. (2017) 21(4):648–57. 10.1111/jcmm.13008
12.
Latunde-Dada GO . Ferroptosis: role of lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta Gen Subj. (2017) 1861(8):1893–900. 10.1016/j.bbagen.2017.05.019
13.
Tsvetkov P Coy S Petrova B Dreishpoon M Verma A Abdusamad M et al Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. (2022) 375(6586):1254–61. 10.1126/science.abf0529
14.
Yang W Wang Y Huang Y Yu J Wang T Li C et al 4-octyl itaconate inhibits aerobic glycolysis by targeting GAPDH to promote cuproptosis in colorectal cancer. Biomed Pharmacother. (2023) 159:114301. 10.1016/j.biopha.2023.114301
15.
Locatelli M Farina C . Role of copper in central nervous system physiology and pathology. Neural Regen Res. (2025) 20(4):1058–68. 10.4103/NRR.NRR-D-24-00110
16.
Li Y Liu J Chen Y Weichselbaum RR Lin W . Nanoparticles synergize ferroptosis and cuproptosis to potentiate cancer immunotherapy. Adv Sci (Weinh). (2024) 11(23):e2310309. 10.1002/advs.202310309
17.
Tong J Lan XT Zhang Z Liu Y Sun DY Wang XJ et al Ferroptosis inhibitor liproxstatin-1 alleviates metabolic dysfunction-associated fatty liver disease in mice: potential involvement of PANoptosis. Acta Pharmacol Sin. (2023) 44(5):1014–28. 10.1038/s41401-022-01010-5
18.
Liao P Wang W Wang W Kryczek I Li X Bian Y et al CD8(+) T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell. (2022) 40(4):365–78.e6. 10.1016/j.ccell.2022.02.003
19.
Zheng J Conrad M . The metabolic underpinnings of ferroptosis. Cell Metab. (2020) 32(6):920–37. 10.1016/j.cmet.2020.10.011
20.
Bersuker K Hendricks JM Li Z Magtanong L Ford B Tang PH et al The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. (2019) 575(7784):688–92. 10.1038/s41586-019-1705-2
21.
Yang F Xiao Y Ding JH Jin X Ma D Li DQ et al Ferroptosis heterogeneity in triple-negative breast cancer reveals an innovative immunotherapy combination strategy. Cell Metab. (2023) 35(1):84–100.e8. 10.1016/j.cmet.2022.09.021
22.
Liu X Li Y Chen S Yang J Jing J Li J et al Dihydromyricetin attenuates intracerebral hemorrhage by reversing the effect of LCN2 via the system Xc- pathway. Phytomedicine. (2023) 115:154756. 10.1016/j.phymed.2023.154756
23.
Jiang X Stockwell BR Conrad M . Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. (2021) 22(4):266–82. 10.1038/s41580-020-00324-8
24.
Grzes KM Sanin DE Kabat AM Stanczak MA Edwards-Hicks J Matsushita M et al Plasmacytoid dendritic cell activation is dependent on coordinated expression of distinct amino acid transporters. Immunity. (2021) 54(11):2514–30.e7. 10.1016/j.immuni.2021.10.009
25.
Zhou B Liu J Kang R Klionsky DJ Kroemer G Tang D . Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. (2020) 66:89–100. 10.1016/j.semcancer.2019.03.002
26.
Zhang H Shi H Li X Zhou S Song X Ma N et al Quercetin alleviates LPS/iE-DAP-induced liver injury by suppressing ferroptosis via regulating ferritinophagy and intracellular iron efflux. Redox Biol. (2025) 81:103557. 10.1016/j.redox.2025.103557
27.
El-Kersh K Hopkins CD Wu X Rai SN Cave MC Smith MR et al Metallomics in pulmonary arterial hypertension patients. Pulm Circ. (2023) 13(1):e12202. 10.1002/pul2.12202
28.
Lakhal-Littleton S Crosby A Frise MC Mohammad G Carr CA Loick PAM et al Intracellular iron deficiency in pulmonary arterial smooth muscle cells induces pulmonary arterial hypertension in mice. Proc Natl Acad Sci U S A. (2019) 116(26):13122–30. 10.1073/pnas.1822010116
29.
Song X Liu J Kuang F Chen X Zeh HJ 3rd Kang R et al PDK4 dictates metabolic resistance to ferroptosis by suppressing pyruvate oxidation and fatty acid synthesis. Cell Rep. (2021) 34(8):108767. 10.1016/j.celrep.2021.108767
30.
White AR Cappai R . Neurotoxicity from glutathione depletion is dependent on extracellular trace copper. J Neurosci Res. (2003) 71(6):889–97. 10.1002/jnr.10537
31.
Du C Feng W Dai X Wang J Geng D Li X et al Cu(2+) -chelatable and ROS-scavenging MXenzyme as NIR-II-triggered blood-brain barrier-crossing nanocatalyst against Alzheimer’s disease. Small. (2022) 18(39):e2203031. 10.1002/smll.202203031
32.
Dixon SJ Lemberg KM Lamprecht MR Skouta R Zaitsev EM Gleason CE et al Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149(5):1060–72. 10.1016/j.cell.2012.03.042
33.
Cheff DM Huang C Scholzen KC Gencheva R Ronzetti MH Cheng Q et al The ferroptosis inducing compounds RSL3 and ML162 are not direct inhibitors of GPX4 but of TXNRD1. Redox Biol. (2023) 62:102703. 10.1016/j.redox.2023.102703
34.
Yan B Ai Y Sun Q Ma Y Cao Y Wang J et al Membrane damage during ferroptosis is caused by oxidation of phospholipids catalyzed by the oxidoreductases POR and CYB5R1. Mol Cell. (2021) 81(2):355–69.e10. 10.1016/j.molcel.2020.11.024
35.
