Abstract
Background:
Hypertrophic cardiomyopathy (HCM) is an autosomal dominant cardiovascular disease characterised by myocardial hypertrophy with a prevalence of approximately 0.2%–0.5%. Recently, in addition to mutations in genes encoding sarcomeric proteins, which have traditionally been implicated in the development of HCM, mutations in genes encoding non-sarcomeric proteins have also been found to be associated with the development of HCM.This report details the first documented case in China of severe HCM caused by compound heterozygous mutations in the non-sarcomeric proteins Alpha-kinase 3 (ALPK3) gene.
Case presentation:
This article reports the case of an 18-year-old female patient with HCM, who presented to hospital with sudden transient loss of consciousness while hiking,and the diagnosis was confirmed by echocardiography and genetic testing. Whole exome sequencing revealed a novel compound heterozygous variant in the ALPK3 gene (c.4234C > T nonsense mutation and c.3491G > A missense mutation) in the proband, which was reported for the first time in China. The patient presented with severe myocardial hypertrophy, biventricular involvement, occult biventricular obstruction, simian crease, history of syncope and high risk of sudden death. After ineffective conservative pharmacological treatment, the patient underwent the first international percutaneous intramyocardial septal radiofrequency ablation (PIMSRA, Liwen procedure), which resulted in complete remission of clinical symptoms 6 months after the procedure. It strongly supports the consideration of the Liwen procedure as an effective therapeutic strategy for similar patients harboring pathogenic ALPK3 variants.
Conclusions:
This case suggests that compound heterozygosity for nonsense mutations combined with missense mutations in the ALPK3 gene can lead to early-onset severe HCM, enriches the mutation spectrum of the ALPK3 gene, reveals high frequency mutation sites in exons 4 and 10 specific to East Asian populations, suggesting potential racial genetic heterogeneity, and that the Liwen procedure is a safe and effective treatment for HCM.
1 Introduction
Hypertrophic cardiomyopathy(HCM) is the most common autosomal dominant cardiovascular disease, mainly due to a pathogenic variant of the gene encoding sarcomeric proteins, or a cardiomyopathy characterised by myocardial hypertrophy of unknown etiology, the need to exclude other cardiovascular diseases or systemic, metabolic diseases caused by ventricular wall thickening, echocardiography or magnetic resonance examination of the left ventricle at end-diastole in any part of the ventricular wall thickness ≥15 mm. The diagnosis can be confirmed by a positive test for the causative gene or by examining members of genetically affected families with LV wall thickness ≥13 mm (1), and the prevalence of the disease is approximately 1/500–1/200 (2, 3), with causative or potentially causative genetic variants present in approximately 60% of cases and no clear causative gene identified in approximately 40% of cases of HCM (4).
It is now increasingly recognised that HCM has a complex genetic aetiology, in HCM patients, over 90% of the harmful genetic variants are due to the eight core genes that encode myelin. However, variants in genes encoding non-sarcomeric proteins with diverse functions, such as ACTN2, ALPK3, CSRP3, FHOD3, and FLNC, have been identified as causative in a minority of patients (5).
The gene has been mapped to chromosome 15q25.2 and contains 14 exons, ALPK3 is an atypical protein kinase family that recognises phosphorylation sites in the alpha-helix. It can broadly regulate cell migration, adhesion and proliferation, vesicular transport and protein translation (6). ALPK3 gene mutations tend to be autosomal recessive (7), Lopes et al. first reported the existence of dominant inheritance of the ALPK3 gene in 2021, and the autosomal dominant mode of inheritance is often dominated by truncating variants (8), patients who carry pathogenic variants of the ALPK3 gene may present with varying degrees of cardiac hypertrophy or dilated cardiomyopathy, heart failure and, in some cases, a combination of non-cardiac symptoms such as short stature, peculiar facial features, cleft palate, short neck, scoliosis, knee and shoulder contractures (9, 10), heterozygous mutations are characterised by significant phenotypic heterogeneity, they are particularly rare in East Asian populations (11).
Recently, our centre found the first case of ALPK3 compound heterozygous mutation (c.4234C > T nonsense mutation and c.3491G > A missense mutation) in HCM in China, and the patient underwent percutaneous intramyocardial septal radiofrequency ablation (PIMSRA, Liwen procedure). The patient recovered well after the procedure, and the 6-month follow-up showed that the septal thickness decreased from 29 mm to 15 mm, and the left ventricular end-diastolic diameter (LVDd) increased to 45 mm, and the clinical symptoms were completely relieved. This case broadens the mutational spectrum of the ALPK3 gene and provides compelling evidence supporting the efficacy of the Liwen procedure (a pioneering minimally invasive technique developed in China) for treating drug-refractory HCM. Notably, this intervention significantly improved the clinical outcome in this patient with severe ALPK3-related HCM. Additionally, analysis of ALPK3 pathogenic variant distribution identified exons 4 and 10 as frequent mutation sites in East Asian populations, suggesting a possible population-specific pattern in ALPK3-related pathogenicity.
2 Case presentation
2.1 History of illness and physical examination
The patient is a female, 18 years old, with symptoms of chest tightness and shortness of breath during activities in June 2023, and sudden loss of consciousness while hiking in December 2023 (lasting 1 min), and in June 2024, she was seen at the Department of Cardiology, First Hospital of Handan City. Physical examination: height 156 cm, weight 54.5 kg, normal development, normal intelligence, simian crease (Figure 1A), no enlargement of cardiac borders, no pathological murmur in the precordial region, and no other extracardiac signs:such as short neck, scoliosis, knee and shoulder contractures. There was no family history of sudden death or cardiomyopathy.
