 Maria Valero-Muñoz1
Maria Valero-Muñoz1 Hannah L. Cooper1Shanpeng Li1
Hannah L. Cooper1Shanpeng Li1 Eng Leng Saw1Richard M. Wilson1
Eng Leng Saw1Richard M. Wilson1 Christine M. Kusminski2
Christine M. Kusminski2 Philipp E. Scherer2
Philipp E. Scherer2 Flora Sam1*
Flora Sam1*
- 1Whitaker Cardiovascular Institute, Boston University Chobanian & Avedisian School of Medicine, Boston, MA, United States
- 2Touchstone Diabetes Center, University of Texas Southwestern Medical Center, Dallas, TX, United States
Background: Obesity and hypertension are among the most prevalent comorbidities in heart failure with preserved ejection fraction (HFpEF). In addition to its relationship with hypertension in HFpEF, obesity is also strongly associated with insulin resistance (IR) and type 2 diabetes (T2D). However, the exact cardiac effects underlying this relationship are unknown. We sought to differentiate the cardiac phenotype associated with increased adiposity in the presence or absence of IR in obese HFpEF. We utilized adipose tissue-specific MitoNEET transgenic mice, which develop chronic, metabolically healthy adipose tissue expansion (obese non–insulin resistant, OB-NIR), and compared them with their wild-type, insulin-resistant littermates (OB-IR).
Methods: OB-NIR MitoNEET and OB-IR wildtype mice were fed a high-fat diet for 16 weeks, at which time HFpEF was induced via uninephrectomy, d-aldosterone infusion, and 1.0% sodium chloride drinking water for 4 additional weeks while maintained on the same diet.
Results: OB-NIR HFpEF mice exhibited reduced cardiac fibrosis without changes in hypertrophy. This reduction was accompanied by increased cardiac expression of SIRT3. Upregulation of several downstream mitochondrial targets of SIRT3 was also observed. These included mitochondrial fission protein 1 (Fis1), a critical regulator of mitochondrial dynamics, and the antioxidant enzyme heme oxygenase-1 (Hmox1). In contrast, levels of hydroxy-3-methylglutaryl coenzyme A (CoA) synthase 2 (HMGCS2) were decreased, while both 3-hydroxybutyrate dehydrogenase 1 (Bdh1) and succinyl-CoA:3-ketoacid CoA transferase (Oxct1) were elevated. Furthermore, genes involved in the electron transport chain, such as ubiquinol-cytochrome C reductase hinge protein (Uqcrh, Complex III) and mitochondrially encoded cytochrome c oxidase I (Mt-Co1, Complex IV), were upregulated.
Discussion: Distinct alterations in cardiac mitochondrial function were observed depending on the presence or absence of IR in obese HFpEF mice. These findings suggest that SIRT3 may play a central role in mediating mitochondrial adaptations in the heart and could represent a promising therapeutic target in HFpEF.
1 Introduction
Obesity has reached epidemic proportions worldwide and is increasingly an extremely common finding in patients with heart failure (HF) with preserved ejection fraction (HFpEF) (1). Currently, HFpEF accounts for over 50% of all HF cases. With an aging population and the rising prevalence of metabolic disorders—such as obesity, type 2 diabetes (T2D), and hypertension—the number of HFpEF cases is expected to increase further. Although obesity-related HFpEF is recognized as a distinct clinical phenotype (2), the exact mechanisms by which obesity directly contributes to its development remain unclear. It had been purported that symptoms in patients with obesity HFpEF were simply due to excess adiposity and body weight rather than other mechanisms, such as underlying cardiac abnormalities (3). A retrospective, case-control bariatric surgery study demonstrated that removal of excess adiposity significantly reduced hospitalizations in “diastolic heart failure” (i.e., HFpEF) (4). However, other mechanisms are also propose to play a role, including systemic inflammation, insulin resistance (IR), neurohormonal dysregulation, or skeletal muscle (SkM) dysfunction, in addition to impaired hemodynamic loading (5). Notably, in lean (non-obese) patients with HFpEF, intramyocardial fat deposition is greater than in lean patients with HFrEF (HF with reduced ejection fraction) and in non-HF control subjects. Moreover, the volume of intramyocardial fat is independently associated with echocardiographic indicators of left ventricular (LV) diastolic dysfunction (6), suggesting that fat content may contribute to myocardial stiffness, altered energetics, and the pathophysiology of HFpEF independently of overall obesity. Furthermore, increased adiposity associated with obesity contributes to adverse cardiovascular remodeling, diastolic dysfunction, and to the progression to HF, particularly in HFpEF (7). Finally, secreted hormones (e.g., leptin and gut-derived peptides), the anatomical distribution of adipose depots, and the presence of visceral adiposity likely also play pathogenic roles in obesity-related HFpEF.
IR and T2D are implicated as causal contributors to adverse cardiovascular outcomes seen in obesity-associated HFpEF. T2D is particularly relevant in the context of HFpEF, as it is linked to impaired hemodynamics, increased symptom burden, and reduced functional capacity compared to patients without T2D (8, 9). Individuals with T2D exhibit more pronounced mitochondrial dysfunction and SkM metabolic impairments than those without T2D and these deficits are further exacerbated in diabetic HFpEF (5, 10). Moreover, systemic IR and hyperglycemia contribute to secondary cardiac IR, leading to oxidative stress and imbalances in neurohumoral, sympathetic, and cytokine signaling (11). These disturbances promote cardiomyocyte hypertrophy, interstitial fibrosis, and altered collagen turnover—hallmarks commonly observed in HFpEF (12). Notably, a recent clinical trial in obesity HFpEF patients, demonstrated that tirzepatide, a long-acting agonist of glucose-dependent insulinotropic polypeptide and glucagon-like peptide-1 receptors, significantly reduced body mass index (BMI), LV mass, and pericardial adipose tissue while also improving HbA1c levels and reduced the composite of death from cardiovascular causes or worsening HF with improved functional status (13).
To determine the precise contribution of increased adiposity-independent of IR or T2D on the cardiac phenotype in obesity-associated HFpEF, the present study sought to delineate the individual contributions of increased adiposity vs. T2D using a preclinical model of obese HFpEF. In rodents, obesity can result from naturally occurring mutations, genetic manipulation or dietary interventions. Among these, diet-induced obesity (DIO) models are particularly relevant, as they recapitulate key features of human obesity, including the frequent co-development of IR and T2D (14). In the present study, a transgenic mouse model overexpressing the adipose tissue (AT)-specific mitochondrial membrane protein MitoNEET was utilized. When challenged with a high-fat diet (HFD), these MitoNEET mice undergo chronic, yet “healthy,” expansion of adipose depots without developing IR or T2D, thereby representing a model of the “metabolically healthy” obese phenotype (15, 16). Given that excessive adiposity contributes to ∼65%–75% of primary hypertension in humans (17), this murine model of obesity—independent of IR and T2D—was subjected to hypertension-associated HFpEF using the SAUNA (SAlty drinking water, UNilateral Nephrectomy, and Aldosterone) model. The SAUNA model induces a HFpEF phenotype characterized by lung congestion, preserved LV ejection fraction, LV hypertrophy, and impaired diastolic function in the setting of moderate hypertension within four weeks (18–23).
Post-translational acetylation of mitochondrial proteins is increasingly recognized as a key contributor to impaired cardiac energetics and adverse cardiac remodeling (24, 25). Within the mitochondria, the acetylation status of proteins involved in mitochondrial dynamics, metabolic flexibility, and antioxidant defense is tightly regulated by the NAD⁺-dependent deacetylase Sirtuin-3 (SIRT3) (26). Reduced SIRT3 activity has been linked to obesity, insulin resistance, and cardiac dysfunction (27–29), suggesting it may serve as a mechanistic link between metabolic dysregulation and HFpEF pathophysiology. In this study, we investigated SIRT3 regulation in a murine model of obese HFpEF, with a particular focus as to how IR may influence its expression and downstream functional consequences.
