Abstract
Cardiocerebral vascular disease has long been the leading cause of morbidity and mortality worldwide. Although there are many effective avenues for preventing and treating cardiocerebral vascular disease, further research is still needed to identify more novel molecular targets for therapeutic intervention. Anoctamin-1 (ANO1), also known as transmembrane protein 16A (TMEM16A), is the molecular identity of calcium-activated chloride channels (CaCCs) and is widely distributed in myocardial cells and the vasculature, including but not limited to the thoracic aorta, mesenteric artery, cerebral artery, and portal vein. ANO1 has many functions in the cardiocerebral vascular system, including cardiac excitability, vascular smooth muscle contraction, and epithelial cell secretion. Aberrant expression or dysfunction of ANO1 is associated with several cardiocerebral vascular diseases, including myocardial ischaemia/reperfusion injury (MIRI), arrhythmias, cardiac fibrosis, hypertension, and stroke. Therefore, this review provides an overview of ANO1, including its structure, distribution, and activation mechanism, and highlights the current knowledge of ANO1 in the pathophysiological process of heart diseases, hypertension, and stroke. We also summarise the pharmacological regulatory target of ANO1, providing promising insights for applying ANO1 inhibitors as cardiac and cerebrovascular therapeutic agents.
Graphical Abstract
1 Introduction
The anoctamin (ANO)/transmembrane protein 16 (TMEM16) family of membrane proteins consists of ten members, ranging from ANO1 (TMEM16A) to ANO10 (TMEM16K), which play key roles in diverse biological processes, including ion transport, phospholipid scrambling and regulation of other membrane proteins (1, 2). ANO1 was first identified as the gene encoding calcium-activated chloride channels (CaCC) (3–5). Since then, numerous experiments have shown that ANO1 is a homodimer with a quaternary structure comprising two pores (6, 7) and that each subunit consists of cytoplasmic N- and C-terminal domains, ten transmembrane α-helix, and extracellular components (8). In addition, a pore is formed in the homodimeric α3-α7 helices, which contain two conserved calcium (Ca2+) binding sites, and the inner half of the α6 helix acts as a gating element (2, 6). Specifically, certain acidic residues (E654, E702, E705, E734, D738, N650, N651 and N730) are critical for coordination with Ca2+ (7–9). The binding of Ca2+ to a site located within the transmembrane domain triggers an allosteric process of an α-helix, altering the pore conduction and leading to the conduction of an outwardly rectifying current, mainly the efflux of chloride ions (Cl−) (as shown in Figure 1) (8).
Figure 1
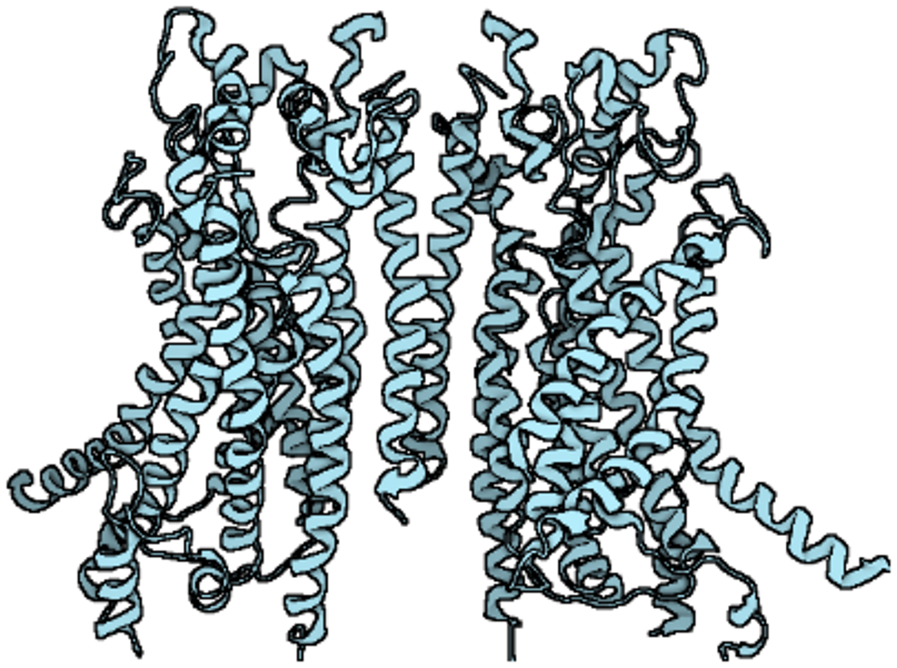
The structure of ANO1.
ANO1, the significant component of the CaCCs in the cardiovascular system, is abundantly distributed in various vascular endothelial cells (ECs) and smooth muscle cells (SMCs) (10). Previous research has shown that activation of ANO1 is gated by cytosolic Ca2+ as well as calmodulin, membrane voltage, and external permeant anions (11, 12). Upon exposure to vasoconstrictors (e.g., endothelin-1, angiotensin II), Gq-protein coupled receptors (GqPCRs) are activated, and phosphatidylinositol 4,5-bisphosphate (PIP2) is rapidly converted to inositol trisphosphate (IP3) and diacylglycerol (DAG). It is generally accepted that ANO1 is directly triggered by Ca2+ release from the sarcoplasmic reticulum (SR) via the IP3 receptors (13), leading to Cl− efflux and depolarisation of the membrane potential. Voltage-gated L-type calcium channels are then activated, and large amounts of Ca2+ enter the cell, causing smooth muscle to contract (14).
