Abstract
Introduction:
Exercise-induced physiological cardiac hypertrophy (PCH) plays a significant role in cardiovascular health. Although substantial progress has been made in recent years, the precise regulatory mechanisms underlying this adaptive remodeling remain incompletely elucidated and warrant further investigation.
Methods:
The literature retrieval and selection process in this study adhered to the PRISMA guidelines. Databases such as Web of Science, PubMed, Embase, and the Cochrane Library were searched, with the retrieval period covering from the establishment of the respective databases up to August 2025. Keywords used in the search included “exercise”, “physiological cardiac hypertrophy”, “assessment methods”, “regulatory mechanisms”, and “cardiovascular health”. Inclusion criteria were: (1) studies exploring the regulatory mechanisms or health effects of exercise on physiological cardiac hypertrophy; (2) studies involving healthy adults (≥18 years) or wild-type animal models (e.g., C57BL/6 mice); (3) studies employing quantitative imaging, laboratory, or electrophysiological methods to assess physiological cardiac hypertrophy. Exclusion criteria included studies focused solely on pathological cardiac hypertrophy, experimental studies lacking a control group, and studies assessed as having a high risk of bias. Literature selection was independently performed by two researchers, and the final eligible studies were systematically summarized.
Results:
This review first outlines the definitions, characteristics, and clinical evaluation methods of PCH. It then examines the impact of different exercise modalities on cardiac remodeling and summarizes the underlying regulatory mechanisms, including transcriptional pathways (e.g., IGF-1/PI3K/Akt, NRG1/ErbB signaling), post-transcriptional processes (e.g., RNA m6A methylation and noncoding RNA regulation), and metabolic adaptations (e.g., fatty acid oxidation and glucose utilization).The beneficial effects of exercise-induced physiological cardiac hypertrophy on cardiovascular health are also thoroughly analyzed.
Discussion:
Despite its benefits, several challenges remain. Distinguishing PCH from pathological cardiac hypertrophy (PMH) remains difficult, given the limitations of current imaging techniques and biomarkers. Moreover, excessive exercise may precipitate cardiac decompensation, arrhythmias, or dysfunction. Future research should therefore prioritize the development of personalized exercise prescriptions, refinement of diagnostic technologies, and elucidation of the molecular mechanisms driving cardiac decompensation. Such efforts will not only deepen the scientific understanding of exercise-related cardiac remodeling but also provide practical guidance for athlete training and cardiovascular disease prevention.
1 Introduction
Cardiovascular health is a fundamental determinant of overall human health and quality of life, serving as the cornerstone of normal cardiac and vascular function. It encompasses multiple dimensions, including myocardial contractile capacity, vascular regulation, and circulatory efficiency. Maintaining optimal cardiovascular health not only ensures adequate perfusion and organ function but also substantially lowers the risk of cardiovascular disease (CVD), which remains the leading cause of morbidity and mortality worldwide (1). Moderate exercise can promote cardiovascular health, enhance skeletal muscle function, and slow aging, with its effects depending on the body's adaptation to regular physical activity (2, 3). Lately, exercise has surfaced as a non-pharmacological method to improve heart health, with adaptive cardiac remodeling has become a key research focus in cardiovascular health (4).
Myocardial hypertrophy (MH) is the myocardium's compensatory response to overload from one or more stimuli, including PMH and PCH. PMH is most commonly induced by conditions such as hypertension or aortic valve stenosis. It is characterized by disorganized cardiomyocyte proliferation, interstitial fibrosis, and impaired contractile function. This maladaptive process is typically accompanied by progressive deterioration of cardiac performance, ultimately predisposing patients to adverse events including arrhythmia and heart failure. Imaging-based diagnostic criteria often reveal abnormal thickening of the left ventricular wall and ventricular dilatation (5). PCH represents an adaptive response of the myocardium to stimuli such as growth, exercise, or pregnancy. Rather than being associated with dysfunction, PCH reflects optimized cardiac performance and structural remodeling that enhances cardiovascular health and, in athletes, improves exercise capacity (6). Diagnostic evaluation of PCH generally relies on multimodal approaches, including advanced imaging, laboratory testing, and electrophysiological assessment. Characteristic findings include left ventricular wall thickening and chamber enlargement without evidence of fibrosis, diastolic dysfunction, or adverse remodeling (7).
As sports medicine research progresses, notable strides have been achieved in comprehending exercise-induced PCH. Yet, its regulatory mechanisms remain incompletely understood. Through a systematic analysis of how different exercise modes impact cardiac structural and functional remodeling, this article delivers an extensive summary of the molecular regulatory mechanisms underlying exercise-induced PCH. It also delves into the benefits of exercise-induced PCH for cardiovascular health and the current challenges in this area. The goal is to offer theoretical guidance for athlete training and CVD prevention and treatment.
2 Concepts and characteristics of PCH
2.1 Overview of PCH
PCH is primarily characterized by moderate ventricular wall thickening, enlarged cardiac chamber volumes, and enhanced pumping function. It typically lacks cardiomyocyte apoptosis or cardiac dysfunction, thus exhibiting cardioprotective effects (6–8). Histologically, PCH is marked by enlarged cardiomyocyte size, orderly arrangement of myocardial fibers, increased sarcomere numbers, and possibly moderate extracellular matrix (ECM) remodeling (9). This physiological adaptation was first termed “athlete's heart” in the late 19th century by Swedish researcher Henschen through clinical observations of cross-country skiers. Subsequent studies have identified three key features: exercise-induced bradycardia, exercise induced PCH, and compensatory enhancement of cardiac function (10). Extensive research has demonstrated that PCH fundamentally differs from PMH, with their distinguishing characteristics summarized in Table 1.
Table 1
| Classification dimension | Feature type | PCH | PMH | References |
|---|---|---|---|---|
| Etiology | Triggering Mechanism | Growth and development, exercise, pregnancy, etc. | Hypertension, aortic valve stenosis, etc. | (6, 11–14) |
| Morphology | Hypertrophy Pattern | Uniform symmetry | Asymmetry | (15–18) |
| Myocyte Arrangement | Ordered arrangement | Disordered arrangement | (19–22) | |
| Interstitial Fibrosis | None | Significant | (23–25) | |
| Mass and Volume Changes | Increased | Increased | (6, 26, 27) | |
| Cell Dynamics | Calcium Ion Regulation | enhanced | dysfunction | (6, 14) |
| Cell Apoptosis | Inhibited | Activated | (6, 13, 23) | |
| Contractile Function | Enhanced | Decreased | (6, 14, 26) | |
| Metabolism | FAO | Increased | Decreased | (23, 26, 28) |
| Glucose Oxidation | Increased | Increased | (23, 26, 28–30) | |
| Gene Expression | Embryonic gene reprogramming | Normal | Upregulation | (29, 31, 32) |
Comparative characteristics of PCH and PMH.
2.2 Clinical evaluation of PCH
2.2.1 Radiological evaluation
Imaging assessment of PCH can be performed using advanced modalities such as three-dimensional echocardiography (3DE), tissue Doppler imaging (TDI), and cardiac magnetic resonance imaging (CMR), which allow for accurate evaluation of cardiac structure, function, and remodeling. 3DE offers a robust approach reconstructing cardiac anatomy by capturing volumetric data in real time. One of its key advantages is the ability to accurately measure left ventricular ejection fraction (LVEF), a critical parameter in the assessment of PCH (33). In healthy individuals, LVEF typically falls within the range of 55%–70%. During PCH, the left ventricular end-diastolic volume (LVEDV) increases significantly, often accompanied by a compensatory rise in stroke volume (SV). Despite the volumetric expansion, LVEF remains within the normal range or may even exhibit a mild increase, indicating preserved or augmented systolic performance (13, 34, 35). This coordinated volume expansion is associated with the sustained activation of the Insulin-like Growth Factor 1 (IGF-1)/Phosphatidylinositol 3-Kinase (PI3K)/Protein Kinase B (Akt) signaling pathway, which in turn leads to uniform myocardial thickening rather than localized hypertrophy (36). In addition to LVEF, the left ventricular mass (LVM) and the left ventricular mass index (LVMI) also serve as crucial indicators for evaluating PCH. Reference values suggest normal LVM ranges from 73 to 115 g in males and 62–95 g in females, with normal LVMI thresholds of ≤115 g/m2 and ≤95 g/m2, respectively. In individuals with PCH, both LVM and LVMI show mild increases that remain below the diagnostic thresholds for PMH, though considerable inter-individual variability may be observed. The molecular basis involves increased protein synthesis regulated by the mammalian target of rapamycin complex 1 (mTORC1) and limited myocardial cell proliferation mediated by the Hippo-YAP signaling pathway (35, 37, 38). Despite the advantages of 3DE in providing detailed insights into cardiac structure and function, the technique is subject to several limitations. These include susceptibility to patient-specific factors, greater computational and storage requirements, and relatively lower temporal and spatial resolution compared to other imaging methods.
TDI is an echocardiographic technique that quantifies myocardial motion by detecting Doppler frequency shifts generated by tissue movement, thereby enabling the construction of energy, velocity, and acceleration maps. This method allows for accurate assessment of both systolic and diastolic cardiac function (39). Key functional parameters derived from TDI include the systolic s' wave and the diastolic E/e' ratio. The s' wave reflects the peak velocity of myocardial contraction during systole and exhibits a strong correlation with the LVEF. In healthy individuals, the lateral mitral annular s' wave typically ranges between 7 and 10 cm/s, although this lower boundary may be as low as 5.4 cm/s depending on factors such as age, sex, and equipment calibration (40–42). In case of exercise-induced PCH, the s' wave generally remains within normal levels or shows a modest increase (43). The E/e’ ratio serves as a surrogate marker for estimating the left ventricular filling pressure (LVFP). An E/e’ ratio below 8 is indicative of normal LVFP, whereas values between 8 and 15 fall into an intermediate zone that necessitates supplementary clinical parameters—such as left atrial volume (LAV)—for accurate interpretation. In the setting of PCH, this ratio may exhibit a slight elevation, reflecting adaptive modifications in diastolic function (44–46). This phenomenon reflects adaptive remodeling through the Calcineurin (CaN)/Nuclear Factor of Activated T-cells (NFAT) signaling pathway, which facilitates the rapid Ca2+ reuptake process. When the E/e’ ratio is in the “gray zone” accompanied by a decrease in the s' wave, it indicates dysfunction of the PI3K/Akt-m6A methylation axis, suggesting potential pathological risks (17). TDI offers several advantages, including high temporal resolution, real-time quantitative assessment, and broad clinical applicability. Nonetheless, its limitations—such as angle dependency and the need for high frame rates—must be taken into account when interpreting results.
CMR is based on the principle of nuclear magnetic resonance, whereby hydrogen protons within myocardial tissue resonate under a strong magnetic field and radiofrequency pulses. The resulting signals are reconstructed into high-resolution, multi-plane, and multi-parameter images, enabling detailed assessment of cardiac morphology and function. Owing to its superior spatial resolution and tissue characterization, CMR is regarded as the gold standard for evaluating myocardial structure and function, and it plays a pivotal role in the clinical assessment of PCH (47). CMR allows precise quantification of parameters such as myocardial thickness, ventricular volumes, and ejection fraction, thereby providing robust diagnostic support. For example, measurements of left ventricular wall thickness (LVWT) are commonly used to evaluate the extent of myocardial hypertrophy (MH). In healthy adults, LVWT is typically 9–10 mm and interventricular septal thickness (LVST) 10–12 mm. In PCH, LVWT and LVST may increase to approximately 13–16 mm, usually accompanied by enhanced diastolic function (E/e’ ≤8), preserved myocardial tissue integrity (absence of fibrosis), and left ventricular dilation (≥10% increase in end-diastolic volume) (48, 49). Comparative studies using CMR have demonstrated that elite endurance athletes exhibit greater LVWT, ventricular diameters, and volumes than non-athletes (50). Gender differences have also been observed, with male athletes generally showing larger cardiac mass and volume than females, while age and ethnicity further influence the degree of PCH (51). Importantly, CMR can detect subtle myocardial alterations, including fibrosis and edema, which are essential for distinguishing PCH from PMH. Despite these advantages, limitations such as high cost, long examination times, and the requirement for patient cooperation restrict its application in large-scale population screening (52).
2.2.2 Laboratory evaluation
Laboratory assessment of PCH can be performed using myocardial biomarkers, which provide insights into cardiomyocyte function and remodeling. Commonly evaluated biomarkers include cardiac troponin (cTn), B-type natriuretic peptide (BNP), galectin-3 (Gal-3), and markers of ECM turnover. These indicators are valuable for detecting subtle changes in myocardial stress, injury, and remodeling, thereby complementing imaging techniques in the evaluation of PCH.
cTn comprises three subunits: troponin T (cTnT), troponin I (cTnI), and troponin C (cTnC). Of these, cTnT and cTnI exhibit high specificity and sensitivity for detecting myocardial injury or functional alterations. Under resting physiological conditions, circulating levels of cTnT and cTnI are minimal. However, during physical exertion, increased oxidative stress and changes in myocardial membrane permeability may facilitate the transient release of these proteins into the bloodstream. This phenomenon is attributed to reversible cellular stress rather than irreversible cardiomyocyte damage (53–55). Reported reference values for cTnT and cTnI in healthy individuals typically range from 0.001 to 0.05 ng/ml, although variations can occur depending on the analytical method, age, and sex (56).
For instance, a study examining cTnT concentrations in cyclists following varying exercise intensities demonstrated that moderate and high-intensity exertion led to a significant elevation in cTnT levels, whereas low-intensity activity produced no notable change. Importantly, cTnT levels returned to baseline after a recovery period, suggesting that such elevations reflect physiological responses to exercise rather than pathological myocardial injury (53). During moderate-to-high-intensity exercise, sustained mechanical stress on the cellular phospholipid bilayer results in localized membrane injury. The synthesis of membrane repair protein driven by IGF-1/PI3K/Akt pathway cannot fully keep pace with the rate of damage, leading to transient release of cTn. Concurrently, heightened energy demand excessively activates fatty acid oxidation (FAO) pathways, promoting the influx of free fatty acids (FFAs) into mitochondria and triggering excessive production of reactive oxygen species (ROS). ROS oxidize phospholipids such as phosphatidylcholine, destabilizing membrane integrity and facilitating the efflux of cTnT and cTnI through disrupted membrane gaps, ultimately leading to marked elevation of their serum concentrations (57, 58). It is noteworthy that cTn levels typically peak within 1–4 h following exercise and gradually return to baseline within 24–72 h. The magnitude of elevation and recovery kinetics, however, vary according to exercise modality. Endurance training is often associated with a pronounced rise in cTn and a prolonged normalization period, whereas resistance training generally induces a more modest elevation with a faster return to baseline (59, 60).
BNP is an endogenous hormone predominantly secreted by ventricular cardiomyocytes in response to increased wall tension. It exerts natriuretic, diuretic, and vasodilatory effects, with its dynamic changes regulated by the concurrent modulation of the CaN/NFAT pathway and FAO metabolic pathways activated by exercise. These changes reflect the cardiac adaptation to exercise (29, 61). In healthy individuals at rest, BNP levels are generally below 100 pg/ml. During exercise-induced PCH, increased ventricular wall tension leads to elevated intracellular Ca2+ concentrations, which bind to calmodulin (CaM) and activate CaN. CaN catalyzes the dephosphorylation of NFAT, which translocated to the nucleus and forms a complex with GATA binding protein 4 (GATA4), significantly enhancing BNP gene expression. Additionally, adrenergic signaling, mitogen-activated protein kinase (MAPK) pathways, and FAO-related signaling also contribute to this regulatory process. Under these signals, BNP secretion increases. This transient elevation, typically remaining below 200 pg/ml, is considered a normal physiological response and is not associated with adverse clinical outcomes in most cases. However, sustained BNP levels ≥300 pg/ml, especially when accompanied by clinical symptoms, may warrant further evaluation for potential cardiac dysfunction (62, 63). For example, Hosseini et al. reported elevated BNP concentrations in soccer players following competitive matches, although the levels remained well below thresholds indicative of heart failure (64). Interestingly, longitudinal observations have shown that serum BNP concentrations tend to decrease with increased cumulative training duration, suggesting an adaptive remodeling process in cardiac function in response to prolonged athletic activity (65). Furthermore, BNP typically normalizes more rapidly than cTn after exercise. Evidence indicates that BNP levels peak within 1–4 h post-exercise and return to baseline within 24 h. Notably, endurance exercise induces a greater elevation in BNP compared with resistance training, largely due to its volume overload stimulus. Despite differences in recovery kinetics between exercise modalities, these responses remain within the spectrum of physiological regulation (66, 67).
Gal-3 is both a fibrotic mediator and an inflammatory regulator, playing a pivotal role in diverse physiological and pathological processes (68). In healthy adults, normal serum Gal-3 levels typically range from 11.8 to 16.3 ng/ml, though values vary with detection method, age, and sex (69, 70). In older adults, downregulation of miR-222 reduces its inhibitory effect on Homeobox Containing 1 (Hmbox1), leading to the activation of the Transforming Growth Factor-β (TGF-β)/Smad pathway and the accumulation of inflammation and fibrosis, resulting in slight elevation of serum Gal-3 (71, 72). Research has shown that during exercise-induced PCH, the IGF-1/PI3K/Akt pathway activation results in phosphorylation of specific serine/threonine residues on Fork head Box O (FOXO), which helps suppress pro-inflammatory cytokine release, thereby reducing macrophage recruitment and activation and subsequently lowering Gal-3 production. Moreover, upregulation of miR-222 during exercise inhibits Hmbox1 mRNA expression, further suppressing TGF-β/Smad pathway activation and reducing Gal-3 production in fibroblasts. In PCH, serum Gal-3 levels remain normal or slightly reduced. In contrast, PMH, with insufficient IGF-1/PI3K/Akt pathway activation, leads to substantial secretion of Gal-3 from macrophages and fibroblasts, resulting in significantly elevated serum levels (73–76).
ECM turnover markers are biological indicators reflecting the dynamic balance between ECM synthesis and degradation. These markers are critical for assessing tissue fibrosis, inflammatory status, and related pathological conditions (77). In the context of PCH, ECM turnover markers aid in distinguishing physiological cardiac adaptation from pathological fibrosis. Commonly measured biomarkers include Procollagen Type I N-Terminal Propertied (PINP), Procollagen Type III N-Terminal Propertied (PIIINP), and Matrix Metalloproteinase-9 (MMP-9) (78). For reference, serum PINP and PIIINP levels in healthy adults are approximately 22–75 ng/ml (electrochemiluminescence immunoassay) and 2.5–5.5 ng/ml (radioimmunoassay), respectively, though values may vary with age, exercise, and assay methodology (79, 80). In PCH, PINP and PIIINP levels typically remain within the normal range or exhibit only mild, transient increases following exercise (81). Slight increases in PINP and PIIINP levels are likely due to the activation of signaling pathways such as Hippo-YAP, IGF-1/PI3K/Akt, and RNA m6A methylation, promoting protein synthesis and cellular growth. These pathways may also prevent pathological fibrosis by blocking TGF-β/Smad signaling and downregulating ANP/BNP expression. As a result, fibroblasts maintain a moderate activation state, synthesizing type I and III collagen, which ultimately leads to a mild elevation in serum PINP and PIIINP levels (81–83).
These elevations generally peak within hours post-exercise and return to baseline within 24–72 h, indicating that ECM remodeling during PCH is reversible and represents a normal physiological adaptation. By contrast, in PMH, PINP and PIIINP levels remain persistently elevated, reflecting sustained fibrosis and impaired diastolic function (84, 85). Notably, the response of ECM turnover markers to exercise differs from that of cTn and BNP, as ECM markers show a more moderate increase with a longer recovery period, suggesting that ECM remodeling is a sustained, rather than acute, process. These dynamic changes provide a valuable molecular-level tool for differentiating PCH from PMH.
