 Yanxia Lin
Yanxia Lin Huanrui Zhang
Huanrui Zhang Yuqi Jiang
Yuqi Jiang Wen Tian
Wen Tian- Department of Geriatric Cardiology, The First Hospital of China Medical University, Shenyang, Liaoning, China
Vascular calcification (VC) refers to the pathological deposition of hydroxyapatite within the arterial wall and is characterized by the transdifferentiation of vascular smooth muscle cells (VSMCs) into osteogenic phenotypes. Emerging evidence indicates that oxidative stress plays a pivotal role in the initiation and progression of vascular calcification. Excessive production of reactive oxygen species (ROS) not only activates the expression of calcification-related genes but also promotes VSMC phenotypic switching through diverse epigenetic mechanisms. In this review, we summarize current advances in understanding the interplay between oxidative stress and epigenetic regulation in VC, to provide novel theoretical perspectives on the pathogenesis of this complex vascular disorder.
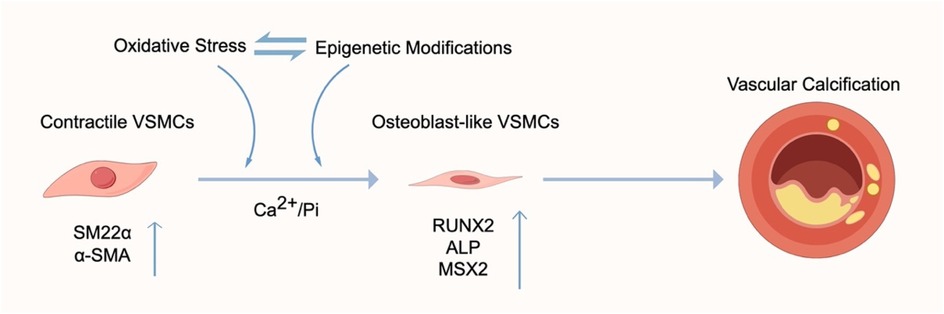
Graphical Abstract.
1 Introduction
Vascular calcification (VC) is defined as the pathological deposition of hydroxyapatite crystals within the arterial wall. It is recognized as a hallmark of advanced vascular disease and a strong predictor of adverse cardiovascular outcomes (1–3). VC contributes to increased arterial stiffness, systolic blood pressure, and pulse wave velocity (4), thereby exacerbating the morbidity and mortality of cardiovascular diseases (5, 6). Mechanistically, VC mirrors physiological bone formation, with the phenotypic switch of vascular smooth muscle cells (VSMCs) from a contractile to an osteoblast-like phenotype serving as the central process (7). This transition is characterized by the downregulation of contractile marker genes and the upregulation of osteogenic transcription factors, including runt-related transcription factor 2 (RUNX2), Msh homeobox 2 (MSX2), and alkaline phosphatase (ALP), among others (8, 9). Beyond these phenotypic changes, dysregulated biological processes linked to oxidative stress—such as VSMC apoptosis, impaired autophagy, and endoplasmic reticulum stress—also play critical roles in the pathogenesis of VC (10).
Recent evidence highlights the significance of oxidative stress and epigenetic changes in VC development (11, 12). Although the direct interactions between these two processes remain insufficiently explored, their synergistic effects on VSMC function and vascular homeostasis are increasingly recognized. This review aims to clarify how oxidative stress and its related epigenetic changes contribute to VC, offering a comprehensive understanding of this complex condition.
2 Oxidative stress and vascular calcification
Oxidative stress occurs when excessive reactive oxygen species (ROS) accumulate and overwhelm the body's natural antioxidant defenses, resulting in damage to DNA, proteins, and lipids (13). ROS can be broadly classified into free radicals—such as the superoxide anion (O2•−) and hydroxyl radical (•OH)—and non-radical oxidants, including hydrogen peroxide (H2O2) and peroxynitrite (ONOO−) (14, 15). These reactive molecules are usually neutralized by antioxidant defense systems such as superoxide dismutase (SOD), catalase, and glutathione peroxidase (GPx) (14, 16). Two main sources of ROS in vascular cells are nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX) and mitochondria (17, 18). Mitochondria are the primary sources of cellular ROS (mtROS), generated as byproducts of electron transport chain (ETC) activity (15, 19, 20) (Figure 1). Under physiological conditions, redox balance is maintained through dynamic regulation between ROS production and antioxidant defenses. However, mitochondrial dysfunction leads to excessive ROS accumulation, thereby aggravating oxidative stress (21). Similarly, increased NOX activity and impaired ROS clearance synergistically contribute to vascular oxidative damage and calcification (18, 22). Accumulating evidence highlights oxidative stress as a key driver of VC. ROS overproduction not only promotes VSMC transdifferentiation into osteogenic-like phenotypes but also accelerates the progression of calcification (23, 24). Conversely, interventions that suppress oxidative stress, including antioxidants and ROS inhibitors, have been shown to attenuate VC development (25).

Figure 1. Generation and clearance of ROS. Mitochondrial ETC complexes I and III and NADPH oxidases are major sources of O2•−. NOX catalyze the oxidation of NADPH to NADP+, generating O2•−. NO reacts with O2•− to form ONOO−. O2•− is rapidly converted to H2O2 by SOD. H2O2 is further decomposed into H2O and O2 by catalase, or reduced to H2O by GPX using GSH as a substrate, producing GSSG. Meanwhile, GR reduces oxidized GSSG to GSH using NADPH as an electron donor. In the presence of Fe2+, H2O2 undergoes the Fenton reaction to form •OH, which exerts strong oxidative damage. ETC, electron transport chain; NADPH, nicotinamide adenine dinucleotide phosphate; NOX, NADPH oxidases; O2•−, superoxide anion; NO, nitric oxide; ONOO−, peroxynitrite; H2O2, hydrogen peroxide; •OH, hydroxyl radical; SOD, superoxide dismutase; GSH, glutathione (reduced form); GSSG, glutathione disulfide (oxidized form); GR, glutathione reductase.
Mitochondria undergo dynamic fission and fusion to preserve their functional integrity. Excessive fission causes fragmentation, reduced bioenergetics, and increased ROS production (26). Dynamin-related protein 1 (DRP1), a key mediator of fission, promotes mitochondrial fragmentation, membrane depolarization, and oxidative stress when overexpressed (27). DRP1 has been implicated in the osteogenic phenotypic switch of VSMCs, and its enrichment at calcified vascular sites has been confirmed. Pharmacological or genetic inhibition of DRP1 attenuates oxidative stress-induced VSMC calcification (28, 29). Notably, quercetin, an antioxidant flavonoid, reduces DRP1 expression and prevents phosphate (Pi)-induced calcification in renal failure rat models, further linking mitochondrial dynamics and oxidative stress to VC (28).
Mitochondrial DNA (mtDNA) is highly vulnerable to oxidative damage because of its proximity to sources of ROS and the absence of protective histones and introns (30). Alterations in mtDNA copy number are considered sensitive biomarkers of oxidative stress (31). Accumulation of oxidative mtDNA damage has been observed in VC and other vascular pathologies (1). DNA polymerase γ (PolG), the only mitochondrial DNA polymerase, is crucial for mtDNA replication, proofreading, and repair. Its exonuclease activity maintains genomic accuracy and prevents mutations. Recent studies show that PolG, along with p53, helps preserve mitochondrial function, reduces oxidative stress, and alleviates VC. Conversely, the loss of this repair ability, as seen in the PolG D257A mutation, accelerates oxidative damage and vascular calcification (32).
The mitochondrial permeability transition pore (MPTP) also plays a crucial role in mitochondrial homeostasis. Elevated Ca2+ and oxidative stress promote MPTP opening (33). Transient openings enable solute exchange, while prolonged openings trigger ROS bursts, mitochondrial swelling, Ca2+ release, and cell death (34). Inorganic polyphosphate-induced VC is primarily mediated by mitochondrial dysfunction, ATP depletion, and sustained MPTP opening (24, 35, 36). Concurrent accumulation of Ca2+ and Pi in the cytoplasm and mitochondria aggravates oxidative stress and drives VC progression (10).