Bai T Li M Liu Y Qiao Z Wang Z . Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Radic Biol Med. (2020) 160:92–102. 10.1016/j.freeradbiomed.2020.07.026
36.
Roemhild K von Maltzahn F Weiskirchen R Knuchel R von Stillfried S Lammers T . Iron metabolism: pathophysiology and pharmacology. Trends Pharmacol Sci. (2021) 42(8):640–56. 10.1016/j.tips.2021.05.001
37.
Nemeth E Tuttle MS Powelson J Vaughn MB Donovan A Ward DM et al Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. (2004) 306(5704):2090–3. 10.1126/science.1104742
38.
Yanatori I Kishi F . DMT1 and iron transport. Free Radic Biol Med. (2019) 133:55–63. 10.1016/j.freeradbiomed.2018.07.020
39.
Song J Liu T Yin Y Zhao W Lin Z Yin Y et al The deubiquitinase OTUD1 enhances iron transport and potentiates host antitumor immunity. EMBO Rep. (2021) 22(2):e51162. 10.15252/embr.202051162
40.
Wang H An P Xie E Wu Q Fang X Gao H et al Characterization of ferroptosis in murine models of hemochromatosis. Hepatology. (2017) 66(2):449–65. 10.1002/hep.29117
41.
Wu M Wang C Ke L Chen D Qin Y Han J . Correlation between copper speciation and transport pathway in caco-2 cells. J Sci Food Agric. (2023) 103(4):1895–900. 10.1002/jsfa.12292
42.
Yang L Yang P Lip GYH Ren J . Copper homeostasis and cuproptosis in cardiovascular disease therapeutics. Trends Pharmacol Sci. (2023) 44(9):573–85. 10.1016/j.tips.2023.07.004
43.
Chen J Jiang Y Shi H Peng Y Fan X Li C . The molecular mechanisms of copper metabolism and its roles in human diseases. Pflugers Arch. (2020) 472(10):1415–29. 10.1007/s00424-020-02412-2
44.
Xue Q Kang R Klionsky DJ Tang D Liu J Chen X . Copper metabolism in cell death and autophagy. Autophagy. (2023) 19(8):2175–95. 10.1080/15548627.2023.2200554
45.
Qin Y Liu Y Xiang X Long X Chen Z Huang X et al Cuproptosis correlates with immunosuppressive tumor microenvironment based on pan-cancer multiomics and single-cell sequencing analysis. Mol Cancer. (2023) 22(1):59. 10.1186/s12943-023-01752-8
46.
Cui L Gouw AM LaGory EL Guo S Attarwala N Tang Y et al Mitochondrial copper depletion suppresses triple-negative breast cancer in mice. Nat Biotechnol. (2021) 39(3):357–67. 10.1038/s41587-020-0707-9
47.
Barceloux DG . Copper. J Toxicol Clin Toxicol. (1999) 37(2):217–30. 10.1081/clt-100102421
48.
Tsvetkov P Detappe A Cai K Keys HR Brune Z Ying W et al Mitochondrial metabolism promotes adaptation to proteotoxic stress. Nat Chem Biol. (2019) 15(7):681–9. 10.1038/s41589-019-0291-9
49.
Saporito-Magrina CM Musacco-Sebio RN Andrieux G Kook L Orrego MT Tuttolomondo MV et al Copper-induced cell death and the protective role of glutathione: the implication of impaired protein folding rather than oxidative stress. Metallomics. (2018) 10(12):1743–54. 10.1039/c8mt00182k
50.
Tong X Tang R Xiao M Xu J Wang W Zhang B et al Targeting cell death pathways for cancer therapy: recent developments in necroptosis, pyroptosis, ferroptosis, and cuproptosis research. J Hematol Oncol. (2022) 15(1):174. 10.1186/s13045-022-01392-3
51.
Brancaccio D Gallo A Piccioli M Novellino E Ciofi-Baffoni S Banci L . [4Fe-4S] cluster assembly in mitochondria and its impairment by copper. J Am Chem Soc. (2017) 139(2):719–30. 10.1021/jacs.6b09567
52.
Schulz V Basu S Freibert SA Webert H Boss L Muhlenhoff U et al Functional spectrum and specificity of mitochondrial ferredoxins FDX1 and FDX2. Nat Chem Biol. (2023) 19(2):206–17. 10.1038/s41589-022-01159-4
53.
Metsla K Kirss S Laks K Sildnik G Palgi M Palumaa T et al alpha-lipoic acid has the potential to normalize copper metabolism, which is dysregulated in alzheimer’s disease. J Alzheimers Dis. (2022) 85(2):715–28. 10.3233/JAD-215026
54.
Yang S Li Y Zhou L Wang X Liu L Wu M . Copper homeostasis and cuproptosis in atherosclerosis: metabolism, mechanisms and potential therapeutic strategies. Cell Death Discov. (2024) 10(1):25. 10.1038/s41420-023-01796-1
55.
Wu CY Cilic A Pak O Dartsch RC Wilhelm J Wujak M et al CEACAM6 as a novel therapeutic target to boost HO-1-mediated antioxidant defense in COPD. Am J Respir Crit Care Med. (2023) 207(12):1576–90. 10.1164/rccm.202208-1603OC
56.
Morgan RW Reeder RW Ahmed T Bell MJ Berger JT Bishop R et al Outcomes and characteristics of cardiac arrest in children with pulmonary hypertension: a secondary analysis of the ICU-RESUS clinical trial. Resuscitation. (2023) 190:109897. 10.1016/j.resuscitation.2023.109897
57.