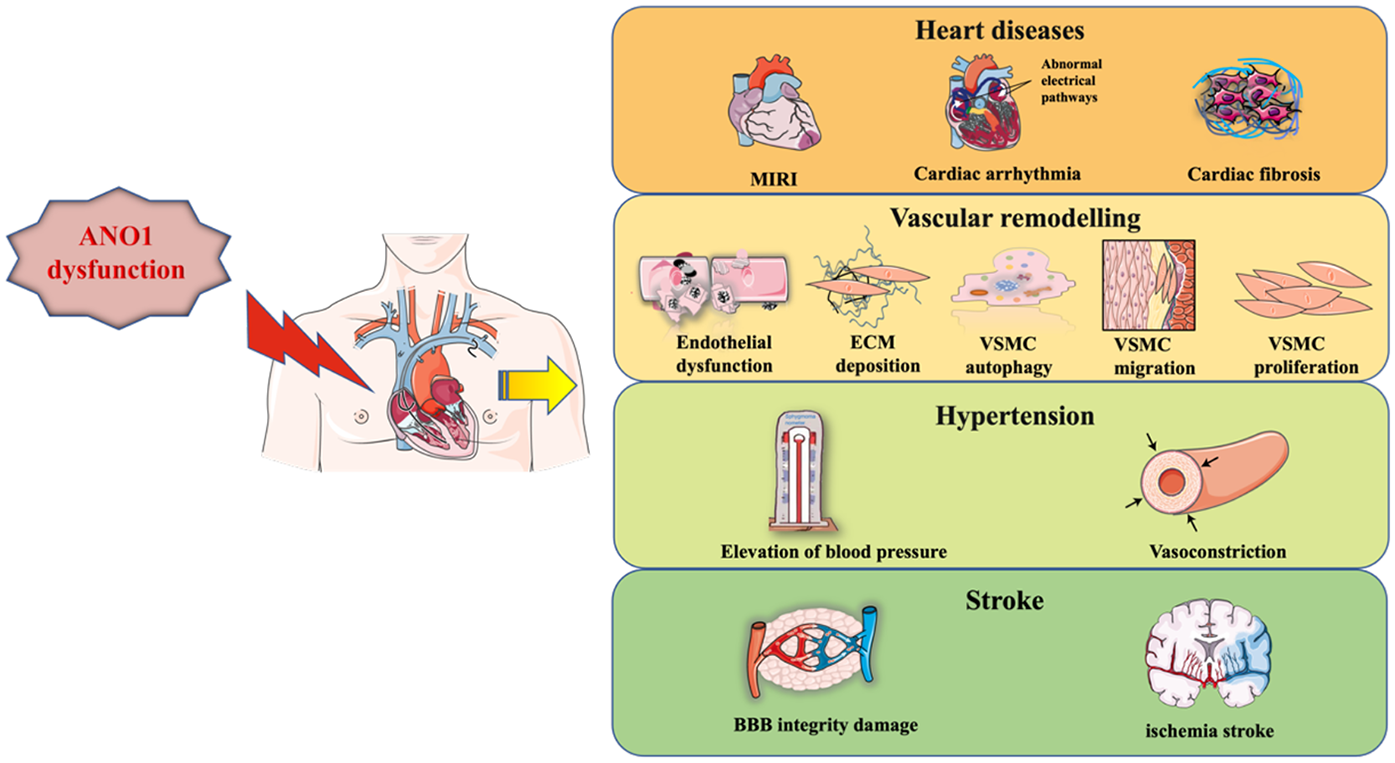
Numerous studies have shown that ANO1 is involved in several pathological processes in the cardiocerebral vascular system: myocardial ischaemia/reperfusion injury (MIRI), arrhythmia, myocardial fibrosis, hypertension and stroke (as shown in Table 1). This review will present a detailed elucidation of the role of ANO1 in cardiocerebral vascular disease (as shown in Figures 2–4), providing a theoretical basis for the therapeutic targeting of ANO1 in treating these diseases (as shown in Table 2).
Table 1
| Role | Model | Abnormal ANO1 expression | Results and mechanism | Reference |
|---|---|---|---|---|
| MIRI | Mouse I/R injury model and H9c2 H/R model | Upregulated | Upregulation of ANO1 induced MIRI via the miR-144-3p/ANO1/NLRP3 inflammasome axis | (26) |
| Arrhythmias | myocardial ischemia mice | Upregulated | Upregulation of ANO1 induced cardiac arrhythmia by accelerating phase 1 repolarization of action potentials and influencing cardiac action potential duration | (17) |
| Cardiac fibrosis | Rat cardiac tissues after MI and cardiac fibroblasts after hypoxia | Upregulated | Upregulation of ANO1 alleviated cardiac fibrosis by inhibiting TGF-β/smad3 pathway | (20) |
| Pressure-overload mice (a model of thoracic aortic constriction) and ANO1 transgenic mice | Downregulated | Downregulation of ANO1 induced cardiac fibrosis, but overexpression of ANO1 alleviated it by inhibiting TGF-β/smad3 pathway | (21) | |
| Rat MI model | First upregulated and then decreased | Upregulation of ANO1 induced cardiac fibrosis via the AT1R-mediated MAPK signaling pathway | (31) | |
| Systemic hypertension | SHR | Upregulated | Upregulation of ANO1 promoted VSMCs in arteries growth and proliferation and blood vessel constriction and induced hypertension | (50) |
| AngII-infused hypertensive mice | Downregulated | Downregulation of ANO1 induced human aortic SMC proliferation and vascular remodeling by disrupting AngII-mediated KLF5/myocardin/SRF pathway | (51) | |
| AngII-infused hypertensive mice and SMC-specific ANO1 transgenic mice | Downregulated | Downregulation of ANO1 induced mice aortic SMC autophagy and exacerbated vascular remodeling by disrupting the formation of p62/Bcl-2/Beclin-1/VPS34 complex | (52) | |
| ANO1 endothelial-specific transgenic and knockout mice with AngII-infused hypertension | Not clear | Upregulation of ANO1 elevated BP and induced endothelial dysfunction by increasing Nox2-containing NADPH oxidase-derived reactive oxygen species, whereas the knockout of ANO1 produced opposite effects | (38) | |
| Pressure overload-induced myocardium remodelling of heart of mice | Upregulated | Upregulation of ANO1 promoted migration and angiogenesis of HUVECs | (53) | |
| HAECs | Downregulated | Downregulation of ANO1 promoted HAEC proliferation, migration, and angiogenesis | (55) | |
| Pulmonary arterial hypertension | MCT-induced PH rats | Upregulated | Upregulation of ANO1 induced an elevation in Ca2+-activated Cl− current | (57) |
| IPAH and two animal models of PAH (the chronic hypoxic-induced mice and monocrotaline treated rats) | Upregulated | Upregulation of ANO1 stimulated vasoconstriction and proliferation of PASMC via enhanced c-fos phosphorylation | (36) | |
| MCT-treated PAH rats | Upregulated | Upregulation of ANO1 promoted PASMC proliferation, remodeling of pulmonary arterioles, and right ventricular hypertrophy by activating ERK1/2 | (60) | |
| High-flow-induced pulmonary hypertensive rats | Upregulated | Upregulation of ANO1 promoted the proliferation of PASMCs via p38MAPK/ERK signal pathway | (59) | |