Figure 1
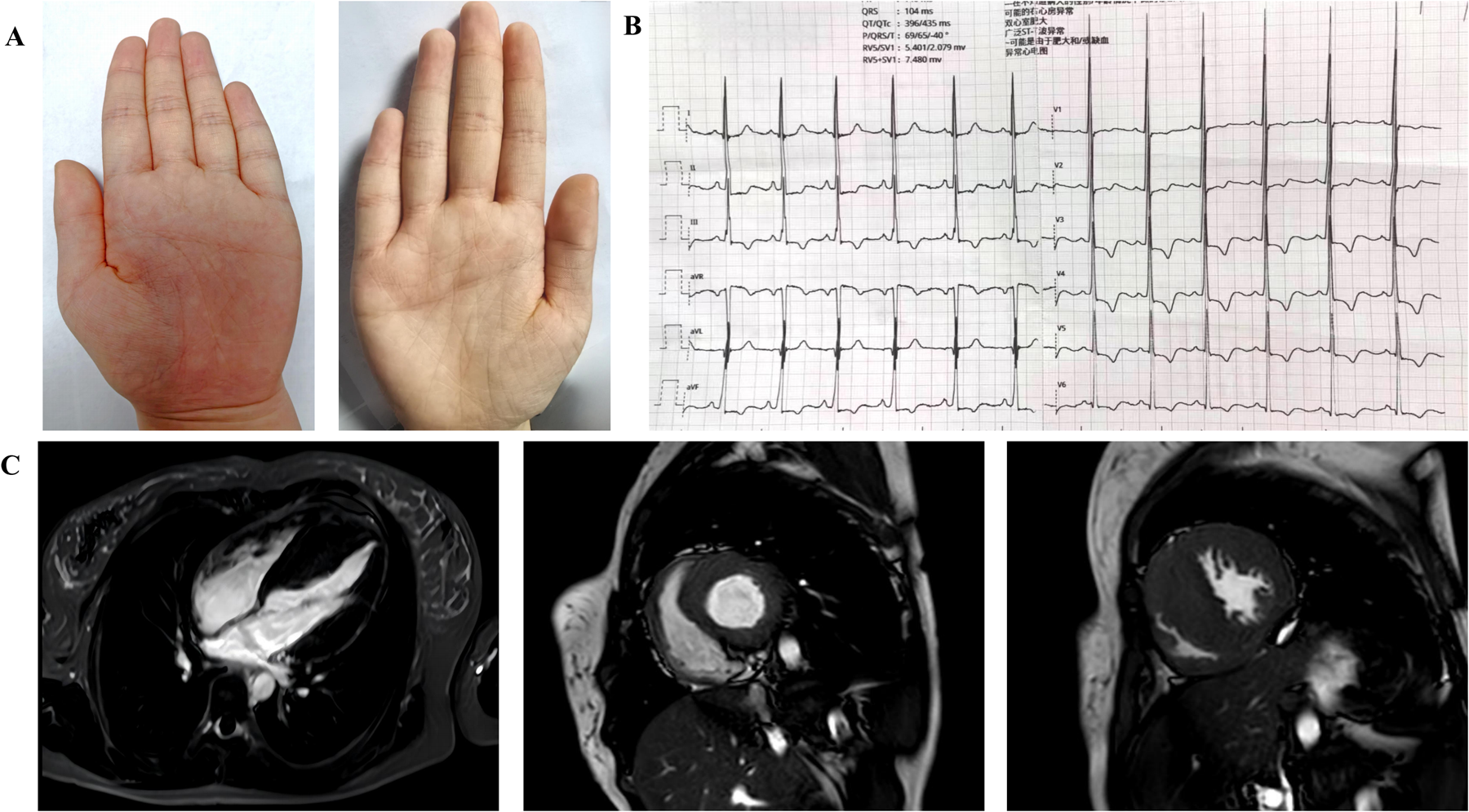
(A) Extracardiac manifestations in patients with compound heterozygous mutations in the ALPK3 gene: palms with simian creases in both hands. (B) Electrocardiogram. The electrocardiogram showed left ventricular hypervoltage and myocardial ischemia. (C) Preoperative cardiac magnetic resonance: Four-chamber view of the heart; Short-axis views of the left ventricle (end diastole). Diffuse hypertrophy of the left ventricle and thickening of the RVFW were observed. End-diastolic measurements revealed an IVS thickness of 29 mm and an RVFW thickness of 8 mm. Late gadolinium enhancement was absent in the septum and the mid-to-distal segments of the left ventricle.
2.2 Laboratory and imaging evaluation
Laboratory tests: N-terminal proB-type natriuretic peptide (NT-proBNP) level 4,015 pg/mL (reference value <125 pg/mL); Cardiac troponin T level 12 pg/mL (reference value <14 pg/mL); and no abnormalities were seen in blood routine; urine routine;liver function; kidney function; coagulation function; thyroid function;and cardiac enzymes.
Electrocardiogram: A 12-lead electrocardiogram suggests left ventricular hypervoltage and myocardial ischemia (Figure 1B). A 24-hour ambulatory electrocardiogram was performed, but no malignant arrhythmias were detected.
Transthoracic echocardiography demonstrated increased myocardial thickness with an interventricular septum (IVS) of 29 mm and a right ventricular free wall (RVFW) measuring 8 mm. The LVDd was 37 mm with a left ventricular end-diastolic volume (LVEDV) of 56.9 mL. At rest, the left ventricular outflow tract pressure gradient (LVOT-PG) is 5.6 mmHg (Vmax 119 cm/s) and in the left ventricular intracavitary compartment pressure gradient (LVIC-PG) is 4.3 mmHg (Vmax 104 cm/s), the right ventricular outflow tract pressure gradient (RVOT-PG) exhibited elevated resting gradients at 18 mmHg (Vmax 214 cm/s). Following provocation with exercise echocardiography, significant dynamic increases were observed: LVOT-PG escalated to 20 mmHg (Vmax 224 cm/s), LVIC-PG rose to 31 mmHg (Vmax 280 cm/s), and RVOT-PG reached 28 mmHg (Vmax 261 cm/s). These findings collectively support the diagnosis of occult biventricular obstructive HCM.