Phosphate transporters (PiT-1/-2) mediate Pi entry into VSMCs via sodium-dependent cotransport, while mitochondrial phosphate carriers (PiC) facilitate intramitochondrial Pi uptake (23). Excessive Pi uptake leads to mitochondrial hyperpolarization and superoxide overproduction (23). H2O2, a key ROS in atherosclerosis, induces VSMC osteogenic differentiation by upregulating RUNX2 (37). ROS accumulation further damages the mitochondrial outer membrane, causing Ca2+ overload and DNA injury (38). In addition, advanced glycation end products (AGEs) and their receptor (RAGE) significantly contribute to VC by amplifying oxidative stress (39). Pi-induced RAGE ligand production enhances oxidative stress, upregulates Pit-1 transcription, and increases RUNX2 expression (40). Meanwhile, NOX-derived ROS participate in AGE-mediated VSMC apoptosis, a critical mechanism in chronic kidney disease and diabetes-associated VC (41) (Figure 2).

Figure 2. NOX drives excessive ROS production and contributes to oxidative stress. Ca2+ and Pi overload promote mitochondrial damage through DRP1-mediated pathways, opening of the MPTP, Pi transport via the PiC, and mtDNA strand breaks. The rapid production of RAGE ligands in response to Pi induces the activation of RAGE signalling. These events enhance oxidative stress and ultimately accelerate vascular calcification. NOX, NADPH oxidases; RAGE, receptor for advanced glycation end products; DRP1, dynamin-related protein 1; MPTP, mitochondrial permeability transition pore; mtDNA, mitochondrial DNA.
Antioxidant systems are essential for maintaining vascular health. Dietary antioxidants, particularly polyphenols, can modulate the uncoupling of endothelial nitric oxide synthase (eNOS). In vascular diseases, eNOS uncoupling favors the generation of superoxide radicals rather than nitric oxide. Polyphenols mitigate oxidative stress and improve vascular endothelial dysfunction (VED) by scavenging free radicals or inhibiting radical-generating pathways (42).
3 Epigenetic regulation and oxidative stress in vascular calcification
Current research on oxidative stress and epigenetics in VC emphasizes their complex interaction. In aging, studies reveal that oxidative stress and epigenetic changes—including DNA methylation, histone modifications, and non-coding RNAs—play a role in the molecular mechanisms behind age-related decline (43). In cancer biology, more focus is being placed on how oxidative stress alters the epigenetic machinery, thereby encouraging tumor initiation, progression, and chemoresistance. Understanding these relationships may lead to new therapeutic strategies (44).
Epigenetics refers to heritable changes in gene expression without alterations in the DNA sequence, including DNA methylation, histone modifications, and regulation by non-coding RNAs (45, 46). Accumulating evidence links ROS with epigenetic modifications in VC.
3.1 DNA methylation
DNA methylation is regulated by DNA methyltransferases (DNMTs) and ten–eleven translocation (TET) family dioxygenases. Usually, CpG island methylation in gene promoters is linked to transcriptional silencing. Typically, CpG island methylation in gene promoters is associated with transcriptional silencing (47). TET proteins are Fe (II)/α-ketoglutarate (α-KG)-dependent dioxygenases that oxidize 5-methylcytosine (5mC) into 5-hydroxymethylcytosine (5hmC) and subsequent products (48). TET activity can be inhibited by 2-hydroxyglutarate (2-HG). α-KG, a tricarboxylic acid (TCA) cycle intermediate, is generated by isocitrate dehydrogenases (IDHs). Their activity is inhibited by 2-HG, which is produced by mutant IDHs through aberrant oxidation of isocitrate. This results in TET inhibition, DNA hypermethylation, increased ROS generation, and enhanced oxidative stress sensitivity (49, 50).
3.1.1 ROS-mediated DNA methylation changes in VC
TET2 overexpression promotes VSMC differentiation by enhancing contractile gene expression and reducing DNA methylation (51). Recent studies also show that the α-KG mitigates VC by activating TET2, which in turn suppresses NLR family pyrin domain containing 3 (NLRP3) inflammasome signaling (52). High-phosphate conditions upregulate DNMTs, increase smooth muscle 22α (SM22α) promoter methylation, downregulate SM22α expression, and enhance RUNX2 expression and mineral deposition. These findings underscore epigenetic SM22α methylation as an early event in VC (53). Numerous studies confirm that oxidative stress activates and upregulates DNMTs (54, 55). Consistently, Li et al. demonstrated that H2O2 enhances osteogenic transdifferentiation of VSMCs by reducing ALP and RUNX2 methylation, an effect reversible by DNMT3a overexpression (56).
3.1.2 DNA methylation-mediated ROS changes in VC
DNA methylation can also affect ROS homeostasis. Folate supplementation prevents atherosclerosis by lowering homocysteine levels, increasing the S-adenosylmethionine (SAM)/S-adenosylhomocysteine (SAH) ratio, and enhancing DNMT activity (29). Folate further protects against oxidative damage and apoptosis in ApoE−/− mice by promoting DNMT activity, increasing methylation of the vascular peroxidase 1 (VPO1) promoter, and reducing VPO1 expression, thus providing vascular protection (57). MtDNA methylation also plays a role in redox regulation. Studies suggest that DNMTs can methylate mtDNA when SAM enters mitochondria (58). Liu et al. demonstrated that platelet-derived growth factor (PDGF)-BB stimulation causes DNMT1 to translocate into mitochondria, where it methylates the mtDNA D-loop. This process suppresses mtDNA transcription, impairs mitochondrial function, reduces ATP production, and results in VSMC dedifferentiation and loss of the contractile phenotype (59). These findings strengthen the link between DNA methylation dynamics, ROS, and VC (60, 61).
3.2 Histone modifications
Histone modifications have significant effects on vascular cells. In eukaryotes, nucleosomes are made up of DNA wrapped around histone octamers, with histone “tails” extending outward that undergo diverse post-translational modifications. These include acetylation, methylation, phosphorylation, ubiquitination, and sumoylation, collectively known as histone modifications. Increasing evidence suggests that histone modifications are closely associated with vascular calcification (62).
3.2.1 Histone acetylation
Histone acetylation is a dynamic and reversible process regulated by histone acetyltransferases (HATs), which add acetyl groups, and histone deacetylases (HDACs), which remove them. Acetyl-CoA acts as the donor of acetyl groups and functions both as a metabolic intermediate and as a signaling molecule in maintaining homeostasis (63). HATs and HDACs regulate the expression of genes involved in VSMC contractility, differentiation, extracellular matrix deposition, and responses to vasoactive stimuli such as angiotensin II (64).
3.2.1.1 ROS-Mediated histone acetylation changes in VC
HDACs are essential in controlling the osteogenic transition of VSMCs (65, 66). For example, HDAC1 suppresses lysine-specific demethylase 1 (LSD1) transcription via H3K9ac modification at the LSD1 promoter, activating autophagy through the mechanistic target of rapamycin (mTOR) pathway and ultimately attenuating VC (67). Similarly, HDAC8 inhibits osteogenic differentiation by suppressing H3K9 acetylation and RUNX2 expression (68, 69). Accumulating evidence suggests that oxidative stress alters HDAC activity (70). Wu et al. reported that H2O2 can inhibit HDACs such as HDAC1 and HDAC6, thereby affecting downstream gene acetylation (71–73). Moreover, NOX4-mediated oxidative stress promotes oxidative modification and nuclear translocation of HDAC4, reducing its inhibitory effect on transcription (74). In VSMCs, cytosolic HDAC4 interacts with cytoskeletal proteins such as ENIGMA (Pdlim7), a process essential for VC development (65). Thus, oxidative stress may promote VC progression by controlling HDAC4 localization and activity.
Acetyl-CoA, derived from glucose, fatty acids, or acetate, not only fuels ATP production but also serves as the primary substrate for histone acetylation. It serves as the primary substrate for histone acetylation. The majority of cytosolic acetyl-CoA is supplied by mitochondrial metabolism through ATP citrate lyase (ACLY), while acetyl-CoA synthetase 2 (ACSS2) provides an additional source from acetate (63). Consequently, the availability of acetyl-CoA establishes a link between cellular energy metabolism and epigenetic regulation, presenting a vital mechanism through which metabolic states can influence vascular calcification. Shao et al. show that the inhibition of acyl-CoA synthetase blocks the mineralization of VSMC (75).