2.2.3 Electrophysiological assessment
The electrocardiogram (ECG) is a non-invasive diagnostic method that captures the heart's electrical activity via surface electrodes, translating the depolarization and repolarization of myocardial cells into characteristic waveform patterns. It serves as a crucial tool for assessing physiological and pathological changes in PCH (86, 87).
During exercise-induced PCH, several characteristic ECG alterations can be observed, among which increased QRS complex amplitude is particularly common (88). In the precordial leads, voltage augmentation may be evident in RV5 or RV6, exceeding the normal upper limit of 2.5 mV. In the limb leads, the R wave amplitude in lead aVL can surpass 12 mm, the R or S wave in lead I may exceed 15 mm, and the R wave in lead aVF may also be greater than 15 mm. A distinctive pattern is when the voltage in RV6 exceeds that in RV5 (S-type), which is attributed to an enhanced myocardial depolarization vector associated with PCH (89–91). Studies analyzing ECG data from endurance athletes with prolonged training have shown that increased QRS amplitude is common and significantly correlates with both the duration and intensity of training (92). ST-T segment abnormalities constitute another frequent ECG feature of PCH. In leads predominantly displaying R waves, T waves may appear deeply inverted, biphasic, or upright, while the ST segment can demonstrate horizontal or upward sloping elevation (typically >0.1 mV) or downward sloping depression. Marked elevation of V3 and V4, accompanied by upward sloping ST segments and upright T waves, reflects altered myocardial repolarization induced by PCH (93, 94). Observations in elite athletes following high-intensity training indicate that ST segment elevation with increased T wave amplitude in leads V3–V5 is common, and these training-induced changes typically normalize after a period of rest (95).
It is important to recognize the limitations of ECG in the assessment of PCH. Firstly, the ECG alterations described above are not specific to PCH; similar increases in QRS complex amplitude can also occur in conditions such as hypertensive heart disease and hypertrophic cardiomyopathy, necessitating careful differential diagnosis (96, 97). Secondly, in mild cases of PCH, the ECG may appear normal, potentially resulting in missed diagnoses. Moreover, although ECG reflects myocardial electrical conduction, it does not directly quantify left ventricular wall thickness, and its accuracy can be affected by factors such as heart rate, body size, and comorbid cardiac conditions (98). Consequently, the evaluation of PCH should be comprehensive, integrating medical history, clinical symptoms, physical findings, and complementary diagnostic tests (Table 2).
Table 2
| Category | Evaluation technique/method | Parameter | Normal range | Changes observed in PCH | Relevant findings/remarks | References |
|---|---|---|---|---|---|---|
| Radiologic evaluation | 3DE | LVEF | 55%–70% | Normal or slightly elevated | LV cavity dilation leads to increased LVEDV | (13, 34, 35) |
| LVM | Male: 73–115 g; Female: 62–95 g | Slightly above normal range | Considerable individual variation; does not reach PMH criteria | (35, 37, 38) | ||
| LVMI | Male: ≤115 g/m2; Female: ≤95 g/m2 | Slightly above normal range | Considerable individual variation; does not reach PMH criteria | (35, 37, 38) | ||
| TDI | s’ wave | 7–10 cm/s | Normal or slightly increased | Significantly correlated with LVEF | (40, 43) | |
| E/e’ ratio | <8 (normal); 8–15 (borderline) | Slightly elevated due to compensatory mechanisms | Possibly associated with enhanced myocardial contractility | (44–46) | ||
| CMR | LVWT,LVST | LVWT: 9–10 mm, LVST: 10–12 mm | Mild increase | May be related to increased hemodynamics and metabolic adaptations | (48–50) | |
| Laboratory evaluation | Myocardial biomarkers | cTnT, cTnI | 0.001–0.05 ng/ml | Temporarily elevated after moderate to high-intensity exercise | Increased membrane permeability of myocardial cells | (53–56, 99) |
| BNP | <100 pg/ml | Elevated post-exercise (generally ≤200 pg/ml) | Elevated ventricular wall stress | (62) | ||
| Gal-3 | 11.8–16.3 ng/ml | Normal or mildly reduced | Reduced macrophage recruitment and activation, enhanced antioxidant capacity | (69, 70, 74, 75) | ||
| PINP、PIIINP | PINP: 22–75 μg/L; PIIINP: 2.5–5.5 ng/ml | Normal or mildly increased | Rapid recovery after rest and adjustment | (79, 80, 84, 85) | ||
| Electrophysiological assessment | ECG | Amplitude of the QRS complex | RV5/RV6 ≤2.5 mV; R wave in lead aVL ≤12 mm; R/S ratio in lead I ≤15 mm; R wave in lead AVF ≤15 mm | RV5/RV6 >2.5 mV; R wave in lead aVL >12 mm; R/S ratio in lead I >15 mm; R wave in lead AVF >15 mm | This may be associated with an increase in myocardial depolarization vector, and is also related to the intensity and duration of training | (88–91) |
| ST-T segment changes | No elevation of the ST segment; upright T wave | ST segment: upward sloping elevation or downward sloping depression; T wave: deeply inverted, biphasic, or upright | Associated with abnormalities in the myocardial repolarization process, which may be alleviated with rest and adjustment | (93, 94) |
Evaluation indicators of PCH.
3 The relationship between exercise modalities and PCH
3.1 Single exercise modality-induced PCH
3.1.1 Endurance exercise
Endurance exercise is characterized by sustained, moderate-intensity physical activity performed over an extended duration, requiring the integrated function of the cardiovascular and skeletal muscle systems (100). During prolonged endurance activities such as running, cycling, or swimming, the primary hemodynamic adaptations include increased heart rate and SV, resulting in a marked elevation of cardiac output. Simultaneously, enhanced skeletal muscle contraction and respiratory effort augment venous return to the heart. In response to this chronic volume overload, the heart undergoes structural remodeling, manifested primarily as chamber dilation and the progressive development of eccentric hypertrophy (101, 102). Parry-Williams et al. reported that athletes engaged in long-term endurance training exhibit cardiac changes including thickening of the left ventricular wall and chamber enlargement, with these adaptations being more prominent in older athletes (103). Similarly, research by the American Physiological Society demonstrated that one year of continuous endurance training resulted in increased the LVEDV and the LVM, along with enhanced myocardial contractility, while ventricular wall thickness remained largely unchanged. These findings suggest that endurance training promotes eccentric hypertrophic remodeling rather than concentric thickening of the myocardium (104). Experimental studies in animal models support these findings; for instance, rats subjected to daily swimming (60 min per session, five times per week) for 10 weeks developed eccentric cardiac hypertrophy, with the degree of remodeling positively correlated with exercise frequency (105). Mechanistically, the sustained elevation in venous return imposes chronic diastolic wall tension, triggering elongation of sarcomeres—the fundamental contractile units of cardiomyocytes—along the longitudinal axis. This leads to an increase in cardiomyocyte length without a corresponding increase in cell diameter, characteristic of eccentric hypertrophy (106–108).
Although long-term endurance exercise confers numerous cardiovascular benefits, the relationship between exercise intensity, duration, and the threshold for maladaptive cardiac remodeling remains controversial. Increasing evidence indicates that prolonged excessive endurance training may exert detrimental effects on cardiac structure and function. In a study of 40 athletes participating in high-intensity endurance events, such as marathons and triathlons, post-race assessments revealed a marked reduction in right ventricular ejection fraction (RVEF) alongside elevated circulating cardiac troponin (cTn) levels, with a linear correlation between the two. Further myocardial evaluation showed that 12% of these athletes exhibited myocardial fibrosis, and this subgroup had accumulated more years of endurance training compared to their counterparts (109). These findings underscore that excessive endurance exercise can negatively impact cardiac health, particularly affecting the right ventricle. A study in rats reported that 16 weeks of high-intensity training induced eccentric hypertrophy, accompanied by left ventricular diastolic dysfunction and significant myocardial fibrosis in the atria and right ventricle. However, these changes were reversible after the cessation of exercise, suggesting that an excessive volume load may surpass the heart's adaptive capacity, potentially triggering a transition from physiological remodeling to pathological cardiac changes (107). Moreover, when exercise load consistently exceeds the physiological tolerance of the body, it leads to sustained elevation of mTORC1 activity, resulting in endoplasmic reticulum (ER) homeostasis imbalance. This condition also triggers a reduction in miR-222 expression, thereby weakening its inhibitory effect on downstream Cyclin-dependent kinase inhibitor 1B (p27) and Hmbox1. This shift signifies the transition of the heart from adaptive remodeling to pathological remodeling, laying the molecular foundation for myocardial fibrosis and electrical activity abnormalities (110, 111). Therefore, quantifying these molecular biomarkers and dynamically adjusting exercise protocols based on the results ensures that cardiac remodeling remains within physiological adaptation limits, helping to prevent adverse cardiac remodeling due to overtraining. This approach forms the foundation for scientific management of athlete cardiac health.
3.1.2 Resistance exercise
Resistance exercise involves active muscle contractions against external loads, aiming to enhance muscular strength, endurance, or hypertrophy. This form of exercise exerts beneficial effects not only on skeletal muscle but also on myocardial structure and function (112). During resistance training, sustained muscular contraction—often accompanied by relative hypoxia—leads to a marked increase in peripheral vascular resistance. This results in acute elevation in blood pressure and significantly augments left ventricular afterload. Over time, these hemodynamic changes stimulate myocardial remodeling, characterized by increased ventricular wall thickness without substantial chamber dilation—a process defined as concentric hypertrophy (113). Research has shown that after a 24-week resistance training program, consisting of three one-hour sessions per week, participants showed an increase in left ventricular mass (LVM) accompanied by ventricular wall thickening compared to baseline. These adaptations resulted in the development of concentric cardiac hypertrophy (114). In an animal study, 12 weeks of resistance training led to progressive increases in LVM, with observed increments of 8%, 12%, and 16% in weeks 4, 8, and 12, respectively. Importantly, these structural adaptations were accompanied by improved left ventricular systolic function, whereas changes in the LVEDV remained minimal. These findings support the development of concentric hypertrophy, with morphological changes becoming more pronounced over time (115). It is noteworthy that, while substantial evidence supports the induction of concentric hypertrophy by long-term resistance training, some human studies indicate that structural myocardial changes may be minimal even under significant afterload. For instance, a study involving participants with a mean age of approximately 68 years reported no significant alterations in ventricular size or wall thickness after 16 weeks of resistance training compared with baseline measurements. This observation may be attributed to age-related decline in cardiac autonomic regulation, which could diminish the myocardium's responsiveness to sympathetic stimulation (51, 114). Mechanistically, the primary driver of ventricular wall thickening in resistance exercise-induced concentric hypertrophy is the parallel addition of sarcomeres within cardiomyocytes. This pattern contrasts with the longitudinal sarcomere elongation seen in eccentric hypertrophy associated with endurance exercise (115).
During resistance exercise, sustained pressure overload induces adaptive cardiovascular remodeling, often manifested as transient yet substantial elevations in arterial blood pressure. Studies have shown that during maximal-intensity training, such as heavy weightlifting, arterial pressures can rise markedly compared to resting values (116). While this form of pressure overload is physiological, it bears resemblance to pathological pressure overload seen in conditions such as hypertension, which can also lead to concentric hypertrophy. However, pathologically induced hypertrophy is typically accompanied by systolic and/or diastolic dysfunction, as well as asymmetric thickening of the interventricular septum and the left ventricular posterior wall (117). The morphological similarities between physiological concentric hypertrophy resulting from resistance training and pathological forms of hypertrophy may pose diagnostic challenges and risk clinical misinterpretation (Figure 1).
Figure 1
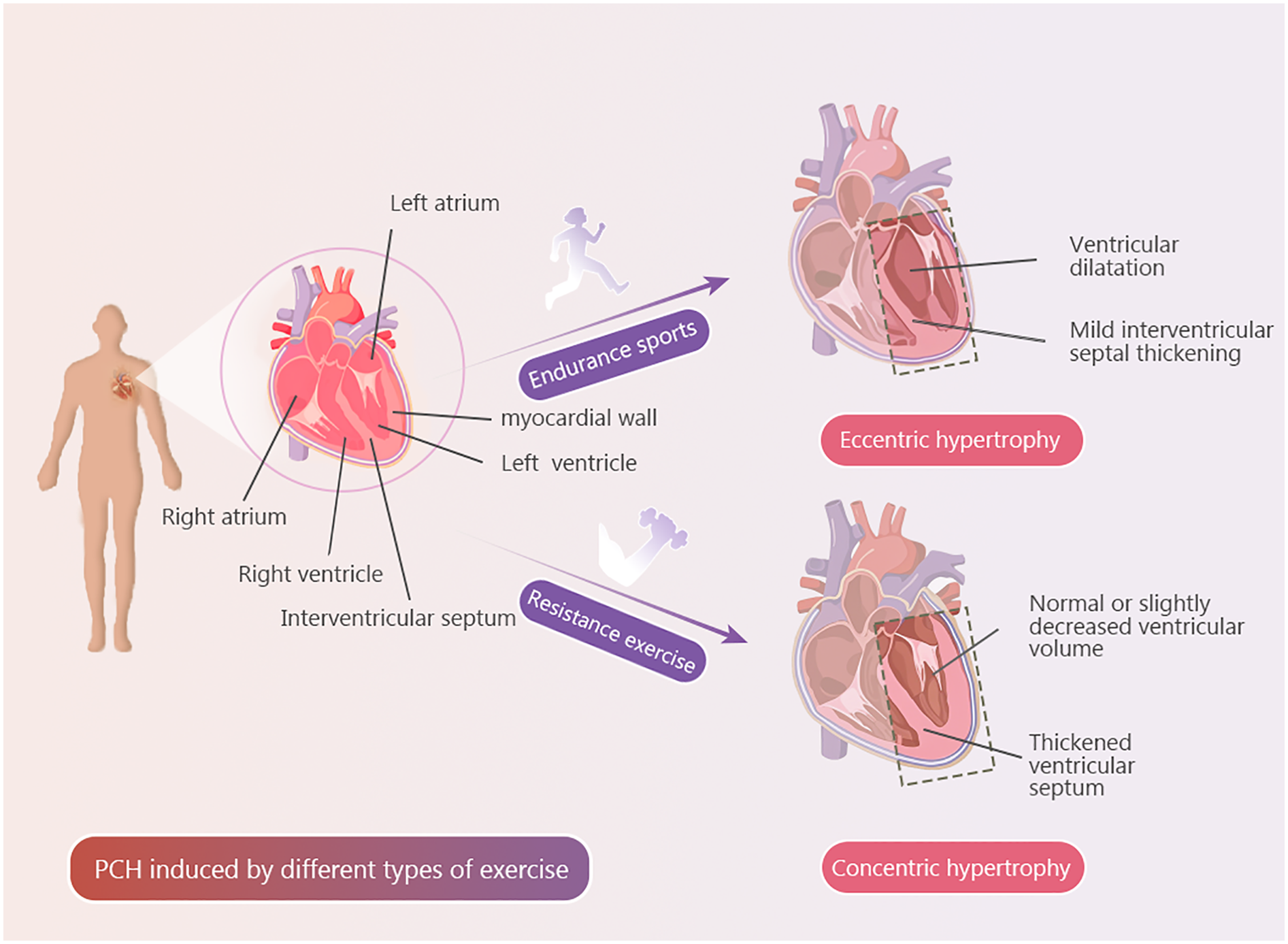
Exercise types and PCH. Explanatory text PCH induced by a single type of exercise.
3.2 Mixed exercise modality-induced PCH
Mixed exercise, which combines endurance and resistance training—such as high-intensity interval training (HIIT) and ball sports—provides alternating stimulation of distinct energy systems, thereby exerting a synergistic effect on the development of PCH. During the high-intensity resistance phases of HIIT, cardiac pressure load increases rapidly, promoting myocardial cell growth and hypertrophy. In contrast, the low-intensity endurance phases enhance blood circulation, ensuring sufficient delivery of oxygen and nutrients to myocardial cells. Additionally, metabolic by-products, such as lactate generated through anaerobic metabolism, are effectively cleared during these phases, supporting myocardial function and metabolic homeostasis (118, 119).
Given that most exercise or fitness programs incorporate both endurance and resistance training, exercise-induced PCH typically manifests as a combination of ventricular dilation and wall thickening, resulting in mixed hypertrophy. This represents an adaptive response of the heart to the imposed exercise load. Scharf et al. utilized magnetic resonance imaging (MRI) to evaluate the cardiac structure of elite triathletes, revealing significantly higher left ventricular end-diastolic volume (LVEDV) and left ventricular mass (LVM) compared with controls, while left atrial volume was inversely correlated with heart rate. The morphological and functional adaptations observed exhibited characteristics of both concentric and eccentric hypertrophy (120). Moreover, the relative proportion of endurance and resistance training within a mixed regimen differentially influences the pattern of PCH. Predominance of endurance exercise enhances myocardial aerobic capacity and cardiac endurance, improves diastolic function, and tends to produce an eccentric hypertrophy–like effect. In contrast, a greater emphasis on resistance exercise promotes myocardial contractility and increases ventricular wall thickness, favoring a concentric hypertrophy–like adaptation (121, 122). Supporting this, a longitudinal study of eight elite sailors (mean age 18) undergoing seven weeks of high-intensity HIIT demonstrated increased LVM and left ventricular wall thickness with minimal ventricular dilation, indicating the development of concentric hypertrophy. The extent of hypertrophy was significantly associated with total training volume and duration (123). Finally, the type and magnitude of exercise-induced PCH are modulated by individual factors, including age, sex, training history, and ethnicity. These findings highlight the importance of optimizing the ratio of endurance to resistance training in mixed exercise programs to maximize adaptive cardiac remodeling.
.3.3 Exercise intensity and PCH
Exercise intensity is commonly classified as low, moderate, or high, with each level exerting distinct physiological effects on the development of PCH. Low-intensity exercise is generally defined as physical activity that maintains heart rate at 50%–60% of an individual's maximum heart rate. This level of exertion is unlikely to induce significant cardiac remodeling and thus does not typically lead to the development of PCH. It is, however, considered appropriate for older adults or individuals with limited exercise tolerance and reduced physical capacity (124).
Moderate-intensity exercise is typically defined as activity that elevates heart rate to 60%–75% of an individual's maximum. This level of exertion increases venous return and cardiac preload, promoting volume overload and consequently inducing eccentric hypertrophy—characterized primarily by ventricular chamber dilation with minimal wall thickening. Moderate-intensity exercise is widely regarded as optimal for enhancing aerobic capacity and cardiorespiratory function and is considered the most effective stimulus for physiological eccentric hypertrophy (125, 126). In a study involving 12 participants (7 males and 5 females), a year-long moderate-intensity endurance training program was implemented. Cardiac magnetic imaging (CMI) was performed every three months to assess LVM, right ventricular mass (RVM), LVEDV, and right ventricular end-diastolic volume (RVEDV). The results demonstrated progressive increases in both cardiac mass and volume over the training period, indicating the development of eccentric hypertrophy (127). Experimental studies support this notion. After 8 weeks of moderate-intensity endurance training, rats in the exercise group exhibited significant increases in left and right ventricular chamber volumes and the LVM, with minimal changes in ventricular wall thickness—indicating the development of eccentric hypertrophy (128). Similarly, Fernandes et al. reported that rats subjected to 10 weeks of moderate-intensity swimming displayed load-induced eccentric remodeling of the myocardium (126). The underlying mechanism is believed to involve enhanced myocardial protein synthesis and suppressed protein degradation, leading to increased accumulation of myofibrils and cellular organelles within cardiomyocytes. These changes contribute to the enlargement of myocardial cells and the development of eccentric hypertrophy (129, 130). Additionally, moderate-intensity exercise promotes myocardial angiogenesis, improving coronary perfusion and facilitating the delivery of oxygen and nutrients to cardiomyocytes—thereby synergistically supporting cardiac growth and remodeling (131).