Cheng F Loscalzo J . Pulmonary comorbidity in lung cancer. Trends Mol Med. (2018) 24(3):239–41. 10.1016/j.molmed.2018.01.005
58.
Lin CH Chin Y Zhou M Sobol RW Hung MC Tan M . Protein lipoylation: mitochondria, cuproptosis, and beyond. Trends Biochem Sci. (2024) 49(8):729–44. 10.1016/j.tibs.2024.04.002
59.
Zhang Y Zhou Q Lu L Su Y Shi W Zhang H et al Copper induces cognitive impairment in mice via modulation of cuproptosis and CREB signaling. Nutrients. (2023) 15(4):972. 10.3390/nu15040972
60.
Mizushima N Levine B Cuervo AM Klionsky DJ . Autophagy fights disease through cellular self-digestion. Nature. (2008) 451(7182):1069–75. 10.1038/nature06639
61.
Ren X Pan C Pan Z Zhao S Wu C Chen W et al Knowledge mapping of copper-induced cell death: a bibliometric study from 2012 to 2022. Medicine (Baltimore). (2022) 101(45):e31133. 10.1097/MD.0000000000031133
62.
Zhu S Niu Y Zhou W Liu Y Liu J Liu X et al Mitochondrial copper overload promotes renal fibrosis via inhibiting pyruvate dehydrogenase activity. Cell Mol Life Sci. (2024) 81(1):340. 10.1007/s00018-024-05358-1
63.
Garza NM Swaminathan AB Maremanda KP Zulkifli M Gohil VM . Mitochondrial copper in human genetic disorders. Trends Endocrinol Metab. (2023) 34(1):21–33. 10.1016/j.tem.2022.11.001
64.
Shim D Han J . Coordination chemistry of mitochondrial copper metalloenzymes: exploring implications for copper dyshomeostasis in cell death. BMB Rep. (2023) 56(11):575–83. 10.5483/BMBRep.2023-0172
65.
Zhu SY Zhou WQ Niu YY Zheng C Liu X Zhang YY et al COX17 restricts renal fibrosis development by maintaining mitochondrial copper homeostasis and restoring complex IV activity. Acta Pharmacol Sin. (2023) 44(10):2091–102. 10.1038/s41401-023-01098-3
66.
Wei J Wang S Zhu H Cui W Gao J Gao C et al Hepatic depletion of nucleolar protein mDEF causes excessive mitochondrial copper accumulation associated with p53 and NRF1 activation. iScience. (2023) 26(7):107220. 10.1016/j.isci.2023.107220
67.
Leary SC Cobine PA Nishimura T Verdijk RM de Krijger R de Coo R et al COX19 mediates the transduction of a mitochondrial redox signal from SCO1 that regulates ATP7A-mediated cellular copper efflux. Mol Biol Cell. (2013) 24(6):683–91. 10.1091/mbc.E12-09-0705
68.
Dreishpoon MB Bick NR Petrova B Warui DM Cameron A Booker SJ et al FDX1 regulates cellular protein lipoylation through direct binding to LIAS. J Biol Chem. (2023) 299(9):105046. 10.1016/j.jbc.2023.105046
69.
Lai S Chen Y Yang F Xiao W Liu Y Wang C . Quantitative site-specific chemoproteomic profiling of protein lipoylation. J Am Chem Soc. (2022) 144(23):10320–9. 10.1021/jacs.2c01528
70.
Li C Wu Y Li H Wang H Liu JX . Lipid-related metabolism during zebrafish embryogenesis under unbalanced copper homeostasis. Fish Physiol Biochem. (2022) 48(6):1571–86. 10.1007/s10695-022-01127-8
71.
Zhong CC Zhao T Hogstrand C Chen F Song CC Luo Z . Copper (cu) induced changes of lipid metabolism through oxidative stress-mediated autophagy and Nrf2/PPARgamma pathways. J Nutr Biochem. (2022) 100:108883. 10.1016/j.jnutbio.2021.108883
72.
Zhang M Qiu H Mao L Wang B Li N Fan Y et al Ammonium tetrathiomolybdate triggers autophagy-dependent NRF2 activation in vascular endothelial cells. Cell Death Dis. (2022) 13(8):733. 10.1038/s41419-022-05183-z
73.
Piper B Bogamuwa S Hossain T Farkas D Rosas L Green AC et al RAB7 deficiency impairs pulmonary artery endothelial function and promotes pulmonary hypertension. J Clin Invest. (2024) 134(3):e169441. 10.1172/JCI169441
74.
Yang D Xiao P Qiu B Yu HF Teng CB . Copper chaperone antioxidant 1: multiple roles and a potential therapeutic target. J Mol Med (Berl). (2023) 101(5):527–42. 10.1007/s00109-023-02311-w
75.
Chen Z Li YY Liu X . Copper homeostasis and copper-induced cell death: novel targeting for intervention in the pathogenesis of vascular aging. Biomed Pharmacother. (2023) 169:115839. 10.1016/j.biopha.2023.115839
76.
Xiang P Chen Q Chen L Lei J Yuan Z Hu H et al Metabolite Neu5Ac triggers SLC3A2 degradation promoting vascular endothelial ferroptosis and aggravates atherosclerosis progression in ApoE(-/-)mice. Theranostics. (2023) 13(14):4993–5016. 10.7150/thno.87968
77.
Cui S Ghai A Deng Y Li S Zhang R Egbulefu C et al Identification of hyperoxidized PRDX3 as a ferroptosis marker reveals ferroptotic damage in chronic liver diseases. Mol Cell. (2023) 83(21):3931–9.e5. 10.1016/j.molcel.2023.09.025
78.