3.2.1.2 Histone acetylation-mediated ROS changes in VC
Conversely, histone acetylation can control ROS production. Sirtuins (SIRTs), a class of NAD+-dependent lysine deacetylases, serve as important redox signaling molecules. Mitochondria play an important role in regulating the cellular NAD+/NADH ratio, which in turn controls the activities of sirtuins. By deacetylating transcription factors, SIRTs regulate the expression of enzymes that generate ROS and antioxidant defenses (76, 77). Multiple studies have identified SIRTs as key effectors in oxidative stress signaling (78–80). Overexpression of SIRT1 protects against H2O2-induced vascular dysfunction and premature aging by deacetylating p53, which results in decreased plasminogen activator inhibitor-1 (PAI-1) expression and increased eNOS activity (81, 82). SIRT1 activation has also been shown to reduce NOX-derived ROS, thus providing antioxidant and anti-aging benefits in the cardiovascular system (83, 84). Notably, SIRT1 reverses H2O2-induced DNA damage and calcification, highlighting its role in counteracting oxidative stress (1). Luteolin, a natural tetrahydroxyl flavonoid, can protect against vascular calcification by modulating the Sirtuin1 (SIRT1)/CXC Chemokine Receptor 4 (CXCR4) signaling pathway and promoting autophagy. In rats, luteolin significantly improved vascular calcification induced by a high-fat diet and vitamin D3. In vitro, it repressed the formation of mineralized nodules and ALP activity in H2O2-treated VSMCs (85). Thus, luteolin may inhibit oxidative stress-induced vascular calcification by activating SIRT1-mediated regulation. HDACs also interact with oxidative stress during VSMC osteogenic differentiation. For example, Bai et al. reported that HDAC5 inhibition reduced angiotensin II–induced oxidative stress in VSMCs (86).
Collectively, these findings indicate that histone acetylation not only mediates ROS-induced transcriptional changes but also provides feedback to control oxidative stress, thereby supporting the epigenetic–redox interaction in VC.
3.2.2 Histone methylation
Histone methylation, a major epigenetic modification, is controlled by histone methyltransferases (HMTs) and reversed by histone demethylases (HDMs). Lysine methylation is the main type in eukaryotes, with common methylation sites including H3K4, H3K9, H3K27, H3K36, H3K79, and H4K20 (87). Two major HDM families regulate histone demethylation: the LSD family and the Jumonji C (JmjC) domain-containing family (JMJD) (88). For example, Kang et al. reported that JMJD2B/KDM4B promotes osteogenic differentiation of VSMCs by lowering H3K9me2 levels at the RUNX2 promoter (89). Similar to DNA methylation, histone methylation requires SAM as the methyl donor, linking mitochondrial function to histone methylation through SAM synthesis. JMJD enzymes depend on Fe (II), oxygen, and α-KG, and are inhibited by fumarate and succinate. Mitochondrial dysfunction can cause excessive histone methylation, partly due to increased ROS, which impairs HDM activity (90). Cumulative evidence shows that histone methylation is closely linked to the initiation and progression of VC, affecting processes such as metabolic reprogramming, apoptosis, oxidative stress, and multiple signaling pathways (91).
3.2.2.1 ROS-Mediated histone methylation changes in VC
SET domain–containing 7 (SETD7), a histone methyltransferase, promotes nuclear factor kappa-B (NF-κB) activation and pro-inflammatory cytokine production via H3K4me1-dependent transcription in response to ROS (92). NF-κB signaling, in turn, plays a central role in phosphate-induced VC (93–95). Intracellular ROS can activate NF-κB, which regulates genes involved in atherosclerosis and inflammation, including interleukin-6 (IL-6) (96). Kurozumi et al. showed that IL-6 recruits JMJD2B to the RUNX2 promoter, reducing H3K9me3 and promoting VSMC calcification (97). Moreover, adenosine-mediated activation of AMP-activated protein kinase (AMPK), a central regulator of cellular energy balance (98), inhibits DNMT3b and leads to hypomethylation of the H19 promoter and decreases RUNX2 expression, thereby mitigating VSMC osteogenic differentiation (99). Since AMPK is a vital energy sensor in cellular metabolism, especially during metabolic stresses like oxidative stress, these findings imply a mechanistic connection between oxidative stress, histone methylation, and VC (100).
3.2.2.2 Histone methylation-mediated ROS changes in VC
Histone methylation can also regulate oxidative stress. Hypoxia-inducible factor-1α (HIF-1α) stabilization depends on mitochondrial ROS (101), and its activation promotes RUNX2 expression and VC (101, 102). N-acetylcysteine, a ROS scavenger, inhibits extracellular matrix calcification by suppressing HIF-1α expression (103). SETD7 has been identified as a negative regulator of HIF-1α transcriptional activity (104, 105), and Liu et al. demonstrated that SETD7 inhibits HIF-1α-mediated genes involved in metabolic reprogramming. The knockdown of SETD7 increases glucose uptake and intracellular ATP levels (104). Furthermore, SETD7 regulates ROS signaling by inhibiting peroxisome proliferator-activated receptor-γ coactivator 1α (PGC1α) and antioxidant enzymes such as SOD2 and catalase (92). Together, these findings suggest that histone methylation not only responds to oxidative stress but also actively regulates ROS production, thereby contributing to VC (Figure 3).

Figure 3. Mitochondrial metabolism tightly links oxidative stress to epigenetic regulation. Pyruvate-derived acetyl-CoA (supplied to the cytosol by ACLY) promotes histone acetylation, whereas SAM produced by folate metabolism serves as the methyl donor for DNA and histone methylation. α-KG is a co-substrate of TET and JMJD demethylases, which is generated by IDHs. It follows that metabolic changes driven by oxidative stress shape the epigenetic landscape. ACLY, ATP citrate lyase; α-KG, alpha-Ketoglutarate; 2-HG, 2-hydroxyglutarate; IDH, isocitrate dehydrogenase; FH, fumarate hydratase; SAM, S-adenosylmethionine; JMJD, Jumonji C domain-containing; TET, ten-eleven translocation; DNMT, DNA methyltransferase; HMT, histone methyltransferase; LSD, Lys-specific demethylase; HDAC, histone deacetylase.
3.3 MicroRNAs (miRNAs)
MicroRNAs (miRNAs) are small noncoding RNAs, about 20–24 nucleotides long, that suppress target gene expression by binding to the 3′ untranslated regions (UTRs) of messenger RNAs (mRNAs). Depending on their genomic location, miRNA genes are categorized as intronic, exonic, or intergenic (106). As post-transcriptional regulators, miRNAs are crucial for mRNA degradation and repression of translation (107). They are increasingly recognized as biomarkers and regulators in cardiovascular diseases, including VC.
Oxidative stress significantly impacts miRNA expression, affecting VSMC function and phenotype. For instance, miR-4463 regulates VSMC phenotypic switching under oxidative stress. When miR-4463 is downregulated, it increases osteopontin (OPN) expression while decreasing smooth muscle actin (SMA) and F-actin, thereby promoting calcification (108). Basic fibroblast growth factor (bFGF), a potential miR-4463 target, promotes VSMC migration through ROS production (109). Similarly, downregulation of miR-92b-3p reduces hypoxia-induced VSMC proliferation by inhibiting the mTOR pathway (110). Poly (ADP-ribose) polymerase 1 (PARP1) also interacts with miRNAs in VC. PARP1 suppresses miR-204 expression, thereby enhancing RUNX2 expression and promoting VSMC osteogenic transformation (111). Excessive PARP1 activation during oxidative stress leads to mitochondrial membrane depolarization (112). Therefore, the PARP1–miR-204–RUNX2 axis is a crucial connection between oxidative stress, miRNA regulation, and VC. More generally, oxidative stress influences the expression of many miRNAs, which then regulate redox sensors and adjust antioxidant defenses (113) (Figure 4).