High-intensity exercise is defined as an activity that raises the heart rate above 75% of the individual's maximum, typically involving short-duration, high-energy expenditure efforts. This type of exercise exerts a dual effect on the heart, inducing both adaptive and potentially maladaptive changes (132). One prominent physiological adaptation is the development of concentric hypertrophy, marked by increased left ventricular wall thickness with minimal or no change in chamber volume (7). A study conducted by the Cellular Medicine Research Institute at Newcastle University examined the impact of HIIT on cardiac structure and function in patients with type 2 diabetes. After 12 weeks of HIIT or pharmacologic intervention, patients in the HIIT group exhibited greater left ventricular wall thickening, increased end-diastolic diameter, and improved myocardial contractility and relaxation compared to the pharmacologic treatment group, indicating the induction of concentric hypertrophy by HIIT (133). High-intensity exercise places substantial demands on cardiac output, stimulating robust protein synthesis in cardiomyocytes, which facilitates cellular growth and suppresses apoptosis (134). Furthermore, this training modality enhances myocardial energy metabolism, thereby contributing to the development of PCH (26). Nevertheless, prolonged or excessive acute high-intensity exercise may lead to electrical remodeling and right ventricular diastolic dysfunction, which could predispose individuals to atrial and ventricular arrhythmias (135) (Table 3).
Table 3
| Exercise intensity | Characteristics | Structural alteration | Physiological function changes | References |
|---|---|---|---|---|
| Low Intensity | Heart rate = 50–60% of maximum heart rate | No significant changes | Improved cardiovascular function, increased pumping capacity | (124, 136, 137) |
| Moderate Intensity | Heart rate = 60%–75% of maximum heart rate | Eccentric hypertrophy | Increased cardiac reserve, improved myocardial contractility | (125, 126, 129, 130) |
| High Intensity | Heart rate ≥75% of maximum heart rate | Concentric hypertrophy | Increased energy metabolism efficiency, significant improved pumping capacity in a short time | (7, 26, 132, 134) |
PCH induced by different exercise intensities.
4 Mechanisms of exercise-induced PCH
4.1 Transcriptional regulation
4.1.1 The IGF-1/PI3K/Akt signaling pathway
During exercise-induced PCH, the IGF-1/ PI3K/Akt signaling pathway plays a central regulatory role. Activation of this pathway is associated with increased myofibril number, improved stroke volume (SV), and enhanced maximal oxygen uptake (VO2 max) (36). IGF-1, a polypeptide hormone structurally analogous to insulin, is primarily synthesized and secreted by the liver in response to growth hormone stimulation. It exerts multiple systemic effects, including the reduction of blood glucose and lipid levels (138). Studies have demonstrated that myocardial IGF-1 levels are elevated in athletes with PCH compared to those without hypertrophy, and serum IGF-1 concentrations increase following exercise, implicating IGF-1 as a key mediator in exercise-induced cardiac remodeling (139). During mechanical loading from exercise, mechanoreceptors in cardiomyocytes are activated, promoting IGF-1 binding to Insulin-like Growth Factor 1 Receptor (IGF-1R). This interaction triggers receptor autophosphorylation and subsequent recruitment of Insulin Receptor Substrate (IRS) proteins, which form a signaling complex that initiates PI3K activation (140).
The PI3K family comprises enzymes that phosphorylate phosphoinositide and are essential regulators of various cellular processes, including proliferation and apoptosis (141). In a study involving four weeks of swimming training in mice, those with reduced PI3K expression exhibited significantly attenuated PCH compared to controls, highlighting PI3K's critical role in exercise-induced cardiac remodeling (8). PI3K catalyzes the conversion of Phosphatidylinositol 4,5-bisphosphate (PIP2) to Phosphatidylinositol (3,4,5)-trisphosphate (PIP3) at the cell membrane. As a secondary messenger, PIP3 facilitates the membrane recruitment of Akt and Pyruvate Dehydrogenase Kinase 1 (PDK1) (142). Akt activation requires phosphorylation at T308 by PDK1 and at S473 by Mammalian target of rapamycin protein complex 2 (mTORC2), representing a pivotal step in the downstream signaling cascade. Evidence from an 8-week swimming protocol in rats showed a marked increase in phosphorylated Akt levels in myocardial tissue compared to sedentary controls. This activation of the PI3K/Akt/mTORC signaling axis was associated with the induction of PCH in response to exercise (143).
After Akt activation, it phosphorylates Tuberous Sclerosis Complex 2 (TSC2), thereby inhibiting its suppressive effect on Ras Homolog Enriched in Brain-GTP (Rheb-GTP). This disinhibition allows Rheb-GTP to bind and activate the mTORC1 (144, 145). mTORC1 is a central serine/threonine kinase complex that phosphorylates key downstream effectors, including ribosomal protein S6 kinase 1 (S6K1) and eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1) (146). Phosphorylated S6K1 enhances ribosomal biogenesis and protein translation, thereby increasing intracellular protein synthesis, promoting cardiomyocyte hypertrophy, and facilitating the progression of PCH (147). Xu et al. further demonstrated that mTORC1 promotes protein biosynthesis primarily through 4E-BP1 phosphorylation (148). Once phosphorylated, 4E-BP1 detaches from eukaryotic translation initiation factor 4E (eIF4E), allowing eIF4E to interact with additional initiation factors to form the translation initiation complex. This process accelerates protein synthesis and leads to an accumulation of myofibrils and organelles within myocardial cells, laying the structural foundation for PCH development (144, 149).It is noteworthy that the activation level of the IGF-1/PI3K/Akt signaling pathway in response to exercise load is influenced by gender differences. Reports indicate that during exercise-induced PCH, female athletes typically exhibit lower activation levels of the IGF-1/PI3K/Akt pathway compared to their male counterparts, though the activation in females is more sustained (150, 151). This may be due to differences in myocardial cell responses to mechanical load and hormonal stimuli between genders.
4.1.2 The NRG1/ErbB signaling pathway
Neuregulin 1 (NRG1) is an epidermal growth factor-like protein that promotes cardiomyocyte proliferation and hypertrophy while inhibiting apoptosis, playing a critical role in cardiac development and the maintenance of cardiac function (152, 153). Studies have shown that long-term aerobic exercise increases blood shear stress, which stimulates endothelial cells in the heart to secrete NRG1. The EGF-like domain of NRG1 binds to Erythroblastic Leukemia Viral Oncogene Homolog 4 (ErbB4) on the cell membrane, promoting the formation of ErbB4/ Erythroblastic Leukemia Viral Oncogene Homolog 2 (ErbB2) heterodimers (154). This process recruits downstream molecules such as Akt to the membrane and activates them. Through the PI3K/Akt/mTORC1 signaling pathway, it promotes the biosynthesis of proteins and ribosomes, facilitating myocardial cell hypertrophy and limited proliferation, thereby contributing to the development of PCH (155, 156). Additionally, studies have shown that, compared to younger athletes, older athletes exhibit significantly reduced expression of NRG1 and lower activity of the ErbB4 receptor in myocardial cells, which impacts hypertrophy and proliferation. This reduction is likely due to aging-related decline in myocardial cell responsiveness to load stimuli (157, 158).
Glycogen synthase kinase-3 beta (GSK-3β) is a multifunctional serine/threonine kinase involved in diverse cellular signaling pathways (159, 160). Following Akt activation, phosphorylation of GSK-3β at the Ser9 residue leads to suppression of its kinase activity, thereby attenuating the degradation of Beta-catenin (β-cat). The stabilized β-catenin accumulates in the cytoplasm and subsequently translocases into the nucleus, where it interacts with transcription factors such as T-cell factor (TCF) and lymphoid enhancer-binding factor (LEF). This complex activates transcription of downstream genes including Cellular Myelocytomatosis oncogene (c-Myc) and Cyclin D1 (CCND1), thereby facilitating G1/S phase cell cycle progression and promoting cardiomyocyte growth and proliferation. This signaling cascade plays a pivotal role in exercise-induced PCH (161–163). Moreover, Akt has been shown to phosphorylate FOXO transcription factors at specific serine/threonine sites, promoting their association with 14-3-3 proteins and consequent nuclear export. This translocation suppresses the transcription of pro-apoptotic genes such as Bcl-2 interacting cell death mediator (Bim) and Fas ligand (FasL), thereby inhibiting apoptosis and contributing to the preservation of cardiomyocyte viability. Together, these mechanisms support adaptive cardiac remodeling in response to exercise stimuli (73, 164).
4.1.3 The Hippo-YAP signaling pathway
The biological output of the Hippo-YAP signaling pathway is primarily mediated through the transcriptional regulation of downstream target genes by Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ). This pathway is critically involved in the development of exercise-induced PCH (165, 166). Under homeostatic conditions, the Hippo pathway remains constitutively active. Specifically, mammalian sterile 20-like kinases 1 and 2 (MST1/2) form a complex with Salvador homolog 1 (SAV1), which facilitates the phosphorylation and activation of large tumor suppressor kinases 1 and 2 (LATS1/2). Activated LATS1/2 subsequently phosphorylates YAP and TAZ, promoting their interaction with 14-3-3 proteins and sequestration in the cytoplasm. This cytoplasmic retention prevents YAP/TAZ from entering the nucleus and engaging in transcriptional activation of genes associated with cell proliferation and survival, thereby inhibiting cardiomyocyte growth and proliferation under non-stress conditions (167, 168).
In response to exercise-induced mechanical and biochemical cues, cardiomyocytes activate Rho-associated coiled-coil containing protein kinase (ROCK). Phosphorylated ROCK negatively regulates the Hippo pathway by inhibiting MST1/2 activity, thereby attenuating the activation of LATS1/2 kinases (169). Furthermore, exercise-induced mechanical stress, such as cardiomyocyte stretching, triggers cytoskeletal remodeling and the release of mechanosensitive signals that reduce LATS1/2 phosphorylation levels (170). As a result, the inactivation of LATS1/2 decreases phosphorylation at key regulatory sites on YAP (Ser127) and TAZ (Ser89), facilitating their nuclear translocation (171). Once translocated into the nucleus, YAP and TAZ interact with specific transcription factors to initiate the expression of target genes associated with cardiomyocyte growth and proliferation, including connective tissue growth factor (CTGF) and cysteine-rich angiogenic inducer 61 (CYR61) (172). These YAP/TAZ downstream effectors play distinct yet complementary roles in cardiac remodeling. CTGF contributes to creating a supportive extracellular environment for cardiomyocyte expansion, while CYR61 modulates cellular processes such as proliferation and survival, collectively promoting hypertrophic and proliferative responses in myocardial tissue and thereby facilitating the development of PCH (173, 174). Furthermore, exercise-induced activation of the IGF-1/PI3K/Akt signaling pathway has been reported to enable Akt to phosphorylate MST1/2 or inhibit LATS1/2 activity, resulting in decreased phosphorylation of YAP. This modulation initiates a cascade of downstream signaling events, which promotes myocardial cell growth and enhances cardiac contractility, thereby contributing to the development of PCH (84). It is important to note that with aging, the sensitivity of myocardial cells to mechanical stress gradually decreases, leading to diminished nuclear translocation of YAP. Consequently, older individuals often require higher-intensity load stimulation to induce PCH comparable to that of younger individuals (175).
4.1.4 The C/EBP β-CITED4 signaling pathway
Exercise, through mechanical stress and metabolic regulation, activates intracellular signaling pathways such as IGF-1/PI3K/Akt and MAPK. These pathways subsequently activate downstream transcription factors, including cAMP response element binding protein (CREB). The cascade of downstream signaling molecules triggered by these transcription factors promotes protein synthesis within myocardial cells and increases myofibril number, playing a crucial role in the development of PCH (176). CREB has been reported to bind to the promoter region of the CCAAT/enhancer-binding protein beta (C/EBPβ) gene, leading to transcriptional repression of C/EBPβ mRNA expression (177). C/EBPβ is a transcription factor known to modulate genes involved in cell proliferation and differentiation (178). Experimental data indicate that aerobic exercise reduces C/EBPβ expression in cardiac tissue. For instance, after two weeks of aerobic training, mice exhibited significant downregulation of cardiac C/EBPβ. Moreover, cardiomyocyte-specific knockout of C/EBPβ in neonatal rat models promoted cardiomyocyte hypertrophy and proliferation (131). Comparable gene expression alterations have also been documented in zebrafish, mirroring the responses observed in murine models. In particular, Molkentin and colleagues demonstrated that swimming exercise suppressed C/EBPβ expression in zebrafish hearts, thereby enhancing cardiomyocyte growth and proliferation, promoting mild PCH, and increasing cardiomyocyte numbers following targeted C/EBPβ deletion (179). Similarly, endurance exercise-induced PCH in mice was associated with reduced C/EBPβ expression (180). Collectively, these findings support the notion that exercise-mediated downregulation of C/EBPβ serves as a critical mechanism contributing to cardiomyocyte proliferation and the adaptive remodeling characteristic of PCH.
Under resting conditions, C/EBPβ suppresses the transcriptional activity of the CBP/p300-interacting trans activator with ED-rich tail 4 (CITED4) gene by directly binding to its promoter region or by recruiting transcriptional repressors such as histone deacetylases (HDACs) (13, 181). CITED4 functions as a transcriptional co-activator, interacting with co-activators like CBP/p300 to enhance the expression of downstream target genes (182). Exercise-induced downregulation of C/EBPβ expression has been shown to alleviate this repression, thereby facilitating the upregulation of CITED4. The increased expression of CITED4 subsequently promotes cardiomyocyte proliferation and hypertrophy, inhibits apoptotic signaling pathways, and contributes to the development of PCH (107, 180). Furthermore, the regulation of the IGF-1/PI3K/Akt pathway has been shown to exhibit racial differences. Compared to European populations, East Asian populations have a lower activation threshold of CITED4 under exercise load due to genetic factors, requiring only moderate-intensity exercise to induce cardiac chamber enlargement. In contrast, African populations require higher-intensity exercise to effectively downregulate C/EBP β expression and produce significant regulatory effects (183–185). These differences highlight the need for personalized exercise protocols that consider genetic background to optimize cardiac adaptive remodeling.
4.1.5 The CaN/NFAT signaling pathway
Mechanical stress imposed on cardiomyocytes during exercise leads to membrane depolarization, which activates voltage-gated L-type calcium channel (VGCC). This facilitates the influx of extracellular Ca2+, subsequently triggering ryanodine receptor (RyR) activation on the sarcoplasmic reticulum and promoting a substantial release of stored Ca2+ into the cytoplasm. As a result, intracellular Ca2+ concentrations rise markedly (186, 187). This calcium surge serves as a central initiator of the calcineurin–nuclear factor of activated T-cells (CaN/NFAT) signaling pathway (188). Ca2+ binds to CaM, forming a Ca2+/CaM complex that activates CaN, a calcium/calmodulin-dependent phosphatase. Activated CaN dephosphorylates serine residues on NFAT proteins, inducing a conformational change that reveals the nuclear localization signal (NLS). This modification enables NFAT translocation into the nucleus, where it regulates the transcription of target genes involved in cardiac remodeling and hypertrophy (189, 190).
Evidence indicates that NFAT, in cooperation with the transcription factor GATA4, binds to the promoter region of the β-myosin heavy chain (β-MHC) gene to enhance its transcriptional activity. This interaction facilitates an increase in cardiomyocyte volume while preserving contractile function, thereby contributing to adaptive cardiac remodeling without inducing fibrosis—a hallmark of exercise-induced PCH (191, 192). Moreover, atrial natriuretic peptide (ANP) and BNP are cardiac-derived peptide hormones with established roles in promoting natriuresis, diuresis, and vasodilation, thereby maintaining cardiovascular and fluid homeostasis (61, 193). Interestingly, studies have shown that nuclear-localized NFAT can bind to the promoter regions of the ANP and BNP genes and suppress their transcription. This inhibitory regulation is thought to help preserve myocardial metabolic balance and structural integrity, reducing the risk of ECM accumulation, fibrosis, and pathological remodeling under conditions of physiological stress such as exercise (194).
4.2 Post-transcriptional regulation
4.2.1 RNA m6A methylation modification
During exercise-induced PCH, RNA N6-methyladenosine (m6A) methylation serves as a key regulatory mechanism. This regulation involves methyltransferases, demethylases, and downstream signaling cascades, which collectively enhance protein synthesis in myocardial cells, increase myofibril number, and promote metabolic reprogramming. m6A methylation involves the addition of a methyl group to the nitrogen at the sixth position of adenosine residues within RNA transcripts, forming N6-methyladenosine. This epitranscriptomic modification plays a fundamental role in RNA metabolism, influencing processes such as RNA stability, splicing, translation, and degradation (195, 196).
During exercise-induced PCH, regulatory pathways such as mRNA degradation and transcriptional repression contribute to the downregulation of methyltransferase like 14 (METTL14) mRNA expression, resulting in decreased METTL14 synthesis (197). Moreover, exercise modulates METTL14 protein levels via mechanisms including translational repression and enhanced protein degradation, collectively leading to an overall reduction in m6A methylation. Notably, reduced m6A levels have been shown to facilitate the degradation of Pleckstrin homology domain leucine-rich repeat protein phosphatase 2 (PHLPP2) mRNA through the YTH domain family protein 2 (YTHDF2) reader, thereby suppressing its expression (198). The downregulation of Phlpp2 alleviates its inhibitory effect on Akt activity, consequently activating the IGF-1/PI3K/Akt signaling pathway. This activation promotes myocardial hypertrophy and proliferation while inhibiting apoptosis, culminating in coordinated ventricular wall thickening and cardiac dilation, which supports the development of PCH (197). Furthermore, previous studies have demonstrated that Akt can inhibit the activity of GSK-3β. This inhibition reduces the phosphorylation of NFAT, enabling NFAT to expose its nuclear localization signal and translocate into the nucleus. Nuclear NFAT binds to DNA and upregulates the expression of pro-survival genes, including interleukin-2 (IL-2) and B-cell lymphoma-extra large (Bcl-xL), as well as factors related to myocardial proliferation such as CITED4. This cascade enhances myocardial protein synthesis and proliferation while suppressing apoptosis, playing a critical role in the physiological remodeling associated with PCH (188, 199).
In the context of exercise-induced PCH, RNA m6A methylation modification exhibits significant sex- and age-related differences in regulation. Studies have shown that, due to estrogen influence, female athletes have reduced METTL14 mRNA expression in myocardial cells at baseline, along with enhanced demethylase activity. As a result, baseline m6A methylation levels are lower in females compared to males. Through this regulatory mechanism, females are able to more effectively downregulate PHLPP2 expression after exercise, thus more efficiently activating the IGF-1/PI3K/Akt pathway to induce PCH (197). However, in older individuals, the downregulation of METTL14 mRNA during exercise is less pronounced, and compared to younger individuals, the overall reduction in m6A methylation is smaller. This limits the degradation efficiency of PHLPP2 mRNA, restricting Akt activation and downstream signal molecule expression (200). These differences in regulation suggest that females exhibit stronger metabolic adaptability, while older individuals require higher load thresholds to achieve equivalent cardiac remodeling effects as younger individuals. This finding provides molecular-level theoretical support for the development of personalized exercise programs.