| PAECs isolated from IPAH | Upregulated | Upregulation of ANO1 promoted human PAEC apoptosis but suppressed PAEC proliferation via mtROS-p38-caspase-3 pathway | (45) | |
| PAECs isolated from IPAH | Upregulated | Upregulation of ANO1 inhibited human PAEC proliferation but unaffected HPAEC apoptosis and caused endothelial dysfunction via ERK1/2 pathway | (46) | |
| Portal hypertension | BDL rat with PHT and PVSMC isolation and transfection | Downregulated in the portal vein of PHT rats in vivo | p-ERK1/2 was increased in vivo and ANO1 promoted PVSMC proliferation in vitro | (62) |
| BDL mice with cirrhotic PHT and PPVL mice with non-cirrhotic PHT | downregulated in BDL-PVSMCs whereas not in PPVL-PVSMCs | Downregulation of ANO1 attenuated spontaneous contraction of the BDL-portal vein and alleviated PHT | (61) | |
| Stroke | 2k2c renohypertensive rats | Downregulated | ANO1 involved in AngII-induced cerebral vasoconstriction via the RhoA/ROCK signalling pathway | (65) |
| Middle cerebral artery occlusion mice; OGD/R model | Upregulated | ANO1 expression was elevated in BCECs via lncRNA ENST00000530525 though recruiting transcription factors or enhancers after ischaemic stroke | (67) | |
| Middle cerebral artery occlusion mice | Upregulated | Upregulated ANO1 exacerbated brain injury via accumulating intracellular adhesion molecule-1 in a nuclear factor-kappaB-dependent manner | (66) | |
| BCECs isolated from mice | Not clear | ANO1 dysfunction stimulated BCEC proliferation and transendothelial permeability | (54) | |
| Mice stroke model | Not clear | Ischaemia-activated ANO1 increases pericyte contraction and reduces cerebral blood flow | (70) | |
| 2-kidney,2-clip hypertensive rats | Downregulated | Downregulation of ANO1 induced BASMC proliferation and cerebrovascular remodeling by promoting cell cycle transition at the G0/G1 phase via the overexpression of cyclin D1 and cyclin E | (71) | |
| AngII-infused hypertensive mice | Downregulated | Downregulation of ANO1 by CaMKIIγ-mediated phosphorylation induced BASMC proliferation and cerebrovascular remodeling | (72) | |
| Cultured rat BASMCs and VSMC-specific ANO1 transgenic mice | Not clear | ANO1 dysfunction promoted H2O2-induced BASMC apoptosis by increasing mitochondrial membrane permeabilization via interaction with CypD | (74) | |
| VSMC-specific ANO1 transgenic mice with AngII-induced hypertension | Not clear | ANO1 dysfunction inhibited AngII-induced BASMC migration and protected the basilar artery against remodeling by suppressing WNK1, RhoA/ROCK2/MLCP/MLC20, and integrinb3/FAK pathways | (75) | |
| VSMC-specific ANO1 transgenic mice with AngII-induced hypertension | Not clear | ANO1 dysfunction inhibited extracellular matrix deposition and AngII-induced cerebrovascular remodeling by reducing levels of MMPs/TIMPs and the activity of WNK1, TGF-β1/Smad3, ERK, and JNK | (76) |
Different roles of ANO1 in the cardiovascular disease.
BASMC, basilar artery smooth muscle cell; BCEC, brain capillary endothelial cell; BDL, bile duct ligation; CVEC, cardiac vascular endothelial cell;H2O2, hydrogen peroxide; H/R, hypoxia/reoxygenation; IPAH, patients with idiopathic PAH; I/R, ischemia/reperfusion; KLF5, Kruppel-like factor 5; MASMC, mouse aortic smooth muscle cell; MCT, monocrotaline; MIRI, myocardial ischemia/reperfusion injury; PAEC, pulmonary arterial endothelial cell; PASMC, pulmonary arterial smooth muscle cell; PHT, portal hypertension; PPVL, partial portal vein ligation; PVSMC, portal vein smooth muscle cell; SRF, serum response factor; SHR, spontaneously hypertensive rat; VSMC, vascular smooth muscle cell.