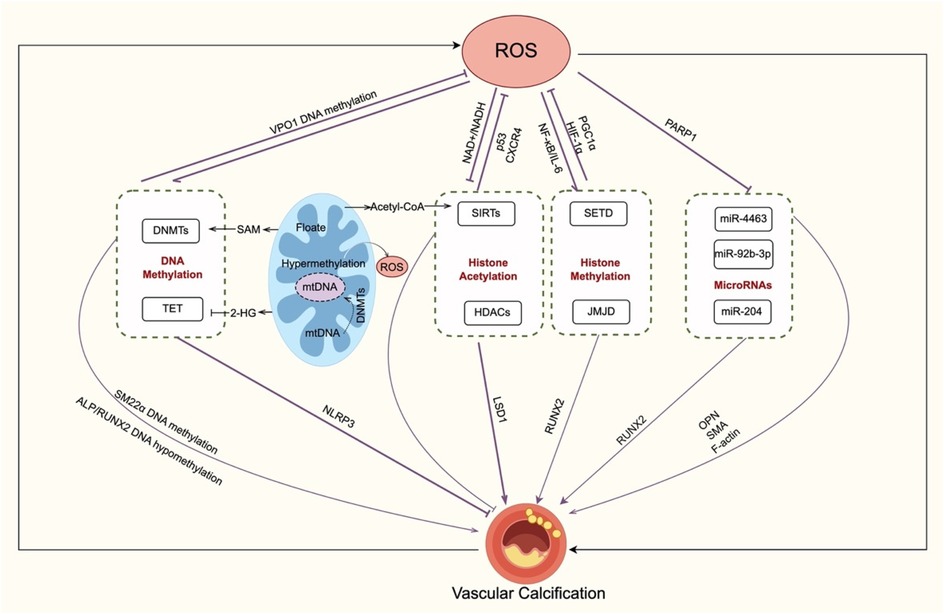
Figure 4. Crosstalk between oxidative stress and epigenetic regulation in VC. Oxidative stress and epigenetic mechanisms create a regulatory loop in VC. DNA methylation, ROS enhance DNMT activity, leading to the methylation of VSMC contractile genes (e.g., SM22α) and osteogenic activation (RUNX2, ALP). Additionally, DNA methylation influences ROS through folate–SAM pathways, VPO1 suppression, and mitochondrial DNA methylation. Histone modifications, ROS change HDAC localization and activity and suppress LSD1 transcription through histone acetylation, reducing vascular calcification; sirtuins (e.g., SIRT1) counteract oxidative stress and inhibit VC. Histone methylation (e.g., SETD, JMJD2B) connects ROS signals with NF-κB, HIF-1α, PGC-1α, and RUNX2 regulation, linking inflammation, energy metabolism, and calcification. MicroRNAs, PARP1 suppresses miR-204 during oxidative stress. ROS-sensitive miRNAs (e.g., miR-4463, miR-92b-3p, miR-204) regulating VSMC osteogenic transformation. transformation. Overall, oxidative stress not only induces but is also modulated by DNA methylation, histone modifications, and non-coding RNAs, driving VC progression. VC, vascular calcification; ROS, reactive oxygen species; DNMT, DNA methyltransferase; TET, ten–eleven translocation protein; SAM, S-adenosylmethionine; 2-HG, 2-hydroxyglutarate; VSMC, vascular smooth muscle cell; ALP, alkaline phosphatase; RUNX2, runt-related transcription factor 2; NLRP3, NLR family pyrin domain containing 3; VPO1, vascular peroxidase 1; mtDNA, mitochondrial DNA; HAT, histone acetyltransferase; HDAC, histone deacetylase; JMJD, Jumonji C domain-containing; SETD, SET domain–containing; CXCR4, CXC chemokine receptor 4; SIRT, sirtuin; AMPK, AMP-activated protein kinase; NF-κB, nuclear factor kappa-B; IL-6, interleukin-6; PGC1α, peroxisome proliferator-activated receptor-γ coactivator 1α; HIF-1α, hypoxia-inducible factor-1α; LSD, lys-specific demethylase; miRNA, microRNA; PARP1, Poly (ADP-ribose) polymerase 1; OPN, osteopontin; SMA, smooth muscle actin;.
4 Summary
Vascular calcification is a hallmark of advanced cardiovascular disease, caused by VSMC phenotypic switching from a contractile to an osteogenic state marked by RUNX2, MSX2, and ALP expression (8, 9). Oxidative stress and epigenetic reprogramming serve as central mechanisms in this process. Phosphate overload increases TCA cycle activity, leading to higher mitochondrial ROS production and connecting metabolic intermediates like acetyl-CoA and SAM to epigenetic regulation (114, 115). DNA methylation, influenced by ROS-regulated DNMT and TET activity, modifies key genes such as SM22α and RUNX2, while mtDNA methylation exacerbates mitochondrial dysfunction (51, 53–61). Histone acetylation/deacetylation (via HDACs and SIRTs) and histone methylation (e.g., H3K9, H3K4) regulate RUNX2, HIF-1α, and NF-κB pathways (67–73, 89–92, 94–98, 116–119). Additionally, miRNAs modulate the VSMC phenotype and oxidative stress responses, thereby reinforcing the feedback loop between ROS and epigenetic changes.
Considering the essential physiological roles of epigenetic mechanisms, non-specific inhibitors present therapeutic challenges. Future research should clarify how ROS, chromatin modifications (such as H3K9me3 and H3K4me1), and non-coding RNAs interact in VC, with focus on metabolic intermediates that connect energy status to epigenetic programming. Targeting this redox–epigenetic axis could lead to new strategies for preventing VC and associated cardiovascular diseases.
