Abstract
In the cardiovascular system, elastic fibres exert a fundamental role providing the long-range elasticity required for physiological functions. Elastic fibres are complex in composition and structure containing, in addition to elastin, a wide range of matrix components, such as microfibrillar proteins, calcium-binding proteins and glycosaminoglycans. Changes in composition and/or structure can affect the biomechanics of the tissue as well as the intrinsic affinity of elastin for Ca2+ ions. Mineralization of elastic fibres can occur in genetic as well as in age-related chronic diseases. In cardiovascular diseases, for instance, calcification represents an integral part of the pathogenetic process, although the regulatory mechanisms are not completely understood. Therefore, a focus is given on elastin synthesis and assembly, on elastic fibre components and on elastin degradation. Moreover, the role and the impact of altered composition and supramolecular organization of elastic fibres are described in the context of the calcified cardiovascular system. Finally, some in vitro and in vivo models of elastic fibres calcification are presented and discussed.
1 Introduction
Elastin is the typical component of the elastic fibres and is the major responsible for soft connective tissue elasticity. The number and size of elastic fibres/lamellae are strictly related to the capacity of tissues to get back to their original shape after stretching. Indeed, elastic fibres are made of a wide range of proteins, glycoproteins and glycosaminoglycans (GAGs) whose amount and composition vary depending on the functional requirements of the tissue (1).
In vertebrates, elastin is mainly produced by fibroblasts and vascular smooth muscle cells (VSMC) during the second half of gestation until the end of childhood; thereafter the elastin gene is almost completely silent with a turnover evaluated to be approximately about 70 years (2). Therefore, elastin is one of the most resistant proteins, being present even in very old subjects, although with lower efficiency. Over the lifetime, elastin accumulates damages as the result of the aging processes and of environmental noxae, which cause progressive and irreversible loss of function especially in the cardiovascular system (3).
Cardiovascular calcification is an integral part of aging and of many cardiovascular diseases such as atherosclerotic microcalcifications, vascular and heart valve mineralization (4). Calcification in blood vessels occurs in the intima (in the context of atherosclerosis and in association with lipids, macrophages and VSMC), and in the media (involving elastin and VSMC). In both cases, areas of mineralization are associated with matrix vesicles and with changes in the expression of mineralization-regulatory proteins (5). Density of arterial calcification exhibit an age-dependent increase contributing to progressive vascular stiffness.
Calcific aortic valve disease comprises either aortic valve (AoV) sclerosis, with mild AoV thickening and/or calcification without obstruction of blood flow, and AoV stenosis with more severe calcification and impaired leaflet function. The intrinsic calcification mainly found at the leaflet hinge is characterized by abnormal collagen and proteoglycan (PG) deposition, whereas from the middle to the tip regions, in more severely affected leaflets and surgical specimens, nodular calcification and increased elastin fragmentation are more frequently observed (6). The elastin calcification propensity seems to directly depend on the size of elastin aggregates and the Ca2+ concentration allowing stronger interactions compared to collagen (7). These data may explain the prevalent occurrence of elastocalcinosis in the cardiovascular system and also in the bioprosthetic valves which are known to be at high risk of calcification and replacement after few years (8).
Calcification of the cardiovascular system depends also on the active role of many cells, such as VSMC and endothelial cells that can transdifferentiate in pro-osteogenic cells (9, 10), as well as VSMC and platelets that release extracellular vesicles enriched in pro-mineralizing factors capable to trigger calcification of the elastic component (11–13).
Since mineral deposits are not simply due to passive precipitation of calcium phosphate but are rather the result of a more complex biomineralization process, in this context, the extracellular matrix (ECM), and in particular elastic fibres are important players in the formation of ectopic calcified structures, because of their affinity for calcium ions, even in the absence of cells (14).
2 Elastin and elastic fibres
Elastin is the main component of elastic fibres, confers elasticity and contributes to tissue biomechanical properties of tissues such as vessels, skin, lungs as well as cartilage and intervertebral discs (15–17). Indeed, elastic fibres are composed of many proteins and glycoproteins and are structurally organized in fibres of different sizes, in larger networks or in lamellae depending on the functional requirements of the tissue.
Elastic fibres can be visualized (Figure 1) by light microscopy also using specific stains such as Verhoeff-Van Gieson or Weighert's (20), by electron microscopy (21, 22), by fluorescence and confocal microscopy after haematoxylin-eosin/phloxine staining (23) and even in vivo by multiphoton microscopy due to its intrinsic fluorescence properties (24).
Figure 1
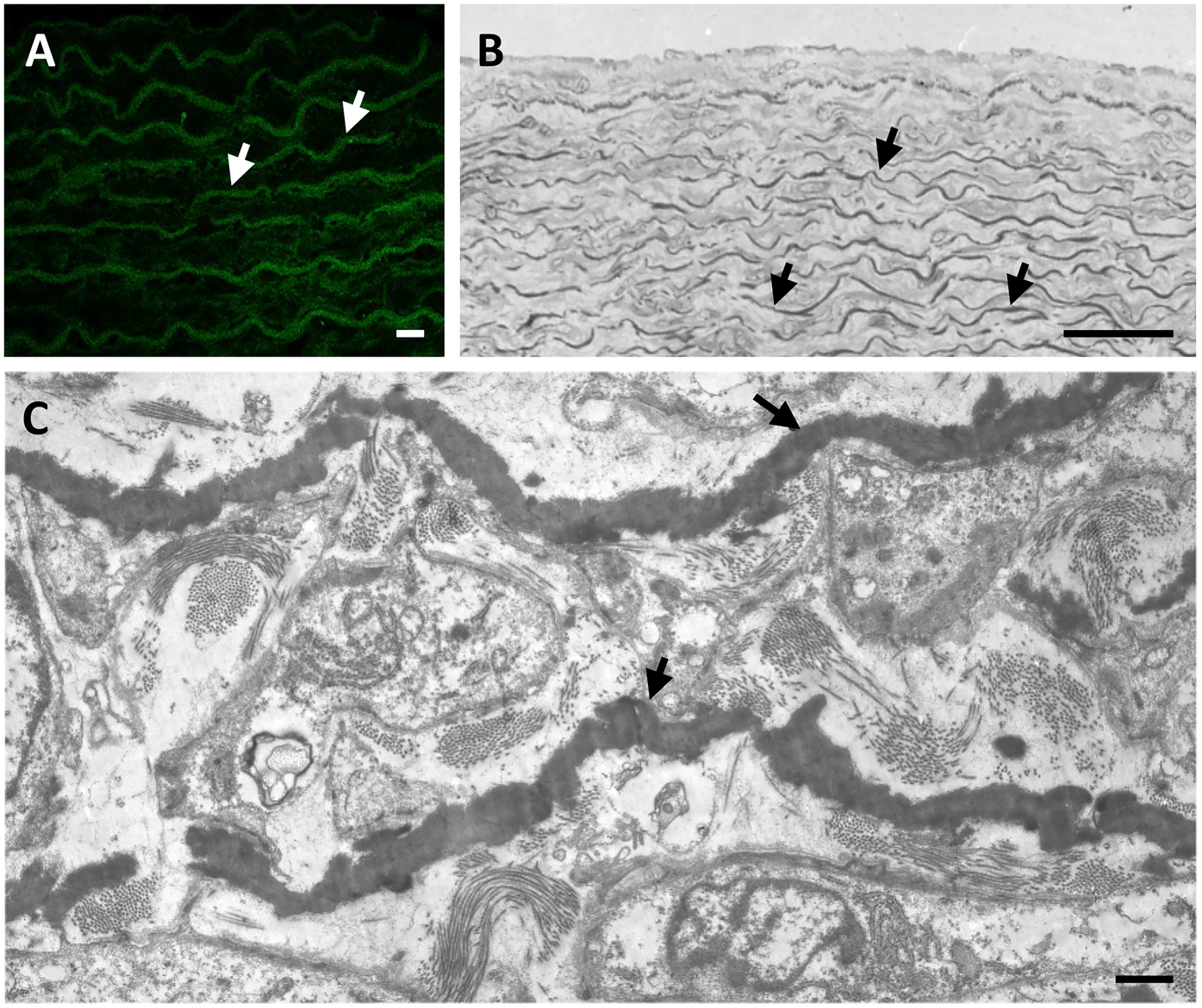
The elastic component (arrows) in arteries is typically organized in form of lamellae parallel to the lumen and alternated with smooth muscle cells in animal models such as rat (A) and in humans (B,C). Elastic fibres in the rat aorta are shown by fluorescence microscopy after staining with haematoxylin and eosin H (A). Human foetal aorta is visualized by light microscopy after toluidine blue staining of semithin sections (B) and by transmission electron microscopy of ultrathin sections (C). Samples have been routinely processed for morphological analyses (18, 19) and figures are unpublished original images from the Authors' archive. Scale bar: 10 μm (A); 100 μm (B); 1 μm (C).
Elastin represents approximately 90% of elastic fibres but, to date, more than 30 molecules have been shown to associate with fibres (Table 1) contributing to their formation and maintenance in vivo (66).
Table 1
| Elastic fibre components | ||||
|---|---|---|---|---|
| Description | Accessiona | Protein ID | Gene | References |
| Basement membrane-specific heparan sulphate proteoglycan core protein (Perlecan) | P98160 | PGBM | HSPG2 | (25) |
| Biglycan | P21810 | PGS1 | BGN | (26, 27) |
| Collagen alpha-1(VI) chain | P12109 | CO6A1 | COL6A1 | (28) |
| Collagen alpha-2(VI) chain | P12110 | CO6A2 | COL6A2 | |
| Collagen alpha-3(VI) chain | P12111 | CO6A3 | COL6A3 | |
| Collagen alpha-6(VI) chain | A6NMZ7 | CO6A6 | COL6A6 | |
| Collagen alpha-1(VIII) chain | P27658 | CO8A1 | COL8A1 | (28) |
| Collagen alpha-2(VIII) chain | P27658 | CO8A2 | COL8A2 | |
| Collagen alpha-1(XVI) chain | Q07092 | COGA1 | COL16A1 | (28) |
| Chondroitin sulphate (CS) | – | – | – | (26, 27, 29) |
| Decorin | P07585 | PGS2 | DCN | (26, 29) |
| EGF-containing fibulin-like extracellular matrix protein 1 (Fibulin-3) | Q12805 | FBLN3 | EFEMP1 | (30, 31) |
| EGF-containing fibulin-like extracellular matrix protein 2 (Fibulin-4) | O95967 | FBLN4 | EFEMP2 | (30, 32–34) |
| Elastin | P15502 | ELN | ELN | (28) |
| Emilin-1 | Q9Y6C2 | EMIL1 | EMILIN1 | (28) |
| Emilin-2 | Q9BXX0 | EMIL2 | EMILIN2 | (28) |
| Fibrillin-1 | P35555 | FBN1 | FBN1 | (35, 36) |
| Fibrillin-2 | P35556 | FBN2 | FBN2 | (35) |
| Fibrillin-3 | Q75N90 | FBN3 | FBN3 | (37) |
| Fibulin-1 | P23142 | FBLN1 | FBLN1 | (30) |
| Fibulin-2 | P98095 | FBLN2 | FBLN2 | (30) |
| Fibulin-5 | Q9UBX5 | FBLN5 | FBLN5 | (32, 38–40) |
| Heparan sulphate (HS) | – | – | – | (25) |
| Latent-transforming growth factor beta-binding protein 1 | Q14766 | LTBP1 | LTBP-1 | (41, 42) |
| Latent-transforming growth factor beta-binding protein 2 | Q14767 | LTBP2 | LTBP-2 | (43, 44) |
| Latent-transforming growth factor beta-binding protein 3 | Q9NS15 | LTBP3 | LTBP-3 | (42) |
| Latent-transforming growth factor beta-binding protein 4 | Q8N2S1 | LTBP4 | LTBP-4 | (42) |
| Lysyl oxidase homolog 1 | Q08397 | LOXL1 | LOXL1 | (45) |
| Microfibril-associated glycoprotein 3 | P55082 | MFAP3 | MFAP3 | (28) |
| Microfibril-associated glycoprotein 4 | P55083 | MFAP4 | MFAP4 | (28) |
| Microfibrillar-associated protein 1 | P55081 | MFAP1 | MFAP1 | (28) |
| Microfibrillar-associated protein 2 | P55001 | MFAP2 | MAGP-1 | (46, 47) |
| Microfibrillar-associated protein 5 | Q13361 | MFAP5 | MAGP-2 | (48, 49) |
| Osteopontin | P10451 | OSTP | SPP1 | (50) |
| Protein-lysine 6-oxidase | P28300 | LYOX | LOX | (45) |
| Transforming growth factor-beta-induced protein ig-h3 | Q15582 | BGH3 | TGFBI | (51) |
| Versican | P13611 | CSPG2 | VCAN | (52) |
| Vitronectin | P04004 | VTNC | VTN | (28) |
| Elastic fibre associated molecules | ||||
| 67-kD elastin-binding protein (EBP), spliced variant of β-galactosidase | – | – | – | (53, 54) |
| ADAMTS-like protein 3 | P82987 | ATL3 | ADAMTSL3 | (55) |
| ADAMTS-like protein 4 | Q6UY14 | ATL4 | ADAMTSL4 | (56) |
| ADAMTS-like protein 5 | Q6ZMM2 | ATL5 | ADAMTSL5 | (57) |
| Clusterin | P10909 | CLUS | CLU | (58) |
| Endostatin - C-terminal fragment of collagen XVIII | – | – | – | (59) |
| Fibronectin | P02751 | FINC | FN1 | (60) |
| Integrin-binding sialoprotein | P21815 | SIAL | IBSP | (61) |
| Matrix Gla protein | P08493 | MGP | MGP | (62) |
| Protein-glutamine gamma-glutamyltransferase 2 | P21980 | TGM2 | TGM2 | (63) |
| SPARC (osteonectin) | P09486 | SPRC | SPARC | (61) |
| Tenascin | P24821 | TENA | TNC | (64) |
| Tenascin-X | P22105 | TENX | TNXB | (65) |
Elastic fibre components and associated molecules.
Accession numbers were retrieved from the Protein Database UniProtKB.
In blood vessels, elastin is present either in form of circumferentially aligned lamellae alternating in the media with syncytia of VSMC, or as a mesh of thin and loose fibres and sheets (i.e., valves) conferring the extensibility and the elastic recoil typical of these tissues. The number of lamellae decreases along the arterial tree, with larger elastic arteries having up to 60 layers in humans, whereas peripheral muscular arteries having less than 3 layers (67). In fact, the amount of elastin in blood vessels is highly dependent on their size and functional role related to the tensile cyclic forces acting radially and longitudinally on the vascular wall. The elastin content in large arteries is one and a half times that of collagen. Medium and small vessels, containing more VSMCs and less elastic tissue, are characterized by lower stretching capabilities. Veins have a similar structure, but exhibit a lower wall thickness compared to arteries and their elastic content is one third of that of collagen (68). The presence of the elastic component in blood vessels is fundamental for a proper function of the heart since elastin allows vessels to store a portion of the stroke volume with each systole and discharging that volume with diastole (Windkessel effect), thus decreasing the load on the heart, minimizing the systolic flow, and maximizing diastolic flow in the arterioles throughout the cardiac cycle (69).
In the valves, elastic fibres, although represented in a lower percentage (13%) compared to collagen (50%), are responsible for the extension of cusps beyond 50% and for the subsequent recoil. This finding indicates that mechanical properties are not dependent on the elastin content per se, but more likely to the organization and distribution of the fibres that are different in the fibrosa (the side of the valve that is more under compression and is more fibrotic with densely aligned collagen fibrils) and in the ventricularis (the side of the valve that is more under tension and is characterized by higher elasticity and radially oriented collagen fibrils) (70).
2.1 Elastin synthesis and assembly
Elastin is a single gene-copy protein, although multiple isoforms can be produced by alternative splicing (71), and consistent differences in amino acid composition have been observed between vertebrate species and even between tissues in the same organism (72).
Elastin is synthesized as a precursor of 60–70 kDa named tropoelastin (TE) that starts to be assembled in form of small globules within cells or at the cell surface before undergoing assembly and crosslinking by enzymes of the lysyl oxidase (LOX) family. TE is characterized by alternation of highly hydrophobic and hydrophilic domains, which are responsible for aggregation and formation of cross-links, respectively. The aggregation rate, the size, and other properties of the globules are modulated by several factors such as temperature, pH, ionic strength, concentration of individual TE monomers, and by the presence of elastin associated molecules. TE is characterized by high hydroxyproline content, although with differences according to tissues, being lower, for instance, in blood vessels compared to lung (1). Once synthesized, TE binds to an elastin binding protein that protects TE from self-aggregation and from proteolytic degradation during elastogenesis and acts as a chaperone to the cell membrane, where, together with two other membrane-bound components, it becomes part of the elastin-receptor complex (73). In the extracellular environment, an important step in elastin assembly is represented by the deposition of TE spherules onto a microfibrillar scaffold (Figure 2A) through the interaction with several molecules, most of them remaining entrapped within the elastic fibres (75). Mature elastic fibres are characterized by a typical amorphous structure although, by different morphological approaches, they exhibit a filamentous structure whose supramolecular assembly confers the elastic properties of the fibres (Figures 2B–D).
