 Isabelle Lafosse1,†Romuald Mentaverri1,2,†Carine Avondo1
Isabelle Lafosse1,†Romuald Mentaverri1,2,†Carine Avondo1 Youssef Bennis1,3
Youssef Bennis1,3 Christophe Tribouilloy1,4
Christophe Tribouilloy1,4 Lucie Hénaut1*
Lucie Hénaut1*
- 1UR UPJV 7517 MP3CV, CURS, Amiens, France
- 2Department of Biochemistry and Endocrine Biology, Amiens University Hospital, Amiens, France
- 3Department of Pharmacology, Amiens University Hospital, Amiens, France
- 4Department of Cardiology, Amiens University Hospital, Amiens, France
Calcific aortic valve disease (CAVD) is characterized by a fibrocalcific remodeling of the aortic valve. This pathology is the most prevalent valvular heart disease worldwide and is associated with a poor prognosis. Despite extensive research, no pharmacological treatments are available to slow or reverse valvular degeneration, making aortic valve replacement the only current therapeutic option. This lack of clinical success may stem from an incomplete understanding of the disease's mechanisms and the limitations of current preclinical models, which do not fully replicate the complexity of CAVD and its associated risk factors and comorbidities. Indeed, while existing models offer valuable insights, a deeper understanding of CAVD requires incorporating comorbidities, gender-specific mechanisms, and dynamic cellular and tissue-level changes. This review aims to provide the reader with an overview of preclinical models developed in recent years to study CAVD, assessing their strengths and limitations. We review how these models can be used to mimic and/or investigate the cellular and molecular mechanisms involved in CAVD development, and highlight how key risk factors and comorbidities can be incorporated to enhance the translational potential of research. We hope that this approach will help guide researchers in selecting the most appropriate model for their studies, with the goal of advancing the identification of effective therapeutic candidates.
1 Introduction
Calcific aortic valve disease (CAVD) is the most common valvular disease worldwide, affecting approximately 10% of individuals over 65 years old (1, 2). It begins with aortic sclerosis, a mild thickening of the aortic valve (AV) leaflets without significant blood flow obstruction, and may progress to severe calcification, restricting leaflet motion. Over time, this remodeling narrows the AV opening, leading to aortic stenosis (AS), which impedes outflow from the left ventricle, increases afterload, and disrupts hemodynamics. Untreated AS causes left ventricular hypertrophy and dysfunction, resulting in symptoms such as dyspnea, angina, and syncope, ultimately leading to heart failure (HF) and death.
Transthoracic Doppler echocardiography is the standard method for assessing AS severity. Hallmark features include reduced aortic valve area (AVA), increased peak aortic transvalvular velocity, and elevated mean pressure gradient. Based on these parameters, AS is classified as mild, moderate, or severe (3). Computed tomography of aortic valve calcification (CT-AVC) complements echocardiography by enabling quantification and grading of calcification, with sex-specific thresholds for severe AS set at >1,300 Agatston units (AU) in women and >2,000 AU in men (3–5). Once symptomatic, severe AS has a ∼50% two-year mortality if left untreated (6). To date, aortic valve replacement (AVR), either surgical (SAVR) or transcatheter (TAVR), remains the only curative therapy for CAVD (6–8), as no pharmacological treatment can prevent its onset or progression. While preclinical studies have yielded encouraging results, none have translated into clinically effective interventions. This persistent translational gap likely reflects the limitations of current experimental models, which do not fully capture the multifactorial nature of CAVD or its frequent association with comorbidities, thereby hampering the clinical applicability of preclinical findings.
This review provides an overview of the current preclinical models used to study CAVD, emphasizing their strengths, limitations, and ability to replicate key cellular mechanisms, risk factors, and comorbidities. Our goal is to share perspectives that may help researchers select suitable models and enhance the relevance of preclinical findings, ultimately accelerating therapeutic development.
2 Pathophysiology of CAVD
2.1 Structure of the aortic valve
The healthy AV is an avascular structure, composed of three individual leaflets, located at the junction between the left ventricle and the aorta. Each leaflet contains three distinct extracellular matrix (ECM) layers—fibrosa, spongiosa and ventricularis—lined by valvular endothelial cells (VECs). The fibrosa (∼40% of valve volume), on the aortic side, is rich in fibronectin (FN) and densely packed type I and type III collagen fibrils, providing resistance to mechanical stress and pressure. The spongiosa (∼30% of valve volume), is the central layer. It contains glycosaminoglycans (GAGs), proteoglycans (PGs), and collagen, allowing for shock absorption and efficient cusp opening during systole. The ventricularis (∼20%–30% of valve volume), adjacent to the left ventricle, is enriched in elastin for flexibility and collagen types I, II and III for structural reinforcement (Figure 1). The outer layer of VECs, continuous with the endothelium of the aorta and the left ventricular myocardium, regulates paracrine signaling and exerts antithrombotic effects, maintaining proper valvular function. The three layers are primarily populated by valvular interstitial cells (VICs), whose phenotype and activity are crucial to maintaining structural integrity (9).
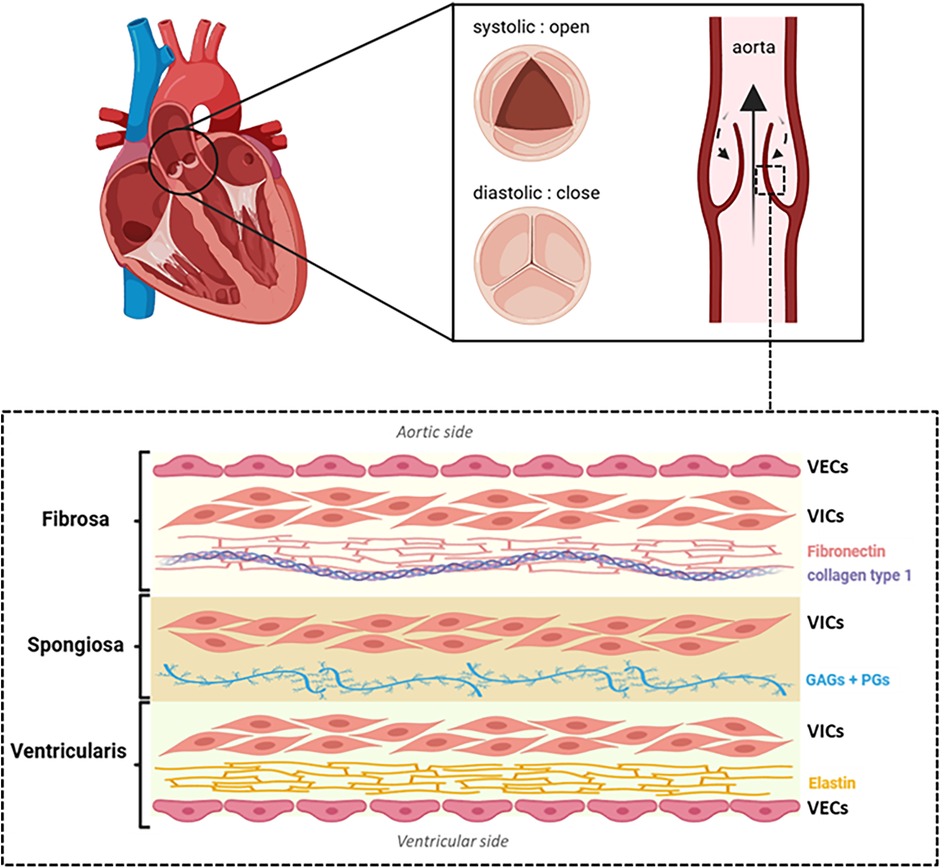
Figure 1. Schematic representation of aortic valve structure. Each leaflet consists of three distinct layers: the fibrosa (rich in fibronectin and collagen type 1 fibers), the spongiosa [composed of glycosaminoglycans [GAGs] and proteoglycans [PGs]] and the ventricularis (rich in elastin). Valvular endothelial cells (VECs) cover both the aortic and ventricular surfaces, while valvular interstitial cells (VICs) are distributed throughout all layers. Illustration created with BioRender.
2.2 Mechanistic insights into CAVD development
2.2.1 Overview of the main mechanisms driving CAVD
Early CAVD begins with endothelial injury or dysfunction, triggered by mechanical or metabolic stress. This injury promotes lipid deposition and upregulation of adhesion molecules like E-Selectin, VCAM-1 and ICAM-1 on VECs, facilitating immune cell adhesion, rolling and infiltration (10). Cytokines and growth factors released by both immune cells and dysfunctional VECs drive VICs activation (9, 11–13). In particular, TGF-β induces quiescent VICs (qVICs) to differentiate into activated VICs (aVICs), characterized by α-smooth muscle actin (α-SMA) expression and a myofibroblastic phenotype. Inflammation further promotes their proliferation, migration, and secretion of matrix metalloproteinases (MMPs), contributing to leaflet fibrosis, thickening, and stiffening. In early CAVD, elevated TGF-β also promotes the formation of myofibroblasts from VECs via endothelial-to-mesenchymal transition (EndMT) (14, 15). During EndMT, VECs downregulate endothelial markers (e.g., CD31 and VE-Cadherin) and upregulate α-SMA. This process can also be triggered by inflammatory cytokines (IFNγ, IL-6, TNF-α, or LPS) (15), disturbed flow (16), or metabolic factors (oxLDL, HDL, hyperglycaemia) (17). Over time, aVICs and aVECs can transition into osteoblast-like cells (obVICs and obVECs), characterized by decreased α-SMA expression and upregulation of osteogenic markers such as bone morphogenetic protein-2 (BMP2), Runt-related transcription factor-2 (Runx2), and alkaline phosphatase (ALP), thereby promoting mineralization. This cell-mediated fibro-calcific remodeling ultimately stiffens the valve and leads to AS. The main mechanisms driving CAVD are illustrated in Figure 2.
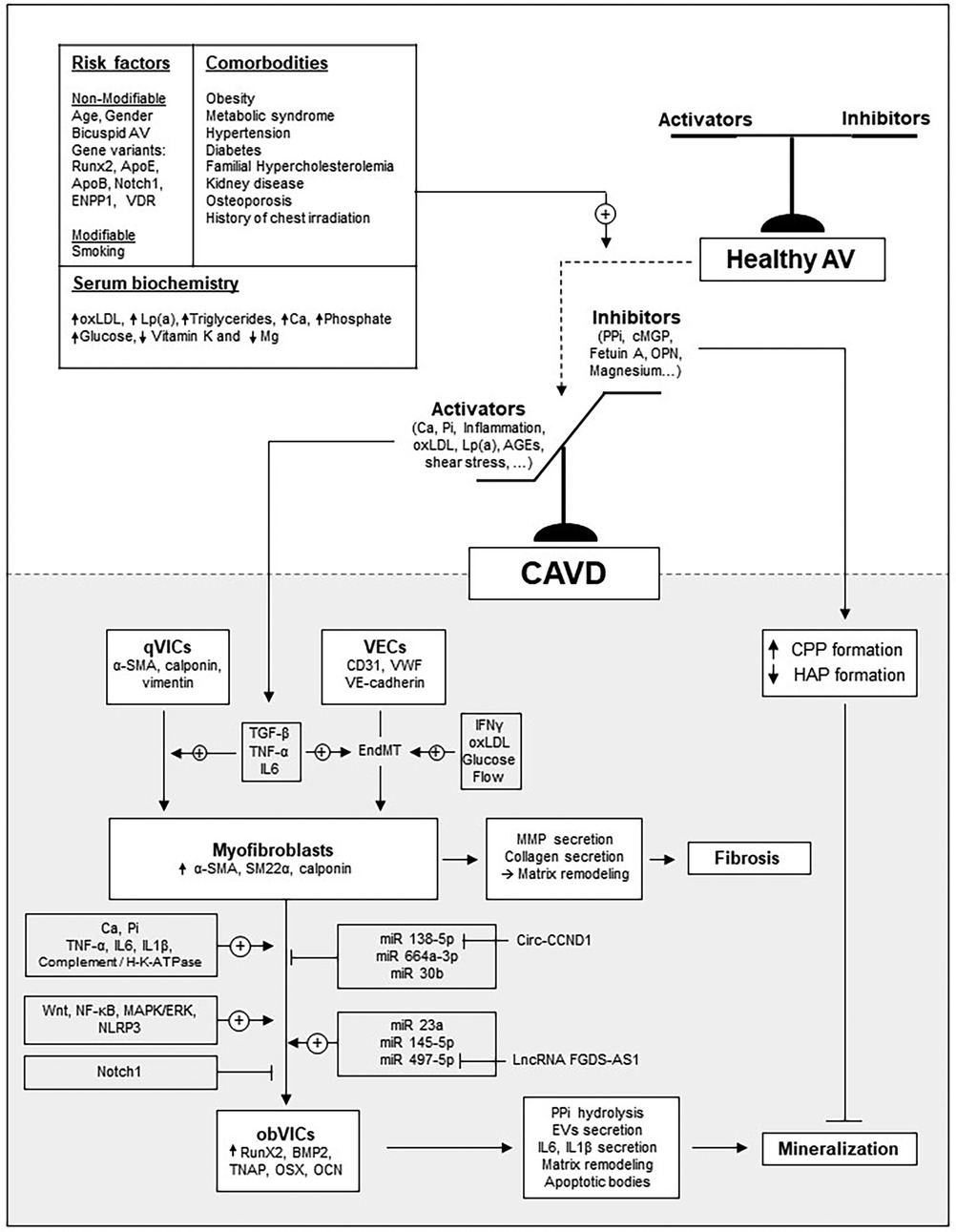
Figure 2. Overview of the main mechanisms driving CAVD development. The schematic illustrates how major risk factors and comorbidities influence the balance between inducers and inhibitors of the mineralization process.
2.2.2 Mechanisms driving mineral deposition
Aortic valve calcification generally reflects an imbalance between inhibitors that prevent calcium-phosphate deposition (mineral phase) and activators that promote VIC/VEC osteogenic transition (cellular phase) (Figure 2).
Among the inhibitors, pyrophosphate (PPi) prevents mineralization by directly interfering with the physicochemical process of hydroxyapatite formation. PPi can be hydrolyzed by ALP, making ALP activity a key regulator of PPi availability. Calcifying VICs show increased ALP activity and decreased PPi levels (18). Other inhibitors include matrix Gla protein (MGP) and fetuin A, which bind calcium and stabilize nascent calcium-phosphate clusters into amorphous, proteinaceous spherical structures known as primary calciprotein particles (CPP), which facilitate calcium-phosphate clearance and prevent ectopic calcification (19–21). MGP expression is significantly reduced in diseased VICs (22) and stenotic AV (23), and circulating Fetuin-A levels are lower in patients with CAVD (24).
On the cellular side, two distinct cell-driven mechanisms are recognized in valve calcification: osteogenic and dystrophic calcification. Osteogenic calcification occurs when VICs and VECs acquire an osteoblast-like phenotype, expressing bone-related markers such as RUNX2, BMP2, and ALP, and producing an osteoid-like ECM that subsequently mineralizes. RUNX2 is a key transcription factor driving osteoblastic commitment by regulating genes such as ALP, osteopontin (OPN), type I collagen, and osteocalcin (OCN). BMP2 promotes osteogenic differentiation by enhancing RUNX2 expression and acetylation, thereby increasing its stability and transcriptional activity. Akin to bone, obVICs release small extracellular vesicles (EVs) enriched in ectonucleotidases that promote calcium-phosphate nucleation (25, 26). Several of these ectonucleotidases, including ALP, are overexpressed in human CAVD samples (27–29). In contrast, dystrophic calcification involves the activation of VICs and VECs into a myofibroblast-like phenotype followed by apoptosis, leading to the formation of apoptotic bodies that closely resemble calcifying EVs and serve as initial nucleation sites for nodule formation. This apoptosis-dependent process is strongly influenced by the mechanical stiffness and ECM composition of the microenvironment, which modulate cytoskeletal tension and thereby the extent of calcification.
Over the years, multiple signaling pathways have been identified as regulators of VIC osteogenic reprogramming. NF-κB signaling induces BMP2 (30) and RUNX2 (31) expression in VICs. This effect involves NF-κB–mediated upregulation of TERT, which binds STAT5 to activate the RUNX2 promoter and drive osteogenic reprogramming (32). The MAPK/ERK pathway also contributes to RUNX2 induction, either through TNF-α stimulation (33, 34) or via complement crosstalk with H-K-ATPase (35, 36). Similarly, NLRP3 activation promotes RUNX2 and ALP expression in VICs (37). Wnt signaling enhances both RUNX2 expression and ALP activity (38), and several of its key components—including the receptor LRP5, the agonist WNT3a, and the nuclear effector β-catenin—are overexpressed in CAVD samples (39). By contrast, activation of Notch1 suppresses BMP2 and RUNX2 expression in cultured VICs (40, 41), consistent with the inverse correlation between Notch1 and RUNX2 observed in CAVD samples (42).
In addition to these signaling pathways, recent studies have demonstrated that dysregulation of miRNAs also contributes to the osteogenic reprogramming of VICs in AS (43). For instance, miR-664a-3p, which is downregulated in mineralized AV, inhibits VICs osteogenic differentiation by directly binding to BMP2 and repressing its expression (44). MiR-30b, which expression is also downregulated in mineralized AV, suppresses VICs osteogenic differentiation by directly inhibiting RUNX2 expression (45, 46). Conversely, miR-23a, which is upregulated in mineralized AV, promotes VIC mineralization by enhancing RUNX2 activation through the suppression of Notch1 expression (42). Recently, Goody et al. observed that miR-145-5p is one of the most strongly upregulated miRNAs in CAVD, with its vesicular content increased in the disease. In vitro calcification experiments demonstrated that EV-mediated transfer of miR-145-5p promotes ALP expression by suppressing ZEB2, a negative regulator of the ALPL gene (47). Interestingly, circ-CCND1, whose expression is upregulated in CAVD samples, was shown to promote osteogenic transition of VICs by sponging miR-138-5p, thereby activating the osteogenic CCND1/P53/P21 pathway (48). By contrast, LncRNA FGD5-AS1, which is downregulated in CAVD patients, sponges miR-497-5p to regulate BIRC5, thereby repressing osteogenic differentiation and alleviating CAVD (49). The main signaling pathways involved in VIC phenotypic changes are summarized in Table 1.
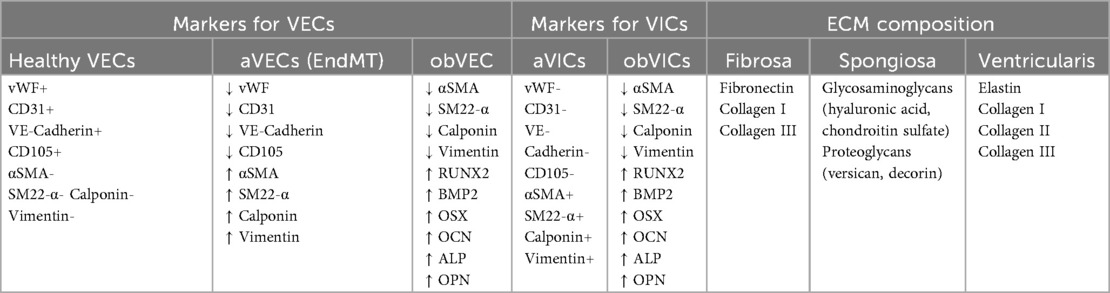
Table 1. Main markers used to identify the phenotypic states of VICs and VECs.
2.3 Risk factors and comorbidities
Older age is the strongest risk factors for AS. Indeed, severe AS affects approximately 3.4% of individuals aged 75 years and older, with symptoms present in 75% of cases (1, 2). Bicuspid aortic valve (BAV), which is a common congenital defect in which the AV has two leaflets rather than three, represents a significant risk factors for AS. Patients with BAV typically develop AS 10–20 years earlier than those with a tricuspid AV, and their lifetime risk of developing the disease is approximately 50%. Although the prevalence of BAV in the general population is estimated at 0.5%–1.0%, it accounts for nearly half of all AVs surgically replaced due to AS in the United States (50). The increased susceptibility to calcification observed in BAVs can be attributed to a combination of hemodynamic and structural factors. Abnormal, turbulent, and asymmetric blood flow generates altered shear stress patterns that promote VIC activation. In parallel, structural abnormalities such as thickened or disorganized valve leaflets and the presence of a raphe—an incomplete separation between cusps—further predispose BAVs to pathological calcification.