Cardiac magnetic resonance showed asymmetric hypertrophy of the IVS(thickness of 29 mm) and thickening of the RVFW (8 mm), and late gadolinium enhancement was negative(Figure 1C).
Coronary CTA: There is no stenosis or malformation of the coronary arteries.
2.3 Whole exome sequencing
A peripheral blood sample was collected from the patient in an EDTA anticoagulant tube and kept at 4°C for under 6 h. DNA was extracted using the Blood Genome Column Medium Extraction Kit (Tiangen Biotech, Beijing, China) as per the instructions. Using the SureSelectXT Clinical Research Exome V4, the protein-coding exome was enriched. Whole exome sequencing (WES) was conducted on the Illumina Hiseq X10 platform (Illumina, San Diego, CA, USA), with initial quality control carried out using FastP to process raw data and eliminate low-quality reads.Variants were annotated based on minor allele frequencies from databases and the American College of Medical Genetics’ practical guidelines on pathogenicity. The pathogenicity of variants was predicted using MutationTaster software and CADD scaled c-scores, with the GRCh37 reference genome serving for alignment. We examined databases such as gnomAD, ExAC, and 1,000 G to determine the prevalence of variants.
Genetic analysis revealed that the proband carried a compound heterozygous mutation in the ALPK3 gene. His immediate family members underwent Sanger sequencing for the paternally derived nonsense mutation ALPK3: c.4234C > T (p.Arg1412Ter) and the maternal missense mutation c.3491G > A (p.Arg1164Gln). According to the American College of Medical Genetics (ACMG) guidelines for mutation classification, the mutations c.4234C > T (p.Arg1412Ter) were categorized as pathogenic variants (PVS1 + PM2 + PM3) and c.3491G > A (p.Arg1164Gln) was classified as a variant of undetermined significance (PM2 + PP3). However, the c.3491G > A variant has since been reported in a Turkish HCM patient, where it was identified in a compound heterozygous state with the p.Ser653Ter variant (12). This evidence bolsters the plausibility that the c.3491G > A variant, likely resulting in a loss of function, contributes to the disease phenotype in a compound heterozygous state with the p.Arg1412Ter variant. The brother carries the ALPK3 c.3491G > A (p.Arg1164Gln), the parents and brother are single heterozygous carriers with a normal phenotype, the proband was compound heterozygous for both variants and manifested HCM, the grandparents refused the genetic test (Figures 2A,B).
Figure 2
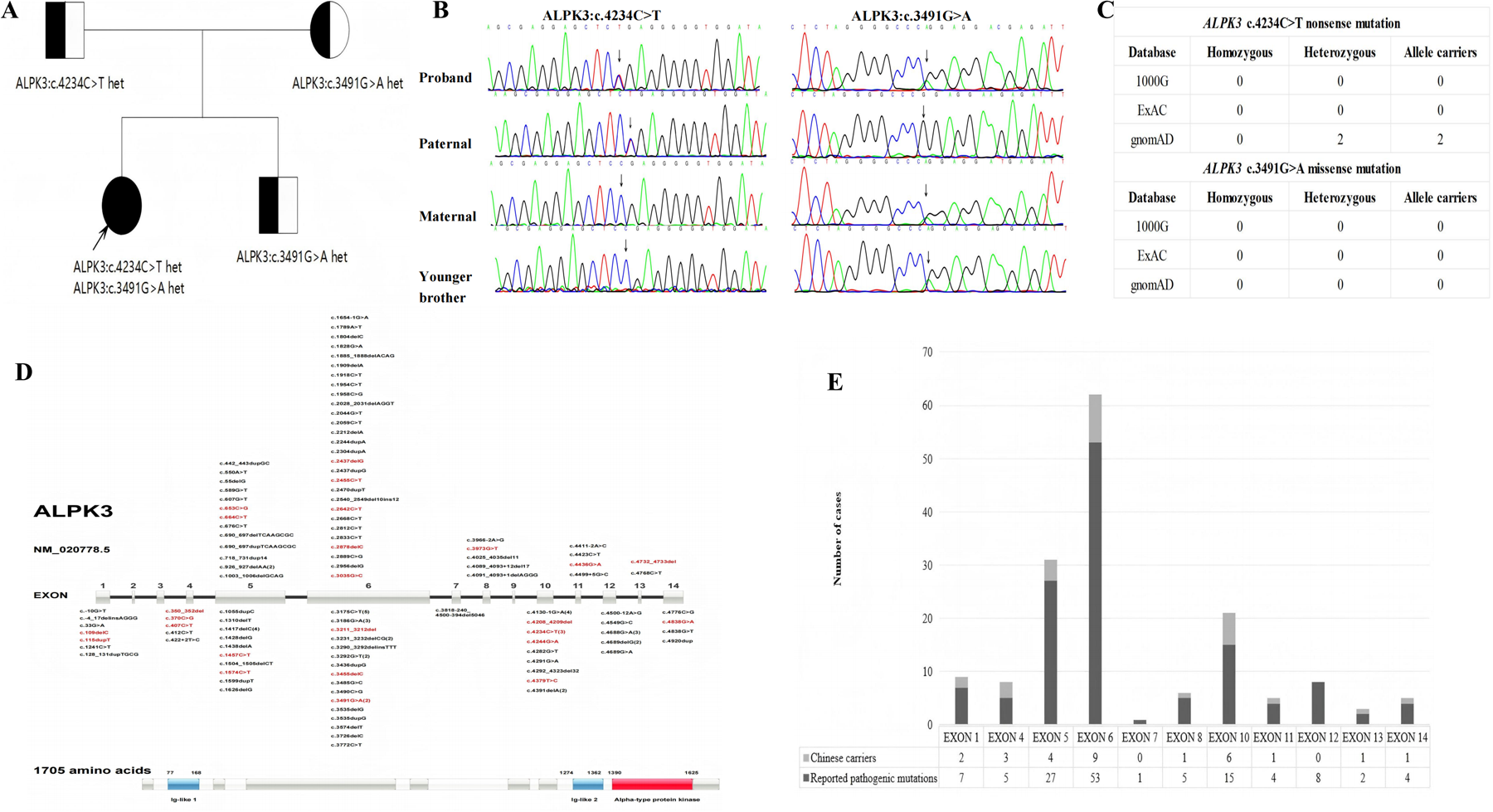
(A) family tree of ALPK3 gene mutation. □ Male, ○ Female, ● Patient,  Carrier,
Carrier,  Proband. (B) Sanger sequencing of the proband's first-degree relatives.(C) Variant frequency analysis of the ALPK3 mutations (c.4234C > T nonsense and c.3491G > A missense) across three major population genomic databases (gnomAD, ExAC, 1,000 Genomes Project) demonstrated.(D) The domain architecture and mutation distribution of ALPK3:Distribution of pathogenic variants in ALPK3 across populations in China and abroad. Red indicates carriers in the Chinese population, while black indicates carriers in non-Chinese populations.(E) Pathogenic variants in the ALPK3 gene exhibit specific distribution characteristics within human populations. Pathogenic mutations in this gene primarily occur in exons 5, 6, and 10. Variations in these three exons account for 72.52% of all reported cases. Variations in exon 4 (60%) and exon 10 (40%) are more prevalent in the Chinese population than the global average, suggesting potential racial specificity.