4.2.2 Non-coding RNA regulation
Circular Uridine (circUtrn), a circular RNA associated with cardioprotective effects, is significantly upregulated in response to exercise-induced stress. CircUtrn enhances the activity of protein phosphatase 5 (PP5) by directly interacting with its catalytic domain (201). PP5, a serine/threonine phosphatase, is involved in critical cellular processes such as DNA damage repair and cell cycle regulation (202). Recent studies have demonstrated that PP5 activates downstream mammalian target of mTORC1, thereby relieving its inhibitory effect on CCND1. This promotes cardiomyocyte cell cycle re-entry, supporting myocardial repair and regeneration (203). This mechanism closely parallels the adaptive mTORC1 activation observed during PCH, enhancing cardiac reserve while mitigating pathological fibrosis. In addition, circUtrn functions as a molecular sponge for microRNA-195-5p (miR-195-5p), a pro-apoptotic microRNA known to suppress anti-apoptotic genes such as F-box and WD repeat domain-containing protein 7 (FBXW7) and Mitofusin 2 (MFN2) (204). By sequestering miR-195-5p, circUtrn alleviates its repression of FBXW7 and MFN2, thereby reducing cardiomyocyte apoptosis, promoting mitochondrial biogenesis, and enhancing energy metabolism. This multi-target regulatory mechanism aligns closely with the fundamental features of exercise-induced PCH, contributing to improved myocardial adaptability and function (205).
p27 negatively regulates cell cycle progression by inhibiting the Cyclin-dependent kinase 2 (CDK2)/Cyclin E complex, thereby blocking the G1-to-S phase transition (206). Studies have shown that miR-222 can bind to the 3ʹ untranslated region (3ʹ UTR) of p27 mRNA, suppressing its translation and leading to reduced p27 protein levels. This alleviation of CDK2/Cyclin E inhibition facilitates G1/S transition, promotes cardiomyocyte proliferation, and contributes to the enhancement of cardiac function associated with PCH (207). In addition to its role in cell cycle regulation, miR-222 also targets Hmbox1, a transcription factor that activates the Transforming Growth Factor-β (TGF-β)/Smad signaling pathway. This pathway promotes fibroblast-to-myofibroblast conversion and excessive ECM deposition, culminating in myocardial fibrosis (208). By binding to the 3ʹ UTR of Hmbox1 mRNA, miR-222 downregulates Hmbox1 expression, thereby inhibiting Transforming Growth Factor-β (TGF-β)/Smad protein (Smad) pathway activation and preventing fibrotic remodeling of the myocardium (209). Moreover, miR-222 indirectly upregulates α-myosin heavy chain (α-MHC) expression while suppressing β-MHC expression through the downregulation of p27 and Hmbox1. This shift increases the α-MHC/β-MHC ratio, thereby enhancing myocardial contractility, inhibiting myocardial fibrosis, and counteracting pathological cardiac remodeling (110).
4.3 Metabolic regulation
4.3.1 Fatty acid oxidation
FAO, a central pathway in myocardial energy metabolism, not only supplies energy during exercise but also modulates gene expression through its metabolic intermediates and associated signaling pathways, forming a complex regulatory network. This mechanism plays a critical role in the development of PCH, enhances exercise capacity, and delays the onset of fatigue. Upon exercise stimulation, epinephrine (EPI) binds to β-adrenergic receptors (βARs), activating adenylate cyclase (AC) and leading to the generation of cyclic adenosine monophosphate (cAMP). cAMP subsequently activates protein kinase A (PKA), a key effector in this cascade (210). Studies have shown that PKA can enhance the activity of the fatty acid transporter CD36, thereby promoting the transmembrane uptake of free fatty acids (FFAs) into cardiomyocytes (211). Once internalized, FFAs are converted into acyl-Coenzyme A (Acyl-CoA) by cytosolic acyl-CoA synthetase (ACS). Acyl-CoA is then transported into mitochondria via carnitine palmitoyl transferase 1 (CPT1), the rate-limiting enzyme of mitochondrial FAO. Within the mitochondria, Acyl-CoA undergoes β-oxidation to generate acetyl-Coenzyme A (Acetyl-CoA), as well as reduced cofactors NADH and FADH2. These products subsequently fuel the tricarboxylic acid (TCA) cycle and oxidative phosphorylation, supplying ATP to meet the high energy demands of cardiomyocytes (149, 150). Enhanced efficiency of this metabolic pathway is essential for supporting the increased energy requirements during PCH induced by exercise (212, 213). Additionally, during exercise-induced PCH, female athletes generally exhibit a higher degree of FAO metabolic dependency compared to male athletes. It has been reported that under the regulation of estrogen, activation of transcription factors such as PPARα in females enhances FAO capacity during exercise, which significantly distinguishes female myocardial cells from the glycolytic metabolism phenotype predominant in males (214). This metabolic difference aids in the enhanced mitochondrial biogenesis capacity of female athletes during long-term endurance exercise.
Additionally, accumulating evidence suggests that acetyl-Coenzyme A (Acetyl-CoA), a key metabolic intermediate and signaling molecule, plays an important role in the epigenetic regulation of gene expression during exercise-induced PCH. Elevated intracellular levels of Acetyl-CoA can activate histone acetyltransferases (HATs), such as p300, which catalyze the acetylation of histone H3 at lysine 9 (H3K9ac) and lysine 27 (H3K27ac). These histone modifications promote chromatin relaxation and transcriptional activation of growth-related genes, including GATA4 and Myocyte Enhancer Factor 2 (MEF2). Consequently, this process downregulates the expression of hypertrophy-related genes such as ANP and BNP, while inhibiting pro-fibrotic pathways like TGF-β. While promoting myocardial cell growth and proliferation, it also induces the development of PCH (215). Moreover, this epigenetic regulation supports cardiomyocyte growth and proliferation while simultaneously suppressing pro-fibrotic signaling pathways, including TGF-β, thereby facilitating the development of a physiological hypertrophic phenotype (197). Peroxisome proliferator-activated receptor alpha (PPARα), a nuclear transcription factor critical for FAO, is highly expressed in myocardial tissue. During exercise, Akt-mediated phosphorylation enhances the transcriptional activity of PPARα. Activated PPARα promotes the expression of key FAO enzymes such as CPT1 and medium-chain acyl-CoA dehydrogenase (MCAD), thereby accelerating mitochondrial FAO and supporting sustained ATP production in cardiomyocytes (216). This metabolic reprogramming shifts substrate preference toward fatty acids, improving energy efficiency while minimizing the accumulation of potentially toxic metabolic byproducts. Collectively, these molecular and metabolic adaptations contribute to the emergence of a PCH phenotype characterized by cardiomyocyte hypertrophy and proliferation, enhanced cardiac output, and resistance to pathological fibrosis.
4.3.2 Glucose metabolism
During exercise-induced PCH, multiple molecular pathways and metabolic regulatory networks are involved in glucose metabolism, supporting the energy supply and adaptive growth of myocardial cells, as well as enhancing exercise capacity. Glucose transporter type 4 (GLUT4), localized in intracellular vesicles, is the principal glucose transporter in cardiomyocytes (217). Wende et al. demonstrated that GLUT4 plays a pivotal role in the cardiac metabolic adaptation to increased hemodynamic load during exercise (218). Notably, activation of Akt signaling during exercise leads to the phosphorylation of Akt substrate of 160kDa (AS160), a Rab GTPase-activating protein. Phosphorylated AS160 loses its GAP activity, allowing GLUT4-containing vesicles to translocate to the plasma membrane. This translocation markedly increases glucose uptake by cardiomyocytes, thereby boosting ATP production to meet the immediate energy demands of the heart under exercise-induced stress (219). Additionally, Akt has been shown to inhibit the activity of GSK-3β, thereby limiting its phosphorylation of glycogen synthase (GS), resulting in the dephosphorylation and activation of GS. This process promotes the conversion of glucose into glycogen, providing a metabolic foundation for myocardial cell proliferation and hypertrophy. It also plays a positive role in protein synthesis and the delay of fatigue onset (220, 221).
Notably, Akt also promotes mitochondrial function by phosphorylating peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), a master regulator of mitochondrial biogenesis. This phosphorylation event enhances the transcription of genes involved in mitochondrial DNA replication and oxidative metabolism, thereby improving mitochondrial efficiency and facilitating the complete oxidation of glycogen-derived substrates such as pyruvate. These processes provide sustained energy support, which is essential for the development of PCH (222, 223). In addition to mitochondrial adaptations, glucose metabolism is tightly regulated during PCH. Under normal physiological conditions, Hmbox1 acts as a negative regulator of glucose metabolism by inhibiting the expression of the glucokinase gene (Gck). However, in a swimming-induced model of PCH, downregulation of Hmbox1 expression was observed during exercise, which led to the upregulation of Gck. Glucokinase catalyzes the phosphorylation of glucose to glucose-6-phosphate (G6P), the first and rate-limiting step in glycolysis. This enhances intracellular glucose uptake and utilization, ensuring a steady supply of energy substrates for cardiomyocyte metabolism. Such metabolic reprogramming is a hallmark of exercise-induced PCH and contributes to its adaptive, non-pathological nature (224) (Figure 2).
Figure 2
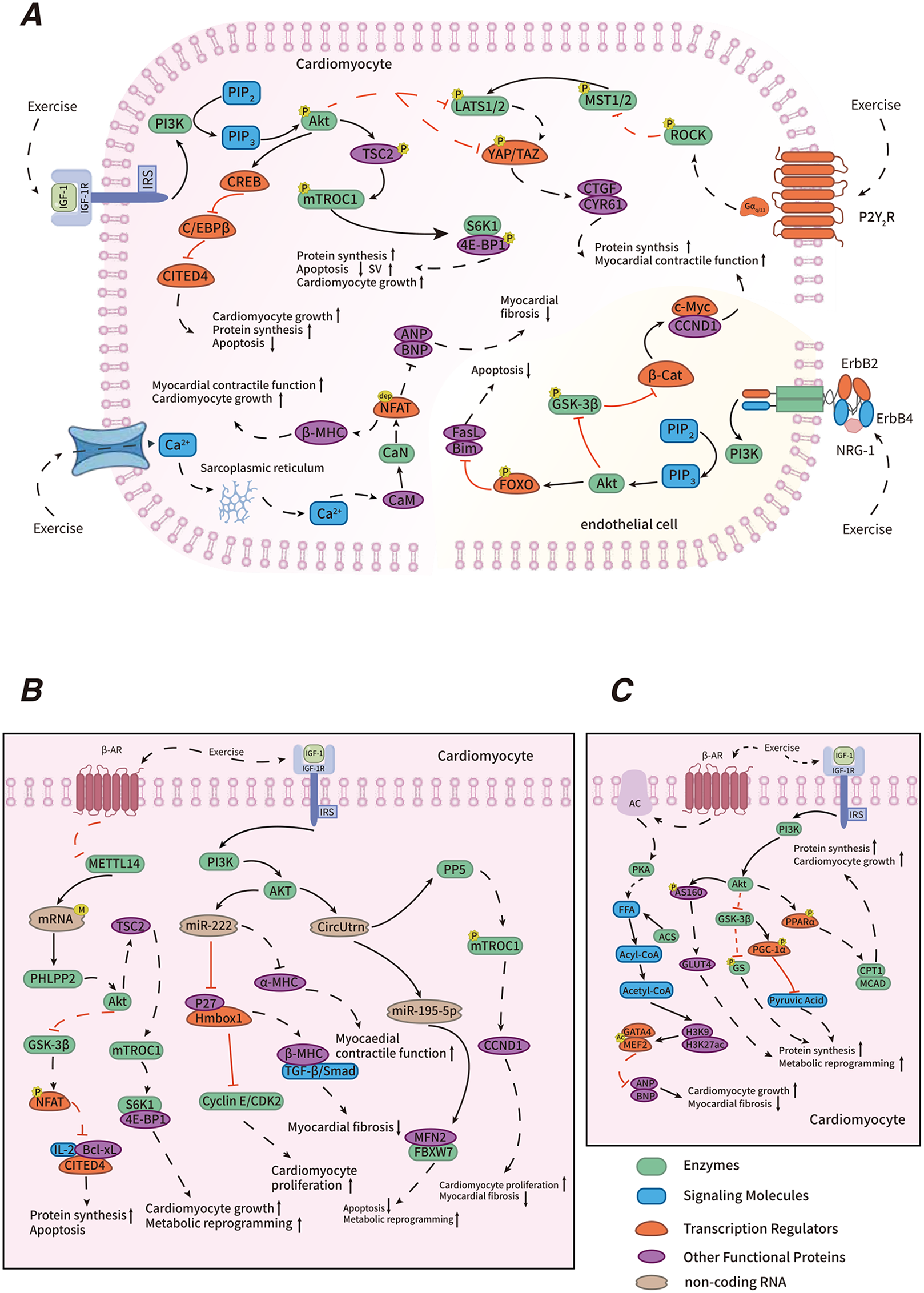
Mechanisms of exercise-induced PCH. Explanatory text: in the regulatory mechanism diagram of exercise-induced physiological cardiac hypertrophy, panel A illustrates transcriptional regulation, including the IGF-1/PI3K/Akt, NRG1/ErbB, Hippo-YAP, C/EBP β-CITED4, and CaN/NFAT signaling pathways. Panel B highlights post-transcriptional regulation, encompassing RNA m6A methylation modification and non-coding RNA regulation. Panel C depicts metabolic regulation, which includes fatty acid oxidation and glucose metabolism. Different colored entities in the diagram represent various functional biomolecules, where “P” denotes phosphorylation, “dep” denotes dephosphorylation, “M” denotes methylation, and “Ac” denotes acetylation. Solid lines indicate events that occur under normal physiological conditions, while dashed lines represent exercise-induced changes. Black lines indicate “promotion,” and red lines represent “suppression.” The symbols “ ” and “
” and “ ” indicate enhancement and attenuation, respectively.
” indicate enhancement and attenuation, respectively.
5 Positive effects of exercise-induced PCH on cardiovascular health
5.1 Improve cardiovascular health
Exercise, as a non-pharmacological intervention, supports cardiovascular health through diverse mechanisms, with PCH playing a pivotal role in this process (104). Exercise-induced PCH enhances cardiac function, a phenomenon closely linked to the remodeling of myocardial structure and the shift in MHC isoform expression. During aerobic exercise, a characteristic feature of PCH is an increased expression of α-MHC and a corresponding decrease in β-MHC expression in cardiomyocytes. Given that α-MHC possesses higher ATPase activity, this isoform switch contributes to improved contractile velocity and force generation (225). These adaptations lead to enhanced SV and a reduced resting heart rate, collectively improving circulatory efficiency and decreasing long-term cardiac workload. Additionally, studies have demonstrated that exercise-induced PCH results in increased cardiomyocyte volume and myofibril content, thereby enhancing both systolic and diastolic function. This structural remodeling enables the heart to more effectively meet the increased metabolic demands during physical activity by improving cardiac output and perfusion efficiency (115, 226). Moreover, PCH contributes to vascular benefits by improving endothelial function and reducing peripheral resistance, which supports the maintenance of stable blood pressure (107). Long-term exercise has been shown to stimulate nitric oxide (NO) release from endothelial cells, elevating intracellular cyclic guanosine monophosphate (cGMP) levels, which promotes vasodilation and decreases peripheral vascular resistance (227, 228). Moderate to high-intensity exercise, through the induction of PCH, is associated with reduced cardiovascular metabolic risk factors. These include lowered blood pressure and blood glucose levels, as well as decreased abdominal fat accumulation—factors that collectively aid in the prevention of cardiovascular diseases (107, 133).
5.2 Treatment of CVDs
Exercise-induced PCH also exerts beneficial effects in the prevention and treatment of CVD. Relevant studies have demonstrated that, after the random allocation of patients with ischemic non-obstructive coronary artery disease into groups, those in the intervention group—who underwent an 8-week personalized exercise program based on cardiopulmonary exercise testing (CPET) data—showed a significant reduction in total myocardial ischemic burden compared to the control group. Furthermore, notable improvements in cardiopulmonary function were observed in the intervention group (14). A key mediator of this protective role is IGF-1, which is essential for cardiovascular health. Clinical studies have reported that elderly individuals with reduced serum IGF-1 levels are at increased risk for heart failure. Notably, exercise-induced PCH has been shown to elevate circulating IGF-1 levels, thereby reducing oxidative stress in cardiomyocytes and improving cardiac function following myocardial infarction (229). In addition, circUtrn has emerged as a critical regulatory molecule in exercise-induced PCH, particularly in the context of myocardial ischemia-reperfusion (I/R) injury. In a mouse swimming training model, elevated circUtrn expression significantly reduced infarct size and enhanced cardiac function post-I/R injury. Conversely, suppression of circUtrn expression attenuated these protective effects, suggesting its potential as a biomarker and therapeutic target for coronary artery disease (201). Exercise-induced PCH also contributes to the modulation of cardiac electrophysiology. During PCH development, the myocardial action potential refractory period undergoes adaptive changes. It has been reported that exercise extends the effective refractory period, thereby reducing the susceptibility to ectopic excitations during this phase and lowering the risk of arrhythmia (230, 231). Furthermore, PCH promotes autonomic balance by modulating sympathetic and parasympathetic nervous system activity, which enhances cardiac rhythm stability and provides additional protection against arrhythmia (232, 233).
6 Challenges and future prospects
6.1 Molecular identification and clinical diagnosis challenges of PCH vs. PMH
Despite considerable advances in research on PCH, distinguishing it from PMH remains a major clinical and scientific challenge due to substantial overlaps in their macroscopic features and molecular profiles. Both PCH and PMH present with similar structural changes, including myocardial thickening and cardiac enlargement, and are often accompanied by comparable clinical symptoms such as palpitations, chest tightness, and shortness of breath. Consequently, differentiation based solely on gross morphology and clinical presentation is unreliable. At the molecular level, PCH and PMH share activation of key signaling pathways such as IGF-1/PI3K/Akt and MAPK, as well as similar alterations in the expression of cell cycle regulators, microRNAs, and other molecular markers. These parallels further complicate differentiation using standard molecular techniques (7, 229).
Current diagnostic modalities also have notable limitations. For instance, echocardiography, the most commonly used non-invasive imaging tool, can be affected by the heart's rhythmic motion, making it difficult to ensure consistent imaging planes and leading to measurement variability. Furthermore, echocardiography has limited spatial resolution for detecting subtle distinctions, such as differences in myocardial fiber orientation or the degree of interstitial fibrosis, which are critical for distinguishing PCH from PMH (234). Similarly, ECG offers limited diagnostic specificity. Both PCH and PMH can manifest as ST-T segment abnormalities, T-wave inversions, and signs of left ventricular hypertrophy, making it challenging to differentiate between them (235). In addition, confounding factors such as electrolyte imbalances and certain medications can induce ECG changes that mimic those seen in hypertrophy, further reducing diagnostic accuracy (236). TDI is primarily employed to evaluate myocardial diastolic function and assist in distinguishing types of MH. In the early stages of PCH, diastolic function at rest often remains normal, and early diastolic parameters in PMH may also appear unremarkable, complicating differentiation between the two. Moreover, TDI image acquisition and analysis require substantial expertise, and inter-operator variability can affect diagnostic accuracy (237). Strain Imaging (SI) also has limitations for early detection of PMH, particularly when ventricular wall thickening is minimal, as abnormal myocardial strain may not be evident. Physiological factors such as age, sex, and exercise level can further influence SI results, increasing the complexity of clinical interpretation. In addition, lack of standardization across different SI systems or manufacturers limits result comparability (238). Cardiac magnetic resonance (CMR) imaging likewise faces challenges in distinguishing MH subtypes. For example, transient metabolic enhancement may occur during early PCH, which can be difficult to differentiate from the metabolic abnormalities observed in early PMH. False-positive or false-negative findings may also arise, particularly in early or borderline cases, complicating accurate differentiation (239). Notably, following cessation of training, athletes typically exhibit gradual reductions in ventricular wall thickness, which can often be detected within a few months using three-dimensional echocardiography (3DE) and CMR. In contrast, in patients with PMH, ventricular wall thickness may remain unchanged or even progress despite exercise cessation, especially in the presence of other pathological factors (15, 240).
In clinical practice, especially when working with specialized populations such as elite athletes, there often exist ambiguous “gray zones” that are difficult to precisely define. For example, some elite athletes may have LVWT measurements in the borderline range of 13–15 mm, often accompanied by a slight increase in plasma BNP levels. This scenario can lead to diagnostic challenges (17). To address this, a novel multi-stage assessment system should be developed. For instance, if a 3DE reveals an LVWT of 13–15 mm, it should be categorized as a “gray zone.” This finding would necessitate the use of additional methods such as biomarkers, molecular markers, comprehensive assessments, and regular follow-up evaluations for multimodal assessment, thereby improving the accuracy of clinical evaluations (241, 242).