Figure 2
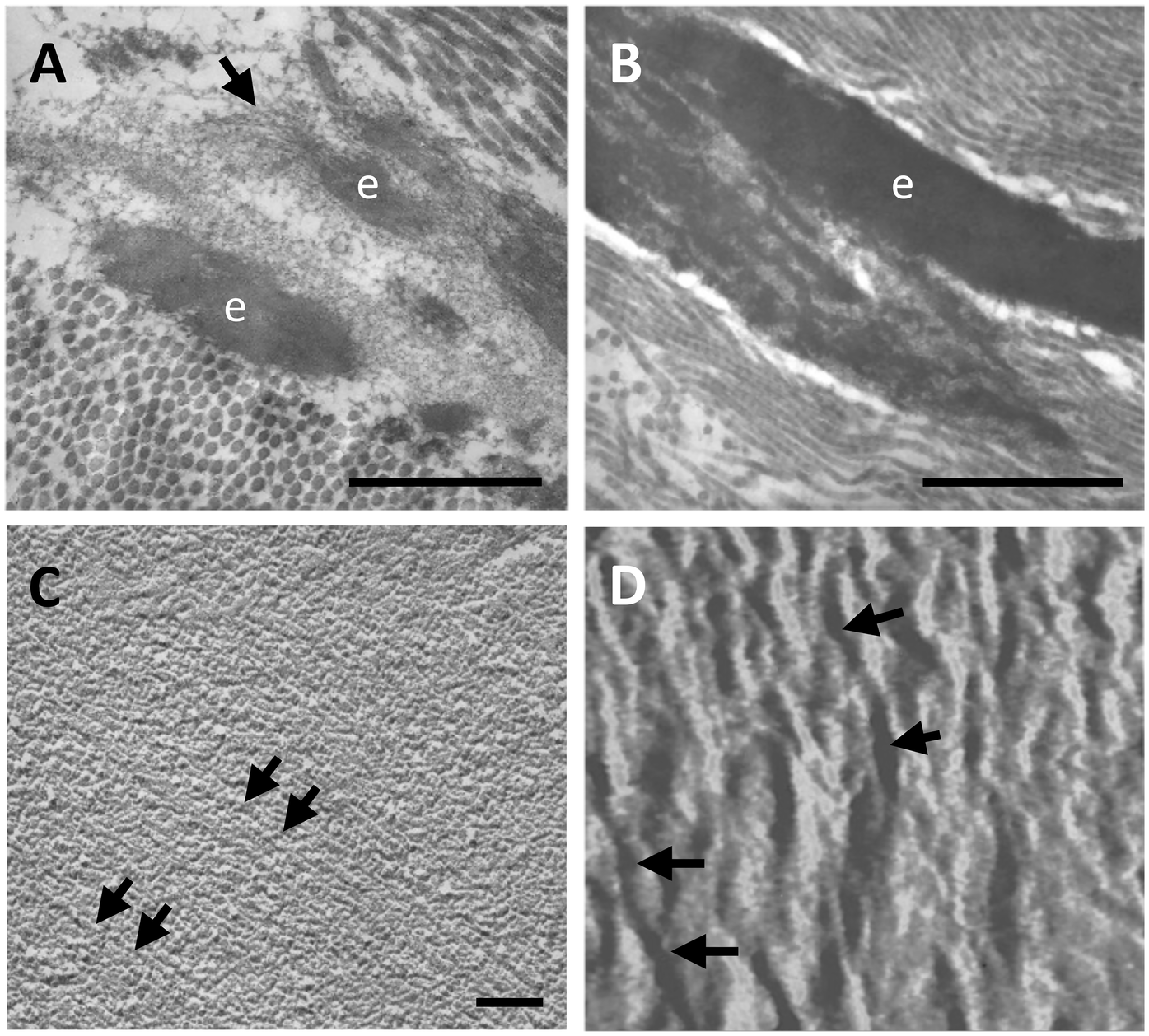
Elastic fibres (e) within the human aorta are shown, by transmission electron microscopy, as amorphous deposits onto a microfibrillar scaffold (arrow) (A) which over time progressively grow to form larger amorphous structures (B). However, the elastic component is not completely amorphous. Indeed, an isolated elastic fibre from bovine nuchal ligament reveals parallel organized fibrils (arrows) both by freeze fractured transmission electron microscopy (C) and by atomic force microscopy (D). Samples have been routinely processed for morphological analyses (19, 74) and figures are unpublished original images from the Authors' archive. Scale bar: 1 μm (A,B), 0.1 μm (C). Scanning range: 700 nm (D).
3 Calcification of the elastic component
Elastin plays a crucial functional role in conferring tissue elasticity; however, it is also susceptible to calcium deposition and tissue stiffening (Figure 3).
Figure 3
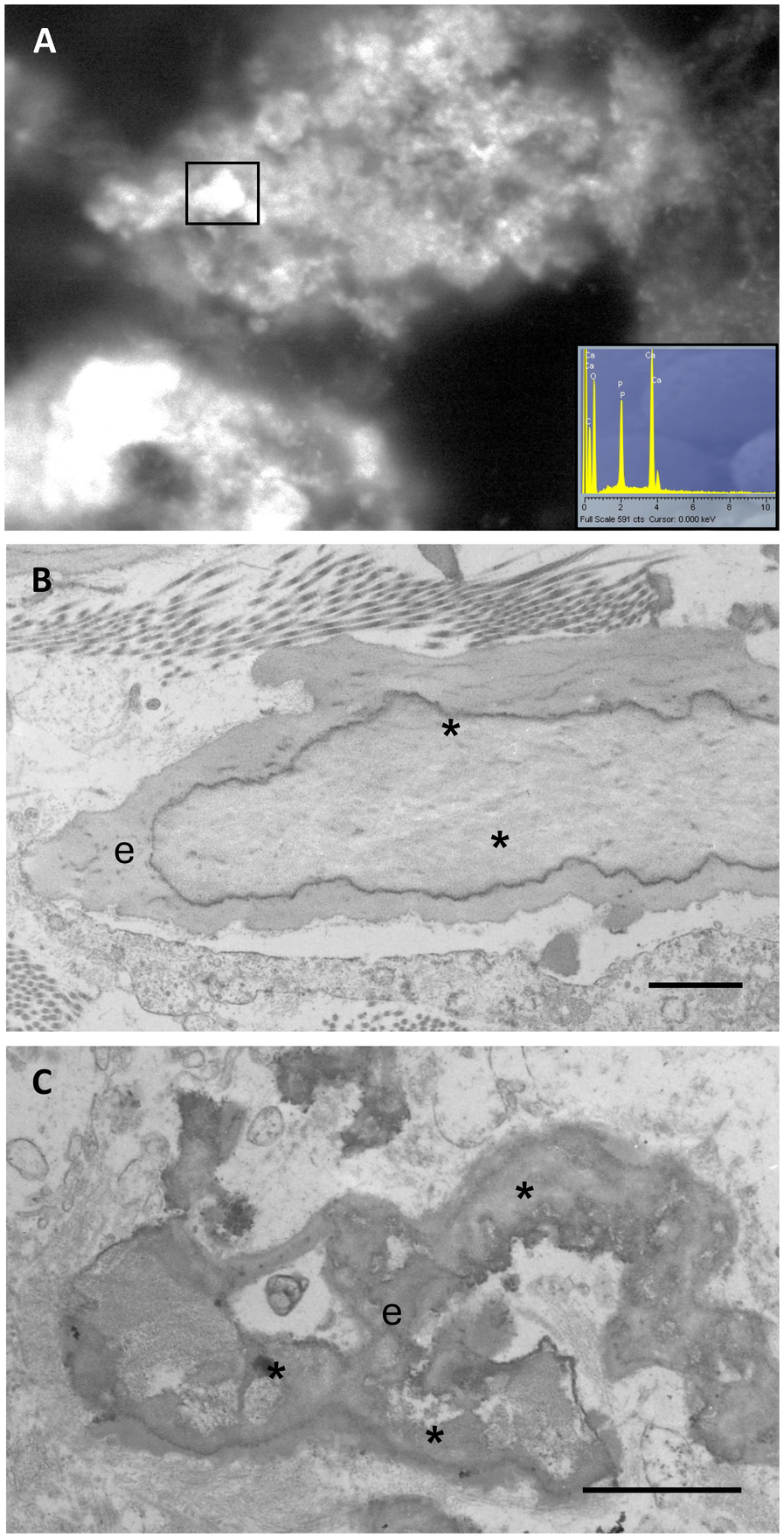
Calcified elastic fibres are observed by wet scanning electron microscopy and x-ray microanalysis (insert), demonstrating that bright mineral deposits (rectangle) consist of calcium and phosphate (A). Calcification (*) in human arteries, observed by transmission electron microscopy, appears within the core of the elastic fibres (e) (B) or in form of deforming aggregates (C). Samples have been routinely processed for morphological analyses (19, 21) and figures are unpublished original images from the Authors' archive. Scale bar: 1 μm (B,C).
Calcium phosphate deposits are usually formed through a multistep process starting from the development of amorphous calcium phosphate minerals [e.g., dicalcium phosphate dihydrate – DCPD - or brushite, CaHPO4⋅2(H2O)] followed by a transition to octacalcium phosphate [OCP, Ca8(HPO4)2(PO4)4⋅5H2O)] and apatite [Ca10(PO4)6(F,OH,Cl)2] or hydroxyapatite [HA, Ca10(PO4)6OH2] accompanied by progressive crystal growth (76). Crystal growth can be inhibited by chemisorption of pyrophosphate that, without attracting Ca as a counter ion, replaces Pi, a necessary counterpart of calcium in mineral deposits (77–79). It is interesting to note that polyphosphates, beside their inhibitory activity, can indirectly promote mineralization being a substrate of alkaline phosphatases that generates two phosphate ions capable of calcium binding (80).
The onset of calcification is not associated with surface precipitation of calcium phosphate, but it is more likely due to a complex biomineralization process that occurs within the bulk of the tissue (81). In addition, mineral–associated vesicles (MVs) from cells undergoing osteoblastic differentiation can nucleate and grow apatite crystals when bound to the elastin component (82, 83). However, evidence of elastin mineralization usually occurs after clinical manifestation or instrumental detection of mineral deposits in later stages of calcification, therefore the initial events triggering the process are still underexplored and the specific initial binding sites for calcium remain unclear.
Based on experimental models, two calcium binding sites have been proposed: neutral backbone carbonyl groups (C = O) and negative carboxyl groups (COO−), either as side groups of the aspartic acid and glutamic acid amino acids within elastin or as terminal groups of elastin-derived peptides. In the early 1970s, Urry proposed that backbone carbonyl groups act as calcium-binding sites, demonstrating that the binding occurred also in highly acid conditions (84, 85). Typically, elastin binds calcium through uncharged coordinating groups (i.e., neutral sites) progressively becoming positively charged and therefore attracting negatively charged ions such as phosphate, facilitating mineral deposition and further calcium ion binding (86).
Interestingly, coacervation and cross-linking, two key processes that in in vivo lead to elastic fibre formation, enhanced the exposure of backbone carbonyl groups, increasing their availability for calcium binding and subsequent mineralization (87). Indeed, Kaibara et al., investigating ion transport across a coacervate membrane composed of bovine neck ligamental α-elastin, found that calcium ion transport was mediated by specific and selective interactions with elastin neutral sites (88). In particular, the glycine-rich neutral sequences may act as nucleation sites for calcification also after elastin degradation (89).
However, other studies showed that also the carboxyl groups may participate in elastin calcification (90). Indeed, calcium ions bind to purified elastin in a manner dependent on both calcium concentration and pH, supporting a mechanism based on electrostatic interactions between Ca2+ and COO− (91). Consistently, studies in non-calcified and calcified insoluble bovine elastin revealed that elastolysis caused a greater abundance of carboxyl groups in mineralized samples (92). Similar findings were also obtained using coacervated insoluble elastin fibrillar structures, which, even when they were only partially degraded by elastases, exhibited increased calcification due to the amount of negatively charged COO− groups formed during elastin degradation (14). Therefore, the density of COO− groups can influence not only the extent and the rate of elastin calcification but also the transformation of the CaP mineral phase (93).
Very recently, Lau and coworkers proposed that, during elastin calcification, backbone carbonyl groups represent the primary calcium binding sites (94), although the carboxyl groups may also participate (90). This hypothesis is supported by the high density of backbone carbonyl groups in elastin (95), the low turnover rate of elastin (3, 96), and the diminished electrostatic interactions of carboxyl groups in the aqueous environment (97, 98).
Overall, these data underline the complexity of the intermolecular interactions of elastin and indicate that the involvement of backbone carbonyl or of carboxyl groups may depend on the experimental conditions such as the use of elastin or of elastin-derived peptides, coacervated or not, degraded or non-degraded, and of the micro-environmental characteristics (e.g., pH, ion concentration and ion type).
Moreover, elastin calcification is associated not only with degradation but also with alterations in amino acid composition, which occur during aging (99) and/or in pathological conditions (e.g., atherosclerotic plaque progression) (100). These changes include an increase of polar amino acids, a decrease of non-polar amino acids, and a reduction of desmosine and isodesmosine content, leading to structural weakening of elastin (Figure 4), altered interactions with the numerous molecules exhibiting qualitative and quantitative changes with time and during disease, enhancing the susceptibility to an in vivo slowly occurring calcification process.
Figure 4
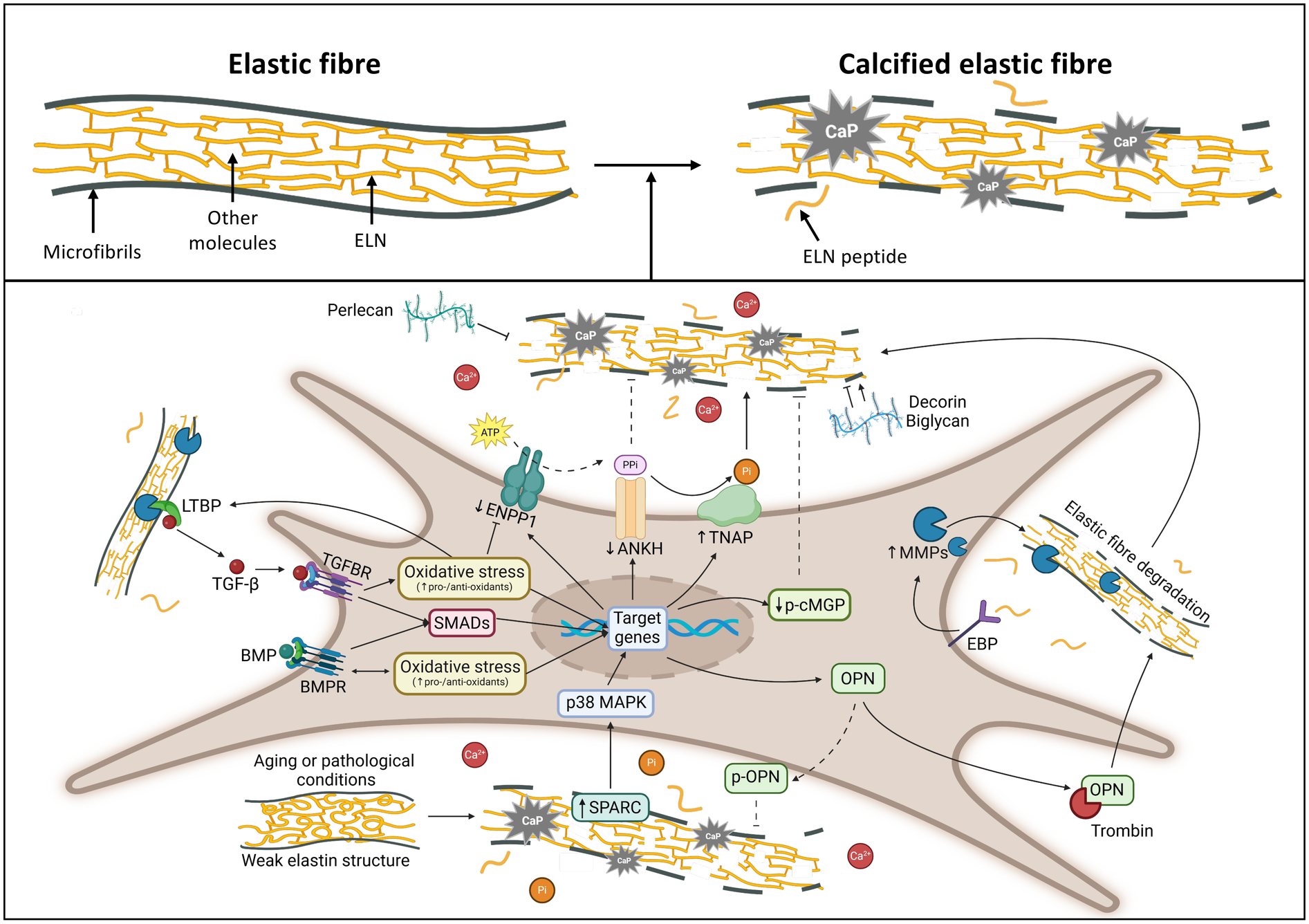
Drawing highlighting the major molecules and pathways involved in elastic fibre calcification. Created with https://www.BioRender.com. ANKH, progressive ankylosis protein homolog; BMPs, bone morphogenetic proteins; BMPRs, bone morphogenetic protein receptors; EBP, elastin binding protein; ELN, elastin; ENPP1, ectonucleotide pyrophosphatase/phosphodiesterase family member 1; LTBPs, latent transforming growth factor-beta binding proteins; MAPK, mitogen-activated protein kinase; p-cMGP, phosphorylated-carboxylated Matrix gla protein; MMPs, matrix metalloproteinases; OPN, osteopontin; p-OPN, phosphorylated osteopontin; Pi, inorganic phosphate; PPi, inorganic pyrophosphate; SMADs, small mother against decapentaplegic; SPARC, secreted protein acidic and cysteine rich; TGF-β, transforming growth factor beta; TGFBRs, transforming growth factor beta receptors; TNAP, tissue-nonspecific alkaline phosphatase.
4 Elastic fibres components and associated molecules and their involvement in the calcification process
Elastic fibres are composed of several components but only for few of them it is known their involvement in the calcification process.
4.1 Fibrillins
As already mentioned, TE is progressively deposited onto a microfibrillar scaffold mainly composed of fibrillins (FBNs). FBNs (1–3) are cysteine-rich glycoproteins characterized by an extensible periodic beaded organization providing both structural and regulatory roles depending on the tissue and the developmental stage. In the vascular system, for instance, VSMC start to synthesize elastin, FBN1 and FBN2 from the mid gestational stage up to the neonatal life (101), although FBN2 seems to be required during development, whereas FBN1 appears mostly important in adults to maintain connective tissue homeostasis and integrity. FBN1 variants are associated with the Marfan syndrome phenotype and, more generally, with cardiovascular, ocular and skeletal defects (102, 103), whereas mutations in FBN2 affect the musculoskeletal system with only occasional cardiovascular signs (104). Interestingly, FBN3 is restricted to embryonic/foetal tissues and has been suggested to play a role in the female reproductive organs, although it is not expressed in animals such as mice and rats (105). In addition to their role as structural components of the microfibrillar scaffolds, FBNs influence the bioavailability of transforming growth factor-beta (TGF-β) and interact with cells through integrin-associated RGD sequences, as well as with cell surface heparan sulphate (HS) proteoglycans, fibronectin and collagen type VI (106).