Proband. (B) Sanger sequencing of the proband's first-degree relatives.(C) Variant frequency analysis of the ALPK3 mutations (c.4234C > T nonsense and c.3491G > A missense) across three major population genomic databases (gnomAD, ExAC, 1,000 Genomes Project) demonstrated.(D) The domain architecture and mutation distribution of ALPK3:Distribution of pathogenic variants in ALPK3 across populations in China and abroad. Red indicates carriers in the Chinese population, while black indicates carriers in non-Chinese populations.(E) Pathogenic variants in the ALPK3 gene exhibit specific distribution characteristics within human populations. Pathogenic mutations in this gene primarily occur in exons 5, 6, and 10. Variations in these three exons account for 72.52% of all reported cases. Variations in exon 4 (60%) and exon 10 (40%) are more prevalent in the Chinese population than the global average, suggesting potential racial specificity.
Variant frequency analysis of the ALPK3 mutations (c.4234C > T nonsense and c.3491G > A missense) across three major population genomic databases (gnomAD, ExAC, 1,000 Genomes Project) demonstrated: The c.4234C > T nonsense variant was observed in 2 heterozygous carriers exclusively in gnomAD, with no occurrences in other databases. No homozygous or heterozygous carriers of the c.3491G > A missense variant were detected in any database. These robust findings provide compelling evidence that the compound heterozygous genotype (c.4234C > T and c.3491G > A) represents the first reported pathogenic variant combination associated with early-onset severe HCM in China. However, it requires further follow-up observation to determine whether the ALPK3 gene c.4234C > T nonsense mutation or the ALPK3 gene c.3491G > A missense mutation alone leads to HCM (Figure 2C).
2.4 The domain architecture and mutation distribution of ALPK3
Distribution of ALPK3 disease-causing variants: A database search using the terms “ALPK3 gene” (database established until April 2025) revealed that 131 ALPK3 mutation sites have been reported worldwide, of these, 72.52% are concentrated in exons 5, 6, and 10. The distribution of mutations in the Chinese population was consistent with the global pattern; however, the detection rates for exons 4 and 10 were significantly higher (60% and 40%, respectively), suggesting potential race specificity. However, due to the small number of cases in China (n = 28), regional differences in detection rates require verification by increasing the sample size (Figures 2D,E).
2.5 Treatment and follow-up
In this case, the patient presented with biventricular hypertrophy, early-onset heart failure, chest tightness during daily activities, and cardiac function class III (New York Heart Association, NYHA) classification, and the patient also had a history of syncope. During exercise echocardiography, the patient achieved the target heart rate (171 bpm) using a standardized protocol (3 stages with a 25W/3 min increment). The peak gradient was observed at 1 min of recovery, hemodynamic measurements demonstrated a LVOT-PGmax of 20 mmHg, an LVIC-PGmax of 31 mmHg, and a RVOT-PGmax of 28 mmHg, based on these hemodynamic findings during physiological provocation, the patient was diagnosed with occult left ventricular intracavitary obstruction accompanied by concurrent RVOT obstruction.
Given the proband's status as a young female with high functional demand, the patient and family jointly opted for the Liwen procedure to minimize disruption to daily activities and academic pursuits. The Liwen procedure™ was performed under real-time echocardiographic guidance without cardioplegic arrest, a radiofrequency ablation needle was percutaneously advanced through the intercostal space and cardiac apex to target hypertrophic septal myocardium, high-frequency alternating current from the needle tip induced localized hyperthermia, myocardial dehydration, and irreversible coagulative necrosis, concurrent ablation of septal branches interrupted vascular supply to hypertrophic tissue, reducing septal thickness and relieving obstruction. On July 10, 2024, the patient underwent this procedure under general anesthesia in the left lateral decubitus position (right shoulder elevated 30°), with continuous electrocardiographic and echocardiographic monitoring, following ultrasound-guided localization of the puncture site, the Cool-tip™ ACT-2020 radiofrequency needle was advanced along the long axis of the interventricular septum to hypertrophic regions, sequential ablation was performed at: the basal septum (Zones I, II, III), mid-ventricular septum (Zones I, II, III) and the apical region, using maximum power outputs of 70W, 80W, and 60W for each respective zone(mean duration: 10 min/zone), a total of 7 ablations were performed (13, 14). The procedure was completed without any intraoperative or postoperative complications.