Figure 2
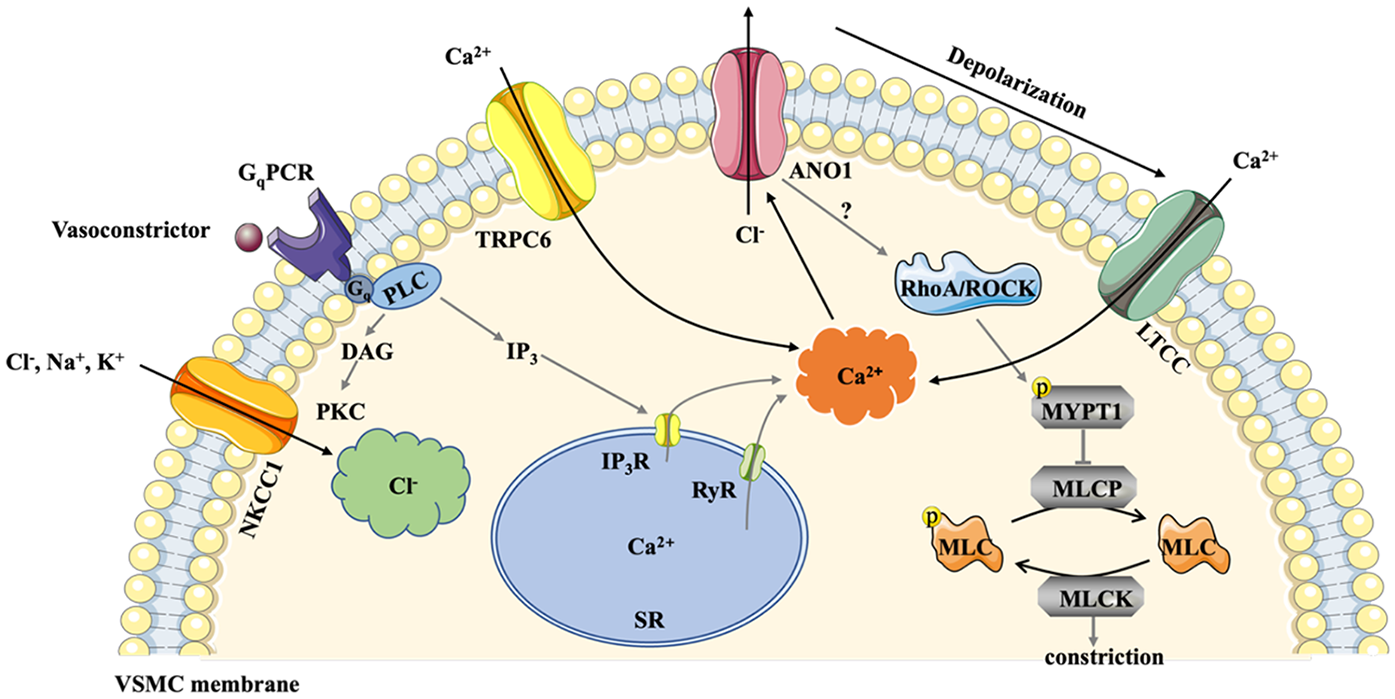
Schematic mechanism of ANO1 activation in VSMC. NKCC1 accumulates substantial Cl− in VSMCs and elevates the driving force for Cl− efflux. Exposed to vasoconstrictors, GqPCR is activated, and PIP2 is quickly converted to IP3 and DAG. ANO1 is directly activated by Ca2+ releasing from the SR via the IP3R or RyR. TRPC6 channel in cerebral arteries also contributes to elevated Ca2+ levels. Activation of ANO1 triggers Cl− outflow and depolarizes the membrane potential, leading to LTCC opening, which causes higher levels of intracellular Ca2+. The Ca2+- calmodulin complex induces phosphorylation of MLCK to constrict smooth muscle. Additionally, ANO1 is involved in Ang II-induced cerebral constriction via activating RhoA/ROCK signaling pathway, elevating the phosphorylation of MYPT1 and subsequent inhibition of MLCP. ANO1, Anoctamin-1; DAG, diacylglycerol; GqPCR, Gq protein coupled receptors; IP3, inositol trisphosphate; IP3R, IP3 receptors; LTCC, L-type calcium channels; MLCK, myosin light chain protein kinase; MLCP, myosin light chain phosphorylase; MYPT1:myosin phosphatase targeting subunit 1; NKCC1, Na+-K+-2Cl− cotransporter 1; PIP2, phosphatidylinositol 4,5-bisphosphate; RyR, ryanodine receptors; SR, sarcoplasmic reticulum; VSMC, vascular smooth muscle cells.
Figure 3