Additional clinical factors associated with AS largely overlap with atherosclerosis risk and include male sex, smoking, and metabolic syndrome (e.g., hypercholesterolemia, hypertension, obesity, and diabetes).
Indeed, the risk of AS is twice higher in men compared to women (51, 52). Moreover, men with AS tend to exhibit greater calcification (53), whereas women show increased fibrosis despite similar disease severity (54), highlighting the importance of considering sex as a biological variable in preclinical research. Oxidized LDL (OxLDL) levels correlate with AV inflammation and fibro-calcific remodelling (55, 56), and recent studies implicate the LPA gene, encoding apolipoprotein(a), in AS pathogenesis (57–59). Besides, elevated lipoprotein(a) [Lp(a)] and oxidized phospholipids (Ox-PL) are independently associated with faster AS progression (60). Secondary hyperparathyroidism and renal failure are also associated with AS progression (61).
3 In vitro modeling of CAVD
To investigate the mechanisms driving CAVD, researchers commonly use in vitro models based on VICs and VECs from various species. These cells can be cultured independently or in co-culture, in 2D or 3D systems. The selection of cell type and culture conditions should be carefully tailored to each study's objectives to ensure relevant and reproducible results.
3.1 Main sources of VICs/VECs for in vitro studies
3.1.1 Human VICs/VECs
3.1.1.1 Main sources and challenges
Most primary VICs and VECs are isolated from AVs of patients undergoing SAVR. However, these tissues are often extensively remodeled, and the resident cells already display features of the disease. To better investigate early mechanisms of AS, valvular cells can instead be isolated from AVs of patients with idiopathic dilated cardiomyopathy or post-infarction heart failure, as well as from non-transplantable donor hearts or accident victims. When working with such primary cultures, it is important to keep in mind that variations in sex, age, and clinical background, along with donor-specific genetic and epigenetic factors, can lead to substantial heterogeneity in cellular responses. While often viewed as a limitation, this variability accurately reflects population diversity and thus enhances the physiological relevance and translational value of in vitro findings. In such contexts, access to key donor information (such as sex, age, and comorbidities) is particularly valuable for conducting comparative studies (e.g., male vs. female, young vs. old, tricuspid vs. bicuspid, or diabetic vs. non-diabetic).
3.1.1.2 hVICs/hVECs isolation and culture
Valve retrieval should ideally be performed rapidly to preserve cell viability. Leaflets should be placed immediately in cold saline, PBS, or DMEM, stored at 4 °C, and processed within 12 h. Interestingly, Cuevas et al. demonstrated that, if valve samples are stored in a cold storage solution suitable for organ transplantation, viable VECs and VICs can still be obtained from leaflets processed between 24 and 61 h post-extraction. This information is particularly relevant for centers that do not have immediate access to the medical facilities where valve excision is performed (62).
Although there is some variability in the methods used by researchers to isolate valvular cells, the general approach typically involves a brief enzymatic digestion to remove human VECs (hVECs), followed by a longer digestion to release human VICs (hVICs) (62–66). Most protocols begin with a 10-min incubation of valve tissue in collagenase at 37 °C. The hVECs are then collected by gentle scraping or vortexing, followed by centrifugation and filtration before being seeded onto FN-coated T25 flasks (64, 65). In culture, VECs form rosette-like colonies, reach confluence in about a week, and are passaged at a 1:3 ratio. For VIC isolation, the remaining valve fragments are rinsed with saline, cut into ∼2 mm2 pieces, and incubated with collagenase under gentle agitation. Two main protocol variations are described in the literature: short incubations (45 min to 3 h) with high collagenase concentrations (∼1,000 U/ml) (66–68), and longer or overnight digestions with lower concentrations (250–600 U/ml) (64, 65, 69). The strained cell suspension is then centrifuged, and seeded at 800,000 cells per 75 cm2 flask. VICs are typically cultured in DMEM supplemented with 10%–15% FBS and antibiotics, displaying a fibroblast-like morphology after 24 h. They are passaged at 90%–95% confluence and seeded at a 1:2 ratio, with medium changes twice weekly. According to Ground et al., successful hVIC culture is more likely when starting with a sufficient amount of tissue, typically greater than 500 mg, and when the initial digestion yields a substantial number of cells, generally exceeding 100,000. They also demonstrated that patient pathology is not predictive of cell culture success, and that a seeding density of 10,000 cells/cm2 is ideal for experiments lasting less than 5 days (66).
The purity of cultured cells is a key concern in valvular cell studies, given its impact on experimental outcomes. Cell purity is usually assessed using immunohistochemistry or flow cytometry. VECs should be αSMA-negative and positive for endothelial markers [CD31, VE-cadherin, von Willebrand factor (vWf)], while VICs should show the opposite profile. The presence of αSMA-positive cells in VEC cultures indicates VIC contamination, and double-positive cells (endothelial markers and αSMA) suggests EndMT. To ensure purity, freshly isolated VECs may be labeled with anti-CD31 or anti-CD105 magnetic beads and sorted via Magnetic-Activated Cell Sorting, before first seeding (54, 55). The presence of endothelial markers in VIC cultures suggests VEC contamination. Additional markers like calponin (myofibroblast) and vimentin (mesenchymal stem cell) may help refine phenotypic identification (64, 65, 70, 71). Over successive passages, VICs tend to differentiate into myofibroblasts, characterized by an increased expression of αSMA (70), a process likely driven by the stiffness of plastic culture surfaces (72). Therefore, experiments on VICs are best conducted between passages 2 and 6. According to Ground et al., optimizing the VIC isolation protocol using 1,000 U/ml collagenase for 2 h resulted in the highest number of viable VICs while minimizing aberrant aVIC differentiation (66). The main markers used to identify the different phenotypic states of VICs and VECs are summarized in Table 1.
Until recently, most VICs used in research were derived from primary cultures. However, in 2025, Wang et al. established and characterized a novel immortalized hVIC line (73). They achieved immortalization of primary VICs through lentiviral transduction with SV40 large T antigen (pGMLV-SV40T-PURO), followed by puromycin selection to establish stable cell lines. Compared with primary VICs, these immortalized cells showed higher viability, reduced senescence, stable transcriptomic profiles across multiple passages, and preserved responsiveness to several osteogenic inducers. RNA-seq analyses further revealed the central role of inflammation-related pathways in driving their osteogenic transformation, suggesting that this model may be ideally suited to investigate the contribution of inflammation to VIC phenotypic changes. In the future, this immortalized VIC line could become a valuable and standardized in vitro tool for studying AV calcification, particularly for laboratories without easy access to surgical AV specimens.
3.1.2 VICs/VECs from other species
Cells from porcine AVs, which closely resemble human AVs in structure and composition, are a valuable alternative to human cells. Being free of disease, porcine AVs provide an interesting source of cells for studying early CAVD events. Moreover, swine naturally develop atherosclerotic valve lesions, mirroring early human valvular calcification (74). Their large size allows for efficient enzymatic digestion and isolation of pure cell populations. Porcine AVs can be sourced from slaughterhouses, where swine hearts are typically discarded, aligning with the ethical principle of reduction in animal experimentation. Porcine VICs and VECs (pVICs/pVECs) can be isolated using the same protocol as for human cells (64). While porcine and human valvular cells share similarities, there are also notable differences between them (64). For instance, vimentin expression is higher in pVECs than in pVICs, which is the opposite of what is seen in humans. Compared to hVECs, pVECs also show reduced migratory capacity. Additionally, while pVECs respond to high doses of TNF-α by undergoing myofibroblastic transition (15), low doses—effective in hVECs—fail to induce α-SMA and vimentin expression in pVECs (64). Similarly, TGF-β, which induces α-SMA in hVECs, has no such effect in pVECs (75). These interspecies differences, along with the limited availability of molecular tools for the porcine models, should be carefully considered when using swine-derived cells.
In recent years, rodent AVs have gained interest as a source of VICs due to several advantages, including low cost, ease of handling, short lifespan, genetic manipulability of rodents, and access to extensive molecular tools. However, isolation of VICs and VECs from rodents is not yet a gold standard, likely due to the technical challenges associated with working on small tissue samples. Lin et al. showed that VICs can be isolated from rat AV using careful valve dissection followed by enzymatic digestion with collagenase II (76). However, due to the small size of rat AV, approximately 30 leaflets were required to seed a T25 flask, raising ethical concerns. To address this, the authors developed immortalized rat VIC (RAVIC) cell lines via lentiviral transduction with Simian virus (SV40) large T antigen (77). This model enables mechanistic studies of CAVD, which can later be validated in primary cultures to reduce animal use. Primary RAVICs can also be obtained via explant culture, where cusps stripped of endothelium are incubated in growth medium until VICs migrate out (78, 79). To date, few studies have been conducted on RAVICs, so that their phenotypic similarity to human cells remains difficult to evaluate. More recently, VICs have been successfully isolated from mice (80), with only 3 AVs (9 leaflets) required to initiate the culture, opening the door to studies using VICs from genetically modified models. However, rodent valves differ markedly from those of humans and pigs. Their cusps are only ∼5–10 cells thick and lack the distinct trilaminar architecture seen in larger animals (81–83). Moreover, wild-type rodents on standard diets do not develop age-related AV calcification (81), suggesting that AV remodeling mechanisms in mice and rats may not fully replicate those in humans. Further investigation is needed to clarify these interspecies differences.
In 2022, Tao et al. successfully isolated rabbit VICs and used them to investigate how ox-LDL promotes their osteogenic transformation (84). Rabbits are easy to raise, have a moderate lifespan, and their size and anatomy make them suitable for short-term, cost-effective experiments. Despite these advantages, rabbit VICs remain underused in in vitro studies. Early studies also used VICs from sheep (85) and dogs (86), but these models have largely been abandoned, mostly due to limited accessibility and ethical concerns.
3.2 Inducing phenotypic switching of VICs and VECs in vitro
3.2.1 Mimicking VIC myofibrobastic transition
In vitro, VIC myofibroblastic differentiation is typically assessed by monitoring the gradual upregulation of α-SMA, calponin, and SM22 (87–89). While TGFβ-1 is the most commonly used inducer of myofibroblastic differentiation in vitro (90–92), the response to TGFβ-1 varies across species. Indeed, α-SMA expression increases within 24 h in ovine VICs (93), after 4 days in hVICs (13), and up to 5 days in pVICs (94). In 2004, Walker et al. showed that culturing pVICs at high density and treating them with TGF-β1 induced a myofibroblast-like phenotype and promoted the formation of multicellular aggregates (90). Apoptosis occurred in the central region of these aggregates, where calcium deposits subsequently formed. Fisher et al. later demonstrated that combining mechanical stretch with TGF-β1 rapidly produced pVIC aggregates; central apoptosis again preceded the formation of calcific nodules (95). Inhibition of apoptosis using Z-VAD, a pan-caspase inhibitor that irreversibly blocks caspase activity, markedly reduced the number of calcific nodules, confirming the essential role of apoptosis in initiating dystrophic calcification.
3.2.2 Mimicking VIC osteogenic transition and mineralization
Osteogenic differentiation and mineralization are induced in vitro by culturing cells in a medium typically supplemented with β-glycerophosphate (β-GP) and/or elevated calcium and inorganic phosphate (Pi) levels. This medium is commonly referred to as osteogenic medium (OM).
β-GP has been widely used because it can be hydrolyzed by ALP expressed by VICs, releasing Pi that promotes the expression of osteogenic markers such as RUNX2, ALP, OPN, and OCN, as well as mineralization. Its efficacy depends on several factors, including concentration (typically 10 mM), calcium availability in the medium, and exposure time (usually around 21 days to achieve significant mineralization in DMEM containing 1.8 mM calcium). Osteogenic media using β-GP often include dexamethasone (100 nM) to further promote osteogenic differentiation, and ascorbic acid (50 µg/ml) to support collagen synthesis (9). However, because ALP activity is crucial for β-GP hydrolysis, this method may be ineffective in VICs with low ALP expression. To further enhance mineralization, β-GP-based OM can be supplemented with cytokines such as BMP-2, BMP4, BMP7, TGFβ-1 or TGFβ-3, which boost ALP expression (96, 97).
Alternatively, mineralization can be induced by directly increasing calcium and/or Pi concentrations in the culture medium. Calcium-enriched media typically use calcium chloride (CaCl2) at 2.5–5 mM (98), while Pi-enriched media rely on sodium phosphate (Na2HPO4 and/or NaH2PO4) at 2–5 mM (99, 100) to mimic hyperphosphatemia, as seen in chronic kidney disease (CKD). In this type of system, mineralization becomes detectable within 10–14 days. Combining both ions accelerates the mineralization process, provided their concentrations remain below their solubility threshold (around 2.2 mM each); above this limit, spontaneous precipitation occurs independently of cellular activity. A preliminary test on a cell-free plate is therefore recommended to distinguish active, cell-driven mineralization from passive calcium/phosphate precipitation caused by supersaturation.
In 2D cultures, VIC and VEC mineralization assays are typically conducted in 48-well plates. The OM is selected based on the study's objectives. Mineralization duration depends on both OM composition and donor variability, as cells from different donors show heterogeneous sensitivity to mineralization.
3.2.3 Mimicking VECs phenotypic transition
TGF-β1 is the most commonly used cytokine to induce EndMT in VECs in vitro. Typical concentrations range from 1 to 5 ng/ml for long-term exposure (1–14 days) to 100 ng/ml for short-term treatments (15, 101–103). TGF-β1 rapidly (2–5 days) and persistently (up to 14 days) increases α-SMA while progressively downregulating endothelial markers like VE-Cadherin. After 14 days, it also enhances ALP activity and upregulates osteogenic markers such as OPN, OCN, and RUNX2 (102). Similarly, IL-6 and TNF-α (100 ng/ml) promote EndMT through an Akt/NF-κB-dependent pathway (15). Interestingly, culturing VECs in OM induces a myofibroblastic phenotype within 7 days, followed by a gradual osteogenic transition by day 14 (102).
3.3 Reproducing comorbidities in vitro
In vitro models can integrate patient comorbidities to improve translational relevance. For example, VICs and VECs can be compared based on donor sex (79, 104), age (78), or valve morphology (bicuspid vs. tricuspid) (89, 105) to investigate how these variables influence valvular cell physiology and drug response. In addition, the effects of circulating factors can be evaluated by exposing healthy cells to patient-derived serum (106). Indeed, we observed that serum from AS patients increases VIC calcification compared to non-stenotic controls, thereby providing a relevant system to evaluate the protective effects of new drug candidates (100). This strategy is particularly valuable for modeling comorbidities like CKD or diabetes, where circulating toxic compounds present in the serum—such as uremic toxins, phosphate, glucose, or advanced glycation end-products—contribute to valvular degeneration. In our hands, adding 1%–2% human serum to OM supports mineralization, while exceeding 10% compromises cell viability. The specific effects of pathological factors—including inflammatory cytokines (100), advanced glycation end-products (107), glucose (108), uremic toxins (109), hypoxia (110), neutrophil extracellular traps (NETs) (111) and oxidized lipoproteins (112)—can be evaluated by exposing VICs or VECs to each factor, with or without OM.
3.4 Mimicking cellular interactions
3.4.1 VICs-VECs interactions
VICs-VECs communication is essential for maintaining leaflet homeostasis. Using a transwell co-culture system (without direct contact), Hjortnaes et al. showed that VICs attenuate TGF-β1-induced EndMT in VECs, evidenced by reduced α-SMA expression. Similar results were observed when VECs were exposed to VIC-conditioned medium (CM) (102). When co-cultured in Transwell systems, VICs also inhibited the osteogenic transition of VECs under OM, suggesting a protective role in VEC physiology. Conversely, VECs not only failed to inhibit, but actually promoted OM-induced osteogenic differentiation and calcification in VICs (102). These findings highlight the value of in vitro systems that incorporate intercellular communication for advancing our understanding of AS pathophysiology.
3.4.2 VICs-macrophages interactions
Co-culture systems and CM have also helped to elucidate VIC-macrophage communication. In 2017, Li et al. showed that CM from pro-inflammatory macrophages enhances VIC osteogenic transition. This effect was blocked when antibodies targeting TNF-α or IL-6 were added to the system; demonstrating the role of inflammation in this process (113). In 2020, Raddatz et al. demonstrated that direct co-culture with macrophages elevated RUNX2 expression in VICs compared to transwell co-cultures, highlighting the role of physical contact between cells in AS pathogenesis (114). More recently, Xia et al. found that EVs from pro-inflammatory macrophages, once internalized by VICs promote osteogenic transition and mineralization more strongly than EVs from control macrophages (115). These models collectively demonstrate how macrophages influence VIC phenotype via cytokines, contact, and EVs and how in vitro systems can dissect these complex interactions.
3.5 Three-dimensional (3D) models
Mechanical cues from the ECM (including its composition, stiffness and stretch) influence VIC phenotype and disease progression (116). VICs sense stiffness via focal adhesions, adjusting integrin expression and cytoskeletal organization to maintain force balance (117, 118). These mechanosensing processes also modulate intracellular signaling, making VICs highly responsive to their mechanical environment (119). While widely used, 2D cultures fail to replicate the complexity of the native tissue microenvironment, particularly mechanotransduction and cell-ECM interactions, which are critical for VIC regulation. In addition, culturing VICs on polystyrene triggers spontaneous activation and pathological differentiation, limiting the physiological relevance of 2D models in CAVD research. To address these shortcomings, 3D culture systems-with or without scaffolds-have been developed (Table 2).
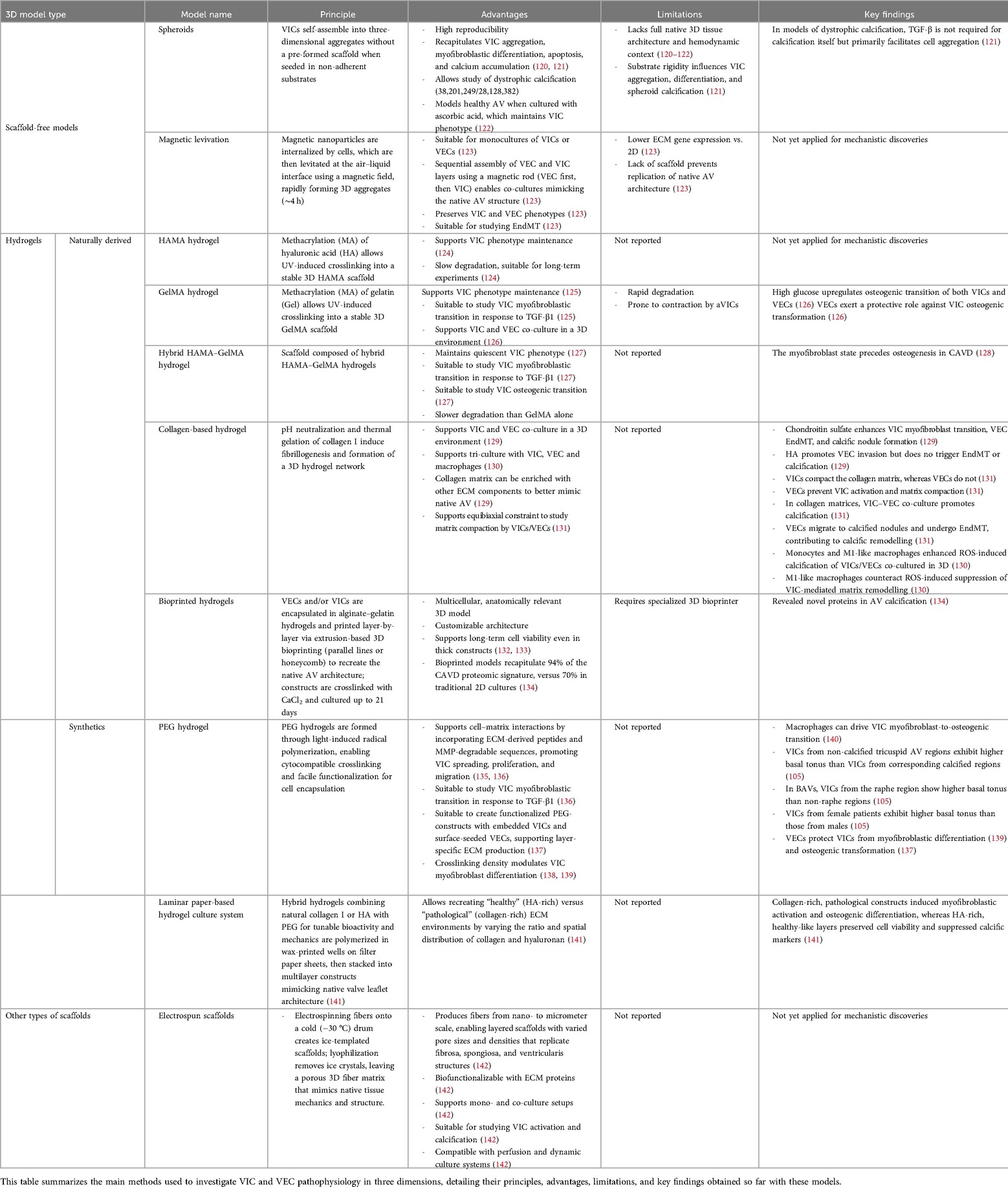
Table 2. Overview of key 3D cellular models currently available to study CAVD.