2 Material and methods
A detailed Material and Methods is available in the Supplementary Material.
2.1 Ethics
This investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health and was approved by the Institutional Animal Care and Use Committee at Boston University School of Medicine (IACUC: PROTO201800310).
2.2 AT-specific MitoNEET transgenic mice
AT-specific MitoNEET transgenic mice were donated by Drs. Kusminski and Scherer and used as a murine model of obesity without IR (OB-NIR). MitoNEET is a transmembrane protein located in the outer mitochondrial membrane and named after a conserved amino acid sequence, part of which includes Asn-Glu-Glu-Thr (NEET). These mice were initially generated by subcloning the MitoNEET gene into a plasmid containing the 5.4 kb aP2-promoter and a conventional 3′ untranslated region. Following linearization, the construct was injected into FVB-derived blastocysts (15, 16). FVB wild-type littermates were obese with IR (OB-IR).
2.3 Statistical analysis
All statistical analyses were performed using GraphPad Prism (GraphPad Software, Inc). The normality of distributions was verified by D’Agostino & Pearson omnibus normality test verified the normality of distributions. Differences between the two groups were analyzed by unpaired Student t-test or Mann–Whitney test as parametric and nonparametric tests, respectively. Statistical outliers were calculated using the ROUT testing. P < 0.05 was considered statistically significant.
3 Results
3.1 Cardiac structure and function in obese MitoNEET mice without insulin resistance (OB-NIR) and wild-type mice with insulin resistance (OB-IR) after HFD feeding for 16 weeks
We initially investigated cardiac changes associated to obesity in OB-IR and OB-NIR mice fed a HFD for 16 weeks prior to HFpEF induction. Systolic blood pressure, LV structure and LV systolic and diastolic functions were comparable between groups (Supplementary Table S1). Both OB-IR and OB-NIR mice had comparable degrees of cardiac hypertrophy, with a significant increase in LV mass, when compared to lean wild-type mice of similar age (108.2 ± 2.9 mg, P < 0.0001 for both). Hematoxylin-eosin staining of the LV of both OB-IR and OB-NIR mice showed similar cardiomyocyte size (Supplementary Figure S2A). Additionally, collagen deposition in the LV as measured by Picrosirius red staining, was no different between OB-IR and OB-NIR (Supplementary Figure S2B).
3.2 Metabolic characteristics in obese HFpEF mice with and without insulin resistance
This obesogenic diet followed by HFpEF induction resulted in a murine model of obesity plus hypertension-associated HFpEF with IR in wild-type mice (OB-IR HFpEF) and without IR in the littermate mice overexpressing MitoNEET in AT (OB-NIR HFpEF) (Supplementary Figure S1). Both obese HFpEF groups of mice had similar body weight at the end of the 20 experimental weeks (40.8 ± 1.8 g in OB-IR vs. 41.9 ± 2.5 g in OB-NIR, P = 0.8884; Table 1) but, as expected, OB-IR HFpEF mice had impaired insulin sensitivity as demonstrated by increased fasting basal glucose levels (140.5 ± 7.1 mg/dl vs. 100.3 ± 5.2 mg/dl in OB-NIR; P = 0.0003), as well as an elevated area under the curve (AUC) for the glucose tolerance test (30,064 ± 1,722 vs. 24,378 ± 2,083 in OB-NIR; P = 0.0279), and a significantly increased HOMA-insulin resistance index (13.48 ± 4.9 vs. 2.86 ± 1.4 in OB-NIR; P = 0.0315; Table 1). Adipokine measurements showed that circulating leptin levels were no different between OB-IR and OB-NIR HFpEF mice. Nonetheless circulating adiponectin levels were increased in OB-NIR (60.50 ± 13.76 µg/ml) compared to OB-IR HFpEF mice (16.01 ± 1.95 µg/ml, P = 0.0329). There were no significant differences in triglycerides and total cholesterol circulating levels between the two groups (Table 1).
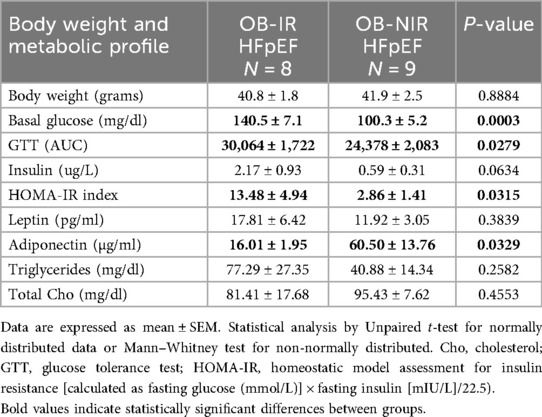
Table 1. Body weight and metabolic profile in obese HFpEF mice with (OB-IR) and without (OB-NIR) insulin resistance.
3.3 Physiological and cardiac characteristics in obese HFpEF mice with and without insulin resistance
The presence or absence of chronic IR made no difference in systolic blood pressure elevation or lung congestion in either group of obese HFpEF mice (OB-IR and OB-NIR) (Table 2). Similarly, LV structure and systolic function were comparable between the two groups, but there were slight changes in diastolic function between OB-IR and OB-NIR HFpEF mice, with the latter showing a significantly increased mitral E velocity (796.2 ± 24.9 mm/s) vs. OB-IR HFpEF mice (664.5 ± 38.4 mm/s; P = 0.0116; Table 2). Mitral E-wave velocity reflects the left atrial (LA)-LV pressure gradient during early diastole and in humans is affected by changes in the rate of LV relaxation and LA pressure.

Table 2. Physiological and echocardiographic parameters in obese HFpEF mice with (OB-IR) and without (OB-NIR) insulin resistance.
Although cardiac hypertrophy was evident, there were no differences in LV mass (Table 2) nor LV weight relative to tibial length between OB-IR and OB NIR HFpEF mice (58.9 ± 3.4 mg/cm vs. 57.9 ± 2.5 mg/cm). Cardiomyocyte size was increased but similarly comparable between OB-IR and OB NIR HFpEF mice (410 ± 19 μm2 vs. 437 ± 21 μm2; Figures 1A,B). However, collagen deposition by Picrosirius red staining showed that OB-IR HFpEF mice had increased fibrosis (0.99% ± 0.03%) compared to OB-NIR HFpEF mice (0.49% ± 0.07%, P = 0.0002; Figures 1C,D). At the molecular level, mRNA expression of cardiac remodeling transcripts atrial natriuretic peptide (anp, namely Nppa), and brain natriuretic peptide (bnp, namely Nppb), collagen 1a (Col1a) and 3a (Col3a), and titin isoforms N2b and N2ba were no different between OB-IR and OB NIR HFpEF mice (Figure 2).