Transthoracic echocardiography was performed during the 6-month follow-up period after the procedure,septal thickness decreased from 29 mm to 15 mm, the LVDd increased from 37 mm to 45 mm and an increased from 56.9 mL to 101 mL was seen in the LVEDV (Figures 3A,B), left ventricular mass and hemodynamic measurements were significantly reduced, NT-proBNP was 2,233 pg/mL, the patient exhibited excellent clinical recovery with complete resolution of exertional symptoms, including chest tightness and dyspnea, there was also an absence of syncopal episodes, detailed data are shown in Table 1.
Figure 3
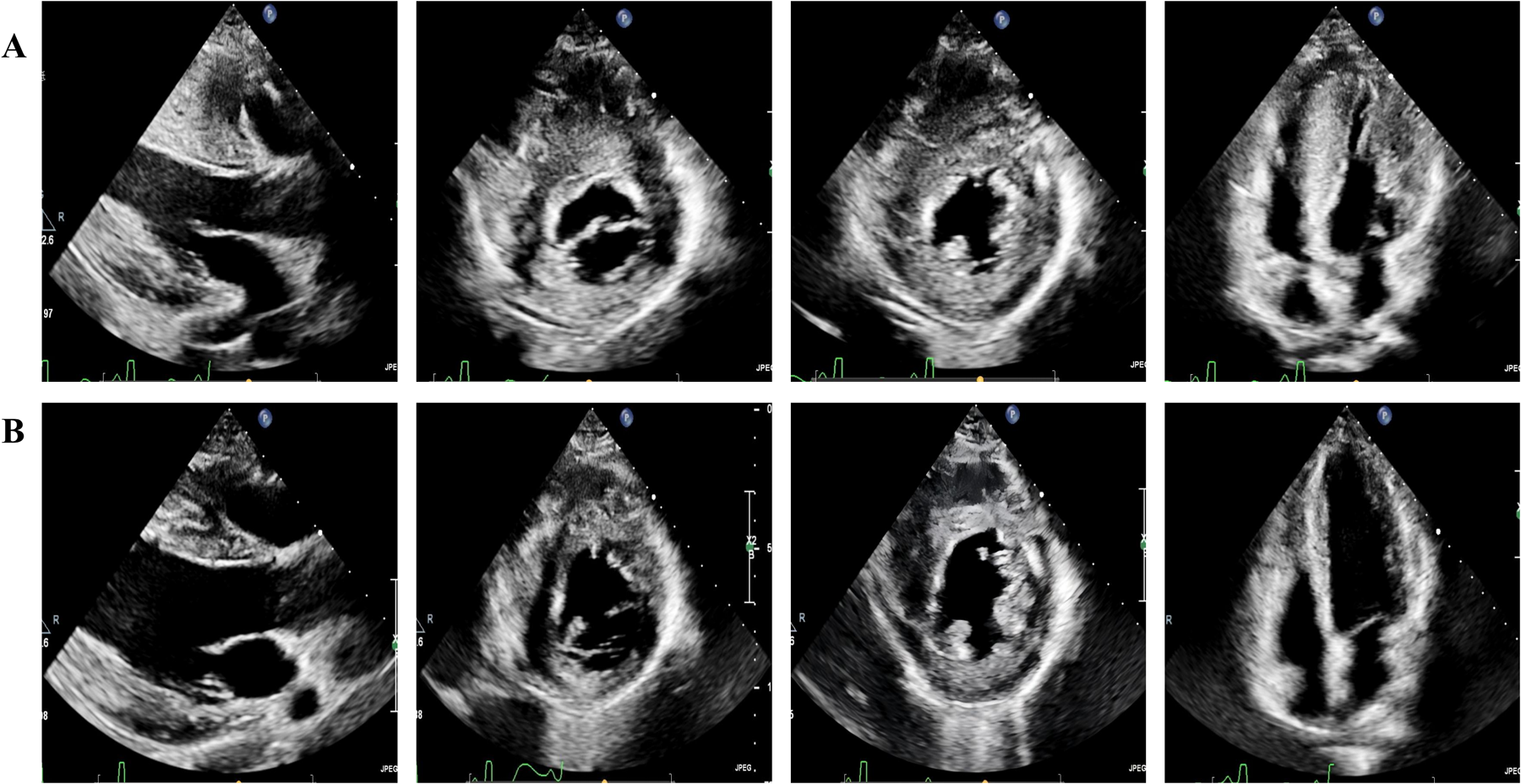
Transthoracic echocardiography. (A) Preoperative transthoracic echocardiography demonstrates severe left ventricular hypertrophy, right ventricular free wall hypertrophy, and a small LVEDV in parasternal long axis of left ventricule, parasternal short axis, and apical four-chamber view. (B) A 6-month postoperative transthoracic echocardiography, compared with (A), reveals significant thinning of both the left ventricular and right ventricular free wall myocardium, alongside a marked increase in LVEDV.
Table 1
| Variable | Pre-operative | 6-month post operative |
|---|---|---|
| IVS (thickness)(mm) | 29 | 15 |
| RVFW (mm) | 8 | 7 |
| LVDd (mm) | 37 | 45 |
| LVEDV (mL) | 56.9 | 101 |
| LVM (g) | 182 | 120 |
| LVMI (g/㎡) | 130 | 79.2 |
| LVOT (rest) (mmHg) | 5.6 | 8.2 |
| LVOT (provocation) (mmHg) | 20 | 4.9 |
| LVIC (rest) (mmHg) | 4.8 | 7 |
| LVIC (provocation) (mmHg) | 31 | 1.8 |
| RVOT (rest) (mmHg) | 18 | 4.4 |
| RVOT (provocation) (mmHg) | 28 | 4.7 |
| NT-proBNP (pg/mL) | 4,457 | 2,233 |
Comparison of patient parameters.