3.5.1 Scaffold-free 3D models
3.5.1.1 Spheroids
Spheroids are three-dimensional, self-assembled cell aggregates that mimic tissue-like interactions. In recent years, these systems have been used to study how VIC myofibroblastic differentiation and apoptosis drive dystrophic calcification. In 2017, Roosen et al. created spheroid structures by seeding porcine VICs into non-adherent agarose microwells, prepared using a 3% agarose solution cast into biocompatible silicone molds, and cultured them in standard medium (122). With this initial protocol, aggregates rapidly degenerated, showing early signs of cell death and mineralization. To address this, the authors supplemented the culture medium with 250 μM ascorbic acid, an essential nutrient and antioxidant, which enabled the formation of viable, high-quality aggregates with no signs of degeneration or calcification. ECM analysis revealed significant increases in GAG, elastin, reticular fibers, and collagen I over the culture period. Aggregates also showed enhanced mRNA expression of Col I/III/V, elastin, hyaluronan, biglycan, decorin, versican, MMP-1/2/3/9, and TIMP-2 compared to monolayer cultures. VICs in aggregates displayed lower α-SMA expression, while osteogenic and chondrogenic markers (OCN, Egr-1, Sox-9, Runx2) remained unchanged, demonstrating that this 3D approach overcomes VIC activation in 2D and promotes a quiescent VIC state. In 2024, Coutts et al. used this model to study the process of dystrophic calcification (120). They applied the protocol developed by Roosen et al. without supplementing the medium with ascorbic acid and observed the formation of calcium nodules within the spheroids after only a few days. Inhibition of apoptosis using Z-VAD markedly reduced calcification, confirming that the process was dystrophic rather than osteogenic. In 2017, Cirka et al. used a different technique to form spheroids, seeding VICs onto collagen-coated micro-contact printed areas on polyacrylamide gels, where the cells self-assembled into aggregates with diameters ranging from 50 to 400 μm (121). These aggregates exhibited myofibroblastic markers, apoptosis, and calcium accumulation. Their exposure to the pan-caspase inhibitor Z-VAD-FMK reduced calcification by approximately 75%, confirming the dystrophic nature of the process. Using this system, the authors showed that TGF-β treatment was not required for calcification itself but primarily facilitated cell aggregation. Interestingly, the authors observed that calcification occurred when aggregates were formed on polyacrylamide gels with stiffness ranging from 9.6 to 76.8 kPa, highlighting the importance of substrate rigidity as a tunable support influencing VIC aggregation, myofibroblastic differentiation, and calcification in this model.
3.5.1.2 Magnetic levitation
In 2014, Tseng et al. used magnetic levitation (Bio-Assembler Kit, Nano3D Biosciences) on VICs, VECs, and their co-cultures to form multilayered cellular constructs, introducing a novel 3D model for AV research (123). Confluent monolayers were incubated with magnetic nanoparticles, detached, and then seeded into ultra-low attachment plates. A magnetic driver positioned above the plate levitated cells to the air-liquid interface, forming 3D cultures within 4 h. VIC and VEC layers were sequentially assembled using a magnetic rod–the VEC layer first, followed by the VIC layer–to create a co-culture mimicking AV structure. The construct was stabilized in VEC medium, then transferred and re-levitated in 24-well plates. Immunohistochemistry and qRT-PCR confirmed preservation of cell phenotype, with CD31 (VEC) and αSMA (VIC) expression. ECM proteins such as collagen type I, laminin, and FN were detected, though gene expression was lower than in 2D cultures. Reduced expression of collagen type I, lysyl oxidase, and αSMA in co-cultures suggested VICs quiescence. Co-localization of CD31 and α-SMA hinted at potential EndMT, suggesting that this model may provide a robust platform for studying AV biology and disease mechanisms. However, the absence of a defined scaffold limits its ability to replicate the AV's complex structure. Hydrogels partly address this limitation by offering a tunable 3D matrix that supports cell interactions and ECM biofunctionalization.
3.5.2 Hydrogels
Over the last decade, photopolymerizable hydrogels have gained attention in the field of 3D culture due to their elastic structure, which allows efficient VIC encapsulation, and their composition, which provides molecular cues essential for cell behavior and differentiation. Both natural and synthetic hydrogels have been explored as cell carriers, considering that an effective scaffold for valvular cells must support adhesion, proliferation, ECM production, and allow investigation of VIC/VEC phenotypic transitions.
3.5.2.1 Naturally derived hydrogels
Naturally derived hydrogels based on gelatin (Gel) and hyaluronic acid (HA), both key factors for VIC adhesion and proliferation (124), have been widely used for AV tissue engineering. Gelatin is a denatured form of collagen, and HA is the predominant GAG in AV ECM, known for its elasticity and specific interactions with FN. In their native form, Gel and HA are soluble and non-crosslinked, limiting their application as scaffolds. To overcome this, researchers methacrylated (MA) them into GelMA (143) and HAMA (124), allowing UV-induced crosslinking into stable 3D structures that preserve their bioactivity. VICs seeded on HAMA spread, proliferated, and formed a confluent monolayer within four days. HAMA preserved HA's ability to bind FN, enabling the design of gels containing both HA and FN, which enhanced ECM production and supported VIC phenotype maintenance (124). Its slow degradation rate (144) makes it suitable for long-term experiments, including those focused on calcifications. Similarly, VICs seeded in GelMA regained their native morphology within 2 weeks, a process accelerated by TGF-β1 (125), which also promotes aVICs formation and collagen-1 synthesis. However, GelMA alone degrades rapidly, limiting its use in extended studies. To address this, a hybrid HAMA-GelMA hydrogel was developed, improving stability and better mimicking the native AV ECM (127). In this system, VICs remain quiescent unless stimulated with TGF-β1 and differentiated into first aVICs and then obVICs when exposed to an OM (128). Silencing α-SMA reduced both osteogenic differentiation and calcification, suggesting that the myofibroblast state precedes osteogenesis in CAVD.
3.5.2.2 Synthetic hydrogels
Natural 3D matrices closely mimic physiological environments, supporting VIC viability, remodeling, and acting as reservoirs for bioactive molecules. However, their complexity may obscure specific cell-matrix interactions.
To overcome this, Benton et al. introduced in 2009 synthetic hydrogels based on polyethylene glycol (PEG), a synthetic, non-toxic, non-immunogenic, polymer approved by the FDA, which can be easily functionalized for cytocompatible encapsulation via light irradiation. PEG hydrogels are highly hydrated and mimic the mechanical properties of soft tissues. To enhance cell-matrix interactions, they incorporated RGD peptide (derived from FN) and crosslinked the PEG with a MMP-degradable sequence (GPQGIWGQ), enabling cell-driven remodeling. This system supported integrin αvβ3 binding, as well as cell spreading, proliferation, migration, and TGF-β1-induced myofibroblast differentiation (136). In 2016, Gould et al. encapsulated VICs in PEG hydrogels functionalized with ligands derived from FN (RGDS), elastin (VGVAPG), or collagen-1 (P15). VICs cultured in FN-functionalized hydrogels showed higher MMP activity at day 2 and exhibited elongation by day 14 compared to those in elastin or collagen-1 gels. The highest proportion of αSMA + VICs was observed in elastin gels (56%), compared to FN (33%) or collagen (38%); along with higher collagen-X:collagen-I ratio, a marker associated with stenotic valves (135). In 2020, Grim et al. demonstrated using PEG hydrogels that CM from pro-inflammatory macrophages promotes the osteogenic transition of activated VICs cultured in 3D (140). They reported that this effect was mediated by the secretion of TNF-α, IL-1β, and IL-6, suggesting that macrophages may drive a myofibroblast-to-osteogenic shift in VICs, thereby linking fibrosis to calcification in AS. In 2023, Tuscher et al. used PEG hydrogels to characterize the basal contractile behavior of VICs from tricuspid AVs. To do so, they tracked VIC-induced gel displacements and shape changes after treatment with Cytochalasin D, an actin polymerization inhibitor that depolymerizes VIC stress fibers. They demonstrated that VICs from the non-calcified region of tricuspid AV were significantly more activated than those from the corresponding calcified regions. Moreover, when studying VICs from bicuspid AVs, they showed that cells from the raphe region were more activated than those from non-raphe regions. Changes in VIC morphology following Cytochalasin D treatment indicated that cells from tricuspid valves and BAVs possess distinct cytoskeletal structures, providing new insights into the cellular mechanics underlying CAVD progression in BAV patients (105). This model also revealed that VICs from female patients exhibited significantly higher basal tonus levels than those from male patients.
3.5.2.3 Co-cultures in hydrogels
Over the years, hydrogel-based co-culture models have been developed to better mimic the native AV environment. In 2015, Puperi et al. created an endothelialized AV model using PEG-RGDS and PEG-PQ (MMP-2/MMP-9 degradable) hydrogels seeded with VICs. The surface was functionalized with RKR (a laminin-derived syndecan-binding peptide) to support VEC adhesion. In this model, VECs formed a CD31+ monolayer with minimal αSMA activation within 7 days. VICs in 3D PEG-PQ showed reduced αSMA expression compared to 2D culture, an effect amplified in co-culture. By day 28, VECs secreted basement membrane components (laminin, perlecan, and collagen type IV), while VICs produced collagen and FN, suggesting layer-specific ECM production (137). In 2022 Bramsen et al. co-cultured VICs and VECs in collagen I hydrogels enriched with chondroitin sulfate (CS) and HA, two GAGs typical of the spongiosa (129). CS enhanced VIC myofibroblast transition, VEC EndMT, and calcific nodule formation, while HA promoted VEC invasion without triggering EndMT or calcification. This observation underscores the influence of ECM composition on valvular cell fate, further supporting the relevance of 3D models for preclinical studies.
In 2014, Gould et al. showed that increasing PEG hydrogel stiffness by raising crosslinking density promoted VIC myofibroblast differentiation, an effect abolished by VEC co-culture due to paracrine NO signaling (139). In contrast, Mabry et al. found that increasing hydrogel stiffness suppressed VIC activation (138). This inverse relationship suggests that while matrix stiffness is a key regulator of VIC phenotype, its effects are context-dependent and strongly influenced by the method used to modulate stiffness. Unlike Gould, Mabry et al. increased stiffness using a secondary non-degradable network in a thiol-ene hydrogel system, which enabled to decouple stiffness from network density. This approach provided independent control over mechanical properties without altering mesh size or cell morphology—two factors often affected when stiffness is tuned by increasing crosslinking. By isolating mechanical cues from structural variables, their system allowed a more precise investigation of how stiffness influences VIC behaviour and differentiation. In 2021, Gee et al. studied porcine VEC and VIC responses under tension using equibiaxially constrained collagen hydrogels. VICs compacted the matrix, while VECs did not. Co-culture reduced VIC activation and matrix compaction, though OM reversed these effects. OM also induced calcified nodule formation, further enhanced by VEC co-culture. Inhibiting canonical NFκB reduced calcification but not fibrosis, indicating its specific role in osteogenic remodeling. Immunofluorescence revealed that VECs clustered on calcified nodules and expressed EndMT markers, suggesting that EndMT contributes to calcific remodeling (131).
Finally, GelMA constructs with embedded VICs and surface-seeded VECs were recently used to model CAVD under diabetic conditions (126). The use of these models allowed researchers to demonstrate that high glucose upregulates osteogenic markers through the TGF-β and BMP-2 pathways in both cell types, leading to increased calcium deposition. Exposure to OM further enhanced calcification in VIC-only constructs compared to VIC–VEC constructs, confirming the protective role of VECs against VIC osteogenic transformation.
Going further in complexity, Salemizadehparizi et al. established in 2025 a tri-culture model of the fibrosa, in which THP-1 monocytes or M1-like macrophages were seeded atop collagen-I constructs containing embedded VICs and surface-seeded VECs (130). The VIC/VEC model was pretreated with H2O2 for 7 days before the addition of monocytes or M1-like macrophages, and was then maintained for 14 days, allowing assessment of the combined effects of ROS and immune cells while avoiding H2O2 toxicity to monocytes/macrophages. Using this system, the authors showed that monocytes and M1-like macrophages enhanced ROS-induced calcification compared to co-cultures without immune cells. Monocyte-tri-cultures formed smaller, more circular nodules, whereas M1-tri-cultures formed nodules of intermediate size and morphology, indicating that inflammatory macrophages modulate calcified deposit structure. Moreover, while H2O2 inhibited hydrogel contraction in VIC-only, co-culture, and monocyte-tri-culture models, contraction persisted in M1-tri-cultures, suggesting that inflammatory macrophages can counteract ROS-induced suppression of VIC-mediated matrix remodeling.
Data obtained with these co-culture models underscore how interactions between cells and the ECM shape VEC–VIC communication and contribute to CAVD development.
3.5.2.4 Toward tri-layered constructs
The tunability of 3D hydrogels offers precise control over ligand and peptide incorporation, cell–material interactions, stiffness and matrix degradability. However, most models lack the AV's trilayered structure and dynamic complexity. In this context, Monroe et al. developed in 2019 a 3D laminar paper-based culture system to dissect how ECM composition directs VIC behavior in CAVD (141). Porcine VICs were encapsulated in hydrogels containing either collagen I or hyaluronan, with both matrices functionalized with PEG-linked peptides and engineered to have equivalent shear mechanics to isolate biochemical effects. The gels were polymerized in wax-printed wells on filter paper sheets, which were then stacked into multilayer constructs mimicking native leaflet architecture. By varying the ratio and spatial distribution of collagen and hyaluronan, the authors recreated “healthy” (HA-rich) vs. “pathological” (collagen-rich) ECM environments. VICs remained viable and proliferative under both conditions; however, collagen-rich, pathological constructs induced higher α-SMA and RunX2 expression, indicating myofibroblastic activation and osteogenic differentiation. In contrast, HA-rich, healthy-like layers preserved cell viability and suppressed the expression of calcific markers, highlighting the critical influence of ECM composition on VIC phenotype. This system demonstrates the power of 3D biomimetic platforms to model both normal and disease-like valve microenvironments.
More recently, Immohr et al. introduced a 3D bioprinting strategy to engineer multicellular, anatomically accurate AV constructs, providing a promising platform for modeling CAVD and drug screening (132, 133). To achieve this, they encapsulated porcine VICs and/or VECs within alginate- and gelatin-based hydrogels. These cell-laden hydrogels were then deposited layer-by-layer using extrusion-based 3D bioprinting according to defined patterns (parallel lines or honeycomb structures) to recreate the native three-dimensional architecture of the AV. After printing, the constructs were stabilized by crosslinking in a calcium chloride solution and subsequently cultured for up to 21 days under standard conditions to maintain cell viability and enable cellular interactions within this multicellular 3D model. Good long-term cell viability was confirmed even in thick, multilayered multicellular constructs, providing proof of principle that 3D bioprinting of VEC- and VIC-based hydrogels is a feasible approach to design constructs that mimic the native AV. In 2024, Clift et al. used this type of 3D bioprinting approach to encapsulate human VICs within GelMA/HAMA-based hydrogels and compare their cellular proteome and vesiculome with those of human CAVD tissues (134). Liquid chromatography–tandem mass spectrometry analyses showed that the bioprinted model recapitulated 94% of the CAVD proteomic signature, vs. 70% in traditional 2D cultures. Integration of cellular and vesicular datasets revealed both known and previously unrecognized proteins linked to AV calcification. This study confirms that 3D bioprinted cellular models more faithfully reproduce human disease biology than 2D systems, offering a robust platform for high-throughput multiomics studies and drug discovery.
3.5.3 Electospun scaffolds
In addition to hydrogels, cryogenic electrospinning has recently emerged as a powerful method for creating 3D tissue models. This technique involves electrospinning fibers onto a cold drum (−30 °C), where water vapor forms ice crystals that serve as a temporary void template. Lyophilisation removes the ice, leaving a loosely packed fiber structure. The resulting scaffold mimics native tissue mechanics and cell distribution, offering a more physiologically relevant environment for studying processes like valve calcification.
In 2022, Stadelmann et al. developed a bi-layered cryogenic electrospun scaffold using polylactic acid, a biodegradable, biocompatible polymer commonly used in tissue engineering (142). The technique produced fibers ranging from nanometers to micrometers, forming two layers: a bottom infiltration layer (IL) with large pores, mimicking the spongiosa and promoting VIC infiltration, and the top non-infiltration layer (n-IL) with dense nanofibers, replicating the fibrosa and supporting VEC adhesion. Both layers were biofunctionalized with ECM proteins (FN, laminin, collagen type I, and fibrin) to support cell-specific needs. In co-culture, VICs were seeded on the IL and VECs on the n-IL, with cultures maintained for 3–6 days and medium changes every 2–3 days. This scaffold showed excellent stability for up to 4 weeks. VICs adhered, maintained viable, migrated into the IL, and displayed fibroblast-like morphology. VECs retained a cobblestone morphology and stayed on the surface of the n-IL. By day 6, both cell types formed confluent layers and expressed appropriate adhesion and cell-contact markers, with only minor variations compared to monocultures. Under osteogenic stimulation, VICs formed calcific nodules and upregulated markers like RUNX2, highlighting the model's suitability for studying VIC osteogenic transition and calcification. This bilayer scaffold, which is compatible with perfused and dynamic systems, provides a promising platform for studying both early and long-term mechanisms of the disease.
4 Ex vivo modelling of CAVD
While in vitro models have advanced our understanding of CAVD, they fall short of replicating the multicellular and matrix complexity of the AV. Ex vivo AV cultures provide a valuable intermediate, preserving native architecture while eliminating systemic variables inherent to in vivo models.
4.1 Porcine models
Most ex vivo models use porcine AV due to their structural and compositional similarities to human valves, their healthy state, and their larger size, which facilitates gene and protein expression analysis. Porcine AV mineralization can be induced ex vivo by culturing AVs for at least 8 days in medium supplemented with 3.8 mM Pi. The addition of inorganic pyrophosphatase, which degrades PPi, further enhances mineralization (145). These culture conditions preserve the structural integrity of the valves and maintain cell viability, supporting the model's relevance. This model was used in 2021 to evaluate whether etidronate, a PPi analogue, could prevent mineralization. The study demonstrated that etidronate effectively inhibited mineralization, confirming the model's suitability for pharmacological screening (146). In 2014, Rodriguez et al. used porcine AV cultured ex vivo to evaluate the role played by the ECM in VIC physiology. To do so, they enzymatically degraded leaflet collagen and studied the impact on VIC phenotype. They observed that ECM disruption triggers VIC proliferation, apoptosis, and the expression of markers like α-SMA, ALP, and OCN, all associated with increased calcification (147).
A key limitation of ex vivo models is maintaining tissue viability for extended periods, which is often challenged by limited nutrient diffusion, ECM degradation, and leaflet contraction caused by myofibroblast activation. This is why, in 2020, Zabirnyk et al. chose to optimize the model by culturing porcine leaflets in an anti-myofibroblastic medium. They demonstrated that this medium preserved tissue structure and prevented the typical ball-like contraction observed with standard media (148), making it suitable to study β-GP-induced mineralization. Using this model, they showed that SNF472, an agent inhibiting the formation and growth of hydroxyapatite crystals, effectively prevents AV mineralization.
