 Zhuo-Han Li
Zhuo-Han Li Xin-Yao Wang1,†
Xin-Yao Wang1,† Qi Luo
Qi Luo- 1Queen Mary School, Jiangxi Medical College, Nanchang University, Nanchang, China
- 2School of Basic Medical Sciences, Jiangxi Medical College, Nanchang University, Nanchang, China
Advanced glycation end products (AGEs) are deleterious to tissues in vivo, arising from the process of non-enzymatic glycation (NEG), also referred to as the Maillard Reaction, which facilitates the non-enzymatic modification of biomolecules by saccharides. AGEs are integral to the physiological and pathophysiological processes associated with senescence, cardiovascular diseases (CVDs), neurodegenerative and neuroinflammatory diseases, diabetes mellitus (DM) and its complications, autoimmune and rheumatic inflammatory diseases, bone-degenerative diseases, and chronic renal diseases. Both endogenous AGEs and exogenous dietary AGEs can affect the structures and functions of proteins and lipids in cardiovascular tissues and the extracellular matrix of cardiovascular cells by inducing oxidative stress and inflammatory responses, causing direct cell and tissue dysfunction, and activating subsequent signaling pathways mediated by the AGE-RAGE axis. This review focuses on the roles and mechanisms of AGEs in CVDs, from cardiovascular tissues to concrete diseases like heart failure, valvular heart disease, and so on, together with the corresponding treatment and prevention strategies, aiming to provide a comprehensive overview of the roles of AGEs in CVDs and corresponding therapeutic measures.
1 Introduction
In developed nations, cardiovascular disease (CVD) is the leading cause of mortality, responsible for up to one-third of global deaths until 2015 (1, 2). The incidence of CVDs has nearly doubled, increasing from 271 million cases in 1990 to 523 million in 2019. Concurrently, mortality and disability-adjusted life years have also increased from 12.1 million to 18.6 million and from 279.8 million to 393.1 million, respectively (3). This trend is particularly pronounced in regions with low (108.3%), low-middle (114.81%), and middle (117.85%) sociodemographic indices (4). Although high-income countries have seen a decline in age-standardised cardiovascular mortality rates over the past three decades, approximately 80% of CVD-related deaths occur in low- and middle-income countries, often affecting younger individuals, with age-standardised mortality rates remaining relatively unchanged (5, 6). Additionally, the age of individuals at high risk of developing CVDs is decreasing, as evidenced by a significant rise in the global age-standardised incidence and prevalence rates of CVDs among youths and young adults from 1990 to 2019 (7). There is also a gender disparity in CVD epidemiology, with women experiencing a higher prevalence rate than men, yet having lower DALY and mortality rates (7). It is evident that CVDs pose a significant threat to global health security. Although some risk factors for CVDs, such as high systolic blood pressure, high body mass index, poor diet, elevated fasting plasma glucose, and high low-density lipoprotein levels, are well-known (6, 7), the molecular pathogenesis of CVDs remains unclear, underscoring the urgent need for further research into their pathogenesis and prevention strategies.
Advanced glycation end products (AGEs) are stable compounds formed through a complex series of non-enzymatic glycation (NEG) reactions, which begins when sugars react with proteins, lipids, or nucleic acids without enzymatic involvement, initially producing transient intermediates such as Schiff bases and Amadori products (8). This reaction was initially discovered by the French scientist Louis Camille Maillard in 1912, who termed it The Maillard Reaction. NEG can occur both inside and outside the body. In food processing, it is used to enhance flavour and texture through high-temperature cooking methods such as grilling, roasting, and deep-frying (8). Within the body, NEG is prevalent and linked to various diseases, with the accumulation of AGEs in tissues being particularly detrimental. Sullivan et al. identified AGEs and their receptor, RAGE, as a crucial factor in aging, as it causes irreversible modifications to proteins in vivo (9). Ahmad et al. found a connection between AGE and neurodegenerative diseases due to the accumulation of AGEs during disease progression (237). The accumulation of AGE is also associated with numerous other conditions, including neuroinflammatory diseases, diabetes mellitus (DM) and its complications, autoimmune/rheumatic inflammatory diseases, bone-degenerative diseases, and chronic kidney diseases (10).
Additionally, CVDs are another major category of illnesses attributed to the harmful effects of AGEs on cardiovascular tissues, involving atherosclerosis, hypertension, heart failure, and cardiovascular complications of diabetes, among others (11–14). For instance, Chen et al. proposed that AGEs might contribute to CVD by playing a role in atherosclerosis and arterial stiffness (15). Therefore, it is crucial to investigate the pathogenic mechanisms of NEG in CVDs, the effects of AGEs on specific cardiovascular tissues, and related therapeutic strategies.
2 Generation of AGEs
2.1 Generation process of AGEs
Advanced glycation end products (AGEs) are stable molecules generated through the non-enzymatic glycation of proteins, lipids, or nucleic acids. This process is initiated by the interaction of reducing sugars with these biomolecules, resulting in the formation of early glycation intermediates such as Schiff bases and Amadori products (16).
According to studies, this reaction process can be roughly divided into early, intermediate, and advanced stages, with different products and characteristics at each stage (17). The carbonyl group on the open-chain form of glucose, fructose, or ribose reacts with the free amino group of biomacromolecules in the early stage with a reversible condensation reaction to form intermediate products aldimines, a type of unstable Schiff base that is susceptible to undergo rearrangement with the consequence of formation of more stable Amadori products (18, 19). In the intermediate stage, Amadori products degrade into dicarbonyl compounds through dehydration, oxidation, fission, and other reactions in an acid-base environment, containing various reactive fission products ready for further reactions (16). Finally, in the advanced stage, an extensive range of reactions, including dehydration, enolization, oxidation, fragmentation, rearrangement, isomerization, and further condensation, act on these dicarbonyl compounds, facilitating the production of irreversible AGEs, including hydroimidazolone, Nε-carboxymethyl-lysine (CML), pentosidine, and glucosepane (17, 20–22). The entire process is usually applied in food processing to control and limit critical steps (19). The overall NEG procedure is illustrated in Figure 1.
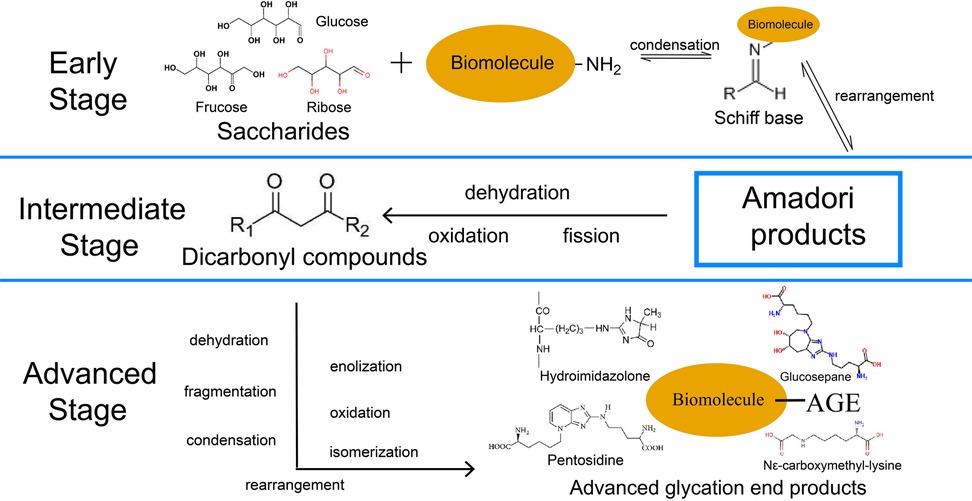
Figure 1. Three-stage procedure of NEG. The solid arrows represent “generation”. NEG, non-enzymatic glycation; AGE, advanced glycation end products.
2.2 Factors affecting the generation of AGEs
The generation of AGEs occurs extensively in our life activities and metabolic processes, determining the functions of biological macromolecules, such as proteins, with the rate and extent of this reaction being affected by various factors within different physiological and pathological conditions.
2.2.1 Blood sugar level
As the main component serving in carrying and transporting oxygen in the erythrocyte, haemoglobin (Hb) can be glycated to HbA1c under high blood sugar conditions as a measurement for monitoring blood sugar levels. Moreover, researchers have illustrated that AGE were generated in HbA1c as a precursor, can serve as a better biomarker of glycaemia in the detection of diabetic patients (23). Therefore, in the blood circulation environment, high blood sugar levels as the increased substrate and internal temperature as the appropriate temperature can enhance AGE level. Furthermore, the reaction between Hb and sugar is also found in circulation because not only the neutral α-amino groups from N-terminal residues of Hb has sugar molecules with high reactivity and strong affinity, the ε-amino groups of Lys residues in the side chain of Hb also have higher values of pH accelerating the unfolding of sugar chains and unprotonated form of the amino group, ultimately trigger the formation of AGE (17, 24). Moreover, it has been proved that the extent of reaction between proteins and carbohydrates tends to diminish with the increase of molecular weight, which means monosaccharide tend to generate more AGEs than oligosaccharide (25). Therefore, increased glucose levels in the blood are conducive to the AGE serum levels.
2.2.2 Oxidative stress (OS)
The intracellular OS level significantly influences AGE production through hyperglycaemia, due to an imbalance in prooxidant-antioxidant equilibrium that favours prooxidant effects, potentially causing tissue damage (26). Research shows hyperglycaemia triggers excessive mitochondrial reactive oxygen species (ROS) production, suppressing glyceraldehyde-3 phosphate dehydrogenase (GAPDH) activity, a crucial glycolysis enzyme. This increases glyceraldehyde-3-phosphate concentration, a precursor of AGEs upstream of GAPDH, triggering AGE production (27). Oxidative stress increases AGE production and facilitates ROS generation by promoting AGE-modified proteins binding to receptors for advanced glycation end products (RAGE), a multiligand immunoglobulin superfamily member present in tissues (20, 28). Elevated AGE production under hyperglycaemia imposes various metabolic challenges in different diseases.
2.2.3 Inflammatory response
Studies showed that in the myeloperoxidase system of activated neutrophils, Nε-(carboxymethyl) lysine (CML), a well-characterized AGE product, generates (29), indicating AGE production may increase at inflammation sites in diseases. Furthermore, binding AGEs with RAGE upregulates inflammatory cytokines, including tumour necrosis factor α, interleukin-6, and C-reactive protein, through NF-κB signalling pathways to increase inflammatory responses in CVD (30).
In addition to common pathological factors affecting NEG velocity and extent, environmental and lifestyle factors can accelerate AGE production. There are two primary AGE sources: endogenous AGEs generated in the body and exogenous AGEs ingested from foods. A high carbohydrate and calorie diet and sedentary lifestyle promote endogenous AGE production via hyperglycaemia, resulting in metabolic burdens in CVD and diabetes mellitus (DM). Exogenous AGEs form when food is processed at high temperatures, such as deep-frying, broiling, roasting, and grilling, making high-temperature cooking harmful (20). The epigenetic effects of AGEs are induced by RAGE, referring to AGEs' induction of gene expression and phenotype variation due to genetic modifications without altering DNA sequence (31). Transient alterations from short-term environmental changes in AGEs could facilitate permanent epigenetic effects (32). Long-term AGE stimulation contributes to metabolic memory, leading to adverse effects in CVD, DM, and other metabolic diseases. In DM, metabolic memory formed by increased AGE production under hyperglycaemia causes inflammatory response reinforcement, maintaining chromatin remodelling changes and histone modifications in promoters, even when blood glucose levels normalize (31). Studies indicate that increased OS and inflammatory mediators with aging are attributed to dietary AGE accumulation, having pathological effects in common diseases of older people (20, 33). Therefore, unhealthy lifestyles like sedentary behavior, high sugar diet, and high heat diet trigger endogenous and exogenous AGE accumulation, which stimulates OS and inflammatory responses, forming metabolic memory through AGEs' epigenetic effects, leading to metabolic burdens in many diseases.
2.3 Measurement of AGEs In Vivo
AGEs serve as biomarkers in CVDs and diabetes and various in vivo measurement methods for AGEs have been employed in clinical studies to investigate their associations with these diseases.
Due to the inherent immunogenicity of AGEs, immunochemical methods including particularly enzyme-linked immunosorbent assay (ELISA) have been widely used in clinical studies to measure AGE levels in serum or plasma (34–36). Using this approach, studies have demonstrated significantly higher serum AGE concentrations in diabetic patients compared to healthy individuals (37).
Certain AGEs exhibit spontaneous fluorescence upon excitation, enabling their quantification through fluorescence intensity measurements. This property allows the bioanalytical method for detection of AGEs in serum and urine using fluorescence spectrophotometry (FS) (38, 39), as well as noninvasive assessment of skin AGEs via skin autofluorescence spectroscopy (SAF) (40, 41). Clinical studies utilizing these methods have revealed elevated AGE levels in various disease states. For instance, Galler et al. reported significantly higher serum levels of fluorescent AGEs in type 1 diabetes patients compared to healthy controls (38), while Suehiro et al. observed increased urinary AGE concentrations in metabolic syndrome patients at high cardiovascular risk relative to healthy individuals (39). For the noninvasive way, some clinical studies have demonstrated that T2D patients had higher value of SAF compared to controls (42), and the SAF results positively correlated with coronary artery calcification and microvascular complications (43, 44). SAF was also found independently associated with all-cause mortality and fatal or nonfatal major adverse cardiovascular events in patients with peripheral artery disease (45). These findings collectively highlight the widespread application of fluorescence-based bioanalytical technologies in clinical research investigating the biomarker potential of AGEs in cardiovascular diseases and metabolic disorders.
Chromatography-mass spectrometry techniques, including high-performance liquid chromatography (HPLC), liquid chromatography-mass spectrometry (LC-MS), and gas chromatography-mass spectrometry (GC-MS), represent another analytical approach for quantifying AGEs in serum and tissue samples. However, the clinical application of these methods remains limited due to their high operational costs and time-intensive procedures (46).
3 Different AGE products in CVDs
3.1 Accumulation of AGEs in proteins
Modifications of proteins in cardiovascular tissues, including myocardial cells and vascular endothelial cells, and in plasma, will change their structures and functions, resulting in pathological effects in cardiovascular tissues.
3.1.1 Structural proteins in cardiovascular tissues
Collagen and laminin are essential structural proteins in the extracellular matrix (ECM) of blood vessels and the myocardium. They are responsible for the mechanical properties of cardiovascular tissues and facilitate cell attachment, adhesion, and migration within the basement membrane (47, 48). As aging advances, there is an increased formation of AGEs on collagen and laminin within the vascular wall and myocardium. This process promotes the development of covalent crosslinks in collagen and laminin fibre, altering the matrix architecture by increasing its pore size and stiffness (47, 49, 50). Consequently, the elasticity of the ECM diminishes, resulting in decreased arterial and myocardial compliance, which increases the risk of hypertension, atherosclerosis, and cardiac failure.
Elastin is another crucial mechanical component found in the media of large and medium-sized arteries. It forms lamellae and connects the fibrils of these arteries, providing ductility (51, 52). Elastin can be viewed as a polymer composed of linear polypeptide chains (tropoelastin) stabilized by lysine-derived crosslinks (52), making it susceptible to NEG. Therefore, age-related production and accumulation of AGEs reduce elastin crosslinks, contributing to the weakening of arterial elasticity (53).
3.1.2 Functional proteins in plasma
Albumin is a plasma protein that maintains osmotic pressure and transports molecules in the body. Studies show that high glycated albumin promotes mononuclear cell adhesion to endothelial monolayers for inflammatory effects (54, 55), indicating that AGEs on albumin may contribute to vascular endothelial inflammation and dysfunction in CVD. As a marker of short-term glycaemic control, glycated albumin and HbA1c are used in blood sugar monitoring (56, 57). Fibrinogen plays a vital role in forming fibrin clots and performs coagulation functions, with lysine residues being crucial (58). The level of AGEs in fibrinogen under hyperglycemia affects fibrin clot modulation and fibrinolysis, resulting in reinforced coagulation activity and resistance to fibrinolysis, increasing the propensity of fibrinogen to form clots and risk for thrombotic events in cardiovascular diseases (59–61). Therefore, fibrinogen serves as a risk marker for CVD (62) and an independent marker for vascular complications, particularly atherosclerosis (63). Myosin is a contractile protein found in cardiac and vascular smooth muscle cells (VSMC). In the context of elevated blood glucose levels, the formation of AGEs in plasma activates RAGE, which is known to engage in complex signal transduction pathways that impair the function of cardiac muscle cells and VSMCs. The mechanisms involved include an increase in overall cell rigidity through the amplification of myosin activity and a reduction in muscle contraction efficiency due to detrimental effects on the contractile capacity of the cells (64).
3.2 Accumulation of AGEs in lipids
In addition to the formation of AGEs in proteins, glycated lipids under hyperglycaemia or dietary levels also harm cardiovascular tissues in other ways.
3.2.1 Phospholipids in cardiovascular tissues
Phospholipids are the main components of the cell membrane and are responsible for membrane fluidity. Some amino phospholipids, particularly phosphatidylethanolamine (PE) and phosphatidylserine (PS), can be glycated under conditions of hyperglycaemia or heated at high temperatures from the diet, with glycated phospholipids promoting the production of ROS and embodying angiogenic activity on endothelial cells which further exacerbates lipid peroxidation and vascular damage (65–67). Furthermore, hydrolysis of phospholipids is initiated by AGEs (68), which impairs membrane fluidity and disrupts membrane integrity and function.
3.2.2 Lipoproteins in cardiovascular tissues
Lipoproteins modulate cholesterol transportation in the blood, with the two main types, low-density lipoprotein (LDL) and high-density lipoprotein (HDL), playing the roles of aggravation and resistance to atherosclerosis, respectively. It has been found that glycation of LDL will change its structure and inhibit its clearance from circulation, which provides more opportunities for its uptake by monocytes and macrophages, eliciting the generation of foam cells and the occurrence of atherosclerosis (69). Moreover, researchers have demonstrated that AGEs within both LDL and HDL under hyperglycaemia induces endothelial cell dysfunction and further atherosclerosis, with mechanisms of glycated LDL containing reduction of nitric oxide bioavailability that results in damage to the vascular system, induction of OS, decrease of fibrinolytic activity that leads to thrombus, in vivo atheroma formation, induction of apoptosis of endothelial cells, stimulation of inflammation like adhesion of monocytes and formation of foam cells, and activation of endoplasmic reticulum stress (70, 71). Meanwhile, AGEs impair HDL function for glycated HDL, which protects against atherosclerosis through mechanisms similar to those of glycated LDL (70).