3 Discussion
Sarcomeric gene mutations predominate in patients with HCM, with pathogenic variants in MYBPC3 (60.5%) and MYH7 (14.1%) collectively accounting for 74.6% of mutation-positive cases (15), recent advances have identified ALPK3, a non-sarcomeric protein-coding gene, as a novel pathogenic contributor to HCM. However, the precise molecular mechanisms remain incompletely characterized. Current evidence suggests that ALPK3 variants may induce cardiomyopathic changes through four distinct pathophysiological mechanisms: (1) disruption of myosin localization, leading to structural and functional impairment of cardiomyocytes that culminates in cellular hypertrophy (16); (2) dysregulation of HEY2 transcription factor phosphorylation and consequent abnormalities in cardiomyocyte differentiation (17); (3) induction of calcium homeostasis dysregulation, thereby elevating arrhythmogenic risk (5); (4) manifestation of sarcomeric disarray and aberrant intercalated disc morphology, resulting in contractile dysfunction (18).
Multicenter international have demonstrated that ALPK3 truncating variants account for 1%–2% of HCM cases. These variants are characterized by later disease onset and predominantly non-obstructive phenotypes at rest, these variants typically present with apical or concentric hypertrophy and are often accompanied by extracardiac manifestations. Notably, heterozygotes exhibit significant clinical heterogeneity (8), and a Portuguese cohort study confirmed that ALPK3 mutations constitute 2.7% of HCM etiologies (19), interestingly, East Asian populations have distinct genetic profiles in HCM pathogenesis (11). Both the first Chinese case reported by our center and the East Asian study found that compound heterozygous mutations (e.g., in this case, c.4234C > T nonsense mutation and c.3491G > A missense mutation) could lead to a more severe clinical phenotype manifested by early-onset biventricular hypertrophy, multilevel outflow tract obstruction, and simian crease.
The ALPK3 gene encodes nuclear alpha-protein kinase 3, which is critical for cardiomyocyte development and structural homeostasis, reported pathogenic variants primarily consist of nonsense and missense mutations and splice-site variations, with biallelic or compound heterozygous configurations predominantly observed (12, 20). Studies indicate that nonsense mutations induce premature protein truncation, potentially exerting pathogenic effects through haploinsufficiency or dominant-negative mechanisms; Meanwhile, missense mutations are postulated to compromise kinase domain functionality by disrupting critical structural motifs (12); additionally, splice-site variants may impair protein expression through aberrant mRNA splicing processes that alter transcriptional integrity (21). Wei-Feng et al. demonstrated that ALPK3 double allele deletion can trigger lethal cardiomyopathy in an animal model study, approximately 75% of germline knockout mice die within one month of birth, and survivors exhibit a transformation from dilated cardiomyopathy to HCM, which is highly consistent with the clinical manifestations of compound heterozygous mutations in humans. Studies have demonstrated that ALPK3 acts as a scaffolding protein that regulates thick filament protein turnover (22). In animal models, ALPK3 knockout mice exhibit a mixed phenotype of hypertrophic and dilated cardiomyopathy, suggesting that ALPK3 plays a critical role in the development and function of cardiomyocytes (23), these findings imply that ALPK3 gene mutations may cause structural and functional abnormalities in cardiomyocytes and subsequently lead to cardiomyopathy.
This HCM case presents compound heterozygosity for an ALPK3 nonsense mutation (c.4234C > T) coupled with a missense mutation (c.3491G > A), compared to ALPK3 heterozygotes, compound heterozygous carriers exhibit an aggressive clinical profile characterized by the following: (1) early disease onset, (2) biventricular hypertrophy with multilevel obstruction, (3) an elevated risk of sudden cardiac death, and (4) accelerated heart failure progression. Notably, the proband exhibited bilateral simian creases, a distinctive extracardiac feature, genetic segregation analysis revealed unaffected heterozygous parents and younger brother with normal echocardiographic parameters, this inheritance pattern strongly supports the biallelic dosage effect theory, and the observed genotype-phenotype correlation is particularly prominent in East Asian cohorts.
The therapeutic objectives for HCM focus on alleviating symptoms, improving cardiac function, and slowing disease progression, although conventional pharmacotherapy (beta-blockers, calcium channel blockers and disopyramide) provides symptomatic relief, it does not address the underlying pathological substrate of myocardial hypertrophy. Current management algorithms incorporate 3 main approaches: (1) pharmacological optimisation for diastolic dysfunction and arrhythmia control, (2) surgical myectomy (Morrow procedure) for left ventricular cavity augmentation and septal thickness reduction, and (3) interventional approaches to diminish LVOT gradients, decelerate heart failure progression and mitigate the risk of sudden cardiac death (4). The Liwen procedure is a novel, ultrasound-guided, percutaneous, intramyocardial, radiofrequency ablation technique that enables real-time, targeted ablation of hypertrophied myocardium, achieving gradient reduction and functional improvement. A multicentre trial enrolled 244 refractory HCM patients who met stringent criteria: resting/provoked LVOT gradients ≥50 mmHg, persistent symptoms of NYHA Grade II or higher, and failure to respond to pharmacotherapy, among the 200 cases in which the procedure was completed, with a median follow-up of 19 months, the Liwen procedure approach demonstrated comparable safety and efficacy profiles to the Morrow procedure, with a significantly lower incidence of arrhythmias: there was a 2.5% incidence of permanent right bundle branch block and no patients with permanent pacemaker implantation (14). Chinese multicentre data further established the superiority of the Liwen procedure over percutaneous endocardial septal radiofrequency ablation (14, 24), with significantly lower recurrence rates compared to alcohol septal ablation (2% vs. 10%) (7).
The Liwen procedure is an option for patients who have not responded well to medication and who still have a high risk of sudden death, such as those with dyspnea, chest pain, or syncope. Compared with traditional surgery, the Liwen procedure has technical advantages such as not opening the chest, a continuous beating heart, precise localization, unrestricted target blood vessels, less trauma, faster recovery, fewer complications, a significant reduction in IVS thickness, and reduced conduction system damage and other complications. The successful treatment of this case further confirms the Liwen procedure's potential and advantages in treating HCM, this procedure has significant clinical value and broad application prospects.