Author contributions
YL: Writing – original draft, Writing – review & editing, Conceptualization, Funding acquisition. HZ: Conceptualization, Writing – review & editing. YJ: Conceptualization, Writing – review & editing. WT: Writing – review & editing, Funding acquisition, Writing – original draft, Conceptualization, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by funds from the Applied Basic Research Foundation of Liaoning Province (2022JH2/101300061), Shenyang Science and Technology Project (2024)—Special Project for Public Health Research and Development (24-214-3-10), and the Scientific Research Fund of Liaoning Provincial Education Department (LJ232410159032).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. ChatGPT (OpenAI) was used solely to improve linguistic clarity, such as correcting grammar, smoothing transitions, reducing redundancy, and harmonizing spelling and terms. It wasn't used to generate ideas, draft sections, extract data, or change scientific claims.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bartoli-Leonard F, Wilkinson FL, Schiro A, Serracino Inglott F, Alexander MY, Weston R. Loss of SIRT1 in diabetes accelerates DNA damage-induced vascular calcification. Cardiovasc Res. (2021) 117(3):836–49. doi: 10.1093/cvr/cvaa134
2. Yu C, Zhang C, Kuang Z, Zheng Q. The role of NLRP3 inflammasome activities in bone diseases and vascular calcification. Inflammation. (2021) 44(2):434–49. doi: 10.1007/s10753-020-01357-z
3. Khetarpal SA, Honigberg MC, Natarajan P. Implications of premature coronary artery calcification in primary and secondary prevention of atherosclerotic cardiovascular disease. JAMA Cardiology. (2021) 6(11):1233–4. doi: 10.1001/jamacardio.2021.3393
4. Schlieper G, Aretz A, Verberckmoes SC, Krüger T, Behets GJ, Ghadimi R, et al. Ultrastructural analysis of vascular calcifications in uremia. J Am Soc Nephrol. (2010) 21(4):689–96. doi: 10.1681/ASN.2009080829
5. van der Toorn JE, Rueda-Ochoa OL, van der Schaft N, Vernooij MW, Ikram MA, Bos D, et al. Arterial calcification at multiple sites: sex-specific cardiovascular risk profiles and mortality risk-the rotterdam study. BMC Med. (2020) 18(1):263. doi: 10.1186/s12916-020-01722-7
6. Zhang H, Li G, Yu X, Yang J, Jiang A, Cheng H, et al. Progression of vascular calcification and clinical outcomes in patients receiving maintenance dialysis. JAMA Netw Open. (2023) 6(5):e2310909. doi: 10.1001/jamanetworkopen.2023.10909
7. Skenteris NT, Seime T, Witasp A, Karlöf E, Wasilewski GB, Heuschkel MA, et al. Osteomodulin attenuates smooth muscle cell osteogenic transition in vascular calcification. Clin Transl Med. (2022) 12(2):e682. doi: 10.1002/ctm2.682
8. Abbasian N. Vascular calcification mechanisms: updates and renewed insight into signaling pathways involved in high phosphate-mediated vascular smooth muscle cell calcification. Biomedicines. (2021) 9(7):804. doi: 10.3390/biomedicines9070804
9. Sutton NR, Malhotra R, St. Hilaire C, Aikawa E, Blumenthal RS, Gackenbach G, et al. Molecular mechanisms of vascular health: insights from vascular aging and calcification. Arterioscler Thromb Vasc Biol. (2023) 43(1):15–29. doi: 10.1161/ATVBAHA.122.317332
10. Lee SJ, Lee I-K, Jeon J-H. Vascular calcification-new insights into its mechanism. Int J Mol Sci. (2020) 21(8):2685. doi: 10.3390/ijms21082685
11. Huang J, Hao J, Wang P, Xu Y. The role of mitochondrial dysfunction in CKD-related vascular calcification: from mechanisms to therapeutics. Kidney Int Rep. (2024) 9(9):2596–607. doi: 10.1016/j.ekir.2024.05.005
12. Lian Y, Xie C, Feng M, Zhu H, Chen X, Liu X, et al. Identification of epigenetic regulators of vascular calcification with a CRISPR-based screen. J Am Soc Nephrol. (2025). doi: 10.1681/ASN.0000000793
13. Boulghobra D, Grillet P-E, Laguerre M, Tenon M, Fauconnier J, Fança-Berthon P, et al. Sinapine, but not sinapic acid, counteracts mitochondrial oxidative stress in cardiomyocytes. Redox Biol. (2020) 34:101554. doi: 10.1016/j.redox.2020.101554
14. Dai Y, Guo Y, Tang W, Chen D, Xue L, Chen Y, et al. Reactive oxygen species-scavenging nanomaterials for the prevention and treatment of age-related diseases. J Nanobiotechnol. (2024) 22(1):252. doi: 10.1186/s12951-024-02501-9
15. Kozlov AV, Javadov S, Sommer N. Cellular ROS and antioxidants: physiological and pathological role. Antioxidants (Basel). (2024) 13(5):602. doi: 10.3390/antiox13050602
16. Napolitano G, Fasciolo G, Venditti P. Mitochondrial management of reactive oxygen species. Antioxidants (Basel). (2021) 10(11):1824. doi: 10.3390/antiox10111824
17. Zhang Y, Murugesan P, Huang K, Cai H. NADPH Oxidases and oxidase crosstalk in cardiovascular diseases: novel therapeutic targets. Nat Rev Cardiol. (2020) 17(3):170–94. doi: 10.1038/s41569-019-0260-8
18. Pecchillo Cimmino T, Ammendola R, Cattaneo F, Esposito G. NOX Dependent ROS generation and cell metabolism. Int J Mol Sci. (2023) 24(3):2086. doi: 10.3390/ijms24032086
19. Mailloux RJ. An update on mitochondrial reactive oxygen species production. Antioxidants (Basel). (2020) 9(6):472. doi: 10.3390/antiox9060472
20. Kuznetsov AV, Margreiter R, Ausserlechner MJ, Hagenbuchner J. The Complex interplay between mitochondria, ROS and entire cellular metabolism. Antioxidants (Basel). (2022) 11(10):1995. doi: 10.3390/antiox11101995
21. Halliwell B. Understanding mechanisms of antioxidant action in health and disease. Nat Rev Mol Cell Biol. (2024) 25(1):13–33. doi: 10.1038/s41580-023-00645-4
22. Lacerda-Abreu MA, Meyer-Fernandes JR. Hyperphosphataemia and NADPH oxidase regulation in pathophysiological processes: implications for oxidative stress and disease progression. Antioxidants (Basel). (2025) 14(4):461. doi: 10.3390/antiox14040461
23. Nguyen NT, Nguyen TT, Park K-S. Oxidative stress related to plasmalemmal and mitochondrial phosphate transporters in vascular calcification. Antioxidants (Basel). (2022) 11(3):494. doi: 10.3390/antiox11030494
24. Phadwal K, Vrahnas C, Ganley IG, MacRae VE. Mitochondrial dysfunction: cause or consequence of vascular calcification? Front Cell Dev Biol. (2021) 9:611922. doi: 10.3389/fcell.2021.611922
25. Fu Z-J, Wang Z-Y, Xu L, Chen X-H, Li X-X, Liao W-T, et al. HIF-1α-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol. (2020) 36:101671. doi: 10.1016/j.redox.2020.101671
26. Liu D, Qin H, Gao Y, Sun M, Wang M. Cardiovascular disease: mitochondrial dynamics and mitophagy crosstalk mechanisms with novel programmed cell death and macrophage polarisation. Pharmacol Res. (2024) 206:107258. doi: 10.1016/j.phrs.2024.107258
27. Aishwarya R, Alam S, Abdullah CS, Morshed M, Nitu SS, Panchatcharam M, et al. Pleiotropic effects of mdivi-1 in altering mitochondrial dynamics, respiration, and autophagy in cardiomyocytes. Redox Biol. (2020) 36:101660. doi: 10.1016/j.redox.2020.101660
28. Rogers MA, Maldonado N, Hutcheson JD, Goettsch C, Goto S, Yamada I, et al. Dynamin-Related protein 1 inhibition attenuates cardiovascular calcification in the presence of oxidative stress. Circ Res. (2017) 121(3):220–33. doi: 10.1161/CIRCRESAHA.116.310293