Moreover, as the lipid composition of lipoproteins, glycated cholesterol was found to contribute to alterations in LDL clearance and increased susceptibility of LDL to OS (72), which means the enhancement of atherogenic properties of lipoproteins that are averse to atherosclerosis.
3.3 Accumulation of AGEs in DNA
Apart from the formation of AGEs in proteins and lipids, the glycated DNA contributes to cardiovascular tissue damage and other pathologies through mutagenesis and genomic instability.
The major nonenzymatic glycation product of DNA, N2-(1-carboxyethyl)-2′-deoxyguanosine (CEdG) (73, 74), has been shown to induce single-strand breaks and increase mutation frequencies in oncogenes and tumor suppressor genes (74), promote mutagenesis by altering base-pairing preferences due to its distinct syn/anti conformations during replication (75), and cause DNA damage when the nucleotide excision repair efficiency is compromised (76). These mechanisms collectively contribute to genetic instability and elevated cancer risk associated with metabolic diseases.
Acetoacetate, a ketone body increased during ketosis in diabetic patients has been found can activate effect on glucose-mediated DNA glycation during hyperglycemia because acetoacetate can induce ROS and ROS elicits DNA glycation (77). Clinical studies have found that CEdG was significantly elevated in diabetes as well, which implies the effects of DNA-AGE on diabetes and its potential on serving as biomarker of diabetes (78). Due to DNA damage resulting from many mechanisms like strand breaks induced by CEdG in the metabolic syndrome, the risk of various cancers and cardiovascular diseases will be elevated (79). However, it is worth mentioning that inhibition of glyoxylase 1 (GLO1) in cancers can increase levels of DNA-AGE and RAGE that is cytotoxic to glioma cells (80), implying the dialectical roles of CEdG in cancers. DNA damage also accompanies with aging and aggravates the incidence of neurodegenerative disease like Alzheimer disease and Parkinson disease (81).
Other structures of cardiovascular tissues impacted by AGEs are summarized in Table 1.
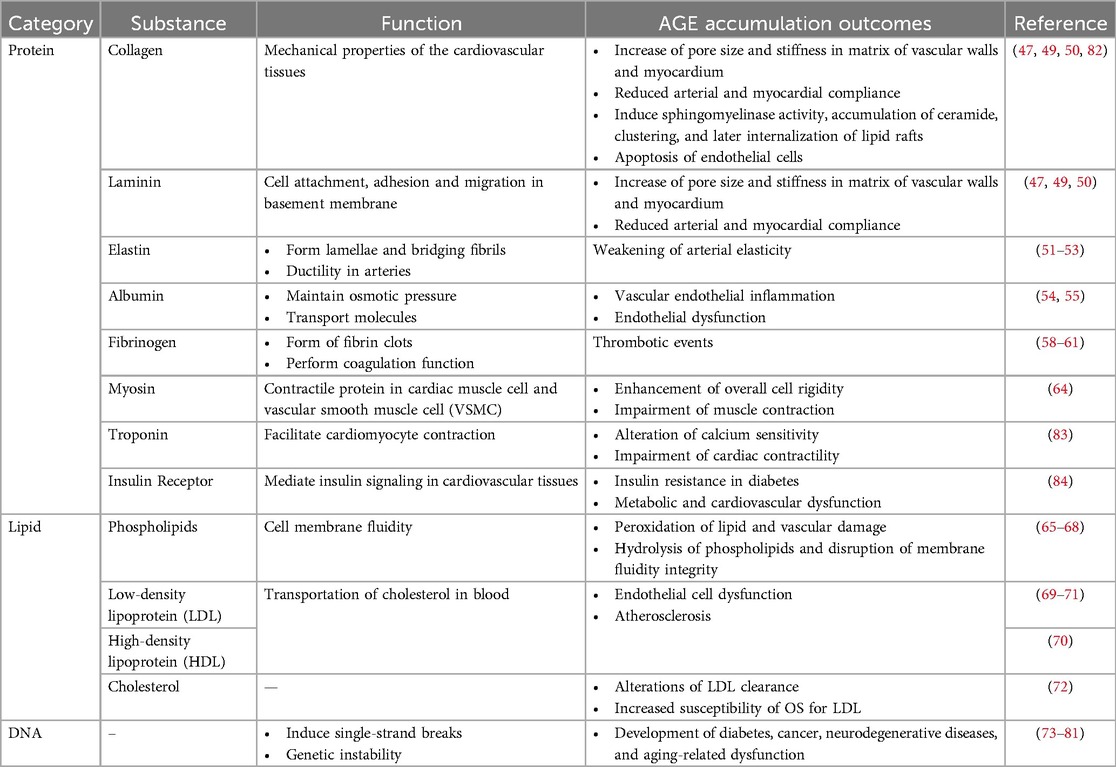
Table 1. Specific structures within cardiovascular tissues that are susceptible to glycation and their resultant effects.
4 The mechanisms of AGE accumulation in CVDs
The mechanism of AGE deposition in CVDs have been investigated. It has been shown that the harmful effects of AGEs in CVDs depend on their interaction with receptors, which can be categorized as either intracellular or extracellular. Intracellular AGE receptors include RAGE, a type I cell surface receptor that is part of the immunoglobulin (Ig) superfamily. This group also includes scavenger receptors such as class H, class A, class B, and class E, as well as the AGE receptor complex (AGE-R) comprising OST-48, 80K-H, and galectin-3 (85). The soluble receptor for advanced glycation end products (sRAGE) is an extracellular receptor for AGEs that exists freely outside the cells. It has two main forms: endogenous secretory RAGE (esRAGE), a splicing variant of the RAGE gene, and cleaved RAGE (cRAGE), which results from the breakdown of membrane RAGE (86). Among the various AGE receptors, the RAGE and AGE-RAGE axis has been extensively studied, allowing for a detailed exploration of the mechanisms by which AGEs act in CVDs.
4.1 OS and activation of inflammatory reaction
OS is linked to the levels of ROS, which include superoxide anion radicals (O2−), hydrogen peroxide (H2O2), and hydroxyl radicals (HO−). This is sometimes accompanied by reactive nitrogen species (RNS), such as nitric oxide (NO−), nitrogen dioxide (NO2), and peroxynitrite (ONOO−). Several mechanisms through which AGEs lead to OS and inflammation have been identified. First, AGEs create irreversible crosslinks with proteins and lipids, altering their structure and function, promoting ROS production and OS, as discussed in Sections 3.1, 3.2. Second, AGEs cause mitochondrial dysfunction by disrupting the respiratory chain and increasing mitochondrial stress, leading to excessive ROS production and OS (87). Third, AGEs cause OS and ROS production by interacting with RAGE. AGE-RAGE interaction activates nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX), resulting in ROS production (88, 89). Excessive ROS generation leads to abnormal oxidation and dysfunction of biomacromolecules, triggering the expression of inflammatory mediators, such as tumour necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), cyclooxygenase 2 (COX-2), and prostaglandin E2 (PGE2) (88). Fourth, signalling pathways activated by the AGE-RAGE axis induce OS and inflammatory responses. The nuclear factor kappa B (NF-κB) signalling pathway is the main pro-inflammatory pathway in ROS and OS production, leading to vascular injury. NF-κB is a transcription factor sensitive to free radicals that regulates the transcription of endothelin-1, VCAM-1, tissue factor, and thrombomodulin, which relate to vascular functions (90). ROS production in endothelial cells stimulated by AGEs is mediated by NF-κB signaling, where NF-κB induces TNF-α expression, mediating ROS generation (91). Additionally, Rho/Rho-associated protein kinase (Rho/ROCK) expression mediated by AGEs is involved in NF-κB signalling pathway and ROS production (92, 93). The Janus kinase-signal transducers and activators of transcription (JAK-STAT) signalling pathway is activated by AGE-RAGE, leading to JAK phosphorylation and STATs activation. Phosphorylated STATs form dimers and move into the nucleus to regulate transcription of inflammatory cytokines, such as interleukin-6 (IL-6), TNF-α, and MCP-1, and inducible enzymes like iNOS and COX2 (94). Moreover, RNS production is mediated by the AGE-RAGE axis, contributing to OS (95).
After OS and inflammatory reactions induced by AGEs, further increased production of AGEs can be triggered by OS and inflammatory reactions themselves, as mentioned in Sections 2.2.2, 2.2.3. This is how the vicious circle is generated, which gradually aggravates the implications of OS and inflammatory reactions. Furthermore, it was proven that the enhancement of NOX activity and activation of NF-κB induced by AGE-RAGE interactions will facilitate the expression and production of iNOS and ONOO−, namely the overproduction of RNS that also results in OS (95). RNS is involved in the production of AGEs (96), which also serves as a part of the vicious circle.
The occurrence of OS and accumulation of inflammatory cytokines such as C-reactive protein (CRP), IL-1β, IL-6, TNF-α and so on will subsequently trigger the oxidative damage of biomacromolecules in vivo and aggregation of inflammatory cells, which ultimately leads to the impairment of cardiovascular structures and development of CVDs.
4.2 Cell dysfunction
AGE can directly initiate injury in cardiovascular cells involving myocardial cells, vascular endothelial cells, smooth muscle cells, and so on to affect their normal physiological functions. Endogenous AGEs produced inside myocardial cells and cardiac fibroblasts are proven to embody cytotoxic and cause CVDs. It was indicated that AGEs in cardiomyocytes could decrease pulse rate and cell activity and induce cell death, which concerns the suppression of autophagy. It is an essential mechanism for maintaining cell homeostasis via the degradation of aging and damaged proteins and organelles (97). AGEs in cardiac fibroblasts exert cytotoxicity and inhibit their roles in regulating myocardial ECM homeostasis and restoring damaged cardiac functions. Furthermore, with the expression of RAGE in cardiomyocytes and cardiac fibroblasts, the AGE-RAGE axis may also induce cytotoxic reactions and dysfunction in these cells (98). Extracellular AGEs also facilitate CVDs through the AGE-RAGE axis, which activates the production of ROS and, subsequently, the NF-κB signalling pathway in vascular wall cells, contributing to atherosclerosis (99).
Recent studies have suggested that the interaction between AGEs and RAGE can destroy the balance of osteogenesis and osteoclastic in VSMCs to promote vascular calcification and pathological bone remodelling (100, 101), contributing to atherosclerosis development. Another mechanism that elicits calcium deposition in VSMCs is Nox-derived OS, which is involved in AGE-induced apoptosis of VSMCs (102). Moreover, it was proven that the interaction between AGEs and RAGE could induce the expression of the pro-sclerotic cytokine TGF-β, which mediates the trans-differentiation of smooth muscle cells to myofibroblasts (103), which may promote vascular stiffness through the destruction of VSMCs.
Studies have demonstrated that AGEs boost vascular endothelial growth factor (VEGF) production and activate its autocrine function in endothelial cells, resulting in angiogenesis. Overexpression of RAGE can trigger reactions in endothelial cells, highlighting the AGE-RAGE axis (104). Profilin-1, an intracellular protein associated with actin, regulates endothelial cell contraction and vascular permeability. AGEs elevate profilin-1 levels in endothelial cells, causing cytoskeletal changes and cell damage, evident from increased endothelial cell permeability, apoptosis initiation, and enhanced cell migration, adhesion, and focal contact formation (105). Additionally, myosin light chain (MLC) phosphorylation by MLC kinase (MLCK) promotes cytoskeletal contractile activity and disrupts tight junctions, increasing endothelial permeability. AGEs in endothelial cells can decrease miR-1-3p expression, a microRNA with therapeutic potential in treating endothelial dysfunction. This decrease amplifies MLCK signalling, leading to cytoskeletal contraction, increased vascular permeability, endothelial cell aging, and barrier dysfunction (106). Endothelial progenitor cells (EPCs), particularly late EPCs, vital for endothelial maintenance, repair, and postnatal angiogenesis, are negatively impacted by AGEs in a concentration-dependent manner, disrupting proliferation, migration, adhesion, and inducing apoptosis (107).
4.3 Extracellular matrix remodeling
In addition to direct impairment of cardiovascular cells by AGEs, modification of ECM components by AGEs leads to alterations in structure and function that contribute to CVD pathological effects. As mentioned in Section 3.1, AGEs modify essential ECM proteins, such as collagen and elastin, affecting their physiological functions by changing crosslink structures. Ligation of AGEs and RAGE may alter the production of these ECM proteins, leading to changes in elastic fibres and increased carotid diameter (108). This is associated with arterial stiffness and atherosclerosis risk. Furthermore, studies show that exercise training can inhibit advanced glycation by reducing CML and RAGE expression, which attenuates aortic stiffening and prevents endothelial dysfunction marked by decreased collagen levels, increased elastin levels, and reduced pulse wave velocity (PWV), with RAGE inhibitor FPS-ZM1 achieving similar effects (109). This indicates the dynamic relationship between collagen and elastin relates to vascular physiological conditions. AGEs may attenuate elastin levels and increase collagen levels to aggravate aortic stiffness, contributing to CVD pathological responses. Beyond glycated collagen that can attenuate vascular wall flexibility and arterial and myocardial compliance, as noted in Section 3.1.1, glycated structural extracellular proteins contribute to myocardial stiffness, causing impaired relaxation and diastolic dysfunction (110). Glycated type IV collagen and laminin can reduce endothelial cell adhesion and spreading by disrupting cell attachment sites (111), destroying endothelial cell integrity and functions.
Glycation of other ECM components, such as fibrinogen and LDL, as mentioned in Section 3.1.2, generally contributes to thrombogenesis, reduction of fibrinolysis, and risk of atherosclerosis. Other studies have shown that irreversible AGE-induced modifications of ECM components, such as type IV collagen, laminin, and vitronectin, can facilitate the overproduction and accumulation of ECM and vasoconstriction (112), which contributes to the accumulation of fibrous tissue, resulting in myocardial fibrosis and hypertension. Furthermore, the AGEs–RAGE axis induces autophagy, which can activate cardiac fibroblasts, which are responsible for the secretion of collagenous extracellular matrix proteins, such as collagen I and collagen III. Activated cardiac fibroblasts transform into myofibroblasts and accumulate in the myocardium. Increased ECM component deposition and myofibroblast accumulation contribute to myocardial fibrosis in heart failure (113). AGEs also increase the expression of KCa3.1 channels in a RAGE-dependent manner by activation of ERK1/2, p38-MAPK, and PI3K/Akt signalling, thereby promoting the proliferation of cardiac fibroblasts and ECM (114), which is another mechanism of deposition of ECM components that correlates to myocardial fibrosis. The small GTPase Rap1a modifies and activates downstream events of the AGE/RAGE signalling cascade to increase cardiac fibroblast collagen gel contraction and ECM remodelling (115). TGF-β signalling interacts with RAGE signalling or independently causes ECM remodelling (116). Consequently, myocardial fibrosis is exacerbated.
4.4 Effects on CVDs-related signaling pathways
After discussing the direct effects of AGEs on OS, inflammation, cell dysfunction, and ECM remodelling that elicit pathological responses in CVDs, we will illustrate the mechanisms by which glycation disrupts normal transduction of crucial signalling pathways in CVDs to microscopically explore the impairment of AGEs on cell metabolism and survival in the development of CVDs. A related illustration is shown in Figure 2.
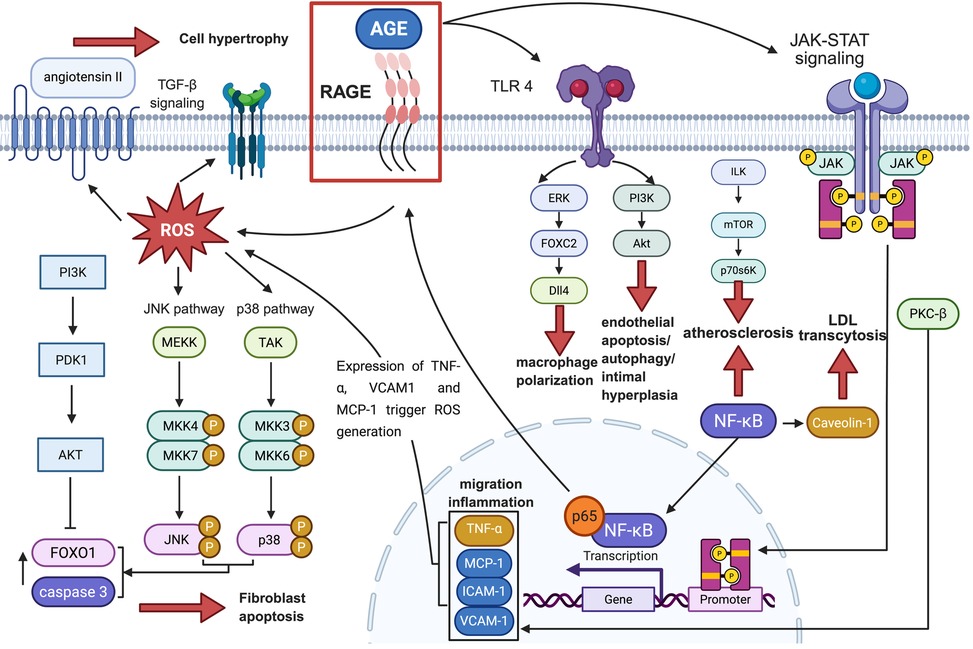
Figure 2. The signaling pathways associated with CVD that are mediated by the AGE-RAGE axis, along with their downstream factors and ultimate effects on pathological conditions. The black or red solid arrows represent “facilitation”, the dashed arrows represent “combination”, and the solid “T” lines represent “inhibition”. OS, oxidative stress; AGE, advanced glycation end products; RhoA, Ras homolog family member A; ROS, reactive oxygen species; Ang II, angiotensin II; TGF-β, transforming growth factor-β; Smad, suppressor of mothers against decapentaplegic; PKC-β, protein kinase C-β; MCP-1, monocyte chemotactic protein-1; ICAM-1, intercellular adhesion molecule-1; fVCAM-1, vascular cell adhesion molecule-1; JNK, c-Jun N-terminal kinase; FOXO1, forkhead box protein O1; TNF-α, tumor necrosis factor-α; NF-κB, nuclear factor kappa-B; NADPH, triphosphopyridine nucleotide; LDL, low-density lipoprotein; TLR4, toll-like receptor 4; ERK, extracellular regulated protein kinases; FOXC2, forkhead box C2; DII4, delta like ligand 4; PI3K, phosphoinositide 3-kinase; AKT, protein kinase B; ERK1/2, extracellular regulated protein kinases 1/2; JAK, Janus kinase; STAT, signal transducer and activator of transcription; ILK, integrin linked kinase; mTOR, mammalian target of rapamycin; p70S6K, p70 ribosomal protein S6 kinase.