4 Patient perspective
When diagnosed with hypertrophic obstructive cardiomyopathy, I felt crushed by despair. This disease severely limited my daily activities and imposed a constant threat of sudden death. I feared it would disrupt my education and future career. After thorough discussion with my family, I opted for the Liwen procedure. Currently, I can engage in routine activities and attend school normally. This intervention has restored my hope for the future.Written informed consent was obtained for publication.
5 Conclusions
This case is the first report of early-onset, severe HCM caused by compound heterozygous mutations (c.4234C > T nonsense mutation and c.3491G > A missense mutation) in the ALPK3 gene in the Chinese population, this finding enriches the mutation spectrum of the ALPK3 gene and reveals high-frequency mutation loci in exons 4 and 10 that are unique to the East Asian population, these results suggest potential ethnogenetic heterogeneity, the patient presented with severe hypertrophy of the IVS, biventricular involvement, occult multilevel outflow tract obstruction, and simian creases, confirming the “dose effect” theory of ALPK3 double-allele mutation.
Following the Liwen procedure, the patient experienced complete resolution of symptoms within 6 months, demonstrating significant clinical improvement in this case of severe, drug-refractory ALPK3-related HCM. This outcome supports the consideration of the Liwen procedure as an effective therapeutic strategy for complex obstructive ALPK3-related HCM, but the 6-month follow-up is a recognized limitation in evaluating long-term outcomes for this potentially progressive cardiomyopathy. To address this, the patient is enrolled in a dedicated long-term surveillance program. This program will track myocardial thickness, key hemodynamic measures, clinical symptoms, and NT-proBNP levels to elucidate the long-term disease course and treatment response.
Statements
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by The First Hospital of Handan Ethics Committee. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
W-JL: Writing – original draft, Investigation, Funding acquisition, Visualization, Writing – review & editing, Methodology, Formal analysis. HY: Formal analysis, Writing – review & editing, Writing – original draft. F-HJ: Writing – original draft, Writing – review & editing, Investigation, Funding acquisition. H-YH: Writing – review & editing, Formal analysis, Data curation. W-JX: Writing – review & editing, Data curation. Y-NW: Data curation, Writing – original draft. L-PD: Data curation, Writing – review & editing. X-FZ: Writing – review & editing, Formal analysis. R-JZ: Data curation, Writing – review & editing. CC: Formal analysis, Visualization, Investigation, Data curation, Validation, Resources, Project administration, Supervision, Writing – review & editing, Methodology, Conceptualization, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the Health Commission of Hebei Province under grants 20242165 and 20241256.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2025.1671882/full#supplementary-material
Abbreviations
HCM, right ventricular outflow tract pressure gradient; ALPK3, Alpha-kinase 3; PIMSRA, percutaneous intramyocardial septal radiofrequency ablation (Liwen procedure); LVDd, left ventricular end-diastolic diameter; NT-proBNP, N-terminal proB-type natriuretic peptide; IVS, Interventricular septum; RVFW, right ventricular free wall; LVEDV, left ventricular end-diastolic volume; LVOT-PG, left ventricular outflow tract pressure gradient; LVIC-PG, left ventricular intracavitary compartment pressure gradient; RVOT-PG, right ventricular outflow tract pressure gradient.
References
1.
Ommen SR Mital S Burke MA Day SM Deswal A Elliott P et al 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology/American Heart Association joint committee on clinical practice guidelines. Circulation. (2020) 142(25):e533–57. 10.1161/CIR.0000000000000938
2.
Ommen SR Ho CY Asif IM Balaji S Burke MA Day SM et al 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR guideline for the management of hypertrophic cardiomyopathy: a report of the American Heart Association/American College of Cardiology joint committee on clinical practice guidelines. Circulation. (2024) 149(23):e1239–311. 10.1161/CIR.0000000000001250
3.
Semsarian C Ingles J Maron MS Maron BJ . New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. (2015) 65(12):1249–54. 10.1016/j.jacc.2015.01.019
4.
Expert group of the National Cardiovascular Disease Center Cardiomyopathy Specialty Alliance and the Cardiovascular Precision Medicine Branch of the China Association for the Promotion of International Exchanges of Healthcare. The 2023 guidelines for the diagnosis and treatment of hypertrophic cardiomyopathy in adults in China. Chin J Mol Cardiol. (2023)23(2):1–35. 10.16563/j.cnki.1671-6272.2023.02.001
5.
Walsh R Offerhaus JA Tadros R Bezzina CR . Minor hypertrophic cardiomyopathy genes, major insights into the genetics of cardiomyopathies. Nat Rev Cardiol. (2022) 19(3):151–67. 10.1038/s41569-021-00608-2
6.
Almomani R Verhagen JM Herkert JC Brosens E van Spaendonck-Zwarts KY Asimaki A et al Biallelic truncating mutations in ALPK3 cause severe pediatric cardiomyopathy. J Am Coll Cardiol. (2016) 67(5):515–25. 10.1016/j.jacc.2015.10.093
7.
Meng X Wang WY Gao J Zhang K Zheng J Wang JJ et al Hypertrophic obstructive cardiomyopathy: comparison of outcomes after myectomy or alcohol ablation. Front Cardiovasc Med. (2022) 9:755376. 10.3389/fcvm.2022.755376
8.
Lopes LR Garcia-Hernández S Lorenzini M Futema M Chumakova O Zateyshchikov D et al Alpha-protein kinase 3 (ALPK3) truncating variants are a cause of autosomal dominant hypertrophic cardiomyopathy. Eur Heart J. (2021) 42(32):3063–73. 10.1093/eurheartj/ehab424
9.
Jaouadi H Kraoua L Chaker L Atkinson A Delague V Levy N et al Novel ALPK3 mutation in a Tunisian patient with pediatric cardiomyopathy and facio-thoraco-skeletal features. J Hum Genet. (2018) 63(10):1077–82. 10.1038/s10038-018-0492-1
10.