4.2 Other models (human, ovine and murine)
While porcine AVs remain the standard for ex vivo culture, human and ovine valves can also be used. As with porcine models, their structure is preserved in antimyofibroblastic media, and mineralization can be induced with OM and reversed pharmacologically, highlighting their potential for drug testing (149, 150). To our knowledge, no standardized model currently exists for studying the remodeling of rodent AV cultured ex vivo under static conditions. This may be related to the small size of murine AV, which complicates dissection, culture, and downstream gene and protein analyses, thereby limiting their utility for mechanistic studies. Nevertheless, developing such models would be useful for mechanistic investigations and the screening of new therapeutic molecules. The study by Jenke et al., published in 2020, provides an elegant example of how ex vivo models can advance our understanding of CAVD pathophysiology (151). In this study, the authors exposed ovine AV leaflets, mounted and cultured under passive tension on synthetic rubber rings, to TGF-β1, in the presence or absence of OM. Using this protocol, they showed that in a 3D leaflet model, TGF-β1 completely suppresses OM-induced mineralization while promoting fibrosis. This effect was associated with downregulation of osteocalcin and ALP expression and upregulation of α-SMA and collagen I expression. By contrast, they reported that in ovine VICs cultured in 2D, TGF-β1 promoted calcification, demonstrating that cellular responses differ profoundly between 2D cultures and native-like 3D tissue environment.
5 Replicating mechanical and hemodynamic conditions
The AV is a dynamic structure that opens and closes each cardiac cycle, exposed to a complex mechanical environment involving cyclic stretch, bending, pressure, and shear stress. Alterations in these forces contribute to the onset and progression of AV dysfunction and remodeling. While static in vitro and ex vivo models have advanced our understanding of CAVD, they fail to replicate physiological mechanical and hemodynamic conditions. To address this, researchers have developed preclinical systems allowing investigation of mechanical and hemodynamic influences on valvular cells and tissues (Table 3).
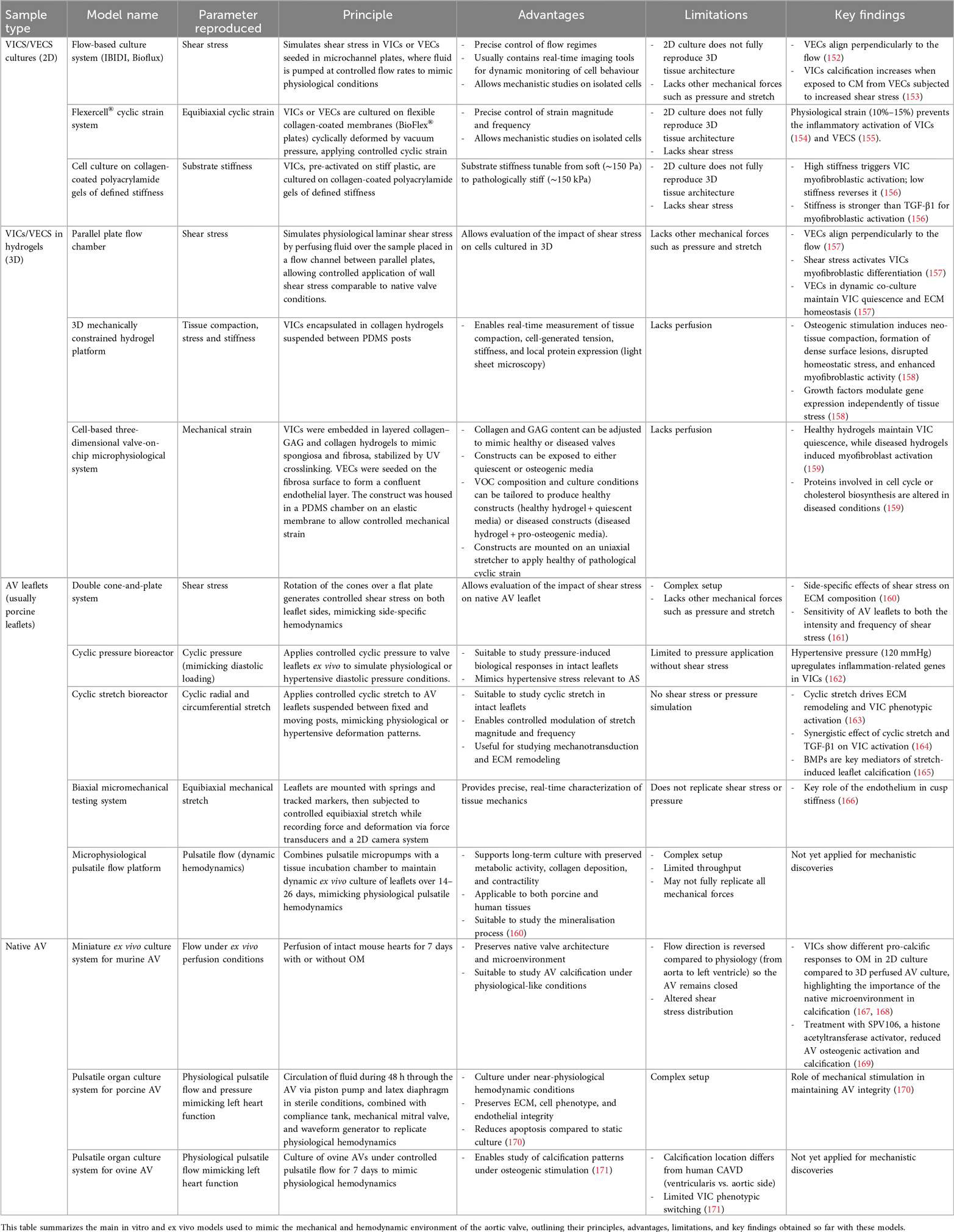
Table 3. Experimental platforms to reproduce aortic valve mechanical and flow conditions in vitro and ex vivo.
5.1 In vitro approaches
To simulate hemodynamic forces, especially shear stress, on 2D cultures of VICs and VECs, flow-based culture systems such as Bioflux (Fluxion Biosciences) and IBIDI (IBIDI GmbH), have been developed. These platforms use microchannel plates through which fluid is pumped at controlled flow rates, generating shear stress that mimics physiological conditions. They offer precise control over flow regimes and often integrate real time imaging tools for dynamic monitoring of cell behavior. This enables investigation into how mechanical forces affect cell proliferation, apoptosis, differentiation, and response to pathophysiological stimuli.
Using this type of system, Butcher et al. showed that aortic endothelial cells exposed to a steady shear stress of 20 dynes/cm2—approximating average physiological levels (172)—aligned parallel to the direction of flow, while VECs aligned perpendicularly, revealing distinct mechanical phenotypes adapted to their respective function (152). Transcriptomic analyses further confirmed that VECs differ from aortic endothelial cells, notably in their higher proliferative capacity (173). More recently, Hsu et al. used this system to investigate how shear stress (1 dyne/cm2—mimicking an atherogenic environment) affects VIC calcification (153). To do so, VECs were exposed for 48 h to static, steady, or oscillatory flow (0.25 or 0.5 OSI), and their CM were then added to VICs cultured in an OM. They found that VIC calcification was significantly increased in the 0.5 OSI group, highlighting the pro-calcific effect of oscillatory shear stress. To increase system complexity, Butcher et al. developed 3D collagen hydrogel-based AV models to study VEC/VIC interactions under flow (157). One model contained only VICs, while the other included a VEC layer on the luminal surface. Both were subjected to 20 dynes/cm2 shear stress for 96 h. As in 2D cultures, VECs aligned perpendicularly to flow. Shear stress activated VICs myofibroblastic differentiation. In this dynamic co-culture model, VECs reduced VIC proliferation, preserved GAGs, and enhanced protein synthesis, promoting a quiescent VIC phenotype and maintaining matrix homeostasis.
In 2012, Quinlan et al. developed a high-throughput system using collagen-coated polyacrylamide gels with defined stiffness values ranging from very soft (∼150 Pa) to levels exceeding those of diseased aortic valves (∼150 kPa), to study how substrate stiffness influences VIC behavior in 2D (156). Porcine VICs, pre-activated on stiff plastic, were seeded on these substrates and cultured with or without TGF-β1, and cell spreading, morphology, and αSMA expression were quantified. They found that increasing substrate stiffness led to larger cell spread area, more elongated morphology, and a higher fraction of VICs displaying pronounced αSMA-positive stress fibers, indicative of myofibroblastic activation. By contrast, relatively low stiffness levels were sufficient to “deactivate” VICs. Although TGF-β1 slightly modulated αSMA expression, substrate stiffness was the dominant cue driving myofibroblastic activation. This study highlights the importance of controlling substrate stiffness in in vitro models of valve physiology and tissue engineering.
In addition, it is also possible to reproduce in vitro the mechanical strains experienced by AV leaflets to study their impact on VICs. Using the Flexercell® system, Smith et al. cultured VICs on flexible collagen-coated membranes (BioFlex® plates) that can be cyclically deformed by vacuum pressure (154). This setup applies controlled, cyclic strain (e.g., 5%–20% at 1 Hz), mimicking physiological or pathological mechanical loading, and allows investigation of downstream effects on gene expression, protein production, and calcification. Using this approach, they showed that applying physiological cyclic strain (15%) to porcine VICs reduced inflammation, as evidenced by decreased expression of MCP-1, VCAM-1, and GM-CSF (154). The system also demonstrated that 10% cyclic strain preserved the integrity of porcine VEC monolayers and limited pro-inflammatory protein expression, whereas both lower (5%) and higher (20%) strain levels were detrimental (155).
In 2024, Shih et al. developed a compact 3D mechanically constrained platform to study how VICs remodel their ECM in response to biochemical and biomechanical cues (158). To do so, they encapsulated VICs in collagen hydrogels suspended between polydimethylsiloxane (PDMS) posts, allowing real-time measurements of tissue compaction, stress (the tension generated by cell contraction and matrix remodelling on the posts), and stiffness, as well as visualization of local protein expression via light sheet microscopy. Osteogenic stimulation induced accelerated neo-tissue compaction, formation of dense surface lesions, and disrupted homeostatic stress levels. Both porcine and human VICs exhibited increased myofibroblastic activity (ACTA2, TGFB1, CNN1) under osteogenic conditions, with protein expression localized in banding patterns at the neo-tissue surface and positively correlated with mechanical stress. The addition of growth factors (EGF and FGF) modulated gene expression independently of tissue stress, demonstrating that differentiation can be biochemically altered without changing mechanical state. This platform, which allows simultaneous analysis of mechanical and molecular drivers of pathological remodeling, offers a high-throughput biologically relevant system to study CAVD mechanisms and potential pharmacological interventions.
In recent years, the field of preclinical modeling has been transformed by the emergence of organ-on-chip technology. An organ-on-chip is a microfluidic device that replicates the structure and function of a human organ by integrating living cells within a controlled, dynamic environment, enabling the study of physiology and disease in vitro. To date, few of these microfluidic models have been developed to replicate the AV physiology. In 2024, Tandon et al. developed what currently represents the most advanced cell-based three-dimensional valve-on-chip (VOC) microphysiological system (159). The VOC platform consisted of a rectangular PDMS chamber mounted on an elastic membrane, which allowed the application of controlled mechanical strain. Within this chamber, the valvular construct was formed by sequentially assembling hydrogel layers to reproduce the valve's native architecture. Quiescent pVICs (1.5 × 106 cells/ml) were first embedded in a collagen–GAG hydrogel to form a spongiosa-like layer and allowed to gel overnight. A second collagen-only layer containing quiescent pVICs was then added to mimic the fibrosa and left to set for another night. After polymerization, the bilayer construct was UV-crosslinked for 1 min to stabilize the matrix. pVECs were then seeded on the fibrosa surface (300,000 cells/cm2) and cultured for 48 h to form a confluent endothelial monolayer. Healthy and diseased valve compositions were simulated by varying the collagen and GAG content. Healthy constructs contained ∼90% collagen with GAGs restricted to the spongiosa layer, whereas diseased constructs contained 50% collagen and fourfold higher GAG concentrations, reproducing the ECM remodeling characteristic of CAVD. Once assembled, the VOCs were cultured in either quiescent or pro-osteogenic medium (supplemented with β-glycerophosphate, dexamethasone, ascorbic acid, and TGF-β1) to induce calcification. The complete construct was then mounted on a custom uniaxial stretcher applying 10% cyclic strain for 48 h, reproducing physiological mechanical forces and enabling the study of dynamic 3D valve responses under healthy and disease-like conditions. While the healthy hydrogel promoted vimentin expression, maintaining VICs in a quiescent phenotype, the diseased hydrogel induced VIC activation into a myofibroblast-like phenotype, as evidenced by positive αSMA expression. Proteins involved in cellular processes such as cell cycle progression, cholesterol biosynthesis, and protein homeostasis were significantly altered and correlated with metabolic changes in diseased VOCs, suggesting that these constructs may serve as valuable tools to study the early, adaptive stages of disease initiation.
5.2 Ex vivo approaches on isolated leaflets
5.2.1 Assessing the impact of shear stress
The biological response of AV leaflets to controlled shear stress can be assessed by a cone-and-plate system. This setup, composed of a flat plate and a rotating cone positioned just above it, generates well-defined shear forces by moving fluid between the two surfaces and allows for the application of steady or pulsatile flow while minimizing secondary flow artefacts (174). A more advanced version—the double cone-and-plate system—allows simultaneous exposure of both leaflet surfaces (aortic and ventricular), better replicating in vivo side-specific hemodynamics. In 2018, Mongkoldhumrongkul et al. used this system to study the effect of side-specific flow on ECM dynamics in porcine AV leaflets (160). Leaflets were subjected to oscillatory or laminar flow for 48 h. Laminar flow promoted elastin synthesis on both sides, while oscillary flow increased collagen and GAG content specifically on the aortic side, underscoring the importance of side-dependent flow in ECM regulation. In 2013, Sun et al. used a double cone-and-plate system to evaluate the sensitivity of AV leaflets to both the intensity and frequency of shear stress, and their role in initiating CAVD-related remodeling (161). They reported that elevated shear magnitude at normal frequency increased BMP-4 and TGF-β1 expression and triggered ECM degradation in porcine AVs. Abnormal frequency at physiological magnitude also induced matrix remodeling. The most pathological response was observed under sustained supra-physiologic shear, peaking at 48 h and persisting through 72 h.
5.2.2 Assessing the impact of pressure
Hypertension is associated with AS in vivo (175). To mimic hypertensive conditions ex-vivo, AVs can be cultured ex vivo in specialized pressure chambers (176). In 2011, Warnock et al. cultured porcine AV leaflets for 24 h under cyclic pressures of 80 mmHg (physiological) or 120 mmHg (hypertensive) in a pressure chamber mimicking diastolic loading (162). Transcriptomic analysis revealed 56 inflammation-related genes differentially expressed under hypertensive condition, including TNF-α, IL-1α, IL-1β, and a striking 41-fold upregulation of PTX3. These findings suggest that pressure-induced activation of inflammatory pathways in VICs could represent a potential therapeutic target in hypertensive AS.
5.2.3 Biomechanical testing systems (stretch, strain and stiffness)
Under physiological conditions, AV leaflets experience ∼10% circumferential and up to 30% radial stretch, which can increase under elevated pressure. In 2006, Balachandran et al. used an ex vivo bioreactor with two chambers containing fixed and moving posts to apply controlled cyclic stretch via an actuator. Porcine AV leaflets were suspended between the posts and subjected to 15% stretch for 48 h. Compared to fresh or statically cultured leaflets, stretched tissues exhibited increased collagen, reduced GAGs and elevated α-SMA expression, indicating a contractile, fibrotic VIC phenotype. These findings suggest that cyclic stretch drives ECM remodeling and VIC phenotypic activation (163). Using the same setup, Merryman et al. demonstrated a synergistic effect of cyclic stretch and TGF-β1 on VIC activation (164). In 2010, Balachandran et al. showed that the induction of a pathological stretch in porcine leaflets cultured in an OM promoted apoptosis, ALP activity and calcification, along with early upregulation of BMP-2, BMP-4, and Runx2 on the fibrosa surface. In this model, inhibition of BMP signaling dose-dependently reduced calcification and ALP levels, highlighting BMPs as key mediators of stretch-induced AV calcification (165). In 2009, El-Hamamsy et al. used a biaxial micromechanical testing system to investigate how the valve endothelium regulates aortic cusp mechanics. Valve samples were mounted in a Krebs bath at 37 °C with continuous O2/CO2 gassing to mimic physiological conditions. Stainless steel springs were threaded through each side of the cusp to preserve endothelial integrity, and four markers tracked deformation during equibiaxial stretching. Strain was measured using force transducers and a 2D camera system. Serotonin (5-HT) reduced cusps stiffness by 25%, an effect reversed by endothelial removal or L-NAME (a nitric oxide synthase inhibitor). In contrast, endothelin-1 increased stiffness by 34%, which was blocked by cytochalasin-B (an actin polymerization inhibitor). These findings underscore the endothelium's key role in modulating mechanical properties essential for valve function (166).
5.2.4 Toward organ-on-chips
In 2023, Dittfeld et al. developed a microphysiological platform integrating pulsatile micropumps with a tissue incubation chamber to enable long-term ex vivo culture of porcine and human AV tissues. When cultured in this device for 14–26 days under dynamic pulsatile conditions, the tissue displayed increased metabolic activity, collagen deposition, and contractility, which are characteristic of early stages of CAVD. GAGs, endothelial and smooth muscle markers, and calcium deposition remained stable (160), supporting the relevance of this platform for preclinical studies.
5.3 Reproducing hemodynamics in native AV
While earlier models focused on isolated leaflets, recent advances have enabled ex vivo culture of whole native murine, porcine, or ovine AVs under flow conditions mimicking pathophysiological states. In 2021, Kruithof et al. established an ex vivo calcification model for intact wild-type murine AV using a miniature tissue culture system. To do so, they perfused mouse hearts for 7 days, with or without OM. In this model, calcification occurred exclusively when the AV leaflets were cultured in an OM supplemented with Pi, whereas treatment with a cocktail composed of β-GP, ascorbic acid, and dexamethasone did not induce calcification. By contrast, they observed that murine VICs cultured in vitro exhibited calcification under both Pi and the β-GP–ascorbic acid–dexamethasone cocktail. This study revealed that significant disparities exist between in vitro and ex vivo responses of VICs, highlighting the added value of investigating CAVD in cells embedded within their native microenvironment (167, 168). However, in this setup, flow was reversed compared to physiological conditions (i.e., directed from the aorta to the left ventricle), keeping the valve closed and continuously exposing the aortic side to hemodynamic stress, which represents a limitation. In 2025, Garoffolo et al. used this model to show that SPV106, a histone acetyltransferase activator, significantly reduced AV calcification and osteogenic marker expression (ALP, RUNX1/2/3), demonstrating the model's utility for preclinical evaluation of anti-calcific strategies (169).
Interestingly, whole AV culture under flow has also been investigated in larger animal models. Konduri et al. cultured native porcine AVs for 48 h in a sterile, pulsatile organ culture system simulating physiological conditions (120/80 mmHg, 4.2 L/min). A piston pump circulated fluid through the valve, separated by a latex diaphragm to maintain sterility. The system replicated left heart function using a compliance tank, mechanical mitral valve and programmable waveform generator. Continuous monitoring of flow and pressure ensured physiological accuracy. Compared to fresh valves, cultured tissues retained ECM composition (collagen, GAGs, elastin), leaflet morphology, and cell phenotype. Endothelial integrity was preserved, and apoptosis levels remained low—comparable to fresh tissue and significantly lower than in static culture—highlighting the crucial role of mechanical stimulation in maintaining valve viability (170). Whole AV culture under flow was also assessed by Niazy et al. using ovine samples. In their system, native ovine AVs were cultured for 7 days in a bioreactor providing pulsatile flow with controlled pressure, temperature, and pH (171). Flow passed from the ventricular to the aortic side. Under dynamic conditions, type I collagen expression was maintained, unlike in static culture. Interestingly, exposure to an OM induced marked calcification in the ventricularis, with smaller deposits in the fibrosa, whereas in human CAVD, calcification typically occurs on the aortic side of the leaflet, rather than in the fibrosa. This suggests that despite pulsatile flow, shear stress distribution may not fully replicate native patterns in this model. Besides, OM exposure did not alter COL1A1, ALP, or αSMA expression, indicating limited VICs phenotypic switching in this model.