Activation of the RAGE/NF-κB signalling pathway by AGEs is a prevalent mechanism that contributes to the progression of cardiovascular diseases. Recent research has shown that AGEs trigger the RAGE-NF-κB signalling cascade, leading to increased NF-κB signalling. Concurrently, the NF-κB subunit p65 is drawn to the RAGE promoter, which boosts RAGE transcription and establishes a positive feedback loop between NF-κB signalling and RAGE expression, further amplifying the NF-κB signalling pathway. Consequently, various downstream effectors of this pathway are activated to perform their respective roles. Caveolin-1, a key membrane-bound scaffolding protein found in caveolae, acts as a downstream effector of the RAGE/NF-κB signalling pathway and is involved in LDL transcytosis in endothelial cells. Enhanced NF-κB signalling leads to the upregulation of Caveolin-1, facilitating LDL transcytosis and causing LDL to deposit beneath the endothelium, which ultimately contributes to the development of atherosclerotic plaques (117).
Protein kinase C (PKC) is a collection of enzymes within the AGC family (cAMP-dependent protein kinase/protein kinase G/protein kinase C) that function as serine/threonine-related protein kinases and play a role in cell signalling (118). The RAGE/PKC-β pathway is another signaling mechanism implicated in vascular dysfunction, where AGEs trigger the increased production of monocyte chemotactic protein-1 (MCP-1) in monocytes and endothelial cells, as well as intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) in endothelial cells during inflammatory conditions and oxidative stress. This process leads to macrophage adhesion to endothelial cells, contributing to endothelial dysfunction (119, 120). Additionally, AGEs influence the RAGE/PKC-β signalling pathway by enhancing the expression and phosphorylation of PKC-β1/2 in cardiac microvascular endothelial cells (CMECs), promoting CMEC proliferation and reducing early apoptosis, potentially playing a role in the development of cardiomyopathy (121).
Research shows that the RAGE/phosphatidylinositol 3-kinase (PI3K)/AKT signalling pathway is activated by AGEs and regulates cellular functions. AGEs increase the expression of proteins related to cell proliferation and migration, as well as filamentous actin, in human aortic vascular smooth muscle cells (HASMCs). This leads to HASMC proliferation and migration, causing intimal hyperplasia and lumen stenosis, conditions linked to arteriosclerosis development (122). The mechanism behind VSMC proliferation involves autophagy triggered by AGEs through the ERK signaling pathway, a member of the mitogen-activated protein kinase (MAPK) family, and AKT signaling pathways. AGEs promote ERK phosphorylation while inhibiting AKT phosphorylation (123). Studies suggest that inhibiting AGE-induced apoptosis in endothelial cells can be achieved by activating the ERK1/2 and PI3K/AKT signalling pathways (124). This indicates that AGEs' impact on apoptosis and endothelial cell death may involve the ERK1/2 and PI3K/AKT pathways.
Rho is an essential small GTP-binding protein that functions as a molecular switch in signal transduction and plays a crucial role in remodelling the actin cytoskeleton across various cell types. Research has shown that the RAGE/Rho signalling pathway can be triggered by AGEs, leading to the expression of RhoA in endothelial cells. This process enhances microvascular hyperpermeability through actin polymerization and the reorganization of the actin cytoskeleton (125).
Research has demonstrated that the interaction between AGEs and the receptor for advanced glycation end-products (RAGE) can trigger Toll-like receptor 4 (TLR4) signalling. This activation promotes ERK phosphorylation, which in turn increases the expression of FOXC2 and subsequently delta-like ligand 4 (Dll4) in macrophages during M1 polarization. FOXC2, a transcription factor from the forkhead family, is essential for vascular development and integrity, whereas Dll4 can activate the Notch signalling pathway, which is vital for the phenotypic conversion of vascular smooth muscle cells (VSMCs) from a contractile to a synthetic state. Consequently, within the RAGE/TLR4/FOXC2 signalling pathway, the high expression of Dll4 induced by AGEs in macrophages through direct cell-to-cell interaction mediates the Notch signalling pathway, leading to the formation and progression of atherosclerotic plaques characterized by VSMCs transitioning from contractile to synthetic phenotypes (126). The RAGE/ILK/mTOR/p70S6K signalling pathway is another key player in the advancement of atherosclerosis, with ILK acting as a versatile intracellular serine/threonine kinase that contributes to the pathogenesis of the disease. Within this pathway, AGE activation triggers the expression of proteins in the ILK/mTOR/p70S6K signalling pathway, leading to VSMC-to-osteoblast trans-differentiation and cell proliferation, which in turn fosters the progression of atherosclerosis (127).
Certain signalling pathways are triggered by ROS, which is facilitated by AGEs. AGE-RAGE-mediated production of ROS activates the Ang II-TGF-β–Smad pathway. In this pathway, ROS-induced autocrine production of Ang II in mesangial cells leads to TGF-β expression and Smad phosphorylation, resulting in mesangial cell hypertrophy and fibronectin synthesis (128). ROS production through AGE-RAGE interactions stimulates p38 and JNK within the MAPK signalling pathway, initiating cascades that activate cascade-3 and FOXO1, promoting fibroblast apoptosis (129). AGEs may also induce vascular adventitial fibroblasts (AFs) migration and inflammatory mediator release by upregulating RAGE expression via MAPK signalling and NF-κB activation, contributing to early atherosclerosis development (130).
5 The role of AGEs in different CVDs
The accumulation of AGEs is a key promotive factor for a range of CVDs (Figure 3). The manifestations of the above-mentioned mechanisms of AGE formation in some specific CVD types will be described separately as follows.
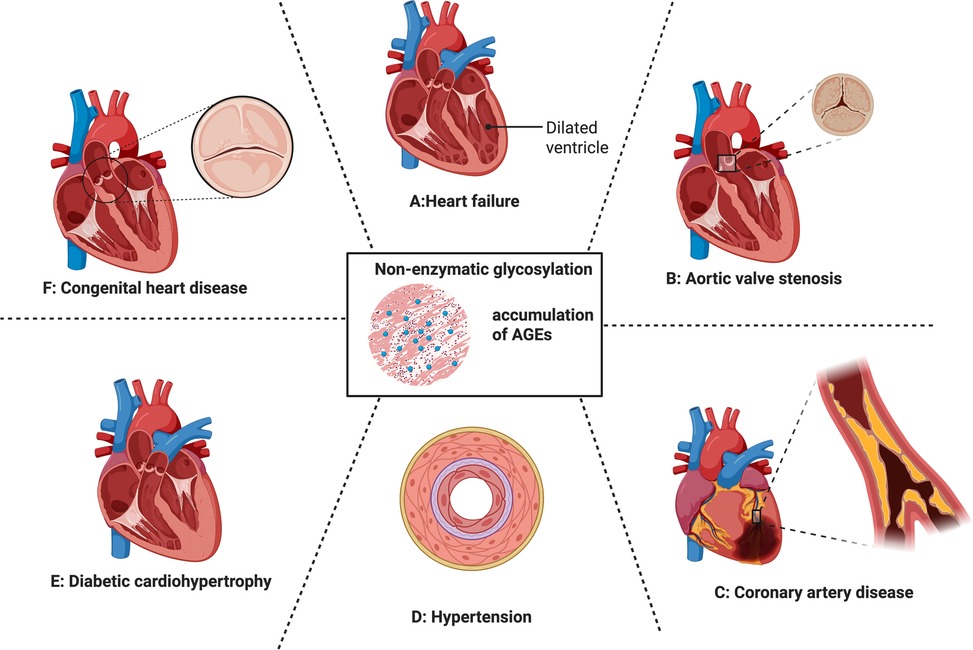
Figure 3. Cardiovascular impact of non-enzymatic glycation and the accumulation of AGEs. From (A–F) heart failure, valvular heart disease, coronary artery disease, hypertension, diabetic cardio hypertrophy, congenital heart disease. Each of them were discussed in 5.1–5.6.
5.1 AGEs and heart failure
Heart failure is a progressive clinical syndrome resulting from diverse cardiovascular abnormalities, such as coronary artery disease, myocarditis, hypertension, and cardiomyopathy, ultimately leading to impaired atrial or ventricular ejection of blood (131). Plentiful evidence acknowledged that AGEs is related to pathophysiological mechanisms of heart failure (11). Both the accumulation of AGEs in the ECM and activation of AGE-RAGE signaling affects cardiac dysfunction by two central mechanisms (132–134).
Extracellular matrix (ECM) remodeling is a hallmark of heart failure, where pressure overload promotes excessive ECM synthesis, contributing to both diastolic and systolic dysfunction (135, 136). AGE formation manipulates ECM remodeling by altering their structures and functions, especially collagen. AGEs in collagen leads to crosslinks in ECM that contribute to myocardial stiffness and left ventricular ejection fraction (LVEF) (137, 138). In mouse models of heart failure, a reduction in collagen type III alongside the accumulation of advanced glycation end products (AGEs) has been observed. Treatment with crosslink breakers was shown to restore collagen function while simultaneously reducing AGE formation (110). As the most critical AGE precursor, Methylglyoxal (MG) overexpression in transgenic mice increased ECM remodeling in scarring myocardium (139). In the study, Methylglyoxal (MG) was observed with co-localization of collagen I in cardiac tissues after myocardial infarction, suggesting non-enzymatic glycation in cardiac tissue was linked to increased fibrosis and hypertrophy. From a clinical perspective, Willemsen et al. demonstrated that patients with heart failure exhibit elevated tissue levels of AGEs, which are associated with diastolic dysfunction and reduced aerobic exercise capacity (140). While increased AGEs were independently associated with reduced exercise capacity, another study found that either continuous or interval exercise training regimens prevented artery stiffness via mitigation of AGE level (141). Collectively, these studies emphasized the adverse effect of AGEs on heart failure and implied possible therapeutic options of exercises for heart failure patients. AGEs exert their effects mainly by binding to RAGEs and initiating several signaling transduction in the cardiac tissue (142, 143). For example, activation of RAGE enhanced NF-κB activation and promoted myofibroblast transition and oxidative stress in heart failure (144, 145). TGF-β1/Smad pathway was another downstream signaling cascade of the AGE-RAGE axis in myocardial fibrosis, an essential heart failure process. Pretreatment of anti-RAGE antibody substantially decreased TGF-β1 and p-smad2/3 expression of cardiac fibroblasts, suggesting the presence of RAGE/TGF-β signaling pathway in heart failure (146).
5.2 AGEs and valvular heart disease
The four major valves in the heart includes three kinds of valvular problems: atresia (block of the valve), regurgitation (backflow of blood through the valve), and stenosis (narrowness of the valve) (147–149). Chronic glycation of proteins has been in many valvular heart diseases, such as stenotic aortic valves, mitral valve disease, and congenital valvular disease (bicuspid aortic valve). By contrast, the accumulation of AGEs is related to disease severity and progression (150–152).
Aortic valve stenosis (AVS), the most common aortic valve disorder, is driven by fibrocalcific remodeling of valve leaflets. Chronic inflammation contributes to calcification (153), and AGE–RAGE signaling promotes valve interstitial cell trans differentiation into osteoblast-like cells with immune cell infiltration (154). A diet rich in carbohydrates and fats enhances AGE–LDL formation, which in turn promotes aortic valve lesions in hamsters. In human aortic valve interstitial cells, AGE–LDL induces the expression of inflammatory mediators, including ICAM-1, IL-6, and ALP, and facilitates calcific nodule formation. Conversely, silencing the RAGE gene attenuates these effects by suppressing NF-κB signaling (155). The sex difference of AGE synthesis was reported in SD rat models, and male SD rats have higher AGEs than female rats, indicating that males are more likely to suffer from CVDs (156). In a study of patients with kidney failure, male patients are more prone to aortic valve calcification with higher AGE accumulation (157).
Heart valve replacement is the only treatment for symptomatic severe valve diseases (158). However, the increasing prevalence of valve degeneration is expected to emerge as a leading cause of cardiovascular morbidity and a significant healthcare burden in the coming decades (159). Several causes contribute to valve degeneration: calcification of the bioprosthetic valve, inflammatory reactions, tissue thickening, and collagen network (160). Immunohistochemistry on 45 clinical explants revealed that AGE and HSA accumulation are standard features in failed BHVs, exhibiting collagen crosslink regardless of calcification severity. Moreover, in vitro infiltration of bovine pericardium demonstrated that this effect of AGEs accumulates mainly in lysine and arginine. The process occurs as early as implantation starts, that affects the effective orifice area of the bioprosthetic valve, indicating AGEs as one cause of implantation failure (161).
5.3 AGEs and coronary artery disease
Coronary Artery Disease (CAD) is a chronic condition characterized by the progressive narrowing or blockage of the coronary arteries, leading to a complex interplay of endothelial dysfunction, foam cell accumulation, inflammation, and thrombosis (162). Two RNA-binding proteins, nuclear factor 90 (NF90) and nuclear factor 110 (NF110), mediate AGE accumulation in vascular smooth muscle cells (VSMCs) via stimulating the degradation of AGE receptor 1 (AGER1) (163). Atherosclerosis is initiated when LDL becomes retained within the sub-endothelial layer of medium and large arteries. This upregulation of AGE-RAGE cascade in macrophages facilitates increased binding and internalization of modified low-density lipoproteins (LDL), such as oxidized LDL (ox-LDL), into the macrophages. Internalized cholesterol forms cholesterol esters in the macrophage stored as lipid droplets, contributing to foam cell formation in the early stage of atherosclerotic lesions (164). LDL cross endothelial cells (ECs) via transcellular transcytosis, which is mainly mediated by Caveolin-1 protein (165, 166). Interaction between AGE and RAGE on the ECs robustly upregulated Caveolin-1. AGE-RAGE signaling promoted phosphorylation of transcription factor NF-κB, which, in turn, translocated into the nucleus and increased expression of upstream RAGE and Caveolin-1. Therefore, AGE-RAGE-NF-κB forms a positive feedback loop in the LDL transcytosis (117).
As one of the most prevalent and studied AGEs, CML arising through the nonenzymatic glycation and oxidative modification of monosaccharides (such as glucose) and protein-bound lysine residues (167). Notably, in vivo experiments suggested that CML accelerates the formation of atherosclerosis via the migration of foam cells into regional lymph nodes. Injection of exogenous CML into diabetic apoE−/− mice promoted plaque formation and cholesterol crystal deposition in the plaques. Moreover, CML upregulates the expression of CD36, a signal molecule expressed by macrophage-derived foam cells, indicating the accumulation of foam cells in atherosclerosis formation. in vitro analysis demonstrated that CML inhibited foam cell migration via ROS generation and FAK signaling, which are responsible for macrophage migration (167). A comparative study assesses CML levels as a predictor factor for coronary artery disease in diabetic patients (168). The result showed that patients with more severe coronary artery diseases have higher CML accumulation. Additionally, CML is related to several inflammatory factors up-regulation, such as TNF-α and IL-6, which is highly correlated with early atherosclerotic lesion formation and increased long-term cardiovascular mortality (169).
5.4 AGEs and hypertension
Systemic arterial hypertension, commonly referred to as hypertension, is a significant CVD characterized by persistently elevated pressure within the arteries (170). Arterial stiffness is a leading factor in the development of hypertension. Elastic arteries enhance the effects of cardiac contraction during the diastolic stage by storing potential energy when they expand elastically during the systolic stage. However, they become less able to absorb the force of blood ejected from the heart as artery stiffen, leading to higher systolic blood pressure and an increased pulse pressure (171).
AGEs in cardiomyocytes is related to arterial stiffness, demonstrating that higher pulse wave velocity (PWV) and pulse pressure were positively related to elevated plasma AGEs (172). Aging is markedly associated with arterial stiffness due to structural and functional changes in the vascular system (173). One comparative study showed that older participants exhibited a notable increase in PWV with higher plasma AGE levels in an age-dependent manner (174). The relationship between arterial stiffness and AGEs is restricted to patients with vascular diseases and induces early vascular changes before the disease. In men and younger individuals, vascular stiffness was significantly associated with AGE accumulation, implying it as a non-invasive marker for arterial stiffness (175).
Considering the potential involvement of AGEs in arterial stiffness, it would be anticipated that elevated AGE levels correlate with hypertension. Recently, a study investigated the role of elevated AGE levels and CVD in an elderly cohort; however, In the survey, AGEs demonstrated no correlation with hypertension in the cohort, and diastolic blood pressure was even negatively associated with skin autofluorescence (AGE markers) (15). This debate may be explained by age difference in diastolic blood pressure: earlier studies conducted in all age ranges demonstrated that AGE levels were positively related to blood pressure in men and children (176).
Hypertension substantially elevates the risk of mortality associated with cardiovascular events (177). A large-scale meta-analysis examined the association between hypertension and mortality across 67 distinct causes of death. The findings revealed that hypertension was associated with an increased risk of all-cause mortality and elevated risks for 17 specific causes, predominantly circulatory diseases. HGI is a measure that quantifies the difference between observed and predicted HbA1c levels based on blood glucose concentration, reflecting individual variability in of hemoglobin (178). Notably, cardiovascular events in female hypertensive patients increased with higher HGI levels, demonstrating a J-shaped relationship (14).
5.5 AGEs and diabetic heart disease
In the past decades, diabetic-induced heart failure, cardiomyopathy, hypertension, and coronary diseases have received markedly research attention to relative mechanisms (179). People with diabetes have two sources of AGE initiation: exogenous and endogenous sources (180). Diabetic cardiomyopathy (DCM) is a distinct pathological condition in which heart failure develops independently of coronary artery disease, hypertension, or valvular abnormalities. It typically manifests first as diastolic dysfunction and progresses over time to heart failure with reduced ejection fraction. In diabetic patients, insulin resistance, compensatory hyperinsulinemia, and persistent hyperglycemia collectively contribute to the heightened susceptibility to DCM (181).
AGEs are generated in DCM development through promoting ferroptosis: AGEs were found to induce ferroptosis in cardiomyocytes via alteration in the expression of key ferroptosis markers, such as Ptgs2, ferritin, and SLC7A11, which in turn, contributing to the development of DCM. Also, AGE increases oxidative stress in the cell via activation of AMPK signaling, aggravating cell viability in DCM mice. These changes further led to cardiomyocyte dysfunction and structural remodeling. Furthermore, the study found that activating the AMPK/NRF2 signaling by sulforaphane attenuates ferroptosis in the AGE-treated group, allowing cardiomyocyte survival and providing potential therapeutic options to activate NRF2 in DCM treatment (182). AGEs bound to human serum albumin in cardiac cells downregulated Nrf-2 expression and its target antioxidant genes Keap1, HMOX1, and NQO1 expression, reducing cellular defense against oxidative damage (183).