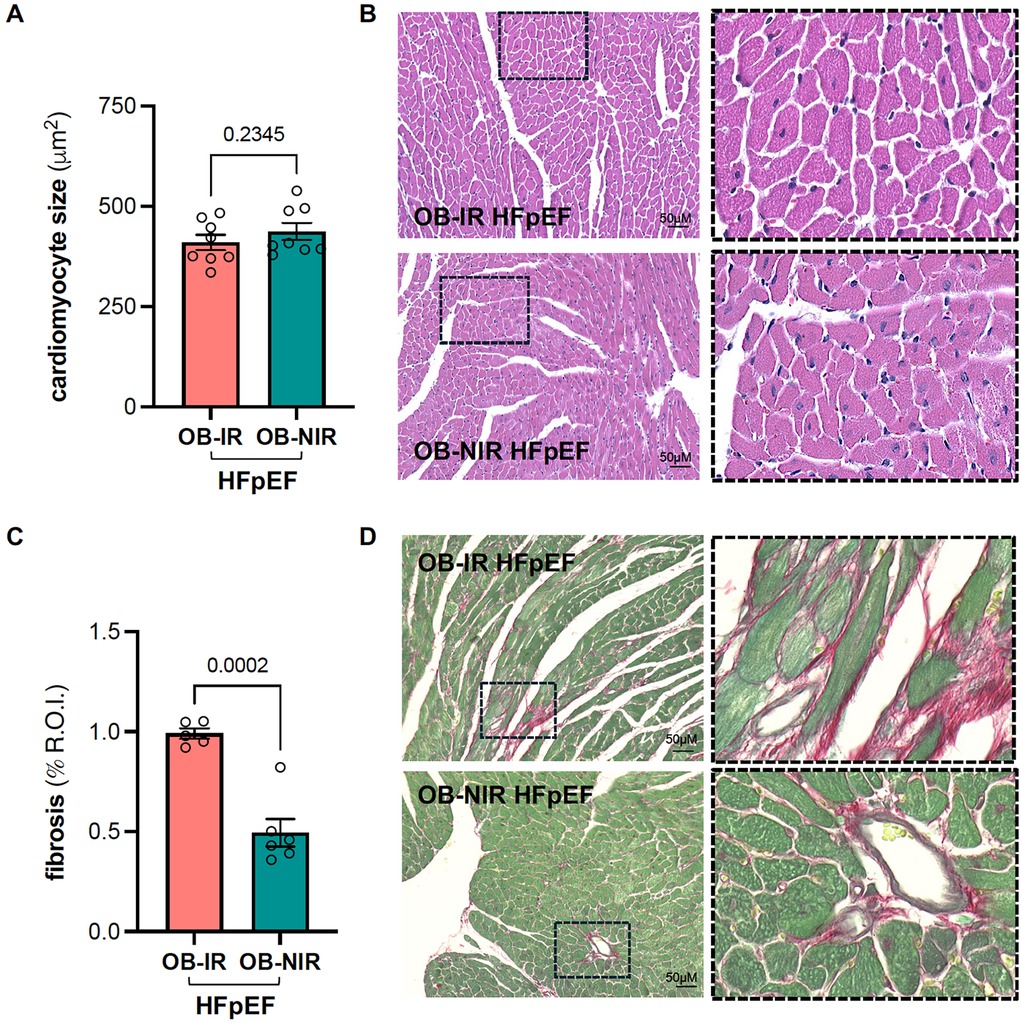
Figure 1. Left ventricular cardiomyocyte size and cardiac fibrosis in obese HFpEF mice with and without insulin resistance (A) cardiomyocyte size and (B) representative hematoxylin-eosin staining images and magnification (right panel, 20x). (C) Quantification of cardiac fibrosis using Picrosirius Red staining and (D) representative microscopic images and magnification (right panel, 20x; R.O.I. indicates region of interest). Data are represented as mean ± SEM. Statistical analysis by unpaired t-test for normally distributed data or Mann–Whitney test for non-normally distributed. OB-IR, obese insulin-resistant HFpEF mice; OB-NIR, obese non-insulin-resistant HFpEF mice. N = 5-8 mice/group.

Figure 2. Cardiac remodeling phenotype in obese HFpEF mice with and without insulin resistance. Gene expression of Nppa, Nppb, Col1a, Col3a, N2b and N2ba relative to OB-IR. Data are represented as mean ± SEM. Statistical analysis by unpaired t-test for normally distributed data or Mann–Whitney test for non-normally distributed. Col1a, collagen 1a; Col3a, collagen 3a; Nppa, natriuretic peptide type A, aka atrial natriuretic peptide; Nppb, natriuretic peptide type b, i.e., brain natriuretic peptide; N2b, titin transcript variant N2b; N2ba, titin transcript variant N2ba. OB-IR. OB-IR, obese insulin-resistant HFpEF mice; OB-NIR, obese non-insulin-resistant HFpEF mice. N = 7-9/group.
3.4 SIRT3 protein expression in the LV of obese HFpEF mice with and without insulin resistance
SIRT3 is a mitochondrial deacetylase that mediates the activity of many metabolic enzymes involved in mitochondrial glycolysis, fatty acid metabolism, tricarboxylic acid (TCA) cycle, electron transport chain (ETC), and ATP synthesis. We previously showed evidence of decreased SIRT3 protein expression in the LV of HFpEF mice (21). SIRT3 protein expression was therefore determined in the LV of obese HFpEF mice with and without IR. In the present study, LV SIRT3 protein expression levels in OB-IR HFpEF mice were comparable to those of Lean-IR HFpEF mice (0.79 ± 0.09 vs. 0.71 ± 0.07 relative expression to Lean Sham-CT, Supplementary Figure S2). However, SIRT3 LV protein expression in OB-NIR HFpEF was increased 1.5-fold vs. OB-IR HFpEF (P = 0.0279; Figure 3A). This 1.5-fold increase restored SIRT3 expression in the LV back to levels comparable to lean Sham controls (Supplementary Figure S3).
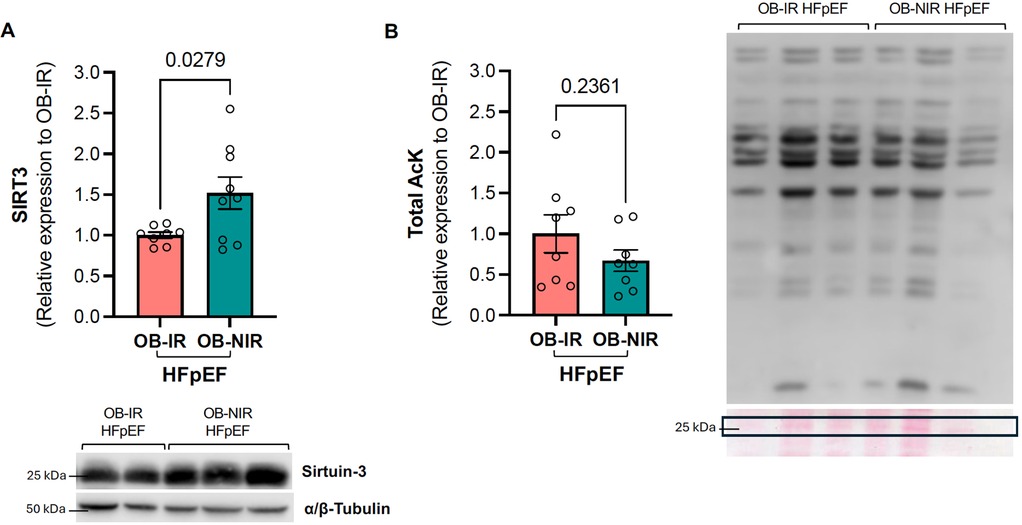
Figure 3. Left ventricular SIRT3 protein expression and total lysine acetylation in obese HFpEF mice with and without insulin resistance. (A) SIRT3 protein expression and (B) Total lysine acetylation in the left ventricle of obese insulin-resistant HFpEF mice (OB-IR) and obese non-insulin-resistant HFpEF mice (OB-NIR). Data are represented as mean ± SEM. Statistical analysis by unpaired t-test for normally distributed data or Mann–Whitney test for non-normally distributed. SIRT3, sirtuin 3; Total ACK, total lysine acetylation relative to 25KDa Ponceau Red band. N = 8-9 mice/group.
Since SIRT3 modulates the enzymatic activity of key proteins involved in energy homeostasis in the heart by regulating their acetylation status, total cardiac lysine acetylation (acK) was also determined, but showed an non-significant decreasing trend in OB-NIR HFpEF mice compared to OB-IR HFpEF (Figure 3B).
3.5 Mitochondrial biogenesis and dynamics in the LV of obese HFpEF mice with and without insulin resistance
SIRT3 plays a central role in regulating the acetylation and deacetylation of mitochondrial proteins in the heart (30) and is involved in several critical mitochondrial processes, including (i) mitochondrial biogenesis and dynamics, (ii) redox homeostasis, and (iii) energy metabolism (Figure 4). Accordingly, selected mitochondrial targets of SIRT3 were further determined in the present study.