6 Assessing mineralization
Accurate assessment of mineralization is essential for characterizing VIC and VEC osteogenic activity and evaluating the effects of therapeutic interventions. A wide range of techniques has been developed and adapted to monitor calcification across experimental settings, including cells cultured in 2D or 3D, as well as tissue explants. These methods differ in sensitivity, specificity, quantification capacity, and applicability to in vitro or ex vivo studies. An overview of the main techniques—including Von Kossa staining, Alizarin Red staining, OsteoSense, o-cresolphthalein complexone, and ⁴⁵Ca liquid scintillation counting—along with their principles, quantification potential, and limitations, is presented in Table 4.
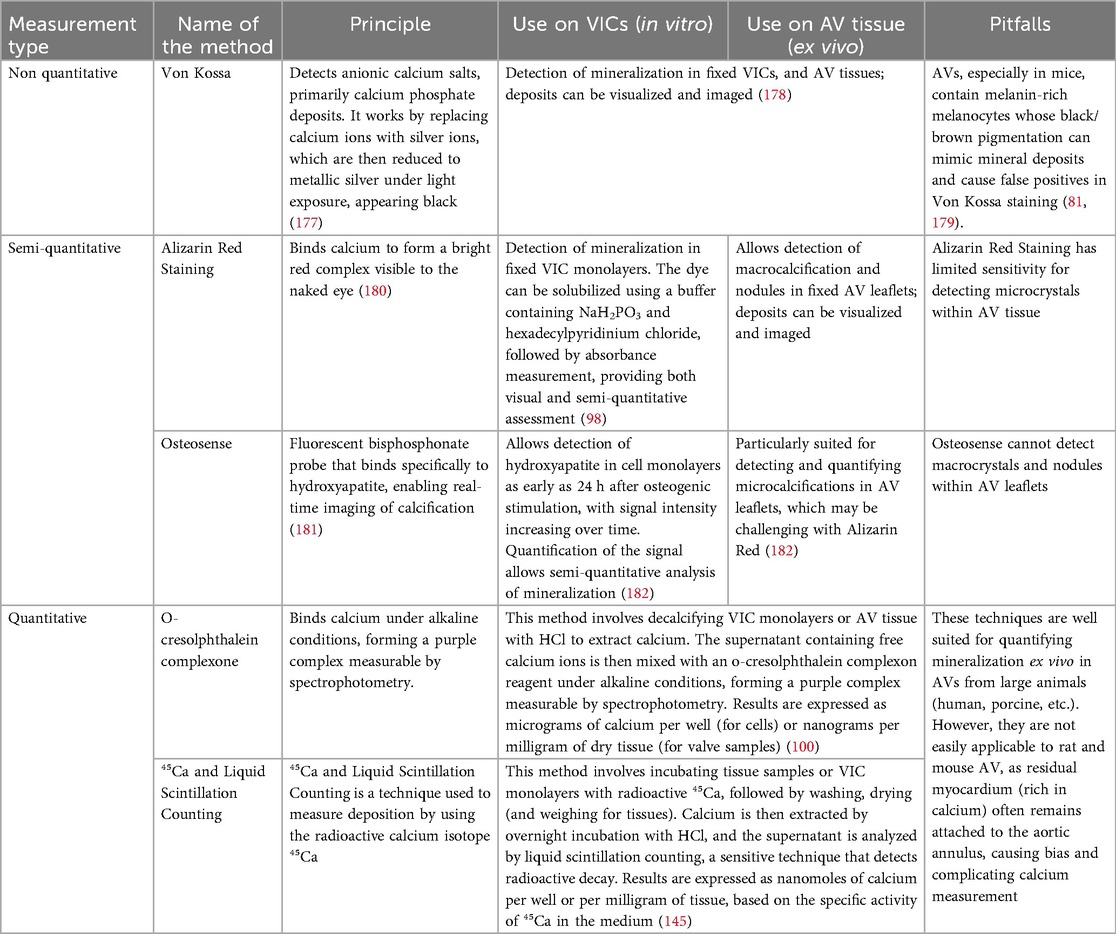
Table 4. Overview of the main techniques used to detect and quantify calcification in vitro and ex vivo, with emphasis on their specificity, sensitivity, and limitations depending on the experimental model.
7 In vivo modeling of CAVD
Unlike in vitro models, which isolate cellular mechanisms, or ex vivo models, which lack systemic interactions, in vivo studies capture the full complexity of the disease within an integrated biological system. These models, which allow the study of CAVD in the presence of key comorbidities such as hypercholesterolemia, diabetes, and CKD, are essential for developing effective therapies. They also provide insights into the progressive nature of AV fibrocalcic remodeling and pathological changes that short-term experiments may miss, and allow evaluation of the functional impact of AV dysfunction on hemodynamic parameters and ventricular remodeling and function via echocardiography. These in vivo models are essential for evaluating the efficacy and safety of potential therapies, offering a physiologically relevant platform for testing pharmacological and interventional strategies before clinical trials.
7.1 Overview of main models
Numerous animal models have been developed to replicate the pathological conditions leading to CAVD. These models account for key risk factors and comorbidities, enabling the investigation of mechanisms such as lipid metabolism, inflammation, hemodynamics, and genetic predispositions. They can be naturally occurring, genetically engineered, diet- or surgically induced, or triggered by pharmacological agents. An overview of these models and their characteristics is provided in Table 5.
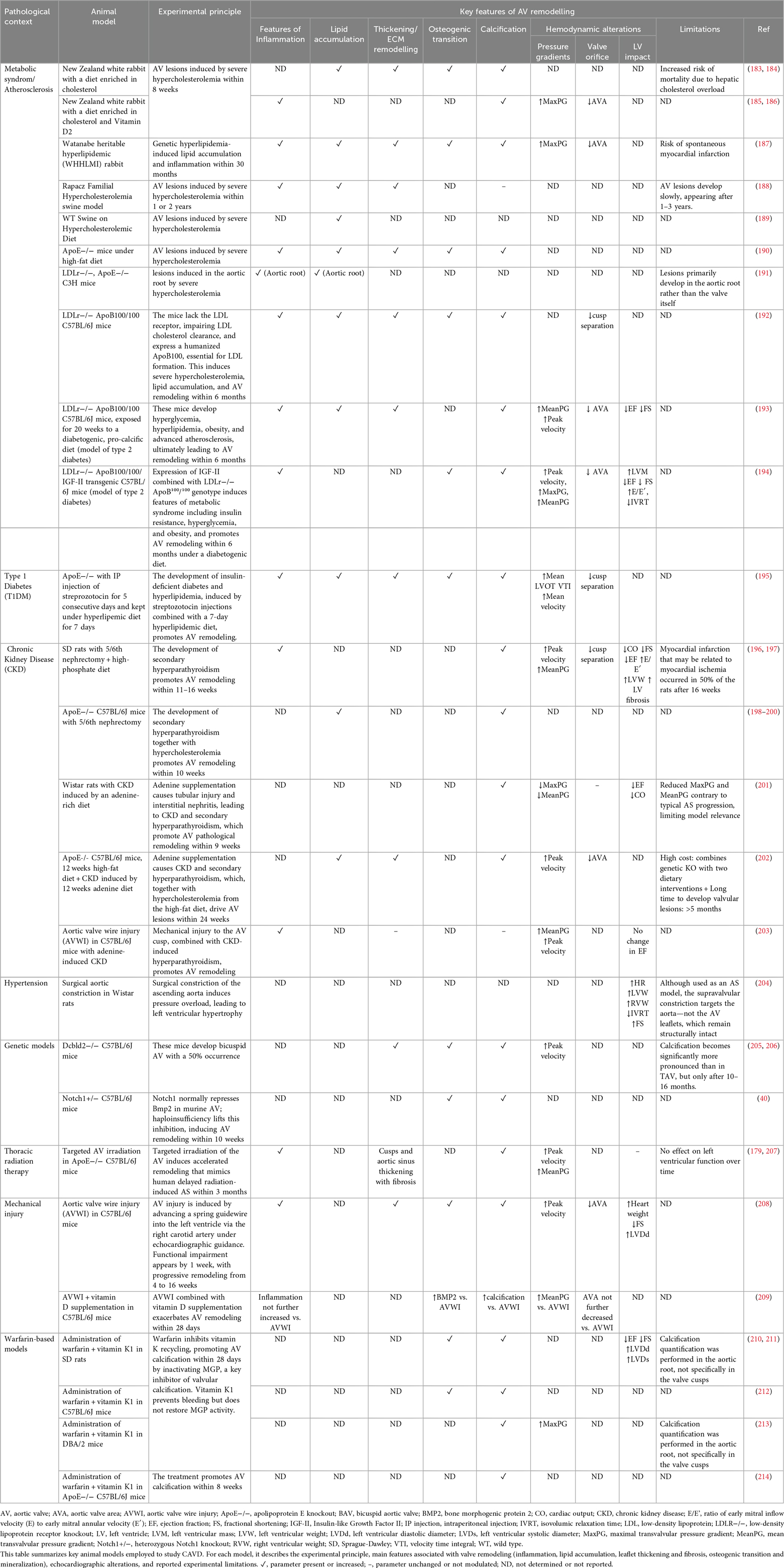
Table 5. Overview of the main animal models currently used to investigate CAVD pathophysiology.
7.2 Relevance of in vivo models
Several critical factors must be considered when using animal models, starting with species selection. Porcine models are anatomically and hemodynamically close to humans, making them highly relevant for CAVD research. However, their use entails high costs, specialized housing, and often limits sample sizes, impacting statistical power. Rodents, widely used for their availability and genetic manipulability, differ fundamentally from humans in key aspects. For example, mice have lymphocyte-dominant white blood cells, whereas humans and pigs have neutrophil-dominant profiles, influencing immune responses (215, 216). Moreover, certain human cytokines relevant to CAVD, such as IL-8 (100), are not expressed in mice or rats, limiting the translational potential of these models. Rodent models also pose technical challenges: their tiny AV yield limited tissue for molecular analyses (e.g., Western blot, PCR), and valve isolation without myocardial contamination is difficult. Nevertheless, they remain valuable for studying cardiovascular calcification in vivo, including through Osteosense injection to visualize global cardiovascular calcification. Regardless of species, in vivo studies increasingly rely on functional imaging such as echocardiography, enabling clinically translatable assessment of hemodynamic and functional parameters. This underscores the importance of interdisciplinary collaboration with clinicians and imaging specialists to ensure accurate and meaningful data interpretation. Despite significant advances, Table 5 shows that no animal model to date fully recapitulates the complex pathophysiology of CAVD—including fibrosis, lipid accumulation, calcification, elevated transvalvular gradients, valve narrowing, and left ventricular remodeling. As a result, we still lack a reliable preclinical model for testing therapeutic interventions aimed at preventing the onset or halting the progression of AS, despite considerable research efforts in this field. Developing such a model remains a critical need to improve our understanding of disease mechanisms and to enable robust therapeutic screening. To develop reliable models of valvular calcification, critical details—such as animal strain, sex, and diet composition—should be systematically reported in every published study. To this end, researchers are encouraged to follow the ARRIVE guidelines, which provide a comprehensive checklist of essential information for in vivo studies. Adherence to these guidelines allows evaluation of methodological rigor, facilitates experiment reproducibility, and ensures accurate interpretation of results. It also promotes complete reporting of experimental design, randomization, blinding, sample size justification, and outcome measures, thereby enhancing study quality and transparency (217). A more standardized approach will not only enhance reporting consistency but also aid in selecting appropriate methodologies, ultimately accelerating the identification of suitable animal models for CAVD.
8 Conclusion
Although no model fully replicates the native AV environment and the mechanisms driving CAVD, significant efforts have been made to develop robust experimental systems (Figure 3). Two-dimensional in vitro models offer precise control of the cellular environment, making them useful for studying signaling pathways and cell–cell interactions. However, they fail to mimic the native valve's 3D architecture and cell–matrix interactions. Three-dimensional models—based on aggregates, scaffolds, leaflet fragments, or whole aortic valves—better address these limitations. The incorporation of hemodynamic flow and mechanical forces into these in vitro and ex vivo systems represents a major advance. Despite these improvements, in vivo models remain essential for studying CAVD progression within an integrated biological context. In this regard, the development of a zebrafish model, which would enable high-throughput compound screening, is highly anticipated (218, 219).
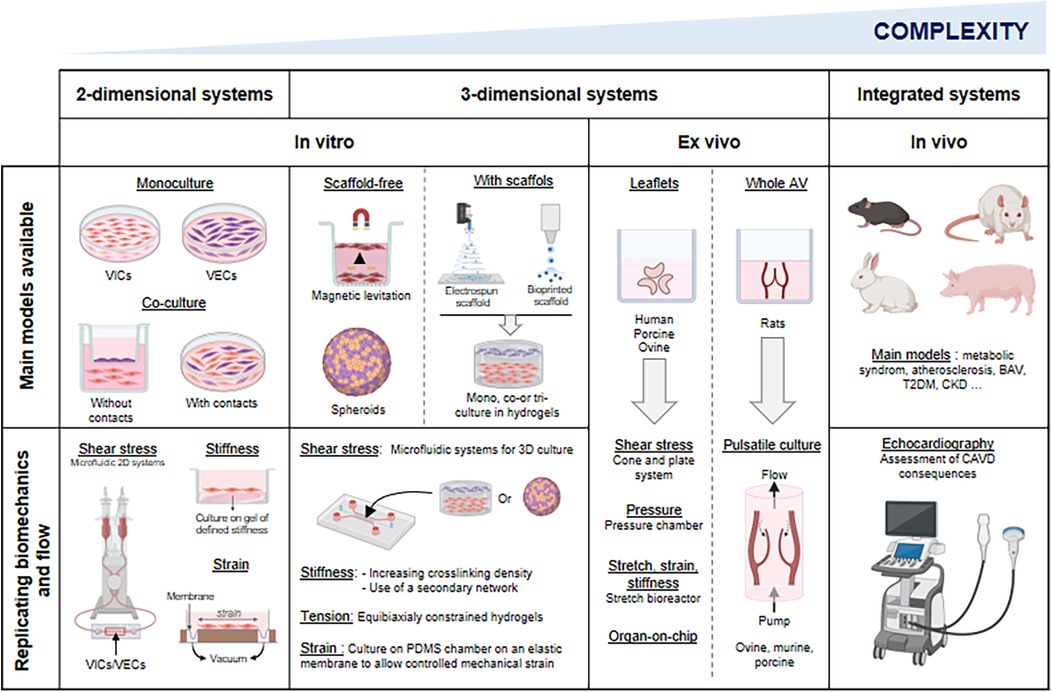
Figure 3. Overview of the main models currently available to study CAVD. In this figure, models are organized by increasing complexity and physiological relevance from left to right. The upper row illustrates static models, where mechanical and hemodynamic stimuli are absent. The lower row presents dynamic models, incorporating mechanical forces and flow conditions to better mimic the physiological environment of the aortic valve. This figure highlights the progressive refinement of experimental systems, from simple 2D in vitro cultures to complex in vivo models, to guide model selection based on research objectives. Illustration created with BioRender.
When modeling CAVD, it is important to keep in mind that each model provides only a partial perspective on disease mechanisms; thus, combining complementary approaches is often the most effective strategy for addressing complex research questions. However, the multifactorial nature of CAVD, which often requires a focus on specific risk factors, continues to limit the generalizability of findings and remains a major obstacle to fully replicate the disease. Among these factors, BAV represents a major risk for early-onset and accelerated CAVD. Yet, few preclinical systems have been specifically developed to modelize this morphological variant. Indeed, the vast majority of existing models rely on tricuspid valve anatomy, limiting their ability to reflect the unique hemodynamic and mechanical conditions associated with BAV. This remains an important gap in the field and underscores the need for future models tailored to this clinically significant phenotype. In the future, integrating spatial transcriptomics, scRNA-seq, and other high-resolution techniques into studies of bicuspid and tricuspid valves, as well as key comorbidities, is expected to enhance our understanding of the mechanisms driving AS. The standardization of trilayered in-flow organoid systems and AV-on-chip platforms still remain key challenges for the coming years and could significantly enhance translational potential.