To address these challenges, future research should prioritize the development and refinement of diagnostic technologies with enhanced precision and specificity. The integration of artificial intelligence, machine learning, and big data analytics holds great promise for advancing diagnostic algorithms. These innovations could facilitate the creation of a more robust and accurate classification system for myocardial hypertrophy, ultimately supporting more effective and individualized clinical decision-making (Table 4).
Table 4
| Technique | Sensitivity | Specificity | Pattern of Manifestation | References |
|---|---|---|---|---|
| 3DE | Moderate-High | Moderate | PCH: Thickened myocardial walls, enlarged heart chamber; PMH: Non-thickened myocardial walls(e.9., significant increase in endocardial thickness) | (34, 243) |
| ECG | Low-Moderate | Low | PCH: Increase in QRS wave amplitude with no abnormalities; PMH: ST-T changes, QRS wave modification, etc., prominent abnormalities | (9, 92, 244) |
| TDI | High | Moderate-High | PCH: Normal or enhanced myocardial function. | (43, 245) |
| PMH: Decreased myocardial function. | ||||
| SI | High | High | PCH: Significant residual increase. | (238, 246, 247) |
| PMH: Decrease in response to changes, especially when compared to alterations. | ||||
| CMI | Extremely High | Extremely High | PCH: Increased serum albumin or increased egg white protein. | (48, 239) |
| PMH: Changes in collagen synthesis or degradation. |
Comparative diagnosis of different testing techniques.
6.2 Overexercise-induced decompensation and its prevention
While exercise-induced PCH offers substantial cardiovascular benefits, its potential adverse effects should not be underestimated. Emerging evidence suggests that, in the early stages of PCH, the heart undergoes adaptive remodeling through compensatory mechanisms such as ventricular wall thickening and chamber dilation. During this phase, SV and ejection fraction may increase slightly. However, these compensatory adaptations are inherently limited. When they are no longer sufficient to meet the body's demands for oxygen and circulation, the heart may enter a decompensate state. This transition is marked by pathological remodeling, including an imbalance between collagen synthesis and degradation in the ECM, leading to myocardial fibrosis and impaired diastolic function. Without timely regulation or intervention, prolonged decompensation may ultimately lead to heart failure, posing a significant threat to health and survival (248). Additionally, cardiac decompensation disrupts the distribution and function of ion channels in cardiomyocytes, altering both the velocity and pattern of intercellular electrical conduction. These disturbances can impair myocardial excitability, automaticity, and conductivity, thereby increasing susceptibility to arrhythmias (249, 250).
Preventing the progression of cardiac decompensation is therefore of paramount importance. Effective strategies include the development of individualized exercise programs, pre-exercise assessments, real-time physiological monitoring during training, and structured recovery protocols (251, 252). In the development of exercise protocols, data from CPET should be systematically analyzed and incorporated into individualized exercise load assessments. Key indicators, such as VO2 max and anaerobic threshold, along with individual factors including age, health status, exercise capacity, and fitness goals, can be used to establish precise exercise intensity thresholds. Integration of genetic and metabolomic analyses may further inform individual responses to different exercise modalities, allowing for the precise tailoring of exercise programs. Exercise interventions should follow recommended frequencies and durations, with progressive increases in load to ensure safe adaptation (253, 254). Prior to implementation, a comprehensive evaluation of the individual's physical condition—including ECG, echocardiography, and other diagnostic assessments—provides a baseline understanding of cardiac function and overall health, forming the foundation for scientifically designed exercise protocols. During training, dynamic monitoring using wearable devices, which track lactate concentration measurement and continuous blood pressure, can provide real-time insights into autonomic nervous system function and cardiovascular load. This approach enables optimization of exercise intensity and mitigates the risk of cardiac decompensation (255, 256). Recent research has indicated that within wearable device-based health management systems, integrating physiological models with neural network methodologies for the development of personalized exercise programs can accurately predict the cardiovascular load responses of the body. This approach also allows for the quantification of environmental factors impacting heart health, such as elevated heart rate due to high-temperature and high-humidity environments. By providing real-time monitoring and assessment, this system can significantly contribute to preventing the onset of cardiac decompensation, thus enhancing overall cardiovascular health management (257). Following exercise, standardized cool-down protocols—including stretching and relaxation—are recommended to promote recovery and reduce fatigue (258). Moreover, lifestyle interventions such as optimizing nutritional intake and maintaining adequate sleep quality, in combination with pharmacological strategies, when necessary, can further mitigate the risk of decompensation (259).
Looking ahead, the implementation of personalized exercise prescriptions tailored to individual characteristics and needs will be essential for maximizing the cardiovascular benefits of physical activity while minimizing the risks. Concurrently, deeper investigation into the molecular mechanisms underlying cardiac decompensation will be critical for identifying novel therapeutic targets and developing targeted pharmacologic interventions, thereby advancing the precision prevention and management of exercise-induced cardiac injury.
7 Conclusion
Exercise-induced PCH represents an adaptive remodeling process reflecting the coordinated optimization of cardiac structure and function in response to regular exercise. This review systematically delineates the differential impacts of various exercise modalities on cardiac morphology and performance: endurance exercise predominantly induces eccentric hypertrophy via volume overload, whereas resistance exercise promotes concentric hypertrophy through pressure overload. The development of PCH is orchestrated by a complex interplay of regulatory mechanisms, including transcriptional, post-transcriptional, and metabolic pathways. Importantly, PCH exhibits both preventive and therapeutic potential for CVDs, such as hypertension and heart failure, by enhancing cardiac pump efficiency, optimizing energy metabolism, and mitigating myocardial fibrosis.
Despite these advances, several challenges remain. First, distinguishing PCH from PMH requires overcoming the limitations of current imaging modalities and biomarkers, necessitating the creation of precise, stratified diagnostic frameworks based on multimodal data integration. Second, the potential risk of cardiac decompensation induced by excessive exercise underscores the urgent need for a personalized, dynamic evaluation system for exercise intensity. Such a system should integrate genetic profiling, metabolic phenotyping, and real-time biosensor data to prevent adverse consequences like myocardial fibrosis and arrhythmias. Future research should prioritize elucidating the dose–response thresholds of exercise on cardiac adaptive remodeling and dissecting the molecular pathways that precipitate cardiac decompensation. These efforts may facilitate a paradigm shift from generalized “personalized exercise prescriptions” toward strategies aimed at “maximizing heart health,” thereby advancing cardiovascular wellness on a broader scale.
Statements
Author contributions
PC: Writing – review & editing, Writing – original draft, Investigation. XZ: Investigation, Conceptualization, Writing – original draft, Supervision, Writing – review & editing, Project administration. YS: Writing – original draft, Investigation. QY: Investigation, Writing – review & editing. LC: Supervision, Writing – review & editing. QR: Supervision, Writing – review & editing. CC: Supervision, Writing – review & editing. HX: Writing – review & editing. LL: Investigation, Writing – review & editing. HR: Writing – review & editing, Investigation. ZZ: Writing – review & editing. YW: Conceptualization, Writing – review & editing, Project administration, Funding acquisition, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Scientific Research Project of Hubei Provincial Department of Education (Grant No. T2024036).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
- α-MHC
α-Myosin heavy chain
- β-cat
β-catenin
- β-MHC
β-Myosin heavy chain
- βAR
β adrenergic receptor
- 3'UTR
3' untranslated region
- 4E-BP1
Eukaryotic translation initiation factor 4E binding protein 1
- AC
Adenyl cyclase
- Acetyl-coA
Acetyl-Coenzyme A
- ACS
Acyl-coA synthetase
- Acyl-CoA
Acyl-coenzyme A
- Akt
protein kinase B
- ANP
atrial natriuretic peptide
- AS160
Akt substrate of 160 kDa
- Bcl-xL
B-cell iymphoma-extra large
- Bim
B-cell lymphoma 2-interacting cell death mediator
- BNP
brain natriuretic peptide
- c-Myc
cellular myelocytomatosis oncogene
- C/EBP β
CCAAT/enhancer binding protein beta
- CaM
Calmodulin
- cAMP
cyclic adenosine monophosphate
- CaN
Calcineurin
- CCND1
Cyclin D1
- CDK2
cyclin-dependent kinase 2
- circUtrn
circular uridine
- CITED4
CBP/p300-interacting trans activator with ED-rich tail 4
- CMR
cardiac magnetic resonance imaging
- CPET
cardiopulmonary exercise testing.
- CPT1
carnitine palmitoyltransferase 1
- CREB
cAMP response element binding protein
- CTGF
connective tissue growth factor
- cTnC
Troponin C
- cTnI
Troponin I
- cTnT
Troponin T
- CVD
cardiovascular diseases
- CYR61
Cysteine-Rich angiogenic inducer 61
- ECG
electrocardiogram
- ECM
extracellular matrix
- eIF4E
Eukaryotic translation initiation factor 4E
- EPI
epinephrine
- ErbB2
erythroblastic leukemia viral oncogene homolog 2
- ErbB4
erythroblastic leukemia viral oncogene homolog 4
- FAO
fatty acid oxidation
- FasL
Fas Ligand
- FBXW7
F-box and WD repeat domain-containing protein 7
- FFA
free fatty acids
- FOXO
forkhead box O
- G6P
Glucose-6-Phosphate
- Gal-3
Galectin-3
- GATA4
GATA binding protein 4
- Gck
glucokinase gene
- GH
growth hormone
- GLUT4
glucose transporter type 4
- GS
glycogen synthase
- GSK-3β
glycogen synthase kinase-3 beta
- H3K27ac
acetylation of histone H3 at lysine 27
- H3K9ac
acetylation of histone H3 at lysine 9
- HATs
histone acetyltransferase
- HDACs
histone deacetylases
- HIIT
High-intensity Interval Training
- Hmbox1
homeobox containing 1
- IGF-1
insulin-like growth factor 1
- IGF-1R
insulin-like growth factor 1 receptor
- IL-2
Interleukin-2
- IRS
insulin receptor substrate
- LATS1/2
large tumor suppressor kinases 1 and 2
- LEF
lymphoid enhancer factor
- LVEDV
left ventricular end-diastolic volume
- LVEF
left ventricular ejection fraction
- LVFP
left ventricular filling pressure
- LVM
left ventricular mass
- LVMI
left ventricular mass index
- LVST
interventricular septal thickness
- LVWT
left ventricular wall thickness
- m6A
N6-methyladenosine
- MAPK
mitogen activated protein kinase
- MCAD
medium-chain acyl-coA dehydrogenase
- MEF2
myocyte enhancer factor 2
- METTL14
Methyltransferase like 14
- MFN2
Mitofusin 2
- MH
myocardial hypertrophy
- miR-195-5p
MicroRNA-195-5p
- MMP-9
matrix metalloproteinase-9
- MST1/2
mammalian sterile 20-like kinase 1/2
- mTORC1
mammalian target of rapamycin protein complex 1
- mTORC2
mammalian target of rapamycin protein complex 2
- NFAT
nuclear factor of activated T-cells
- NLS
nuclear localization signal
- NRG1
Neuregulin 1
- p27
cyclin-dependent kinase inhibitor 1B
- PCH
physiological cardiac hypertrophy
- PDK1
pyruvate dehydrogenase kinase 1
- PGC-1α
peroxisome proliferator-activated receptor gamma coactivator 1-Alpha
- PHLPP2
Pleckstrin homology domain leucine - rich repeat protein phosphatase 2
- PI3K
Phosphatidylinositol 3-Kinase
- PIIINP
procollagen type III N-Terminal propeptide
- PINP
procollagen type I N-Terminal propeptide
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PIP3
phosphatidylinositol (3,4,5)-trisphosphate
- PKA
protein kinase A
- PMH
pathological myocardial hypertrophy
- PP5
protein phosphatase 5
- PPARα
peroxisome proliferator-activated receptor alpha
- Rheb GTP
Ras homolog enriched in brain guanosine triphosphate
- ROCK
rho-associated coiled-coil containing protein kinase
- RVEF
right ventricular ejection fraction
- RyR
ryanodine receptor
- S6K1
ribosomal protein S6 kinase 1
- SAV1
Salvador homolog 1
- SI
strain imaging
- SV
stroke volume
- TAZ
transcriptional coactivator with PDZ-binding motif
- TCF
transcription factor 4
- TF
transcription factor
- TGF-β
transforming growth factor-β
- TSC2
Tuberous Sclerosis Complex 2
- VGCC
voltage-gated L-type calcium channel
- YAP
yes-associated protein
- YTHDF2
YTH domain family member 2
References
1.
Dunstan DW Dogra S Carter SE Owen N . Sit less and move more for cardiovascular health: emerging insights and opportunities. Nat Rev Cardiol. (2021) 18(9):637–48. 10.1038/s41569-021-00547-y
2.
D'Onofrio G Kirschner J Prather H Goldman D Rozanski A . Musculoskeletal exercise: its role in promoting health and longevity. Prog Cardiovasc Dis. (2023) 77:25–36. 10.1016/j.pcad.2023.02.006
3.
Isath A Koziol KJ Martinez MW Garber CE Martinez MN Emery MS et al Exercise and cardiovascular health: a state-of-the-art review. Prog Cardiovasc Dis. (2023) 79:44–52. 10.1016/j.pcad.2023.04.008
4.
Kunutsor SK Laukkanen JA . Physical activity, exercise and adverse cardiovascular outcomes in individuals with pre-existing cardiovascular disease: a narrative review. Expert Rev Cardiovasc Ther. (2024) 22(1–3):91–101. 10.1080/14779072.2024.2328644
5.
Tham YK Bernardo BC Ooi JY Weeks KL McMullen JR . Pathophysiology of cardiac hypertrophy and heart failure: signaling pathways and novel therapeutic targets. Arch Toxicol. (2015) 89(9):1401–38. 10.1007/s00204-015-1477-x
6.
Oldfield CJ Duhamel TA Dhalla NS . Mechanisms for the transition from physiological to pathological cardiac hypertrophy. Can J Physiol Pharmacol. (2020) 98(2):74–84. 10.1139/cjpp-2019-0566
7.
Nakamura M Sadoshima J . Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. (2018) 15(7):387–407. 10.1038/s41569-018-0007-y
8.
Bass-Stringer S Tai CMK McMullen JR . IGF1-PI3K-induced physiological cardiac hypertrophy: implications for new heart failure therapies, biomarkers, and predicting cardiotoxicity. J Sport Health Sci. (2021) 10(6):637–47. 10.1016/j.jshs.2020.11.009
9.
D’Ascenzi F Fiorentini C Anselmi F Mondillo S . Left ventricular hypertrophy in athletes: how to differentiate between hypertensive heart disease and athlete’s heart. Eur J Prev Cardiol. (2021) 28(10):1125–33. 10.1177/2047487320911850
10.
Lovic D Narayan P Pittaras A Faselis C Doumas M Kokkinos P . Left ventricular hypertrophy in athletes and hypertensive patients. J Clin Hypertens. (2017) 19(4):413–7. 10.1111/jch.12977
11.
Barry SP Davidson SM Townsend PA . Molecular regulation of cardiac hypertrophy. Int J Biochem Cell Biol. (2008) 40(10):2023–39. 10.1016/j.biocel.2008.02.020
12.
da Rocha AL Teixeira GR Pinto AP de Morais GP Oliveira LD de Vicente LG et al Excessive training induces molecular signs of pathologic cardiac hypertrophy. J Cell Physiol. (2018) 233(11):8850–61. 10.1002/jcp.26799
13.
Shimizu I Minamino T . Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol. (2016) 97:245–62. 10.1016/j.yjmcc.2016.06.001
14.
Yang D Liu HQ Liu FY Guo Z An P Wang MY et al Mitochondria in pathological cardiac hypertrophy research and therapy. Front Cardiovasc Med. (2022) 8:822969. 10.3389/fcvm.2021.822969
15.
Danielian A Shah AB . Differentiating physiology from pathology the gray zones of the athlete’s heart. Clin Sports Med. (2022) 41(3):425–40. 10.1016/j.csm.2022.02.005
16.
Palmisano A Darvizeh F Cundari G Rovere G Ferrandino G Nicoletti V et al Advanced cardiac imaging in athlete’s heart: unravelling the grey zone between physiologic adaptation and pathology. Radiol Med. (2021) 126(12):1518–31. 10.1007/s11547-021-01411-2
17.
Rashdan L Hodovan J Masri A . Imaging cardiac hypertrophy in hypertrophic cardiomyopathy and its differential diagnosis. Curr Opin Cardiol. (2023) 38(5):397–404. 10.1097/Hco.0000000000001070
18.
Shahzadi SK Naidoo N Alsheikh-Ali A Rizzo M Rizvi AA Santos RD et al Reconnoitering the role of long-noncoding RNAs in hypertrophic cardiomyopathy: a descriptive review. Int J Mol Sci. (2021) 22((17):9378. 10.3390/ijms22179378
19.
Qian JN Zhang JJ Cao J Wang X Zhang W Chen XF . The regulatory effect of receptor-interacting protein kinase 3 on CaMKIIδ in TAC-induced myocardial hypertrophy. Int J Mol Sci. (2023) 24(19):14529. 10.3390/ijms241914529
20.
Yuan ML Zhao B Jia HP Zhang C Zuo XW . Sinomenine ameliorates cardiac hypertrophy by activating Nrf2/ARE signaling pathway. Bioengineered. (2021) 12(2):12778–88. 10.1080/21655979.2021.2000195
21.
Zhang FL Zhou HX Xue JF Zhang YM Zhou LP Leng JW et al Deficiency of transcription factor Sp1 contributes to hypertrophic cardiomyopathy. Circ Res. (2024) 134(3):290–306. 10.1161/Circresaha.123.323272
22.
Zhao LN Cao HJ Yuan Y Liao CY Huang D Li XY et al Annexin A5 knockdown inhibits cardiomyocyte apoptosis and alleviates cardiac hypertrophy via activating the PI3K/AKT/bcl-2 signaling pathway. Sci Rep. (2024) 14(1):31915. 10.1038/s41598-024-83244-3
23.
Caturano A Vetrano E Galiero R Salvatore T Docimo G Epifani R et al Cardiac hypertrophy: from pathophysiological mechanisms to heart failure development. Rev Cardiovasc Med. (2022) 23(5):165. 10.31083/j.rcm2305165
24.
Weeks KL Tham YK Yildiz SG Alexander Y Donner DG Kiriazis H et al Foxo1 is required for physiological cardiac hypertrophy induced by exercise but not by constitutively active PI3K. Am J Physiol Heart Circ Physiol. (2021) 320(4):H1470–85. 10.1152/ajpheart.00838.2020
25.
Zhu FY Li P Sheng YH . Treatment of myocardial interstitial fibrosis in pathological myocardial hypertrophy. Front Pharmacol. (2022) 13:1004181. 10.3389/fphar.2022.1004181
26.
Xiang KF Qin Z Zhang HM Liu X . Energy metabolism in exercise-induced physiologic cardiac hypertrophy. Front Pharmacol. (2020) 11:1133. 10.3389/fphar.2020.01133
27.
Kavazis AN . Pathological vs. physiological cardiac hypertrophy. J Physiol. (2015) 593(17):3767. 10.1113/JP271161
28.
Kolwicz SC Tian R . Glucose metabolism and cardiac hypertrophy. Cardiovasc Res. (2011) 90(2):194–201. 10.1093/cvr/cvr071
29.
Martin TG Juarros MA Leinwand LA . Regression of cardiac hypertrophy in health and disease: mechanisms and therapeutic potential. Nat Rev Cardiol. (2023) 20(5):347–63. 10.1038/s41569-022-00806-6
30.
Tran DH Wang ZV . Glucose metabolism in cardiac hypertrophy and heart failure. J Am Heart Assoc. (2019) 8(12):e012673. 10.1161/JAHA.119.012673
31.
Hou JL Kang YJ . Regression of pathological cardiac hypertrophy: signaling pathways and therapeutic targets. Pharmacol Ther. (2012) 135(3):337–54. 10.1016/j.pharmthera.2012.06.006
32.
Shi XM Qiu HY . New insights into energy substrate utilization and metabolic remodeling in cardiac physiological adaption. Front Physiol. (2022) 13:831829. 10.3389/fphys.2022.831829
33.