Liao J Xie SS Deng Y Wu DD Meng H Lan WF et al PRDX6-mediated pulmonary artery endothelial cell ferroptosis contributes to monocrotaline-induced pulmonary hypertension. Microvasc Res. (2023) 146:104471. 10.1016/j.mvr.2022.104471
79.
Xie SS Deng Y Guo SL Li JQ Zhou YC Liao J et al Endothelial cell ferroptosis mediates monocrotaline-induced pulmonary hypertension in rats by modulating NLRP3 inflammasome activation. Sci Rep. (2022) 12(1):3056. 10.1038/s41598-022-06848-7
80.
Xue C Senchanthisai S Sowden M Pang J White RJ Berk BC . Endothelial-to-Mesenchymal transition and inflammation play key roles in cyclophilin A-induced pulmonary arterial hypertension. Hypertension. (2020) 76(4):1113–23. 10.1161/HYPERTENSIONAHA.119.14013
81.
Chen Y Yi X Huo B He Y Guo X Zhang Z et al BRD4770 functions as a novel ferroptosis inhibitor to protect against aortic dissection. Pharmacol Res. (2022) 177:106122. 10.1016/j.phrs.2022.106122
82.
Che H Yi J Zhao X Yu H Wang X Zhang R et al Characterization of PKCalpha-rutin interactions and their application as a treatment strategy for pulmonary arterial hypertension by inhibiting ferroptosis. Food Funct. (2024) 15(2):779–93. 10.1039/d3fo01306e
83.
Festa RA Thiele DJ . Copper: an essential metal in biology. Curr Biol. (2011) 21(21):R877–83. 10.1016/j.cub.2011.09.040
84.
Yang Y Wu J Wang L Ji G Dang Y . Copper homeostasis and cuproptosis in health and disease. MedComm (2020). (2024) 5(10):e724. 10.1002/mco2.724
85.
Cai D Chen SY . Nanoparticle endothelial delivery of PGC-1alpha attenuates hypoxia-induced pulmonary hypertension by attenuating EndoMT-caused vascular wall remodeling. Redox Biol. (2022) 58:102524. 10.1016/j.redox.2022.102524
86.
Ghadimi K Cappiello JL Wright MC Levy JH Bryner BS DeVore AD et al Inhaled epoprostenol compared with nitric oxide for right ventricular support after major cardiac surgery. Circulation. (2023) 148(17):1316–29. 10.1161/CIRCULATIONAHA.122.062464
87.
Mo N Tai C Yang Y Ling C Zhang B Wei L et al MT2A promotes angiogenesis in chronically ischemic brains through a copper-mitochondria regulatory mechanism. J Transl Med. (2025) 23(1):162. 10.1186/s12967-025-06163-5
88.
Farrant J Dodd S Vaughan C Reid A Schmitt M Garratt C et al Rationale and design of a randomised trial of trientine in patients with hypertrophic cardiomyopathy. Heart. (2023) 109(15):1175–82. 10.1136/heartjnl-2022-322271
89.
Zimnicka AM Tang H Guo Q Kuhr FK Oh MJ Wan J et al Upregulated copper transporters in hypoxia-induced pulmonary hypertension. PLoS One. (2014) 9(3):e90544. 10.1371/journal.pone.0090544
90.
Bogaard HJ Mizuno S Guignabert C Al Hussaini AA Farkas D Ruiter G et al Copper dependence of angioproliferation in pulmonary arterial hypertension in rats and humans. Am J Respir Cell Mol Biol. (2012) 46(5):582–91. 10.1165/rcmb.2011-0296OC
91.
Poels EM Bitsch N Slenter JM Kooi ME de Theije CC de Windt LJ et al Supplementing exposure to hypoxia with a copper depleted diet does not exacerbate right ventricular remodeling in mice. PLoS One. (2014) 9(4):e92983. 10.1371/journal.pone.0092983
92.
Ferrian S Cao A McCaffrey EF Saito T Greenwald NF Nicolls MR et al Single-cell imaging maps inflammatory cell subsets to pulmonary arterial hypertension vasculopathy. Am J Respir Crit Care Med. (2024) 209(2):206–18. 10.1164/rccm.202209-1761OC
93.
Bai J Zhang X Zhao Z Sun S Cheng W Yu H et al Cuo nanozymes catalyze cysteine and glutathione depletion induced ferroptosis and cuproptosis for synergistic tumor therapy. Small. (2024) 20(40):e2400326. 10.1002/smll.202400326
94.
Chen K Zhou A Zhou X He J Xu Y Ning X . Cellular trojan horse initiates bimetallic Fe-Cu MOF-mediated synergistic cuproptosis and ferroptosis against malignancies. Sci Adv. (2024) 10(15):eadk3201. 10.1126/sciadv.adk3201
95.
Meng X Shen Y Zhao H Lu X Wang Z Zhao Y . Redox-manipulating nanocarriers for anticancer drug delivery: a systematic review. J Nanobiotechnology. (2024) 22(1):587. 10.1186/s12951-024-02859-w
96.
Gao Y Jin F Zhang P Zheng C Zheng X Xie J et al Elesclomol-copper synergizes with imidazole ketone erastin by promoting cuproptosis and ferroptosis in myelodysplastic syndromes. Biomed Pharmacother. (2024) 175:116727. 10.1016/j.biopha.2024.116727
97.
Sun X Zhang X Yan H Wu H Cao S Zhao W et al Protective effect of curcumin on hepatolenticular degeneration through copper excretion and inhibition of ferroptosis. Phytomedicine. (2023) 113:154539. 10.1016/j.phymed.2022.154539
98.