Jorholt J Formicheva Y Vershinina T Kiselev A Muravyev A Demchenko E et al Two new cases of hypertrophic cardiomyopathy and skeletal muscle features associated with ALPK3 homozygous and compound heterozygous variants. Genes (Basel). (2020) 11(10):1201. 10.3390/genes11101201
11.
Dai J Li K Huang M Sun Y Liu H Li Z et al The involvement of ALPK3 in hypertrophic cardiomyopathy in east Asia. Front Med (Lausanne). (2022) 9:915649. 10.3389/fmed.2022.915649
12.
Liu L Liu B Li J Zhang Y . Percutaneous intramyocardial septal radiofrequency ablation of hypertrophic obstructive cardiomyopathy: a novel minimally invasive treatment for reduction of outflow tract obstruction. EuroIntervention. (2018) 13(18):e2112–3. 10.4244/EIJ-D-17-00657
13.
Zhou M Ta S Hahn RT Hsi DH Leon MB Hu R et al Percutaneous intramyocardial septal radiofrequency ablation in patients with drug-refractory hypertrophic obstructive cardiomyopathy. JAMA Cardiol. (2022) 7(5):529–38. 10.1001/jamacardio.2022.0259
14.
Alimohamed MZ Johansson LF Posafalvi A Boven LG van Dijk KK Walters L et al Diagnostic yield of targeted next generation sequencing in 2002 Dutch cardiomyopathy patients. Int J Cardiol. (2021) 332:99–104. 10.1016/j.ijcard.2021.02.069
15.
Agarwal R Wakimoto H Paulo JA Zhang Q Reichart D Toepfer C et al Pathogenesis of cardiomyopathy caused by variants in ALPK3, an essential pseudokinase in the cardiomyocyte nucleus and sarcomere. Circulation. (2022) 146(22):1674–93. 10.1161/CIRCULATIONAHA.122.059688
16.
Hosoda T Monzen K Hiroi Y Oka T Takimoto E Yazaki Y et al A novel myocyte-specific gene midori promotes the differentiation of P19CL6 cells into cardiomyocytes. J Biol Chem. (2001) 276(38):35978–89. 10.1074/jbc.M100485200
17.
Phelan DG Anderson DJ Howden SE Wong RC Hickey PF Pope K et al ALPK3-deficient Cardiomyocytes generated from patient-derived induced pluripotent stem cells and mutant human embryonic stem cells display abnormal calcium handling and establish that ALPK3 deficiency underlies familial cardiomyopathy. Eur Heart J. (2016) 37(33):2586–90. 10.1093/eurheartj/ehw160
18.
Duarte F Oliveira L Baixia M Mota-Vieira L Machado C . Unique genetic profiles in hypertrophic cardiomyopathy patients from São Miguel Island (Azores, Portugal). Clin Genet. (2025) 107(4):434–40. 10.1111/cge.14656
19.
Chumakova OS Milovanova NV Bychkov IO Zakharova EY Mershina EA Sinitsin VE et al Overlapping phenotype of adult-onset ALPK3-cardiomyopathy in the setting of two novel variants. Cardiol Res. (2022) 13(6):398–404. 10.14740/cr1449
20.
Ding WW Wang BZ Han L Li ZP Zhang W Wang H et al ALPK3 gene-related pediatric cardiomyopathy with craniofacial-skeletal features: a report and literature review. Zhonghua Er Ke Za Zhi. (2021) 59(9):787–92. 10.3760/cma.j.cn112140-20210222-00150
21.
Bukaeva A Myasnikov R Kulikova O Meshkov A Kiseleva A Petukhova A et al A rare coincidence of three inherited diseases in a family with cardiomyopathy and multiple extracardiac abnormalities. Int J Mol Sci. (2024) 25(14):7556. 10.3390/ijms25147556
22.
Feng W Wang L Bogomolovas J Zhang Z Huang T Chang CW et al Αprotein kinase 3 is essential for neonatal and adult cardiac function. J Am Heart Assoc. (2025) 14(7):e039464. 10.1161/JAHA.124.039464
23.
Van Sligtenhorst I Ding ZM Shi ZZ Read RW Hansen G Vogel P . Cardiomyopathy in α-kinase 3 (ALPK3)-deficient mice. Vet Pathol. (2012) 49(1):131–41. 10.1177/0300985811402841
24.
Xie X Chen S Cui Y Zhou Z Lu J Du Z et al Midterm outcomes of percutaneous intramyocardial septal radiofrequency ablation for hypertrophic cardiomyopathy: a single-center, observational study. J Am Heart Assoc. (2024) 13(15):e034080. 10.1161/JAHA.123.034080
Summary
Keywords
hypertrophic cardiomyopathy, occult biventricular obstructive hypertrophic cardiomyopathy, ALPK3 gene, domain architecture and mutation distribution of ALPK3, liwen procedure
Citation
Liu W-J, Hua Y, Jiao F-H, Hu H-Y, Xu W-J, Wang Y-N, Duan L-P, Zhao X-F, Zhang R-J and Chang C (2025) Novel compound heterozygous ALPK3 mutations (c.4234C>T and c.3491G>A), causing hypertrophic cardiomyopathy treated with the liwen procedure: case report. Front. Cardiovasc. Med. 12:1671882. doi: 10.3389/fcvm.2025.1671882
Received
23 July 2025
Revised
05 November 2025
Accepted
24 November 2025
Published
08 December 2025
Volume
12 - 2025
Edited by
Peter Martin Wenaweser, Heart Clinic Zurich, Switzerland
Reviewed by
Dolina Gencheva, Plovdiv Medical University, Bulgaria
Lei Zuo, Air Force Medical University, China
Updates
Copyright
© 2025 Liu, Hua, Jiao, Hu, Xu, Wang, Duan, Zhao, Zhang and Chang.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
* Correspondence: Chao Chang 2215127856@qq.com
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.