Addetia K Miyoshi T Amuthan V Citro R Daimon M Fajardo PG et al Normal values of left ventricular size and function on three-dimensional echocardiography: results of the world alliance societies of echocardiography study. J Am Soc Echocardiogr. (2022) 35((5):449–59. 10.1016/j.echo.2021.12.004
34.
Chotalia M Ali M Hebballi R Singh H Parekh D Bangash MN et al Hyperdynamic left ventricular ejection fraction in ICU patients with sepsis. Crit Care Med. (2022) 50(5):770–9. 10.1097/Ccm.0000000000005315
35.
Okamoto C Tsukamoto O Hasegawa T Hitsumoto T Matsuoka K Takashima S et al Lower B-type natriuretic peptide levels predict left ventricular concentric remodelling and insulin resistance. ESC Heart Fail. (2022) 9(1):636–47. 10.1002/ehf2.13700
36.
Bo B Zhou Y Zheng QY Wang GD Zhou K Wei JS . The molecular mechanisms associated with aerobic exercise-induced cardiac regeneration. Biomolecules. (2021) 11(1):19. 10.3390/biom11010019
37.
De Bosscher R Claeys M Dausin C Goetschalckx K Claus P Herbots L et al Three-dimensional echocardiography of the athlete’s heart: a comparison with cardiac magnetic resonance imaging. Int J Cardiovasc Imaging. (2023) 39(2):295–306. 10.1007/s10554-022-02726-5
38.
Egbe AC Miranda WR Anderson JH Pellikka PA Stephens EH Andi K et al Left ventricular adaptation to aortic regurgitation in adults with repaired coarctation of aorta. Int J Cardiol. (2023) 383:62–9. 10.1016/j.ijcard.2023.04.061
39.
Peixoto AB Bravo-Valenzuela NJ Rocha LA Araujo Junior E . Spectral Doppler, tissue Doppler, and speckle-tracking echocardiography for the evaluation of fetal cardiac function: an update. Radiol Bras. (2021) 54(2):99–106. 10.1590/0100-3984.2020.0052
40.
Kadappu KK Thomas L . Tissue Doppler imaging in echocardiography: value and limitations. Heart Lung Circ. (2015) 24(3):224–33. 10.1016/j.hlc.2014.10.003
41.
Lancellotti P Badano LP Lang RM Akhaladze N Athanassopoulos GD Barone D et al Normal reference ranges for echocardiography: rationale, study design, and methodology (NORRE study). Eur Heart J Cardiovasc Imaging. (2013) 14(4):303–8. 10.1093/ehjci/jet008
42.
Nagueh SF Smiseth OA Appleton CP Byrd BF Dokainish H Edvardsen T et al Recommendations for the evaluation of left ventricular diastolic function by echocardiography: an update from the American society of echocardiography and the European association of cardiovascular imaging. Eur Heart J Cardiovasc Imaging. (2016) 17(12):1321–60. 10.1093/ehjci/jew082
43.
Demirelli S Sam CT Ermis E Degirmenci H Sen I Arisoy A et al Long-term cardiac remodeling in elite athletes: assessment by tissue Doppler and speckle tracking echocardiography. Echocardiography. (2015) 32(9):1367–73. 10.1111/echo.12860
44.
Albaeni A Davis JW Ahmad M . Echocardiographic evaluation of the athlete’s heart. Echocardiogr J Cardiovasc Ultrasound and All Tech. (2021) 38(6):1002–16. 10.1111/echo.15066
45.
de Gregorio C Speranza G Magliarditi A Pugliatti P Andò G Coglitore S . Detraining-related changes in left ventricular wall thickness and longitudinal strain in a young athlete likely to have hypertrophic cardiomyopathy. J Sports Sci Med. (2012) 11(3):557–61. Available online at:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3737922/
46.
Zhou D Huang YQ Fu M Cai AP Tang ST Feng YQ . Prognostic value of tissue Doppler E/e’ ratio in hypertension patients with preserved left ventricular ejection fraction. Clin Exp Hypertens. (2018) 40(6):554–9. 10.1080/10641963.2017.1407332
47.
Rajiah PS Francois CJ Leiner T . Cardiac MRI: state of the art. Radiology. (2023) 307(3):e223008. 10.1148/radiol.223008
48.
Gati S Sharma S Pennell D . The role of cardiovascular magnetic resonance imaging in the assessment of highly trained athletes. JACC Cardiovasc Imaging. (2018) 11(2):247–59. 10.1016/j.jcmg.2017.11.016
49.
Prakken NH Velthuis BK Teske AJ Mosterd A Mali WP Cramer MJ . Cardiac MRI reference values for athletes and nonathletes corrected for body surface area, training hours/week and sex. Eur J Cardiovasc Prev Rehabil. (2010) 17(2):198–203. 10.1097/HJR.0b013e3283347fdb
50.
Missenard O Gabaudan C Astier H Desmots F Garnotel E Massoure PL . Absence of cardiac damage induced by long-term intensive endurance exercise training: a cardiac magnetic resonance and exercise echocardiography analysis in masters athletes. Am J Prev Cardiol. (2021) 7:100196. 10.1016/j.ajpc.2021.100196
51.
Szabo L Brunetti G Cipriani A Juhasz V Graziano F Hirschberg K et al Certainties and uncertainties of cardiac magnetic resonance imaging in athletes. J Cardiovasc Dev Dis. (2022) 9(10):361. 10.3390/jcdd9100361
52.
Aquaro GD Corsi E Todiere G Grigoratos C Barison A Barra V et al Magnetic resonance for differential diagnosis of left ventricular hypertrophy: diagnostic and prognostic implications. J Clin Med. (2022) 11(3):651. 10.3390/jcm11030651
53.
Aengevaeren VL Baggish AL Chung EH George K Kleiven O Mingels AMA et al Exercise-induced cardiac troponin elevations: from underlying mechanisms to clinical relevance. Circulation. (2021) 144(24):1955–72. 10.1161/Circulationaha.121.056208
54.
Stewart GM Yamada A Haseler LJ Kavanagh JJ Chan J Koerbin G et al Influence of exercise intensity and duration on functional and biochemical perturbations in the human heart. J Physiol. (2016) 594(11):3031–44. 10.1113/JP271889
55.
Wang X Li S Xia C Meng X Li Y Weng S et al Exercise-induced cardiac troponin elevations and cardiac ventricular dysfunction assessed by tissue Doppler echocardiography and speckle tracking among non-elite runners in Beijing marathon. J Sci Med Sport. (2024) 27(8):508–14. 10.1016/j.jsams.2024.04.005
56.
Katrukha IA Katrukha AG . Myocardial injury and the release of troponins I and T in the blood of patients. Clin Chem. (2021) 67(1):124–30. 10.1093/clinchem/hvaa281
57.
Carbone A D’Andrea A Riegler L Scarafile R Pezzullo E Martone F et al Cardiac damage in athlete’s heart: when the “supernormal” heart fails!. World J Cardiol. (2017) 9(6):470–80. 10.4330/wjc.v9.i6.470
58.
Shave R Baggish A George K Wood M Scharhag J Whyte G et al Exercise-induced cardiac troponin elevation: evidence, mechanisms, and implications. J Am Coll Cardiol. (2010) 56(3):169–76. 10.1016/j.jacc.2010.03.037
59.
Baker P Leckie T Harrington D Richardson A . Exercise-induced cardiac troponin elevation: an update on the evidence, mechanism and implications. Int J Cardiol Heart Vasc. (2019) 22:181–6. 10.1016/j.ijcha.2019.03.001
60.
Li F Yi L Yan H Wang X Nie J Zhang H et al High-sensitivity cardiac troponin T release after a single bout of high-intensity interval exercise in experienced marathon runners. J Exerc Sci Fit. (2017) 15(2):49–54. 10.1016/j.jesf.2017.08.001
61.
Alcidi G Goffredo G Correale M Brunetti ND Iacoviello M . Brain natriuretic peptide biomarkers in current clinical and therapeutic scenarios of heart failure. J Clin Med. (2022) 11(11):3192. 10.3390/jcm11113192
62.
Kuwahara K . The natriuretic peptide system in heart failure: diagnostic and therapeutic implications. Pharmacol Ther. (2021) 227:107863. 10.1016/j.pharmthera.2021.107863
63.
Pahlavani HA . Exercise-induced signaling pathways to counteracting cardiac apoptotic processes. Front Cell Dev Biol. (2022) 10:950927. 10.3389/fcell.2022.950927
64.
Hosseini SM Azizi M Samadi A Talebi N Gatterer H Burtscher M . Impact of a soccer game on cardiac biomarkers in adolescent players. Pediatr Exerc Sci. (2018) 30(1):92–7. 10.1123/pes.2017-0060
65.
Pagourelias ED Giannoglou G Kouidi E Efthimiadis GK Zorou P Tziomalos K et al Brain natriuretic peptide and the athlete’s heart: a pilot study. Int J Clin Pract. (2010) 64(4):511–7. 10.1111/j.1742-1241.2009.02184.x
66.
Cirer-Sastre R Legaz-Arrese A Corbi F George K Nie J Carranza-Garcia LE et al Cardiac biomarker release after exercise in healthy children and adolescents: a systematic review and meta-analysis. Pediatr Exerc Sci. (2019) 31(1):28–36. 10.1123/pes.2018-0058
67.
Donnellan E Phelan D . Biomarkers of cardiac stress and injury in athletes: what do they mean?Curr Heart Fail Rep. (2018) 15(2):116–22. 10.1007/s11897-018-0385-9
68.
Liu H Hwang SY Lee SS . Role of galectin in cardiovascular conditions including cirrhotic cardiomyopathy. Pharmaceuticals (Basel). (2023) 16(7):971–87. 10.3390/ph16070978
69.
Cao ZQ Yu X Leng P . Research progress on the role of gal-3 in cardio/cerebrovascular diseases. Biomed Pharmacother. (2021) 133:111066. 10.1016/j.biopha.2020.111066
70.
Gajovic N Markovic SS Jurisevic M Jovanovic M Arsenijevic N Mijailovic Z et al Galectin-3 as an important prognostic marker for COVID-19 severity. Sci Rep. (2023) 13(1):1460. 10.1038/s41598-023-28797-5
71.
Le Goff C Farre Segura J Dufour P Kaux JF Cavalier E . Intense sport practices and cardiac biomarkers. Clin Biochem. (2020) 79:1–8. 10.1016/j.clinbiochem.2020.02.008
72.
Zhang Y Xiong X Wang J Guo F . Prognostic value of serum IGF-1, gal-3, and PTX-3 levels in elderly patients with chronic heart failure. Am J Transl Res. (2024) 16(4):1393–400. 10.62347/ZOMD7815
73.
Guo SC Mangal R Dandu C Geng XK Ding YC . Role of forkhead box protein O1 (FoxO1) in stroke: a literature review. Aging Dis. (2022) 13(2):521–33. 10.14336/Ad.2021.0826
74.
Hattasch R Spethmann S de Boer RA Ruifrok WP Schattke S Wagner M et al Galectin-3 increase in endurance athletes. Eur J Prev Cardiol. (2014) 21 (10):1192–9. 10.1177/2047487313492069
75.
Seropian IM Cassaglia P Miksztowicz V González GE . Unraveling the role of galectin-3 in cardiac pathology and physiology. Front Physiol. (2023) 14:1304735. 10.3389/fphys.2023.1304735.
76.
Sharma UC Mosleh W Chaudhari MR Katkar R Weil B Evelo C et al Myocardial and serum galectin-3 expression dynamics marks post-myocardial infarction cardiac remodelling. Heart Lung Circ. (2017) 26(7):736–45. 10.1016/j.hlc.2016.11.007
77.
Musale V Wasserman DH Kang L . Extracellular matrix remodelling in obesity and metabolic disorders. Life Metab. (2023) 2(4):155–69. 10.1093/lifemeta/load021
78.
Takawale A Sakamuri SSVP Kassiri Z . Extracellular matrix communication and turnover in cardiac physiology and pathology. Compr Physiol. (2015) 5(2):687–719. 10.1002/cphy.c140045
79.
Conings N Santens B De Meester P Troost E Claus P Moons P et al Biomarkers in transposition of the great arteries after arterial switch operation: a pilot trial with deep phenotyping. Int J Cardiol. (2024) 397:131652. 10.1016/j.ijcard.2023.131652
80.
Pettersson-Pablo P Samyn D Wasim J Vink M . Reference interval for type III procollagen (PIIINP) using the advia centaur PIIINP assay in adults and elderly. Scand J Clin Lab Invest. (2021) 81(8):649–52. 10.1080/00365513.2021.2001045
81.
Ding Y Wang Y Zhang W Jia Q Wang X Li Y et al Roles of biomarkers in myocardial fibrosis. Aging Dis. (2020) 11(5):1157–74. 10.14336/AD.2020.0604
82.
Gholipour M Tabrizi A . The role of Hippo signaling pathway in physiological cardiac hypertrophy. Bioimpacts. (2020) 10(4):251–7. 10.34172/bi.2020.32
83.
Nelson AE Ho KK . A robust test for growth hormone doping–present status and future prospects. Asian J Androl. (2008) 10(3):416–25. 10.1111/j.1745-7262.2008.00395.x
84.
Nikolov A Popovski N . Extracellular matrix in heart disease: focus on circulating collagen type I and III derived peptides as biomarkers of myocardial fibrosis and their potential in the prognosis of heart failure: a concise review. Metabolites. (2022) 12(4):297. 10.3390/metabo12040297
85.
Nishida M Mi X Ishii Y Kato Y Nishimura A . Cardiac remodeling: novel pathophysiological mechanisms and therapeutic strategies. J Biochem. (2024) 176(4):255–62. 10.1093/jb/mvae031
86.
Moreno-Sanchez PA Garcia-Isla G Corino VDA Vehkaoja A Brukamp K van Gils M et al ECG-based data-driven solutions for diagnosis and prognosis of cardiovascular diseases: a systematic review. Comput Biol Med. (2024) 172:108235. 10.1016/j.compbiomed.2024.108235
87.
Van Diepen MA Daems JJN Verwijs SM Van Hattum JC Boekholdt SM Van Randen A et al Sex-specific performance of electrocardiographic criteria for left ventricular hypertrophy in elite athletes. Heart Rhythm. (2025). 10.1016/j.hrthm.2025.05.004
88.
Lutfullin IY Kim ZF Bilalova RR Tsibulkin NA Almetova RR Mudarisova RR et al A 24-hour ambulatory ECG monitoring in assessment of QT interval duration and dispersion in rowers with physiological myocardial hypertrophy. Biol Sport. (2013) 30(4):237–41. 10.5604/20831862.1077547
89.
Abela M Sharma S . Abnormal ECG findings in athletes: clinical evaluation and considerations. Curr Treat Options Cardiovasc Med. (2019) 21(12):95. 10.1007/s11936-019-0794-4
90.
Fanale V Segreti A Fossati C Di Gioia G Coletti F Crispino SP et al Athlete’s ECG made easy: a practical guide to surviving everyday clinical practice. J Cardiovasc Dev Dis. (2024) 11(10):303. 10.3390/jcdd11100303
91.
Yang P Ge ZX Gao JM Liu X Xu M Ke HY . Evaluation of the electrocardiogram RV/V criteria in the diagnosis of left ventricular hypertrophy in marathon runners. J Clin Hypertens. (2023) 25(7):638–46. 10.1111/jch.14692
92.
De Bosscher R Moeyersons J Dausin C Claeys M Janssens K Claus P et al Relating QRS voltages to left ventricular mass and body composition in elite endurance athletes. Eur J Appl Physiol. (2023) 123(3):547–59. 10.1007/s00421-022-05080-5
93.
Grazioli G Usín D Trucco E Sanz M Montserrat S Vidal B et al Differentiating hypertrophic cardiomyopathy from athlete’s heart: an electrocardiographic and echocardiographic approach. J Electrocardiol. (2016) 49(4):539–44. 10.1016/j.jelectrocard.2016.03.005
94.
Krysztofiak H Dimitrow PP . Differentiating physiology from pathology in elite athletes. Left ventricular hypertrophy versus hypertrophic cardiomyopathy. Kardiol Pol. (2016) 74(8):705–16. 10.5603/KP.a2016.0084
95.
Machado Leite S Freitas J Campelo M Maciel MJ . Electrocardiographic evaluation in athletes: “normal” changes in the athlete’s heart and benefits and disadvantages of screening. Rev Port Cardiol. (2016) 35(3):169–77. 10.1016/j.repc.2015.09.024
96.
Pelliccia A Tatangelo M Borrazzo C Zampaglione D Mango F Fedele E et al Low QRS voltages and left ventricular hypertrophy: a risky association. Eur J Prev Cardiol. (2023) 30(11):1132–8. 10.1093/eurjpc/zwad035
97.
Sharma S Merghani A Mont L . Exercise and the heart: the good, the bad, and the ugly. Eur Heart J. (2015) 36(23):1445. 10.1093/eurheartj/ehv090
98.
Johnson C Sculthorpe N George K Stout M Procter W Cooper RM et al Concentric and eccentric remodelling of the left ventricle and its association to function in the male athletes heart: an exploratory study. J Cardiovasc Dev Dis. (2023) 10(7):269. 10.3390/jcdd10070269
99.
Clerico A Zaninotto M Aimo A Cardinale DM Dittadi R Sandri MT et al Variability of cardiac troponin levels in normal subjects and in patients with cardiovascular diseases: analytical considerations and clinical relevance. Clin Chem Lab Med. (2023) 61(7):1209–29. 10.1515/cclm-2022-1285
100.
Morrison BN George K Kreiter E Dixon D Rebello L Massarotto RJ et al Effects of endurance exercise training on left ventricular structure in healthy adults: a systematic review and meta-analysis. Eur J Prev Cardiol. (2023) 30(9):772–93. 10.1093/eurjpc/zwad023
101.
Mihl C Dassen WRM Kuipers H . Cardiac remodelling: concentric versus eccentric hypertrophy in strength and endurance athletes. Neth Heart J. (2008) 16(4):129. 10.1007/Bf03086131
102.
Seo DY Kwak HB Kim AH Park SH Heo JW Kim HK et al Cardiac adaptation to exercise training in health and disease. Pflugers Archiv Eur J Physiol. (2020) 472(2):155–68. 10.1007/s00424-019-02266-3
103.
Parry-Williams G Sharma S . The effects of endurance exercise on the heart: panacea or poison?Nat Rev Cardiol. (2020) 17(7):402–12. 10.1038/s41569-020-0354-3
104.
Hellsten Y Nyberg M . Cardiovascular adaptations to exercise training. Compr Physiol. (2016) 6(1):1–32. 10.1002/cphy.c140080
105.
Fernandes T Soci UPR Oliveira EM . Eccentric and concentric cardiac hypertrophy induced by exercise training: microRNAs and molecular determinants. Braz J Med Biol Res. (2011) 44(9):836–47. 10.1590/S0100-879 ( 2011007500112
106.
Bishop SP Zhang JY Ye L . Cardiomyocyte proliferation from fetal- to adult- and from normal- to hypertrophy and failing hearts. Biology-Basel. (2022) 11(6):880. 10.3390/biology11060880
107.
Chen HH Chen C Spanos M Li GP Lu R Bei YH et al Exercise training maintains cardiovascular health: signaling pathways involved and potential therapeutics. Signal Transduct Target Ther. (2022) 7(1):30610. 10.38/s41392-022-01153-1
108.
Dalen H Letnes JM Hoydal MA Wisloff U . Diastolic function and dysfunction in athletes. Eur Heart J Cardiovasc Imaging. (2024) 25(11):1537–45. 10.1093/ehjci/jeae155
109.
Ito S . High-intensity interval training for health benefits and care of cardiac diseases—the key to an efficient exercise protocol. World J Cardiol. (2019) 11(7):171–88. 10.4330/wjc.v11.i7.171
110.