Yegambaram M Sun X Lu Q Jin Y Ornatowski W Soto J et al Mitochondrial hyperfusion induces metabolic remodeling in lung endothelial cells by modifying the activities of electron transport chain complexes I and III. Free Radic Biol Med. (2024) 210:183–94. 10.1016/j.freeradbiomed.2023.11.008
99.
Antigny F Crottes D Vandier C Capuano V Gueguinou M . Pulmonary arterial hypertension and cancer: exploring their resemblance as channelopathies. Trends Mol Med. (2025). 10.1016/j.molmed.2025.03.002
100.
Zhang P Zhou C Ren X Jing Q Gao Y Yang C et al Inhibiting the compensatory elevation of xCT collaborates with disulfiram/copper-induced GSH consumption for cascade ferroptosis and cuproptosis. Redox Biol. (2024) 69:103007. 10.1016/j.redox.2023.103007
101.
Liang S Yegambaram M Wang T Wang J Black SM Tang H . Mitochondrial metabolism, redox, and calcium homeostasis in pulmonary arterial hypertension. Biomedicines. (2022) 10(2):341. 10.3390/biomedicines10020341
102.
Xue Q Yan D Chen X Li X Kang R Klionsky DJ et al Copper-dependent autophagic degradation of GPX4 drives ferroptosis. Autophagy. (2023) 19(7):1982–96. 10.1080/15548627.2023.2165323
103.
Liu L Pang J Qin D Li R Zou D Chi K et al Deubiquitinase OTUD5 as a novel protector against 4-HNE-triggered ferroptosis in myocardial ischemia/reperfusion injury. Adv Sci (Weinh). (2023) 10(28):e2301852. 10.1002/advs.202301852
104.
Tsang T Davis CI Brady DC . Copper biology. Curr Biol. (2021) 31(9):R421–R7. 10.1016/j.cub.2021.03.054
105.
Chen L Min J Wang F . Copper homeostasis and cuproptosis in health and disease. Signal Transduct Target Ther. (2022) 7(1):378. 10.1038/s41392-022-01229-y
106.
Yang S Li X Yan J Jiang F Fan X Jin J et al Disulfiram downregulates ferredoxin 1 to maintain copper homeostasis and inhibit inflammation in cerebral ischemia/reperfusion injury. Sci Rep. (2024) 14(1):15175. 10.1038/s41598-024-64981-x
107.
Liu J Tang H Chen F Li C Xie Y Kang R et al NFE2L2 and SLC25A39 drive cuproptosis resistance through GSH metabolism. Sci Rep. (2024) 14(1):29579. 10.1038/s41598-024-81317-x
108.
Cobine PA Moore SA Leary SC . Getting out what you put in: copper in mitochondria and its impacts on human disease. Biochim Biophys Acta Mol Cell Res. (2021) 1868(1):118867. 10.1016/j.bbamcr.2020.118867
109.
Olsson KM Corte TJ Kamp JC Montani D Nathan SD Neubert L et al Pulmonary hypertension associated with lung disease: new insights into pathomechanisms, diagnosis, and management. Lancet Respir Med. (2023) 11(9):820–35. 10.1016/S2213-2600(23)00259-X
110.
Liu R Xu C Zhang W Cao Y Ye J Li B et al FUNDC1-mediated Mitophagy and HIF1alpha activation drives pulmonary hypertension during hypoxia. Cell Death Dis. (2022) 13(7):634. 10.1038/s41419-022-05091-2
111.
Jiang Y Guo Y Feng X Yang P Liu Y Dai X et al Iron metabolism disorder regulated by BMP signaling in hypoxic pulmonary hypertension. Biochim Biophys Acta Mol Basis Dis. (2023) 1869(2):166589. 10.1016/j.bbadis.2022.166589
112.
Hu P Xu Y Jiang Y Huang J Liu Y Wang D et al The mechanism of the imbalance between proliferation and ferroptosis in pulmonary artery smooth muscle cells based on the activation of SLC7A11. Eur J Pharmacol. (2022) 928:175093. 10.1016/j.ejphar.2022.175093
113.
Docherty KF Welsh P Verma S De Boer RA O'Meara E Bengtsson O et al Iron deficiency in heart failure and effect of dapagliflozin: findings from DAPA-HF. Circulation. (2022) 146(13):980–94. 10.1161/CIRCULATIONAHA.122.060511
114.
Wang E Zhang B Huang L Li P Han R Zhou S et al LncRNA MIR210HG promotes phenotype switching of pulmonary arterial smooth muscle cells through autophagy-dependent ferroptosis pathway. Apoptosis. (2024) 29(9-10):1648–62. 10.1007/s10495-024-01963-4
115.
Yang M Wu X Hu J Wang Y Wang Y Zhang L et al COMMD10 inhibits HIF1alpha/CP loop to enhance ferroptosis and radiosensitivity by disrupting cu-fe balance in hepatocellular carcinoma. J Hepatol. (2022) 76(5):1138–50. 10.1016/j.jhep.2022.01.009
116.
Gao W Huang Z Duan J Nice EC Lin J Huang C . Elesclomol induces copper-dependent ferroptosis in colorectal cancer cells via degradation of ATP7A. Mol Oncol. (2021) 15(12):3527–44. 10.1002/1878-0261.13079
117.
Graul EL Nordon C Rhodes K Marshall J Menon S Kallis C et al Temporal risk of nonfatal cardiovascular events after chronic obstructive pulmonary disease exacerbation: a population-based study. Am J Respir Crit Care Med. (2024) 209(8):960–72. 10.1164/rccm.202307-1122OC
118.