Author contributions
IL: Conceptualization, Writing – review & editing, Writing – original draft. RM: Writing – review & editing. CA: Writing – review & editing. YB: Writing – review & editing. CT: Writing – review & editing. LH: Writing – original draft, Writing – review & editing, Conceptualization.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
The authors are grateful to the Fédération Hospitalo-Universitaire program “CArdiac Research Network on Aortic VAlve and heart faiLure” (GCS G4 FHU CARNAVAL) and to the Investissements d'Avenir program (ANR-16-RHUS-0003_STOP-AS, managed by the French National Research Agency), for supporting this work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. Basic AI tools were used to evaluate English grammar.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Osnabrugge RLJ, Mylotte D, Head SJ, Van Mieghem NM, Nkomo VT, LeReun CM, et al. Aortic stenosis in the elderly: disease prevalence and number of candidates for transcatheter aortic valve replacement: a meta-analysis and modeling study. J Am Coll Cardiol. (2013) 62(11):1002–12. doi: 10.1016/j.jacc.2013.05.015
2. Danielsen R, Aspelund T, Harris TB, Gudnason V. The prevalence of aortic stenosis in the elderly in Iceland and predictions for the coming decades: the AGES-reykjavík study. Int J Cardiol. (2014) 176(3):916–22. doi: 10.1016/j.ijcard.2014.08.053
3. Tastet L, Shen M, Capoulade R, Arsenault M, Bédard É, Ternacle J, et al. Sex differences in the progression of aortic valve calcification and clinical outcomes: the PROGRESSA study. JACC Cardiovasc Imaging. (2022) 15(7):1349–51. doi: 10.1016/j.jcmg.2022.02.015
4. Pawade T, Sheth T, Guzzetti E, Dweck MR, Clavel MA. Why and how to measure aortic valve calcification in patients with aortic stenosis. JACC Cardiovasc Imaging. (2019) 12(9):1835–48. doi: 10.1016/j.jcmg.2019.01.045
5. Pawade T, Clavel MA, Tribouilloy C, Dreyfus J, Mathieu T, Tastet L, et al. Computed tomography aortic valve calcium scoring in patients with aortic stenosis. Circ Cardiovasc Imaging. (2018) 11(3):e007146. doi: 10.1161/CIRCIMAGING.117.007146
6. Vahanian A, Beyersdorf F, Praz F, Milojevic M, Baldus S, Bauersachs J, et al. 2021 ESC/EACTS guidelines for the management of valvular heart disease. G Ital Cardiol (Rome). (2022) 23(5 Suppl 1):e1–75. doi: 10.1714/3787.37722
7. Huang B, Yan H, Li Y, Zhou Q, Abudoureyimu A, Cao G, et al. Transcatheter aortic valve replacement in elderly patients: opportunities and challenges. J Cardiovasc Dev Dis. (2023) 10(7):279. doi: 10.3390/jcdd10070279
8. Lauck SB, Baron SJ, Irish W, Borregaard B, Moore KA, Gunnarsson CL, et al. Temporal changes in mortality after transcatheter and surgical aortic valve replacement: retrospective analysis of US medicare patients (2012–2019). J Am Heart Assoc. (2021) 10(20):e021748. doi: 10.1161/JAHA.120.021748
9. Rutkovskiy A, Malashicheva A, Sullivan G, Bogdanova M, Kostareva A, Stensløkken KO, et al. Valve interstitial cells: the key to understanding the pathophysiology of heart valve calcification. J Am Heart Assoc. (2017) 6(9):e006339. doi: 10.1161/JAHA.117.006339
10. Dweck MR, Boon NA, Newby DE. Calcific aortic stenosis: a disease of the valve and the myocardium. J Am Coll Cardiol. (2012) 60(19):1854–63. doi: 10.1016/j.jacc.2012.02.093
11. Leopold JA. Cellular mechanisms of aortic valve calcification. Circ Cardiovasc Interv. (2012) 5(4):605–14. doi: 10.1161/CIRCINTERVENTIONS.112.971028
12. Sathyamurthy I, Alex S. Calcific aortic valve disease: is it another face of atherosclerosis? Indian Heart J. (2015) 67(5):503–6. doi: 10.1016/j.ihj.2015.07.033
13. Bogdanova M, Zabirnyk A, Malashicheva A, Semenova D, Kvitting JPE, Kaljusto ML, et al. Models and techniques to study aortic valve calcification in vitro, ex vivo and in vivo. An overview. Front Pharmacol. (2022) 13:835825. doi: 10.3389/fphar.2022.835825
14. Ma X, Zhao D, Yuan P, Li J, Yun Y, Cui Y, et al. Endothelial-to-Mesenchymal transition in calcific aortic valve disease. Acta Cardiol Sin. (2020) 36(3):183–94. doi: 10.6515/ACS.202005_36(3).20200213A
15. Mahler GJ, Farrar EJ, Butcher JT. Inflammatory cytokines promote mesenchymal transformation in embryonic and adult valve endothelial cells. Arterioscler Thromb Vasc Biol. (2012) 33(1):121. doi: 10.1161/ATVBAHA.112.300504
16. Krenning G, Barauna VG, Krieger JE, Harmsen MC, Moonen JRAJ. Endothelial plasticity: shifting phenotypes through force feedback. Stem Cells Int. (2016) 2016:9762959. doi: 10.1155/2016/9762959
17. Jackson AO, Zhang J, Jiang Z, Yin K. Endothelial-to-mesenchymal transition: a novel therapeutic target for cardiovascular diseases. Trends Cardiovasc Med. (2017) 27(6):383–93. doi: 10.1016/j.tcm.2017.03.003
18. Rattazzi M, Bertacco E, Iop L, D’Andrea S, Puato M, Buso G, et al. Extracellular pyrophosphate is reduced in aortic interstitial valve cells acquiring a calcifying profile: implications for aortic valve calcification. Atherosclerosis. (2014) 237(2):568–76. doi: 10.1016/j.atherosclerosis.2014.10.027
19. Kutikhin AG, Feenstra L, Kostyunin AE, Yuzhalin AE, Hillebrands JL, Krenning G. Calciprotein particles: balancing mineral homeostasis and vascular pathology. Arterioscler Thromb Vasc Biol. (2021) 41(5):1607–24. doi: 10.1161/ATVBAHA.120.315697
20. O’Young J, Liao Y, Xiao Y, Jalkanen J, Lajoie G, Karttunen M, et al. Matrix gla protein inhibits ectopic calcification by a direct interaction with hydroxyapatite crystals. J Am Chem Soc. (2011) 133(45):18406–12. doi: 10.1021/ja207628k
21. Heiss A, Pipich V, Jahnen-Dechent W, Schwahn D. Fetuin-A is a mineral carrier protein: small angle neutron scattering provides new insight on fetuin-A controlled calcification inhibition. Biophys J. (2010) 99(12):3986–95. doi: 10.1016/j.bpj.2010.10.030
22. Venardos N, Bennett D, Weyant MJ, Reece TB, Meng X, Fullerton DA. Matrix gla protein regulates calcification of the aortic valve. J Surg Res. (2015) 199(1):1–6. doi: 10.1016/j.jss.2015.04.076
23. Fiedler J, Park DH, Hobuß L, Anaraki PK, Pfanne A, Just A, et al. Identification of miR-143 as a Major contributor for human stenotic aortic valve disease. J Cardiovasc Transl Res. (2019) 12(5):447–58. doi: 10.1007/s12265-019-09880-7
24. Larik MO. Fetuin-A levels in association with calcific aortic valve disease: a meta-analysis. Atheroscler Plus. (2023) 54:27–9. doi: 10.1016/j.athplu.2023.09.004
25. Bertazzo S, Gentleman E, Cloyd KL, Chester AH, Yacoub MH, Stevens MM. Nano-analytical electron microscopy reveals fundamental insights into human cardiovascular tissue calcification. Nat Mater. (2013) 12(6):576–83. doi: 10.1038/nmat3627
26. Bouchareb R, Boulanger MC, Fournier D, Pibarot P, Messaddeq Y, Mathieu P. Mechanical strain induces the production of spheroid mineralized microparticles in the aortic valve through a RhoA/ROCK-dependent mechanism. J Mol Cell Cardiol. (2014) 67:49–59. doi: 10.1016/j.yjmcc.2013.12.009
27. Côté N, El Husseini D, Pépin A, Guauque-Olarte S, Ducharme V, Bouchard-Cannon P, et al. ATP Acts as a survival signal and prevents the mineralization of aortic valve. J Mol Cell Cardiol. (2012) 52(5):1191–202. doi: 10.1016/j.yjmcc.2012.02.003
28. Mahmut A, Boulanger MC, Bouchareb R, Hadji F, Mathieu P. Adenosine derived from ecto-nucleotidases in calcific aortic valve disease promotes mineralization through A2a adenosine receptor. Cardiovasc Res. (2015) 106(1):109–20. doi: 10.1093/cvr/cvv027
29. Mathieu P, Voisine P, Pépin A, Shetty R, Savard N, Dagenais F. Calcification of human valve interstitial cells is dependent on alkaline phosphatase activity. J Heart Valve Dis. (2005) 14(3):353–7.15974530
30. Wang D, Zeng Q, Song R, Ao L, Fullerton DA, Meng X. Ligation of ICAM-1 on human aortic valve interstitial cells induces the osteogenic response: a critical role of the Notch1-NF-κB pathway in BMP-2 expression. Biochim Biophys Acta. (2014) 1843(11):2744–53. doi: 10.1016/j.bbamcr.2014.07.017
31. Éva Sikura K, Combi Z, Potor L, Szerafin T, Hendrik Z, Méhes G, et al. Hydrogen sulfide inhibits aortic valve calcification in heart via regulating RUNX2 by NF-κB, a link between inflammation and mineralization. J Adv Res. (2021) 27:165–76. doi: 10.1016/j.jare.2020.07.005
32. Cuevas RA, Hortells L, Chu CC, Wong R, Crane A, Boufford CK, et al. Non-Canonical TERT activity initiates osteogenesis in calcific aortic valve disease. Circ Res. (2025) 136(4):403–21. doi: 10.1161/CIRCRESAHA.122.321889
33. Félix Vélez NE, Tu K, Guo P, Reeves RR, Aguado BA. Secreted cytokines from inflammatory macrophages modulate sex differences in valvular interstitial cells on hydrogel biomaterials. J Biomed Mater Res A. (2025) 113(3):e37885. doi: 10.1002/jbm.a.37885
34. Gonzalez Rodriguez A, Schroeder ME, Grim JC, Walker CJ, Speckl KF, Weiss RM, et al. Tumor necrosis factor-α promotes and exacerbates calcification in heart valve myofibroblast populations. FASEB J. (2021) 35(3):e21382. doi: 10.1096/fj.202002013RR
35. Deng XS, Meng X, Fullerton D, Stone M, Jaggers J. Complement upregulates runx-2 to induce profibrogenic change in aortic valve interstitial cells. Ann Thorac Surg. (2021) 112(6):1962–72. doi: 10.1016/j.athoracsur.2020.12.058
36. Deng XS, Meng X, Fullerton D, Stone M, Iguidbashian J, Jaggers J. Complement cross talks with H-K-ATPase to upregulate Runx2 in human aortic valve interstitial cells. J Surg Res. (2023) 286:118–26. doi: 10.1016/j.jss.2022.12.028
37. The E, de Graaf DM, Zhai Y, Yao Q, Ao L, Fullerton DA, et al. Interleukin 38 alleviates aortic valve calcification by inhibition of NLRP3. Proc Natl Acad Sci U S A. (2022) 119(36):e2202577119. doi: 10.1073/pnas.2202577119
38. Li SJ, Kao YH, Chung CC, Cheng WL, Chen YJ. HDAC I inhibitor regulates RUNX2 transactivation through canonical and non-canonical wnt signaling in aortic valvular interstitial cells. Am J Transl Res. (2019) 11(2):744–54.30899376
39. Caira FC, Stock SR, Gleason TG, McGee EC, Huang J, Bonow RO, et al. Human degenerative valve disease is associated with up-regulation of low-density lipoprotein receptor-related protein 5 receptor-mediated bone formation. J Am Coll Cardiol. (2006) 47(8):1707–12. doi: 10.1016/j.jacc.2006.02.040
40. Nigam V, Srivastava D. Notch1 represses osteogenic pathways in aortic valve cells. J Mol Cell Cardiol. (2009) 47(6):828–34. doi: 10.1016/j.yjmcc.2009.08.008
41. Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. (2005) 437(7056):270–4. doi: 10.1038/nature03940
42. Toshima T, Watanabe T, Narumi T, Otaki Y, Shishido T, Aono T, et al. Therapeutic inhibition of microRNA-34a ameliorates aortic valve calcification via modulation of Notch1-Runx2 signalling. Cardiovasc Res. (2020) 116(5):983–94. doi: 10.1093/cvr/cvz210
43. Krauze A, Procyk G, Gąsecka A, Garstka-Pacak I, Wrzosek M. The role of MicroRNAs in aortic stenosis-lessons from recent clinical research studies. Int J Mol Sci. (2023) 24(17):13095. doi: 10.3390/ijms241713095
44. Jiang Y, Ji W, Zhu J, Shen Z, Chen J. Upregulation of miR-664a-3p ameliorates calcific aortic valve disease by inhibiting the BMP2 signaling pathway. Dis Markers. (2022) 2022:2074356. doi: 10.1155/2022/2074356
45. Oba E, Aung NY, Ohe R, Sadahiro M, Yamakawa M. The distribution of macrophage subtypes and their relationship to bone morphogenetic protein 2 in calcified aortic valve stenosis. Am J Transl Res. (2020) 12(5):1728–40.32509172
46. El Husseini D, Boulanger MC, Mahmut A, Bouchareb R, Laflamme MH, Fournier D, et al. P2y2 receptor represses IL-6 expression by valve interstitial cells through akt: implication for calcific aortic valve disease. J Mol Cell Cardiol. (2014) 72:146–56. doi: 10.1016/j.yjmcc.2014.02.014
47. Goody PR, Christmann D, Goody D, Hildebrand S, Billig H, Nehl D, et al. Calcific aortic valve disease augments vesicular microRNA-145-5p to regulate the calcification of valvular interstitial cells via cellular crosstalk. Basic Res Cardiol. (2025) 120(5):991–1010. doi: 10.1007/s00395-025-01133-w
48. Yan F, Xie X, Huo Q, Zhang W, Wu T, Shi L. circ-CCND1 regulates the CCND1/P53/P21 pathway through sponging miR-138-5p in valve interstitial cells to aggravate aortic valve calcification. J Physiol Biochem. (2022) 78(4):845–54. doi: 10.1007/s13105-022-00907-3
49. Wei J, Zhu X, Ay S, Yan X, Meng X, Ge S. Long non-coding RNA FGD5 antisense RNA 1 targets baculovirus inhibitor 5 via microRNA-497-5p to alleviate calcific aortic valve disease. Clin Hemorheol Microcirc. (2024) 86(3):285–302. doi: 10.3233/CH-221692
50. Roberts WC, Ko JM. Frequency by decades of unicuspid, bicuspid, and tricuspid aortic valves in adults having isolated aortic valve replacement for aortic stenosis, with or without associated aortic regurgitation. Circulation. (2005) 111(7):920–5. doi: 10.1161/01.CIR.0000155623.48408.C5
51. Stewart BF, Siscovick D, Lind BK, Gardin JM, Gottdiener JS, Smith VE, et al. Clinical factors associated with calcific aortic valve disease fn1fn1This study was supported in part by contracts NO1-HC85079 through HC-850086 from the national heart, lung, and blood institute, national institutes of health, Bethesda, Maryland. J Am Coll Cardiol. (1997) 29(3):630–4. doi: 10.1016/S0735-1097(96)00563-3
52. Owens DS, Katz R, Takasu J, Kronmal R, Budoff MJ, O’Brien KD. Incidence and progression of aortic valve calcium in the multi-ethnic study of atherosclerosis (MESA). Am J Cardiol. (2010) 105(5):701–8. doi: 10.1016/j.amjcard.2009.10.071
53. Aggarwal SR, Clavel MA, Messika-Zeitoun D, Cueff C, Malouf J, Araoz PA, et al. Sex differences in aortic valve calcification measured by multidetector computed tomography in aortic stenosis. Circ Cardiovasc Imaging. (2013) 6(1):40–7. doi: 10.1161/CIRCIMAGING.112.980052
54. Simard L, Côté N, Dagenais F, Mathieu P, Couture C, Trahan S, et al. Sex-Related discordance between aortic valve calcification and hemodynamic severity of aortic stenosis. Circ Res. (2017) 120(4):681–91. doi: 10.1161/CIRCRESAHA.116.309306
55. Mohty D, Pibarot P, Després JP, Côté C, Arsenault B, Cartier A, et al. Association between plasma LDL particle size, valvular accumulation of oxidized LDL, and inflammation in patients with aortic stenosis. Arterioscler Thromb Vasc Biol. (2008) 28(1):187–93. doi: 10.1161/ATVBAHA.107.154989
56. Côté C, Pibarot P, Després JP, Mohty D, Cartier A, Arsenault BJ, et al. Association between circulating oxidised low-density lipoprotein and fibrocalcific remodelling of the aortic valve in aortic stenosis. Heart. (2008) 94(9):1175–80. doi: 10.1136/hrt.2007.125740
57. Thanassoulis G, Campbell CY, Owens DS, Smith JG, Smith AV, Peloso GM, et al. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. (2013) 368(6):503–12. doi: 10.1056/NEJMoa1109034
58. Kamstrup PR, Tybjærg-Hansen A, Nordestgaard BG. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol. (2014) 63(5):470–7. doi: 10.1016/j.jacc.2013.09.038
59. Arsenault BJ, Boekholdt SM, Dubé MP, Rhéaume E, Wareham NJ, Khaw KT, et al. Lipoprotein(a) levels, genotype, and incident aortic valve stenosis: a prospective Mendelian randomization study and replication in a case-control cohort. Circ Cardiovasc Genet. (2014) 7(3):304–10. doi: 10.1161/CIRCGENETICS.113.000400
60. Capoulade R, Chan KL, Yeang C, Mathieu P, Bossé Y, Dumesnil JG, et al. Oxidized phospholipids, lipoprotein(a), and progression of calcific aortic valve stenosis. J Am Coll Cardiol. (2015) 66(11):1236–46. doi: 10.1016/j.jacc.2015.07.020
61. Hekimian G, Boutten A, Flamant M, Duval X, Dehoux M, Benessiano J, et al. Progression of aortic valve stenosis is associated with bone remodelling and secondary hyperparathyroidism in elderly patients--the COFRASA study. Eur Heart J. (2013) 34(25):1915–22. doi: 10.1093/eurheartj/ehs450
62. Cuevas RA, Chu CC, Moorhead WJ, Wong R, Sultan I, St Hilaire C. Isolation of human primary valve cells for in vitro disease modeling. J Vis Exp. (2021) (170). doi: 10.3791/62439
63. Goto S, Rogers MA, Blaser MC, Higashi H, Lee LH, Schlotter F, et al. Standardization of human calcific aortic valve disease in vitro modeling reveals passage-dependent calcification. Front Cardiovasc Med. (2019) 6:49. doi: 10.3389/fcvm.2019.00049
64. Nehl D, Goody PR, Maus K, Pfeifer A, Aikawa E, Bakthiary F, et al. Human and porcine aortic valve endothelial and interstitial cell isolation and characterization. Front Cardiovasc Med. (2023) 10:1151028. doi: 10.3389/fcvm.2023.1151028
65. Gould RA, Butcher JT. Isolation of valvular endothelial cells. J Vis Exp. (2010) (46):2158. doi: 10.3791/2158
66. Ground M, Park YE, Waqanivavalagi S, Callon K, Walker R, Milsom P, et al. Generating robust human valvular interstitial cell cultures: protocol and considerations. J Mol Cell Cardiol. (2022) 173:118–26. doi: 10.1016/j.yjmcc.2022.10.008
67. Sohier J, Carubelli I, Sarathchandra P, Latif N, Chester AH, Yacoub MH. The potential of anisotropic matrices as substrate for heart valve engineering. Biomaterials. (2014) 35(6):1833–44. doi: 10.1016/j.biomaterials.2013.10.061
68. Colazzo F, Sarathchandra P, Smolenski RT, Chester AH, Tseng YT, Czernuszka JT, et al. Extracellular matrix production by adipose-derived stem cells: implications for heart valve tissue engineering. Biomaterials. (2011) 32(1):119–27. doi: 10.1016/j.biomaterials.2010.09.003
69. Chen Z, Gordillo-Martinez F, Jiang L, He P, Hong W, Wei X, et al. Zinc ameliorates human aortic valve calcification through GPR39 mediated ERK1/2 signalling pathway. Cardiovasc Res. (2021) 117(3):820–35. doi: 10.1093/cvr/cvaa090
70. Latif N, Quillon A, Sarathchandra P, McCormack A, Lozanoski A, Yacoub MH, et al. Modulation of human valve interstitial cell phenotype and function using a fibroblast growth factor 2 formulation. PLoS One. (2015) 10(6):e0127844. doi: 10.1371/journal.pone.0127844
71. Bogdanova M, Zabirnyk A, Malashicheva A, Enayati KZ, Karlsen TA, Kaljusto ML, et al. Interstitial cells in calcified aortic valves have reduced differentiation potential and stem cell-like properties. Sci Rep. (2019) 9(1):12934. doi: 10.1038/s41598-019-49016-0
72. Yip CYY, Chen JH, Zhao R, Simmons CA. Calcification by valve interstitial cells is regulated by the stiffness of the extracellular matrix. Arterioscler Thromb Vasc Biol. (2009) 29(6):936–42. doi: 10.1161/ATVBAHA.108.182394