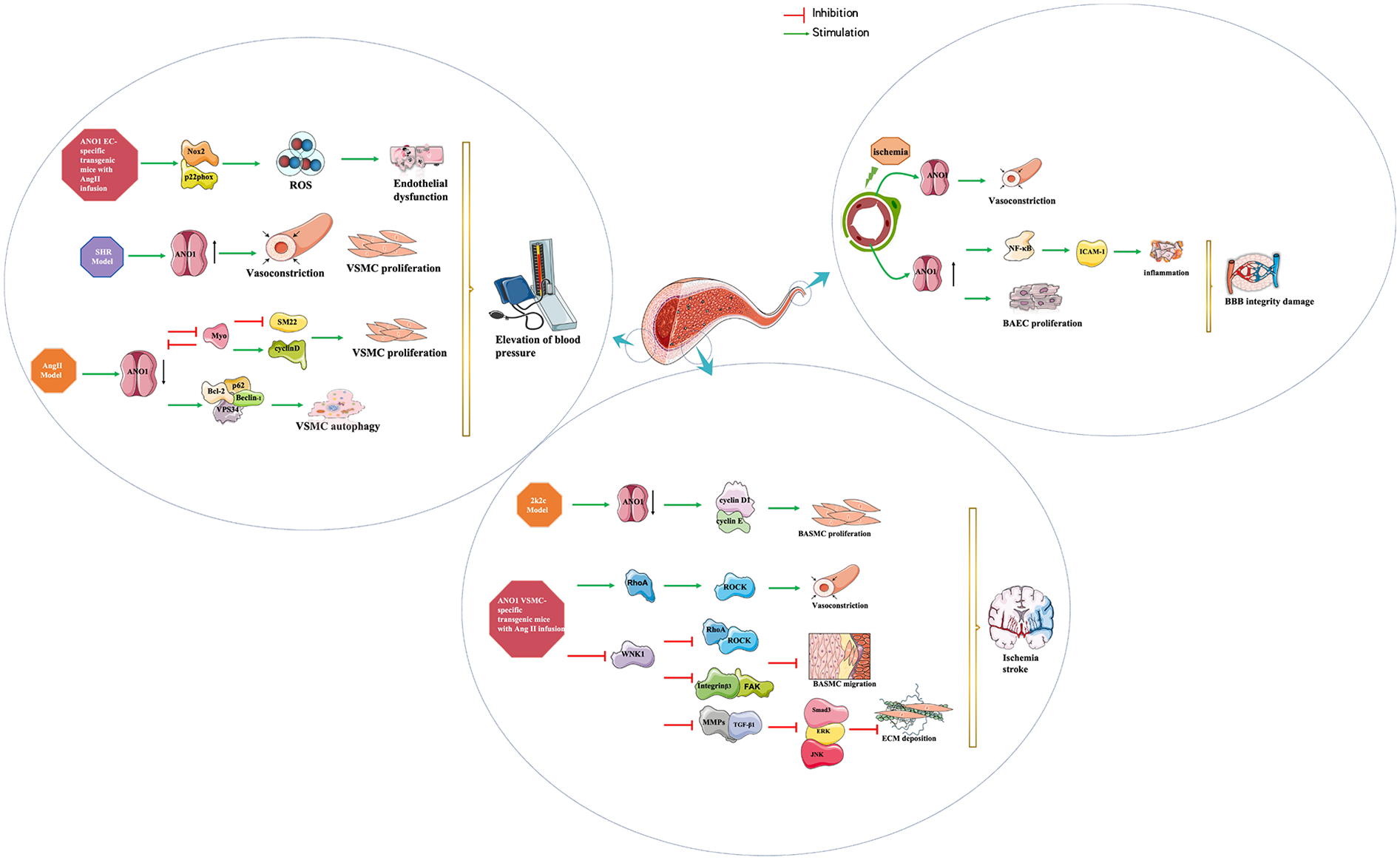
Diagram summarizing the findings on the role of ANO1 in systemic hypertension and stroke. ANO1 plays an important role in the pathophysiology of systemic hypertension, not only in vascular contraction but also in vascular remodelling, including changes in endothelial and VSMC function (left panel). ANO1 also has a pathological effect in cerebrovascular remodelling during the development of hypertension and blood brain barrier injury after stroke. (middle panel and right panel) ANO1, Anoctamin-1;BBB:blood brain barrier; BCEC, brain capillary endothelial cell; BASMC, basilar artery smooth muscle cell; ROS, reactive oxygen species; SHR, spontaneously hypertensive rat; VSMC, vascular smooth muscle cell.
Figure 4
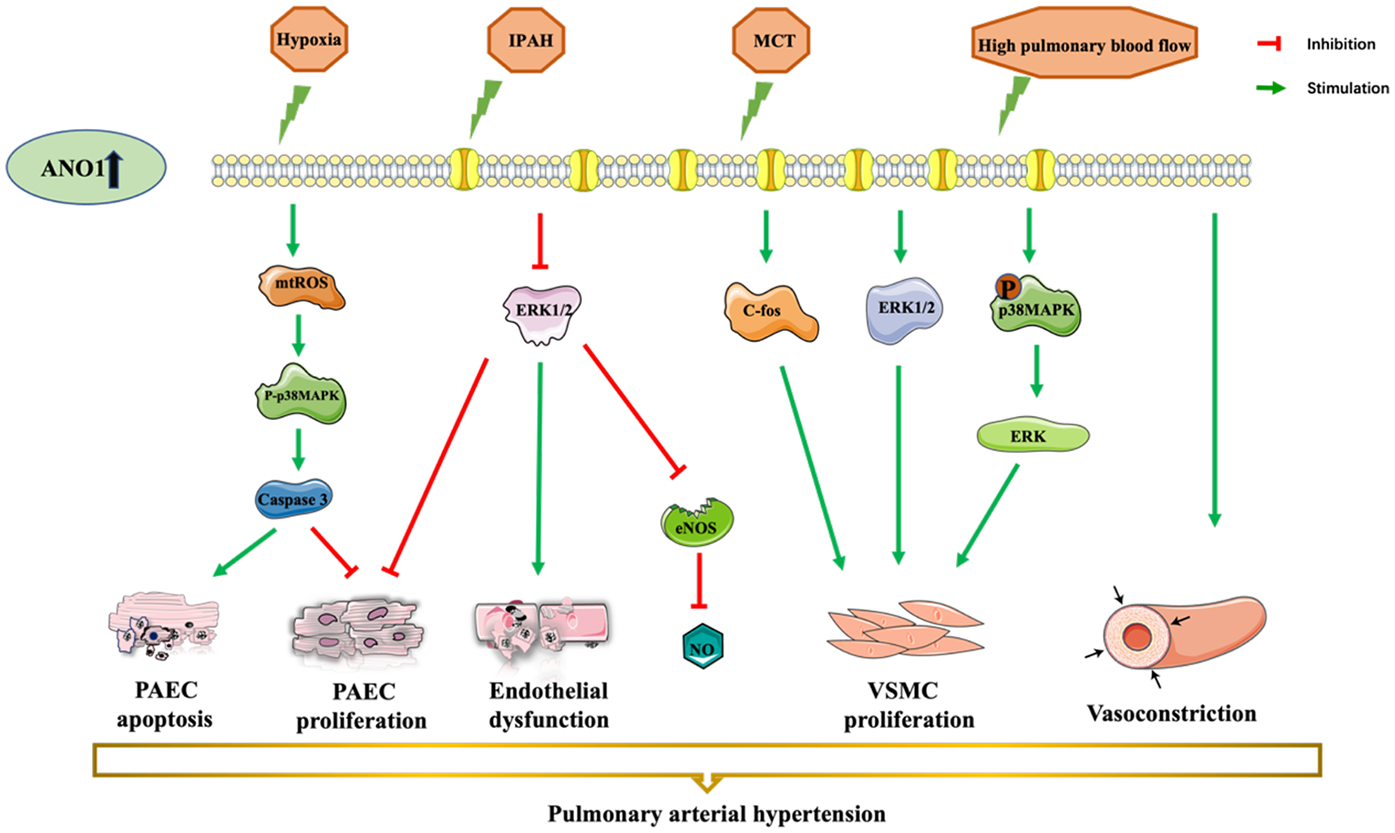
ANO1 dysfunction-induced vascular disorders and pulmonary arterial hypertension. ANO1 is upregulated in different models, in MCT- or chronic hypoxia-induced or high-flow-induced rat PAH model and human idiopathic PAH. There is a consensus that overexpression of ANO1 leads to endothelial dysfunction, PAEC apoptosis, PAEC proliferation, VSMC proliferation, and VSMC constriction. ANO1, Anoctamin-1; MCT, monocrotaline; NO, nitric oxide; PAEC, pulmonary artery endothelial cell; PASMCs, pulmonary artery smooth muscle cells; VSMC, vascular smooth cell.