Hadzic S Wu CY Gredic M Pak O Loku E Kraut S et al Fibroblast growth factor 10 reverses cigarette smoke- and elastase-induced emphysema and pulmonary hypertension in mice. Eur Respir J. (2023) 62(5):2201606. 10.1183/13993003.01606-2022
119.
Qian Y Shi Q Zhou W He B Xu H Liu B et al FGF10 protects against particulate matter-induced lung injury by inhibiting ferroptosis via Nrf2-dependent signaling. Int Immunopharmacol. (2024) 134:112165. 10.1016/j.intimp.2024.112165
120.
Liu L Zhang Y Wang L Liu Y Chen H Hu Q et al Scutellarein alleviates chronic obstructive pulmonary disease through inhibition of ferroptosis by chelating iron and interacting with arachidonate 15-lipoxygenase. Phytother Res. (2023) 37(10):4587–606. 10.1002/ptr.7928
121.
Gunes Gunsel G Conlon TM Jeridi A Kim R Ertuz Z Lang NJ et al The arginine methyltransferase PRMT7 promotes extravasation of monocytes resulting in tissue injury in COPD. Nat Commun. (2022) 13(1):1303. 10.1038/s41467-022-28809-4
122.
Li Y Yang Y Guo T Weng C Yang Y Wang Z et al Heme oxygenase-1 determines the cell fate of ferroptotic death of alveolar macrophages in COPD. Front Immunol. (2023) 14:1162087. 10.3389/fimmu.2023.1162087
123.
Liu L Qin Y Cai Z Tian Y Liu X Li J et al Effective-components combination improves airway remodeling in COPD rats by suppressing M2 macrophage polarization via the inhibition of mTORC2 activity. Phytomedicine. (2021) 92:153759. 10.1016/j.phymed.2021.153759
124.
Li C Liu P Zhu H Yang H Zha J Yao H et al T follicular helper cell is essential for M2 macrophage polarization and pulmonary vascular remodeling in hypoxia-induced pulmonary hypertension. Respir Res. (2024) 25(1):428. 10.1186/s12931-024-03058-9
125.
Gredic M Wu CY Hadzic S Pak O Savai R Kojonazarov B et al Myeloid-cell-specific deletion of inducible nitric oxide synthase protects against smoke-induced pulmonary hypertension in mice. Eur Respir J. (2022) 59(4):2101153. 10.1183/13993003.01153-2021
126.
Han L Zhu W Qi H He L Wang Q Shen J et al The cuproptosis-related gene glutaminase promotes alveolar macrophage copper ion accumulation in chronic obstructive pulmonary disease. Int Immunopharmacol. (2024) 129:111585. 10.1016/j.intimp.2024.111585
127.
Bu N Du Q Xiao T Jiang Z Lin J Chen W et al Mechanism of S-palmitoylation in polystyrene nanoplastics-induced macrophage cuproptosis contributing to emphysema through alveolar epithelial cell pyroptosis. ACS Nano. (2025) 19(19):18708–28. 10.1021/acsnano.5c02892
128.
El-Mahdy MA Abdelghany TM Hemann C Ewees MG Mahgoup EM Eid MS et al Chronic cigarette smoke exposure triggers a vicious cycle of leukocyte and endothelial-mediated oxidant stress that results in vascular dysfunction. Am J Physiol Heart Circ Physiol. (2020) 319(1):H51–65. 10.1152/ajpheart.00657.2019
129.
Sevilla-Montero J Munar-Rubert O Pino-Fadon J Aguilar-Latorre C Villegas-Esguevillas M Climent B et al Cigarette smoke induces pulmonary arterial dysfunction through an imbalance in the redox status of the soluble guanylyl cyclase. Free Radic Biol Med. (2022) 193(Pt 1):9–22. 10.1016/j.freeradbiomed.2022.09.026
130.
Sumi MP Tupta B Roychowdhury S Comhair S Asosingh K Stuehr DJ et al Hemoglobin resident in the lung epithelium is protective for smooth muscle soluble guanylate cyclase function. Redox Biol. (2023) 63:102717. 10.1016/j.redox.2023.102717
131.
Xia H Wu Y Zhao J Cheng C Lin J Yang Y et al N6-Methyladenosine-modified circSAV1 triggers ferroptosis in COPD through recruiting YTHDF1 to facilitate the translation of IREB2. Cell Death Differ. (2023) 30(5):1293–304. 10.1038/s41418-023-01138-9
132.
Read AD Bentley RE Archer SL Dunham-Snary KJ . Mitochondrial iron-sulfur clusters: structure, function, and an emerging role in vascular biology. Redox Biol. (2021) 47:102164. 10.1016/j.redox.2021.102164
133.
Ren X Yan J Zhao Q Bao X Han X Zheng C et al The Fe-S cluster assembly protein IscU2 increases alpha-ketoglutarate catabolism and DNA 5mC to promote tumor growth. Cell Discov. (2023) 9(1):76. 10.1038/s41421-023-00558-8
134.
Du J Huang Z Li Y Ren X Zhou C Liu R et al Copper exerts cytotoxicity through inhibition of iron-sulfur cluster biogenesis on ISCA1/ISCA2/ISCU assembly proteins. Free Radic Biol Med. (2023) 204:359–73. 10.1016/j.freeradbiomed.2023.05.017
135.
Dwivedi K Sharkey M Delaney L Alabed S Rajaram S Hill C et al Improving prognostication in pulmonary hypertension using AI-quantified fibrosis and radiologic severity scoring at baseline CT. Radiology. (2024) 310(2):e231718. 10.1148/radiol.231718
136.