73. Wang Z, Rao Z, Wang Y, Dong N. Establishment and characterization of a novel immortalized human aortic valve interstitial cell line. Sci Rep. (2025) 15(1):10917. doi: 10.1038/s41598-025-85909-z
74. Bowler MA, Merryman WD. In vitro models of aortic valve calcification: solidifying a system. Cardiovasc Pathol. (2015) 24(1):1–10. doi: 10.1016/j.carpath.2014.08.003
75. Pho M, Lee W, Watt DR, Laschinger C, Simmons CA, McCulloch CA. Cofilin is a marker of myofibroblast differentiation in cells from porcine aortic cardiac valves. Am J Physiol Heart Circ Physiol. (2008) 294(4):H1767–1778. doi: 10.1152/ajpheart.01305.2007
76. Lin C, Zhu D, Markby G, Corcoran BM, Farquharson C, Macrae VE. Isolation and characterization of primary rat valve interstitial cells: a new model to study aortic valve calcification. J Vis Exp. (2017) 129:56126. doi: 10.3791/56126
77. Tsang HG, Cui L, Farquharson C, Corcoran BM, Summers KM, Macrae VE. Exploiting novel valve interstitial cell lines to study calcific aortic valve disease. Mol Med Rep. (2018) 17(2):2100–6. doi: 10.3892/mmr.2017.8163
78. Seya K, Yu Z, Kanemaru K, Daitoku K, Akemoto Y, Shibuya H, et al. Contribution of bone morphogenetic protein-2 to aortic valve calcification in aged rat. J Pharmacol Sci. (2011) 115(1):8–14. doi: 10.1254/jphs.10198FP
79. Masjedi S, Lei Y, Patel J, Ferdous Z. Sex-related differences in matrix remodeling and early osteogenic markers in aortic valvular interstitial cells. Heart Vessels. (2017) 32(2):217–28. doi: 10.1007/s00380-016-0909-8
80. Bouchareb R, Lebeche D. Isolation of mouse interstitial valve cells to study the calcification of the aortic valve in vitro. J Vis Exp. (2021) (171). doi: 10.3791/62419
81. Hinton RB, Alfieri CM, Witt SA, Glascock BJ, Khoury PR, Benson DW, et al. Mouse heart valve structure and function: echocardiographic and morphometric analyses from the fetus through the aged adult. Am J Physiol Heart Circ Physiol. (2008) 294(6):H2480–2488. doi: 10.1152/ajpheart.91431.2007
82. Grauss RW, Hazekamp MG, van Vliet S, Gittenberger-de Groot AC, DeRuiter MC. Decellularization of rat aortic valve allografts reduces leaflet destruction and extracellular matrix remodeling. J Thorac Cardiovasc Surg. (2003) 126(6):2003–10. doi: 10.1016/S0022-5223(03)00956-5
83. Guerraty M, Mohler Iii ER. Models of aortic valve calcification. J Investig Med. (2007) 55(6):278–83. doi: 10.2310/6650.2007.00012
84. Tao Y, Geng Y, Dang W, Xu X, Zhao H, Zou L, et al. Mechanism of endoplasmic Reticulum stress pathway in the osteogenic phenotypic transformation of aortic valve interstitial cells. Front Endocrinol (Lausanne). (2022) 13:856331. doi: 10.3389/fendo.2022.856331
85. Jian B, Narula N, Li QY, Mohler ER, Levy RJ. Progression of aortic valve stenosis: tGF-beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg. (2003) 75(2):457–65; discussion 465–466. doi: 10.1016/S0003-4975(02)04312-6
86. Mohler ER, Chawla MK, Chang AW, Vyavahare N, Levy RJ, Graham L, et al. Identification and characterization of calcifying valve cells from human and canine aortic valves. J Heart Valve Dis. (1999) 8(3):254–60.10399657
87. Latif N, Sarathchandra P, Chester AH, Yacoub MH. Expression of smooth muscle cell markers and co-activators in calcified aortic valves. Eur Heart J. (2015) 36(21):1335–45. doi: 10.1093/eurheartj/eht547
88. Porras AM, van Engeland NCA, Marchbanks E, McCormack A, Bouten CVC, Yacoub MH, et al. Robust generation of quiescent porcine valvular interstitial cell cultures. J Am Heart Assoc. (2017) 6(3):e005041. doi: 10.1161/JAHA.116.005041
89. Kostina A, Shishkova A, Ignatieva E, Irtyuga O, Bogdanova M, Levchuk K, et al. Different notch signaling in cells from calcified bicuspid and tricuspid aortic valves. J Mol Cell Cardiol. (2018) 114:211–9. doi: 10.1016/j.yjmcc.2017.11.009
90. Walker GA, Masters KS, Shah DN, Anseth KS, Leinwand LA. Valvular myofibroblast activation by transforming growth factor-beta: implications for pathological extracellular matrix remodeling in heart valve disease. Circ Res. (2004) 95(3):253–60. doi: 10.1161/01.RES.0000136520.07995.aa
91. Kennedy JA, Hua X, Mishra K, Murphy GA, Rosenkranz AC, Horowitz JD. Inhibition of calcifying nodule formation in cultured porcine aortic valve cells by nitric oxide donors. Eur J Pharmacol. (2009) 602(1):28–35. doi: 10.1016/j.ejphar.2008.11.029
92. Cheek JD, Wirrig EE, Alfieri CM, James JF, Yutzey KE. Differential activation of valvulogenic, chondrogenic, and osteogenic pathways in mouse models of myxomatous and calcific aortic valve disease. J Mol Cell Cardiol. (2012) 52(3):689–700. doi: 10.1016/j.yjmcc.2011.12.013
93. Gwanmesia P, Ziegler H, Eurich R, Barth M, Kamiya H, Karck M, et al. Opposite effects of transforming growth factor-β1 and vascular endothelial growth factor on the degeneration of aortic valvular interstitial cell are modified by the extracellular matrix protein fibronectin: implications for heart valve engineering. Tissue Eng Part A. (2010) 16(12):3737–46. doi: 10.1089/ten.tea.2010.0304
94. Gu X, Masters KS. Regulation of valvular interstitial cell calcification by adhesive peptide sequences. J Biomed Mater Res A. (2010) 93(4):1620. doi: 10.1002/jbm.a.32660
95. Fisher CI, Chen J, Merryman WD. Calcific nodule morphogenesis by heart valve interstitial cells is strain dependent. Biomech Model Mechanobiol. (2013) 12(1):5–17. doi: 10.1007/s10237-012-0377-8
96. Osman L, Yacoub MH, Latif N, Amrani M, Chester AH. Role of human valve interstitial cells in valve calcification and their response to atorvastatin. Circulation. (2006) 114(1 Suppl):I547–552. doi: 10.1161/CIRCULATIONAHA.105.001115
97. Zhang XW, Zhang BY, Wang SW, Gong DJ, Han L, Xu ZY, et al. Twist-related protein 1 negatively regulated osteoblastic transdifferentiation of human aortic valve interstitial cells by directly inhibiting runt-related transcription factor 2. J Thorac Cardiovasc Surg. (2014) 148(4):1700–8.e1. doi: 10.1016/j.jtcvs.2014.02.084
98. Issa H, Hénaut L, Abdallah JB, Boudot C, Lenglet G, Avondo C, et al. Activation of the calcium-sensing receptor in human valvular interstitial cells promotes calcification. J Mol Cell Cardiol. (2019) 129:2–12. doi: 10.1016/j.yjmcc.2019.01.021
99. Kessler JR, Bluemn TS, DeCero SA, Dutta P, Thatcher K, Mahnke DK, et al. Exploring molecular profiles of calcification in aortic vascular smooth muscle cells and aortic valvular interstitial cells. J Mol Cell Cardiol. (2023) 183:1–13. doi: 10.1016/j.yjmcc.2023.08.001
100. Dhayni K, Chabry Y, Hénaut L, Avondo C, Boudot C, Ouled-Haddou H, et al. Aortic valve calcification is promoted by interleukin-8 and restricted through antagonizing CXC motif chemokine receptor 2. Cardiovasc Res. (2023) 119(13):2355–67. doi: 10.1093/cvr/cvad117
101. Yang JH, Wylie-Sears J, Bischoff J. Opposing actions of Notch1 and VEGF in post-natal cardiac valve endothelial cells. Biochem Biophys Res Commun. (2008) 374(3):512. doi: 10.1016/j.bbrc.2008.07.057
102. Hjortnaes J, Shapero K, Goettsch C, Hutcheson JD, Keegan J, Kluin J, et al. Valvular interstitial cells suppress calcification of valvular endothelial cells. Atherosclerosis. (2015) 242(1):251. doi: 10.1016/j.atherosclerosis.2015.07.008
103. Paranya G, Vineberg S, Dvorin E, Kaushal S, Roth SJ, Rabkin E, et al. Aortic valve endothelial cells undergo transforming growth factor-β-mediated and non-transforming growth factor-β-mediated transdifferentiation in vitro. Am J Pathol. (2001) 159(4):1335. doi: 10.1016/S0002-9440(10)62520-5
104. Simon LR, Scott AJ, Figueroa Rios L, Zembles J, Masters KS. Cellular-scale sex differences in extracellular matrix remodeling by valvular interstitial cells. Heart Vessels. (2023) 38(1):122–30. doi: 10.1007/s00380-022-02164-2
105. Tuscher R, Khang A, West TM, Camillo C, Ferrari G, Sacks MS. Functional differences in human aortic valve interstitial cells from patients with varying calcific aortic valve disease. Front Physiol. (2023) 14:1168691. doi: 10.3389/fphys.2023.1168691
106. Rogers JD, Aguado BA, Watts KM, Anseth KS, Richardson WJ. Network modeling predicts personalized gene expression and drug responses in valve myofibroblasts cultured with patient sera. Proc Natl Acad Sci U S A. (2022) 119(8):e2117323119. doi: 10.1073/pnas.2117323119
107. Yang X, Zeng J, Xie K, Su S, Guo Y, Zhang H, et al. Advanced glycation end product-modified low-density lipoprotein promotes pro-osteogenic reprogramming via RAGE/NF-κB pathway and exaggerates aortic valve calcification in hamsters. Mol Med. (2024) 30(1):76. doi: 10.1186/s10020-024-00833-8
108. Kopytek M, Mazur P, Ząbczyk M, Undas A, Natorska J. Diabetes concomitant to aortic stenosis is associated with increased expression of NF-κB and more pronounced valve calcification. Diabetologia. (2021) 64(11):2562–74. doi: 10.1007/s00125-021-05545-w
109. Delgado-Marin M, Sánchez-Esteban S, Cook-Calvete A, Jorquera-Ortega S, Zaragoza C, Saura M. Indoxyl sulfate-induced valve endothelial cell endothelial-to-mesenchymal transition and calcification in an integrin-linked kinase-dependent manner. Cells. (2024) 13(6):481. doi: 10.3390/cells13060481
110. Csiki DM, Ababneh H, Tóth A, Lente G, Szöőr Á, Tóth A, et al. Hypoxia-inducible factor activation promotes osteogenic transition of valve interstitial cells and accelerates aortic valve calcification in a mice model of chronic kidney disease. Front Cardiovasc Med. (2023) 10:1168339. doi: 10.3389/fcvm.2023.1168339
111. Liu L, Lan Z, Liu X, Chen Y, Chen Z, Cheng L, et al. NETs accelerate aortic valve calcification by promoting M1 macrophage polarization through the TLR9 signaling pathway. Mol Cell Biochem. (2025). doi: 10.1007/s11010-025-05375-z
112. Li F, Zhao Z, Cai Z, Dong N, Liu Y. Oxidized low-density lipoprotein promotes osteoblastic differentiation of valvular interstitial cells through RAGE/MAPK. Cardiology. (2015) 130(1):55–61. doi: 10.1159/000369126
113. Li G, Qiao W, Zhang W, Li F, Shi J, Dong N. The shift of macrophages toward M1 phenotype promotes aortic valvular calcification. J Thorac Cardiovasc Surg. (2017) 153(6):1318–27.e1. doi: 10.1016/j.jtcvs.2017.01.052
114. Raddatz MA, Huffstater T, Bersi MR, Reinfeld BI, Madden MZ, Booton SE, et al. Macrophages promote aortic valve cell calcification and Alter STAT3 splicing. Arterioscler Thromb Vasc Biol. (2020) 40(6):e153–65. doi: 10.1161/ATVBAHA.120.314360
115. Xia H, Gao M, Chen J, Huang G, Xiang X, Wang Y, et al. M1 macrophage-derived extracellular vesicle containing tsRNA-5006c promotes osteogenic differentiation of aortic valve interstitial cells through regulating mitophagy. PeerJ. (2022) 10:e14307. doi: 10.7717/peerj.14307
116. Discher DE, Janmey P, Wang YL. Tissue cells feel and respond to the stiffness of their substrate. Science. (2005) 310(5751):1139–43. doi: 10.1126/science.1116995
117. Choquet D, Felsenfeld DP, Sheetz MP. Extracellular matrix rigidity causes strengthening of integrin-cytoskeleton linkages. Cell. (1997) 88(1):39–48. doi: 10.1016/S0092-8674(00)81856-5
118. Wang HB, Dembo M, Hanks SK, Wang Y. Focal adhesion kinase is involved in mechanosensing during fibroblast migration. Proc Natl Acad Sci U S A. (2001) 98(20):11295–300. doi: 10.1073/pnas.201201198
119. Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. (2002) 3(5):349–63. doi: 10.1038/nrm809
120. Coutts CW, Baldwin AM, Jebeli M, Jolin GE, Mungai RW, Billiar KL. The role of apoptosis and oxidative stress in a cell spheroid model of calcific aortic valve disease. Cells. (2023) 13(1):45. doi: 10.3390/cells13010045
121. Cirka HA, Uribe J, Liang V, Schoen FJ, Billiar KL. Reproducible in vitro model for dystrophic calcification of cardiac valvular interstitial cells: insights into the mechanisms of calcific aortic valvular disease. Lab Chip. (2017) 17(5):814–29. doi: 10.1039/C6LC01226D
122. Roosens A, Puype I, Cornelissen R. Scaffold-free high throughput generation of quiescent valvular microtissues. J Mol Cell Cardiol. (2017) 106:45–54. doi: 10.1016/j.yjmcc.2017.03.004
123. Tseng H, Balaoing LR, Grigoryan B, Raphael RM, Killian TC, Souza GR, et al. A three-dimensional co-culture model of the aortic valve using magnetic levitation. Acta Biomater. (2014) 10(1):173–82. doi: 10.1016/j.actbio.2013.09.003
124. Masters KS, Shah DN, Walker G, Leinwand LA, Anseth KS. Designing scaffolds for valvular interstitial cells: cell adhesion and function on naturally derived materials. J Biomed Mater Res A. (2004) 71(1):172–80. doi: 10.1002/jbm.a.30149
125. Benton JA, DeForest CA, Vivekanandan V, Anseth KS. Photocrosslinking of gelatin macromers to synthesize porous hydrogels that promote valvular interstitial cell function. Tissue Eng Part A. (2009) 15(11):3221–30. doi: 10.1089/ten.tea.2008.0545
126. Vadana M, Cecoltan S, Ciortan L, Macarie RD, Tucureanu MM, Mihaila AC, et al. Molecular mechanisms involved in high glucose-induced valve calcification in a 3D valve model with human valvular cells. J Cell Mol Med. (2020) 24(11):6350–61. doi: 10.1111/jcmm.15277
127. Hjortnaes J, Camci-Unal G, Hutcheson JD, Jung SM, Schoen FJ, Kluin J, et al. Directing valvular interstitial cell myofibroblast-like differentiation in a hybrid hydrogel platform. Adv Healthc Mater. (2015) 4(1):121–30. doi: 10.1002/adhm.201400029
128. Hjortnaes J, Goettsch C, Hutcheson JD, Camci-Unal G, Lax L, Scherer K, et al. Simulation of early calcific aortic valve disease in a 3D platform: a role for myofibroblast differentiation. J Mol Cell Cardiol. (2016) 94:13–20. doi: 10.1016/j.yjmcc.2016.03.004
129. Bramsen JA, Alber BR, Mendoza M, Murray BT, Chen MH, Huang P, et al. Glycosaminoglycans affect endothelial to mesenchymal transformation, proliferation, and calcification in a 3D model of aortic valve disease. Front Cardiovasc Med. (2022) 9:975732. doi: 10.3389/fcvm.2022.975732
130. Salemizadehparizi F, Huang P, Chen MH, Mahler GJ. The contributions of increased reactive oxygen species and inflammatory macrophages to the development of aortic valve calcification. Free Radic Biol Med. (2025) 241:64–75. doi: 10.1016/j.freeradbiomed.2025.09.017
131. Gee TW, Richards JM, Mahmut A, Butcher JT. Valve endothelial-interstitial interactions drive emergent complex calcific lesion formation in vitro. Biomaterials. (2021) 269:120669. doi: 10.1016/j.biomaterials.2021.120669
132. Immohr MB, Teichert HL, dos Santos Adrego F, Schmidt V, Sugimura Y, Bauer SJ, et al. Three-Dimensional bioprinting of ovine aortic valve endothelial and interstitial cells for the development of multicellular tissue engineered tissue constructs. Bioengineering (Basel). (2023) 10(7):787. doi: 10.3390/bioengineering10070787
133. Immohr MB, Dos Santos Adrego F, Teichert HL, Schmidt V, Sugimura Y, Bauer S, et al. 3D-bioprinting Of aortic valve interstitial cells: impact of hydrogel and printing parameters on cell viability. Biomed Mater. (2022) 18(1):015004. doi: 10.1088/1748-605X/ac9f91
134. Clift CL, Blaser MC, Gerrits W, Turner ME, Sonawane A, Pham T, et al. Intracellular proteomics and extracellular vesiculomics as a metric of disease recapitulation in 3D-bioprinted aortic valve arrays. Sci Adv. (2024) 10(9):eadj9793. doi: 10.1126/sciadv.adj9793
135. Gould ST, Anseth KS. Role of cell-matrix interactions on VIC phenotype and tissue deposition in 3D PEG hydrogels. J Tissue Eng Regen Med. (2016) 10(10):E443–53. doi: 10.1002/term.1836
136. Benton JA, Fairbanks BD, Anseth KS. Characterization of valvular interstitial cell function in three dimensional matrix metalloproteinase degradable PEG hydrogels. Biomaterials. (2009) 30(34):6593–603. doi: 10.1016/j.biomaterials.2009.08.031
137. Puperi DS, Balaoing LR, O’Connell RW, West JL, Grande-Allen KJ. 3-dimensional Spatially organized PEG-based hydrogels for an aortic valve co-culture model. Biomaterials (2015) 67:354–64. doi: 10.1016/j.biomaterials.2015.07.039
138. Mabry KM, Lawrence RL, Anseth KS. Dynamic stiffening of poly(ethylene glycol)-based hydrogels to direct valvular interstitial cell phenotype in a three-dimensional environment. Biomaterials. (2015) 49:47–56. doi: 10.1016/j.biomaterials.2015.01.047
139. Gould ST, Matherly EE, Smith JN, Heistad DD, Anseth KS. The role of valvular endothelial cell paracrine signaling and matrix elasticity on valvular interstitial cell activation. Biomaterials. (2014) 35(11):3596–606. doi: 10.1016/j.biomaterials.2014.01.005
140. Grim JC, Aguado BA, Vogt BJ, Batan D, Andrichik CL, Schroeder ME, et al. Secreted factors from pro-inflammatory macrophages promote an osteoblast-like phenotype in valvular interstitial cells. Arterioscler Thromb Vasc Biol. (2020) 40(11):e296–308. doi: 10.1161/ATVBAHA.120.315261
141. Monroe MN, Nikonowicz RC, Grande-Allen KJ. Heterogeneous multi-laminar tissue constructs as a platform to evaluate aortic valve matrix-dependent pathogenicity. Acta Biomater. (2019) 97:420–7. doi: 10.1016/j.actbio.2019.07.046
142. Stadelmann K, Weghofer A, Urbanczyk M, Maulana TI, Loskill P, Jones PD, et al. Development of a bi-layered cryogenic electrospun polylactic acid scaffold to study calcific aortic valve disease in a 3D co-culture model. Acta Biomater. (2022) 140:364–78. doi: 10.1016/j.actbio.2021.11.030
143. Nichol JW, Koshy ST, Bae H, Hwang CM, Yamanlar S, Khademhosseini A. Cell-laden microengineered gelatin methacrylate hydrogels. Biomaterials. (2010) 31(21):5536–44. doi: 10.1016/j.biomaterials.2010.03.064
144. Ifkovits JL, Tous E, Minakawa M, Morita M, Robb JD, Koomalsingh KJ, et al. Injectable hydrogel properties influence infarct expansion and extent of postinfarction left ventricular remodeling in an ovine model. Proc Natl Acad Sci U S A. (2010) 107(25):11507–12. doi: 10.1073/pnas.1004097107
145. Rathan S, Yoganathan AP, O’Neill WC. The role of inorganic pyrophosphate in aortic valve calcification. J Heart Valve Dis. (2014) 23(4):387–94.25803964
146. Chester AH, Sarathchandra P, McCormack A, Yacoub MH. Organ culture model of aortic valve calcification. Front Cardiovasc Med. (2021) 8:734692. doi: 10.3389/fcvm.2021.734692