Table 2
| Name (alias) | Structure | IC50 (μmol/L) | Mechanism | Pharmacological effect | Special effect |
|---|---|---|---|---|---|
| T16Ainh-A01 |
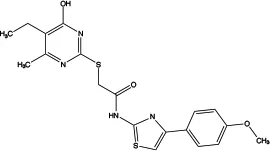
|
1.8 | Inhibits Cl− influx into nuclear and inhibits the activation of ERK1/2 | Relaxes VSMC constriction (16, 50, 65); reduces BP (50); improves vascular remodeling and right ventricular hypertrophy (60); anti-cardiac fibrosis (22) | Concentration- and dose-dependent |
| CaCCinh-A01 |
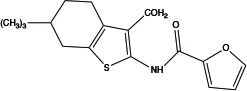
|
2.1 | Inhibits ANO1 expression and causes a global decrease of Cl− content; binding with ANO1 causes the collapse of the ion conduction pore (84);indirectly alters intracellular Ca2+ signalling (92) | Relaxes VMC constriction (83); anti-cardiac fibrosis (22) | Concentration- and dose-dependent; good pharmacokinetic characteristics; poor selectivity among ANO1 family proteins |
| MONNA |
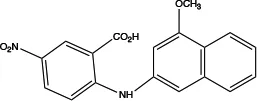
|
0.08 | Inhibits Ca2+-activated ANO1 chloride currents; indirectly alters intracellular Ca2+ signalling (92) | Relaxes VSMC constriction (16, 83) | Concentration-and dose-dependent; in the presence and absence of Cl− |
| TMinh-23 |
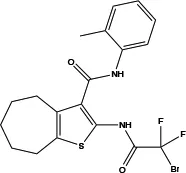
|
0.03 | Inhibits Ca2+-activated ANO1 chloride currents ex vivo and in vivo | Reduces VSMC vasoconstriction and lowers BP (37) | Good selectivity and reversibility |
| Ani9 |
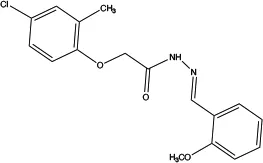
|
0.077 | Inhibits ANO1 Cl− current (85) | Attenuates VSMC contraction (16) | Dose-dependent; highly potency and selectivity; does not affect the intracellular Ca2+ signaling and CFTR Cl− channel activity and ANO2 |
| Tannic acid |
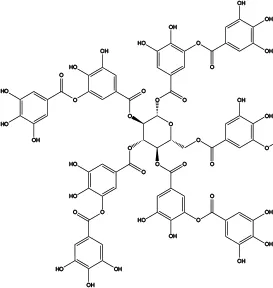
|
6 | Inhibits Ca2+-activated ANO1 chloride currents | Relaxes aortic smooth muscle contraction (79) | Concentration- and dose-dependent; inhibits CaCCs in various cells but does not affect CFTR Cl− channels and ENaC Na+ channels |
| Benzbromarone |
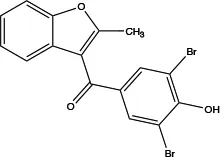
|
not clear | Inhibits Ca2+-activated ANO1 chloride currents ex vivo and in vivo | Vasodilates; reduces right ventricular pressure; attenuates remodeling of established PAH (36) | Dose-dependent; non-specific inhibitor |
| Nimodipine |
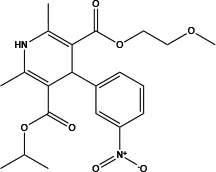
|
5 | Decreases intracellular Ca2+ concentration and partly directly inhibits ANO1 | Relaxes aortic smooth muscle (82) | Concentration-dependent |
| Niclosamide |
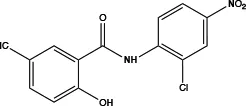
|
not clear | At positive membrane potentials and under conditions of elevated intracellular Ca2+ >2 μM inhibits ANO1 | Relaxes VSMC constriction (86) | Concentration- and dose-dependent, strongly dependent on the membrane potential and the intracellular calcium concentration,inhibits CaV1.2 |
Profiling inhibitors of ANO1 and their potential pharmacological effects.
BP, blood pressure; IC50, the 50% inhibitory concentration; PAH, pulmonary arterial hypertension; VSMC, vascular smooth muscle cell.
2 The role of ANO1 in heart diseases
ANO1 is endogenously present in cardiac vascular endothelial cells (15), coronary VSMCs (16), ventricular myocytes (17, 18), atrial fibroblasts (19), and cardiac fibroblasts (20). Overexpression of ANO1 enhances depolarisation, increases the potential for Ca2+ channel opening, vasoconstriction, and ultimately decreases coronary perfusion, which can lead to ischaemic heart disease (16). Evidence suggests that ANO1 dysfunction is an emerging pathogenesis of MIRI, cardiac arrhythmias, and cardiac fibrosis (17, 20–22).
MIRI refers to increased damage to the ischaemic myocardium after restoration of blood flow (23, 24). The pathogenesis of MIRI is currently attributed to oxidative stress, Ca2+ overload, mitochondrial damage, and neutrophil activation (25). Initially, it was investigated that ANO1 was upregulated and miR-144-3p was downregulated, and then the NLRP3 inflammasome was activated during MIRI (26). Further study found that the upregulation of miR-144-3p inversely regulated ANO1 via binding to the 3′-untranslated region of ANO1 and suppressed the activation of NLRP3 inflammasome, which in turn attenuated NLRP3-mediated myocardial apoptosis and MIRI in vivo and in vitro (26). Therefore, the miR-144-3p/ANO1 axis may be a promising therapeutic target in myocardial ischaemia.