Microtubule (MT) dysfunction was identified as a driver for contractile ventricular diseases (184). Increased stability of cardiac microtubules plays a significant role in the cardiac dysfunction observed in STZ-induced type 1 diabetic rats (185). Excessive AGEs cultured cardiomyocytes reduced SIRT2 expression, a gene responsible for cytoskeleton stability and the deacetylation of microtubules in the progression of diabetic cardiomyopathy (DCM). Injection of AGE neonatal rat ventricular myocytes enhances leakage of Ca2 + from the sarcoplasmic reticulum (SR), leading to partial depletion of SR Ca2+ stores and a subsequent reduction in systolic Ca2+ transients, finally causing contractile dysfunction in diabetic cardiomyopathy (186).
Diabetes is universally acknowledged as an inflammatory disorder, as the impairment in insulin secretion and sensitivity is attributed to factors such as proinflammatory proteins and immune infiltration in DCM patients (187). Previous experimental evidence found that myeloid differentiation 2 (MD2), a co-receptor for toll-like receptor 4 (TLR4), could induce inflammatory response in DCM development. AGEs and MD2-TLR4 form a complex that upregulates MAPK/ NF-κB and other proinflammatory factors (188). Macrophage polarization is crucial in immune responses, inflammation, and tissue remodeling. This plasticity allows macrophages to switch between two central polarization states, M1 (proinflammatory) and M2 (anti-inflammatory or tissue-repairing) (189). Excessive AGE accumulation induces the expression of microRNA miR-471-3p in bone marrow–derived macrophages (BMDMs) from diabetic mice as well as in RAW264.7 macrophage cells. Mechanistically, miR-471-3p directly binds to silent information regulator 1 (SIRT1), which was reported to have an anti-inflammatory role in switching macrophage polarization from M1 to M2 in DCM development (190).
5.6 AGEs and congenital heart diseases
AGEs may exert detrimental effects on congenital heart malformations starting from embryo development. For example, the accumulation of CML and MG upregulates smad proteins phosphorylation, thus leading to more borderer myocardium in the cushions of the outflow tract and more likely to develop congenital heart diseases (191). The bicuspid aortic valve (BAV) is the most prevalent congenital heart abnormality and is often linked to abnormalities in the proximal aorta (192). BAV encompasses a range of morphological abnormalities where the aortic valve consists of two functional cusps and has fewer than three distinct zones of parallel cusp apposition (193). One study investigated the lifetime health outcomes of individuals with BAV over a median follow-up of 19.1 years. It showed that a significant portion of deaths in BAV patients were linked to cardiovascular complications, particularly aneurysm formation, aortic dilatation and aortic dissection or rupture (192). BAV patients possess higher levels of AGEs that play a crucial role in cell apoptosis, thus increasing the risk of structural changes in the development of aortic aneurysms and the likelihood of acute aortic dissection. Therefore, the sRAGE may serve as a tool to predict the risk of BAV and BAV-associated aortopathies (194). Mechanistically, higher sRAGE levels correlate with increased activation of the NF-κB pathway, a key regulator of inflammation and cellular stress responses. This activation may contribute to the structural remodeling of aortic aneurysms in BAV patients (195). Increased levels of AGE also enhanced vimentin expression, which is a 48 kDa fragment cleaved by caspase-3 in cell apoptosis. Additionally, vimentin may function as a mechanotransducive regulator of Notch signaling, contributing to arterial wall remodeling (150).
6 Therapeutic strategies for CVDs targeting AGE production
In recent years, experimental evidence supported that target AGEs could offer therapeutic options for patients with CVDs (Figure 4).
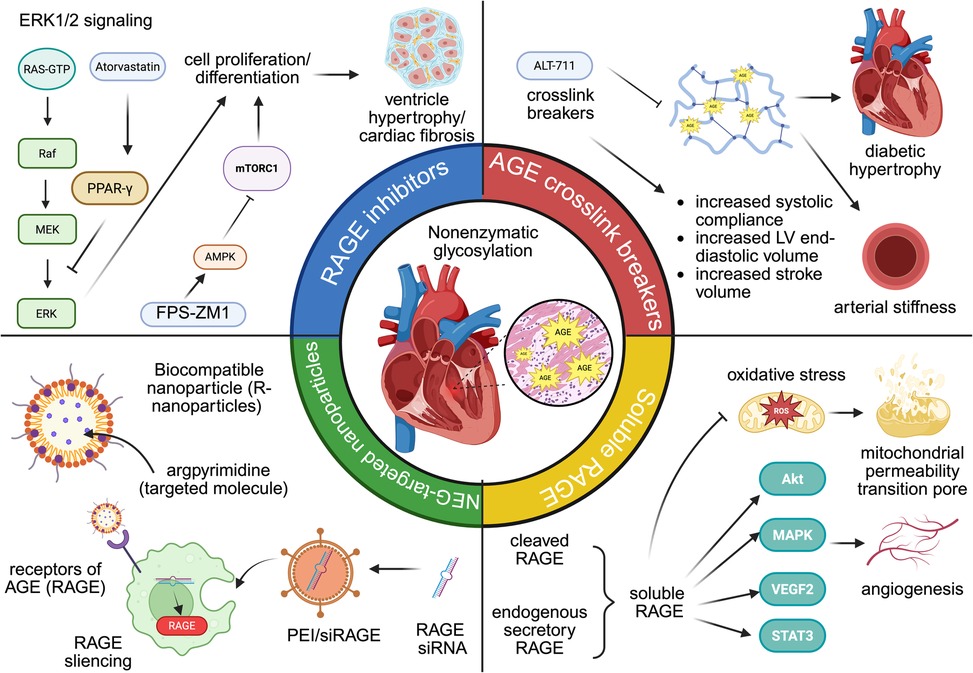
Figure 4. The role of AGE-targeted therapy in cardiovascular diseases. From left to right: RAGE inhibitors (atorvastatin and FPS-ZM1), AGE crosslink breakers (ALT-711), AGE-targeted nanoparticles (R-nanoparticles and PEI/siRAGE), soluble RAGE.
6.1 RAGE inhibitors
Inhibitors of RAGE can reduce cardiac fibrosis and enhance diastolic function by downregulating AGE-RAGE signaling. Atorvastatin (Ator), originally identified for its lipid-lowering properties, has also been reported to activate peroxisome proliferator-activated receptor gamma (PPAR-γ) signaling. PPAR-γ could exert inhibiting effects on the AGE-RAGE axis, leading to the downregulation of ERK1/2 phosphorylation in rat fibroblast. Previous studies have proved the effects ERK1/2 in cell proliferation in many CVD models, such as left ventricle hypertrophy and cardiac fibrosis (196). Similarly, FPS-ZM1, another RAGE-specific inhibitor, upregulates AMPK phosphorylation and downregulates mTOR/NF-κB in cardiac tissue. Activation of AMPK mediates protective effects against systolic overload-induced cardiac dysfunction, whereas activation of mTOR/ NF-κB has detrimental effects that act as pro-inflammation and pro-apoptotic drivers. Consequently, the blocking effects of FPS-ZM1 suppressed inflammation and cardiac remodeling in heart failure (197). Although RAGE inhibition holds promising therapeutic potentials in AGE-mediated cardiovascular injury, several safety concerns in the application of RAGE inhibitors in other diseases warrant more investigation in future clinical trials. In early Phase I and II trials of Alzheimer disease, high doses of azeliragon (small-molecule RAGE antagonist) could increase falls, confusion, and cognitive decline, leading to early discontinuation of the high-dose arm (198).
6.2 AGE crosslink breaker
AGE-crosslink breakers restore cardiovascular function by disturbing the formation of glycogen-dependent crosslinks. ALT-711 is a phenyl-4,5-dimethyl thiazolium chloride derivative used in many types of CVDs, including systolic hypertension, heart failure, and other aging-related heart dysfunction (199, 200). Mechanistically, ALT-711 effectively reverses significant artery stiffness in diabetic animals, restoring vascular properties toward normal levels. Diabetic mice treated with ALT-117 for three weeks demonstrated increased systemic arterial compliance (0.87 ± 0.08 ml/mmHg than 0.56 ± 0.05 ml/mmHg in untreated diabetic mice). Similarly, carotid artery compliance rose from 0.28 ± 0.03 mm2/mmHg in untreated diabetics to 0.45 ± 0.05 mm2/mmHg post-treatment (201). Another study investigated the effect of ALT-711 on left ventricle stiffness. After one month of daily ALT-711 administration, a significant reduction of approximately 40% in LV stiffness was observed, with measurements decreasing to 33.1 ± 4.6 mm Hg·m2/ml. This decrease in stiffness was accompanied by improvements in cardiac function, including increased LV end-diastolic volume and stroke volume, without significant changes in systolic blood pressure or heart rate (202).
6.3 Soluble RAGE
Soluble forms of RAGE (sRAGE) could be generated from two primary sources: first, cleaved RAGE (cRAGE) is generated through the proteolytic shedding of membrane-bound RAGE by matrix metalloproteinases; second, endogenous secretory RAGE (esRAGE) is synthesized from RAGE gene. sRAGE has protective effects for cardiomyocytes via suppression of cell apoptosis and mitigation of angiogenesis, making it one of the hot points of therapy strategy in CVDs. sRAGE significantly restored left ventricular end-diastolic volume (LVEDV) and left ventricular end-systolic volume (LVESV) in I/R mice. Immunohistochemical staining for CD31 revealed increased capillary density in sRAGE-treated hearts, suggesting that sRAGE promotes angiogenesis following I/R injury. Meanwhile, the expression of pro-angiogenic genes, such as Akt, MAPK signals, VEGFR2, and STAT3, were significantly increased in sRAGE-treated cardiac microvascular endothelial cells (CMECs). Collectively, these angiogenic effects lead to improved cardiac function and reduced tissue damage post-injury (203). sRAGE treatment could also downregulate the expression of apoptosis markers and autophagy-related proteins. Therefore, I/R injured myocardial tissues observed reduced TUNEL-positive cells, lower caspase-3 activity, and decreased autophagosome formation (204). Cardiac dysfunction can lead to the opening of the mitochondrial permeability transition pore (mPTP), subsequent generation of reactive oxygen species, and mitochondrial calcium overload. Exogenous administration of sRAGE inhibits mPTP opening and mitochondria-mediated apoptosis by suppression of mitochondria cytochrome c, caspase 3, and caspase nine release (205).
6.4 Nanoparticles that target AGEs
In recent years, nanoparticles (NP) have been developed as an innovative drug delivery system, utilizing 1–100 nm biomaterials to transport therapeutic agents into diseased tissues (206). NPs targeting the AGE accumulation are an emerging class of agents for therapeutic interventions and diagnostic imaging in other diseases. For example, one study constructs a novel biocompatible nanoparticle encapsulated with argpyrimidine, an AGE that targets the nanoparticle to cell surfaces that express RAGE. The results showed that compared to free soluble anti-hyperglycemia drugs, anti-hyperglycemia drugs encapsulated inside argpyrimidine are more effective in controlling hyperglycemia in diabetic rats. Moreover, rats treated with nanoparticles demonstrated better structural integrity in heart integrity and less cardiac fibrosis formation (207). As discussed above, AGE/RAGE is associated with inflammation status in ischemia injury. PEI/siRAGE is a small interfering RNA nanoparticle that selected RAGE as the target gene to assess its in vivo efficacy. Compared with a control group treated with saline, PEI/siRAGE reduces more than 10% of the infarcted size in cardiac tissue, exhibiting a beneficial effect against myocardial infarction (208). Curcumin and aged garlic extract are modified herbal medicines that demonstrated anti-oxidative and anti-glycation impacts in treating CVDs (209, 210). However, limited solubility and low bioavailability impede their efficacy for more cardiovascular applications. A study investigated the impact of nanomedicine based on these two herbal medicines targets both AGEs and ROS in streptozotocin-induced diabetic rats: Both nano-curcumin and AGE demonstrated downregulation of RAGE gene level, and histopathological results also exhibited higher structural integrity in DCM tissue (211). The study by Lu et al. reveals a novel advantage of engineered macrophage membrane-coated siRNA nanoparticle (MMM/RNA NP) platform with overexpression of RAGE in the surface of MMM/RNA NP, through which the nanoparticle effectively achieved higher cellular internalization to the site of myocardial ischemia–reperfusion injury, leading to improved left ventricular ejection fraction (LVEF) and fractional shortening (LVFS) in ischemic mice (212).
7 Clinical translation of AGE targeted therapy
Over the past decades, extensive preclinical evaluations of various AGE-derived therapeutics have demonstrated their impressive potential for clinical use. The first clinical trial that demonstrated the feasibility of AGE-targeted therapy recruited 93 participants with elevated pulse pressures were assigned to receive either 210 mg of ALT-711 or a placebo daily for 56 days. The results demonstrated that ALT-711 significantly improved total arterial compliance (15% vs. no change) and reduced pulse pressure (−5.3 vs. −0.6 mm Hg) compared to the placebo group (213). In recent years, 11 other clinical trials of ALT-711 under FDA approvals for treating CVDs have been registered (https://clinicaltrials.gov/) (Table 2). Although the elimination of AGE in CVDs holds a promising concept, few documented trials show that AGE-targeted therapy is more favorable than other standard therapy (214). For example, synthetic AGE inhibitors that reached phase I/II clinical trials such as aminoguanidine or alagebrium both faced toxicity issues including major liver/kidney toxicity and autoimmune-like reactions (215). In recent years, nature products have been evaluated for their antiglycation activity. Gallic acid (GA) is a plant-derived polyphenol present in gallnuts, grapes, tea leaves, and oak bark with antioxidant activities. Treatment of GA in cardiac H9C2 (2-1) cells decrease intracellular ROS and oxidant level (P < 0.05), suggesting the cytoprotection efficacy of GA (216). Epigallocatechin-3-gallate (EGCG) is a major catechin found predominantly in green tea, valued for its potent antioxidant, anti-inflammatory, and potential disease-preventive properties. In simulated glycation systems, EGCG inhibited the formation of AGEs by 54.44% and α-dicarbonyl compounds (key intermediates of AGEs) by 84.47%, which is attributed to free radical scavenging and stabilization of protein secondary structure (217). Kaempferol, a polyphenol derived from green tea, has been shown to downregulate RAGE expression and its downstream effector NF-κB p65, thereby suppressing glycation-induced inflammatory signaling. This anti-inflammatory effect mitigates myocardial injury by reducing oxidative stress, apoptosis, and fibrosis, particularly under diabetic conditions where AGE–RAGE activation sustains chronic inflammation. Histological and ultrastructural evidence further supports its cardioprotective role, demonstrating preserved mitochondrial integrity and intact myofibril architecture during ischemia–reperfusion injury (218). Nevertheless, most natural compound of AGE inhibitor/crosslink breakers were still in pre-clinical stage and future clinical studies are required to validate their efficacy in CVD diseases.
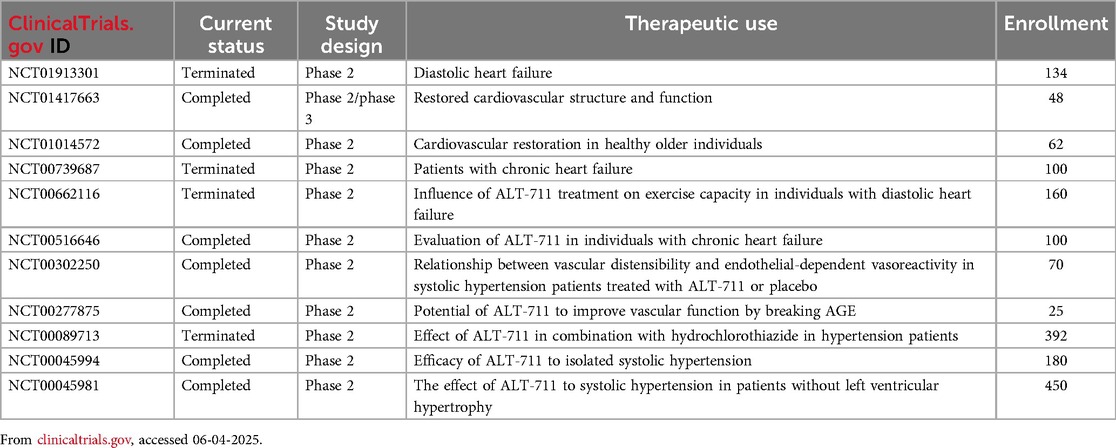
Table 2. FDA-approved clinical trials of AGE-targeted therapy.
In addition to its therapeutic uses, AGE level is also utilized as a prognostic indicator in cardiovascular diseases (CVDs). HbA1c is produced when glucose non-enzymatically and irreversibly attaches to the N-terminal valine of the β-chain of hemoglobin in red blood cells (219). HGI represents individual variations in the correlation between plasma glucose and HbA1c and was initially employed to diagnose diabetes mellitus. Furthermore, several studies have associated high HGI levels with significant diabetes mellitus complications, including CVD (220). One study examined the link between cardiovascular risk in diabetic patients and elevated HGI. Patients with varying HGI levels experienced different outcomes following intensive treatment: both the low and moderate HGI groups (low HGI ≤ −0.520 and moderate −0.520 to 0.202) showed a reduction in MI, stroke, or death from cardiovascular causes. Conversely, high HGI was not linked to treatment outcomes and even increased the overall mortality rate by 41% (221). Exploring the connection between HGI and cardiovascular outcomes reveals a U-shaped relationship, where both low and high HGI levels are associated with heightened risks. Lin et al. concentrated on patients with diabetes and coronary artery diseases, discovering that both low and high HGI levels were tied to increased MACE risks, with low HGI significantly raising the risk of cardiovascular death (222). Among 5,260 patients with coronary artery diseases, high HGI was linked to increased 365-day mortality, while low HGI was associated with 30-day mortality, indicating HGI's correlation with both short-term and long-term mortality in the progression of coronary artery disease (220). Cardiovascular risks and elevated HGI were also suggested in heart failure (223, 224) and myocardial injury (225).