Wu WH Bonnet S Shimauchi T Toro V Grobs Y Romanet C et al Potential for inhibition of checkpoint kinases 1/2 in pulmonary fibrosis and secondary pulmonary hypertension. Thorax. (2022) 77(3):247–58. 10.1136/thoraxjnl-2021-217377
137.
Mahoney-Sanchez L Bouchaoui H Ayton S Devos D Duce JA Devedjian JC . Ferroptosis and its potential role in the physiopathology of Parkinson’s disease. Prog Neurobiol. (2021) 196:101890. 10.1016/j.pneurobio.2020.101890
138.
Pei Z Qin Y Fu X Yang F Huo F Liang X et al Inhibition of ferroptosis and iron accumulation alleviates pulmonary fibrosis in a bleomycin model. Redox Biol. (2022) 57:102509. 10.1016/j.redox.2022.102509
139.
Lu X Zhang J Liu H Ma W Yu L Tan X et al Cannabidiol attenuates pulmonary arterial hypertension by improving vascular smooth muscle cells mitochondrial function. Theranostics. (2021) 11(11):5267–78. 10.7150/thno.55571
140.
Sun Q Hackler J Hilger J Gluschke H Muric A Simmons S et al Selenium and copper as biomarkers for pulmonary arterial hypertension in systemic sclerosis. Nutrients. (2020) 12(6):1894. 10.3390/nu12061894
141.
Gulec S Collins JF . Molecular mediators governing iron-copper interactions. Annu Rev Nutr. (2014) 34:95–116. 10.1146/annurev-nutr-071812-161215
142.
Kitala-Tanska K Socha K Juskiewicz J Krajewska-Wlodarczyk M Majewski M . The effect of an elevated dietary copper level on the vascular contractility and oxidative stress in middle-aged rats. Nutrients. (2024) 16(8):1172. 10.3390/nu16081172
143.
Zhang R Wang L Zhao QH Jiang R Gong SG Jiang X et al Alteration of extracellular superoxide dismutase in idiopathic pulmonary arterial hypertension. Front Med (Lausanne). (2020) 7:509. 10.3389/fmed.2020.00509
144.
Jones RJ De Bie E Groves E Zalewska KI Swietlik EM Treacy CM et al Autoimmunity is a significant feature of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. (2022) 206(1):81–93. 10.1164/rccm.202108-1919OC
145.
Wu S Wang Q Du J Meng Q Li Y Miao Y et al A p-n heterojunction sonosensitizer for improved sono-immunotherapy via induction of multimodal cell death mechanisms. Theranostics. (2025) 15(7):2737–56. 10.7150/thno.106999
146.
Mi XQ Liu BC Qu L Yuan Y Li H Xu AY et al Intranasal iron administration induces iron deposition, immunoactivation, and cell-specific vulnerability in the olfactory bulb of C57BL/6 mice. Zool Res. (2025) 46(1):209–24. 10.24272/j.issn.2095-8137.2024.240
147.
Zhou QY Ren C Li JY Wang L Duan Y Yao RQ et al The crosstalk between mitochondrial quality control and metal-dependent cell death. Cell Death Dis. (2024) 15(4):299. 10.1038/s41419-024-06691-w
148.
James J Zemskova M Eccles CA Varghese MV Niihori M Barker NK et al Single mutation in the NFU1 gene metabolically reprograms pulmonary artery smooth muscle cells. Arterioscler Thromb Vasc Biol. (2021) 41(2):734–54. 10.1161/ATVBAHA.120.314655
149.
Niihori M James J Varghese MV McClain N Lawal OS Philip RC et al Mitochondria as a primary determinant of angiogenic modality in pulmonary arterial hypertension. J Exp Med. (2024) 221(11):e20231568. 10.1084/jem.20231568
150.
Pan M Cheng ZW Huang CG Ye ZQ Sun LJ Chen H et al Long-term exposure to copper induces mitochondria-mediated apoptosis in mouse hearts. Ecotoxicol Environ Saf. (2022) 234:113329. 10.1016/j.ecoenv.2022.113329
151.
Li J Tuo D Guo G Gan J . Aberrant expression of cuproptosis-related gene LIPT1 is associated with metabolic dysregulation of fatty acid and prognosis in hepatocellular carcinoma. J Cancer Res Clin Oncol. (2023) 149(17):15763–79. 10.1007/s00432-023-05325-6
152.
Ni M Solmonson A Pan C Yang C Li D Notzon A et al Functional assessment of lipoyltransferase-1 deficiency in cells, mice, and humans. Cell Rep. (2019) 27(5):1376–86.e6. 10.1016/j.celrep.2019.04.005
153.
Mukha D Fokra M Feldman A Sarvin B Sarvin N Nevo-Dinur K et al Glycine decarboxylase maintains mitochondrial protein lipoylation to support tumor growth. Cell Metab. (2022) 34(5):775–82.e9. 10.1016/j.cmet.2022.04.006
154.
Liu Z Villareal L Goodla L Kim H Falcon DM Haneef M et al Iron promotes glycolysis to drive colon tumorigenesis. Biochim Biophys Acta Mol Basis Dis. (2023) 1869(8):166846. 10.1016/j.bbadis.2023.166846
155.
Michelakis ED Gurtu V Webster L Barnes G Watson G Howard L et al Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci Transl Med. (2017) 9(413):eaao4583. 10.1126/scitranslmed.aao4583
156.
Liu Y Zhu W Ni D Zhou Z Gu JH Zhang W et al Alpha lipoic acid antagonizes cytotoxicity of cobalt nanoparticles by inhibiting ferroptosis-like cell death. J Nanobiotechnology. (2020) 18(1):141. 10.1186/s12951-020-00700-8
157.