In addition, arrhythmia is a common manifestation with abnormal periodicity and regularity of myocardial electromechanical activity, particularly disturbed by various ion channels in the myocardial cell membranes (27). The increased expression and activity of ANO1 in mouse ventricular myocytes under conditions of myocardial ischaemia accelerate phase 1 repolarisation of action potentials and influence cardiac action potential duration, which partially leads to arrhythmias (17). Furthermore, the activation of ANO1 in cardiac fibroblasts affects the electrophysiological activity of cardiomyocytes via gap junctions, ultimately leading to ectopic pacing, which may be an underlying mechanism related to arrhythmia.
Cardiac fibrosis is a complex pathological process of increased myocardial stiffness and decreased systolic ejection (28). Cardiac fibroblasts (CFs), the most abundant cardiac cells, together with cardiomyocytes and vascular smooth muscle cells (VSMCs), make up the myocardium (29). In the progression and development of cardiac fibrosis, cardiac fibroblasts proliferate and differentiate into myofibroblasts under the influence of pathological factors that have enhanced their ability to secrete matrix metalloproteinases, proliferate, and migrate (30). Studies have shown that ANO1 expression is significantly increased during myocardial fibrosis. Gao et al. found that ANO1 expression in rat cardiac tissues increased significantly under hypoxic conditions after myocardial infarction (MI), especially in the border zone of the infarct and in cardiac fibroblasts (CFs) (20). Tian et al. further demonstrated that ANO1 expression peaked at weeks 1–2 after MI and then gradually declined, with these dynamic changes being closely related to the fibrosis process (31). Furthermore, in vitro experiments revealed that ANO1 expression increased in CFs under hypoxic conditions or TGF-β stimulation, indicating a potential role for ANO1 in the activation and differentiation of CFs (20, 32). However, the role of ANO1 in myocardial fibrosis is controversial. Tian et al. reported that ANO1 exacerbates cardiac fibrosis via the AT1R-mediated MAPK signalling pathway (31). ANO1 also affects the function of CFs by regulating intracellular Cl− concentration and inhibition of ANO1 by both T16Ainh-A01 and CaCCinh-A01 could reduce the proliferation, migration, and collagen secretion of CFs in a dose-dependent manner by reducing Cl− (22). Lately Tian et al. found that kockdown of ANO1 can significantly reduce the migration and adhesion ability of CFs, which may be due to the downregulation of integrin expression and reduced activation of focal adhesion kinase (32). But this study only used in vitro models of CFs and did not include in vivo experiments (e.g., animal models of myocardial infarction) to validate the pathological role of ANO1. Nevertheless, Gao et al. and Kong et al. revealed that the overexpression of ANO1 impeded the proliferation and differentiation of CFs, possibly via the inhibition of the TGF-β/smad 3 signalling pathway, which ultimately alleviated cardiac fibrosis in vivo MI modelling (20, 21). The focus of both papers is on myocardial tissue as a whole (comprising cardiomyocytes and fibroblasts, among others) through the construction of myocardial fibrosis model.
All in all, such disparities may be related to the diversity of experimental models, intervention times or microenvironments. We suppose that ANO1 may regulate fibrosis via distinct mechanisms at various pathological stages: initially, it mitigates fibrosis by curbing inflammatory responses; subsequently, it exacerbates fibrosis by stimulating the activation of CFs. This implies that precise modulation of ANO1 as a therapeutic target is imperative in specific pathological scenarios. Further studies are needed to investigate the molecular mechanisms of ANO1 in different cell types, as well as its potential clinical applications.
3 The role of ANO1 in hypertension
Hypertension is characterised by elevated blood pressure (BP) and vascular remodelling that can further damage target organs (heart, kidneys, brain, eyes, vasculature, etc.) and is influenced by environmental and genetic factors. Genetic diversity in ANO1 has been statistically associated with hypertension in humans, especially in males (33), although this has not been demonstrated by genome-wide sequencing studies (34). Accumulating evidence has proven that ANO1 plays an important role in the pathophysiology of hypertension, not only in vascular contraction but also in vascular remodelling, including changes in endothelial and VSMC function (35–38).
The mechanisms underlying the involvement of ANO1 in the initiation and progression of different types of hypertension are intricate and controversial. Substantial Cl− accumulated in VSMCs due to the increased activity of Na+-K+-2Cl− cotransporter 1(NKCC1) in hypertension and elevated the driving force for Cl− efflux (as shown in Figure 2) (39). The cooperation between ANO1 and NKCC1 increased Cl− efflux, potentiated membrane depolarisation and enhanced the open probability of Ca2+ channels, ultimately leading to an increase in BP (40). As for the sources of Ca2+, there are no clear and precise conclusions. Ca2+ from outside the SMCs entered the cell via voltage-gated L-type calcium channels or transient receptor potential canonical 6 channel (TRCP6), and some Ca2+ was released from the SR via IP3 and ryanodine receptors (41). In addition, ANO1 partially regulated arterial contractility by positively modulating the expression of the CACNA1C subunit of the L-type Ca2+ channels in tail arteries from transgenic mice (42). Moreover, TRPC6 and ANO1 channels were spatially localised in close proximity on the plasma membrane of cerebral artery myocytes, and TRPC6 channel activated a local intracellular Ca2+ signal that stimulated ANO1, thereby enhancing vasoconstriction and decreasing blood flow (43). Interestingly, ANO1 knockdown reduced the aorta contraction and potentially induced hypotension but increased tail and saphenous artery contraction by switching smooth cells to a pro-contractile phenotype, indicating that ANO1 is multifunctional for different parts of the vascular walls (44). Moverover, excess expression of ANO1 impaired endothelial function, possibly via the generation of reactive oxygen species (38, 45) and the reduction of nitric oxide (46), triggering vascular remodelling and ensued numerous cardiovascular diseases (different types of hypertension, stroke, etc.). Next, we elaborate on the role of ANO1 in different types of hypertension, including systemic hypertension, pulmonary arterial hypertension, and portal hypertension.