Exome sequencing performed in beta-thalassemia patients with ectopic calcification, disclosing rare sequence variants in genes related to elastic fibre assembly and integrity, led to the hypothesis that altered fibrillins and collagen type VI molecules may affect the microfibrillar scaffold, thus making elastic fibres less resilient and more prone to hydroxyapatite deposition (107).
Since fibrillin microfibrils vary in structure, composition, distribution and possibly functional roles depending on the tissue, they can modify the susceptibility of elastic tissues to calcify, and can differently interact with molecules such as calreticulin and protein-disulfide isomerase (PDI) (108), known to be involved in the vitamin K-dependent carboxylation of matrix gla protein (MGP) (109, 110).
Moreover, fibrillins contain several calcium-binding EGF-like domains (111), thus suggesting that these residues may have a special impact in the calcification process (112), although it has been suggested that fibrillins may not support mineral deposition directly, but though its modulation of TGF-β (transforming growth factor-beta) and BMP (bone morphogenetic protein) signalling (113) (Figure 4).
Indeed, BMPs represent a large family (approximately 20 subfamily members) of secreted signalling molecules belonging to the TGF-β superfamily. Although firstly isolated from bones, thereafter it has been clearly demonstrated their ability to induce soft connective tissue calcification, especially for BMP-2, -4, -5, -6, -7 and -9. These BMPs, except BMP-7 that may have an inhibitory activity, have been localized in sites of vascular calcification (114). Both BMPs and TGF-β transduce signals through SMAD-dependent and -independent pathways (19) requiring complex regulatory mechanisms (e.g., latency control in the matrix, extracellular antagonists, ubiquitination and phosphorylation in the cytoplasm, nucleus-cytoplasm transportation, and transcriptional co-regulation in the nuclei) (115). SMAD-signalling pathways (Figure 4) are crucial for the activation of several pro-osteogenic genes in the context of elastin metabolism and elastocalcinosis (116).
4.2 Glycosaminoglycans and proteoglycans
The interactions between elastin and GAGs have been demonstrated for decades (117, 118). GAGs are negatively charged polysaccharides, which can be either sulphated (heparan sulphate, HS; chondroitin sulphate, CS; keratan sulphate, KS; dermatan sulphate, DS) or non-sulphated (hyaluronic acid), and are covalently attached to a core protein to form a huge variety of PGs with different tissue distribution (119). They are widely present in the ECM, but also in the pericellular areas, on the cellular surface and in intracellular secretory granules (120). According to their characteristics, PGs modulate how cells respond to the ECM and vice versa. Indeed, they contribute to tissue hydration, interact with growth factors, cell surface receptors and ECM molecules regulating not only protein assembly and supramolecular organization, but also modulating cell adhesion, proliferation, migration, differentiation, apoptosis and cell signalling (120). In the extracellular environment, GAGs interact with TE and with FBNs promoting the assembly of TE and of the microfibrillar scaffold (121–123). HS, in particular, although it does not seem to influence elastin gene expression and/or the elastin synthesis, promotes aggregation and assembly of isolated elastin molecules and of elastin peptides and increases the expression of fibulin 5 in cell cultures also from aged donors (124, 125).
GAGs and PGs in vascular disease have been largely investigated due to their role in maintaining vascular architecture and their increases in pathological conditions, thus altering the proportion of other components (e.g., collagen and elastin) as well as their reciprocal interactions with cells (126). It has been proposed that GAGs accumulation in the vessel wall may serve as an adaptive compensatory response to increased mechanical stress and inflammation, since GAGs, attracting water molecules, cause a tissue swelling which may help absorbing mechanical stress as observed in aged and/or in diseased vessels (127).
The importance of GAGs and PGs in the mineralization process has been extensively investigated due to their hydrodynamic properties (depending on molecular weight, molecular size and viscosity) and high charge density interfering with the diffusion of calcium and phosphate ions (128–130). It has been proposed that negatively-charged groups in PGs interact with active growth sites on the HA nuclei to reduce apatite growing or that, depending on their size, may shield the active sites on the HA seed crystals from the availability of calcium phosphate ions (131). Consistently, it has been recently highlighted that pretreatment of bioprosthetic heart valves with a “HPA/NT/HRP” enzyme-oxidative-polymerization strategy exerts anti-calcification properties by improving GAGs stability, thus prolonging the efficiency of these valves substitutes (132).
However, evidence has been also provided for a possible role of some PGs as inducers of calcification in blood vessels (133). For instance, perlecan, a HS-PG, interacts with elastin, FBN 1 (134) collagen type VI and with several growth factors and signalling molecules (e.g., fibroblast growth factors, platelet-derived growth factor, vascular endothelial growth factor and BMPs) (135). It has multiple biological functions, promoting angiogenesis, tissue development and ECM stabilization playing a role as a blood shear-flow endothelial sensor that regulates blood volume and pressure (135) and consequently intercellular crosstalk. Reduced expression of perlecan was demonstrated in calcified atherosclerotic arteries (Figure 4), in contrast with the increased production of CS- and DS-PGs (136). The calcification inhibitory effect of perlecan can be related to the high presence of HS that exerts an important role in elastin coacervation and fibre assembly (125).
On the contrary, aggrecan, a CS-/KS-PG also known as cartilage-specific proteoglycan core protein, is up-regulated in the atherosclerotic plaques, where, by favouring lipoprotein accumulation, acts as an inducer of chronic inflammation promoting VSMC trans-differentiation (137).
Interestingly, decorin (a secreted CS-/DS-PG), and biglycan (a small leucine-rich repeat PG) act both as promoters and inhibitors in the mineralization process (138) (Figure 4). This apparent discrepancy is due to differences in the environmental conditions. In a cell-free model, decorin and biglycan bind to hydroxyl apatite crystals reducing hydroxyapatite crystal growth. By contrast, in a more complex scenario, the presence of growth factors, and of specific electrostatic interactions of PGs with LDL (low density lipoproteins), both in blood vessels and in valve leaflets (139), can favour calcification of the elastic component enhancing the interactions of elastin with fatty acids and lipophilic compounds (140). Indeed, elastin is known to have high affinity for lipids and calcium and to be more susceptible to elastolytic activities if fibres are altered by the enriched presence of lipids (141, 142).
4.3 Tenascin
Tenascin-C is a glycoprotein regulated by mechanical and tensile stress during development and in adult life, and colocalizes with elastin only in mineralized areas. Indeed, calcified elastin displayed a time-dependent pattern of tenascin-C, MMP-12 (matrix metalloproteinase-12), and alkaline phosphatase expression (64, 143). Similarly, tenascin-X, a large glycoprotein of the ECM which harbors binding site for collagens, decorin and elastin, is expressed in the vascular wall and in particular in association with the elastic lamellae (65). It has been implicated in the occurrence of a subset of Ehlers Danlos Syndrome (144) where, beyond collagen abnormalities, also irregular, fragmented and branching elastic fibres can be observed possibly due to altered interactions with collagen type VI (145). It is also worth mentioning that expression of tenascin-X is significantly reduced in calcific aortic valves (146).
4.4 Matrix Gla protein
Matrix Gla Protein (MGP) is expressed by VSMC in the intima and the media of vessels and undergoes post-translational modifications important for its activation and function (i.e., vitamin K-dependent carboxylation of five γ-glutamic acid residues and serine phosphorylation). For instance, MGP-derived peptides containing both post-translational modifications, added into calcium and phosphate solutions, significantly influence the nucleation and growth of HA in contrast to unmodified peptides. This model allowed to demonstrate that phosphorylation is more effective than carboxylation in modulating mineralization (147). Reduced expression of MGP has been associated with elastin calcification in Monckeberg's sclerosis (148), in Pseudoxanthoma elasticum (109), in thalassemia patients (149), in Singleton-Merten syndrome (150) and in knockout mice (151), demonstrating that p-cMGP (phosphorylated-carboxylated MGP) acts as an inhibitor of calcification (152) (Figure 4). It has been hypothesized that MGP can inhibit trans-differentiation of VSMCs and/or the formation of matrix vesicles (153). In the calcification process matrix vesicles have a functional role not only as a reservoir of HA and matrix components acting as intercellular signalling modules, but also as a source of MMP-2 that, by closely interacting with elastic fibres, favours their degradation and subsequent calcification (83). Mutations of the MGP gene cause Keutel syndrome characterized by a complex phenotype, including extensive arterial calcification, as clearly shown in Mgp−/− mice that die before 2 months of age (154, 155), exhibiting pronounced vascular calcification (151). The observation that MGP is colocalized with elastin (62) led to the hypothesis that the elastin content may be a key regulator of arterial calcification in Mgp–/– mice. Indeed, elastin haploinsufficiency can decrease arterial mineral accumulation (152).
4.5 Osteonectin
Osteonectin, also known as SPARC (secreted protein acidic and rich in cysteine) or BM40 (basement membrane 40), is one of the most abundant non-collagenous proteins expressed in mineralized tissues with high affinity for HA. It is localized in calcified areas on stromal cells and within elastic fibres and occasionally in association with lipids (61, 143). It has been suggested that osteonectin regulates the extracellular matrix mineralization through the p38 MAPK signal transmission pathway (Figure 4) that has a broad regulatory effect on alkaline phosphatase activity, on the response to osteogenic signals driven by BMP-2, TGF-β, parathyroid hormone, Wnt proteins, mechanical stimuli and pro-osteogenic transcription factors such as Runx2 (156).
4.6 Osteopontin
Osteopontin (OPN) is a highly phosphorylated sialoprotein found in mineralized extracellular matrices where it mediates HA binding through polyaspartic acid sequence and sites of Ser/Thr phosphorylation (157), although, more recently, it has been demonstrated to play multiple modulatory roles including vascular remodelling (158), supporting the observations that it is present in normal elastic fibres (61). While osteopontin, in its full-length phosphorylated form, has been shown to inhibit calcium phosphate production (Figure 4), other sialoproteins, such as bone sialoprotein, can act as a nucleator of calcium phosphate formation. This behaviour can be related to the type of interactions with mineral crystal nuclei depending on the primary structures of the sialoproteins and to the extent to which they are phosphorylated. More specifically, polyglutamate-containing bone sialoprotein acts as a nucleator, while the polyaspartate-containing osteopontin inhibits calcium phosphate formation and growth (159). Although osteopontin is relatively protease-resistant, it has been shown that it can be hydrolysed by thrombin (Figure 4) exposing a cryptic α4β1/α9β1 integrin-binding motif (SVVYGLR), thus exerting pro-inflammatory, pro-angiogenic and pro-osteogenic effects favouring aortic MMP-9 activation (160).
5 Elastin degradation and the calcification process
Due to its almost absent turnover, elastin is characterized by high resistance and durability. However, there are several elastolytic enzymes, belonging to the class of metallo-, cysteine- and serine-endopeptidases, that can degrade elastic fibres (i.e., TE, elastin and FBNs) (161) (Figure 4), lowering their functions, and at the same time releasing bioactive peptides called elastokines (3). These fragments, depending on their amino acid sequence, can interact with different types of receptors inducing a variety of biological activities (e.g., angiogenesis, cell proliferation, vasorelaxation), which may lead to the development of age-related morpho-functional alterations and/or to the progression of pathological conditions (162, 163). Overproduction of elastolytic enzymes and/or a decrease of some of their inhibitors are frequently associated with inflammatory conditions where the release of elastin fragments with chemotactic properties for monocyte contributes to structural deterioration (e.g., atherosclerosis), enlargement and eventual rupture of large vessels (e.g., aortic aneurysms) and promote osteogenic differentiation and subsequent calcification (1, 23, 164, 165). Metallopeptidases activity is regulated either at the mRNA level or by proenzyme activation depending on substrate affinity/specificity, on the presence of specific inhibitors or on binding to other matrix molecules such as GAGs, PGs and other glycoproteins (e.g., tenascin-C) (143, 166, 167).
In particular, MMP-1, MMP-2, MMP-3, MMP-7, MMP-9 and MMP-12 have been demonstrated to bind to and to digest insoluble elastin (168, 169), their activation being associated with cardiac valve diseases, atherosclerosis, aortic aneurysms, and restenosis (7). Indeed, enhanced elastin calcification is directly associated with increased MMP expression (i.e., MMP-2 and -9), and also with up-regulated expression of osteogenic genes (e.g., ALP) (169, 170) (Figure 4).
Similarly, it has been reported, both in vitro and in vivo, that the elastolytic activity of cathepsin K, S and V may have a role in the progression of atherosclerosis as well as in vascular and valve calcification (89, 171). Since cathepsin activation can be regulated by interactions with GAGs and/or PGs (172), variations of tissue composition in terms of GAGs/PGs, and possibly other matrix components, may contribute to the different susceptibility to ectopic calcification of tissues and/or of areas within the same tissue (173).
6 Functional consequences of calcification in the cardiovascular system
Elastic fibres are present in all soft connective tissue and, with age, dermal, cardiovascular, and pulmonary tissues become less able to recoil, leading to the hypothesis that age-related failure of elastic fibres may dictate the apparent 100–120-year limit on human life expectancy (174). Nevertheless, the major clinical relevance of elastin calcification is observed in the cardiovascular system causing stiffening of arterial walls, higher values of systolic pressure, and increased incidence of cardiovascular events (175). Similarly, aortic valve calcification is the most common valve disease in the developed countries, frequently requiring heart surgery and/or transcatheter valve replacement since prolonged valve calcification may alter the heart's structure (e.g., enlargement or thickening of the heart walls) affecting cardiac function (176). Aortic valves leaflets are susceptible to risk factors similar to those of atherosclerosis, moreover they are constantly subjected to mechanical stress but if these stimuli exceed the physiological range or if the capability to respond to mechanical stress is insufficient, valves undergo calcification with increased expression of osteogenic markers (e.g., BMP2 and RUNX2), changes in the behaviour of valve interstitial cells and altered composition/organization of the extracellular components (177, 178).
Vascular calcification is a typical complication occurring in patients with diabetes, atherosclerosis, chronic kidney disease, hypertension, thus causing a progressive stiffening of blood vessels with altered blood flow and altered mechanical signals provided to endothelial and smooth muscle cells and their crosstalk (179).
Indeed, blood vessels are typically exposed to the pulsatile pressure formed by heart contraction, however, their morphological structure and organization exert different mechanical and hemodynamic properties due to blood flow velocity gradient on the surface of the vessel wall and blood viscosity (180). Indeed, the wall shear stress (WSS) acting on the vessel wall ranges from 1 dyne/cm2 in the venous endothelium to 40 dyne/cm2 in the calcified arteries (181). Within this context, the ratio between collagen and elastin and tissue elasticity are crucial for maintaining a dynamic balance between growth and remodelling under a certain range of blood pressure and consequently modulating the organization and alignment of endothelial cells. Compared to arteries, despite the significantly lower WSS, veins exhibit lower levels of calcification, possibly due to less organized layers of elastic lamellae and smooth muscle cells that are also characterized by lower osteogenic trans differentiation. Furthermore, the lower venous pressure and the presence of venous valves, maintaining unidirectional blood flow and preventing blood reflux, can play a role in preventing calcification (181).
Within arteries, calcifications occur in the different layers of the wall depending on the disease context. For instance, intimal calcification, that is mainly involved in atherosclerosis, begins with microcalcifications (less than 20 μm in size), originating from the lipid pool and the necrotic core, and then progressively developing into sheet, nodular or plaque calcifications representing predictors of unstable plaque at the risk of rupture, whereas extensive calcification is associated with plaque stability. On the other side, medial calcification is a chronic systemic vascular disease commonly seen in diabetes, chronic kidney disease and is characterized by spread calcium phosphate deposits, leading to decreased compliance of the whole vessel wall and to increased incidence of cardiovascular complications. Therefore, medial calcification is considered a strong and independent risk factor for cardiovascular death and predictor of risk of future coronary events in patients with diabetes (182).
Indeed, calcification in peripheral artery disease (PAD) has multiple consequences accelerating pulse wave velocity from heartbeats, impairing microcirculation and reducing the artery's capacity to adjust blood flow according to physiological demands thus impairing tissue perfusion (183). In the lower extremity arteries, calcification poses additional challenges due to the deformations of blood vessels and of blood flow during limb flexion and eventually to microfractures of mineralized plates causing the release of cellular debris, calcium crystals, matrix peptides and lipid residues (184).
In agreement with these finding coronary artery calcium content is regarded as an arterial manifestation with prognostic value, and extra coronary calcification (e.g., calcification of the aorta or mitral valve) is also independently associated with cardiovascular disease (185). The ARIC (Atherosclerosis Risk in Communities) Study reported that Pulse Wave Velocity (PWV) is strongly associated with descending aorta calcification and that among PWV measures, cfPWV (carotid femoral PWV) was most strongly associated with vascular and valvular calcification, followed by hfPWV (heart-femoral PWV) and haPWV (heart-ankle PWV). Interestingly, faPWV (femoral-ankle PWV) was inversely associated with vascular calcification (186).
Interestingly, microcalcifications have been described also in thoracic aneurysms of various aetiologies and these events are specifically associated with elastin fragmentation, vascular smooth muscle cell phenotypic switching, and increased aortic wall rupture risk on biomechanical testing. Higher microcalcifications have been observed associated with mild and moderate histopathologic thoracic aortic aneurysm severity, whereas severe pathological disease was associated with low levels of microcalcification, suggesting a nonlinear pathobiological course. While elastin fragmentation is known to have an augmenting effect on microcalcification, however total elastin destruction or loss seen in the end-stage disease have the opposite effect. This finding must be taken into account when assessing histological severity by microcalcification (187).
7 Models of elastin calcification
Given the complexity of the ectopic calcification process and the difference in the morpho-functional characteristics within the cardiovascular system, experimental models are crucial for understanding mechanisms, testing hypotheses, and developing new treatments. These models range from simple acellular systems to in vitro models of increased complexity and to animal models either treated with pro-osteogenic stimuli of genetically manipulated to better dissect the role of specific molecules and/or pathways. Each model has advantages and disadvantages (Table 2), although they are always capable to provide insightful information for translational perspectives.