Figure 4. Schematic of SIRT3 mitochondrial targets. Aco1, aconitase 1; Bdh1, 3-hydroxybutyrate dehydrogenase 1; Cat, catalase; ETC, electron transport chain; Fis1, fission protein 1; Gpx1, glutathione peroxidase 1; HMGCS2, hydroxy-3-methylglutary coenzyme A (CoA) synthase 2; Hmox1, heme oxygenase-1; Mfn, mitofusin; Mnsod2, manganese superoxide dismutase 2; Mt-Co1, mitochondrially encoded cytochrome c oxidase I; Ndufa: NADH:ubiquinone oxidoreductase subunit A; Nrf2, nuclear factor erythroid 2-related factor 2; Opa1, optic atrophy 1; Oxct1, succinyl-CoA:3-ketoacid CoA transferase; OXPHOS, oxidative phosphorylation; Ppargc1a, peroxisome proliferator-activated receptor gamma coactivator 1-alpha; Sdha, succinate dehydrogenase complex flavoprotein subunit A; Slc16a1, solute carrier family 16 member 1; TCA, tricarboxylic acid; Uqcrh, ubiquinol-cytochrome c reductase hinge protein. Created using Servier Medical Art, licensed under CC BY 4.0.
The number, morphology, and distribution of mitochondria are regulated via a process called mitochondrial dynamics, which ensures that the energy demands of the cell are met. This involves proteins such as the mitochondrial master regulator peroxisome proliferator-activated receptor gamma coactivator 1-alpha (Ppargc1a); mitofusins (Mfn1 and Mfn2), which are essential for the fusion of the outer mitochondrial membrane; optic atrophy 1 (Opa1) which facilitates the fusion of the inner mitochondrial membrane, and fission protein 1 (Fis1), that conversely plays a role in regulating mitochondrial division (Figure 4) (27). In the present study, LV mRNA expression of Ppargc1a, Mfn1 and 2, and Opa1 remained unchanged, but there was a significant increase of Fis1 transcripts by 1.4-fold in OB-NIR HFpEF mice vs. OB-IR HFpEF mice (P = 0.0165; Figure 5), suggesting increased mitochondrial division in the LV of obese HFpEF mice lacking IR (OB-NIR).

Figure 5. Cardiac gene expression of mitochondrial biogenesis and dynamics regulators in obese HFpEF mice with and without insulin resistance. Data are represented as mean ± SEM. Statistical analysis by unpaired t-test for normally distributed data or Mann–Whitney test for non-normally distributed. Fis1, fission protein; Mfn, mitofusin; OB-IR, obese insulin-resistant HFpEF mice; OB-NIR, obese non-insulin-resistant HFpEF mice; Opa1, optic atrophy 1; Ppargc1a, peroxisome proliferator-activated receptor gamma coactivator 1-alpha. N = 7-9 mice/group.
3.6 Mitochondrial redox balance in the LV of obese HFpEF mice with and without insulin resistance
The elevated metabolic rate in the heart, sustained mainly by mitochondrial respiration, leads to the production of reactive oxygen species (ROS), from which cells are protected by antioxidant enzymes, such as catalases, peroxidases, superoxide dismutases (SODs), etc. The mRNA expression of antioxidant enzymes regulated by SIRT3 (Figure 4), including catalase (Cat), glutathione peroxidase 1 (Gpx1), heme oxygenase-1 (Hmox1), and nuclear factor erythroid 2-related factor 2 (Nrf2) were also measured. There were no changes in Cat, Gpx1, or Nrf2 transcript levels between OB-NIR HFpEF mice and OB-IR HFpEF, but there was a significant increase in Hmox1 mRNA expression by 1.6-fold in OB-NIR HFpEF mice compared to OB-IR HFpEF mice (P = 0.0265; Figure 6A). Moreover, protein expression analysis demonstrated a trend towards reduction of manganese superoxide dismutase 2 (MnSOD2) acetylation at lysine 68 in OB-NIR HFpEF mice compared to OB-IR HFpEF mice (Figure 6B).

Figure 6. Cardiac gene expression of mitochondrial redox balance regulators and MnSOD2 acetylation levels at lysine68 in obese HFpEF mice with and without insulin resistance. (A) mRNA expression of Cat, Gpx1, Hmox1 and NFr2 in the left ventricle of obese insulin-resistant HFpEF mice (OB-IR) and obese non-insulin-resistant HFpEF mice (OB-NIR). (B) Cardiac MnSOD2 acetylation at lysine 68 (AcK68) relative to total MnSOD2 protein expression in OB-IR and OB-NIR HFpEF mice. Data are represented as mean ± SEM. Statistical analysis by unpaired t-test for normally distributed data or Mann–Whitney test for non-normally distributed. Cat, catalase; Gpx1, glutathione peroxidase 1; Hmox1, heme oxygenase-1; Nrf2, nuclear factor erythroid 2-related factor 2; MNSOD2, manganese superoxide dismutase 2. N = 7-9 mice/group.
3.7 Mitochondrial metabolism in the LV of obese HFpEF mice with and without insulin resistance
Given that mitochondrial metabolism is tightly regulated by protein acetylation, it is notable that over 60% of mitochondrial proteins involved in energy metabolism display acetylation sites. These include proteins associated with (i) ketone body metabolism, (ii) the tricarboxylic acid (TCA) cycle, and (iii) the electron transport chain (ETC) (30). In the present study, the expression of key SIRT3 targets involved in these processes were investigated (Figure 4).
(i) Ketone body metabolism: Hydroxy-3-methylglutary coenzyme A (CoA) synthase 2 (HMGCS2) protein expression was decreased in OB-NIR HFpEF mice vs. OB-IR HFpEF mice (0.60 ± 0.16 vs. 1.00 ± 0.21, P = 0.0392; Figure 7A). This was accompanied by an increase in the mRNA expression of 3-hydroxybutyrate dehydrogenase 1 (Bdh1) and succinyl-CoA:3-ketoacid CoA transferase (Oxct1) by 1.5- and 1.3-fold in OB-NIR HFpEF, respectively (P = 0.0499 and P = 0.0450), while transcript levels of the ketone bodies transporter solute carrier family 16 member 1 (Slc16a1) remained unchanged (Figure 7B). Altogether, this suggests increased ketone body utilization in OB-NIR HFpEF mice.
(ii) TCA cycle: there were no changes in mRNA expression of aconitase 1 (Aco1) between OB-NIR HFpEF mice and OB-IR HFpEF mice (Figure 7B).
(iii) ETC: there was an increasing but insignificant trend in the mRNA expression of NADH:ubiquinone oxidoreductase subunits A9 (Ndufa9) and A11 (Ndufa11) (Complex I components), as well as succinate dehydrogenase complex flavoprotein subunit A (Sdha, a key component of Complex II), in OB-NIR HFpEF mice compared to OB-IR HFpEF mice. This was accompanied by increased transcript levels of ubiquinol-cytochrome C reductase hinge protein (Uqcrh, a Complex III subunit) and mitochondrially encoded cytochrome c oxidase I (Mt-Co1, mitochondrial-encoded subunit of Complex IV) by 1.7- and 1.3-fold in OB-NIR HFpEF mice compared to OB-IR HFpEF mice (P = 0.0031 and P = 0.0404, respectively; Figure 7B), suggesting enhanced mitochondrial respiratory chain function.