147. Rodriguez KJ, Piechura LM, Porras AM, Masters KS. Manipulation of valve composition to elucidate the role of collagen in aortic valve calcification. BMC Cardiovasc Disord. (2014) 14:29. doi: 10.1186/1471-2261-14-29
148. Zabirnyk A, Perez MDM, Blasco M, Stensløkken KO, Ferrer MD, Salcedo C, et al. A novel ex vivo model of aortic valve calcification. A preliminary report. Front Pharmacol. (2020) 11:568764. doi: 10.3389/fphar.2020.568764
149. Kachanova OS, Boyarskaya NV, Docshin PM, Scherbinin TS, Zubkova VG, Saprankov VL, et al. Ex vivo model of pathological calcification of human aortic valve. Front Cardiovasc Med. (2024) 11:1411398. doi: 10.3389/fcvm.2024.1411398
150. Weber A, Pfaff M, Schöttler F, Schmidt V, Lichtenberg A, Akhyari P. Reproducible in vitro tissue culture model to study basic mechanisms of calcific aortic valve disease: comparative analysis to valvular interstitials cells. Biomedicines. (2021) 9(5):474. doi: 10.3390/biomedicines9050474
151. Jenke A, Kistner J, Saradar S, Chekhoeva A, Yazdanyar M, Bergmann AK, et al. Transforming growth factor-β1 promotes fibrosis but attenuates calcification of valvular tissue applied as a three-dimensional calcific aortic valve disease model. Am J Physiol Heart Circ Physiol. (2020) 319(5):H1123–41. doi: 10.1152/ajpheart.00651.2019
152. Butcher JT, Penrod AM, García AJ, Nerem RM. Unique morphology and focal adhesion development of valvular endothelial cells in static and fluid flow environments. Arterioscler Thromb Vasc Biol. (2004) 24(8):1429–34. doi: 10.1161/01.ATV.0000130462.50769.5a
153. Hsu CPD, Tchir A, Mirza A, Chaparro D, Herrera RE, Hutcheson JD, et al. Valve endothelial cell exposure to high levels of flow oscillations exacerbates valve interstitial cell calcification. Bioengineering (Basel). (2022) 9(8):393. doi: 10.3390/bioengineering9080393
154. Smith KE, Metzler SA, Warnock JN. Cyclic strain inhibits acute pro-inflammatory gene expression in aortic valve interstitial cells. Biomech Model Mechanobiol. (2010) 9(1):117–25. doi: 10.1007/s10237-009-0165-2
155. Metzler SA, Pregonero CA, Butcher JT, Burgess SC, Warnock JN. Cyclic strain regulates pro-inflammatory protein expression in porcine aortic valve endothelial cells. J Heart Valve Dis. (2008) 17(5):571–7; discussion 578.18980092
156. Quinlan AMT, Billiar KL. Investigating the role of substrate stiffness in the persistence of valvular interstitial cell activation. J Biomed Mater Res A. (2012) 100(9):2474–82. doi: 10.1002/jbm.a.34162
157. Butcher JT, Nerem RM. Valvular endothelial cells regulate the phenotype of interstitial cells in co-culture: effects of steady shear stress. Tissue Eng. (2006) 12(4):905–15. doi: 10.1089/ten.2006.12.905
158. Shih JY, Gee T, Scuderi G, Butcher J. Biomechanical remodeling of aortic valve interstitial cells during calcified lesion formation in vitro. Ann Biomed Eng. (2024) 52(5):1270–9. doi: 10.1007/s10439-024-03451-x
159. Tandon I, Woessner AE, Ferreira LA, Shamblin C, Vaca-Diez G, Walls A, et al. A three-dimensional valve-on-chip microphysiological system implicates cell cycle progression, cholesterol metabolism and protein homeostasis in early calcific aortic valve disease progression. Acta Biomater. (2024) 186:167–84. doi: 10.1016/j.actbio.2024.07.036
160. Dittfeld C, Winkelkotte M, Scheer A, Voigt E, Schmieder F, Behrens S, et al. Challenges of aortic valve tissue culture—maintenance of viability and extracellular matrix in the pulsatile dynamic microphysiological system. J Biol Eng. (2023) 17:60. doi: 10.1186/s13036-023-00377-1
161. Sun L, Rajamannan NM, Sucosky P. Defining the role of fluid shear stress in the expression of early signaling markers for calcific aortic valve disease. PLoS One. (2013) 8(12):e84433. doi: 10.1371/journal.pone.0084433
162. Warnock JN, Nanduri B, Pregonero Gamez CA, Tang J, Koback D, Muir WM, et al. Gene profiling of aortic valve interstitial cells under elevated pressure conditions: modulation of inflammatory gene networks. Int J Inflam. (2011) 2011:176412. doi: 10.4061/2011/176412
163. Balachandran K, Konduri S, Sucosky P, Jo H, Yoganathan AP. An ex vivo study of the biological properties of porcine aortic valves in response to circumferential cyclic stretch. Ann Biomed Eng. (2006) 34(11):1655–65. doi: 10.1007/s10439-006-9167-8
164. Merryman WD, Lukoff HD, Long RA, Engelmayr GC, Hopkins RA, Sacks MS. Synergistic effects of cyclic tension and transforming growth factor-beta1 on the aortic valve myofibroblast. Cardiovasc Pathol. (2007) 16(5):268–76. doi: 10.1016/j.carpath.2007.03.006
165. Balachandran K, Sucosky P, Jo H, Yoganathan AP. Elevated cyclic stretch induces aortic valve calcification in a bone morphogenic protein-dependent manner. Am J Pathol. (2010) 177(1):49–57. doi: 10.2353/ajpath.2010.090631
166. El-Hamamsy I, Balachandran K, Yacoub MH, Stevens LM, Sarathchandra P, Taylor PM, et al. Endothelium-dependent regulation of the mechanical properties of aortic valve cusps. J Am Coll Cardiol. (2009) 53(16):1448–55. doi: 10.1016/j.jacc.2008.11.056
167. Kruithof BPT, van de Pol V, Los T, Lodder K, Gourabi BM, DeRuiter MC, et al. New calcification model for intact murine aortic valves. J Mol Cell Cardiol. (2021) 156:95–104. doi: 10.1016/j.yjmcc.2021.03.003
168. Kruithof BPT, Lieber SC, Kruithof-de Julio M, Gaussin V, Goumans MJ. Culturing mouse cardiac valves in the miniature tissue culture system. J Vis Exp. (2015) (104):52750. doi: 10.3791/52750
169. Garoffolo G, Ferrari S, De Martino S, Pizzo E, Candino V, Curini L, et al. Reversion of aortic valve cells calcification by activation of notch signalling via histone acetylation induction. Signal Transduct Target Ther. (2025) 10(1):311. doi: 10.1038/s41392-025-02411-8
170. Konduri S, Xing Y, Warnock JN, He Z, Yoganathan AP. Normal physiological conditions maintain the biological characteristics of porcine aortic heart valves: an ex vivo organ culture study. Ann Biomed Eng. (2005) 33(9):1158–66. doi: 10.1007/s10439-005-5506-4
171. Niazy N, Barth M, Selig JI, Feichtner S, Shakiba B, Candan A, et al. Degeneration of aortic valves in a bioreactor system with pulsatile flow. Biomedicines. (2021) 9(5):462. doi: 10.3390/biomedicines9050462
172. Weston MW, LaBorde DV, Yoganathan AP. Estimation of the shear stress on the surface of an aortic valve leaflet. Ann Biomed Eng. (1999) 27(4):572–9. doi: 10.1114/1.199
173. Farivar RS, Cohn LH, Soltesz EG, Mihaljevic T, Rawn JD, Byrne JG. Transcriptional profiling and growth kinetics of endothelium reveals differences between cells derived from porcine aorta versus aortic valve. Eur J Cardiothorac Surg. (2003) 24(4):527–34. doi: 10.1016/S1010-7940(03)00408-1
174. Sucosky P, Padala M, Elhammali A, Balachandran K, Jo H, Yoganathan AP. Design of an ex vivo culture system to investigate the effects of shear stress on cardiovascular tissue. J Biomech Eng. (2008) 130(3):035001. doi: 10.1115/1.2907753
175. Cuniberti LA, Stutzbach PG, Guevara E, Yannarelli GG, Laguens RP, Favaloro RR. Development of mild aortic valve stenosis in a rabbit model of hypertension. J Am Coll Cardiol. (2006) 47(11):2303–9. doi: 10.1016/j.jacc.2005.12.070
176. Schipke KJ, To SDF, Warnock JN. Design of a cyclic pressure bioreactor for the ex vivo study of aortic heart valves. J Vis Exp. (2011) (54):3316. doi: 10.3791/3316
177. Louvet L, Metzinger L, Büchel J, Steppan S, Massy ZA. Magnesium attenuates phosphate-induced deregulation of a MicroRNA signature and prevents modulation of Smad1 and osterix during the course of vascular calcification. Biomed Res Int. (2016) 2016:7419524. doi: 10.1155/2016/7419524
178. Schneider MR. Von kossa and his staining technique. Histochem Cell Biol. (2021) 156(6):523–6. doi: 10.1007/s00418-021-02051-3
179. Mpweme Bangando H, Simard C, Aize M, Lebrun A, Manrique A, Guinamard R, et al. TRPM4 Participates in irradiation-induced aortic valve remodeling in mice. Cancers (Basel). (2022) 14(18):4477. doi: 10.3390/cancers14184477
180. Lievremont M, Potus J, Guillou B. Use of alizarin red S for histochemical staining of Ca2+ in the mouse; some parameters of the chemical reaction in vitro. Acta Anat (Basel). (1982) 114(3):268–80. doi: 10.1159/000145596
181. Zaheer A, Lenkinski RE, Mahmood A, Jones AG, Cantley LC, Frangioni JV. In vivo near-infrared fluorescence imaging of osteoblastic activity. Nat Biotechnol. (2001) 19(12):1148–54. doi: 10.1038/nbt1201-1148
182. Greco A, Herrmann J, Babic M, Gummi MR, van der Giet M, Tölle M, et al. Molecular imaging and quantification of smooth muscle cell and aortic tissue calcification in vitro and ex vivo with a fluorescent hydroxyapatite-specific probe. Biomedicines. (2022) 10(9):2271. doi: 10.3390/biomedicines10092271
183. Drolet MC, Arsenault M, Couet J. Experimental aortic valve stenosis in rabbits. J Am Coll Cardiol. (2003) 41(7):1211–7. doi: 10.1016/S0735-1097(03)00090-1
184. Drolet MC, Couet J, Arsenault M. Development of aortic valve sclerosis or stenosis in rabbits: role of cholesterol and calcium. J Heart Valve Dis. (2008) 17(4):381–7.18751467
185. Rajamannan NM, Subramaniam M, Caira F, Stock SR, Spelsberg TC. Atorvastatin inhibits hypercholesterolemia-induced calcification in the aortic valves via the Lrp5 receptor pathway. Circulation. (2005) 112(9 Suppl):I229–234. doi: 10.1161/01.CIRCULATIONAHA.104.524306
186. Sarphie TG. Surface responses of aortic valve endothelia from diet-induced, hypercholesterolemic rabbits. Atherosclerosis. (1985) 54(3):283–99. doi: 10.1016/0021-9150(85)90122-4
187. Hara T, Tsukada N, Okano M, Ishida T, Hirata K, Shiomi M. Progression of calcific aortic valve sclerosis in WHHLMI rabbits. Atherosclerosis. (2018) 273:8–14. doi: 10.1016/j.atherosclerosis.2018.03.044
188. Porras AM, Shanmuganayagam D, Meudt JJ, Krueger CG, Hacker TA, Rahko PS, et al. Development of aortic valve disease in familial hypercholesterolemic swine: implications for elucidating disease etiology. J Am Heart Assoc. (2015) 4(10):e002254. doi: 10.1161/JAHA.115.002254
189. Guerraty MA, Grant GR, Karanian JW, Chiesa OA, Pritchard WF, Davies PF. Hypercholesterolemia induces side-specific phenotype changes and PPAR&[gamma] pathway activation in swine aortic valve endothelium. Arterioscler Thromb Vasc Biol. (2010) 30(2):225. doi: 10.1161/ATVBAHA.109.198549
190. Bian JH, Yuan CZ, Gu JX, Lin WF, Xiong JQ, Tang ZW, et al. TREM2 Modulates macrophage pyroptosis and inflammatory responses to ameliorate aortic valve calcification. Int Immunopharmacol. (2025) 149:114161. doi: 10.1016/j.intimp.2025.114161
191. Torikai H, Chen MH, Jin L, He J, Angle JF, Shi W. Atherogenesis in apoe−/− and ldlr−/− mice with a genetically resistant background. Cells. (2023) 12(9):1255. doi: 10.3390/cells12091255
192. Miller JD, Weiss RM, Serrano KM, Brooks RM, Berry CJ, Zimmerman K, et al. Lowering plasma cholesterol levels halts progression of aortic valve disease in mice. Circulation. (2009) 119(20):2693–701. doi: 10.1161/CIRCULATIONAHA.108.834614
193. Scatena M, Jackson MF, Speer MY, Leaf EM, Wallingford MC, Giachelli CM. Increased calcific aortic valve disease in response to a diabetogenic, procalcific diet in the LDLr-/-ApoB100/100 mouse model. Cardiovasc Pathol. (2018) 34:28–37. doi: 10.1016/j.carpath.2018.02.002
194. Le Quang K, Bouchareb R, Lachance D, Laplante MA, El Husseini D, Boulanger MC, et al. Early development of calcific aortic valve disease and left ventricular hypertrophy in a mouse model of combined dyslipidemia and type 2 diabetes mellitus. Arterioscler Thromb Vasc Biol. (2014) 34(10):2283–91. doi: 10.1161/ATVBAHA.114.304205
195. Tucureanu MM, Filippi A, Alexandru N, Ana Constantinescu C, Ciortan L, Macarie R, et al. Diabetes-induced early molecular and functional changes in aortic heart valves in a murine model of atherosclerosis. Diab Vasc Dis Res. (2019) 16(6):562–76. doi: 10.1177/1479164119874469
196. Wang L, Tang R, Zhang Y, Liu Z, Chen S, Song K, et al. A rat model with multivalve calcification induced by subtotal nephrectomy and high-phosphorus diet. Kidney Dis (Basel). (2020) 6(5):346–54. doi: 10.1159/000506013
197. Messaoudi H, Levesque T, Perzo N, Berg E, Feugray G, Dumesnil A, et al. Subtotal nephrectomy associated with a high-phosphate diet in rats mimics the development of calcified aortic valve disease associated with chronic renal failure. J Clin Med. (2023) 12(4):1539. doi: 10.3390/jcm12041539
198. Ivanovski O, Nikolov IG, Joki N, Caudrillier A, Phan O, Mentaverri R, et al. The calcimimetic R-568 retards uremia-enhanced vascular calcification and atherosclerosis in apolipoprotein E deficient (apoE−/−) mice. Atherosclerosis. (2009) 205(1):55–62. doi: 10.1016/j.atherosclerosis.2008.10.043
199. Ivanovski O, Szumilak D, Nguyen-Khoa T, Nikolov IG, Joki N, Mothu N, et al. Effect of simvastatin in apolipoprotein E deficient mice with surgically induced chronic renal failure. J Urol. (2008) 179(4):1631–6. doi: 10.1016/j.juro.2007.11.042
200. Phan O, Ivanovski O, Nguyen-Khoa T, Mothu N, Angulo J, Westenfeld R, et al. Sevelamer prevents uremia-enhanced atherosclerosis progression in apolipoprotein E-deficient mice. Circulation. (2005) 112(18):2875–82. doi: 10.1161/CIRCULATIONAHA105.541854
201. Roosens B, Bala G, Gillis K, Remory I, Droogmans S, Somja J, et al. Echocardiographic integrated backscatter for detecting progression and regression of aortic valve calcifications in rats. Cardiovasc Ultrasound. (2013) 11:4. doi: 10.1186/1476-7120-11-4
202. Zhang J, Lei W, Zhou J, Zhang Y, Huang F, Chen M. Uric acid promotes aortic valve calcification via mediating valve interstitial cell osteogenic differentiation and endothelial dysfunction. FASEB J. (2025) 39(6):e70437. doi: 10.1096/fj.202402831R
203. Düsing P, Göbel I, Ackerschott A, Reese L, Giavalisco P, Dethloff F, et al. The role of uremic toxin indoxyl sulfate in the pathophysiology of aortic valve stenosis. Cardiovasc Res. (2025) 121:cvaf106. doi: 10.1093/cvr/cvaf106
204. Okoshi MP, Cezar MDM, Iyomasa RM, Silva MB, Costa LCO, Martinez PF, et al. Effects of early aldosterone antagonism on cardiac remodeling in rats with aortic stenosis-induced pressure overload. Int J Cardiol. (2016) 222:569–75. doi: 10.1016/j.ijcard.2016.07.266
205. Ahmad AA, Ghim M, Toczek J, Neishabouri A, Ojha D, Zhang Z, et al. Multimodality imaging of aortic valve calcification and function in a murine model of calcific aortic valve disease and bicuspid aortic valve. J Nucl Med. (2023) 64(9):1487–94. doi: 10.2967/jnumed.123.265516
206. Jung JJ, Ahmad AA, Rajendran S, Wei L, Zhang J, Toczek J, et al. Differential BMP signaling mediates the interplay between genetics and leaflet numbers in aortic valve calcification. JACC Basic Transl Sci. (2022) 7(4):333–45. doi: 10.1016/j.jacbts.2021.12.006
207. Rucher G, Prigent K, Simard C, Frelin AM, Coquemont-Guyot M, Elie N, et al. Targeted radiation exposure induces accelerated aortic valve remodeling in ApoE−/− mice. J Clin Med. (2023) 12(18):5854. doi: 10.3390/jcm12185854
208. Honda S. A novel mouse model of aortic valve stenosis induced by direct wire injury. Arterioscler Thromb Vasc Biol. (2014) 34:270–8. doi: 10.1161/ATVBAHA.113.302610
209. Qian N, Wang Y, Hu W, Cao N, Qian Y, Chen J, et al. A novel mouse model of calcific aortic valve stenosis. Animal Model Exp Med. (2024) 7(4):523–32.38372410
210. Fang M, Liu K, Li X, Wang Y, Li W, Li B. AntagomiR-29b inhibits vascular and valvular calcification and improves heart function in rats. J Cell Mol Med. (2020) 24(19):11546–57. doi: 10.1111/jcmm.15770
211. Price PA, Faus SA, Williamson MK. Warfarin causes rapid calcification of the elastic lamellae in rat arteries and heart valves. Arterioscler Thromb Vasc Biol. (1998) 18(9):1400–7. doi: 10.1161/01.atv.18.9.1400
212. Liu J, Liu C, Qian C, Abela G, Sun W, Kong X. Ginkgo Biloba extract EGB761 alleviates warfarin-induced aortic valve calcification through the BMP2/Smad1/5/Runx2 signaling pathway. J Cardiovasc Pharmacol. (2021) 78(3):411–21. doi: 10.1097/FJC.0000000000001082
213. Krüger T, Oelenberg S, Kaesler N, Schurgers LJ, van de Sandt AM, Boor P, et al. Warfarin induces cardiovascular damage in mice. Arterioscler Thromb Vasc Biol. (2013) 33(11):2618–24. doi: 10.1161/ATVBAHA.113.302244
214. Rattazzi M, Faggin E, Bertacco E, Nardin C, Pagliani L, Plebani M, et al. Warfarin, but not rivaroxaban, promotes the calcification of the aortic valve in ApoE-/- mice. Cardiovasc Ther. (2018) 36(4):e12438. doi: 10.1111/1755-5922.12438
215. Doeing DC, Borowicz JL, Crockett ET. Gender dimorphism in differential peripheral blood leukocyte counts in mice using cardiac, tail, foot, and saphenous vein puncture methods. BMC Clin Pathol. (2003) 3:3. doi: 10.1186/1472-6890-3-3
216. Meurens F, Summerfield A, Nauwynck H, Saif L, Gerdts V. The pig: a model for human infectious diseases. Trends Microbiol. (2012) 20(1):50–7. doi: 10.1016/j.tim.2011.11.002
217. Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, et al. The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. Br J Pharmacol. (2020) 177(16):3617–24. doi: 10.1111/bph.15193
218. Kaiser AD, Wang J, Brown AL, Zhu E, Hsiai T, Marsden AL. A fluid-structure interaction model of the zebrafish aortic valve. J Biomech. (2025) 190:112794. doi: 10.1016/j.jbiomech.2025.112794
Keywords: aortic valve, calcific aortic valve disease, preclinical models, valvular interstitial cells, translational potential
Citation: Lafosse I, Mentaverri R, Avondo C, Bennis Y, Tribouilloy C and Hénaut L (2025) Insights into preclinical models of calcific aortic valve disease and their translational potential. Front. Cardiovasc. Med. 12:1621990. doi: 10.3389/fcvm.2025.1621990
Received: 2 May 2025; Accepted: 15 October 2025;
Published: 6 November 2025.
Edited by:
Masanori Aikawa, Brigham and Women's Hospital, United StatesReviewed by:
Knosalla Christoph, German Heart Center Berlin, GermanyCopyright: © 2025 Lafosse, Mentaverri, Avondo, Bennis, Tribouilloy and Hénaut. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
Rolando A. Cuevas, University of Pittsburgh, United States
*Correspondence: Lucie Hénaut, bHVjaWUuaGVuYXV0QHUtcGljYXJkaWUuZnI=
†These authors have contributed equally to this work and share first authorship