Table 2
| Main features | Acellular model | 2D cell culture | 3D cell culture (spheroid model) | 3D cell culture (organoid model) | Invertebrate animal model | Vertebrate animal model |
|---|---|---|---|---|---|---|
| Easy to be established | Yes | yes | Yes | No | Yes | No |
| Easy to maintain for long time | Yes | Yes | Challenging | Challenging | Yes | Yes |
| Specialized facilities and expertise | No | No | Yes | Yes | Yes | Yes |
| Relative cost | Low | Low | High | Higher | Low | Higher |
| In vivo similarities of cell shape and differentiation | No | No | Yes | Yes | Yes | Yes |
| Cell-cell interactions | No | Limited | Yes | Yes | Yes | Yes |
| Cell-matrix interaction | No | Limited | Limited | Yes | Yes | Yes |
| Genetic manipulation | No | Yes | Yes | Yes | Yes | Yes |
| Physiological complexity | No | No | No | Limited | Yes | Yes |
| Recapitulation of human physiology/pathology | No | No | No | Limited | Limited | Partly |
| Ethics Committee approval | No | Depending on the source and type of cell line | Depending on the source and type of cell line | Yes | No | Yes |
Strengths and limits of model systems.
7.1 In vitro models
Elastin synthesis can be induced in vitro by cytokines and growth factors (e.g., interleukin 10, TGF-β, insulin-like growth factor) (188, 189) and/or matrix molecules (e.g., hyaluronan) (190), although elastic fibre development is almost negligible, thus limiting the study of elastogenesis in vitro (191). In fact, to be assembled, elastic fibres require the presence of a microfibrillar scaffold, as demonstrated in immortalized ciliary body pigmented epithelial cells (192) and in cardiovascular tissue equivalents (191); nevertheless, to our knowledge, these models were not used to investigate calcification of the elastin component.
Therefore, most of in vitro cell culture models, even using co-culture with endothelial and VSMC (193), investigate the calcification potential of cells, upon treatment with culture media supplemented with phosphate or with ascorbate, dexamethasone and beta-glycerophosphate (4), looking at mineral deposition of the overlaying extracellular matrix, but not specifically on elastic fibres.
To address the challenges associated with the in vitro development of elastic fibres, alternative models employing formed fibrils or elastin peptides have been proposed. It is well known that calcification entails the formation of different mineral crystal phases, which vary based on the pathological context and the physicochemical characteristics of the extracellular environment (194–197). Therefore, these in vitro systems provide valuable tools for investigating the nature and progression of mineral deposition and for assessing compounds with potential anti-mineralization properties.
For instance, cross-linked elastin-like polypeptide (ELP) membranes are employed to mimic medial arterial calcification processes (198, 199). In particular, Gourgas and collaborators developed synthetic elastin-like polypeptide membranes (ELP3), that recapitulate key motifs found in human tropoelastin (198, 200) and, when immersed in simulated body fluid (SBF), they represent a suitable substrate for mineral nucleation and growth (201) depending on SBF ionic strength and incubation time (200). This model further highlighted that amorphous calcium phosphate (ACP) and OCP act as transient precursor phases during mineralization, in agreement with observations in the Mgp−/− mice, where both ACP and OCP were detected in the medial layer of calcified arteries prior to transformation into more stable apatite forms (201). The model of the ELP3 membranes has also been used to assess the effect of calcification inhibitors such as m3pS, a synthetic peptide corresponding to the N-terminal region of MGP containing three phosphoserine residues (199). Interestingly, ELP3 membranes, incubated for several days in SBF with or without m3pS, showed that, in the absence of the peptide, both OCP and carbonated hydroxyapatite were detected, whereas m3pS-treated samples showed only trace amounts of DCPD and minimal HA formation. These results suggested that the presence of m3pS altered the mineral maturation and that serine phosphorylation in MGP played a direct role in preventing mineral phase maturation in in vitro elastin mineralization process (199).
It is noteworthy that ELPs, due their inverse temperature transition properties (i.e., a phenomenon where a substance becomes more ordered, or folds, as the temperature increases) and biocompatibilities, can be efficiently used for drug delivery, as demonstrated in vitro for the prolonged release of BMPs (up to 14 days) and their enhanced mineralization capabilities (202, 203).
Additionally, intrinsically disordered proteins (IDPs) have recently been suggested to represent an interesting model also for the calcification process (204). IDPs are proteins that, lacking a stable and fixed three-dimensional structure, exist in a dynamic ensemble of conformations, allowing to interact with multiple partners regulating cellular processes like signalling, gene regulation, and molecular recognition (205). Therefore, IDPs can establish intermolecular interactions at the protein–mineral interface, thereby modulating the nucleation and crystal growth (206). For instance, it has been observed that amelogenin, a highly conserved IDP, influences enamel biomineralization by transitioning from a disordered random coils structure to an ordered β-sheet conformation upon binding to forming crystals (207). In the light of these data, it has been recently explored the mineralization potential of IDP elastin-like recombinamers (ELRs) (208) that, due to their biocompatibility and biodegradability, are well-suited for biomimetic applications (209). These ELRs are recombinant polypeptides inspired by the repetitive elastin sequence Val-Pro-Gly-X-Gly (VPGXG), where X represents any amino acid except proline, typically found in the intrinsically disordered regions of tropoelastin which are characterized by a phase transition from a soluble unidimensional disordered chain of amino acids into a stable coacervate capable of complex intermolecular interactions (210). In particular, ELRs, composed by two distinct tropoelastin motifs, a hydrophobic sequence (VPGIG) and a positively charged segment (VPGKG where K serves as cross-linking site), when incubated in supersaturated mineralizing solution, induced the formation of ELR spherulites functioning as nucleation and templating sites for Ca-P crystallization not only on the surface but also throughout the internal structure of assembled ELRs (208).
Similarly, isolated insoluble bovine elastin coacervated in vitro to form fibrillar structures can be used in a reductionist approach to test the mineralization potential and/or to investigate the regulatory role of standard or of calcifying medium, with or without addition of pro- or anti-osteogenic molecules. For instance, chemical and enzymatic fragmentation of the fibrils markedly increased HA deposition. Furthermore, variations in the composition of calcifying medium, such as the use of DMEM enriched with HPO42− compared to HEPES buffer solution supplemented with both Ca2+ and HPO42−, modulate the extent of elastin calcification (14). The same in vitro model was used to test the mineralization inhibitory potential of GAGs. In particular, elastin fibrils were coacervated in the presence or absence of different concentration of HS, enzymatically digested and subsequently incubated in a calcifying medium. The results demonstrated an inverse correlation between HS concentration and the amount of calcium deposition on elastin fibrils (169). These data support the hypothesis that elastin degradation precedes calcification and underscore the importance of ionic environment as well as the type and the amount of extracellular matrix associated molecules, such as GAGs, in modulating mineral deposition on elastin matrices.
In the last years, nanomaterials, and in particular nanoparticles, have raised a huge interest for their physico-chemical properties, lower toxicity and biocompatibility (211). In the field of cardiovascular calcification, innovative and interesting models are represented by a tissue culture set-up using porcine aortic valve leaflets (212) or by murine aortic ring sections (213) that can be stimulated to calcify ex vivo. Samples treated for one week with human serum albumin-based nanoparticles conjugated with the chelator diethylenetriaminepentaacetate (DTPA) and with anti-elastin antibodies to specifically target calcified elastin exhibited reduction of calcium content (38%–46%) either in terms of progression of calcification or of reduction of existing crystals. The main advantage of the valve leaflet model is that valve interstitial cells are preserved maintaining interactions with the extracellular matrix and endothelial cells. However, the disadvantage is the transferability to humans because of the differences in the microstructure of porcine and human valves (212).
7.2 In vivo models
In vivo models are typically comprised by several mice knockout for specific molecules either related to elastic fibres or to the calcification process (155, 214, 215) or a combination of double knockout either in the homozygous or heterozygous condition depending if the genotype is life-compatible (Table 3). However, only in few cases a clear description of the calcification of the elastic component has been clearly reported.
Table 3
| Mouse models | Gene | Role/disease model |
|---|---|---|
| Eln+/− | Elastin (Eln) | Structural component of elastic fibres. Animals have reduced susceptibility to vascular calcification (152). |
| Fbn-1+/mgR | Fibrillin1 (Fbn) | Major component of elastin microfibrillar scaffold. Reduced expression of the monomer (15% of normal allele) is associated with focal calcification of intact elastic laminae (216). |
| Mgp−/− | Matrix gla protein (Mgp) | Vitamin K-dependent protein with anti-mineralization properties. The amount of deposited minerals in Mgp−/− arteries scales with elastin amounts, decreasing from the thoracic to the abdominal aorta (152, 217). |
| Mgp−/−Eln+/− | Matrix gla protein (Mgp) and elastin (Eln) | Animals have markedly reduced arterial mineral deposition compared with Mgp−/−mice indicating that also the elastin content is a critical determinant of arterial medial calcification (152). |
| MMP3 flox+/+ | Matrix Metalloproteases 3 (MMP3) | Matrix metalloproteinase with elastolytic activity. Animals exhibit suppressed phosphate-induced SMC osteogenic transformation and medial artery calcification (218). |
| Spp1−/− | Osteopontin (Spp1 also named Opn) | Inhibitor of mineralization present also within elastic fibres (219, 220). |
| Phospho1−/− | Phosphoethanolamine/Phosphocholine Phosphatase 1 (Phospho1) | Participates in the initiation of hydroxyapatite (HA) deposition inside MVs by scavenging Pi from phosphoethanolamine and phosphocholine (221). |
| Phospho1−/−Spp1−/− | Phosphoethanolamine/Phosphocholine Phosphatase 1 (Phospho1) and Osteopontin (Spp1) | Ablating Spp1 function prevents the development of the skeletal phenotype of Phospho1−/− mice, establishing a primary role for OPN, rather than PPi, in the pathophysiology of the Phospho1−/− defects (221). |
| Enpp1−/− | Ectonucleotide pyrophosphatase/phosphodiesterase (Enpp1) | The enzyme generates PPi, a strong inhibitor of ectopic mineralization. Animals exhibit abnormalities almost identical to those present in the tiptoe walking (ttw/ttw) mice such as altered bone mineralization and arterial calcification. Used model for Generalized Arterial Calcification in Infants (GACI) (222). |
| Enpp1asj/asj | Ectonucleotide pyrophosphatase/phosphodiesterase (Enpp1) | The presence of an inactivating mutation significantly reduces ENPP1 function. Animals have a median lifespan of 58 days and histologic examinations reveal calcification in hearts and aorta (223). |
| Alpl−/− | Alkaline phosphatase (Alpl also named Akp2) | The enzyme promotes mineralization catalysing the hydrolysis of the inhibitor PPi while concomitantly increasing the levels of Pi. It is involved in medial artery calcification. Animals are characterized by elevated levels of OPN whose amount correlates with the severity of hypophosphatasia (221, 224). |
| Ank−/− | Progressive ankylosis protein homolog (Ank also named Ankh) | A multipass transmembrane protein involved in the transport of the inhibitor PPi. (224). |
Mice models deficient for elastic fibres components and/or for promoters/inhibitors of calcification and exhibiting elastic fibres mineralization.
Since aging is frequently associated with an increased susceptibility to pro-calcifying factors (225), the Klotho-deficient mice, a well-known model of premature aging, developing also arterial calcification and elastin fragmentation, is used to investigate anti-calcifying treatments and to further elucidate the signalling pathways connecting aging and elastocalcinosis (226, 227). Indeed, Klotho is an aging-suppressor gene, but it acts also as a co-receptor for fibroblast growth factor-23 (FGF23) establishing the FGF23-Klotho endocrine system required for the maintenance of calcium-phosphate homeostasis (228). Klotho deficiency is correlated, for instance, to high levels of Bmps, Runx2 (an osteoblast transcription factor), phosphorylation of Smad1/5/8 and Smad2/3, osteoprotegerin, osteopontin and alkaline phosphatase (227, 229). Within this context, treatment with acetazolamide, an inhibitor of carbonic anhydrase used to promote diuresis thus lowering hypertension, was demonstrated to reverse the aging phenotype of the Klotho deficient mouse significantly reducing vascular calcification and osteogenic trans-differentiation (229).
In addition, an interesting model is represented by Abcc6−/− mouse resembling the genetic disease Pseudoxanthoma elasticum, where progressive calcification of elastic fibres has been associated to mutations in a gene encoding for a liver transmembrane transporter (230, 231), whose pathogenic role has not yet completely understood, further supporting the complexity of mechanisms leading to ectopic calcification. Therefore, this animal model has been frequently used to support and/or to validate results from studies in cell culture systems. It is the case, for instance, of the role of oxidative stress (110, 232, 233), vitamin K-dependent carboxylation of MGP (109, 110, 234, 235), alkaline phosphatase and pyrophosphate metabolism (236–239). Most importantly, using the Abcc6−/− model, in which the ectopic calcifications can be observed starting from 5 weeks of age, it was possible to highlight which are the alterations due to the gene deletion per se or the consequences of the calcified environment (240). Moreover, in the Abcc6−/− model, it has been demonstrated that progressive ankylosis protein homolog (Ank) and OPN expression are downregulated already before mineral deposition, whereas increased intracellular O2− levels, elevated TNAP activity, and Bmp2 upregulation are observed after the occurrence of calcification (240).
Invertebrate animal model (e.g., Saccharomyces cerevisiae, Drosophila melanogaster, Danio rerio) are widely used by researchers to study specific biological processes, since they have genetic characteristics similar to humans, are easy to maintain and to be reproduced in a laboratory setting, and easy to generate mutants suitable to explore certain traits or diseases.
For instance, in the cardiovascular context, zebrafish represents an interesting invertebrate animal model for its ability to produce elastic fibres. Although zebrafish expresses the abcc6a gene and exhibit morphogenetic changes upon the insertion of specific sequence variants (241), the resulting phenotype only partially mimics that of PXE, as ectopic mineralization is rarely observed in zebrafish mutants (242). In fact, in contrast to the limited expression of the gene in the liver of mammals, abcc6a is expressed in the eyes, heart, and intestines and not in the liver of young adult zebrafish (241). By contrast, the zebrafish expressing enpp1 mutants (i.e., model of the Generalized Arterial Calcification of Infancy) exhibit calcification of cartilage, skin, and cardiovascular system, such as the bulbus arteriosus (243).
A promising in vivo model is represented by subcutaneous implantation of glutaraldehyde-crosslinked vascular tissues into young rats, allowing to develop HA deposition onto elastic laminae (244). Results underline the importance of the environment in the calcification of the elastic component as it occurs in young patients undergoing valves replacement. In fact, children have higher serum phosphate, osteocalcin, parathormone and vitamin D compared to adults and consequently young patients have higher incidence of complications due to mineral deposition (245). The subdermal implant can be used to investigate the modulatory effects of exogenous molecules, such as Al3+ ions that were demonstrated to bind irreversibly to elastin, inducing conformational changes characterized by a reduction of the extent of β sheet structures and an increase in coil-turn structures, thus contributing to a complete inhibition of elastin calcification (246–248).
Supplementation of endogenous/exogenous molecules causing a calcium overload represents an efficient model to demonstrate the impact of inhibitors of calcification. For instance, a combination of vitamin D3 and nicotine (the so called VDN model) can induce a rapid hypercalcemia in rats, whereas the release of catecholamine is responsible for elastocalcinosis in large elastic arteries (249). These effects can be counteracted in rats by endothelin (250) and antioxidant molecules (251, 252).
Furthermore, the severe calcification induced in mice by overproduction of vitamin D3 can be downregulated by inhibitors of carbonic anhydrase (i.e, acetazolamide) since extracellular pH has a profound effect on calcium and phosphate solubility, which is enhanced by acidification and decreased by alkalinization (229).
An additional interesting in vivo model has been proposed by Basalyga and coworkers (167) who used a vascular injury model based on a single periarterial application of CaCl2 solution, which lead to calcium accumulation within elastic fibres and to elastin degradation, further highlighting the role of MMPs as key mediators of the calcification process. Consistently, MMP-deficient mice do not exhibit elastin fragmentation or vascular calcification.
Medial arterial calcification set up in the rat model was used to investigate antibody-target nanoparticles as a potential therapeutic strategy. In particular, chelating agents (e.g., EDTA) have been combined with human serum albumin-based nanoparticles which were functionalized with antibodies that selectively bind to specific epitopes exposed by degraded elastin. This treatment was able to reduce vascular calcification and improve arterial elasticity without side effects in bone or mineral metabolism (253, 254).
Although characterized by their cost-effectiveness, easy maintenance and manipulation and well-established methodologies, a limit of the models described so far is represented by physiological differences in heart rate, blood pressure and metabolism compared to humans. Therefore, large animal model of vascular calcification, such as pigs, can be used more efficiently in translational research and for testing of interventional devices, since they better mimic human cardiovascular physiology and vascular anatomy (255).
Since all models do not completely overlap what is occurring in humans, rare genetic diseases represent unique models for the discovery of fundamental and novel biological mechanisms. Indeed, monogenic defects, their penetrance in early life, and the expanding collection of human mutations and phenotypes make rare diseases ideal targets for exploring evolutionarily conserved biological mechanisms whose alterations may cause also common human diseases. However, also in this case there are some limits, such as the genetic heterogeneity among individuals, and the fact that human mutations can be exceedingly rare, thus preventing large-scale studies. Indeed, cardiovascular calcification is also a complication of several genetic diseases such as Generalized arterial calcification of infancy (GACI) (256), Pseudoxanthoma elasticum (PXE) (257), Singleton-Merten Syndrome (SGMRT) (258), Hutchinson-Gilford Progeria (HGPS) (259), calcification of joints and arteries (CALJA) (260), idiopathic basal ganglia calcification (261), as well as of chronic disease as in atherosclerosis, diabetes and chronic kidney disease (4, 262). Interestingly, in all these diseases, the causative gene is not directly related to elastin or to elastic fibre's components although, in most cases, elastic fibres are the preferential target of mineral deposition and therefore they may represent valuable models to deepen the knowledge on ectopic calcification. Indeed, a general unifying traits are represented by trans-differentiation of mesenchymal cells towards a pro-osteogenic phenotype (10, 263–266), increased expression and activity of tissue-nonspecific alkaline phosphatase (TNAP) and mitochondrial dysfunction, leading to oxidative stress and reduced availability of calcification inhibitors such as extracellular inorganic pyrophosphate (PPi) released upon activation of ectonucleotide pyrophosphatase/phosphodiesterase (ENPP1) and transported by transmembrane ankylosis protein (ANKH) (239, 267–270) (Figure 4).