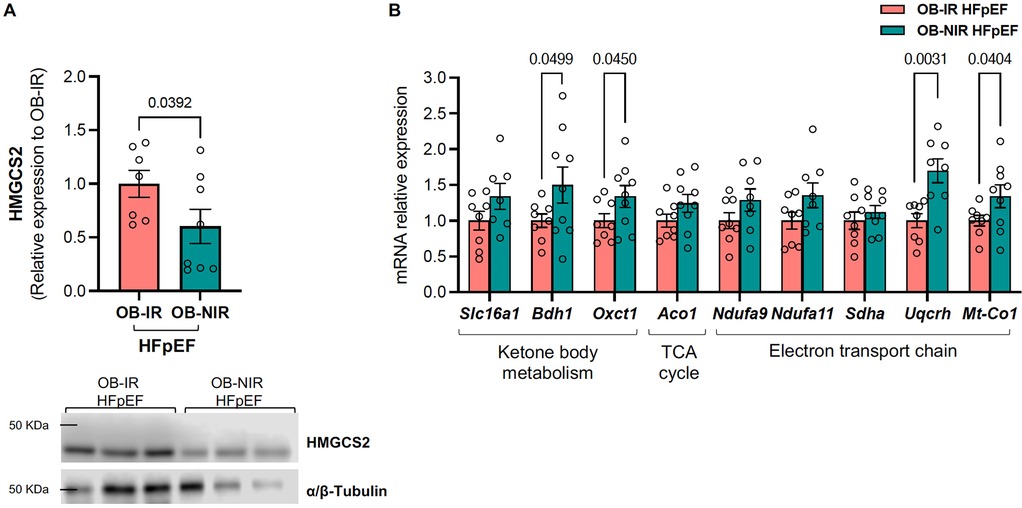
Figure 7. HMGCS2 protein and mitochondrial metabolism mRNA expression from the LV of obese HFpEF mice with and without insulin resistance. (A) Cardiac protein expression of HMGCS2 and (B) mRNA relative expression of ketone body metabolism, tricarboxylic acid (TCA) cycle, and electron transport chain related genes in obese insulin-resistant HFpEF mice (OB-IR) and obese non-insulin-resistant HFpEF mice (OB-NIR). Data are represented as mean ± SEM. Statistical analysis by unpaired t-test for normally distributed data or Mann–Whitney test for non-normally distributed. *P < 0.05, **P < 0.01 vs. OB-IR. Aco1, aconitase 1; Bdh1, 3-hydroxybutyrate dehydrogenase 1; HMGCS2, hydroxy-3-methylglutary coenzyme A (CoA) synthase 2; Mt-Co1, mitochondrially encoded cytochrome c oxidase I; Ndufa, NADH:ubiquinone oxidoreductase subunit A; Oxct1, succinyl-CoA:3-ketoacid CoA transferase; Sdha, succinate dehydrogenase complex flavoprotein subunit A; Slc16a1, solute carrier family 16 member 1; Uqcrh, ubiquinol-cytochrome c reductase hinge protein. N = 7-9 mice/group.
4 Discussion
The present study demonstrates that the LV phenotype of obese HFpEF mice varies according to their IR status. Compared to their obese insulin-resistant (OB-IR) HFpEF littermates, obese non–insulin-resistant (OB-NIR) HFpEF mice exhibited: (i) reduced cardiac fibrosis, (ii) increased cardiac expression of SIRT3 protein, and (iii) altered gene expression of mitochondrial SIRT3 targets, primarily those involved in ketone body metabolism and the ETC.
HFpEF patients often present with significant visceral adiposity, frequently accompanied by IR with or without T2D (31). Although obesity is a major risk factor for HFpEF, the mechanisms by which obesity alone contributes to HFpEF development remain unclear. Is the observed association primarily attributable to coexisting IR and/or T2D? In the present study, we aimed to disentangle the individual contributions of increased adiposity and IR/T2D to the obese HFpEF phenotype in mice. Our findings demonstrate that IR in obese HFpEF mice is associated with increased cardiac hypertrophy and fibrosis compared to lean, wild-type controls (Supplementary Figure S4). Interestingly, obese HFpEF mice without IR also exhibited comparable levels of cardiac hypertrophy; however, their cardiac fibrotic burden was significantly less relative to their OB-IR HFpEF counterparts. Given that cardiac hypertrophy was similar across obese HFpEF mice irrespective of IR status—and is therefore unlikely to be driven solely by fibrosis—these results suggest that extrinsic factors, such as increased adiposity, may contribute to elevated total and central blood volume. This hemodynamic burden may, in turn, promote adverse LV remodeling in obesity-associated HFpEF (32). In contrast, the greater degree of cardiac fibrosis observed in IR-positive obese HFpEF mice may reflect direct pathophysiological effects of IR on the myocardium, including enhanced neurohumoral activation and elevated systemic inflammation, leading to increased extracellular matrix remodeling (33).
Protein acetylation is a reversible post-translational modification process widely prevalent in the heart. Its dysregulation is implicated in many pathological conditions in animal models, including IR (34), obesity (35), hypertension (36), and HFpEF (37, 38). A major regulator of protein acetylation here is SIRT3, a mitochondrial NAD-dependent protein deacetylase that controls the acetylation status of proteins involved in mitochondrial dynamics, oxidative stress response, and metabolism (26, 39). We previously demonstrated that SIRT3 expression was decreased in the LV of non-obese, lean HFpEF mice (21). Similarly, SIRT3 expression is reduced in cardiac biopsies from failing human hearts in obese patients compared to non-obese patients (40), and this reduction is associated with a hyperacetylated mitochondrial profile in obese sucrose-fed rats. In the current study, obese HFpEF without IR (OB-NIR), exhibited increased SIRT3 expression accompanied by a non-significant reduction in LV hyperacetylation compared to their IR obese HFpEF (OB-IR) counterparts. Previous studies have shown that SIRT3 helps preserve cardiac function and capillary density in the context of obesity (41). Additionally, SIRT3 has been reported to mitigate obesity-related cardiac remodeling by attenuating inflammation and fibrosis by modulating the ROS-NF-κB-MCP-1 signaling pathway (42). In addition, the current findings demonstrate differential SIRT3 expression between OB-IR HFpEF and OB-NIR HFpEF, suggesting a partial restoration of mitochondrial protein deacetylation driven by increased SIRT3 levels in the OB-NIR HFpEF group. These molecular changes may contribute to modulation of adverse cardiac remodeling, particularly by influencing myocardial fibrosis in obesity-associated HFpEF.
There are currently no approved medications that directly target or modulate only SIRT3. However, several drugs have been reported to influence SIRT3 activity or expression indirectly. For example, proprotein convertase subtilisin/kexin type-9 (PCSK9) inhibitors may exert some of their pleiotropic effects through SIRT3, as demonstrated in vitro and in observational studies involving patients with atherosclerosis treated with PCSK9 inhibitors (43, 44). Although PCSK9 inhibitors are not approved for the treatment of HFpEF, emerging evidence suggests a potential link between PCSK9 and HFpEF pathophysiology. Notably, studies in PCSK9-deficient mice have shown that loss of PCSK9 expression induces metabolic reprogramming in cardiomyocytes, accompanied by structural remodeling, preserved ejection fraction, and reduced exercise capacity compared to wild-type controls (45). Thus, these findings suggest a possible intersection between PCSK9 signaling and SIRT3-mediated mitochondrial regulation, highlighting the need for further investigation into their interplay as a contributor to adverse cardiac remodeling and HFpEF progression.
Mitochondria undergo continuous fusion and fission, maintaining a balance in structure and function across various physiological and pathological states (27). In this study, transcript levels of proteins regulating mitochondrial dynamics were measured, and revealed a significant increase in Fis1 expression in OB-NIR HFpEF mice, indicative of enhanced mitochondrial division. This aligns with other studies where Fis1 upregulation via the SIRT3-FoxO3 pathway occurred in response to stress and oxidative damage (46). This also coincides with the observed increase in the antioxidant enzyme Hmox1, known to mediate mitochondrial quality control and dynamics in the heart (47). Overall, these findings suggest that the increase in fission preserves mitochondrial reserve capacity in response to oxidative damage, indicating enhanced mitochondrial quality control associated with improved insulin sensitivity in obesity-related HFpEF, potentially linked to SIRT3 expression.