One significant drawback of using the HGI as a prognostic tool is that numerous studies concentrate solely on CVD patients who have diabetes. The scarcity of research on HGI in non-diabetic individuals might undermine the unique pathophysiological roles of AGEs could have in diabetic vs. non-diabetic CVD cases. For instance, a study identified a linear association between HGI levels and the severity of CAD in non-diabetic people. In CAD patients, HbA1c levels rose progressively with the severity of clinical symptoms, with stable angina patients, unstable angina patients, and those with acute myocardial infarction exhibiting the lowest, intermediate, and highest HGI levels, respectively (226). Conversely, another study examined the link between HbA1c levels and both all-cause and CVD mortality in elderly non-diabetic individuals, finding that both low and high HbA1c levels are linked to heightened risks of all-cause mortality in older patients (227). Future studies should investigate the role of HGI in CVDs among both diabetic and non-diabetic groups to gain a better understanding of HGI's function and improve its effectiveness in clinical risk evaluation.
8 Discussion
As the key metabolic byproducts in non-enzymatic glycation, Advanced glycation products (AGEs) can lead to extensive physiological and pathological consequences. The association between AGEs and cardiovascular pathology is well-documented, especially in relation to diabetes mellitus and metabolic syndrome. Elevated glucose levels accelerate the production of AGEs, which irreversibly modify structural proteins and functional proteins in cardiovascular tissues (228, 229).
AGEs primarily exert their harmful effects by engaging the AGE-RAGE axis, a well-known signaling pathway that initiates OS and persistent inflammation (230). When AGEs bind to the RAGE, it sets off a series of intracellular signaling pathways, such as NF-κB, JAK-STAT, MAPK, and PI3K/AKT, which intensify OS and inflammatory reactions (95). In addition to activating proinflammatory signaling, AGEs directly impair cardiovascular cell populations by inhibiting autophagy, inducing apoptosis, disrupting endothelial repair through endothelial progenitor cells (EPCs), and promoting fibrosis via myofibroblast transformation. Moreover, AGE modification of plasma proteins alters lipid metabolism, increasing the atherogenic potential of lipoproteins and thereby accelerating plaque development.
Although oxidative stress (OS), chronic inflammation, and the modification of structural proteins are common mechanisms driving the pathological effects of AGEs, the specific consequences differ based on the cardiovascular condition, such as heart failure, valvular heart disease, coronary heart disease, systolic hypertension, and congenital heart disease. In the context of diabetes mellitus, persistent high blood sugar levels hasten non-enzymatic glycation, a natural reaction between reducing sugars and biological macromolecules like proteins, lipids, and nucleic acids (231). This prolonged hyperglycemia speeds up the production of AGEs from both internal and dietary origins (232). The clinical significance of AGEs has shown that AGE-targeted therapy can slow the progression of CVDs. Apart from drug intervention, emerging evidence suggests diet can modulate the systemic AGE burden and thereby potentiate pharmacologic or nutraceutical AGE-targeted strategies. Several small clinical trials report that restricting dietary AGE intake, even over short to moderate periods, significantly lowers circulating AGE levels and reduces inflammation and oxidative stress in both healthy individuals and those with cardio-kidney-metabolic (CKM) conditions (233). Furthermore, utilizing the hemoglobin glycation index effectively predicts the onset of CVD in both diabetic and non-diabetic individuals (234).
Despite encouraging outcomes in the investigation of AGEs and its therapeutic applications, several significant challenges hinder their complete understanding, clinical translation, and therapeutic targeting (Table 3). In recent years, the development of precision medicine also suggested heterogeneity of AGE level in different cohorts. Therefore, updates in multi-omics association studies may help to identify AGE-targeted treatment options. Adams et al. measured AGE levels in 506 individuals and heritability (h2) of serum AGEs was estimated at 0.628, revealing a substantial heritable component for serum AGE levels. Likewise, the DHS MIND Study reported 8 single nucleotide polymorphisms in the genetic component of AGEs, which have been related to higher serum AGE levels (235). One study collected data from 4,182 individuals in the Long-Life Family Study that identified 17 gene expressions (Bonferroni p < 2.73 × 10−⁶) pleiotropically linked to the eGFR (a measure of kidney function) and sRAGE in kidney aging. These transcriptomic investigations highlight gene-expression architecture in AGE expression suggesting further investigation of AGE variants and heart diseases (236).
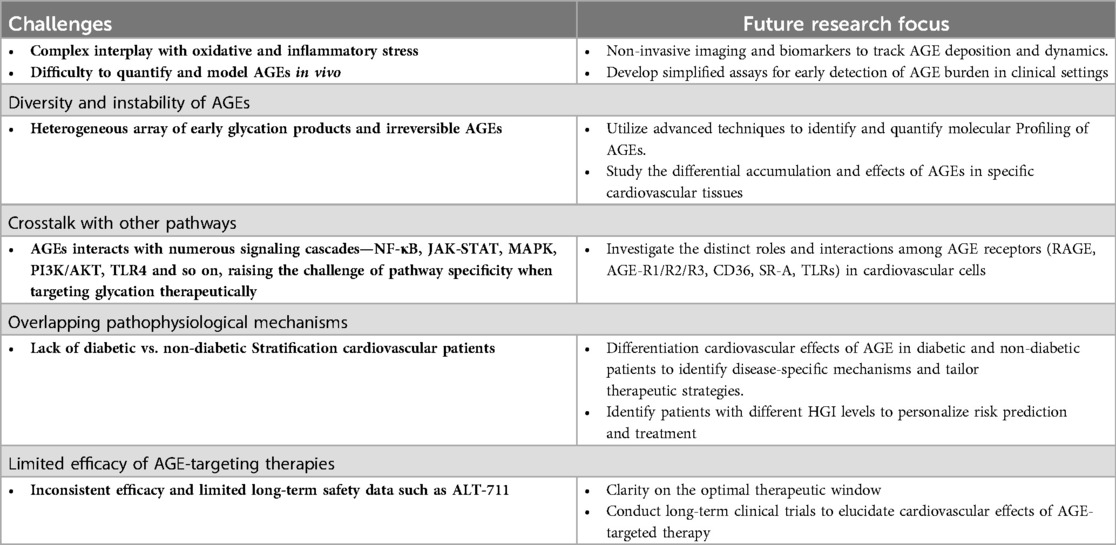
Table 3. The list of current challenges and further research attention of non-enzymatic glycation.
In conclusion, the role of AGEs in cardiovascular pathology is increasingly recognized as a critical mechanistic axis that underlies multiple forms of CVDs. This highlights several promising targets—particularly the inhibition of the AGE–RAGE axis may offer substantial benefits for future CVD treatment.
Author contributions
Z-HL: Methodology, Visualization, Validation, Conceptualization, Writing – review & editing, Writing – original draft. X-YW: Visualization, Methodology, Conceptualization, Writing – review & editing, Writing – original draft. QL: Conceptualization, Writing – review & editing, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by the National Natural Science Foundation of China (No. 81860057).
Acknowledgments
The authors thank Grammarly for its linguistic assistance during the preparation of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, et al. Heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation. (2013) 127:e6–245. doi: 10.1161/CIR.0b013e31828124ad
2. Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the global burden of disease study 2010. Lancet. (2012) 380:2095–128. doi: 10.1016/s0140-6736(12)61728-0
3. Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, et al. Global burden of cardiovascular diseases and risk factors, 1990–2019: update from the GBD 2019 study. J Am Coll Cardiol. (2020) 76:2982–3021. doi: 10.1016/j.jacc.2020.11.010
4. Li Y, Cao GY, Jing WZ, Liu J, Liu M. Global trends and regional differences in incidence and mortality of cardiovascular disease, 1990–2019: findings from 2019 global burden of disease study. Eur J Prev Cardiol. (2023) 30:276–86. doi: 10.1093/eurjpc/zwac285
5. Gersh BJ, Sliwa K, Mayosi BM, Yusuf S. Novel therapeutic concepts: the epidemic of cardiovascular disease in the developing world: global implications. Eur Heart J. (2010) 31:642–8. doi: 10.1093/eurheartj/ehq030
6. Jagannathan R, Patel SA, Ali MK, Narayan KMV. Global updates on cardiovascular disease mortality trends and attribution of traditional risk factors. Curr Diab Rep. (2019) 19:44. doi: 10.1007/s11892-019-1161-2
7. Sun J, Qiao Y, Zhao M, Magnussen CG, Xi B. Global, regional, and national burden of cardiovascular diseases in youths and young adults aged 15–39 years in 204 countries/territories, 1990–2019: a systematic analysis of global burden of disease study 2019. BMC Med. (2023) 21:222. doi: 10.1186/s12916-023-02925-4
8. Gill V, Kumar V, Singh K, Kumar A, Kim JJ. Advanced glycation end products (AGEs) may be a striking link between modern diet and health. Biomolecules. (2019) 9:888. doi: 10.3390/biom9120888
9. Sullivan R. Contributions to senescence: non-enzymatic glycosylation of proteins. Arch Physiol Biochem. (1996) 104:797–806. doi: 10.1076/apab.104.7.797.13107
10. Shen CY, Lu CH, Wu CH, Li KJ, Kuo YM, Hsieh SC, et al. The development of Maillard reaction, and advanced glycation end product (AGE)-receptor for AGE (RAGE) signaling inhibitors as novel therapeutic strategies for patients with AGE-related diseases. Molecules. (2020) 25:5591. doi: 10.3390/molecules25235591
11. Arshi B, Chen J, Ikram MA, Zillikens MC, Kavousi M. Advanced glycation end-products, cardiac function and heart failure in the general population: the Rotterdam study. Diabetologia. (2023) 66:472–81. doi: 10.1007/s00125-022-05821-3
12. Yan SF, Ramasamy R, Naka Y, Schmidt AM. Glycation, inflammation, and RAGE: a scaffold for the macrovascular complications of diabetes and beyond. Circ Res. (2003) 93:1159–69. doi: 10.1161/01.Res.0000103862.26506.3d
13. Vasdev S, Gill V, Singal P. Role of advanced glycation end products in hypertension and atherosclerosis: therapeutic implications. Cell Biochem Biophys. (2007) 49:48–63. doi: 10.1007/s12013-007-0039-0
14. Shangguan Q, Yang J, Li B, Chen H, Yang L. Association of the hemoglobin glycation index with cardiovascular and all-cause mortality in individuals with hypertension: findings from NHANES 1999–2018. Front Endocrinol (Lausanne). (2024) 15:1401317. doi: 10.3389/fendo.2024.1401317
15. Chen J, Arshi B, Waqas K, Lu T, Bos D, Ikram MA, et al. Advanced glycation end products measured by skin autofluorescence and subclinical cardiovascular disease: the Rotterdam study. Cardiovasc Diabetol. (2023) 22:326. doi: 10.1186/s12933-023-02052-7
16. Panda A, Sabnam K, De S, Dasgupta S. Non-enzymatic glycation of human angiogenin: effects on enzymatic activity and binding to hRI and DNA. Biochimie. (2023) 208:151–9. doi: 10.1016/j.biochi.2022.12.020
17. Kutzli I, Weiss J, Gibis M. Glycation of plant proteins via Maillard reaction: reaction chemistry, technofunctional properties, and potential food application. Foods. (2021) 10:376. doi: 10.3390/foods10020376
18. Stitt AW. The role of advanced glycation in the pathogenesis of diabetic retinopathy. Exp Mol Pathol. (2003) 75:95–108. doi: 10.1016/s0014-4800(03)00035-2
19. Zhang Q, Ames JM, Smith RD, Baynes JW, Metz TO. A perspective on the Maillard reaction and the analysis of protein glycation by mass spectrometry: probing the pathogenesis of chronic disease. J Proteome Res. (2009) 8:754–69. doi: 10.1021/pr800858h
20. Semba RD, Nicklett EJ, Ferrucci L. Does accumulation of advanced glycation end products contribute to the aging phenotype? J Gerontol A Biol Sci Med Sci. (2010) 65:963–75. doi: 10.1093/gerona/glq074
21. Koska J, Saremi A, Howell S, Bahn G, De Courten B, Ginsberg H, et al. Advanced glycation end products. oxidation products, and incident cardiovascular events in patients with type 2 diabetes. Diabetes Care. 2018 41:570–6. doi: 10.2337/dc17-1740
22. Cho SJ, Roman G, Yeboah F, Konishi Y. The road to advanced glycation end products: a mechanistic perspective. Curr Med Chem. (2007) 14:1653–71. doi: 10.2174/092986707780830989
23. Gopalkrishnapillai B, Nadanathangam V, Karmakar N, Anand S, Misra A. Evaluation of autofluorescent property of hemoglobin-advanced glycation end product as a long-term glycemic index of diabetes. Diabetes. (2003) 52:1041–6. doi: 10.2337/diabetes.52.4.1041
24. Lin H, Yi J. Current status of HbA1c biosensors. Sensors (Basel). (2017) 17:1798. doi: 10.3390/s17081798
25. ter Haar R, Schols HA, Gruppen H. Effect of saccharide structure and size on the degree of substitution and product dispersity of α-lactalbumin glycated via the Maillard reaction. J Agric Food Chem. (2011) 59:9378–85. doi: 10.1021/jf2027395
26. Niki E. Oxidative stress and antioxidants: distress or eustress? Arch Biochem Biophys. (2016) 595:19–24. doi: 10.1016/j.abb.2015.11.017
27. Du X, Matsumura T, Edelstein D, Rossetti L, Zsengellér Z, Szabó C, et al. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Invest. (2003) 112:1049–57. doi: 10.1172/jci18127
28. Papachristoforou E, Lambadiari V, Maratou E, Makrilakis K. Association of glycemic indices (hyperglycemia, glucose variability, and hypoglycemia) with oxidative stress and diabetic complications. J Diabetes Res. 2020 2020:7489795. doi: 10.1155/2020/7489795
29. Anderson MM, Requena JR, Crowley JR, Thorpe SR, Heinecke JW. The myeloperoxidase system of human phagocytes generates nepsilon-(carboxymethyl)lysine on proteins: a mechanism for producing advanced glycation end products at sites of inflammation. J Clin Invest. (1999) 104:103–13. doi: 10.1172/jci3042
30. Basta G. Receptor for advanced glycation endproducts and atherosclerosis: from basic mechanisms to clinical implications. Atherosclerosis. (2008) 196:9–21. doi: 10.1016/j.atherosclerosis.2007.07.025
31. Perrone A, Giovino A, Benny J, Martinelli F. Advanced glycation end products (AGEs): biochemistry, signaling, analytical methods, and epigenetic effects. Oxid Med Cell Longev. (2020) 2020:3818196. doi: 10.1155/2020/3818196
32. Rodríguez JM, Leiva Balich L, Concha MJ, Mizón C, Bunout Barnett D, Barrera Acevedo G, et al. Reduction of serum advanced glycation end-products with a low calorie Mediterranean diet. Nutr Hosp. (2015) 31:2511–7. doi: 10.3305/nh.2015.31.6.8936
33. Uribarri J, Cai W, Peppa M, Goodman S, Ferrucci L, Striker G, et al. Circulating glycotoxins and dietary advanced glycation endproducts: two links to inflammatory response, oxidative stress, and aging. J Gerontol A Biol Sci Med Sci. (2007) 62:427–33. doi: 10.1093/gerona/62.4.427
34. Regin BS, Sowmiya B, Shiny A, Shanthi Rani CS, Gokulakrishnan K, Mohan V, et al. Association of POC-based measurement of skin AGEs with serum AGEs as well as AGE biomarkers in individuals with and without glucose intolerance. J Assoc Physicians India. (2023) 71:11–2. doi: 10.5005/japi-11001-0188
35. Juranek J, Osowski A, Wojtkiewicz J, Banach M. Plasma levels of soluble RAGE, AGEs and AOPPs at the early stage of amyotrophic lateral sclerosis: a preliminary study. Polim Med. (2023) 53:105–10. doi: 10.17219/pim/175544
36. Fujii EY, Nakayama M. The measurements of RAGE, VEGF, and AGEs in the plasma and follicular fluid of reproductive women: the influence of aging. Fertil Steril. (2010) 94:694–700. doi: 10.1016/j.fertnstert.2009.03.029
37. Mitsuhashi T, Vlassara H, Founds HW, Li YM. Standardizing the immunological measurement of advanced glycation endproducts using normal human serum. J Immunol Methods. (1997) 207:79–88. doi: 10.1016/s0022-1759(97)00110-5
38. Galler A, Müller G, Schinzel R, Kratzsch J, Kiess W, Münch G. Impact of metabolic control and serum lipids on the concentration of advanced glycation end products in the serum of children and adolescents with type 1 diabetes, as determined by fluorescence spectroscopy and nepsilon-(carboxymethyl)lysine ELISA. Diabetes Care. (2003) 26:2609–15. doi: 10.2337/diacare.26.9.2609
39. Suehiro A, Uchida K, Nakanishi M, Wakabayashi I. Measurement of urinary advanced glycation end-products (AGEs) using a fluorescence assay for metabolic syndrome-related screening tests. Diabetes Metab Syndr. (2016) 10:S110–113. doi: 10.1016/j.dsx.2015.10.004
40. Meerwaldt R, Links T, Graaff R, Thorpe SR, Baynes JW, Hartog J, et al. Simple noninvasive measurement of skin autofluorescence. Ann N Y Acad Sci. (2005) 1043:290–8. doi: 10.1196/annals.1333.036
41. Cleary PA, Braffett BH, Orchard T, Lyons TJ, Maynard J, Cowie C, et al. Clinical and technical factors associated with skin intrinsic fluorescence in subjects with type 1 diabetes from the diabetes control and complications trial/epidemiology of diabetes interventions and complications study. Diabetes Technol Ther. (2013) 15:466–74. doi: 10.1089/dia.2012.0316
42. Planas A, Simó-Servat O, Bañeras J, Sánchez M, García E, Ortiz Á M, et al. Usefulness of skin advanced glycation end products to predict coronary artery calcium score in patients with type 2 diabetes. Acta Diabetol. (2021) 58:1403–12. doi: 10.1007/s00592-021-01735-5