Huston JH Shah SJ . Understanding the pathobiology of pulmonary hypertension due to left heart disease. Circ Res. (2022) 130(9):1382–403. 10.1161/CIRCRESAHA.122.319967
158.
Fauvel C Damy T Berthelot E Bauer F Eicher JC de Groote P et al Post-capillary pulmonary hypertension in heart failure: impact of current definition in the PH-HF multicentre study. Eur Heart J. (2024) 45(35):3274–88. 10.1093/eurheartj/ehae467
159.
Yuan HJ Xue YT Liu Y . Cuproptosis, the novel therapeutic mechanism for heart failure: a narrative review. Cardiovasc Diagn Ther. (2022) 12(5):681–92. 10.21037/cdt-22-214
160.
Chen Z Zhu Y Chen S Li Z Fu G Wang Y . Immune patterns of cuproptosis in ischemic heart failure: a transcriptome analysis. J Cell Mol Med. (2024) 28(7):e18187. 10.1111/jcmm.18187
161.
Wang W Lu K Jiang X Wei Q Zhu L Wang X et al Ferroptosis inducers enhanced cuproptosis induced by copper ionophores in primary liver cancer. J Exp Clin Cancer Res. (2023) 42(1):142. 10.1186/s13046-023-02720-2
162.
Ahola S Langer T . Ferroptosis in mitochondrial cardiomyopathy. Trends Cell Biol. (2024) 34(2):150–60. 10.1016/j.tcb.2023.06.002
163.
Vrentzos E Ikonomidis I Pavlidis G Katogiannis K Korakas E Kountouri A et al Six-month supplementation with high dose coenzyme Q10 improves liver steatosis, endothelial, vascular and myocardial function in patients with metabolic-dysfunction associated steatotic liver disease: a randomized double-blind, placebo-controlled trial. Cardiovasc Diabetol. (2024) 23(1):245. 10.1186/s12933-024-02326-8
164.
Sharp J Farha S Park MM Comhair SA Lundgrin EL Tang WH et al Coenzyme Q supplementation in pulmonary arterial hypertension. Redox Biol. (2014) 2:884–91. 10.1016/j.redox.2014.06.010
165.
Marin-Romero S Ballaz-Quincoces A Gomez-Cuervo C Marchena-Yglesias PJ Lopez-Miguel P Francisco-Albesa I et al Symptom-related screening programme for early detection of chronic thromboembolic pulmonary hypertension after acute pulmonary embolism: the SYSPPE study. Thorax. (2024) 79(2):144–52. 10.1136/thorax-2023-220580
166.
An Y Xu M Yan M Zhang H Li C Wang L et al Erythrophagocytosis-induced ferroptosis contributes to pulmonary microvascular thrombosis and thrombotic vascular remodeling in pulmonary arterial hypertension. J Thromb Haemost. (2025) 23(1):158–70. 10.1016/j.jtha.2024.09.011
167.
Wu D Lin Y Yang M Li H Wang W Wu Q et al Tissue factor regulates autophagy in pulmonary artery endothelial cells from chronic thromboembolic pulmonary hypertension rats via the p38 MAPK-FoxO1 pathway. Respir Res. (2024) 25(1):261. 10.1186/s12931-024-02886-z
168.
Yien YY . What does the FOX(O) say? High iron: hepcidin!. Blood. (2024) 144(12):1243–5. 10.1182/blood.2024025595
169.
Thakor P Siddiqui MQ Patel TR . Analysis of the interlink between glucose-6-phosphate dehydrogenase (G6PD) and lung cancer through multi-omics databases. Heliyon. (2024) 10(15):e35158. 10.1016/j.heliyon.2024.e35158
170.
Signoretti C Matsumura S Fatehi S D’Silva M Mathew R Cendali F et al G6pd(N126D) variant increases the risk of developing VEGFR (vascular endothelial growth factor receptor) blocker-induced pulmonary vascular disease. J Am Heart Assoc. (2024) 13(19):e035174. 10.1161/JAHA.123.035174
171.
Lu J Ling X Sun Y Liu L Liu L Wang X et al FDX1 enhances endometriosis cell cuproptosis via G6PD-mediated redox homeostasis. Apoptosis. (2023) 28(7-8):1128–40. 10.1007/s10495-023-01845-1
172.
Johnson S Sommer N Cox-Flaherty K Weissmann N Ventetuolo CE Maron BA . Pulmonary hypertension: a contemporary review. Am J Respir Crit Care Med. (2023) 208(5):528–48. 10.1164/rccm.202302-0327SO
173.
Savale L Tu L Normand C Boucly A Sitbon O Montani D et al Effect of sotatercept on circulating proteomics in pulmonary arterial hypertension. Eur Respir J. (2024) 64(4):2401483. 10.1183/13993003.01483-2024
Summary
Keywords
ferroptosis, cuproptosis, pulmonary hypertension, oxidative stress, metabolic reprogramming, mitochondrial dysfunction
Citation
Yang Y, Liang L, Pei W and Sun Y (2025) Interaction of ferroptosis and cuproptosis in the perspective of pulmonary hypertension. Front. Cardiovasc. Med. 12:1611449. doi: 10.3389/fcvm.2025.1611449
Received
14 April 2025
Accepted
09 June 2025
Published
26 June 2025
Volume
12 - 2025
Edited by
Tatsuya Sato, Sapporo Medical University, Japan
Reviewed by
Stefan Hadzic, University of Giessen, Germany
Andrew J. Murray, University of Cambridge, United Kingdom
Updates
Copyright
© 2025 Yang, Liang, Pei and Sun.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
* Correspondence: Yinhui Sun S33468088@163.com
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.