Mitochondria metabolism is highly regulated by protein acetylation. Analysis of the mitochondrial acetylome have revealed that most of the mitochondrial proteins containing acetylation sites are involved in processes related to fatty acid metabolism, TCA cycle, and ETC (48). A “3-Hit” HFpEF mouse model exhibited increased cardiac hyperacetylation when compared to HFrEF and older control mice, with enrichment in the TCA cycle, oxidative phosphorylation (OXPHOS), and fatty acid oxidation (49). It is known that during obesity, the heart is very dependent on fatty acid oxidation as its primary source of ATP, while the contribution from glucose oxidation significantly decreases. In the current study, there was enhanced ketone body metabolism in OB-NIR HFpEF mice. This was characterized by reduced expression of HMGCS2, responsible for ketogenesis, alongside increased levels of Bdh1 and Oxct1, involved in the oxidation of ketone bodies, altogether suggesting a shift toward greater utilization of ketone bodies as an energy source. Evidence suggests that increased reliance on ketone body oxidation is an adaptive response in HF pathophysiology (50). Ketone bodies generate more energy in the form of heat compared with glucose and are more efficient than fatty acids for ATP production per molecule of oxygen consumed, providing an efficient alternative energy source for the heart especially when glucose and fatty acid oxidation pathways are impaired (51). In preclinical studies, augmentation of cardiac ketone body utilization, via exogenous supplementation, exerts cardioprotective effects such as attenuation of diastolic dysfunction, fibrosis, and pathological remodeling in HFpEF (37, 52). Similarly, SGLT2 inhibitors increased ketone levels in animal models of HF (53, 54) as well as in diabetic and nondiabetic subjects (55), and it has been postulated that the cardiovascular benefits driven by SGLT2 inhibitors may be partially mediated by their ability to increase circulating ketone bodies, thereby supporting myocardial energy metabolism (56). Thus, the increased ketone body metabolism in OB-NIR mice may similarly provide such a cardiac benefit in HFpEF. Lopashuk et al. (35) showed that decreased cardiac SIRT3 expression in murine models of obesity induced by high-fat feeding or genetic deletion leads to hyperacetylation and activation of beta-hydroxy acid dehydrogenase (β-HAD) and long-chain acyl-CoA dehydrogenase (LCAD), promoting increased fatty acid β-oxidation and IR. Conversely, in enhanced insulin sensitivity status, such as OB-NIR HFpEF, cardiac SIRT3 expression is abundant and leads to deacetylation of fatty acid oxidation enzymes and preventing IR in the heart. In the present study, there were no detectable changes in the TCA cycle but rather increased activity in the ETC between OB-IR HFpEF and OB-NIR HFpEF, suggesting that the metabolic environment in OB-NIR HFpEF mice may recapitulate the normal condition as proposed by Lopaschuk et al. (35) OB-NIR HFpEF mice demonstrated enhanced transcript levels of the ETC Complex subunits III and IV (Uqcrh and Mt-Co1, respectively) that may correlate with improved mitochondrial function and cardiac performance, contrasting with mitochondrial impairments typically seen in IR HFpEF models. As respiratory capacity and ATP synthesis have been shown to be decreased in cardiac mitochondria of SIRT3 KO mice (57), these results may also reflect a compensatory increase to optimize OXPHOS efficiency, reducing ROS generation, and maintaining energy production.
In conclusion, obese HFpEF mice without insulin resistance (OB-NIR) exhibit a distinct cardiac phenotype compared to insulin-resistant (OB-IR) counterparts, highlighting the potential independent contributions of adiposity and mitochondrial adaptations in modulating disease severity in obesity-associated HFpEF. The observed reduction in cardiac fibrosis, increased SIRT3 expression, and improved mitochondrial dynamics and function in OB-NIR HFpEF mice suggest the presence of adaptive metabolic responses aimed at preserving energy homeostasis and attenuating oxidative stress. Moreover, the increased reliance on ketone body metabolism in OB-NIR mice may reflect a compensatory mechanism that supports mitochondrial efficiency and cardiac function—an adaptive capacity that appears to be compromised in the setting of obesity with insulin resistance.
These findings underscore the critical role of metabolic flexibility and mitochondrial quality control in the pathophysiology HFpEF, providing valuable insights into the complex interplay between adiposity, insulin sensitivity, and cardiac function. Further studies are warranted to explore the potential of targeting these pathways in HFpEF patients and to determine if preservation of mitochondrial and metabolic adaptations could mitigate the adverse cardiac outcomes typically observed in obesity with IR and T2D. Modulation of protein acetylation represents a promising therapeutic avenue; but studies with rigorous experimental approaches and validated acetylation-modulating agents in clinically relevant disease models -such as obesity-HFpEF- are essential to establish efficacy and translational potential.
4.1 Clinical relevance
Recent studies underscore the heterogeneity of obesity in HFpEF and highlight the critical role of metabolic health in shaping disease progression. Our findings demonstrate that obesity without IR is associated with preserved mitochondrial adaptations, including elevated SIRT3 expression and reduced protein hyperacetylation, which may protect against adverse cardiac remodeling. These findings suggest that the metabolic status of obese HFpEF patients is an active modifier of mitochondrial quality control and myocardial remodeling. The observed upregulation of SIRT3 in insulin-sensitive obesity may confer partial cardiac protection, supporting the concept for phenotype-specific therapeutic approaches in obese HFpEF. While both SIRT3 and ketone metabolism emerge as promising therapeutic targets, translational challenges remain. Future studies should focus on defining how metabolic health modulates HFpEF progression across diverse patient populations, and whether interventions aimed at enhancing SIRT3 activity can mitigate fibrosis and remodeling in obesity-associated HFpEF.
4.2 Limitations
This study is based on gene and protein expression associations, and as such, the findings are limited by the transient nature of gene regulation. While these associations provide valuable insights into potential mechanisms, they do not offer definitive evidence of long-term effects or functional consequences. Further studies are needed to validate these gene expression patterns and explore their functional relevance to better understand the physiological implications of the observed gene expression changes. In particular, studies investigating mitochondrial function—including measurements of mitochondrial respiration, bioenergetics, and dynamics—are essential to determine the physiological implications of the reported transcriptional and proteomic changes. Finally, the current study included only male mice, and thus the sex specificity of these findings cannot be excluded. Future studies incorporating female mice are necessary to confirm and extend these findings across sexes.
Data availability statement
The original contributions presented in this study are included in the article/Supplementary Material. Additional information or resources can be made available upon reasonable request to the corresponding author.
Ethics statement
The animal study was approved by Institutional Animal Care and Use Committee at Boston University School of Medicine (IACUC: PROTO201800310). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
MV-M: Conceptualization, Investigation, Methodology, Project administration, Supervision, Validation, Data curation, Formal analysis, Visualization, Writing – original draft, Writing – review & editing. HC: Data curation, Formal analysis, Investigation, Project administration, Writing – review & editing. SL: Investigation, Methodology, Data curation, Writing – review & editing, Methodology. ES: Investigation, Methodology, Data curation, Formal analysis, Visualization, Writing – original draft, Writing – review & editing. RW: Data curation, Investigation, Methodology, Writing – review & editing. CK: Writing – review & editing. PS: Writing – original draft, Writing – review & editing. FS: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by NIH R01HL145985 to FS. PES was supported by NIH R01DK055758 and R01DK099110.