43. Hangai M, Takebe N, Honma H, Sasaki A, Chida A, Nakano R, et al. Association of advanced glycation end products with coronary artery calcification in Japanese subjects with type 2 diabetes as assessed by skin autofluorescence. J Atheroscler Thromb. (2016) 23:1178–87. doi: 10.5551/jat.30155
44. Kim JJ, Jeong B, Cho Y, Kwon MH, Lee YH, Kang U, et al. The association between skin auto-fluorescence of palmoplantar sites and microvascular complications in Asian patients with type 2 diabetes mellitus. Sci Rep. (2018) 8:6309. doi: 10.1038/s41598-018-24707-2
45. de Vos LC, Mulder DJ, Smit AJ, Dullaart RP, Kleefstra N, Lijfering WM, et al. Skin autofluorescence is associated with 5-year mortality and cardiovascular events in patients with peripheral artery disease. Arterioscler Thromb Vasc Biol. (2014) 34:933–8. doi: 10.1161/atvbaha.113.302731
46. Corica D, Pepe G, Currò M, Aversa T, Tropeano A, Ientile R, et al. Methods to investigate advanced glycation end-product and their application in clinical practice. Methods. (2022) 203:90–102. doi: 10.1016/j.ymeth.2021.12.008
47. Dandia H, Makkad K, Tayalia P. Glycated collagen—a 3D matrix system to study pathological cell behavior. Biomater Sci. (2019) 7:3480–8. doi: 10.1039/c9bm00184k
48. Thompson K, Chen J, Luo Q, Xiao Y, Cummins TR, Bhatwadekar AD. Advanced glycation end (AGE) product modification of laminin downregulates Kir4.1 in retinal müller cells. PLoS One. (2018) 13:e0193280. doi: 10.1371/journal.pone.0193280
49. Aronson D. Cross-linking of glycated collagen in the pathogenesis of arterial and myocardial stiffening of aging and diabetes. J Hypertens. (2003) 21:3–12. doi: 10.1097/00004872-200301000-00002
50. Portillo JC, Pfaff A, Vos S, Weng M, Nagaraj RH, Subauste CS. Advanced glycation end products upregulate CD40 in human retinal endothelial and Müller cells: relevance to diabetic retinopathy. Cells. (2024) 13:429. doi: 10.3390/cells13050429
51. Sawabe M. Vascular aging: from molecular mechanism to clinical significance. Geriatr Gerontol Int. (2010) 10(Suppl 1):S213–220. doi: 10.1111/j.1447-0594.2010.00603.x
52. Konova E, Baydanoff S, Atanasova M, Velkova A. Age-related changes in the glycation of human aortic elastin. Exp Gerontol. (2004) 39:249–54. doi: 10.1016/j.exger.2003.10.003
53. Watanabe M, Sawai T, Nagura H, Suyama K. Age-related alteration of cross-linking amino acids of elastin in human aorta. Tohoku J Exp Med. (1996) 180:115–30. doi: 10.1620/tjem.180.115
54. Paradela-Dobarro B, Rodiño-Janeiro BK, Alonso J, Raposeiras-Roubín S, González-Peteiro M, González-Juanatey JR, et al. Key structural and functional differences between early and advanced glycation products. J Mol Endocrinol. (2016) 56:23–37. doi: 10.1530/jme-15-0031
55. Paradela-Dobarro B, Bravo SB, Rozados-Luís A, González-Peteiro M, Varela-Román A, González-Juanatey JR, et al. Inflammatory effects of in vivo glycated albumin from cardiovascular patients. Biomed Pharmacother. (2019) 113:108763. doi: 10.1016/j.biopha.2019.108763
56. Tang M, Berg AH, Zheng H, Rhee EP, Allegretti AS, Nigwekar SU, et al. Glycated albumin and adverse clinical outcomes in patients with CKD: a prospective cohort study. Am J Kidney Dis. (2024) 84:329–38. doi: 10.1053/j.ajkd.2024.02.006
57. Selvin E, Rawlings AM, Lutsey PL, Maruthur N, Pankow JS, Steffes M, et al. Fructosamine and glycated albumin and the risk of cardiovascular outcomes and death. Circulation. (2015) 132:269–77. doi: 10.1161/circulationaha.115.015415
58. Rehman S, Song J, Faisal M, Alatar AA, Akhter F, Ahmad S, et al. The neoepitopes on methylglyoxal- (MG-) glycated fibrinogen generate autoimmune response: its role in diabetes, atherosclerosis, and diabetic atherosclerosis subjects. Oxid Med Cell Longev. (2021) 2021:6621568. doi: 10.1155/2021/6621568
59. Robinson PW, Jury DR, Langdon AG. The relative clotting activity of glycated and non-glycated forms of fibrinogen. Ann Clin Biochem. (1991) 28 (Pt 6):618–9. doi: 10.1177/000456329102800613
60. Brownlee M, Vlassara H, Cerami A. Nonenzymatic glycosylation reduces the susceptibility of fibrin to degradation by plasmin. Diabetes. (1983) 32:680–4. doi: 10.2337/diab.32.7.680
61. Dunn EJ, Ariëns RA, Grant PJ. The influence of type 2 diabetes on fibrin structure and function. Diabetologia. (2005) 48:1198–206. doi: 10.1007/s00125-005-1742-2
62. Ernst E, Resch KL. Fibrinogen as a cardiovascular risk factor: a meta-analysis and review of the literature. Ann Intern Med. (1993) 118:956–63. doi: 10.7326/0003-4819-118-12-199306150-00008
63. Zhang J, Wang Y, Zhang R, Li H, Han Q, Wu Y, et al. Serum fibrinogen predicts diabetic ESRD in patients with type 2 diabetes mellitus. Diabetes Res Clin Pract. (2018) 141:1–9. doi: 10.1016/j.diabres.2018.04.025
64. Simard E, Söllradl T, Maltais JS, Boucher J, D’Orléans-Juste P, Grandbois M. Receptor for advanced glycation end-products signaling interferes with the vascular smooth muscle cell contractile phenotype and function. PLoS One. (2015) 10:e0128881. doi: 10.1371/journal.pone.0128881
65. Nakagawa K, Oak JH, Higuchi O, Tsuzuki T, Oikawa S, Otani H, et al. Ion-trap tandem mass spectrometric analysis of Amadori-glycated phosphatidylethanolamine in human plasma with or without diabetes. J Lipid Res. (2005) 46:2514–24. doi: 10.1194/jlr.D500025-JLR200
66. Lin Q, Han L, Liu G, Cheng W, Wang L. A preliminary study on the formation pathways of glycated phosphatidylethanolamine of food rich in phospholipid during the heat-processing. RSC Adv. (2018) 8:11280–8. doi: 10.1039/c8ra01072b
67. Colombo S, Criscuolo A, Zeller M, Fedorova M, Domingues MR, Domingues P. Analysis of oxidised and glycated aminophospholipids: complete structural characterisation by C30 liquid chromatography-high resolution tandem mass spectrometry. Free Radic Biol Med. (2019) 144:144–55. doi: 10.1016/j.freeradbiomed.2019.05.025
68. Assero G, Lupo G, Anfuso CD, Ragusa N, Alberghina M. High glucose and advanced glycation end products induce phospholipid hydrolysis and phospholipid enzyme inhibition in bovine retinal pericytes. Biochim Biophys Acta. (2001) 1533:128–40. doi: 10.1016/s1388-1981(01)00151-2
69. Cai W, He JC, Zhu L, Peppa M, Lu C, Uribarri J, et al. High levels of dietary advanced glycation end products transform low-density lipoprotein into a potent redox-sensitive mitogen-activated protein kinase stimulant in diabetic patients. Circulation. (2004) 110:285–91. doi: 10.1161/01.Cir.0000135587.92455.0d
70. Toma L, Stancu CS, Sima AV. Endothelial dysfunction in diabetes is aggravated by glycated lipoproteins; novel molecular therapies. Biomedicines. (2020) 9:18. doi: 10.3390/biomedicines9010018
71. Wani MJ, Arif A, Salman KA, Mahmood R. Glycated LDL generates reactive species that damage cell components, oxidize hemoglobin and alter surface morphology in human erythrocytes. Int J Biol Macromol. (2024) 269:132257. doi: 10.1016/j.ijbiomac.2024.132257
72. Bucala R, Mitchell R, Arnold K, Innerarity T, Vlassara H, Cerami A. Identification of the major site of apolipoprotein B modification by advanced glycosylation end products blocking uptake by the low density lipoprotein receptor. J Biol Chem. (1995) 270:10828–32. doi: 10.1074/jbc.270.18.10828
73. Seidel W, Pischetsrieder M. Immunochemical detection of N2-[1-(1-carboxy)ethylguanosine, an advanced glycation end product formed by the reaction of DNA and reducing sugars or L-ascorbic acid in vitro. Biochim Biophys Acta. (1998) 1425:478–84. doi: 10.1016/s0304-4165(98)00101-9
74. Pischetsrieder M, Seidel W, Münch G, Schinzel R. N(2)-(1-carboxyethyl)deoxyguanosine, a nonenzymatic glycation adduct of DNA, induces single-strand breaks and increases mutation frequencies. Biochem Biophys Res Commun. (1999) 264:544–9. doi: 10.1006/bbrc.1999.1528
75. Wuenschell GE, Tamae D, Cercillieux A, Yamanaka R, Yu C, Termini J. Mutagenic potential of DNA glycation: miscoding by (R)- and (S)-N2-(1-carboxyethyl)-2′-deoxyguanosine. Biochemistry. (2010) 49:1814–21. doi: 10.1021/bi901924b
76. Tamae D, Lim P, Wuenschell GE, Termini J. Mutagenesis and repair induced by the DNA advanced glycation end product N2–1-(carboxyethyl)-2′-deoxyguanosine in human cells. Biochemistry. (2011) 50:2321–9. doi: 10.1021/bi101933p
77. Bagherzadeh-Yazdi M, Bohlooli M, Khajeh M, Ghamari F, Ghaffari-Moghaddam M, Poormolaie N, et al. Acetoacetate enhancement of glucose mediated DNA glycation. Biochem Biophys Rep. (2021) 25:100878. doi: 10.1016/j.bbrep.2020.100878
78. Jaramillo R, Shuck SC, Chan YS, Liu X, Bates SE, Lim PP, et al. DNA advanced glycation end products (DNA-AGEs) are elevated in urine and tissue in an animal model of type 2 diabetes. Chem Res Toxicol. (2017) 30:689–98. doi: 10.1021/acs.chemrestox.6b00414
79. Cowey S, Hardy RW. The metabolic syndrome: a high-risk state for cancer? Am J Pathol. (2006) 169:1505–22. doi: 10.2353/ajpath.2006.051090
80. Jandial R, Neman J, Lim PP, Tamae D, Kowolik CM, Wuenschell GE, et al. Inhibition of GLO1 in glioblastoma multiforme increases DNA-AGEs, stimulates RAGE expression, and inhibits brain tumor growth in orthotopic mouse models. Int J Mol Sci. (2018) 19:406. doi: 10.3390/ijms19020406
81. Mrak RE, Griffin ST, Graham DI. Aging-associated changes in human brain. J Neuropathol Exp Neurol. (1997) 56:1269–75. doi: 10.1097/00005072-199712000-00001
82. Patschan S, Chen J, Polotskaia A, Mendelev N, Cheng J, Patschan D, et al. Lipid mediators of autophagy in stress-induced premature senescence of endothelial cells. Am J Physiol Heart Circ Physiol. (2008) 294:H1119–29. doi: 10.1152/ajpheart.00713.2007
83. Janssens JV, Ma B, Brimble MA, Van Eyk JE, Delbridge LMD, Mellor KM. Cardiac troponins may be irreversibly modified by glycation: novel potential mechanisms of cardiac performance modulation. Sci Rep. (2018) 8:16084. doi: 10.1038/s41598-018-33886-x
84. Rhinesmith T, Turkette T, Root-Bernstein R. Rapid non-enzymatic glycation of the insulin receptor under hyperglycemic conditions inhibits insulin binding in vitro: implications for insulin resistance. Int J Mol Sci. (2017) 18:2602. doi: 10.3390/ijms18122602
85. Twarda-Clapa A, Olczak A, Białkowska AM, Koziołkiewicz M. Advanced glycation end-products (AGEs): formation, chemistry, classification, receptors, and diseases related to AGEs. Cells. (2022) 11:1312. doi: 10.3390/cells11081312
86. Raucci A, Cugusi S, Antonelli A, Barabino SM, Monti L, Bierhaus A, et al. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). FASEB J. (2008) 22:3716–27. doi: 10.1096/fj.08-109033
87. Akhter F, Chen D, Akhter A, Yan SF, Yan SS. Age-dependent accumulation of dicarbonyls and advanced glycation endproducts (AGEs) associates with mitochondrial stress. Free Radic Biol Med. (2021) 164:429–38. doi: 10.1016/j.freeradbiomed.2020.12.021
88. Shen C, Ma Y, Zeng Z, Yin Q, Hong Y, Hou X, et al. RAGE-specific inhibitor FPS-ZM1 attenuates AGEs-induced neuroinflammation and oxidative stress in rat primary microglia. Neurochem Res. (2017) 42:2902–11. doi: 10.1007/s11064-017-2321-x
89. Wautier MP, Chappey O, Corda S, Stern DM, Schmidt AM, Wautier JL. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am J Physiol Endocrinol Metab. (2001) 280:E685–94. doi: 10.1152/ajpendo.2001.280.5.E685
90. Bierhaus A, Hofmann MA, Ziegler R, Nawroth PP. AGEs and their interaction with AGE-receptors in vascular disease and diabetes mellitus. I. The AGE concept. Cardiovasc Res. (1998) 37:586–600. doi: 10.1016/s0008-6363(97)00233-2
91. Morita M, Yano S, Yamaguchi T, Sugimoto T. Advanced glycation end products-induced reactive oxygen species generation is partly through NF-kappa B activation in human aortic endothelial cells. J Diabetes Complications. (2013) 27:11–5. doi: 10.1016/j.jdiacomp.2012.07.006
92. Lu Y, Li H, Jian W, Zhuang J, Wang K, Peng W, et al. The Rho/Rho-associated protein kinase inhibitor fasudil in the protection of endothelial cells against advanced glycation end products through the nuclear factor κB pathway. Exp Ther Med. (2013) 6:310–6. doi: 10.3892/etm.2013.1125
93. Chen J, Sun Z, Jin M, Tu Y, Wang S, Yang X, et al. Inhibition of AGEs/RAGE/rho/ROCK pathway suppresses non-specific neuroinflammation by regulating BV2 microglial M1/M2 polarization through the NF-κB pathway. J Neuroimmunol. (2017) 305:108–14. doi: 10.1016/j.jneuroim.2017.02.010
94. Sun M, Li Y, Bu W, Zhao J, Zhu J, Gu L, et al. DJC suppresses advanced glycation end products-induced JAK-STAT signaling and ROS in mesangial cells. Evid Based Complement Alternat Med. (2017) 2017:2942830. doi: 10.1155/2017/2942830
95. Ott C, Jacobs K, Haucke E, Navarrete Santos A, Grune T, Simm A. Role of advanced glycation end products in cellular signaling. Redox Biol. (2014) 2:411–29. doi: 10.1016/j.redox.2013.12.016
96. Nagai R, Mera K, Nakajou K, Fujiwara Y, Iwao Y, Imai H, et al. The ligand activity of AGE-proteins to scavenger receptors is dependent on their rate of modification by AGEs. Biochim Biophys Acta. (2007) 1772:1192–8. doi: 10.1016/j.bbadis.2007.09.001
97. Takata T, Sakasai-Sakai A, Ueda T, Takeuchi M. Intracellular toxic advanced glycation end-products in cardiomyocytes may cause cardiovascular disease. Sci Rep. (2019) 9:2121. doi: 10.1038/s41598-019-39202-5
98. Takeuchi M, Sakasai-Sakai A, Takata T, Takino JI, Koriyama Y, Kikuchi C, et al. Intracellular toxic AGEs (TAGE) triggers numerous types of cell damage. Biomolecules. (2021) 11:387. doi: 10.3390/biom11030387
99. Takeuchi M, Takino J, Yamagishi S. Involvement of the toxic AGEs (TAGE)-RAGE system in the pathogenesis of diabetic vascular complications: a novel therapeutic strategy. Curr Drug Targets. (2010) 11:1468–82. doi: 10.2174/1389450111009011468
100. Wang ZQ, Jing LL, Yan JC, Sun Z, Bao ZY, Shao C, et al. Role of AGEs in the progression and regression of atherosclerotic plaques. Glycoconj J. (2018) 35:443–50. doi: 10.1007/s10719-018-9831-x
101. Tada Y, Yano S, Yamaguchi T, Okazaki K, Ogawa N, Morita M, et al. Advanced glycation end products-induced vascular calcification is mediated by oxidative stress: functional roles of NAD(P)H-oxidase. Horm Metab Res. (2013) 45:267–72. doi: 10.1055/s-0032-1329965
102. Koike S, Yano S, Tanaka S, Sheikh AM, Nagai A, Sugimoto T. Advanced glycation end-products induce apoptosis of vascular smooth muscle cells: a mechanism for vascular calcification. Int J Mol Sci. (2016) 17:1567. doi: 10.3390/ijms17091567
103. Oldfield MD, Bach LA, Forbes JM, Nikolic-Paterson D, McRobert A, Thallas V, et al. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE). J Clin Invest. (2001) 108:1853–63. doi: 10.1172/jci11951
104. Yamamoto Y, Yonekura H, Watanabe T, Sakurai S, Li H, Harashima A, et al. Short-chain aldehyde-derived ligands for RAGE and their actions on endothelial cells. Diabetes Res Clin Pract. (2007) 77(Suppl 1):S30–40. doi: 10.1016/j.diabres.2007.01.030
105. Li Z, Zhong Q, Yang T, Xie X, Chen M. The role of profilin-1 in endothelial cell injury induced by advanced glycation end products (AGEs). Cardiovasc Diabetol. (2013) 12:141. doi: 10.1186/1475-2840-12-141
106. Cheng M, Yang Z, Qiao L, Yang Y, Deng Y, Zhang C, et al. AGEs induce endothelial cells senescence and endothelial barrier dysfunction via miR-1-3p/MLCK signaling pathways. Gene. (2023) 851:147030. doi: 10.1016/j.gene.2022.147030
107. Li H, Zhang X, Guan X, Cui X, Wang Y, Chu H, et al. Advanced glycation end products impair the migration, adhesion and secretion potentials of late endothelial progenitor cells. Cardiovasc Diabetol. (2012) 11:46. doi: 10.1186/1475-2840-11-46
108. Baumann M, Richart T, Sollinger D, Pelisek J, Roos M, Kouznetsova T, et al. Association between carotid diameter and the advanced glycation end product N-epsilon-carboxymethyllysine (CML). Cardiovasc Diabetol. (2009) 8:45. doi: 10.1186/1475-2840-8-45