8 Conclusions
Elastic fibres, and especially degraded elastin, play a critical role in the development and progression of calcification with several clinical implications for the cardiovascular tissues. This is due to the capabilities of elastic fibres to sequester Ca2+ ions fostering the formation of Ca2+-based mineralized structures that alter tissue elasticity and signalling pathways mediated by cell and cell-matrix interactions. Therefore, changes in the structure and composition of the ECM contribute to develop a suitable environment further triggering and promoting mineralization. Within this context, matrix components, whose amount and physico-chemical characteristics undergo site-specific age-related changes, modulate mesenchymal cell behaviour towards an osteogenic phenotype and regulate the different susceptibility of soft connective tissues to mineral deposition.
Both in vitro and in vivo models are essential for studying mineral composition and calcification mechanisms. In vitro systems, in particular, enable to analyse the rate of mineral formation and its temporal evolution as well as the influence of regulatory factors. In vivo models, mimicking human diseases, offer insights into the role of genetic, biochemical, and environmental factors on elastin calcification and its inhibition.
Statements
Author contributions
FDL: Writing – original draft, Visualization. AM: Writing – original draft. SB: Writing – original draft. DQ: Writing – original draft, Funding acquisition, Conceptualization, Writing – review & editing. FB: Writing – original draft, Visualization, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The author declares that financial support was received for publication of this article from PXE Italia Odv grant n. E93C250003000 (DQ).
Acknowledgments
Authors gratefully acknowledge the support of PXE Italia Odv. DQ is member of the International Network on ectopic Calcification (INTEC).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
Green EM Mansfield JC Bell JS Winlove CP . The structure and micromechanics of elastic tissue. Interface Focus. (2014) 4:20130058. 10.1098/rsfs.2013.0058
2.
Shapiro SD Endicott SK Province MA Pierce JA Campbell EJ . Marked longevity of human lung parenchymal elastic fibers deduced from prevalence of D-aspartate and nuclear weapons-related radiocarbon. J Clin Invest. (1991) 87:1828–34. 10.1172/JCI115204
3.
Heinz A . Elastases and elastokines: elastin degradation and its significance in health and disease. Crit Rev Biochem Mol Biol. (2020) 55:252–73. 10.1080/10409238.2020.1768208
4.
Quaglino D Boraldi F Lofaro FD . The biology of vascular calcification. Int Rev Cell Mol Biol. (2020) 354:261–353. 10.1016/bs.ircmb.2020.02.007
5.
Proudfoot D Shanahan CM . Biology of calcification in vascular cells: intima versus media. Herz. (2001) 26:245–51. 10.1007/PL00002027
6.
Gomez-Stallons MV Tretter JT Hassel K Gonzalez-Ramos O Amofa D Ollberding NJ et al Calcification and extracellular matrix dysregulation in human postmortem and surgical aortic valves. Heart. (2019) 105:1616–21. 10.1136/heartjnl-2019-314879
7.
Radvar E Mehta K D’Ambrosio A Mastroianni G Al-Jawad M Stevens MM et al Investigating the role of elastin and extracellular matrix damage in cardiovascular calcification. J Struct Biol. (2025) 217:108140. 10.1016/j.jsb.2024.108140
8.
Di Minno MND Poggio P Conte E Myasoedova V Songia P Mushtaq S et al Cardiovascular morbidity and mortality in patients with aortic valve calcification: a systematic review and meta-analysis. J Cardiovasc Comput Tomogr. (2019) 13:190–5. 10.1016/j.jcct.2019.06.006
9.
Abbasian N . Vascular calcification mechanisms: updates and renewed insight into signaling pathways involved in high phosphate-mediated vascular smooth muscle cell calcification. Biomedicines. (2021) 9:804. 10.3390/biomedicines9070804
10.
Boraldi F Bartolomeo A De Biasi S Orlando S Costa S Cossarizza A et al Innovative flow cytometry allows accurate identification of rare circulating cells involved in endothelial dysfunction. PLoS One. (2016) 11:e0160153. 10.1371/journal.pone.0160153
11.
Boraldi F Burns JS Bartolomeo A Dominici M Quaglino D . Mineralization by mesenchymal stromal cells is variously modulated depending on commercial platelet lysate preparations. Cytotherapy. (2018) 20:335–42. 10.1016/j.jcyt.2017.11.011
12.
Schurgers LJ Akbulut AC Kaczor DM Halder M Koenen RR Kramann R . Initiation and propagation of vascular calcification is regulated by a concert of platelet- and smooth muscle cell-derived extracellular vesicles. Front Cardiovasc Med. (2018) 5:36. 10.3389/fcvm.2018.00036
13.
Ouyang L Zhang K Chen J Wang J Huang H . Roles of platelet-derived growth factor in vascular calcification. J Cell Physiol. (2018) 233:2804–14. 10.1002/jcp.25985
14.
Boraldi F Moscarelli P Lofaro FD Sabia C Quaglino D . The mineralization process of insoluble elastin fibrillar structures: ionic environment vs degradation. Int J Biol Macromol. (2020) 149:693–706. 10.1016/j.ijbiomac.2020.01.250
15.
Dobrin R . Vascular mechanics. In: BohrDFSmlyoARSparkesHV, editors. Handbook of Physiology Cardiovascular System. Bethesda, MD: American Physiology Society (1983). p. 65–104.
16.
Mansfield J Yu J Attenburrow D Moger J Tirlapur U Urban J et al The elastin network: its relationship with collagen and cells in articular cartilage as visualized by multiphoton microscopy. J Anat. (2009) 215:682–91. 10.1111/j.1469-7580.2009.01149.x
17.
Yu J . Elastic tissues of the intervertebral disc. Biochem Soc Trans. (2002) 30:848–52. 10.1042/bst0300848
18.
de Carvalho HF Taboga SR . Fluorescence and confocal laser scanning microscopy imaging of elastic fibers in hematoxylin-eosin stained sections. Histochem Cell Biol. (1996) 106:587–92. 10.1007/BF02473274
19.
Boraldi F Lofaro FD Losi L Quaglino D . Dermal alterations in clinically unaffected skin of pseudoxanthoma elasticum patients. J Clin Med. (2021) 10:500. 10.3390/jcm10030500
20.
Percival KR Radi ZA . A modified verhoeff-van gieson elastin histochemical stain to enable pulmonary arterial hypertension model characterization. Eur J Histochem. (2016) 60:2588. 10.4081/ejh.2016.2588
21.
Boraldi F Tonelli M Gheduzzi D Ronchetti IP Quaglino D . Identification of mineralized elastic fibers on wet samples by SEM. Microsc Res Tech. (2005) 67:296–9. 10.1002/jemt.20212
22.
Fornieri C Ronchetti IP Edman AC Sjöström M . Contribution of cryotechniques to the study of elastin ultrastructure. J Microsc. (1982) 126:87–93. 10.1111/j.1365-2818.1982.tb00359.x
23.
Ruiz TFR Ferrato LJ de Souza LG Brito-Filho GE Leonel ECR Taboga SR . The elastic system: a review of elastin-related techniques and hematoxylin-eosin/phloxine applicability for normal and pathological tissue description. Acta Histochem. (2024) 126:152209. 10.1016/j.acthis.2024.152209
24.
Li H Yan M Yu J Xu Q Xia X Liao J et al In vivo identification of arteries and veins using two-photon excitation elastin autofluorescence. J Anat. (2020) 236:171–9. 10.1111/joa.13080
25.
Tiedemann K Sasaki T Gustafsson E Göhring W Bätge B Notbohm H et al Microfibrils at basement membrane zones interact with perlecan via fibrillin-1. J Biol Chem. (2005) 280:11404–12. 10.1074/jbc.M409882200
26.
Baccarani-Contri M Vincenzi D Cicchetti F Mori G Pasquali-Ronchetti I . Immunocytochemical localization of proteoglycans within normal elastin fibers. Eur J Cell Biol. (1990) 53:305–12.
27.
Reinboth B Hanssen E Cleary EG Gibson MA . Molecular interactions of biglycan and decorin with elastic fiber components: biglycan forms a ternary complex with tropoelastin and microfibril-associated glycoprotein 1. J Biol Chem. (2002) 277:3950–7. 10.1074/jbc.M109540200
28.
Kielty CM . Elastic fibres in health and disease. Expert Rev Mol Med. (2006) 8:1–23. 10.1017/S146239940600007X
29.
Trask BC Trask TM Broekelmann T Mecham RP . The microfibrillar proteins MAGP-1 and fibrillin-1 form a ternary complex with the chondroitin sulfate proteoglycan decorin. Mol Biol Cell. (2000) 11:1499–507. 10.1091/mbc.11.5.1499
30.
Yanagisawa H Davis EC . Unraveling the mechanism of elastic fiber assembly: the roles of short fibulins. Int J Biochem Cell Biol. (2010) 42:1084–93. 10.1016/j.biocel.2010.03.009
31.
McLaughlin PJ Bakall B Choi J Liu Z Sasaki T Davis EC et al Lack of fibulin-3 causes early aging and herniation, but not macular degeneration in mice. Hum Mol Genet. (2007) 16:3059–70. 10.1093/hmg/ddm264
32.
Kobayashi N Kostka G Garbe JHO Keene DR Bächinger HP Hanisch F-G et al A comparative analysis of the fibulin protein family. Biochemical characterization, binding interactions, and tissue localization. J Biol Chem. (2007) 282:11805–16. 10.1074/jbc.M611029200
33.
McLaughlin PJ Chen Q Horiguchi M Starcher BC Stanton JB Broekelmann TJ et al Targeted disruption of fibulin-4 abolishes elastogenesis and causes perinatal lethality in mice. Mol Cell Biol. (2006) 26:1700–9. 10.1128/MCB.26.5.1700-1709.2006
34.
Horiguchi M Inoue T Ohbayashi T Hirai M Noda K Marmorstein LY et al Fibulin-4 conducts proper elastogenesis via interaction with cross-linking enzyme lysyl oxidase. Proc Natl Acad Sci U S A. (2009) 106:19029–34. 10.1073/pnas.0908268106
35.
Charbonneau NL Dzamba BJ Ono RN Keene DR Corson GM Reinhardt DP et al Fibrillins can co-assemble in fibrils, but fibrillin fibril composition displays cell-specific differences. J Biol Chem. (2003) 278:2740–9. 10.1074/jbc.M209201200
36.
Charbonneau NL Carlson EJ Tufa S Sengle G Manalo EC Carlberg VM et al In vivo studies of mutant fibrillin-1 microfibrils. J Biol Chem. (2010) 285:24943–55. 10.1074/jbc.M110.130021
37.
Corson GM Charbonneau NL Keene DR Sakai LY . Differential expression of fibrillin-3 adds to microfibril variety in human and avian, but not rodent, connective tissues. Genomics. (2004) 83:461–72. 10.1016/j.ygeno.2003.08.023
38.
Yanagisawa H Davis EC Starcher BC Ouchi T Yanagisawa M Richardson JA et al Fibulin-5 is an elastin-binding protein essential for elastic fibre development in vivo. Nature. (2002) 415:168–71. 10.1038/415168a
39.
Hirai M Horiguchi M Ohbayashi T Kita T Chien KR Nakamura T . Latent TGF-beta-binding protein 2 binds to DANCE/fibulin-5 and regulates elastic fiber assembly. EMBO J. (2007) 26:3283–95. 10.1038/sj.emboj.7601768
40.
Lomas AC Mellody KT Freeman LJ Bax DV Shuttleworth CA Kielty CM . Fibulin-5 binds human smooth-muscle cells through alpha5beta1 and alpha4beta1 integrins, but does not support receptor activation. Biochem J. (2007) 405:417–28. 10.1042/BJ20070400
41.
Massam-Wu T Chiu M Choudhury R Chaudhry SS Baldwin AK McGovern A et al Assembly of fibrillin microfibrils governs extracellular deposition of latent TGF beta. J Cell Sci. (2010) 123:3006–18. 10.1242/jcs.073437
42.
Koli K Hyytiäinen M Ryynänen MJ Keski-Oja J . Sequential deposition of latent TGF-beta binding proteins (LTBPs) during formation of the extracellular matrix in human lung fibroblasts. Exp Cell Res. (2005) 310:370–82. 10.1016/j.yexcr.2005.08.008
43.
Gibson MA Hatzinikolas G Davis EC Baker E Sutherland GR Mecham RP . Bovine latent transforming growth factor beta 1-binding protein 2: molecular cloning, identification of tissue isoforms, and immunolocalization to elastin-associated microfibrils. Mol Cell Biol. (1995) 15:6932–42. 10.1128/MCB.15.12.6932
44.
Saharinen J Hyytiäinen M Taipale J Keski-Oja J . Latent transforming growth factor-beta binding proteins (LTBPs)–structural extracellular matrix proteins for targeting TGF-beta action. Cytokine Growth Factor Rev. (1999) 10:99–117. 10.1016/s1359-6101(99)00010-6
45.
Hayashi K Fong KSK Mercier F Boyd CD Csiszar K Hayashi M . Comparative immunocytochemical localization of lysyl oxidase (LOX) and the lysyl oxidase-like (LOXL) proteins: changes in the expression of LOXL during development and growth of mouse tissues. J Mol Histol. (2004) 35:845–55. 10.1007/s10735-004-2340-1
46.
Jensen SA Reinhardt DP Gibson MA Weiss AS . Protein interaction studies of MAGP-1 with tropoelastin and fibrillin-1. J Biol Chem. (2001) 276:39661–6. 10.1074/jbc.M104533200
47.
Craft CS Zou W Watkins M Grimston S Brodt MD Broekelmann TJ et al Microfibril-associated glycoprotein-1, an extracellular matrix regulator of bone remodeling. J Biol Chem. (2010) 285:23858–67. 10.1074/jbc.M110.113019
48.
Zilberberg L Todorovic V Dabovic B Horiguchi M Couroussé T Sakai LY et al Specificity of latent TGF-β binding protein (LTBP) incorporation into matrix: role of fibrillins and fibronectin. J Cell Physiol. (2012) 227:3828–36. 10.1002/jcp.24094
49.
Lemaire R Bayle J Mecham RP Lafyatis R . Microfibril-associated MAGP-2 stimulates elastic fiber assembly. J Biol Chem. (2007) 282:800–8. 10.1074/jbc.M609692200
50.
Baccarani-Contri M Taparelli F Pasquali-Ronchetti I . Osteopontin is a constitutive component of normal elastic fibers in human skin and aorta. Matrix Biol J Int Soc Matrix Biol. (1995) 14:553–60. 10.1016/s0945-053x(05)80004-6
51.
Gibson MA Kumaratilake JS Cleary EG . Immunohistochemical and ultrastructural localization of MP78/70 (betaig-h3) in extracellular matrix of developing and mature bovine tissues. J Histochem Cytochem. (1997) 45:1683–96. 10.1177/002215549704501212
52.
Isogai Z Aspberg A Keene DR Ono RN Reinhardt DP Sakai LY . Versican interacts with fibrillin-1 and links extracellular microfibrils to other connective tissue networks. J Biol Chem. (2002) 277:4565–72. 10.1074/jbc.M110583200
53.
Hinek A Rabinovitch M . 67-kD elastin-binding protein is a protective “companion” of extracellular insoluble elastin and intracellular tropoelastin. J Cell Biol. (1994) 126:563–74. 10.1083/jcb.126.2.563
54.
Privitera S Prody CA Callahan JW Hinek A . The 67-kDa enzymatically inactive alternatively spliced variant of beta-galactosidase is identical to the elastin/laminin-binding protein. J Biol Chem. (1998) 273:6319–26. 10.1074/jbc.273.11.6319
55.
Sengle G Tsutsui K Keene DR Tufa SF Carlson EJ Charbonneau NL et al Microenvironmental regulation by fibrillin-1. PLoS Genet. (2012) 8:e1002425. 10.1371/journal.pgen.1002425
56.
Gabriel LAR Wang LW Bader H Ho JC Majors AK Hollyfield JG et al ADAMTSL4, A secreted glycoprotein widely distributed in the eye, binds fibrillin-1 microfibrils and accelerates microfibril biogenesis. Invest Ophthalmol Vis Sci. (2012) 53:461–9. 10.1167/iovs.10-5955
57.
Bader HL Wang LW Ho JC Tran T Holden P Fitzgerald J et al A disintegrin-like and metalloprotease domain containing thrombospondin type 1 motif-like 5 (ADAMTSL5) is a novel fibrillin-1-, fibrillin-2-, and heparin-binding member of the ADAMTS superfamily containing a netrin-like module. Matrix Biol J Int Soc Matrix Biol. (2012) 31:398–411. 10.1016/j.matbio.2012.09.003
58.
Janig E Haslbeck M Aigelsreiter A Braun N Unterthor D Wolf P et al Clusterin associates with altered elastic fibers in human photoaged skin and prevents elastin from ultraviolet-induced aggregation in vitro. Am J Pathol. (2007) 171:1474–82. 10.2353/ajpath.2007.061064
59.
Miosge N Sasaki T Timpl R . Angiogenesis inhibitor endostatin is a distinct component of elastic fibers in vessel walls. FASEB J. (1999) 13:1743–50. 10.1096/fasebj.13.13.1743
60.
Sabatier L Chen D Fagotto-Kaufmann C Hubmacher D McKee MD Annis DS et al Fibrillin assembly requires fibronectin. Mol Biol Cell. (2009) 20:846–58. 10.1091/mbc.e08-08-0830
61.
Contri MB Boraldi F Taparelli F De Paepe A Ronchetti IP . Matrix proteins with high affinity for calcium ions are associated with mineralization within the elastic fibers of pseudoxanthoma elasticum dermis. Am J Pathol. (1996) 148:569–77.
62.
Gheduzzi D Boraldi F Annovi G DeVincenzi CP Schurgers LJ Vermeer C et al Matrix gla protein is involved in elastic fiber calcification in the dermis of pseudoxanthoma elasticum patients. Lab Invest. (2007) 87:998–1008. 10.1038/labinvest.3700667
63.