29. Cui S, Li W, Lv X, Wang P, Gao Y, Huang G. Folic acid supplementation delays atherosclerotic lesion development by modulating MCP1 and VEGF DNA methylation levels in vivo and in vitro. Int J Mol Sci. (2017) 18(5):990. doi: 10.3390/ijms18050990
30. Li Y, Shen Y, Jin K, Wen Z, Cao W, Wu B, et al. The DNA repair nuclease MRE11A functions as a mitochondrial protector and prevents T cell pyroptosis and tissue inflammation. Cell Metab. (2019) 30(3):477–92. doi: 10.1016/j.cmet.2019.06.016
31. Newman LE, Weiser Novak S, Rojas GR, Tadepalle N, Schiavon CR, Grotjahn DA, et al. Mitochondrial DNA replication stress triggers a pro-inflammatory endosomal pathway of nucleoid disposal. Nat Cell Biol. (2024) 26(2):194–206. doi: 10.1038/s41556-023-01343-1
32. Wang P, Wu B, You S, Lu S, Xiong S, Zou Y, et al. DNA Polymerase gamma recovers mitochondrial function and inhibits vascular calcification by interacted with p53. Int J Biol Sci. (2022) 18(1):409–25. doi: 10.7150/ijbs.65030
33. Godoy JA, Rios JA, Picón-Pagès P, Herrera-Fernández V, Swaby B, Crepin G, et al. Mitostasis, calcium and free radicals in health, aging and neurodegeneration. Biomolecules. (2021) 11(7):1012. doi: 10.3390/biom11071012
34. Greco T, Shafer J, Fiskum G. Sulforaphane inhibits mitochondrial permeability transition and oxidative stress. Free Radical Biol Med. (2011) 51(12):2164–71. doi: 10.1016/j.freeradbiomed.2011.09.017
35. Bernardi P, Rasola A, Forte M, Lippe G. The mitochondrial permeability transition pore: channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol Rev. (2015) 95(4):1111–55. doi: 10.1152/physrev.00001.2015
36. Šileikytė J, Forte M. The mitochondrial permeability transition in mitochondrial disorders. Oxid Med Cell Longevity. (2019) 2019:3403075. doi: 10.1155/2019/3403075
37. Ying KE, Feng W, Ying W-Z, Li X, Xing D, Sun Y, et al. Dietary salt initiates redox signaling between endothelium and vascular smooth muscle through NADPH oxidase 4. Redox Biol. (2022) 52:102296. doi: 10.1016/j.redox.2022.102296
38. Madreiter-Sokolowski CT, Thomas C, Ristow M. Interrelation between ROS and ca(2+) in aging and age-related diseases. Redox Biol. (2020) 36:101678. doi: 10.1016/j.redox.2020.101678
39. Kay AM, Simpson CL, Stewart JA Jr. The role of AGE/RAGE signaling in diabetes-mediated vascular calcification. J Diabetes Res. (2016) 2016:6809703. doi: 10.1155/2016/6809703
40. Belmokhtar K, Ortillon J, Jaisson S, Massy ZA, Boulagnon Rombi C, Doué M, et al. Receptor for advanced glycation end products: a key molecule in the genesis of chronic kidney disease vascular calcification and a potential modulator of sodium phosphate co-transporter PIT-1 expression. Nephrol Dial Transplant. (2019) 34(12):2018–30. doi: 10.1093/ndt/gfz012
41. Koike S, Yano S, Tanaka S, Sheikh A, Nagai A, Sugimoto T. Advanced glycation End-products induce apoptosis of vascular smooth muscle cells: a mechanism for vascular calcification. Int J Mol Sci. (2016) 17(9):1567. doi: 10.3390/ijms17091567
42. Varadharaj S, Kelly OJ, Khayat RN, Kumar PS, Ahmed N, Zweier JL. Role of dietary antioxidants in the preservation of vascular function and the modulation of health and disease. Front Cardiovasc Med. (2017) 4:64. doi: 10.3389/fcvm.2017.00064
43. Guillaumet-Adkins A, Yañez Y, Peris-Diaz MD, Calabria I, Palanca-Ballester C, Sandoval J. Epigenetics and oxidative stress in aging. Oxid Med Cell Longevity. (2017) 2017:9175806. doi: 10.1155/2017/9175806
44. García-Guede Á, Vera O, Ibáñez-de-Caceres I. When oxidative stress meets epigenetics: implications in cancer development. Antioxidants (Basel). (2020) 9(6):468. doi: 10.3390/antiox9060468
45. Shi Y, Zhang H, Huang S, Yin L, Wang F, Luo P, et al. Epigenetic regulation in cardiovascular disease: mechanisms and advances in clinical trials. Signal Transduct Targeted Ther. (2022) 7(1):200. doi: 10.1038/s41392-022-01055-2
46. Kajuluri LP, Guo YY, Lee S, Christof M, Malhotra R. Epigenetic regulation of human vascular calcification. Genes (Basel). (2025) 16(5):506. doi: 10.3390/genes16050506
47. Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet. (2005) 37(8):853–62. doi: 10.1038/ng1598
48. Liang J, Na X, Meng L, He L, Shu T, Fang Y, et al. Ten-Eleven translocation family proteins: structure, biological functions, diseases, and targeted therapy. MedComm (2020). (2025) 6(7):e70245. doi: 10.1002/mco2.70245
49. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. (2010) 18(6):553–67. doi: 10.1016/j.ccr.2010.11.015
50. Lu Y, Kwintkiewicz J, Liu Y, Tech K, Frady LN, Su Y-T, et al. Chemosensitivity of IDH1-mutated gliomas due to an impairment in PARP1-mediated DNA repair. Cancer Res. (2017) 77(7):1709–18. doi: 10.1158/0008-5472.CAN-16-2773
51. Liu R, Jin Y, Tang WH, Qin L, Zhang X, Tellides G, et al. Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity. Circulation. (2013) 128(18):2047–57. doi: 10.1161/CIRCULATIONAHA.113.002887
52. Fu M, Lan Z, Ye Y, Gong Y, Liang Q, Li M, et al. The metabolite alpha-ketoglutarate inhibits vascular calcification partially through modulation of the TET2/NLRP3 inflammasome signaling pathway. Kidney Int. (2025) 108(2):233–52. doi: 10.1016/j.kint.2025.04.016
53. de Oca AM, Madueño JA, Martinez-Moreno JM, Guerrero F, Muñoz-Castañeda J, Rodriguez-Ortiz ME, et al. High-phosphate-induced calcification is related to SM22α promoter methylation in vascular smooth muscle cells. J Bone Miner Res. (2010) 25(9):1996–2005. doi: 10.1002/jbmr.93
54. Maugeri A, Mazzone MG, Giuliano F, Vinciguerra M, Basile G, Barchitta M, et al. Curcumin modulates DNA methyltransferase functions in a cellular model of diabetic retinopathy. Oxid Med Cell Longevity. (2018) 2018:5407482. doi: 10.1155/2018/5407482
55. Shrishrimal S, Kosmacek EA, Oberley-Deegan RE. Reactive oxygen Species drive epigenetic changes in radiation-induced fibrosis. Oxid Med Cell Longevity. (2019) 2019:4278658. doi: 10.1155/2019/4278658
56. Li L, Ling Z, Dong W, Chen X, Vater C, Liao H, et al. Dnmt3a-Mediated DNA methylation changes regulate osteogenic differentiation of hMSCs cultivated in the 3D scaffolds under oxidative stress. Oxid Med Cell Longevity. (2019) 2019:4824209. doi: 10.1155/2019/4824209
57. Cui S, Lv X, Li W, Li Z, Liu H, Gao Y, et al. Folic acid modulates VPO1 DNA methylation levels and alleviates oxidative stress-induced apoptosis in vivo and in vitro. Redox Biol. (2018) 19:81–91. doi: 10.1016/j.redox.2018.08.005
58. Matilainen O, Quirós PM, Auwerx J. Mitochondria and epigenetics—crosstalk in homeostasis and stress. Trends Cell Biol. (2017) 27(6):453–63. doi: 10.1016/j.tcb.2017.02.004
59. Liu Y-F, Zhu J-J, Yu Tian X, Liu H, Zhang T, Zhang Y-P, et al. Hypermethylation of mitochondrial DNA in vascular smooth muscle cells impairs cell contractility. Cell Death Dis. (2020) 11(1):35. doi: 10.1038/s41419-020-2240-7
60. Zhao B, Yang Y, Wang X, Chong Z, Yin R, Song S-H, et al. Redox-active quinones induces genome-wide DNA methylation changes by an iron-mediated and tet-dependent mechanism. Nucleic Acids Res. (2014) 42(3):1593–605. doi: 10.1093/nar/gkt1090