Conflict of interest
FS is a full-time employee of Eli Lilly and Co, Indianapolis.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2025.1678992/full#supplementary-material
References
1. Reddy YNV, Carter RE, Sundaram V, Kaye DM, Handoko ML, Tedford RJ, et al. An evidence-based screening tool for heart failure with preserved ejection fraction: the HFpEF-ABA score. Nat Med. (2024) 30(8):2258–64. doi: 10.1038/s41591-024-03140-1
2. Obokata M, Reddy YNV, Pislaru SV, Melenovsky V, Borlaug BA. Evidence supporting the existence of a distinct obese phenotype of heart failure with preserved ejection fraction. Circulation. (2017) 136(1):6–19. doi: 10.1161/CIRCULATIONAHA.116.026807
3. Caruana L, Petrie MC, Davie AP, McMurray JJ. Do patients with suspected heart failure and preserved left ventricular systolic function suffer from “diastolic heart failure” or from misdiagnosis? A prospective descriptive study. Br Med J. (2000) 321(7255):215–8. doi: 10.1136/bmj.321.7255.215
4. Romero Funes D, Gutierrez Blanco D, Botero-Fonnegra C, Hong L, Lo Menzo E, Szomstein S, et al. Bariatric surgery decreases the number of future hospital admissions for diastolic heart failure in subjects with severe obesity: a retrospective analysis of the US national inpatient sample database. Surg Obes Relat Dis. (2022) 18(1):1–8. doi: 10.1016/j.soard.2021.09.009
5. Hamo CE, DeJong C, Hartshorne-Evans N, Lund LH, Shah SJ, Solomon S, et al. Heart failure with preserved ejection fraction. Nat Rev Dis Primers. (2024) 10(1):55. doi: 10.1038/s41572-024-00540-y
6. Wu CK, Lee JK, Hsu JC, Su MM, Wu YF, Lin TT, et al. Myocardial adipose deposition and the development of heart failure with preserved ejection fraction. Eur J Heart Fail. (2020) 22(3):445–54. doi: 10.1002/ejhf.1617
7. Kitzman DW, Shah SJ. The HFpEF obesity phenotype: the elephant in the room. J Am Coll Cardiol. (2016) 68(2):200–3. doi: 10.1016/j.jacc.2016.05.019
8. Borlaug BA, Jensen MD, Kitzman DW, Lam CSP, Obokata M, Rider OJ. Obesity and heart failure with preserved ejection fraction: new insights and pathophysiological targets. Cardiovasc Res. (2023) 118(18):3434–50. doi: 10.1093/cvr/cvac120
9. Kitzman DW, Nicklas BJ. Pivotal role of excess intra-abdominal adipose in the pathogenesis of metabolic/obese HFpEF. JACC Heart Fail. (2018) 6(12):1008–10. doi: 10.1016/j.jchf.2018.08.007
10. Mishra S, Kass DA. Cellular and molecular pathobiology of heart failure with preserved ejection fraction. Nat Rev Cardiol. (2021) 18(6):400–23. doi: 10.1038/s41569-020-00480-6
11. von Bibra H, Paulus W, St John Sutton M. Cardiometabolic syndrome and increased risk of heart failure. Curr Heart Fail Rep. (2016) 13(5):219–29. doi: 10.1007/s11897-016-0298-4
12. Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. (2013) 62(4):263–71. doi: 10.1016/j.jacc.2013.02.092
13. Packer M, Zile MR, Kramer CM, Baum SJ, Litwin SE, Menon V, et al. Tirzepatide for heart failure with preserved ejection fraction and obesity. N Engl J Med. (2025) 392(5):427–37. doi: 10.1056/NEJMoa2410027
14. King AJ. The use of animal models in diabetes research. Br J Pharmacol. (2012) 166(3):877–94. doi: 10.1111/j.1476-5381.2012.01911.x
15. Kusminski CM, Holland WL, Sun K, Park J, Spurgin SB, Lin Y, et al. MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nat Med. (2012) 18(10):1539–49. doi: 10.1038/nm.2899
16. Kusminski CM, Park J, Scherer PE. MitoNEET-mediated effects on browning of white adipose tissue. Nat Commun. (2014) 5:3962. doi: 10.1038/ncomms4962
17. Hall JE, do Carmo JM, da Silva AA, Wang Z, Hall ME. Obesity, kidney dysfunction and hypertension: mechanistic links. Nat Rev Nephrol. (2019) 15(6):367–85. doi: 10.1038/s41581-019-0145-4
18. Hulsmans M, Sager HB, Roh JD, Valero-Munoz M, Houstis NE, Iwamoto Y, et al. Cardiac macrophages promote diastolic dysfunction. J Exp Med. (2018) 215(2):423–40. doi: 10.1084/jem.20171274
19. Valero-Munoz M, Li S, Wilson RM, Hulsmans M, Aprahamian T, Fuster JJ, et al. Heart failure with preserved ejection fraction induces beiging in adipose tissue. Circ Heart Fail. (2016) 9(1):e002724. doi: 10.1161/CIRCHEARTFAILURE.115.002724
20. Valero-Munoz M, Oh A, Faudoa E, Breton-Romero R, El Adili F, Bujor A, et al. Endothelial-mesenchymal transition in heart failure with a preserved ejection fraction: insights into the cardiorenal syndrome. Circ Heart Fail. (2021) 14(9):e008372. doi: 10.1161/CIRCHEARTFAILURE.121.008372
21. Valero-Munoz M, Saw EL, Hekman RM, Blum BC, Hourani Z, Granzier H, et al. Proteomic and phosphoproteomic profiling in heart failure with preserved ejection fraction (HFpEF). Front Cardiovasc Med. (2022) 9:966968. doi: 10.3389/fcvm.2022.966968
22. Wilson RM, De Silva DS, Sato K, Izumiya Y, Sam F. Effects of fixed-dose isosorbide dinitrate/hydralazine on diastolic function and exercise capacity in hypertension-induced diastolic heart failure. Hypertension. (2009) 54(3):583–90. doi: 10.1161/HYPERTENSIONAHA.109.134932
23. Yoon S, Kim M, Lee H, Kang G, Bedi K, Margulies KB, et al. S-nitrosylation of histone deacetylase 2 by neuronal nitric oxide synthase as a mechanism of diastolic dysfunction. Circulation. (2021) 143(19):1912–25. doi: 10.1161/CIRCULATIONAHA.119.043578
24. Horton JL, Martin OJ, Lai L, Riley NM, Richards AL, Vega RB, et al. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight. (2016) 2(1):e84897. doi: 10.1172/jci.insight.84897
25. Lai L, Leone TC, Keller MP, Martin OJ, Broman AT, Nigro J, et al. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: a multisystems approach. Circ Heart Fail. (2014) 7(6):1022–31. doi: 10.1161/CIRCHEARTFAILURE.114.001469
26. Zhang J, Xiang H, Liu J, Chen Y, He RR, Liu B. Mitochondrial sirtuin 3: new emerging biological function and therapeutic target. Theranostics. (2020) 10(18):8315–42. doi: 10.7150/thno.45922
27. Cao M, Zhao Q, Sun X, Qian H, Lyu S, Chen R, et al. Sirtuin 3: emerging therapeutic target for cardiovascular diseases. Free Radic Biol Med. (2022) 180:63–74. doi: 10.1016/j.freeradbiomed.2022.01.005
28. Hershberger KA, Martin AS, Hirschey MD. Role of NAD(+) and mitochondrial sirtuins in cardiac and renal diseases. Nat Rev Nephrol. (2017) 13(4):213–25. doi: 10.1038/nrneph.2017.5
29. Newsom SA, Boyle KE, Friedman JE. Sirtuin 3: a major control point for obesity-related metabolic diseases? Drug Discov Today Dis Mech. (2013) 10(1-2):e35–40. doi: 10.1016/j.ddmec.2013.04.001
30. Parodi-Rullan RM, Chapa-Dubocq XR, Javadov S. Acetylation of mitochondrial proteins in the heart: the role of SIRT3. Front Physiol. (2018) 9:1094. doi: 10.3389/fphys.2018.01094
31. Kass DA. Understanding HFpEF with obesity: will pigs come to the rescue? JACC Basic Transl Sci. (2021) 6(2):171–3. doi: 10.1016/j.jacbts.2020.12.010