Lockhart-Cairns MP Newandee H Thomson J Weiss AS Baldock C Tarakanova A . Transglutaminase-mediated cross-linking of tropoelastin to fibrillin stabilises the elastin precursor prior to elastic fibre assembly. J Mol Biol. (2020) 432:5736–51. 10.1016/j.jmb.2020.08.023
64.
Bailey M Pillarisetti S Jones P Xiao H Simionescu D Vyavahare N . Involvement of matrix metalloproteinases and tenascin-C in elastin calcification. Cardiovasc Pathol. (2004) 13:146–55. 10.1016/S1054-8807(04)00009-2
65.
Zweers MC Peeters ACTM Graafsma S Kranendonk S van der Vliet JA den Heijer M et al Abdominal aortic aneurysm is associated with high Serum levels of tenascin-X and decreased aneurysmal tissue tenascin-X. Circulation. (2006) 113:1702–7. 10.1161/CIRCULATIONAHA.104.513820
66.
Baldwin AK Simpson A Steer R Cain SA Kielty CM . Elastic fibres in health and disease. Expert Rev Mol Med. (2013) 15:e8. 10.1017/erm.2013.9
67.
Cocciolone AJ Hawes JZ Staiculescu MC Johnson EO Murshed M Wagenseil JE . Elastin, arterial mechanics, and cardiovascular disease. Am J Physiol-Heart Circ Physiol. (2018) 315:H189–205. 10.1152/ajpheart.00087.2018
68.
Camasão DB Mantovani D . The mechanical characterization of blood vessels and their substitutes in the continuous quest for physiological-relevant performances. A critical review. Mater Today Bio. (2021) 10:100106. 10.1016/j.mtbio.2021.100106
69.
Wagenseil JE Mecham RP . Vascular extracellular matrix and arterial mechanics. Physiol Rev. (2009) 89:957–89. 10.1152/physrev.00041.2008
70.
Vesely I . The role of elastin in aortic valve mechanics. J Biomech. (1998) 31:115–23. 10.1016/s0021-9290(97)00122-x
71.
Indik Z Yeh H Ornstein-Goldstein N Sheppard P Anderson N Rosenbloom JC et al Alternative splicing of human elastin mRNA indicated by sequence analysis of cloned genomic and complementary DNA. Proc Natl Acad Sci. (1987) 84:5680–4. 10.1073/pnas.84.16.5680
72.
Grant RA . Variations in the amino acid composition of aortic elastin from different species. Br J Exp Pathol. (1966) 47:163–7.
73.
Ozsvar J Yang C Cain SA Baldock C Tarakanova A Weiss AS . Tropoelastin and elastin assembly. Front Bioeng Biotechnol. (2021) 9:643110. 10.3389/fbioe.2021.643110
74.
Pasquali Ronchetti I Alessandrini A Baccarani Contri M Fornieri C Mori G Quaglino D et al Study of elastic fiber organization by scanning force microscopy. Matrix Biol J Int Soc Matrix Biol. (1998) 17:75–83. 10.1016/s0945-053x(98)90126-3
75.
Wu WJ Vrhovski B Weiss AS . Glycosaminoglycans mediate the coacervation of human tropoelastin through dominant charge interactions involving lysine side chains*. J Biol Chem. (1999) 274:21719–24. 10.1074/jbc.274.31.21719
76.
Carino A Ludwig C Cervellino A Müller E Testino A . Formation and transformation of calcium phosphate phases under biologically relevant conditions: experiments and modelling. Acta Biomater. (2018) 74:478–88. 10.1016/j.actbio.2018.05.027
77.
Fleisch H Russell RG Straumann F . Effect of pyrophosphate on hydroxyapatite and its implications in calcium homeostasis. Nature. (1966) 212:901–3. 10.1038/212901a0
78.
Nitschke Y Yan Y Buers I Kintziger K Askew K Rutsch F . ENPP1-Fc prevents neointima formation in generalized arterial calcification of infancy through the generation of AMP. Exp Mol Med. (2018) 50:139. 10.1038/s12276-018-0163-5
79.
Jung A Bisaz S Bartholdi P Fleisch H . Influence of pyrophosphate on the exchange of calcium and phosphate ions on hydroxyapatite. Calcif Tissue Res. (1973) 13:27–40. 10.1007/BF02015393
80.
Lomashvili KA Garg P Narisawa S Millan JL O’Neill WC . Upregulation of alkaline phosphatase and pyrophosphate hydrolysis: potential mechanism for uremic vascular calcification. Kidney Int. (2008) 73:1024–30. 10.1038/ki.2008.26
81.
Bertazzo S Gentleman E Cloyd KL Chester AH Yacoub MH Stevens MM . Nano-analytical electron microscopy reveals fundamental insights into human cardiovascular tissue calcification. Nat Mater. (2013) 12:576–83. 10.1038/nmat3627
82.
Zazzeroni L Faggioli G Pasquinelli G . Mechanisms of arterial calcification: the role of matrix vesicles. Eur J Vasc Endovasc Surg. (2018) 55:425–32. 10.1016/j.ejvs.2017.12.009
83.
Kapustin AN Davies JD Reynolds JL McNair R Jones GT Sidibe A et al Calcium regulates key components of vascular smooth muscle cell–derived matrix vesicles to enhance mineralization. Circ Res. (2011) 109:e1–12. 10.1161/CIRCRESAHA.110.238808
84.
Geddes AJ Parker KD Atkins ED Beighton E . “Cross-beta” conformation in proteins. J Mol Biol. (1968) 32:343–58. 10.1016/0022-2836(68)90014-4
85.
Urry D Ohnishi M . Spectroscopic Approaches to Biomolecular Conformation. Chicago: Amer. Med. Assoc. Press (1970). p. 263–300.
86.
Urry DW . Neutral sites for calcium Ion binding to elastin and collagen: a charge neutralization theory for calcification and its relationship to atherosclerosis. Proc Natl Acad Sci. (1971) 68:810–4. 10.1073/pnas.68.4.810
87.
Zhang P Gourgas O Lainé A Murshed M Mantovani D Cerruti M . Coacervation conditions and cross-linking determines availability of carbonyl groups on elastin and its calcification. Cryst Growth Des. (2020) 20:7170–9. 10.1021/acs.cgd.0c00759
88.
Kaibara K Akinari Y Okamoto K Uemura Y Yamamoto S Kodama H et al Characteristic interaction of Ca2+ ions with elastin coacervate: ion transport study across coacervate layers of alpha-elastin and elastin model polypeptide, (val-pro-gly-val-gly)n. Biopolymers. (1996) 39:189–98. 10.1002/(sici)1097-0282(199608)39:2%3C189::aid-bip7%3E3.0.co;2-l
89.
Andrault P-M Panwar P Mackenzie NCW Brömme D . Elastolytic activity of cysteine cathepsins K, S, and V promotes vascular calcification. Sci Rep. (2019) 9:9682. 10.1038/s41598-019-45918-1
90.
Yang C Weiss AS Tarakanova A . Changes in elastin structure and extensibility induced by hypercalcemia and hyperglycemia. Acta Biomater. (2023) 163:131–45. 10.1016/j.actbio.2022.03.041
91.
Molinari Tosatti MP Gotte L Moret V . Some features of the binding of calcium ions to elastin. Calcif Tissue Res. (1971) 6:329–34. 10.1007/bf02196213
92.
Schiffmann E Lavender DR Miller EJ Corcoran BA . Amino acids at the nucleating site in mineralizing elastic tissue. Calcif Tissue Res. (1969) 3:125–35. 10.1007/bf02058655
93.
Song T Cerruti M . Unraveling the role of carboxylate groups and elastin particle size in medial calcification. Int J Biol Macromol. (2024) 274:133267. 10.1016/j.ijbiomac.2024.133267
94.
Lau K Sharpe S Cerruti M . Initiation of medial calcification: revisiting calcium Ion binding to elastin. J Phys Chem B. (2024) 128:9631–42. 10.1021/acs.jpcb.4c04464
95.
Schmelzer CEH Hedtke T Heinz A . Unique molecular networks: formation and role of elastin cross-links. IUBMB Life. (2020) 72:842–54. 10.1002/iub.2213
96.
Heinz A . Elastic fibers during aging and disease. Ageing Res Rev. (2021) 66:101255. 10.1016/j.arr.2021.101255
97.
Koskamp JA Ruiz Hernandez SE de Leeuw NH Wolthers M . Recalibrating the calcium trap in amino acid carboxyl groups via classical molecular dynamics simulations. Phys Chem Chem Phys. (2023) 25:1220–35. 10.1039/d2cp02879d
98.
Martinek T Duboué-Dijon E Timr Š Mason PE Baxová K Fischer HE et al Calcium ions in aqueous solutions: accurate force field description aided by ab initio molecular dynamics and neutron scattering. J Chem Phys. (2018) 148:222813. 10.1063/1.5006779
99.
LaBella FS Vivian S Thornhill DP . Amino acid composition of human aortic elastin as influenced by age. J Gerontol. (1966) 21:550–5. 10.1093/geronj/21.4.550
100.
Kramsch DM Franzblau C Hollander W . The protein and lipid composition of arterial elastin and its relationship to lipid accumulation in the atherosclerotic plaque. J Clin Invest. (1971) 50:1666–77. 10.1172/JCI106656
101.
Carta L Pereira L Arteaga-Solis E Lee-Arteaga SY Lenart B Starcher B et al Fibrillins 1 and 2 perform partially overlapping functions during aortic development. J Biol Chem. (2006) 281:8016–23. 10.1074/jbc.M511599200
102.
Pyeritz RE . The Marfan syndrome. Annu Rev Med. (2000) 51:481–510. 10.1146/annurev.med.51.1.481
103.
Loeys BL Dietz HC Braverman AC Callewaert BL De Backer J Devereux RB et al The revised Ghent nosology for the Marfan syndrome. J Med Genet. (2010) 47:476–85. 10.1136/jmg.2009.072785
104.
Gupta PA Putnam EA Carmical SG Kaitila I Steinmann B Child A et al Ten novel FBN2 mutations in congenital contractural arachnodactyly: delineation of the molecular pathogenesis and clinical phenotype. Hum Mutat. (2002) 19:39–48. 10.1002/humu.10017
105.
Davis MR Andersson R Severin J de Hoon M Bertin N Baillie JK et al Transcriptional profiling of the human fibrillin/LTBP gene family, key regulators of mesenchymal cell functions. Mol Genet Metab. (2014) 112:73–83. 10.1016/j.ymgme.2013.12.006
106.
Dingemans KP Teeling P Lagendijk JH Becker AE . Extracellular matrix of the human aortic media: an ultrastructural histochemical and immunohistochemical study of the adult aortic media. Anat Rec. (2000) 258:1–14. 10.1002/(SICI)1097-0185(20000101)258:1%3C1::AID-AR1%3E3.0.CO;2-7
107.
Boraldi F Lofaro FD Romano O Grilli A Losi L Moscarelli P et al Exome sequencing and bioinformatic approaches reveals rare sequence variants involved in cell signalling and elastic fibre homeostasis: new evidence in the development of ectopic calcification. Cell Signal. (2019) 59:131–40. 10.1016/j.cellsig.2019.03.020
108.
Eckersley A Mellody KT Pilkington S Griffiths CEM Watson REB O’Cualain R et al Structural and compositional diversity of fibrillin microfibrils in human tissues. J Biol Chem. (2018) 293:5117–33. 10.1074/jbc.RA117.001483
109.
Boraldi F Annovi G Vermeer C Schurgers LJ Trenti T Tiozzo R et al Matrix gla protein and alkaline phosphatase are differently modulated in human dermal fibroblasts from PXE patients and controls. J Invest Dermatol. (2013) 133:946–54. 10.1038/jid.2012.460
110.
Boraldi F Annovi G Guerra D Paolinelli Devincenzi C Garcia-Fernandez MI Panico F et al Fibroblast protein profile analysis highlights the role of oxidative stress and vitamin K recycling in the pathogenesis of pseudoxanthoma elasticum. Proteomics Clin Appl. (2009) 3:1084–98. 10.1002/prca.200900007
111.
Sengle G Sakai LY . The fibrillin microfibril scaffold: a niche for growth factors and mechanosensation?Matrix Biol J Int Soc Matrix Biol. (2015) 47:3–12. 10.1016/j.matbio.2015.05.002
112.
Handford P Downing AK Rao Z Hewett DR Sykes BC Kielty CM . The calcium binding properties and molecular organization of epidermal growth factor-like domains in human fibrillin-1. J Biol Chem. (1995) 270:6751–6. 10.1074/jbc.270.12.6751
113.
Nistala H Lee-Arteaga S Smaldone S Siciliano G Carta L Ono RN et al Fibrillin-1 and -2 differentially modulate endogenous TGF-β and BMP bioavailability during bone formation. J Cell Biol. (2010) 190:1107–21. 10.1083/jcb.201003089
114.
Schmidmaier G Capanna R Wildemann B Beque T Lowenberg D . Bone morphogenetic proteins in critical-size bone defects: what are the options?Injury. (2009) 40 Suppl 3:S39–43. 10.1016/S0020-1383(09)70010-5
115.
Wu M Wu S Chen W Li Y-P . The roles and regulatory mechanisms of TGF-β and BMP signaling in bone and cartilage development, homeostasis and disease. Cell Res. (2024) 34:101–23. 10.1038/s41422-023-00918-9
116.
Daamen WF Quaglino D . Signaling pathways in elastic tissues. Cell Signal. (2019) 63:109364. 10.1016/j.cellsig.2019.109364
117.
Podrazký V Stokrová S Fric I . Elastin–proteoglycan interaction. Conformational changes of alpha-elastin induced by the interaction. Connect Tissue Res. (1975) 4:51–4. 10.3109/03008207509152197
118.
Contri MB Fornieri CC Ronchetti IP . Elastin-proteoglycans association revealed by cytochemical methods. Connect Tissue Res. (1985) 13:237–49. 10.3109/03008208509152403
119.
Couchman JR Pataki CA . An introduction to proteoglycans and their localization. J Histochem Cytochem. (2012) 60:885–97. 10.1369/0022155412464638
120.
Iozzo RV Schaefer L . Proteoglycan form and function: a comprehensive nomenclature of proteoglycans. Matrix Biol. (2015) 42:11–55. 10.1016/j.matbio.2015.02.003
121.
Sabatier L Djokic J Hubmacher D Dzafik D Nelea V Reinhardt DP . Heparin/heparan sulfate controls fibrillin-1, -2 and -3 self-interactions in microfibril assembly. FEBS Lett. (2014) 588:2890–7. 10.1016/j.febslet.2014.06.061
122.
Gheduzzi D Guerra D Bochicchio B Pepe A Tamburro AM Quaglino D et al Heparan sulphate interacts with tropoelastin, with some tropoelastin peptides and is present in human dermis elastic fibers. Matrix Biol J Int Soc Matrix Biol. (2005) 24:15–25. 10.1016/j.matbio.2004.12.001
123.
Bochicchio B Yeo GC Lee P Emul D Pepe A Laezza A et al Domains 12–16 of tropoelastin promote cell attachment and spreading through interactions with glycosaminoglycan and integrins alphaV and alpha5beta1. FEBS J. (2021) 288:4024–38. 10.1111/febs.15702
124.
Boraldi F Moscarelli P Bochicchio B Pepe A Salvi AM Quaglino D . Heparan sulfates facilitate harmless amyloidogenic fibril formation interacting with elastin-like peptides. Sci Rep. (2018) 8:3115. 10.1038/s41598-018-21472-0
125.
Annovi G Boraldi F Moscarelli P Guerra D Tiozzo R Parma B et al Heparan sulfate affects elastin deposition in fibroblasts cultured from donors of different ages. Rejuvenation Res. (2012) 15:22–31. 10.1089/rej.2011.1182
126.
Wight TN . A role for proteoglycans in vascular disease. Matrix Biol. (2018) 71–72:396–420. 10.1016/j.matbio.2018.02.019
127.
Chen X Chen D Ban E Toussaint KC Janmey PA Wells RG et al Glycosaminoglycans modulate long-range mechanical communication between cells in collagen networks. Proc Natl Acad Sci U S A. (2022) 119:e2116718119. 10.1073/pnas.2116718119
128.
Batko K Krzanowski M Gajda M Dumnicka P Pietrzycka A Fedak D et al Proteoglycan/glycosaminoglycan and collagen content in the arterial wall of patients with end-stage renal disease: new indicators of vascular disease. Pol Arch Intern Med. (2019) 129:781–9. 10.20452/pamw.15022
129.
Borland SJ Morris TG Borland SC Morgan MR Francis SE Merry CLR et al Regulation of vascular smooth muscle cell calcification by syndecan-4/FGF-2/PKCα signalling and cross-talk with TGFβ. Cardiovasc Res. (2017) 113:1639–52. 10.1093/cvr/cvx178
130.
Yan J Stringer SE Hamilton A Charlton-Menys V Götting C Müller B et al Decorin GAG synthesis and TGF-β signaling mediate ox-LDL-induced mineralization of human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. (2011) 31:608–15. 10.1161/ATVBAHA.110.220749
131.
Hao J Shen M Wang C Wei J Wan Q Zhu Y et al Regulation of biomineralization by proteoglycans: from mechanisms to application. Carbohydr Polym. (2022) 294:119773. 10.1016/j.carbpol.2022.119773
132.
Lei Y Ning Q Xia Y Wang Y . Enzyme-oxidative-polymerization method for improving glycosaminoglycans stability and reducing calcification in bioprosthetic heart valves. Biomed Mater Bristol Engl. (2019) 14:025012. 10.1088/1748-605X/aafd7c
133.
Fischer JW Steitz SA Johnson PY Burke A Kolodgie F Virmani R et al Decorin promotes aortic smooth muscle cell calcification and colocalizes to calcified regions in human atherosclerotic lesions. Arterioscler Thromb Vasc Biol. (2004) 24:2391–6. 10.1161/01.ATV.0000147029.63303.28
134.
Hayes AJ Lord MS Smith SM Smith MM Whitelock JM Weiss AS et al Colocalization in vivo and association in vitro of perlecan and elastin. Histochem Cell Biol. (2011) 136:437–54. 10.1007/s00418-011-0854-7
135.