109. Gu Q, Wang B, Zhang XF, Ma YP, Liu JD, Wang XZ. Contribution of receptor for advanced glycation end products to vasculature-protecting effects of exercise training in aged rats. Eur J Pharmacol. (2014) 741:186–94. doi: 10.1016/j.ejphar.2014.08.017
110. Candido R, Forbes JM, Thomas MC, Thallas V, Dean RG, Burns WC, et al. A breaker of advanced glycation end products attenuates diabetes-induced myocardial structural changes. Circ Res. (2003) 92:785–92. doi: 10.1161/01.Res.0000065620.39919.20
111. Haitoglou CS, Tsilibary EC, Brownlee M, Charonis AS. Altered cellular interactions between endothelial cells and nonenzymatically glucosylated laminin/type IV collagen. J Biol Chem. (1992) 267:12404–7. doi: 10.1016/S0021-9258(18)42287-9
112. Brownlee M. Glycation products and the pathogenesis of diabetic complications. Diabetes Care. (1992) 15:1835–43. doi: 10.2337/diacare.15.12.1835
113. Liang B, Zhou Z, Yang Z, Liu J, Zhang L, He J, et al. AGEs-RAGE axis mediates myocardial fibrosis via activation of cardiac fibroblasts induced by autophagy in heart failure. Exp Physiol. (2022) 107:879–91. doi: 10.1113/ep090042
114. Zhao LM, Zhang W, Wang LP, Li GR, Deng XL. Advanced glycation end products promote proliferation of cardiac fibroblasts by upregulation of KCa3.1 channels. Pflugers Arch. (2012) 464:613–21. doi: 10.1007/s00424-012-1165-0
115. Burr SD, Stewart JA Jr. Rap1a regulates cardiac fibroblast contraction of 3D diabetic collagen matrices by increased activation of the AGE/RAGE cascade. Cells. (2021) 10:1286. doi: 10.3390/cells10061286
116. Serban AI, Stanca L, Geicu OI, Munteanu MC, Dinischiotu A. RAGE and TGF-β1 cross-talk regulate extracellular matrix turnover and cytokine synthesis in AGEs exposed fibroblast cells. PLoS One. (2016) 11:e0152376. doi: 10.1371/journal.pone.0152376
117. Shu M, Cheng W, Jia X, Bai X, Zhao Y, Lu Y, et al. AGEs promote atherosclerosis by increasing LDL transcytosis across endothelial cells via RAGE/NF-κB/caveolin-1 pathway. Mol Med. (2023) 29:113. doi: 10.1186/s10020-023-00715-5
118. Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res. (2010) 106:1319–31. doi: 10.1161/circresaha.110.217117
119. Rempel LC, Finco AB, Maciel RA, Bosquetti B, Alvarenga LM, Souza WM, et al. Effect of PKC-β signaling pathway on expression of MCP-1 and VCAM-1 in different cell models in response to advanced glycation end products (AGEs). Toxins (Basel. (2015) 7:1722–37. doi: 10.3390/toxins7051722
120. Xu Y, Wang S, Feng L, Zhu Q, Xiang P, He B. Blockade of PKC-beta protects HUVEC from advanced glycation end products induced inflammation. Int Immunopharmacol. (2010) 10:1552–9. doi: 10.1016/j.intimp.2010.09.006
121. Luan Y, Zhang J, Wang M, Fu G, Zhang W. Advanced glycation end products facilitate the proliferation and reduce early apoptosis of cardiac microvascular endothelial cells via PKCβ signaling pathway: insight from diabetic cardiomyopathy. Anatol J Cardiol. (2020) 23:141–50. doi: 10.14744/AnatolJCardiol.2019.21504
122. Yuan G, Si G, Hou Q, Li Z, Xu K, Wang Y, et al. Advanced glycation end products induce proliferation and migration of human aortic smooth muscle cells through PI3K/AKT pathway. Biomed Res Int. (2020) 2020:8607418. doi: 10.1155/2020/8607418
123. Hu P, Lai D, Lu P, Gao J, He H. ERK and Akt signaling pathways are involved in advanced glycation end product-induced autophagy in rat vascular smooth muscle cells. Int J Mol Med. (2012) 29:613–8. doi: 10.3892/ijmm.2012.891
124. Xiang Y, Li Q, Li M, Wang W, Cui C, Zhang J. Ghrelin inhibits AGEs-induced apoptosis in human endothelial cells involving ERK1/2 and PI3K/Akt pathways. Cell Biochem Funct. (2011) 29:149–55. doi: 10.1002/cbf.1736
125. Hirose A, Tanikawa T, Mori H, Okada Y, Tanaka Y. Advanced glycation end products increase endothelial permeability through the RAGE/rho signaling pathway. FEBS Lett. (2010) 584:61–6. doi: 10.1016/j.febslet.2009.11.082
126. Xing Y, Pan S, Zhu L, Cui Q, Tang Z, Liu Z, et al. Advanced glycation end products induce atherosclerosis via RAGE/TLR4 signaling mediated-M1 macrophage polarization-dependent vascular smooth muscle cell phenotypic conversion. Oxid Med Cell Longev. (2022) 2022:9763377. doi: 10.1155/2022/9763377
127. Xu Z, Liu X, Wang Z, Tao J, Han Z, Gu M, et al. Effect of sirolimus on arteriosclerosis induced by advanced glycation end products via inhibition of the ILK/mTOR pathway in kidney transplantation recipients. Eur J Pharmacol. (2017) 813:1–9. doi: 10.1016/j.ejphar.2017.06.038
128. Fukami K, Ueda S, Yamagishi S, Kato S, Inagaki Y, Takeuchi M, et al. AGEs activate mesangial TGF-beta-Smad signaling via an angiotensin II type I receptor interaction. Kidney Int. (2004) 66:2137–47. doi: 10.1111/j.1523-1755.2004.66004.x
129. Alikhani M, Maclellan CM, Raptis M, Vora S, Trackman PC, Graves DT. Advanced glycation end products induce apoptosis in fibroblasts through activation of ROS, MAP kinases, and the FOXO1 transcription factor. Am J Physiol Cell Physiol. (2007) 292:C850–6. doi: 10.1152/ajpcell.00356.2006
130. Liu Y, Liang C, Liu X, Liao B, Pan X, Ren Y, et al. AGEs increased migration and inflammatory responses of adventitial fibroblasts via RAGE, MAPK and NF-kappaB pathways. Atherosclerosis. (2010) 208:34–42. doi: 10.1016/j.atherosclerosis.2009.06.007
131. Arrigo M, Jessup M, Mullens W, Reza N, Shah AM, Sliwa K, et al. Acute heart failure. Nat Rev Dis Primers. (2020) 6:16. doi: 10.1038/s41572-020-0151-7
132. Raposeiras-Roubín S, Rodiño-Janeiro BK, Grigorian-Shamagian L, Moure-González M, Seoane-Blanco A, Varela-Román A, et al. Soluble receptor of advanced glycation End products levels are related to ischaemic aetiology and extent of coronary disease in chronic heart failure patients, independent of advanced glycation end products levels: new roles for soluble RAGE. Eur J Heart Fail. (2010) 12:1092–100. doi: 10.1093/eurjhf/hfq117
133. Khan MI, Rath S, Adhami VM, Mukhtar H. Hypoxia driven glycation: mechanisms and therapeutic opportunities. Semin Cancer Biol. (2018) 49:75–82. doi: 10.1016/j.semcancer.2017.05.008
134. Sakaguchi T, Yan SF, Yan SD, Belov D, Rong LL, Sousa M, et al. Central role of RAGE-dependent neointimal expansion in arterial restenosis. J Clin Invest. (2003) 111:959–72. doi: 10.1172/JCI17115
135. Barallobre-Barreiro J, Radovits T, Fava M, Mayr U, Lin W-Y, Ermolaeva E, et al. Extracellular matrix in heart failure: role of ADAMTS5 in proteoglycan remodeling. Circulation. (2021) 144:2021–34. doi: 10.1161/CIRCULATIONAHA.121.055732
136. Frangogiannis NG. The extracellular matrix in ischemic and non-ischemic heart failure. Circ Res. (2019) 125:117–46. doi: 10.1161/CIRCRESAHA.119.311148
137. van Heerebeek L, Hamdani N, Handoko ML, Falcao-Pires I, Musters RJ, Kupreishvili K, et al. Diastolic stiffness of the failing diabetic heart: importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation. (2008) 117:43–51. doi: 10.1161/CIRCULATIONAHA.107.728550
138. Falcão-Pires I, Hamdani N, Borbély A, Gavina C, Schalkwijk CG, van der Velden J, et al. Diabetes mellitus worsens diastolic left ventricular dysfunction in aortic stenosis through altered myocardial structure and cardiomyocyte stiffness. Circulation. (2011) 124:1151–9. doi: 10.1161/CIRCULATIONAHA.111.025270
139. Blackburn NJR, Vulesevic B, McNeill B, Cimenci CE, Ahmadi A, Gonzalez-Gomez M, et al. Methylglyoxal-derived advanced glycation end products contribute to negative cardiac remodeling and dysfunction post-myocardial infarction. Basic Res Cardiol. (2017) 112:57. doi: 10.1007/s00395-017-0646-x
140. Willemsen S, Hartog JWL, Hummel YM, van Ruijven MHI, van der Horst ICC, van Veldhuisen DJ, et al. Tissue advanced glycation End products are associated with diastolic function and aerobic exercise capacity in diabetic heart failure patients. Eur J Heart Fail. (2011) 13:76–82. doi: 10.1093/eurjhf/hfq168
141. Ouyang A, Olver TD, Emter CA, Fleenor BS. Chronic exercise training prevents coronary artery stiffening in aortic-banded miniswine: role of perivascular adipose-derived advanced glycation end products. J Appl Physiol (1985). (2019) 127:816–27. doi: 10.1152/japplphysiol.00146.2019
142. Brett J, Schmidt AM, Yan SD, Zou YS, Weidman E, Pinsky D, et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol. (1993) 143:1699–712.8256857
143. Hartog JWL, Voors AA, Bakker SJL, Smit AJ, Veldhuisen DJ Van. Advanced glycation end–products (AGEs) and heart failure: pathophysiology and clinical implications. Eur J Heart Fail. (2007) 9(12):1146–55. doi: 10.1016/j.ejheart.2007.09.009
144. Burr SD, Stewart JA. Rap1a overlaps the AGE/RAGE signaling cascade to Alter expression of α-SMA. p-NF-κB, and p-PKC-ζ in cardiac fibroblasts isolated from type: 2, diabetic mice. Cells. (2021) 10:557. doi: 10.3390/cells10030557
145. Burr SD, Dorroh CC, Stewart JA. Rap1a activity elevated the impact of endogenous AGEs in diabetic collagen to stimulate increased myofibroblast transition and oxidative stress. Int J Mol Sci. (2022) 23:4480. doi: 10.3390/ijms23094480
146. Fang M, Wang J, Li S, Guo Y. Advanced glycation end-products accelerate the cardiac aging process through the receptor for advanced glycation end-products/transforming growth factor-β-Smad signaling pathway in cardiac fibroblasts. Geriatr Gerontol Int. (2016) 16:522–7. doi: 10.1111/ggi.12499
147. Eleid MF, Nkomo VT, Pislaru SV, Gersh BJ. Valvular heart disease: new concepts in pathophysiology and therapeutic approaches. Annu Rev Med. (2023) 74:155–70. doi: 10.1146/annurev-med-042921-122533
148. Hinton RB, Yutzey KE. Heart valve structure and function in development and disease. Annu Rev Physiol. (2011) 73:29–46. doi: 10.1146/annurev-physiol-012110-142145
149. Small AM, Yutzey KE, Binstadt BA, Voigts Key K, Bouatia-Naji N, Milan D, et al.; American Heart Association council on genomic and precision medicine; council on cardiopulmonary, critical care, perioperative and resuscitation; and council on cardiovascular and stroke nursing unraveling the mechanisms of valvular heart disease to identify medical therapy targets: a scientific statement from the American Heart Association. Circulation. (2024) 150:e109–28. doi: 10.1161/CIR.0000000000001254
150. Barnard SJ, Haunschild J, Heiser L, Dieterlen MT, Klaeske K, Borger MA, et al. Apoptotic cell death in bicuspid-aortic-valve-associated aortopathy. Int J Mol Sci. (2023) 24:7429. doi: 10.3390/ijms24087429
151. Yang P-S, Lee SH, Park J, Kim T-H, Uhm J-S, Joung B, et al. Atrial tissue expression of receptor for advanced glycation End-products (RAGE) and atrial fibrosis in patients with mitral valve disease. Int J Cardiol. (2016) 220:1–6. doi: 10.1016/j.ijcard.2016.06.137
152. Kopytek M, Ząbczyk M, Mazur P, Undas A, Natorska J. Accumulation of advanced glycation end products (AGEs) is associated with the severity of aortic stenosis in patients with concomitant type 2 diabetes. Cardiovasc Diabetol. (2020) 19:92. doi: 10.1186/s12933-020-01068-7
153. Basta G, Corciu AI, Vianello A, Del Turco S, Foffa I, Navarra T, et al. Circulating soluble receptor for advanced glycation end-product levels are decreased in patients with calcific aortic valve stenosis. Atherosclerosis. (2010) 210:614–8. doi: 10.1016/j.atherosclerosis.2009.12.029
154. Li F, Cai Z, Chen F, Shi X, Zhang Q, Chen S, et al. Pioglitazone attenuates progression of aortic valve calcification via down-regulating receptor for advanced glycation End products. Basic Res Cardiol. (2012) 107:306. doi: 10.1007/s00395-012-0306-0
155. Yang X, Zeng J, Xie K, Su S, Guo Y, Zhang H, et al. Advanced glycation end product-modified low-density lipoprotein promotes pro-osteogenic reprogramming via RAGE/NF-κB pathway and exaggerates aortic valve calcification in hamsters. Mol Med. (2024) 30:76. doi: 10.1186/s10020-024-00833-8
156. Wang X, Desai K, Juurlink BHJ, Champlain J de, Wu L. Gender-related differences in advanced glycation endproducts, oxidative stress markers and nitric oxide synthases in rats. Kidney Int. (2006) 69:281–7. doi: 10.1038/sj.ki.5000043
157. Ward LJ, Laucyte-Cibulskiene A, Hernandez L, Ripsweden J, Stenvinkel P, Kublickiene K. Coronary artery calcification and aortic valve calcification in patients with kidney failure: a sex-disaggregated study. Biol Sex Differ. (2023) 14:48. doi: 10.1186/s13293-023-00530-x
158. Lindman BR, Clavel M-A, Mathieu P, Iung B, Lancellotti P, Otto CM, et al. Calcific aortic stenosis. Nat Rev Dis Primers. (2016) 2:16006. doi: 10.1038/nrdp.2016.6
159. Cartlidge TRG, Doris MK, Sellers SL, Pawade TA, White AC, Pessotto R, et al. Detection and prediction of bioprosthetic aortic valve degeneration. J Am Coll Cardiol. (2019) 73:1107–19. doi: 10.1016/j.jacc.2018.12.056
160. Rodriguez-Gabella T, Voisine P, Puri R, Pibarot P, Rodés-Cabau J. Aortic bioprosthetic valve durability: incidence, mechanisms, predictors, and management of surgical and transcatheter valve degeneration. J Am Coll Cardiol. (2017) 70:1013–28. doi: 10.1016/j.jacc.2017.07.715
161. Frasca A, Xue Y, Kossar AP, Keeney S, Rock C, Zakharchenko A, et al. Glycation and serum albumin infiltration contribute to the structural degeneration of bioprosthetic heart valves. JACC Basic Transl Sci. (2020) 5:755–66. doi: 10.1016/j.jacbts.2020.06.008
162. Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation. (2005) 11:3484–5. doi: 10.1161/CIRCULATIONAHA.105.537878
163. Xie F, Liu B, Qiao W, He J, Cheng J, Wang Z, et al. Smooth muscle NF90 deficiency ameliorates diabetic atherosclerotic calcification in male mice via FBXW7-AGER1-AGEs axis. Nat Commun. (2024) 15:4985. doi: 10.1038/s41467-024-49315-9
164. Xu L, Wang Y-R, Li P-C, Feng B. Advanced glycation end products increase lipids accumulation in macrophages through upregulation of receptor of advanced glycation end products: increasing uptake, esterification and decreasing efflux of cholesterol. Lipids Health Dis. (2016) 15:161. doi: 10.1186/s12944-016-0334-0
165. Hartley A, Haskard D, Khamis R. Oxidized LDL and anti-oxidized LDL antibodies in atherosclerosis—novel insights and future directions in diagnosis and therapy. Trends Cardiovasc Med. (2019) 29:22–6. doi: 10.1016/j.tcm.2018.05.010
166. Zhao Y, Jia X, Yang X, Bai X, Lu Y, Zhu L, et al. Deacetylation of caveolin-1 by Sirt6 induces autophagy and retards high glucose-stimulated LDL transcytosis and atherosclerosis formation. Metab Clin Exp. (2022) 131:13. doi: 10.1016/j.metabol.2022.155162
167. Xu S, Li L, Yan J, Ye F, Shao C, Sun Z, et al. CML/CD36 accelerates atherosclerotic progression via inhibiting foam cell migration. Biomed Pharmacother. (2018) 97:1020–31. doi: 10.1016/j.biopha.2017.11.041
168. Shrivastav D, Singh DD, Mir R, Mehra P, Mehta V, Dabla PK. Comparative analysis of nε-carboxymethyl-lysine and inflammatory markers in diabetic and non-diabetic coronary artery disease patients. World J Diabetes. (2023) 14:1754–65. doi: 10.4239/wjd.v14.i12.1754
169. Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. (2005) 352:1685–95. doi: 10.1056/NEJMra043430
170. Oparil S, Acelajado MC, Bakris GL, Berlowitz DR, Cífková R, Dominiczak AF, et al. Hypertension. Nat Rev Dis Primers. (2018) 4:18014. doi: 10.1038/nrdp.2018.14
171. Boutouyrie P, Chowienczyk P, Humphrey JD, Mitchell GF. Arterial stiffness and cardiovascular risk in hypertension. Circ Res. (2021) 128:864–86. doi: 10.1161/CIRCRESAHA.121.318061
172. McNulty M, Mahmud A, Feely J. Advanced glycation end-products and arterial stiffness in hypertension*. Am J Hypertens 2007 20:242–7. doi: 10.1016/j.amjhyper.2006.08.009
173. Mitchell GF. Arterial stiffness in aging: does it have a place in clinical practice? Hypertension. (2021) 77:768–80. doi: 10.1161/HYPERTENSIONAHA.120.14515
174. Huang QF, Sheng CS, Liu M, Li FH, Li Y, Wang JG. Arterial stiffness and wave reflections in relation to plasma advanced glycation end products in a Chinese population. Am J Hypertens. (2013) 26(6):754–61. doi: 10.1093/ajh/hpt014