61. Ellison EM, Abner EL, Lovell MA. Multiregional analysis of global 5-methylcytosine and 5-hydroxymethylcytosine throughout the progression of Alzheimer’s disease. J Neurochem. (2017) 140(3):383–94. doi: 10.1111/jnc.13912
62. Ma W, Jia K, Cheng H, Xu H, Li Z, Zhang H, et al. Orphan nuclear receptor NR4A3 promotes vascular calcification via histone lactylation. Circ Res. (2024) 134(11):1427–47. doi: 10.1161/CIRCRESAHA.123.323699
63. Pietrocola F, Galluzzi L, Bravo-San Pedro J, Madeo F, Kroemer G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. (2015) 21(6):805–21. doi: 10.1016/j.cmet.2015.05.014
64. Natarajan R. Drugs targeting epigenetic histone acetylation in vascular smooth muscle cells for restenosis and atherosclerosis. Arterioscler Thromb Vasc Biol. (2011) 31(4):725–7. doi: 10.1161/ATVBAHA.111.222976
65. Abend A, Shkedi O, Fertouk M, Caspi LH, Kehat I. Salt-inducible kinase induces cytoplasmic histone deacetylase 4 to promote vascular calcification. EMBO Rep. (2017) 18(7):1166–85. doi: 10.15252/embr.201643686
66. Malhotra R, Mauer AC, Lino Cardenas CL, Guo X, Yao J, Zhang X, et al. HDAC9 Is implicated in atherosclerotic aortic calcification and affects vascular smooth muscle cell phenotype. Nat Genet. (2019) 51(11):1580–7. doi: 10.1038/s41588-019-0514-8
67. Zhou J, Zhou H, Liu C, Huang L, Lu D, Gao C. HDAC1-mediated Deacetylation of LSD1 regulates vascular calcification by promoting autophagy in chronic renal failure. J Cell Mol Med. (2020) 24(15):8636–49. doi: 10.1111/jcmm.15494
68. Fu Y, Zhang P, Ge J, Cheng J, Dong W, Yuan H, et al. Histone deacetylase 8 suppresses osteogenic differentiation of bone marrow stromal cells by inhibiting histone H3K9 acetylation and RUNX2 activity. Int J Biochem Cell Biol. (2014) 54:68–77. doi: 10.1016/j.biocel.2014.07.003
69. Yang H, Liu Y, Liu X, Gu H, Zhang J, Sun C. Concise review: the regulatory mechanism of lysine acetylation in mesenchymal stem cell differentiation. Stem Cells Int. (2020) 2020:7618506. doi: 10.1155/2020/7618506
70. Du Y, Tang G, Yuan W. Suppression of HDAC2 by sodium butyrate alleviates apoptosis of kidney cells in db/db mice and HG-induced NRK-52E cells. Int J Mol Med. (2020) 45(1):210–22. doi: 10.3892/ijmm.2019.4397
71. Cyr AR, Domann FE. The redox basis of epigenetic modifications: from mechanisms to functional consequences. Antioxid Redox Signal. (2011) 15(2):551–89. doi: 10.1089/ars.2010.3492
72. Wu C-C, Lee P-T, Kao T-J, Chou S-Y, Su R-Y, Lee Y-C, et al. Upregulation of Znf179 acetylation by SAHA protects cells against oxidative stress. Redox Biol. (2018) 19:74–80. doi: 10.1016/j.redox.2018.08.001
73. Man AWC, Chen M, Wu Z, Reifenberg G, Daiber A, Münzel T, et al. Renal effects of fetal reprogramming with pentaerythritol tetranitrate in spontaneously hypertensive rats. Front Pharmacol. (2020) 11:454. doi: 10.3389/fphar.2020.00454
74. Matsushima S, Kuroda J, Ago T, Zhai P, Park JY, Xie L-H, et al. Increased oxidative stress in the nucleus caused by Nox4 mediates oxidation of HDAC4 and cardiac hypertrophy. Circ Res. (2013) 112(4):651–63. doi: 10.1161/CIRCRESAHA.112.279760
75. Ting TC, Miyazaki-Anzai S, Masuda M, Levi M, Demer LL, Tintut Y, et al. Increased lipogenesis and stearate accelerate vascular calcification in calcifying vascular cells. J Biol Chem. (2011) 286(27):23938–49. doi: 10.1074/jbc.M111.237065
76. Wang X, Liu M, Zhu M, Shi L, Liu L, Zhao Y, et al. Resveratrol protects the integrity of alveolar epithelial barrier via SIRT1/PTEN/p-akt pathway in methamphetamine-induced chronic lung injury. Cell Prolif. (2020) 53(3):e12773. doi: 10.1111/cpr.12773
77. Li M, Chiang Y-L, Lyssiotis CA, Teater MR, Hong JY, Shen H, et al. Non-oncogene addiction to SIRT3 plays a critical role in lymphomagenesis. Cancer Cell. (2019) 35(6):916–31.e9. doi: 10.1016/j.ccell.2019.05.002
78. Webster BR, Lu Z, Sack MN, Scott I. The role of sirtuins in modulating redox stressors. Free Radical Biol Med. (2012) 52(2):281–90. doi: 10.1016/j.freeradbiomed.2011.10.484
79. Merksamer PI, Liu Y, He W, Hirschey MD, Chen D, Verdin E. The sirtuins, oxidative stress and aging: an emerging link. Aging. (2013) 5(3):144–50. doi: 10.18632/aging.100544
80. Wu YT, Wu SB, Wei YH. Roles of sirtuins in the regulation of antioxidant defense and bioenergetic function of mitochondria under oxidative stress. Free Radical Res. (2014) 48(9):1070–84. doi: 10.3109/10715762.2014.920956
81. Ota H, Akishita M, Eto M, Iijima K, Kaneki M, Ouchi Y. Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J Mol Cell Cardiol. (2007) 43(5):571–9. doi: 10.1016/j.yjmcc.2007.08.008
82. Cencioni C, Spallotta F, Martelli F, Valente S, Mai A, Zeiher A, et al. Oxidative stress and epigenetic regulation in ageing and age-related diseases. Int J Mol Sci. (2013) 14(9):17643–63. doi: 10.3390/ijms140917643
83. Zarzuelo MJ, López-Sepúlveda R, Sánchez M, Romero M, Gómez-Guzmán M, Ungvary Z, et al. SIRT1 Inhibits NADPH oxidase activation and protects endothelial function in the rat aorta: implications for vascular aging. Biochem Pharmacol. (2013) 85(9):1288–96. doi: 10.1016/j.bcp.2013.02.015
84. Gano LB, Donato AJ, Pasha HM, Hearon CM, Sindler AL, Seals DR. The SIRT1 activator SRT1720 reverses vascular endothelial dysfunction, excessive superoxide production, and inflammation with aging in mice. Am J Physiol Heart Circ Physiol. (2014) 307(12):H1754–63. doi: 10.1152/ajpheart.00377.2014
85. Yu X, Xu L, Su C, Wang C, Wang Z, Wang Y, et al. Luteolin protects against vascular calcification by modulating SIRT1/CXCR4 signaling pathway and promoting autophagy. AAPS J. (2024) 26(6):111. doi: 10.1208/s12248-024-00982-y
86. Bai L, Kee HJ, Choi SY, Seok YM, Kim GR, Kee S-J, et al. HDAC5 Inhibition reduces angiotensin II-induced vascular contraction, hypertrophy, and oxidative stress in a mouse model. Biomed Pharmacother. (2021) 134:111162. doi: 10.1016/j.biopha.2020.111162
87. Li Q, Zou J, Wang M, Ding X, Chepelev I, Zhou X, et al. Critical role of histone demethylase Jmjd3 in the regulation of CD4+ T-cell differentiation. Nat Commun. (2014) 5:5780. doi: 10.1038/ncomms6780
88. Schiffmann I, Greve G, Jung M, Lübbert M. Epigenetic therapy approaches in non-small cell lung cancer: update and perspectives. Epigenetics. (2016) 11(12):858–70. doi: 10.1080/15592294.2016.1237345
89. Kang P, Wu Z, Huang Y, Luo Z, Huo S, Chen Q. Histone H3K9 demethylase JMJD2B/KDM4B promotes osteogenic differentiation of bone marrow-derived mesenchymal stem cells by regulating H3K9me2 on RUNX2. PeerJ. (2022) 10:e13862. doi: 10.7717/peerj.13862
90. Sullivan LB, Martinez-Garcia E, Nguyen H, Mullen A, Dufour E, Sudarshan S, et al. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol Cell. (2013) 51(2):236–48. doi: 10.1016/j.molcel.2013.05.003
91. Cao Y-C, Shan S-K, Guo B, Li C-C, Li F-X-Z, Zheng M-H, et al. Histone lysine methylation modification and its role in vascular calcification. Front Endocrinol (Lausanne). (2022) 13:863708. doi: 10.3389/fendo.2022.863708