Liu X Li H Hastings MH Xiao C Damilano F Platt C et al miR-222 inhibits pathological cardiac hypertrophy and heart failure. Cardiovasc Res. (2024) 120(3):262–72. 10.1093/cvr/cvad184
111.
Ma Z Qi J Gao L Zhang J . Role of exercise on alleviating pressure overload-induced left ventricular dysfunction and remodeling via AMPK-dependent autophagy activation. Int Heart J. (2020) 61(5):1022–33. 10.1536/ihj.19-443
112.
Sharma N Chahal A Balasubramanian K Sanjeevi RR Rai RH Bansal N et al Effects of resistance training on muscular strength, endurance, body composition and functional performance among sarcopenic patients: a systematic review. J Diabetes Metab Disord. (2023) 22(2):1053–71. 10.1007/s40200-023-01283-5
113.
Fulghum K Hill BG . Metabolic mechanisms of exercise-induced cardiac remodeling. Front Cardiovasc Med. (2018) 5:127. 10.3389/fcvm.2018.00127
114.
Spence AL Naylor LH Carter HH Buck CL Dembo L Murray CP et al A prospective randomised longitudinal MRI study of left ventricular adaptation to endurance and resistance exercise training in humans. J Physiol Lond. (2011) 589(22):5443–52. 10.1113/jphysiol.2011.217125
115.
Valenzuela PL Baggish A Castillo-García A Santos-Lozano A Boraita A Lucia A . Strenuous endurance exercise and the heart: physiological versus pathological adaptations. Compr Physiol. (2022) 12(4):4067–85. 10.1002/cphy.c210045
116.
Stout M . Athletes’ heart and echocardiography: athletes’ heart. Echocardiography. (2008) 25(7):749–54. 10.1111/j.1540-8175.2008.00670.x
117.
Salas-Pacheco JL Lomelí-Sánchez O Baltazar-González O Soto ME . Longitudinal systolic dysfunction in hypertensive cardiomyopathy with normal ejection fraction. Echocardiogr J Cardiovasc Ultrasound All Tech. (2022) 39(1):46–53. 10.1111/echo.15267
118.
Coates AM Joyner MJ Little JP Jones AM Gibala MJ . A perspective on high-intensity interval training for performance and health. Sports Med. (2023) 53(Suppl 1):85–96. 10.1007/s40279-023-01938-6
119.
Wisloff U Ellingsen O Kemi OJ . High-Intensity interval training to maximize cardiac benefits of exercise training?Exerc Sport Sci Rev. (2009) 37(3):139–46. 10.1097/JES.0b013e3181aa65fc
120.
Scharf M Brem MH Wilhelm M Schoepf UJ Uder M Lell MM . Atrial and ventricular functional and structural adaptations of the heart in elite triathletes assessed with cardiac MR imaging. Radiology. (2010) 257(1):71–9. 10.1148/radiol.10092377
121.
Hosseini M Piri M Agha-Alinejad H Haj-Sadeghi S . The effect of endurance, resistance and concurrent training on the heart structure of female students. Biol Sport. (2012) 29(1):17–21. 10.5604/20831862.979404
122.
Tan JRC Krasilshchikov O Kuan GRY Hashim HA Aldhahi MI Al-Mhanna SB et al The effects of combining aerobic and heavy resistance training on body composition, muscle hypertrophy, and exercise satisfaction in physically active adults. Healthcare. (2023) 11(17):2443. 10.3390/healthcare11172443
123.
Venckunas T Gumauskiene B Muanjai P Cadefau JA Kamandulis S . High-Intensity interval training improves cardiovascular fitness and induces left-ventricular hypertrophy during off-season. J Funct Morphol Kinesiol. (2025) 10(3):271. 10.3390/jfmk10030271
124.
Jansen J Marshall PW Benatar JR Cross R Lindbom TK Kingsley M . Low-Intensity resistance exercise in cardiac rehabilitation: a narrative review of mechanistic evidence and clinical implications. J Clin Med. (2024) 13(23):7338. 10.3390/jcm13237338
125.
Tsuda T Robinson BW . Beneficial effects of exercise on hypertension-induced cardiac hypertrophy in adolescents and young adults. Curr Hypertens Rep. (2024) 26(11):451–62. 10.1007/s11906-024-01313-4
126.
Yan ZP Zeng N Li JT Liao T Ni GX . Cardiac effects of treadmill running at different intensities in a rat model. Front Physiol. (2021) 12:774681. 10.3389/fphys.2021.774681
127.
Arbab-Zadeh A Perhonen M Howden E Peshock RM Zhang R Adams-Huet B et al Cardiac remodeling in response to 1 year of intensive endurance training. Circulation. (2014) 130(24):2152. 10.1161/Circulationaha.114.010775
128.
Nakao K Fujie S Iemitsu M . Activation patterns of intracellular signaling pathways in cardiac hypertrophy induced by different exercise modes in rats. Eur Heart J. (2024) 45:ehae6662995. 10.1093/eurheartj/ehae666.2995
129.
Bernardo BC Ooi JYY Weeks KL Patterson NL McMullen JR . Understanding key mechanisms of exercise-induced cardiac protection to mitigate disease: current knowledge and emerging concepts. Physiol Rev. (2018) 98(1):419–75. 10.1152/physrev.00043.2016
130.
McMullen JR Izumo S . Role of the insulin-like growth factor 1 (IGF1)/phosphoinositide-3-kinase (PI3K) pathway mediating physiological cardiac hypertrophy. Novartis Found Symp. (2006) 274:90–111. discussion 111–117, 152–115, 272–116. 10.1002/0470029331.ch7
131.
Bellafiore M Battaglia G Bianco A Palma A . Expression pattern of angiogenic factors in healthy heart in response to physical exercise intensity. Front Physiol. (2019) 10:238. 10.3389/fphys.2019.00238
132.
Zheng L Pan D Gu Y Wang R Wu Y Xue M . Effects of high-intensity and moderate-intensity exercise training on cardiopulmonary function in patients with coronary artery disease: a meta-analysis. Front Cardiovasc Med. (2022) 9:961414. 10.3389/fcvm.2022.961414
133.
Cassidy S Thoma C Hallsworth K Parikh J Hollingsworth KG Taylor R et al High intensity intermittent exercise improves cardiac structure and function and reduces liver fat in patients with type 2 diabetes: a randomised controlled trial. Diabetologia. (2016) 59(1):56–66. 10.1007/s00125-015-3741-2
134.
Li B Feng L Wu X Cai M Yu JJ Tian Z . Effects of different modes of exercise on skeletal muscle mass and function and IGF-1 signaling during early aging in mice. J Exp Biol. (2022) 225(21):557–61. 10.1242/jeb.244650
135.
Bo B Li SS Zhou K Wei JS . The regulatory role of oxygen metabolism in exercise-induced cardiomyocyte regeneration. Front Cell Dev Biol. (2021) 9:664527. 10.3389/fcell.2021.664527
136.
El Assar M Alvarez-Bustos A Sosa P Angulo J Rodríguez-Mañas L . Effect of physical activity/exercise on oxidative stress and inflammation in muscle and vascular aging. Int J Mol Sci. (2022) 23(15):8713. 10.3390/ijms23158713
137.
Memme JM Erlich AT Phukan G Hood DA . Exercise and mitochondrial health. J Physiol. (2021) 599(3):803–17. 10.1113/JP278853
138.
Al-Samerria S Radovick S . The role of insulin-like growth factor-1 (IGF-1) in the control of neuroendocrine regulation of growth. Cells. (2021) 10(10):1–15. 10.3390/cells10102664
139.
Zebrowska A Gasior Z Langfort J . Serum IGF-I and hormonal responses to incremental exercise in athletes with and without left ventricular hypertrophy. J Sports Sci Med. (2009) 8(1):67–76. Available online at:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3737784/
140.
Moonesi M Zaka Khosravi S Molaei Ramshe S Allahbakhshian Farsani M Solali S Mohammadi MH et al IGF Family effects on development, stability, and treatment of hematological malignancies. J Cell Physiol. (2021) 236(6):4097–105. 10.1002/jcp.30156
141.
Rathinaswamy MK Burke JE . Class I phosphoinositide 3-kinase (PI3K) regulatory subunits and their roles in signaling and disease. Adv Biol Regul. (2020):75:100657. 10.1016/j.jbior.2019.100657
142.
Xian QY Zhu DX . The involvement of WDHD1 in the occurrence of esophageal cancer as a downstream target of PI3K/AKT pathway. J Oncol. (2022) 2022:5871188. 10.1155/2022/5871188
143.
Ma ZC Qi J Meng S Wen BJ Zhang J . Swimming exercise training-induced left ventricular hypertrophy involves microRNAs and synergistic regulation of the PI3K/AKT/mTOR signaling pathway. Eur J Appl Physiol. (2013) 113(10):2473–86. 10.1007/s00421-013-2685-9
144.
Mir SA Dar A Alshehri SA Wahab S Hamid L Almoyad MAA et al Exploring the mTOR signalling pathway and its inhibitory scope in cancer. Pharmaceuticals. (2023) 16(7):1004. 10.3390/ph16071004
145.
Qiang M Chen Z Liu HY Dong JX Gong KJ Zhang XJ et al Targeting the PI3K/AKT/mTOR pathway in lung cancer: mechanisms and therapeutic targeting. Front Pharmacol. (2025) 16:1516583. 10.3389/fphar.2025.1516583
146.
Jiang JK Zhang LS Zou JL Liu JY Yang J Jiang Q et al Phosphorylated S6K1 and 4E-BP1 play different roles in constitutively active rheb-mediated retinal ganglion cell survival and axon regeneration after optic nerve injury. Neural Regen Res. (2023) 18(11):2526–34. 10.4103/1673-5374.371372
147.
Sciarretta S Forte M Frati G Sadoshima J . The complex network of mTOR signalling in the heart. Cardiovasc Res. (2022) 118(2):424–39. 10.1093/cvr/cvab033
148.
Xu LF Brink M . mTOR, cardiomyocytes and inflammation in cardiac hypertrophy. Biochim Biophys Acta. (2016) 1863(7):1894–903. 10.1016/j.bbamcr.2016.01.003
149.
Yang MM Lu YM Piao WL Jin H . The translational regulation in mTOR pathway. Biomolecules. (2022) 12(6):802. 10.3390/biom12060802
150.
Dworatzek E Mahmoodzadeh S Schubert C Westphal C Leber J Kusch A et al Sex differences in exercise-induced physiological myocardial hypertrophy are modulated by oestrogen receptor beta. Cardiovasc Res. (2014) 102(3):418–28. 10.1093/cvr/cvu065
151.
Foryst-Ludwig A Kintscher U . Sex differences in exercise-induced cardiac hypertrophy. Pflugers Arch. (2013) 465(5):731–7. 10.1007/s00424-013-1225-0
152.
Lemmens K Doggen K De Keulenaer GW . Role of neuregulin-1/ErbB signaling in cardiovascular physiology and disease—implications for therapy of heart failure. Circulation. (2007) 116(8):954–60. 10.1161/Circulationaha.107.690487
153.
Yang X Cheng K Wang LY Jiang JG . The role of endothelial cell in cardiac hypertrophy: focusing on angiogenesis and intercellular crosstalk. Biomed Pharmacother. (2023) 163:114799. 10.1016/j.biopha.2023.114799
154.
Wang Y Wei JL Zhang P Zhang X Wang YF Chen WJ et al Neuregulin-1, a potential therapeutic target for cardiac repair. Front Pharmacol. (2022) 13:945206. 10.3389/fphar.2022.945206
155.
Amemiya Y Maki M Shibata H Takahara T . New insights into the regulation of mTOR signaling via ca-binding proteins. Int J Mol Sci. (2023) 24(4):3923. 10.3390/ijms24043923
156.
Zaryouh H De Pauw I Baysal H Peeters M Vermorken JB Lardon F et al Recent insights in the PI3K/akt pathway as a promising therapeutic target in combination with EGFR-targeting agents to treat head and neck squamous cell carcinoma. Med Res Rev. (2022) 42(1):112–55. 10.1002/med.21806
157.
Schuttler D Clauss S Weckbach LT Brunner S . Molecular mechanisms of cardiac remodeling and regeneration in physical exercise. Cells. (2019) 8(10):1128. 10.3390/cells8101128
158.
Tao L Bei Y Zhang H Xiao J Li X . Exercise for the heart: signaling pathways. Oncotarget. (2015) 6(25):20773–84. 10.18632/oncotarget.4770
159.
Li JY Zhang Y Tang R Liu H Li XY Lei WR et al Glycogen synthase kinase-3β: a multifaceted player in ischemia-reperfusion injury and its therapeutic prospects. J Cell Physiol. (2024) 239(9):e31335. 10.1002/jcp.31335
160.
Turkistani A Al-kuraishy HM Al-Gareeb AI Albuhadily AK Alexiou A Papadakis M et al Therapeutic potential effect of glycogen synthase kinase 3 Beta (GSK-3β) inhibitors in Parkinson disease: exploring an overlooked avenue. Mol Neurobiol. (2024) 61(9):7092–108. 10.1007/s12035-024-04003-z
161.
Chen JS Huang JQ Luo B Dong SH Wang RC Jiang ZK et al PIK3CD induces cell growth and invasion by activating AKT/GSK-3β/β-catenin signaling in colorectal cancer. Cancer Sci. (2019) 110(3):997–1011. 10.1111/cas.13931
162.
Chen Y Liu XD Wang HH Liu SY Hu NN Li X . Akt regulated phosphorylation of GSK-3β/cyclin D1, p21 and p27 contributes to cell proliferation through cell cycle progression from G1 to S/G2M phase in low-dose arsenite exposed HaCat cells. Front Pharmacol. (2019) 10:1176. 10.3389/fphar.2019.01176
163.
Cui SY Liu ZB Tao B Fan SZ Pu Y Meng XP et al miR-145 attenuates cardiac fibrosis through the AKT/GSK-3β/β-catenin signaling pathway by directly targeting SOX9 in fibroblasts. J Cell Biochem. (2021) 122(2):209–21. 10.1002/jcb.29843
164.
Zhang XB Tang NM Hadden TJ Rishi AK . Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta. (2011) 1813(11):1978–86. 10.1016/j.bbamcr.2011.03.010
165.
Chen X Li Y Luo J Hou N . Molecular mechanism of hippo-YAP1/TAZ pathway in heart development, disease, and regeneration. Front Physiol. (2020) 11:389. 10.3389/fphys.2020.00389
166.
Zheng A Chen Q Zhang L . The hippo-YAP pathway in various cardiovascular diseases: focusing on the inflammatory response. Front Immunol. (2022) 13:971416. 10.3389/fimmu.2022.971416
167.
Islam R Hong Z . YAP/TAZ as mechanobiological signaling pathway in cardiovascular physiological regulation and pathogenesis. Mechanobiol Med. (2024) 2(4):100085. 10.1016/j.mbm.2024.100085
168.
Ramaccini D Pedriali G Perrone M Bouhamida E Modesti L Wieckowski MR et al Some insights into the regulation of cardiac physiology and pathology by the hippo pathway. Biomedicines. (2022) 10(3):1–20. 10.3390/biomedicines10030726
169.
Ueda Y Kondo N Kinashi T . MST1/2 balance immune activation and tolerance by orchestrating adhesion, transcription, and organelle dynamics in lymphocytes. Front Immunol. (2020) 11:733. 10.3389/fimmu.2020.00733
170.
Aragona M Panciera T Manfrin A Giulitti S Michielin F Elvassore N et al A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell. (2013) 154(5):1047–59. 10.1016/j.cell.2013.07.042
171.
Thines L Gorisse L Li ZG Sayedyahossein S Sacks DB . Calmodulin activates the hippo signaling pathway by promoting LATS1 kinase-mediated inhibitory phosphorylation of the transcriptional coactivator YAP. J Biol Chem. (2022) 298(5):101839. 10.1016/j.jbc.2022.101839
172.
Jin JY Zhao XX Fu HF Gao Y . The effects of YAP and its related mechanisms in central nervous system diseases. Front Neurosci. (2020) 14:595. 10.3389/fnins.2020.00595
173.
Flores-Vergara R Olmedo I Aránguiz P Riquelme JA Vivar R Pedrozo Z . Communication between cardiomyocytes and fibroblasts during cardiac ischemia/reperfusion and remodeling: roles of TGF-β, CTGF, the renin angiotensin axis, and non-coding RNA molecules. Front Physiol. (2021) 12:716721. 10.3389/fphys.2021.716721
174.
Lan C Cao N Chen CY Qu S Fan C Luo H et al Progesterone, via yes-associated protein, promotes cardiomyocyte proliferation and cardiac repair. Cell Prolif. (2020) 53(11):e12910. 10.1111/cpr.12910
175.
Limyati Y Sanjaya A Lucretia T Gunadi JW Biben V Jasaputra DK et al Potential role of exercise in regulating YAP and TAZ during cardiomyocytes aging. Curr Cardiol Rev. (2022) 18(5):24–33. 10.2174/1573403X18666220404152924
176.
Li P Hu Y Tong LG Bi XC . High-intensity training on CREB activation for improving brain health: a narrative review of possible molecular talks. Front Endocrinol (Lausanne). (2025) 15:1498495. 10.3389/fendo.2024.1498495
177.
Wang Q Xie Y He Q Geng Y Xu JR . LncRNA-Cox2 regulates macrophage polarization and inflammatory response through the CREB-C/EBPβ signaling pathway in septic mice. Int Immunopharmacol. (2021) 101:108347. 10.1016/j.intimp.2021.108347
178.
Xiong J Zhang ZT Ye KQ . C/EBPβ/AEP signaling drives Alzheimer’s disease pathogenesis. Neurosci Bull. (2023) 39(7):1173–85. 10.1007/s12264-023-01025-w
179.
Molkentin JD . The transcription factor C/EBPβ serves as a master regulator of physiologic cardiac hypertrophy. Circ Res. (2011) 108(3):277–8. 10.1161/RES.0b013e31820ff484
180.
Zhang GL Sun ML Zhang XA . Exercise-Induced adult cardiomyocyte proliferation in mammals. Front Physiol. (2021) 12:729364. 10.3389/fphys.2021.729364
181.
Bernardo BC Ooi JYY Weeks KL Patterson NL McMullen JR . Understanding key mechanisms of exercise-induced cardiac protection to mitigate disease: current knowledge and emerging concepts. Physiol Rev. (2018) 98(1):419–75. 10.1152/physrev.00043.2016
182.
Bei YH Huang ZZ Feng X Li L Wei M Zhu YJ et al Lymphangiogenesis contributes to exercise-induced physiological cardiac growth. J Sport Health Sci. (2022) 11(4):466–78. 10.1016/j.jshs.2022.02.005
183.
Ozo U Sharma S . The impact of ethnicity on cardiac adaptation. Eur Cardiol. (2020) 15:e61. 10.15420/ecr.2020.01
184.
Sheikh N Papadakis M Carre F Kervio G Panoulas VF Ghani S et al Cardiac adaptation to exercise in adolescent athletes of African ethnicity: an emergent elite athletic population. Br J Sports Med. (2013) 47(9):585–92. 10.1136/bjsports-2012-091874
185.
Sheikh N Sharma S . Impact of ethnicity on cardiac adaptation to exercise. Nat Rev Cardiol. (2014) 11(4):198–217. 10.1038/nrcardio.2014.15
186.
Blatter LA Kanaporis G Martinez-Hernandez E Oropeza-Almazan Y Banach K . Excitation-contraction coupling and calcium release in atrial muscle. Pflugers Archiv. (2021) 473(3):317–29. 10.1007/s00424-020-02506-x
187.
Nusier M Shah AK Dhalla NS . Structure-Function relationships and modifications of cardiac sarcoplasmic Reticulum ca-transport. Physiol Res. (2021) 70:S443–70. 10.33549/physiolres.934805
188.
Zhang PY Huang CY Liu HY Zhang MT Liu L Zhai YH et al The mechanism of the NFAT transcription factor family involved in oxidative stress response. J Cardiol. (2024) 83(1):30–6. 10.1016/j.jjcc.2023.04.017
189.