32. Alpert MA, Lavie CJ, Agrawal H, Aggarwal KB, Kumar SA. Obesity and heart failure: epidemiology, pathophysiology, clinical manifestations, and management. Transl Res. (2014) 164(4):345–56. doi: 10.1016/j.trsl.2014.04.010
33. Tuleta I, Frangogiannis NG. Fibrosis of the diabetic heart: clinical significance, molecular mechanisms, and therapeutic opportunities. Adv Drug Deliv Rev. (2021) 176:113904. doi: 10.1016/j.addr.2021.113904
34. Williams AS, Koves TR, Davidson MT, Crown SB, Fisher-Wellman KH, Torres MJ, et al. Disruption of acetyl-lysine turnover in muscle mitochondria promotes insulin resistance and redox stress without overt respiratory dysfunction. Cell Metab. (2020) 31(1):131–47.e11. doi: 10.1016/j.cmet.2019.11.003
35. Alrob OA, Sankaralingam S, Ma C, Wagg CS, Fillmore N, Jaswal JS, et al. Obesity-induced lysine acetylation increases cardiac fatty acid oxidation and impairs insulin signalling. Cardiovasc Res. (2014) 103(4):485–97. doi: 10.1093/cvr/cvu156
36. Dikalov SI, Dikalova AE. Crosstalk between mitochondrial hyperacetylation and oxidative stress in vascular dysfunction and hypertension. Antioxid Redox Signal. (2019) 31(10):710–21. doi: 10.1089/ars.2018.7632
37. Deng Y, Xie M, Li Q, Xu X, Ou W, Zhang Y, et al. Targeting mitochondria-inflammation circuit by beta-hydroxybutyrate mitigates HFpEF. Circ Res. (2021) 128(2):232–45. doi: 10.1161/CIRCRESAHA.120.317933
38. Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, Kim SY, et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature. (2019) 568(7752):351–6. doi: 10.1038/s41586-019-1100-z
39. Rardin MJ, Newman JC, Held JM, Cusack MP, Sorensen DJ, Li B, et al. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc Natl Acad Sci U S A. (2013) 110(16):6601–6. doi: 10.1073/pnas.1302961110
40. Castillo EC, Morales JA, Chapoy-Villanueva H, Silva-Platas C, Trevino-Saldana N, Guerrero-Beltran CE, et al. Mitochondrial hyperacetylation in the failing hearts of obese patients mediated partly by a reduction in SIRT3: the involvement of the mitochondrial permeability transition pore. Cell Physiol Biochem. (2019) 53(3):465–79. doi: 10.33594/000000151
41. Zeng H, Vaka VR, He X, Booz GW, Chen JX. High-fat diet induces cardiac remodelling and dysfunction: assessment of the role played by SIRT3 loss. J Cell Mol Med. (2015) 19(8):1847–56. doi: 10.1111/jcmm.12556
42. Guo X, Yan F, Li J, Zhang C, Su H, Bu P. SIRT3 ablation deteriorates obesity-related cardiac remodeling by modulating ROS-NF-kappaB-MCP-1 signaling pathway. J Cardiovasc Pharmacol. (2020) 76(3):296–304. doi: 10.1097/FJC.0000000000000877
43. D'Onofrio N, Prattichizzo F, Marfella R, Sardu C, Martino E, Scisciola L, et al. SIRT3 mediates the effects of PCSK9 inhibitors on inflammation, autophagy, and oxidative stress in endothelial cells. Theranostics. (2023) 13(2):531–42. doi: 10.7150/thno.80289
44. Marfella R, Prattichizzo F, Sardu C, Paolisso P, D'Onofrio N, Scisciola L, et al. Evidence of an anti-inflammatory effect of PCSK9 inhibitors within the human atherosclerotic plaque. Atherosclerosis. (2023) 378:117180. doi: 10.1016/j.atherosclerosis.2023.06.971
45. Da Dalt L, Castiglioni L, Baragetti A, Audano M, Svecla M, Bonacina F, et al. PCSK9 deficiency rewires heart metabolism and drives heart failure with preserved ejection fraction. Eur Heart J. (2021) 42(32):3078–90. doi: 10.1093/eurheartj/ehab431
46. Tseng AH, Shieh SS, Wang DL. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic Biol Med. (2013) 63:222–34. doi: 10.1016/j.freeradbiomed.2013.05.002
47. Hull TD, Boddu R, Guo L, Tisher CC, Traylor AM, Patel B, et al. Heme oxygenase-1 regulates mitochondrial quality control in the heart. JCI Insight. (2016) 1(2):e85817. doi: 10.1172/jci.insight.85817
48. Baeza J, Smallegan MJ, Denu JM. Mechanisms and dynamics of protein acetylation in mitochondria. Trends Biochem Sci. (2016) 41(3):231–44. doi: 10.1016/j.tibs.2015.12.006
49. Liu X, Zhang Y, Deng Y, Yang L, Ou W, Xie M, et al. Mitochondrial protein hyperacetylation underpins heart failure with preserved ejection fraction in mice. J Mol Cell Cardiol. (2022) 165:76–85. doi: 10.1016/j.yjmcc.2021.12.015
50. Matsuura TR, Puchalska P, Crawford PA, Kelly DP. Ketones and the heart: metabolic principles and therapeutic implications. Circ Res. (2023) 132(7):882–98. doi: 10.1161/CIRCRESAHA.123.321872
51. Kodur N, Yurista S, Province V, Rueth E, Nguyen C, Tang WHW. Ketogenic diet in heart failure: fact or fiction? JACC Heart Fail. (2023) 11(7):838–44. doi: 10.1016/j.jchf.2023.05.009
52. Liao S, Tang Y, Yue X, Gao R, Yao W, Zhou Y, et al. Beta-hydroxybutyrate mitigated heart failure with preserved ejection fraction by increasing treg cells via Nox2/GSK-3beta. J Inflamm Res. (2021) 14:4697–706. doi: 10.2147/JIR.S331320
53. Santos-Gallego CG, Requena-Ibanez JA, San Antonio R, Ishikawa K, Watanabe S, Picatoste B, et al. Empagliflozin ameliorates adverse left ventricular remodeling in nondiabetic heart failure by enhancing myocardial energetics. J Am Coll Cardiol. (2019) 73(15):1931–44. doi: 10.1016/j.jacc.2019.01.056
54. Yurista SR, Sillje HHW, Oberdorf-Maass SU, Schouten EM, Pavez Giani MG, Hillebrands JL, et al. Sodium-glucose co-transporter 2 inhibition with empagliflozin improves cardiac function in non-diabetic rats with left ventricular dysfunction after myocardial infarction. Eur J Heart Fail. (2019) 21(7):862–73. doi: 10.1002/ejhf.1473
55. Ferrannini E, Baldi S, Frascerra S, Astiarraga B, Heise T, Bizzotto R, et al. Shift to fatty substrate utilization in response to sodium-glucose cotransporter 2 inhibition in subjects without diabetes and patients with type 2 diabetes. Diabetes. (2016) 65(5):1190–5. doi: 10.2337/db15-1356
56. Lopaschuk GD, Verma S. Mechanisms of cardiovascular benefits of sodium glucose co-transporter 2 (SGLT2) inhibitors: a state-of-the-art review. JACC Basic Transl Sci. (2020) 5(6):632–44. doi: 10.1016/j.jacbts.2020.02.004
Keywords: HFPEF, insulin resistance, obesity, SIRT3, mitochondria metabolism
Citation: Valero-Muñoz M, Cooper HL, Li S, Saw EL, Wilson RM, Kusminski CM, Scherer PE and Sam F (2025) Metabolic dysregulation in the heart in obesity-associated HFpEF. Front. Cardiovasc. Med. 12:1678992. doi: 10.3389/fcvm.2025.1678992
Received: 3 August 2025; Accepted: 10 September 2025;
Published: 26 September 2025.
Edited by:
Tatsuya Sato, Sapporo Medical University, JapanReviewed by:
Pongpan Tanajak, Rehabilitation Center, ThailandNiklas Dörmann, Universitätsmedizin Greifswald, Germany
Copyright: © 2025 Valero-Muñoz, Cooper, Li, Saw, Wilson, Kusminski, Scherer and Sam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Flora Sam, ZmxvcmFzYW1AYnUuZWR1