92. He S, Owen DR, Jelinsky SA, Lin L-L. Lysine methyltransferase SETD7 (SET7/9) regulates ROS signaling through mitochondria and NFE2L2/ARE pathway. Sci Rep. (2015) 5:14368. doi: 10.1038/srep14368
93. Zhao G, Xu M-J, Zhao M-M, Dai X-Y, Kong W, Wilson GM, et al. Activation of nuclear factor-kappa B accelerates vascular calcification by inhibiting ankylosis protein homolog expression. Kidney Int. (2012) 82(1):34–44. doi: 10.1038/ki.2012.40
94. Voelkl J, Tuffaha R, Luong TTD, Zickler D, Masyout J, Feger M, et al. Zinc inhibits phosphate-induced vascular calcification through TNFAIP3-mediated suppression of NF-κB. J Am Soc Nephrol. (2018) 29(6):1636–48. doi: 10.1681/ASN.2017050492
95. Voelkl J, Lang F, Eckardt KU, Amann K, Kuro-o M, Pasch A, et al. Signaling pathways involved in vascular smooth muscle cell calcification during hyperphosphatemia. Cell Mol Life Sci. (2019) 76(11):2077–91. doi: 10.1007/s00018-019-03054-z
96. Dong X, Wu D, Zhang Y, Jia L, Pan X, Sun J, et al. Cathelicidin modulates vascular smooth muscle cell phenotypic switching through ROS/IL-6 pathway. Antioxidants (Basel). (2020) 9(6):491. doi: 10.3390/antiox9060491
97. Kurozumi A, Nakano K, Yamagata K, Okada Y, Nakayamada S, Tanaka Y. IL-6 and sIL-6R induces STAT3-dependent differentiation of human VSMCs into osteoblast-like cells through JMJD2B-mediated histone demethylation of RUNX2. Bone. (2019) 124:53–61. doi: 10.1016/j.bone.2019.04.006
98. Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res. (2018) 114(4):590–600. doi: 10.1093/cvr/cvy010
99. Dai X, Liu S, Cheng L, Huang T, Guo H, Wang D, et al. Epigenetic upregulation of H19 and AMPK inhibition concurrently contribute to S-adenosylhomocysteine hydrolase deficiency-promoted atherosclerotic calcification. Circ Res. (2022) 130(10):1565–82. doi: 10.1161/CIRCRESAHA.121.320251
100. Tanaka M, Inoue H, Takahashi N, Uehara M. AMPK Negatively regulates RANKL-induced osteoclast differentiation by controlling oxidative stress. Free Radical Biol Med. (2023) 205:107–15. doi: 10.1016/j.freeradbiomed.2023.05.033
101. Balogh E, Tóth A, Méhes G, Trencsényi G, Paragh G, Jeney V. Hypoxia triggers osteochondrogenic differentiation of vascular smooth muscle cells in an HIF-1 (hypoxia-inducible factor 1)-dependent and reactive oxygen Species-dependent manner. Arterioscler Thromb Vasc Biol. (2019) 39(6):1088–99. doi: 10.1161/ATVBAHA.119.312509
102. Mokas S, Larivière R, Lamalice L, Gobeil S, Cornfield DN, Agharazii M, et al. Hypoxia-inducible factor-1 plays a role in phosphate-induced vascular smooth muscle cell calcification. Kidney Int. (2016) 90(3):598–609. doi: 10.1016/j.kint.2016.05.020
103. Escobales N, Nuñez RE, Jang S, Parodi-Rullan R, Ayala-Peña S, Sacher JR, et al. Mitochondria-targeted ROS scavenger improves post-ischemic recovery of cardiac function and attenuates mitochondrial abnormalities in aged rats. J Mol Cell Cardiol. (2014) 77:136–46. doi: 10.1016/j.yjmcc.2014.10.009
104. Liu X, Chen Z, Xu C, Leng X, Cao H, Ouyang G, et al. Repression of hypoxia-inducible factor α signaling by Set7-mediated methylation. Nucleic Acids Res. (2015) 43(10):5081–98. doi: 10.1093/nar/gkv379
105. Gu Y, Wang Y, Wang X, Gao L, Yu W, Dong W-F. Opposite effects of SET7/9 on apoptosis of human acute myeloid leukemia cells and lung cancer cells. J Cancer. (2017) 8(11):2069–78. doi: 10.7150/jca.19143
106. Kuosmanen SM, Kansanen E, Kaikkonen MU, Sihvola V, Pulkkinen K, Jyrkkänen H-K, et al. NRF2 Regulates endothelial glycolysis and proliferation with miR-93 and mediates the effects of oxidized phospholipids on endothelial activation. Nucleic Acids Res. (2018) 46(3):1124–38. doi: 10.1093/nar/gkx1155
107. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. (2009) 136(2):215–33. doi: 10.1016/j.cell.2009.01.002
108. Wang X, Li H, Zhang Y, Liu Q, Sun X, He X, et al. Suppression of miR-4463 promotes phenotypic switching in VSMCs treated with ox-LDL. Cell Tissue Res. (2021) 383(3):1155–65. doi: 10.1007/s00441-020-03338-y
109. Schröder K, Helmcke I, Palfi K, Krause K-H, Busse R, Brandes RP. Nox1 mediates basic fibroblast growth factor-induced migration of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. (2007) 27(8):1736–43. doi: 10.1161/ATVBAHA.107.142117
110. Lee J, Heo J, Kang H. miR-92b-3p-TSC1 axis is critical for mTOR signaling-mediated vascular smooth muscle cell proliferation induced by hypoxia. Cell Death Differ. (2019) 26(9):1782–95. doi: 10.1038/s41418-018-0243-z
111. Wang C, Xu W, An J, Liang M, Li Y, Zhang F, et al. Poly(ADP-ribose) polymerase 1 accelerates vascular calcification by upregulating Runx2. Nat Commun. (2019) 10(1):1203. doi: 10.1038/s41467-019-09174-1
112. Yu C, Kim B-S, Kim E. FAF1 Mediates regulated necrosis through PARP1 activation upon oxidative stress leading to dopaminergic neurodegeneration. Cell Death Differ. (2016) 23(11):1873–85. doi: 10.1038/cdd.2016.99
113. Carbonell T, Gomes AV. MicroRNAs in the regulation of cellular redox status and its implications in myocardial ischemia-reperfusion injury. Redox Biol. (2020) 36:101607. doi: 10.1016/j.redox.2020.101607
114. Green HLH, Brewer AC. Dysregulation of 2-oxoglutarate-dependent dioxygenases by hyperglycaemia: does this link diabetes and vascular disease? Clin Epigenetics. (2020) 12(1):59. doi: 10.1186/s13148-020-00848-y
115. Shapiro IM, Risbud MV, Landis WJ. Toward understanding the cellular control of vertebrate mineralization: the potential role of mitochondria. Bone. (2024) 185:117112. doi: 10.1016/j.bone.2024.117112
116. Xiong L, Xiao Q, Chen R, Huang L, Gao J, Wang L, et al. Histone deacetylase 9 promotes osteogenic trans-differentiation of vascular smooth muscle cells via ferroptosis in chronic kidney disease vascular calcification. Renal Fail. (2024) 46(2):2422435. doi: 10.1080/0886022X.2024.2422435
117. Zhong H, Yu H, Chen J, Mok SWF, Tan X, Zhao B, et al. The short-chain fatty acid butyrate accelerates vascular calcification via regulation of histone deacetylases and NF-κB signaling. Vasc Pharmacol. (2022) 146:107096. doi: 10.1016/j.vph.2022.107096
118. Schader T, Löwe O, Reschke C, Malacarne P, Hahner F, Müller N, et al. Oxidation of HDAC4 by Nox4-derived H2O2 maintains tube formation by endothelial cells. Redox Biol. (2020) 36:101669. doi: 10.1016/j.redox.2020.101669
Keywords: oxidative stress, epigenetic modification, vascular calcification, DNA methylation, histone modification, microRNA
Citation: Lin Y, Zhang H, Jiang Y and Tian W (2025) Oxidative stress and its related epigenetic modifications in vascular calcification: mechanisms and advances. Front. Cardiovasc. Med. 12:1662989. doi: 10.3389/fcvm.2025.1662989
Received: 9 July 2025; Accepted: 6 October 2025;
Published: 20 October 2025.
Edited by:
Tatsuya Sato, Sapporo Medical University, JapanReviewed by:
Yanzhong Liu, Henan University of Chinese Medicine of Pharmacy, ChinaLova Kajuluri, Massachusetts General Hospital, United States
Copyright: © 2025 Lin, Zhang, Jiang and Tian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wen Tian, d3RpYW41NkBjbXUuZWR1LmNu