Hayes AJ Farrugia BL Biose IJ Bix GJ Melrose J . Perlecan, a multi-functional, cell-instructive, matrix-stabilizing proteoglycan with roles in tissue development has relevance to connective tissue repair and regeneration. Front Cell Dev Biol. (2022) 10:856261. 10.3389/fcell.2022.856261
136.
Shibata M Shigematsu T Hatamura I Saji F Mune S Kunimoto K et al Reduced expression of perlecan in the aorta of secondary hyperparathyroidism model rats with medial calcification. Ren Fail. (2010) 32:214–23. 10.3109/08860220903367544
137.
Talusan P Bedri S Yang S Kattapuram T Silva N Roughley PJ et al Analysis of intimal proteoglycans in atherosclerosis-prone and atherosclerosis-resistant human arteries by mass spectrometry. Mol Cell Proteomics MCP. (2005) 4:1350–7. 10.1074/mcp.M500088-MCP200
138.
Sugars RV Milan AM Brown JO Waddington RJ Hall RC Embery G . Molecular interaction of recombinant decorin and biglycan with type I collagen influences crystal growth. Connect Tissue Res. (2003) 44 Suppl 1:189–95. 10.1080/713713596
139.
Neufeld EB Zadrozny LM Phillips D Aponte A Yu Z-X Balaban RS . Decorin and biglycan retain LDL in disease-prone valvular and aortic subendothelial intimal matrix. Atherosclerosis. (2014) 233:113–21. 10.1016/j.atherosclerosis.2013.12.038
140.
Tintut Y Hsu JJ Demer LL . Lipoproteins in cardiovascular calcification: potential targets and challenges. Front Cardiovasc Med. (2018) 5:172. 10.3389/fcvm.2018.00172
141.
Saulnier JM Hauck M Fülöp T Wallach JM . Human aortic elastin from normal individuals and atherosclerotic patients: lipid and cation contents; susceptibility to elastolysis. Clin Chim Acta Int J Clin Chem. (1991) 200:129–36. 10.1016/0009-8981(91)90084-p
142.
Guantieri V Tamburro AM Gordini DD . Interactions of human and bovine elastins with lipids: their proteolysis by elastase. Connect Tissue Res. (1983) 12:79–83. 10.3109/03008208309005614
143.
Perrotta I Russo E Camastra C Filice G Di Mizio G Colosimo F et al New evidence for a critical role of elastin in calcification of native heart valves: immunohistochemical and ultrastructural study with literature review. Histopathology. (2011) 59:504–13. 10.1111/j.1365-2559.2011.03977.x
144.
Okuda-Ashitaka E Matsumoto K-I . Tenascin-X as a causal gene for classical-like ehlers-danlos syndrome. Front Genet. (2023) 14:1107787. 10.3389/fgene.2023.1107787
145.
Minamitani T Ariga H Matsumoto K . Deficiency of tenascin-X causes a decrease in the level of expression of type VI collagen. Exp Cell Res. (2004) 297:49–60. 10.1016/j.yexcr.2004.03.002
146.
Matsumoto K-I Satoh K Maniwa T Araki A Maruyama R Oda T . Noticeable decreased expression of tenascin-X in calcific aortic valves. Connect Tissue Res. (2012) 53:460–8. 10.3109/03008207.2012.702818
147.
Goiko M Dierolf J Gleberzon JS Liao Y Grohe B Goldberg HA et al Peptides of matrix gla protein inhibit nucleation and growth of hydroxyapatite and calcium oxalate monohydrate crystals. PLoS One. (2013) 8:e80344. 10.1371/journal.pone.0080344
148.
Shanahan CM Cary NR Salisbury JR Proudfoot D Weissberg PL Edmonds ME . Medial localization of mineralization-regulating proteins in association with mönckeberg’s sclerosis: evidence for smooth muscle cell-mediated vascular calcification. Circulation. (1999) 100:2168–76. 10.1161/01.cir.100.21.2168
149.
Boraldi F Garcia-Fernandez M Paolinelli-Devincenzi C Annovi G Schurgers L Vermeer C et al Ectopic calcification in β-thalassemia patients is associated with increased oxidative stress and lower MGP carboxylation. Biochim Biophys Acta. (2013) 1832:2077–84. 10.1016/j.bbadis.2013.07.017
150.
Gay BB Kuhn JP . A syndrome of widened medullary cavities of bone, aortic calcification, abnormal dentition, and muscular weakness (the singleton-merten syndrome). Radiology. (1976) 118:389–95. 10.1148/118.2.389
151.
Luo G Ducy P McKee MD Pinero GJ Loyer E Behringer RR et al Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. (1997) 386:78–81. 10.1038/386078a0
152.
Khavandgar Z Roman H Li J Lee S Vali H Brinckmann J et al Elastin haploinsufficiency impedes the progression of arterial calcification in MGP-deficient mice. J Bone Miner Res. (2014) 29:327–37. 10.1002/jbmr.2039
153.
Bjørklund G Svanberg E Dadar M Card DJ Chirumbolo S Harrington DJ et al The role of matrix gla protein (MGP) in vascular calcification. Curr Med Chem. (2020) 27:1647–60. 10.2174/0929867325666180716104159
154.
Bak K Parashar A Allgayer R Marulanda J Gourgas O Cerruti M et al An inducible model for medial calcification based on matrix gla protein deficiency. J Struct Biol. (2024) 216:108144. 10.1016/j.jsb.2024.108144
155.
Bäck M Aranyi T Cancela ML Carracedo M Conceição N Leftheriotis G et al Endogenous calcification inhibitors in the prevention of vascular calcification: a consensus statement from the COST action EuroSoftCalcNet. Front Cardiovasc Med. (2018) 5:196. 10.3389/fcvm.2018.00196
156.
Zhu Y-S Gu Y Jiang C Chen L . Osteonectin regulates the extracellular matrix mineralization of osteoblasts through P38 signaling pathway. J Cell Physiol. (2020) 235:2220–31. 10.1002/jcp.29131
157.
Sodek J Ganss B McKee MD . Osteopontin. Crit Rev Oral Biol Med. (2000) 11:279–303. 10.1177/10454411000110030101
158.
Scatena M Liaw L Giachelli CM . Osteopontin: a multifunctional molecule regulating chronic inflammation and vascular disease. Arterioscler Thromb Vasc Biol. (2007) 27:2302–9. 10.1161/ATVBAHA.107.144824
159.
Boskey AL . Osteopontin and related phosphorylated sialoproteins: effects on mineralization. Ann N Y Acad Sci. (1995) 760:249–56. 10.1111/j.1749-6632.1995.tb44635.x
160.
Shao J-S Sierra OL Cohen R Mecham RP Kovacs A Wang J et al Vascular calcification and aortic fibrosis: a bifunctional role for osteopontin in diabetic arteriosclerosis. Arterioscler Thromb Vasc Biol. (2011) 31:1821–33. 10.1161/ATVBAHA.111.230011
161.
Boraldi F Lofaro FD Cossarizza A Quaglino D . The “elastic perspective” of SARS-CoV-2 infection and the role of intrinsic and extrinsic factors. Int J Mol Sci. (2022) 23:1559. 10.3390/ijms23031559
162.
Maurice P Blaise S Gayral S Debelle L Laffargue M Hornebeck W et al Elastin fragmentation and atherosclerosis progression: the elastokine concept. Trends Cardiovasc Med. (2013) 23:211–21. 10.1016/j.tcm.2012.12.004
163.
Antonicelli F Bellon G Debelle L Hornebeck W . Elastin-elastases and inflamm-aging. Curr Top Dev Biol. (2007) 79:99–155. 10.1016/S0070-2153(06)79005-6
164.
Wells JM Gaggar A Blalock JE . MMP Generated matrikines. Matrix Biol. (2015) 44–46:122–9. 10.1016/j.matbio.2015.01.016
165.
Lei Y Sinha A Nosoudi N Grover A Vyavahare N . Hydroxyapatite and calcified elastin induce osteoblast-like differentiation in rat aortic smooth muscle cells. Exp Cell Res. (2014) 323:198–208. 10.1016/j.yexcr.2014.01.011
166.
Vyavahare N Jones PL Tallapragada S Levy RJ . Inhibition of matrix metalloproteinase activity attenuates tenascin-C production and calcification of implanted purified elastin in rats. Am J Pathol. (2000) 157:885–93. 10.1016/S0002-9440(10)64602-0
167.
Basalyga DM Simionescu DT Xiong W Baxter BT Starcher BC Vyavahare NR . Elastin degradation and calcification in an abdominal aorta injury model: role of matrix metalloproteinases. Circulation. (2004) 110:3480–7. 10.1161/01.CIR.0000148367.08413.E9
168.
Van Doren SR . Matrix metalloproteinase interactions with collagen and elastin. Matrix Biol J Int Soc Matrix Biol. (2015) 44–46:224–31. 10.1016/j.matbio.2015.01.005
169.
Lofaro FD Costa S Simone ML Quaglino D Boraldi F . Fibroblasts’ secretome from calcified and non-calcified dermis in pseudoxanthoma elasticum differently contributes to elastin calcification. Commun Biol. (2024) 7:577. 10.1038/s42003-024-06283-6
170.
Lee JS Basalyga DM Simionescu A Isenburg JC Simionescu DT Vyavahare NR . Elastin calcification in the rat subdermal model is accompanied by up-regulation of degradative and osteogenic cellular responses. Am J Pathol. (2006) 168:490–8. 10.2353/ajpath.2006.050338
171.
Aikawa E Aikawa M Libby P Figueiredo J-L Rusanescu G Iwamoto Y et al Arterial and aortic valve calcification abolished by elastolytic cathepsin S deficiency in chronic renal disease. Circulation. (2009) 119:1785–94. 10.1161/CIRCULATIONAHA.108.827972
172.
Tatara Y Suto S Itoh K . Novel roles of glycosaminoglycans in the degradation of type I collagen by cathepsin K. Glycobiology. (2017) 27:1089–98. 10.1093/glycob/cwx083
173.
Taverna D Boraldi F De Santis G Caprioli RM Quaglino D . Histology-directed and imaging mass spectrometry: an emerging technology in ectopic calcification. Bone. (2015) 74:83–94. 10.1016/j.bone.2015.01.004
174.
Robert L Robert AM Fülöp T . Rapid increase in human life expectancy: will it soon be limited by the aging of elastin?Biogerontology. (2008) 9:119–33. 10.1007/s10522-007-9122-6
175.
Stanford W Thompson BH Weiss RM . Coronary artery calcification: clinical significance and current methods of detection. AJR Am J Roentgenol. (1993) 161:1139–46. 10.2214/ajr.161.6.8249716
176.
Beckmann E Grau JB Sainger R Poggio P Ferrari G . Insights into the use of biomarkers in calcific aortic valve disease. J Heart Valve Dis. (2010) 19:441–52.
177.
Bogdanova M Kostina A Zihlavnikova Enayati K Zabirnyk A Malashicheva A Stensløkken K-O et al Inflammation and mechanical stress stimulate osteogenic differentiation of human aortic valve interstitial cells. Front Physiol. (2018) 9:1635. 10.3389/fphys.2018.01635
178.
Chen J-H Simmons CA . Cell-matrix interactions in the pathobiology of calcific aortic valve disease: critical roles for matricellular, matricrine, and matrix mechanics cues. Circ Res. (2011) 108:1510–24. 10.1161/CIRCRESAHA.110.234237
179.
Lin X Shan S-K Xu F Zhong J-Y Wu F Duan J-Y et al The crosstalk between endothelial cells and vascular smooth muscle cells aggravates high phosphorus-induced arterial calcification. Cell Death Dis. (2022) 13:650. 10.1038/s41419-022-05064-5
180.
Randles A Frakes DH Leopold JA . Computational fluid dynamics and additive manufacturing to diagnose and treat cardiovascular disease. Trends Biotechnol. (2017) 35:1049–61. 10.1016/j.tibtech.2017.08.008
181.
Xu S Wang F Mai P Peng Y Shu X Nie R et al Mechanism analysis of vascular calcification based on fluid dynamics. Diagnostics. (2023) 13:2632. 10.3390/diagnostics13162632
182.
Lanzer P Hannan FM Lanzer JD Janzen J Raggi P Furniss D et al Medial arterial calcification: jACC state-of-the-art review. J Am Coll Cardiol. (2021) 78:1145–65. 10.1016/j.jacc.2021.06.049
183.
Rocha-Singh KJ Zeller T Jaff MR . Peripheral arterial calcification: prevalence, mechanism, detection, and clinical implications. Catheter Cardiovasc Interv. (2014) 83:E212–20. 10.1002/ccd.25387
184.
Kamenskiy A Pipinos II Tian Y de Oliveira BB Orton D Liu X et al Vascular calcification amplifies limb flexion-induced arterial deformations and causes intermittent vessel pinching. Ann Surg. (2025) 6. 10.1097/SLA.0000000000006776
185.
Hoffmann U Massaro JM Agostino D Kathiresan RB Fox S S C et al Cardiovascular event prediction and risk reclassification by coronary, aortic, and valvular calcification in the framingham heart study. J Am Heart Assoc. (2016) 5:e003144. 10.1161/JAHA.115.003144
186.
Ejiri K Ding N Kim E Honda Y Cainzos-Achirica M Tanaka H et al Association of segment-specific pulse wave velocity with vascular calcification: the ARIC (atherosclerosis risk in communities) study. J Am Heart Assoc. (2024) 13:e031778. 10.1161/JAHA.123.031778
187.
Fletcher AJ Nash J Syed MBJ Macaskill MG Tavares AAS Walker N et al Microcalcification and thoracic aortopathy: a window into disease severity. Arterioscler Thromb Vasc Biol. (2022) 42:1048–59. 10.1161/ATVBAHA.122.317111
188.
Davidson JM Zoia O Liu J-M . Modulation of transforming growth factor-beta 1 stimulated elastin and collagen production and proliferation in porcine vascular smooth muscle cells and skin fibroblasts by basic fibroblast growth factor, transforming growth factor-α, and insulin-like growth factor-I. J Cell Physiol. (1993) 155:149–56. 10.1002/jcp.1041550119
189.
Reitamo S Remitz A Tamai K Ledo I Uitto J . Interleukin 10 up-regulates elastin gene expression in vivo and in vitro at the transcriptional level. Biochem J. (1994) 302(Pt 2):331–3. 10.1042/bj3020331
190.
Kothapalli CR Taylor PM Smolenski RT Yacoub MH Ramamurthi A . Transforming growth factor beta 1 and hyaluronan oligomers synergistically enhance elastin matrix regeneration by vascular smooth muscle cells. Tissue Eng Part A. (2009) 15:501–11. 10.1089/ten.tea.2008.0040
191.
Long JL Tranquillo RT . Elastic fiber production in cardiovascular tissue-equivalents. Matrix Biol. (2003) 22:339–50. 10.1016/S0945-053X(03)00052-0
192.
Robb BW Wachi H Schaub T Mecham RP Davis EC . Characterization of an in vitro model of elastic fiber assembly. Mol Biol Cell. (1999) 10:3595–605. 10.1091/mbc.10.11.3595
193.
Persiani E Ceccherini E Gisone I Cecchettini A Vozzi F . Protocol to generate an in vitro model to study vascular calcification using human endothelial and smooth muscle cells. STAR Protoc. (2023) 4:102328. 10.1016/j.xpro.2023.102328
194.
Lofaro FD Giuggioli D Bonacorsi S Orlandi M Spinella A De Pinto M et al BMP-4 and fetuin A in systemic sclerosis patients with or without calcinosis. Front Immunol. (2024) 15:1502324. 10.3389/fimmu.2024.1502324
195.
Boraldi F Bartolomeo A Annovi G Debret R Quaglino D . Magnesium modifies the structural features of enzymatically mineralized collagen gels affecting the retraction capabilities of human dermal fibroblasts embedded within this 3D system. Mater Basel Switz. (2016) 9:477. 10.3390/ma9060477
196.
Boraldi F Lofaro FD Quaglino D . Apoptosis in the extraosseous calcification process. Cells. (2021) 10:131. 10.3390/cells10010131
197.
Daculsi G Bouler JM LeGeros RZ . Adaptive crystal formation in normal and pathological calcifications in synthetic calcium phosphate and related biomaterials. Int Rev Cytol. (1997) 172:129–91. 10.1016/s0074-7696(08)62360-8
198.
Gourgas O Muiznieks LD Bello DG Nanci A Sharpe S Cerruti M . Cross-linked elastin-like polypeptide membranes as a model for medial arterial calcification. Biomacromolecules. (2019) 20:2625–36. 10.1021/acs.biomac.9b00417
199.
Parashar A Gourgas O Lau K Li J Muiznieks L Sharpe S et al Elastin calcification in in vitro models and its prevention by MGP’s N-terminal peptide. J Struct Biol. (2021) 213:107637. 10.1016/j.jsb.2020.107637
200.
Gourgas O Cole GB Muiznieks LD Sharpe S Cerruti M . Effect of the ionic concentration of simulated body fluid on the minerals formed on cross-linked elastin-like polypeptide membranes. Langmuir ACS J Surf Colloids. (2019) 35:15364–75. 10.1021/acs.langmuir.9b02542
201.
Gourgas O Marulanda J Zhang P Murshed M Cerruti M . Multidisciplinary approach to understand medial arterial calcification. Arterioscler Thromb Vasc Biol. (2018) 38:363–72. 10.1161/ATVBAHA.117.309808
202.
Machado R Bessa PC Reis RL Rodriguez-Cabello JC Casal M . Elastin-based nanoparticles for delivery of bone morphogenetic proteins. Methods Mol Biol. (2012) 906:353–63. 10.1007/978-1-61779-953-2_29
203.
Bessa PC Machado R Nürnberger S Dopler D Banerjee A Cunha AM et al Thermoresponsive self-assembled elastin-based nanoparticles for delivery of BMPs. J Controlled Release. (2010) 142:312–8. 10.1016/j.jconrel.2009.11.003
204.
Boskey AL Villarreal-Ramirez E . Intrinsically disordered proteins and biomineralization. Matrix Biol J Int Soc Matrix Biol. (2016) 52–54:43–59. 10.1016/j.matbio.2016.01.007
205.
Trivedi R Nagarajaram HA . Intrinsically disordered proteins: an overview. Int J Mol Sci. (2022) 23:14050. 10.3390/ijms232214050
206.