175. Birukov A, Cuadrat R, Polemiti E, Eichelmann F, Schulze MB. Advanced glycation end-products, measured as skin autofluorescence, associate with vascular stiffness in diabetic, pre-diabetic and normoglycemic individuals: a cross-sectional study. Cardiovasc Diabetol. (2021) 20:110. doi: 10.1186/s12933-021-01296-5
176. van Waateringe RP, Slagter SN, van Beek AP, van der Klauw MM, van Vliet-Ostaptchouk JV, Graaff R, et al. Skin autofluorescence, a non-invasive biomarker for advanced glycation End products, is associated with the metabolic syndrome and its individual components. Diabetol Metab Syndr. (2017) 9:42. doi: 10.1186/s13098-017-0241-1
177. Zhou D, Xi B, Zhao M, Wang L, Veeranki SP. Uncontrolled hypertension increases risk of all-cause and cardiovascular disease mortality in US adults: the NHANES III linked mortality study. Sci Rep. (2018) 8:9418. doi: 10.1038/s41598-018-27377-2
178. Lyu L, Yu J, Liu Y, He S, Zhao Y, Qi M, et al. High hemoglobin glycation Index is associated with telomere attrition independent of HbA1c, mediated by TNFα. J Clin Endocrinol Metab. (2022) 107:462–73. doi: 10.1210/clinem/dgab703
179. Ritchie RH, Abel ED. Basic mechanisms of diabetic heart disease. Circ Res. (2020) 126:1501–25. doi: 10.1161/CIRCRESAHA.120.315913
180. Huebschmann AG, Regensteiner JG, Vlassara H, Reusch JEB. Diabetes and advanced glycoxidation end products. Diabetes Care. (2006) 29:1420–32. doi: 10.2337/dc05-2096
181. Jia G, Whaley-Connell A, Sowers JR. Diabetic cardiomyopathy: a hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia. (2018) 61:21–8. doi: 10.1007/s00125-017-4390-4
182. Wang X, Chen X, Zhou W, Men H, Bao T, Sun Y, et al. Ferroptosis is essential for diabetic cardiomyopathy and is prevented by sulforaphane via AMPK/NRF2 pathways. Acta Pharm Sin B. (2022) 12:708–22. doi: 10.1016/j.apsb.2021.10.005
183. Thakur MR, Nachane SS, Tupe RS. Alleviation of albumin glycation-induced diabetic cardiomyopathy by L-arginine: insights into nrf-2 signaling. Int J Biol Macromol. (2024) 264:130478. doi: 10.1016/j.ijbiomac.2024.130478
184. Caporizzo MA, Prosser BL. The microtubule cytoskeleton in cardiac mechanics and heart failure. Nat Rev Cardiol. (2022) 19:364–78. doi: 10.1038/s41569-022-00692-y
185. Shiels H, O’Connell A, Qureshi MA, Howarth FC, White E, Calaghan S. Stable microtubules contribute to cardiac dysfunction in the streptozotocin-induced model of type 1 diabetes in the rat. Mol Cell Biochem. (2007) 294:173–80. doi: 10.1007/s11010-006-9257-9
186. Yan D, Luo X, Li Y, Liu W, Deng J, Zheng N, et al. Effects of advanced glycation end products on calcium handling in cardiomyocytes. Cardiology. (2014) 129:75–83. doi: 10.1159/000364779
187. Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. (2011) 11:98–107. doi: 10.1038/nri2925
188. Wang Y, Luo W, Han J, Khan ZA, Fang Q, Jin Y, et al. MD2 activation by direct AGE interaction drives inflammatory diabetic cardiomyopathy. Nat Commun. (2020) 11:2148. doi: 10.1038/s41467-020-15978-3
189. Li J, Nan W, Huang X, Meng H, Wang S, Zheng Y, et al. Eicosapentaenoic acid induces macrophage mox polarization to prevent diabetic cardiomyopathy. EMBO Rep. (2024) 25:5507–36. doi: 10.1038/s44319-024-00271-x
190. Liu G, Yan D, Yang L, Sun Y, Zhan L, Lu L, et al. The effect of miR-471-3p on macrophage polarization in the development of diabetic cardiomyopathy. Life Sci. (2021) 268:118989. doi: 10.1016/j.lfs.2020.118989
191. Roest PAM, Molin DGM, Schalkwijk CG, van Iperen L, Wentzel P, Eriksson UJ, et al. Specific local cardiovascular changes of nɛ-(carboxymethyl)Lysine, vascular endothelial growth factor, and Smad2 in the developing embryos coincide with maternal diabetes–induced congenital heart defects. Diabetes. (2009) 58:1222–8. doi: 10.2337/db07-1016
192. Yang L-T, Ye Z, Wajih Ullah M, Maleszewski JJ, Scott CG, Padang R, et al. Bicuspid aortic valve: long-term morbidity and mortality. Eur Heart J. (2023) 44:4549–62. doi: 10.1093/eurheartj/ehad477
193. Xiong T-Y, Ali WB, Feng Y, Hayashida K, Jilaihawi H, Latib A, et al. Transcatheter aortic valve implantation in patients with bicuspid valve morphology: a roadmap towards standardization. Nat Rev Cardiol. (2023) 20:52–67. doi: 10.1038/s41569-022-00734-5
194. Branchetti E, Bavaria JE, Grau JB, Shaw RE, Poggio P, Lai EK, et al. Circulating soluble receptor for advanced glycation end product identifies patients with bicuspid aortic valve and associated aortopathies. Arterioscler Thromb Vasc Biol. (2014) 34:2349–57. doi: 10.1161/ATVBAHA.114.303784
195. Jia H, Kang L, Lu S, Chen Z, Shen J, Huang B, et al. Circulating soluble receptor of advanced glycation end product is associated with bicuspid aortic aneurysm progression via NF-κB pathway. Interact Cardiovasc Thorac Surg. (2021) 34:274–82. doi: 10.1093/icvts/ivab242
196. Chen M, Li H, Wang G, Shen X, Zhao S, Su W. Atorvastatin prevents advanced glycation end products (AGEs)-induced cardiac fibrosis via activating peroxisome proliferator-activated receptor gamma (PPAR-γ). Metab Clin Exp. (2016) 65:441–53. doi: 10.1016/j.metabol.2015.11.007
197. Liu Y, Yu M, Zhang Z, Yu Y, Chen Q, Zhang W, et al. Blockade of receptor for advanced glycation end products protects against systolic overload-induced heart failure after transverse aortic constriction in mice. Eur J Pharmacol. (2016) 791:535–43. doi: 10.1016/j.ejphar.2016.07.008
198. Galasko D, Bell J, Mancuso JY, Kupiec JW, Sabbagh MN, van Dyck C, et al. For the Alzheimer’s disease cooperative study clinical trial of an inhibitor of RAGE-aβ interactions in Alzheimer disease. Neurology. (2014) 82:1536–42. doi: 10.1212/WNL.0000000000000364
199. Zieman SJ, Melenovsky V, Clattenburg L, Corretti MC, Capriotti A, Gerstenblith G, et al. Advanced glycation endproduct crosslink breaker (alagebrium) improves endothelial function in patients with isolated systolic hypertension. J Hypertens. (2007) 25:577–83. doi: 10.1097/HJH.0b013e328013e7dd
200. Willemsen S, Hartog JWL, Hummel YM, Posma JL, van Wijk LM, van Veldhuisen DJ, et al. Effects of alagebrium, an advanced glycation end-product breaker, in patients with chronic heart failure: study design and baseline characteristics of the BENEFICIAL trial. Eur J Heart Fail. (2010) 12:294–300. doi: 10.1093/eurjhf/hfp207
201. Wolffenbuttel BHR, Boulanger CM, Crijns FRL, Huijberts MSP, Poitevin P, Swennen GNM, et al. Breakers of advanced glycation end products restore large artery properties in experimental diabetes. Proc Natl Acad Sci U S A. (1998) 95:4630–4. doi: 10.1073/pnas.95.8.4630
202. Asif M, Egan J, Vasan S, Jyothirmayi GN, Masurekar MR, Lopez S, et al. An advanced glycation endproduct cross-link breaker can reverse age-related increases in myocardial stiffness. Proc Natl Acad Sci U S A. (2000) 97:2809–13. doi: 10.1073/pnas.040558497
203. Cao X, Li B, Han X, Zhang X, Dang M, Wang H, et al. Soluble receptor for advanced glycation end-products promotes angiogenesis through activation of STAT3 in myocardial ischemia/reperfusion injury. Apoptosis. (2020) 25:341–53. doi: 10.1007/s10495-020-01602-8
204. Dang M, Zeng X, Chen B, Wang H, Li H, Liu Y, et al. Soluble receptor for advance glycation end-products inhibits ischemia/reperfusion-induced myocardial autophagy via the STAT3 pathway. Free Radic Biol Med. (2019) 130:107–19. doi: 10.1016/j.freeradbiomed.2018.10.437
205. Guo C, Zeng X, Song J, Zhang M, Wang H, Xu X, et al. A soluble receptor for advanced glycation end-products inhibits hypoxia/reoxygenation-induced apoptosis in rat cardiomyocytes via the mitochondrial pathway. Int J Mol Sci. (2012) 13:11923–40. doi: 10.3390/ijms130911923
206. Khan I, Saeed K, Khan I. Nanoparticles: properties, applications and toxicities. Arab J Chem. (2019) 12:908–31. doi: 10.1016/j.arabjc.2017.05.011
207. Bhattacherjee A, Chakraborti AS. Argpyrimidine-tagged rutin-encapsulated biocompatible (ethylene glycol dimers) nanoparticles: application for targeted drug delivery in experimental diabetes (part 2). Int J Pharm. (2017) 528:8–17. doi: 10.1016/j.ijpharm.2017.05.058
208. Yang MJ, Ku SH, Kim D, Kim WJ, Mok H, Kim SH, et al. Enhanced cytoplasmic delivery of RAGE siRNA using bioreducible polyethylenimine-based nanocarriers for myocardial gene therapy. Macromol Biosci. (2015) 15:1755–63. doi: 10.1002/mabi.201500213
209. Serrano JCE, Castro-Boqué E, García-Carrasco A, Morán-Valero MI, González-Hedström D, Bermúdez-López M, et al. Antihypertensive effects of an optimized aged garlic extract in subjects with grade I hypertension and antihypertensive drug therapy: a randomized, triple-blind controlled trial. Nutrients. (2023) 15:3691. doi: 10.3390/nu15173691
210. Li H, Sureda A, Devkota HP, Pittalà V, Barreca D, Silva AS, et al. Curcumin, the golden spice in treating cardiovascular diseases. Biotechnol Adv. (2020) 38:107343. doi: 10.1016/j.biotechadv.2019.01.010
211. Abdel-Mageid AD, Abou-Salem MES, Salaam NMHA, El-Garhy HAS. The potential effect of garlic extract and curcumin nanoparticles against complication accompanied with experimentally induced diabetes in rats. Phytomedicine. (2018) 43:126–34. doi: 10.1016/j.phymed.2018.04.039
212. Lu H, Wang J, Chen Z, Wang J, Jiang Y, Xia Z, et al. Engineered macrophage membrane-coated S100A9-siRNA for ameliorating myocardial ischemia-reperfusion injury. Adv Sci. (2024) 11:2403542. doi: 10.1002/advs.202403542
213. Kass DA, Shapiro EP, Kawaguchi M, Capriotti AR, Scuteri A, de Groof RC, et al. Improved arterial compliance by a novel advanced glycation end-product crosslink breaker. Circulation. (2001) 104:1466–8. doi: 10.1161/hc3801.097806
214. Tang M, Kalim S. Avenues for post-translational protein modification prevention and therapy. Mol Aspects Med. (2022) 86:101083. doi: 10.1016/j.mam.2022.101083
215. Toprak C, Yigitaslan S. Alagebrium and complications of diabetes mellitus. Eurasian J Med. (2019) 51:285–92. doi: 10.5152/eurasianjmed.2019.18434
216. Umadevi S, Gopi V, Simna SP, Parthasarathy A, Yousuf SMJ, Elangovan V. Studies on the cardio protective role of gallic acid against AGE-induced cell proliferation and oxidative stress in H9C2 (2-1) cells. Cardiovasc Toxicol. (2012) 12:304–11. doi: 10.1007/s12012-012-9170-2
217. Zhao W, Cai P, Zhang N, Wu T, Sun A, Jia G. Inhibitory effects of polyphenols from black chokeberry on advanced glycation end-products (AGEs) formation. Food Chem. (2022) 392:133295. doi: 10.1016/j.foodchem.2022.133295
218. Soman S, Rajamanickam C, Rauf AA, Madambath I. Molecular mechanisms of the antiglycative and cardioprotective activities of psidium guajava leaves in the rat diabetic myocardium. Pharm. Biol. (2016) 54:3078–85. doi: 10.1080/13880209.2016.1207090
219. Sacks DB, Kirkman MS, Little RR. Point-of-care HbA1c in clinical practice: caveats and considerations for optimal use. Diabetes Care. (2024) 47:1104–10. doi: 10.2337/dci23-0040
220. Wei X, Chen X, Zhang Z, Wei J, Hu B, Long N, et al. Risk analysis of the association between different hemoglobin glycation index and poor prognosis in critical patients with coronary heart disease-a study based on the MIMIC-IV database. Cardiovasc Diabetol. (2024) 23:113. doi: 10.1186/s12933-024-02206-1
221. Hempe JM, Liu S, Myers L, McCarter RJ, Buse JB, Fonseca V. The hemoglobin glycation index identifies subpopulations with Harms or benefits from intensive treatment in the ACCORD trial. Diabetes Care. (2015) 38:1067–74. doi: 10.2337/dc14-1844
222. Lin Z, He J, Yuan S, Song C, Bian X, Yang M, et al. Hemoglobin glycation Index and cardiovascular outcomes in patients with diabetes and coronary artery disease: insights from a large cohort study. Nutr Diabetes. (2024) 14:69. doi: 10.1038/s41387-024-00318-x
223. Cheng W, Huang R, Pu Y, Li T, Bao X, Chen J, et al. Association between the haemoglobin glycation index (HGI) and clinical outcomes in patients with acute decompensated heart failure. Ann Med. (2024) 56:2330615. doi: 10.1080/07853890.2024.2330615
224. Wang R, Chen C, Xu G, Jin Z. Association of triglyceride glucose-body mass Index and hemoglobin glycation Index with heart failure prevalence in hypertensive populations: a study across different glucose metabolism Status. Lipids Health Dis. (2024) 23:53. doi: 10.1186/s12944-024-02045-9
225. Wang Z, Liu Y, Xie J, Liu N-F. Association between hemoglobin glycation Index and subclinical myocardial injury in the general population free from cardiovascular disease. Nutr Metab Cardiovasc Dis. (2022) 32:469–78. doi: 10.1016/j.numecd.2021.10.018
226. Cai A, Li G, Chen J, Li X, Wei X, Li L, et al. Glycated hemoglobin level is significantly associated with the severity of coronary artery disease in non-diabetic adults. Lipids Health Dis. (2014) 13:181. doi: 10.1186/1476-511X-13-181
227. Huang L, He L, Zeng Q, Huang J, Luo X, Zhong Q. Nonlinear association between glycated hemoglobin levels and mortality in elderly patients with non-diabetic chronic kidney disease: a national health and nutrition examination survey analysis. Front Endocrinol (Lausanne). (2025) 16:1416506. doi: 10.3389/fendo.2025.1416506
228. Chow JP, Simionescu DT, Warner H, Wang B, Patnaik SS, Liao J, et al. Mitigation of diabetes-related complications in implanted collagen and elastin scaffolds using matrix-binding polyphenol. Biomaterials. (2013) 34:685–95. doi: 10.1016/j.biomaterials.2012.09.081
229. Gustavsson CG, Agardh C-D. Markers of inflammation in patients with coronary artery disease are also associated with glycosylated haemoglobin A1c within the normal range. Eur Heart J. (2004) 25:2120–4. doi: 10.1016/j.ehj.2004.09.008
230. Yan SF, Ramasamy R, Naka Y, Schmidt AM. Glycation, inflammation, and RAGE: a scaffold for the macrovascular complications of diabetes and beyond. Circ Res. (2003) 93:1159–69. doi: 10.1161/01.RES.0000103862.26506.3D
231. Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res. (2018) 122:624–38. doi: 10.1161/CIRCRESAHA.117.311586
232. Xu H-H, Hao S-X, Sun H-Y, Dong X-X, Lin Y, Lou H, et al. THBru attenuates diabetic cardiomyopathy by inhibiting RAGE-dependent inflammation. Acta Pharmacol Sin. (2024) 45:2107–18. doi: 10.1038/s41401-024-01307-7
233. Uribarri J, Tuttle KR. Dietary advanced glycation End products and cardiovascular-kidney-metabolic complications. Clin J Am Soc Nephrol. (2025). doi: 10.2215/CJN.0000000859
234. Wang S, Gu L, Chen J, Jiang Q, Sun J, Wang H, et al. Association of hemoglobin glycation index and glycation gap with cardiovascular disease among US adults. Diabetes Res Clin Pract. (2022) 190:109990. doi: 10.1016/j.diabres.2022.109990
235. Adams JN, Raffield LM, Martelle SE, Freedman BI, Langefeld CD, Carr JJ, et al. Genetic analysis of advanced glycation end products in the DHS MIND study. Gene. (2016) 584:173–9. doi: 10.1016/j.gene.2016.02.029
236. Feitosa MF, Lin SJ, Acharya S, Thyagarajan B, Wojczynski MK, Kuipers AL, et al. Discovery of genomic and transcriptomic pleiotropy between kidney function and soluble receptor for advanced glycation end products using correlated meta-analyses: the long life family study. Aging Cell. (2024) 23:e14261. doi: 10.1111/acel.14261
Keywords: advanced glycation end products, non-enzymatic glycation, cardiovascular diseases, heart failure, therapeutic strategies, clinical translation
Citation: Li Z-H, Wang X-Y and Luo Q (2025) The roles of advanced glycation end products in cardiovascular diseases: from mechanisms to therapeutic strategies. Front. Cardiovasc. Med. 12:1637252. doi: 10.3389/fcvm.2025.1637252
Received: 29 May 2025; Accepted: 18 August 2025;
Published: 1 September 2025.
Edited by:
Alessando Mattina, IRRCS ISMETT/UPMC, ItalyReviewed by:
Yansheng Feng, The University of Texas Health Science Center at San Antonio, United StatesMohsen Kerkeni, Higher Institute of Biotechnology of Monastir, Tunisia
Copyright: © 2025 Li, Wang and Luo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qi Luo, anhuY3VscTIwMThAMTYzLmNvbQ==
†These authors have contributed equally to this work