Kaur G Jans DA . Dual nuclear import mechanisms of sex determining factor SRY: intracellular ca as a switch. FASEB J. (2011) 25(2):665–75. 10.1096/fj.10-173351
190.
Murphy JG Crosby KC Dittmer PJ Sather WA Dell’Acqua ML . AKAP79/150 recruits the transcription factor NFAT to regulate signaling to the nucleus by neuronal L-type ca channels. Mol Biol Cell. (2019) 30(14):1743–56. 10.1091/mbc.E19-01-0060
191.
Katanasaka Y Suzuki H Sunagawa Y Hasegawa K Morimoto T . Regulation of cardiac transcription factor GATA4 by post-translational modification in cardiomyocyte hypertrophy and heart failure. Int Heart J. (2016) 57(6):672–5. 10.1536/ihj.16-404
192.
Yoon JJ Tai AL Kim HY Han BH Shin S Lee HS et al Tongguanwan alleviates doxorubicin- and isoproterenol-induced cardiac hypertrophy and fibrosis by modulating apoptotic and fibrotic pathways. Int J Mol Sci. (2024) 25(19):10573. 10.3390/ijms251910573
193.
Zhu NA Li TL Bai YL Sun J Guo JP Yuan HT et al Targeting myocardial inflammation: investigating the therapeutic potential of atrial natriuretic peptide in atrial fibrosis. Mol Biol Rep. (2024) 51(1):506. 10.1007/s11033-024-09393-w
194.
Li XR Lan YH Wang Y Nie MH Lu YH Zhao EY . Telmisartan suppresses cardiac hypertrophy by inhibiting cardiomyocyte apoptosis via the NFAT/ANP/BNP signaling pathway. Mol Med Rep. (2017) 15(5):2574–82. 10.3892/mmr.2017.6318
195.
Fan YS Lv XY Chen ZH Peng YY Zhang MQ . M6a methylation: critical roles in aging and neurological diseases. Front Mol Neurosci. (2023) 16:1102147. 10.3389/fnmol.2023.1102147
196.
Wang HX Han JJ Kong H Ma C Zhang XA . The emerging role of m6A and programmed cell death in cardiovascular diseases. Biomolecules. (2025) 15(2):247. 10.3390/biom15020247
197.
Wang LJ Wang JQ Yu PJ Feng JY Xu GE Zhao X et al METTL14 is required for exercise-induced cardiac hypertrophy and protects against myocardial ischemia-reperfusion injury. Nat Commun. (2022) 13(1):6762. 10.1038/s41467-022-34434-y
198.
Wen TX Li T Xu YQ Zhang YZ Pan H Wang Y . The role of m6A epigenetic modifications in tumor coding and non-coding RNA processing. Cell Commun Signal. (2023) 21(1):355. 10.1186/s12964-023-01385-w
199.
Rudd CE Krueger J Taylor A . Glycogen synthase kinase-3 (GSK-3) inactivation compensates for the lack of CD28 in the priming of CD8 cytotoxic T-cells: implications for anti-PD-1 immunotherapy. J Immunol. (2018) 200(1):1–13. 10.3389/fimmu.2017.01653
200.
Zhang X Cai H Xu H Dong S Ma H . Critical roles of m(6)A methylation in cardiovascular diseases. Front Cardiovasc Med. (2023) 10:1187514. 10.3389/fcvm.2023.1187514
201.
Wang LJ Feng JY Feng X Meng DN Zhao X Wang JQ et al Exercise-induced circular RNA circUtrn is required for cardiac physiological hypertrophy and prevents myocardial ischaemia-reperfusion injury. Cardiovasc Res. (2023) 119(16):2638–52. 10.1093/cvr/cvad161
202.
Campos A Clemente-Blanco A . Cell cycle and DNA repair regulation in the damage response: protein phosphatases take over the reins. Int J Mol Sci. (2020) 21(2):446. 10.3390/ijms21020446
203.
Gergs U Boknik P Buchwalow IB Fabritz L Gründker N Kucerova D et al Modulation of cardiac contractility by serine/threonine protein phosphatase type 5. Int J Cardiol. (2012) 154(2):116–21. 10.1016/j.ijcard.2010.09.009
204.
Lin W Hou LL Tang JLY Huang AW Jia ZY . Mir-195-5p targets Smad7 regulation of the wnt/β-catenin pathway to promote osteogenic differentiation of vascular smooth muscle cells. BMC Cardiovasc Disord. (2024) 24(1):221. 10.1186/s12872-024-03891-2
205.
Wang L Qin DZ Shi HT Zhang YN Li H Han QH . MiR-195-5p promotes cardiomyocyte hypertrophy by targeting MFN2 and FBXW7. BioMed Res Int. (2019) 2019:1580982. 10.1155/2019/1580982
206.
Fagundes R Teixeira LK . Cyclin E/CDK2: DNA replication, replication stress and genomic instability. Front Cell Dev Biol. (2021) 9:774845. 10.3389/fcell.2021.774845
207.
Alissa M Aldurayhim M Abdulaziz O Alsalmi O Awad A Algopishi UB et al From molecules to heart regeneration: understanding the complex and profound role of non-coding RNAs in stimulating cardiomyocyte proliferation for cardiac repair. Curr Probl Cardiol. (2024) 49(12):102857. 10.1016/j.cpcardiol.2024.102857
208.
Huang H Liu Y Su Q Ling JY . Overexpression of homeobox containing1 relieves myocardial fibrosis and inflammation in diabetic cardiomyopathy rats. J Biomed Nanotechnol. (2023) 19(11):2005–12. 10.1166/jbn.2023.3697
209.
Yan BQ Wang HJ Tan Y Fu W . microRNAs in cardiovascular disease: small molecules but big roles. Curr Top Med Chem. (2019) 19(21):1918–47. 10.2174/1568026619666190808160241
210.
Berthouze M Laurent AC Breckler M Lezoualc'h F . New perspectives in cAMP-signaling modulation. Curr Heart Fail Rep. (2011) 8(3):159–67. 10.1007/s11897-011-0062-8
211.
Shu HY Peng YZ Hang WJ Nie JL Zhou N Wang DW . The role of CD36 in cardiovascular disease. Cardiovasc Res. (2022) 118(1):115–29. 10.1093/cvr/cvaa319
212.
Qin JL Ye L Wen XQ Zhang X Di YQ Chen ZH et al Fatty acids in cancer chemoresistance. Cancer Lett. (2023) 572:216352. 10.1016/j.canlet.2023.216352
213.
Wang DT Ye QZ Gu HC Chen ZG . The role of lipid metabolism in tumor immune microenvironment and potential therapeutic strategies. Front Oncol. (2022) 12::984560. 10.3389/fonc.2022.984560
214.
Foryst-Ludwig A Kreissl MC Sprang C Thalke B Bohm C Benz V et al Sex differences in physiological cardiac hypertrophy are associated with exercise-mediated changes in energy substrate availability. Am J Physiol Heart Circ Physiol. (2011) 301(1):H115–22. 10.1152/ajpheart.01222.2010
215.
Sunagawa Y Katayama A Funamoto M Shimizu K Shimizu S Sari N et al The polyunsaturated fatty acids, EPA and DHA, ameliorate myocardial infarction-induced heart failure by inhibiting p300-HAT activity in rats. J Nutr Biochem. (2022) 106:109031. 10.1016/j.jnutbio.2022.109031
216.
Hu YX Qiu SL Shang JJ Wang Z Lai XL . Pharmacological effects of botanical drugs on myocardial metabolism in chronic heart failure. Chin J Integr Med. (2024) 30(5):458–67. 10.1007/s11655-023-3649-5
217.
Chang YC Chan MH Yang YF Li CH Hsiao M . Glucose transporter 4: insulin response mastermind, glycolysis catalyst and treatment direction for cancer progression. Cancer Lett. (2023) 563:216179. 10.1016/j.canlet.2023.216179
218.
Wende AR Kim J Holland WL Wayment BE O’Neill BT Tuinei J et al Glucose transporter 4-deficient hearts develop maladaptive hypertrophy in response to physiological or pathological stresses. Am J Physiol Heart Circ Physiol. (2017) 313(6):H1098–108. 10.1152/ajpheart.00101.2017
219.
Kohler ZM Trencsenyi G Juhasz L Zvara A Szabo JP Dux L et al Tilorone increases glucose uptake in vivo and in skeletal muscle cells by enhancing Akt2/AS160 signaling and glucose transporter levels. J Cell Physiol. (2023) 238(5):1080–94. 10.1002/jcp.30998
220.
Taheri R Mokhtari Y Yousefi AM Bashash D . The PI3K/akt signaling axis and type 2 diabetes mellitus (T2DM): from mechanistic insights into possible therapeutic targets. Cell Biol Int. (2024) 48(8):1049–68. 10.1002/cbin.12189
221.
Wang L Li JJ Di LJ . Glycogen synthesis and beyond, a comprehensive review of GSK3 as a key regulator of metabolic pathways and a therapeutic target for treating metabolic diseases. Med Res Rev. (2022) 42(2):946–82. 10.1002/med.21867
222.
Abu Shelbayeh O Arroum T Morris S Busch KB . PGC-1alpha is a master regulator of mitochondrial lifecycle and ROS stress response. Antioxidants (Basel). (2023) 12(5):1075. 10.3390/antiox12051075
223.
Ji XN Zhang CZ Yang J Tian YR You LJ Yang H et al Kaempferol improves exercise performance by regulating glucose uptake, mitochondrial biogenesis, and protein synthesis via PI3K/AKT and MAPK signaling pathways. Foods. (2024) 13(7):1068. 10.3390/foods13071068
224.
Bei YH Zhu YJ Zhou JW Ai SW Yao JH Yin MM et al Inhibition of Hmbox1 promotes cardiomyocyte survival and glucose metabolism through gck activation in ischemia/reperfusion injury. Circulation. (2024) 150(11):848–66. 10.1161/Circulationaha.123.067592.
225.
Soci UPR Fernandes T Barauna VG Hashimoto NY Mota GDA Rosa KT et al Epigenetic control of exercise training-induced cardiac hypertrophy by. Clin Sci. (2016) 130(22):2005–15. 10.1042/Cs20160480
226.
Moreira JBN Wohlwend M Wisloff U . Exercise and cardiac health: physiological and molecular insights. Nat Metab. (2020) 2(9):829–39. 10.1038/s42255-020-0262-1
227.
Juttner AA Danser AHJ Roks AJM . Pharmacological developments in antihypertensive treatment through nitric oxide-cGMP modulation. Adv Pharmacol. (2022) 94:57–94. 10.1016/bs.apha.2022.01.001
228.
Schüttler D Clauss S Weckbach LT Brunner S . Molecular mechanisms of cardiac remodeling and regeneration in physical Exercise. Cells. (2019) 8(10):1128. 10.3390/cells8101128
229.
Aschar-Sobbi R Izaddoustdar F Korogyi AS Wang QL Farman GP Yang F et al Increased atrial arrhythmia susceptibility induced by intense endurance exercise in mice requires TNFα. Nat Commun. (2015) 6:6018. 10.1038/ncomms7018
230.
Varró A Tomek J Nagy N Virág L Passini E Rodriguez B et al Cardiac transmembrane Ion channels and action potentials: cellular physiology and arrhythmogenic behavior. Physiol Rev. (2021) 101(3):1083–176. 10.1152/physrev.00024.2019
231.
Besnier F Labrunée M Pathak A Traon APL Galès C Sénard JM et al Exercise training-induced modification in autonomic nervous system: an update for cardiac patients. Ann Phys Rehabil Med. (2017) 60(1):27–35. 10.1016/j.rehab.2016.07.002
232.
Guasch E Mont L . Diagnosis, pathophysiology, and management of exercise-induced arrhythmias. Nat Rev Cardiol. (2017) 14(2):88–101. 10.1038/nrcardio.2016.173
233.
Sulovic LS Mahmutovic M Lazic S Sulovic N . The role of echocardiography in the evaluation of cardiac re-modelling and differentiation between physiological and pathological hypertrophy in teenagers engaged in competitive amateur sports. Cardiol Young. (2017) 27(4):706–12. 10.1017/S1047951116001116
234.
Corrado D Biffi A Basso C Pelliccia A Thiene G . 12-lead ECG in the athlete: physiological versus pathological abnormalities. Br J Sports Med. (2009) 43(9):669–76. 10.1136/bjsm.2008.054759
235.
Tisdale JE Chung MK Campbell KB Hammadah M Joglar JA Leclerc J et al Drug-induced arrhythmias: a scientific statement from the American Heart Association. Circulation. (2020) 142(15):E214–33. 10.1161/Cir.0000000000000905
236.
Galanti G Toncelli L Del Furia F Stefani L Cappelli B De Luca A et al Tissue Doppler imaging can be useful to distinguish pathological from physiological left ventricular hypertrophy: a study in master athletes and mild hypertensive subjects. Cardiovasc Ultrasound. (2009) 7:48. 10.1186/1476-7120-7-48
237.
Smiseth OA Rider O Cvijic M Valkovic L Remme EW Voigt JU . Myocardial strain imaging theory, current practice, and the future. Jacc-Cardiovascular Imaging. (2024) 18(3):340–81. 10.1016/j.jcmg.2024.07.011
238.
Czimbalmos C Csecs I Toth A Kiss O Suhai FI Sydo N et al The demanding grey zone: sport indices by cardiac magnetic resonance imaging differentiate hypertrophic cardiomyopathy from athlete’s heart. PLoS One. (2019) 14(2):e0211624. 10.1371/journal.pone.0211624
239.
Pittaras A Faselis C Doumas M Grassos C Kokkinos P . Physical activity and cardiac morphologic adaptations. Rev Cardiovasc Med. (2023) 24(5):142. 10.31083/j.rcm2405142
240.
Augustine DX Howard L . Left ventricular hypertrophy in athletes: differentiating physiology from pathology. Curr Treat Options Cardiovasc Med. (2018) 20(12):96. 10.1007/s11936-018-0691-2
241.
Pagourelias ED Efthimiadis GK Kouidi E Zorou P Giannoglou G Deligiannis A et al Efficacy of various “classic” echocardiographic and laboratory indices in distinguishing the “gray zone” between athlete’s heart and hypertrophic cardiomyopathy: a pilot study. Echocardiography. (2013) 30(2):131–9. 10.1111/echo.12014
242.
Dominguez F González-López E Padron-Barthe L Cavero MA Garcia-Pavia P . Role of echocardiography in the diagnosis and management of hypertrophic cardiomyopathy. Heart. (2018) 104(3):260–72. 10.1136/heartjnl-2016-310559
243.
Bernardini A Crotti L Olivotto I Cecchi F . Diagnostic and prognostic electrocardiographic features in patients with hypertrophic cardiomyopathy. Eur Heart J Suppl. (2023) 25:C173–8. 10.1093/eurheartjsupp/suad074
244.
Weiner RB DeLuca JR Wang F Lin J Wasfy MM Berkstresser B et al Exercise-induced left ventricular remodeling among competitive athletes A phasic phenomenon. Circ Cardiovasc Imaging. (2015) 8(12):e003651. 10.1161/CIRCIMAGING.115.003651
245.
Brady B King G Murphy RT Walsh D . Myocardial strain: a clinical review. Ir J Med Sci. (2023) 192(4):1649–56. 10.1007/s11845-022-03210-8
246.
Frantz S Hundertmark MJ Schulz-Menger J Bengel FM Bauersachs J . Left ventricular remodelling post-myocardial infarction: pathophysiology, imaging, and novel therapies. Eur Heart J. (2022) 43(27):2549. 10.1093/eurheartj/ehac223
247.
Liu Y Chen XW Zhang HG . Editorial: cardiac hypertrophy: from compensation to decompensation and pharmacological interventions. Front Pharmacol. (2021) 12:665936. 10.3389/fphar.2021.665936
248.
Han B Trew ML Zgierski-Johnston CM . Cardiac conduction velocity, remodeling and arrhythmogenesis. Cells. (2021) 10(11):2923. 10.3390/cells10112923
249.
Huang CLH Lei M . Cardiomyocyte electrophysiology and its modulation: current views and future prospects. Philos Trans R Soc B Biol Sci. (2023) 378(1879):20220160. 10.1098/rstb.2022.0160
250.
Gronwald T Rogers B Hoos O . Fractal correlation properties of heart rate variability: a new biomarker for intensity distribution in endurance exercise and training prescription?Front Physiol. (2020) 11:550572. 10.3389/fphys.2020.550572
251.
Wackerhage H Schoenfeld BJ . Personalized, evidence-informed training plans and exercise prescriptions for performance, fitness and health. Sports Med. (2021) 51(9):1805–13. 10.1007/s40279-021-01495-w
252.
Faggian S Centanini A Quinto G Vecchiato M Ermolao A Battista F et al The many faces of exercise intensity: a call to agree on definitions and provide standardized prescriptions. Eur J Prev Cardiol. (2024) 31(12):e89–91. 10.1093/eurjpc/zwae034
253.
Taylor JL Myers J Bonikowske AR . Practical guidelines for exercise prescription in patients with chronic heart failure. Heart Fail Rev. (2023) 28(6):1285–96. 10.1007/s10741-023-10310-9
254.
Wen Y Zhang Y Lv Q Lan W Shu Y Qi Q et al The effect of individual exercise rehabilitation program on ischemic burden and cardiac function in patients with ischemic non-obstructive coronary heart disease: a randomized parallel controlled clinical trial. Front Cardiovasc Med. (2025) 12:1421923. 10.3389/fcvm.2025.1421923
255.
Kabbadj K Taiek N El Hjouji W El Karrouti O El Hangouche AJ . Cardiopulmonary exercise testing: methodology. Interpretation, and role in exercise prescription for cardiac rehabilitation. US Cardiol. (2024) 18:e22. 10.15420/usc.2024.37
256.
Li G Wang Z Hao Y Qian J Hu B Wang Y et al Consensus statement of Chinese experts on exercise prescription (2023). Sports Med Health Sci. (2024) 6(2):200–3. 10.1016/j.smhs.2024.02.003
257.
Nazaret A Tonekaboni S Darnell G Ren SY Sapiro G Miller AC . Modeling personalized heart rate response to exercise and environmental factors with wearables data. NPJ Digit Med. (2023) 6(1):207. 10.1038/s41746-023-00926-4
258.
Noone J Mucinski JM Delany JP Sparks LM Goodpaster BH . Understanding the variation in exercise responses to guide personalized physical activity prescriptions. Cell Metab. (2024) 36(4):702–24. 10.1016/j.cmet.2023.12.025
259.
Jaarsma T Hill L Bayes-Genis A La Rocca HPB Castiello T Celutkiene J et al Self-care of heart failure patients: practical management recommendations from the heart failure association of the European Society of Cardiology. Eur J Heart Fail. (2021) 23(1):157–74. 10.1002/ejhf.2008
Summary
Keywords
exercise, physiological cardiac hypertrophy, regulatory mechanisms, cardiovascular health, research progress
Citation
Cheng P, Zhang X, Si Y, Yin Q, Chen L, Ru Q, Chu C, Xiang H, Liao L, Ran H, Zhang Z and Wu Y (2025) Regulatory mechanisms of exercise-induced physiological cardiac hypertrophy: progress and prospects. Front. Cardiovasc. Med. 12:1657950. doi: 10.3389/fcvm.2025.1657950
Received
02 July 2025
Accepted
04 September 2025
Published
25 September 2025
Volume
12 - 2025
Edited by
DeLisa Fairweather, Mayo Clinic Florida, United States
Reviewed by
Bing Bo, Henan University, China
Amogh Verma, Rama Medical College Hospital and Research Centre, India
Bela Juhász, University of Debrecen, Hungary
Updates
Copyright
© 2025 Cheng, Zhang, Si, Yin, Chen, Ru, Chu, Xiang, Liao, Ran, Zhang and Wu.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
* Correspondence: Yuxiang Wu yxwu@jhun.edu.cn
†These authors have contributed equally to this work and share first authorship
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.