Delak K Harcup C Lakshminarayanan R Sun Z Fan Y Moradian-Oldak J et al The tooth enamel protein, porcine amelogenin, is an intrinsically disordered protein with an extended molecular configuration in the monomeric form. Biochemistry. (2009) 48:2272–81. 10.1021/bi802175a
207.
Beniash E Simmer JP Margolis HC . Structural changes in amelogenin upon self-assembly and mineral interactions. J Dent Res. (2012) 91:967–72. 10.1177/0022034512457371
208.
Elsharkawy S Al-Jawad M Pantano MF Tejeda-Montes E Mehta K Jamal H et al Protein disorder-order interplay to guide the growth of hierarchical mineralized structures. Nat Commun. (2018) 9:2145. 10.1038/s41467-018-04319-0
209.
Girotti A Reguera J Rodríguez-Cabello JC Arias FJ Alonso M Matestera A . Design and bioproduction of a recombinant multi(bio)functional elastin-like protein polymer containing cell adhesion sequences for tissue engineering purposes. J Mater Sci Mater Med. (2004) 15:479–84. 10.1023/b:jmsm.0000021124.58688.7a
210.
González-Obeso C González-Pérez M Mano JF Alonso M Rodríguez-Cabello JC . Complex morphogenesis by a model intrinsically disordered protein. Small Weinh Bergstr Ger. (2020) 16:e2005191. 10.1002/smll.202005191
211.
Joudeh N Linke D . Nanoparticle classification, physicochemical properties, characterization, and applications: a comprehensive review for biologists. J Nanobiotechnology. (2022) 20:262. 10.1186/s12951-022-01477-8
212.
Feldmann A Nitschke Y Linß F Mulac D Stücker S Bertrand J et al Improved reversion of calcifications in porcine aortic heart valves using elastin-targeted nanoparticles. Int J Mol Sci. (2023) 24:16471. 10.3390/ijms242216471
213.
Keuth J Nitschke Y Mulac D Riehemann K Rutsch F Langer K . Reversion of arterial calcification by elastin-targeted DTPA-HSA nanoparticles. Eur J Pharm Biopharm. (2020) 150:108–19. 10.1016/j.ejpb.2020.03.007
214.
Herrmann J Babic M Tölle M van der Giet M Schuchardt M . Research models for studying vascular calcification. Int J Mol Sci. (2020) 21:2204. 10.3390/ijms21062204
215.
Sinha S Eddington H Kalra PA . Vascular calcification: lessons from scientific models. J Ren Care. (2009) 35 Suppl 1:51–6. 10.1111/j.1755-6686.2009.00065.x
216.
Bunton TE Biery NJ Myers L Gayraud B Ramirez F Dietz HC . Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of Marfan syndrome. Circ Res. (2001) 88:37–43. 10.1161/01.res.88.1.37
217.
Behzadi P Wendling AA Cuevas RA Crane A Chu CC Moorhead WJ et al Rapamycin reduces mineral density and promotes beneficial vascular remodeling in a murine model of severe medial arterial calcification. Am J Physiol Heart Circ Physiol. (2025) 329:H191–205. 10.1152/ajpheart.00530.2024
218.
Xie Y Lin T Jin Y Berezowitz AG Wang X-L Lu J et al Smooth muscle cell-specific matrix metalloproteinase 3 deletion reduces osteogenic transformation and medial artery calcification. Cardiovasc Res. (2024) 120:658–70. 10.1093/cvr/cvae035
219.
Persy VP Verhulst A Ysebaert DK De Greef KE De Broe ME . Reduced postischemic macrophage infiltration and interstitial fibrosis in osteopontin knockout mice. Kidney Int. (2003) 63:543–53. 10.1046/j.1523-1755.2003.00767.x
220.
Zhao Y Huang Z Gao L Ma H Chang R . Osteopontin/SPP1: a potential mediator between immune cells and vascular calcification. Front Immunol. (2024) 15:1395596. 10.3389/fimmu.2024.1395596
221.
Yadav MC Huesa C Narisawa S Hoylaerts MF Moreau A Farquharson C et al Ablation of osteopontin improves the skeletal phenotype of phospho1(-/-) mice. J Bone Miner Res. (2014) 29:2369–81. 10.1002/jbmr.2281
222.
Huesa C Staines KA Millán JL MacRae VE . Effects of etidronate on the Enpp1-/- mouse model of generalized arterial calcification of infancy. Int J Mol Med. (2015) 36:159–65. 10.3892/ijmm.2015.2212
223.
Albright RA Stabach P Cao W Kavanagh D Mullen I Braddock AA et al ENPP1-Fc prevents mortality and vascular calcifications in rodent model of generalized arterial calcification of infancy. Nat Commun. (2015) 6:10006. 10.1038/ncomms10006
224.
Harmey D Hessle L Narisawa S Johnson KA Terkeltaub R Millán JL . Concerted regulation of inorganic pyrophosphate and osteopontin by akp2, enpp1, and ank: an integrated model of the pathogenesis of mineralization disorders. Am J Pathol. (2004) 164:1199–209. 10.1016/S0002-9440(10)63208-7
225.
Boraldi F Bartolomeo A Di Bari C Cocconi A Quaglino D . Donor’s age and replicative senescence favour the in vitro mineralization potential of human fibroblasts. Exp Gerontol. (2015) 72:218–26. 10.1016/j.exger.2015.10.009
226.
Lau WL Leaf EM Hu MC Takeno MM Kuro-o M Moe OW et al Vitamin D receptor agonists increase klotho and osteopontin while decreasing aortic calcification in mice with chronic kidney disease fed a high phosphate diet. Kidney Int. (2012) 82:1261–70. 10.1038/ki.2012.322
227.
Lin Y Sun Z . Klotho deficiency-induced arterial calcification involves osteoblastic transition of VSMCs and activation of BMP signaling. J Cell Physiol. (2022) 237:720–9. 10.1002/jcp.30541
228.
Kuro-O M . Aging and FGF23-klotho system. Vitam Horm. (2021) 115:317–32. 10.1016/bs.vh.2020.12.013
229.
Leibrock CB Alesutan I Voelkl J Michael D Castor T Kohlhofer U et al Acetazolamide Sensitive tissue calcification and aging of klotho-hypomorphic mice. J Mol Med Berl Ger. (2016) 94:95–106. 10.1007/s00109-015-1331-x
230.
Klement JF Matsuzaki Y Jiang Q-J Terlizzi J Choi HY Fujimoto N et al Targeted ablation of the abcc6 gene results in ectopic mineralization of connective tissues. Mol Cell Biol. (2005) 25:8299–310. 10.1128/MCB.25.18.8299-8310.2005
231.
Favre G Laurain A Aranyi T Szeri F Fulop K Le Saux O et al The ABCC6 transporter: a new player in biomineralization. Int J Mol Sci. (2017) 18:1941. 10.3390/ijms18091941
232.
Li Q Jiang Q Uitto J . Pseudoxanthoma elasticum: oxidative stress and antioxidant diet in a mouse model (Abcc6-/-). J Invest Dermatol. (2008) 128:1160–4. 10.1038/sj.jid.5701145
233.
Pasquali-Ronchetti I Garcia-Fernandez MI Boraldi F Quaglino D Gheduzzi D De Vincenzi Paolinelli C et al Oxidative stress in fibroblasts from patients with pseudoxanthoma elasticum: possible role in the pathogenesis of clinical manifestations. J Pathol. (2006) 208:54–61. 10.1002/path.1867
234.
Jiang Q Li Q Grand-Pierre AE Schurgers LJ Uitto J . Administration of vitamin K does not counteract the ectopic mineralization of connective tissues in Abcc6 (-/-) mice, a model for pseudoxanthoma elasticum. Cell Cycle Georget Tex. (2011) 10:701–7. 10.4161/cc.10.4.14862
235.
Gorgels TGMF Waarsing JH Herfs M Versteeg D Schoensiegel F Sato T et al Vitamin K supplementation increases vitamin K tissue levels but fails to counteract ectopic calcification in a mouse model for pseudoxanthoma elasticum. J Mol Med Berl Ger. (2011) 89:1125–35. 10.1007/s00109-011-0782-y
236.
Dedinszki D Szeri F Kozák E Pomozi V Tőkési N Mezei TR et al Oral administration of pyrophosphate inhibits connective tissue calcification. EMBO Mol Med. (2017) 9:1463–70. 10.15252/emmm.201707532
237.
Li Q Huang J Pinkerton AB Millan JL van Zelst BD Levine MA et al Inhibition of tissue-nonspecific alkaline phosphatase attenuates ectopic mineralization in the Abcc6-/- mouse model of PXE but not in the Enpp1 mutant mouse models of GACI. J Invest Dermatol. (2019) 139:360–8. 10.1016/j.jid.2018.07.030
238.
Bouderlique E Kervadec J Tang E Zaworski J Coudert A Rubera I et al Oral pyrophosphate protects Abcc6-/- mice against vascular calcification induced by chronic kidney disease. J Mol Med Berl Ger. (2024) 102:1217–27. 10.1007/s00109-024-02468-y
239.
Boraldi F Annovi G Bartolomeo A Quaglino D . Fibroblasts from patients affected by pseudoxanthoma elasticum exhibit an altered PPi metabolism and are more responsive to pro-calcifying stimuli. J Dermatol Sci. (2014) 74:72–80. 10.1016/j.jdermsci.2013.12.008
240.
Boraldi F Bartolomeo A Li Q Uitto J Quaglino D . Changes in dermal fibroblasts from Abcc6(-/-) mice are present before and after the onset of ectopic tissue mineralization. J Invest Dermatol. (2014) 134:1855–61. 10.1038/jid.2014.88
241.
Sun J She P Liu X Gao B Jin D Zhong TP . Disruption of Abcc6 transporter in zebrafish causes ocular calcification and cardiac fibrosis. Int J Mol Sci. (2020) 22:278. 10.3390/ijms22010278
242.
Hoareau M El Kholti N Debret R Lambert E . Zebrafish as a model to study vascular elastic fibers and associated pathologies. Int J Mol Sci. (2022) 23:2102. 10.3390/ijms23042102
243.
Apschner A Huitema LFA Ponsioen B Peterson-Maduro J Schulte-Merker S . Zebrafish enpp1 mutants exhibit pathological mineralization, mimicking features of generalized arterial calcification of infancy (GACI) and pseudoxanthoma elasticum (PXE). Dis Model Mech. (2014) 7:811–22. 10.1242/dmm.015693
244.
Paule WJ Bernick S Strates B Nimni ME . Calcification of implanted vascular tissues associated with elastin in an experimental animal model. J Biomed Mater Res. (1992) 26:1169–77. 10.1002/jbm.820260907
245.
Schoen FJ Levy RJ . Bioprosthetic heart valve failure: pathology and pathogenesis. Cardiol Clin. (1984) 2:717–39.
246.
Levy RJ Schoen FJ Flowers WB Staelin ST . Initiation of mineralization in bioprosthetic heart valves: studies of alkaline phosphatase activity and its inhibition by AlCl3 or FeCl3 preincubations. J Biomed Mater Res. (1991) 25:905–35. 10.1002/jbm.820250802
247.
Webb CL Flowers WE Horton C Schoen FJ Levy RJ . Long-term efficacy of Al3+ for prevention of bioprosthetic heart valve calcification. ASAIO Trans. (1990) 36:M408–10.
248.
Vyavahare N Ogle M Schoen FJ Levy RJ . Elastin calcification and its prevention with aluminum chloride pretreatment. Am J Pathol. (1999) 155:973–82. 10.1016/S0002-9440(10)65197-8
249.
Thorin E Henrion D Oster L Thorin-Trescases N Capdeville C Martin JA et al Vascular calcium overload produced by administration of vitamin D3 and nicotine in rats. Changes in tissue calcium levels, blood pressure, and pressor responses to electrical stimulation or norepinephrine in vivo. J Cardiovasc Pharmacol. (1990) 16:257–66. 10.1097/00005344-199008000-00012
250.
Wu SY Zhang BH Pan CS Jiang HF Pang YZ Tang CS et al Endothelin-1 is a potent regulator in vivo in vascular calcification and in vitro in calcification of vascular smooth muscle cells. Peptides. (2003) 24:1149–56. 10.1016/j.peptides.2003.07.008
251.
Amer AE El-Sheakh AR Hamed MF El-Kashef HA Nader MA Shehatou GSG . Febuxostat attenuates vascular calcification induced by vitamin D3 plus nicotine in rats. Eur J Pharm Sci. (2021) 156:105580. 10.1016/j.ejps.2020.105580
252.
Amer AE Shehatou GSG El-Kashef HA Nader MA El-Sheakh AR . Flavocoxid ameliorates aortic calcification induced by hypervitaminosis D3 and nicotine in rats via targeting TNF-α, IL-1β, iNOS, and osteogenic Runx2. Cardiovasc Drugs Ther. (2022) 36:1047–59. 10.1007/s10557-021-07227-6
253.
Karamched SR Nosoudi N Moreland HE Chowdhury A Vyavahare NR . Site-specific chelation therapy with EDTA-loaded albumin nanoparticles reverses arterial calcification in a rat model of chronic kidney disease. Sci Rep. (2019) 9:2629. 10.1038/s41598-019-39639-8
254.
Lei Y Nosoudi N Vyavahare N . Targeted chelation therapy with EDTA-loaded albumin nanoparticles regresses arterial calcification without causing systemic side effects. J Control Release. (2014) 196:79–86. 10.1016/j.jconrel.2014.09.029
255.
Kamenskiy A de Oliveira BB Heinis F Renavikar P Eberth J MacTaggart J . Large animal model of controlled peripheral artery calcification. Acta Biomater. (2025) 199:301–14. 10.1016/j.actbio.2025.04.055
256.
Baujat G Besançon A . Generalized arterial calcification of infancy (GACI). Arch Pediatr Organe. (2024) 31:4S21–24S26. 10.1016/S0929-693X(24)00153-2
257.
Boraldi F Murro V Lofaro FD Mucciolo DP Costa S Pavese L et al Phenotypic features and genetic findings in a cohort of Italian pseudoxanthoma Elasticum patients and update of the ophthalmologic evaluation score. J Clin Med. (2021) 10:2710. 10.3390/jcm10122710
258.
Rutsch F Buers I Nitschke Y . Hereditary disorders of cardiovascular calcification. Arterioscler Thromb Vasc Biol. (2021) 41:35–47. 10.1161/ATVBAHA.120.315577
259.
De Sandre-Giovannoli A Bernard R Cau P Navarro C Amiel J Boccaccio I et al Lamin a truncation in Hutchinson-Gilford progeria. Science. (2003) 300:2055. 10.1126/science.1084125
260.
St Hilaire C Ziegler SG Markello TC Brusco A Groden C Gill F et al NT5E Mutations and arterial calcifications. N Engl J Med. (2011) 364:432–42. 10.1056/NEJMoa0912923
261.
Westenberger A Balck A Klein C . Primary familial brain calcifications: genetic and clinical update. Curr Opin Neurol. (2019) 32:571–8. 10.1097/WCO.0000000000000712
262.
Kapustin A Shanahan CM . Targeting vascular calcification: softening-up a hard target. Curr Opin Pharmacol. (2009) 9:84–9. 10.1016/j.coph.2008.12.004
263.
Shao J-S Cheng S-L Pingsterhaus JM Charlton-Kachigian N Loewy AP Towler DA . Msx2 promotes cardiovascular calcification by activating paracrine wnt signals. J Clin Invest. (2005) 115:1210–20. 10.1172/JCI24140
264.
Moe SM Duan D Doehle BP O’Neill KD Chen NX . Uremia induces the osteoblast differentiation factor Cbfa1 in human blood vessels. Kidney Int. (2003) 63:1003–11. 10.1046/j.1523-1755.2003.00820.x
265.
Durham AL Speer MY Scatena M Giachelli CM Shanahan CM . Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res. (2018) 114:590–600. 10.1093/cvr/cvy010
266.
Tyson J Bundy K Roach C Douglas H Ventura V Segars MF et al Mechanisms of the osteogenic switch of smooth muscle cells in vascular calcification: WNT signaling, BMPs, mechanotransduction, and EndMT. Bioengineering. (2020) 7:88. 10.3390/bioengineering7030088
267.
Garcia-Fernandez MI Gheduzzi D Boraldi F Paolinelli CD Sanchez P Valdivielso P et al Parameters of oxidative stress are present in the circulation of PXE patients. Biochim Biophys Acta. (2008) 1782:474–81. 10.1016/j.bbadis.2008.05.001
268.
Lofaro FD Boraldi F Garcia-Fernandez M Estrella L Valdivielso P Quaglino D . Relationship between mitochondrial structure and bioenergetics in pseudoxanthoma elasticum dermal fibroblasts. Front Cell Dev Biol. (2020) 8:610266. 10.3389/fcell.2020.610266
269.
Pedriali G Morciano G Patergnani S Cimaglia P Morelli C Mikus E et al Aortic valve stenosis and mitochondrial dysfunctions: clinical and molecular perspectives. Int J Mol Sci. (2020) 21:4899. 10.3390/ijms21144899
270.
Nollet LL Vanakker OM . Mitochondrial dysfunction and oxidative stress in hereditary ectopic calcification diseases. Int J Mol Sci. (2022) 23:15288. 10.3390/ijms232315288
Summary
Keywords
elastic fibre, mineralization, elastocalcinosis, cardiovascular system, extracellular matrix, calcification models
Citation
Lofaro FD, Mazzilli A, Bonacorsi S, Quaglino D and Boraldi F (2025) Calcification of the elastic component: the impact on the cardiovascular system. Front. Cardiovasc. Med. 12:1636812. doi: 10.3389/fcvm.2025.1636812
Received
28 May 2025
Accepted
08 August 2025
Published
25 August 2025
Volume
12 - 2025
Edited by
Georges Leftheriotis, Université Côte d'Azur, France
Reviewed by
Elisa Ceccherini, National Research Council (CNR), Italy
Gilles Faury, Université Grenoble Alpes, France
Updates
Copyright
© 2025 Lofaro, Mazzilli, Bonacorsi, Quaglino and Boraldi.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
* Correspondence: Daniela Quaglino daniela.quaglino@unimore.it Federica Boraldi federica.boraldi@unimore.it
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.