3.1 The role of ANO1 in systemic hypertension
The regulation of systemic arterial BP can be divided into long-term and short-term mechanisms. Long-term control is predominantly mediated by the kidneys through the regulation of sodium excretion and extracellular fluid volume (47). Short-term regulation is mediated by various cardiovascular reflexes that alter myocardial contractiliry and peripheral vascular resistance (48), and local autoregulatory mechanisms, such as the pressure-induced myogenic constriction of resistance vessels, which ensures stable blood flow to critical organs like the kidneys. Yip et al. discovered that ANO1 is expressed in afferent arterioles and is involved in pressure-induced myogenic contraction, indicating a role for ANO1 in short-term renal autoregulation (49). It is important to note, however, that the myogenic response primarily serves to buffer acute pressure fluctuations and has a limited role in setting long-term BP. This is supported by the finding that ANO1 knockout mice remain susceptible to salt-induced hypertension (35), underscoring that ANO1 is not part of the primary, kidney-dependent long-term control system. Beyond the kidney, ANO1 also influences blood pressure through systemic vascular tone. It is expressed in various vessels, including the resistance arteries, aorta, and carotid and mesenteric arteries of SHRs (50). Consistent with this distribution, conditional knockout of ANO1 or applying ANO1 inhibitor TMinh-23 reduced the responsiveness to vasoconstrictor stimuli, resulting in a reduction in systemic BP (35). The specificity of this mechanism is highlighted by the observation that TMinh-23 preferentially suppressed constriction in mesenteric resistance arteries compared to the aorta, conclusively demonstrating that the peripheral vasculature is the primary site of action for this blood pressure-lowering effect (37). It's interesting to note that the function and expression of ANO1 in the aorta has been altered in different animal models (50–52). ANO1 expression and activity are upregulated in aortic SMCs from spontaneously hypertensive rat (SHR) (37, 50), but downregulated in human aortic SMCs and mouse aortic SMCs from the AngII-induced hypertensive models (51, 52). These distinctions highlight the intricate pathophysiology of hypertension. In the SHR model, hypertension develops as a genetically determined primary condition that progresses gradually through vascular remodeling. Here, increased ANO1 expression may represent a functional and structural adaptation of VSMCs to sustained hypertensive stimuli, establishing a positive feedback mechanism that perpetuates elevated blood pressure (37). By contrast, hypertension in the acute AngII infusion model is primarily driven by the potent vasoconstrictor AngII, and the observed downregulation of ANO1 may act as a negative feedback response to mitigate excessive vasoconstriction and calcium overload. Thus, ANO1 expression appears to be highly context-dependent, shaped by the predominant etiological factors and the stage of hypertension development. The schematic mechanism of ANO1 function in vascular remodelling is shown in Figure 3. AngII markedly upregulated ANO1 expression in VSMCs of SHR in a concentration- and time-dependent manner through the AT1R/PI3 K/Akt pathway, which made VSMCs more sensitive to other agonists and exacerbated peripheral resistance and stimulated the growth and proliferation of VSMCs (50). However, in an AngII-induced hypertension model, AngII promoted Kruppel-like factor 5 (KLF5) expression and KLF5-myocardin binding, disrupted the feedback loop between ANO1 and myocardin, and then antagonised ANO1 transcription in human aortic SMCs, subsequently inducing cellular proliferation and vascular remodelling (51). Similary, reduced ANO1 expression in mouse aortic SMCs of AngII-induced hypertensive mice activated cell autophagy and exacerbated vascular remodelling by promoting p62/Bcl-2/Beclin-1/VPS34 complex formation (52).
Endothelial dysfunction, including endothelial proliferation, apoptosis, migration, and angiogenesis, is involved in the pathogenic process of various vascular diseases, in which ANO1 plays a multifaceted role (38, 45, 46, 53–55). It was found that ANO1 induced human umbilical vein EC dysfunction by inhibiting Nox2 degradation and increasing Nox2-containing NADPH oxidase activity, resulting in increased reactive oxygen species generation during AngII-infused hypertension, whereas knockout endothelial-specific ANO1 reverse the hypertensive effect (38). Moreover, Zhang et al. also revealed that ANO1 positively induced the migration and angiogenesis of human umbilical ECs under normal or under stress conditions in vitro (53). In contrast, ANO1 inhibition by cholesterol and cholesterol-induced DNA methyltransferase 1-mediated methylation facilitated human aortic EC proliferation, migration, and angiogenesis, indicating that ANO1 was negatively correlated with human aortic EC activity (55). Thus, activation of ANO1 limited endothelial angiogenesis during high cholesterol, which was beneficial in diseases with high cholesterol pathological states such as atherosclerosis (55).
3.2 The role of ANO1 in pulmonary arterial hypertension
Pulmonary arterial hypertension (PAH) is a progressive multifactorial disease with vasoconstriction and hyperproliferation of pulmonary artery endothelial cells (PAECs) and pulmonary artery smooth muscle cells (PASMCs), ultimately leading to right heart failure (56). ANO1 was upregulated in the PASMCs from monocrotaline- or chronic hypoxia-induced or high-flow-induced rat PAH model and in patients with idiopathic PAH (36, 57–59), contributing to vascular constriction and increased blood pressure (as shown in Figure 4) (36, 57).
ANO1 was positively correlated with PASMC proliferation in PAH (36, 59, 60). Increased expression and activity of ANO1 stimulated human PASMC proliferation during idiopathic PAH by increasing c-fos phosphorylation, which can be suppressed by benzbromarone and blocking or silencing of ANO1 (36). Another research found that ANO1 facilitated PASMC proliferation, pulmonary arterioles remodelling and right ventricular hypertrophy, possibly via activating ERK1/2 in an MCT-treated hypertensive rat model (60). Moreover, further investigation showed that ANO1-induced proliferation of PASMCs may be mediated by the p38MAPK/ERK signalling pathway in high-flow-induced PAH rats (59).
Previous studies have shown that ANO1 was distributed in the mitochondria and plasma membranes in human PAECs (45, 46). The augmented expression and activity of ANO1 promoted mitochondria-dependent PAEC apoptosis via a mtROS-p38-caspase-3 pathway, suppressing PAEC proliferation in idiopathic PAH patients (45). However, Skofic Maurer et al. studied that enhanced ANO1 expression and activity contributed to endothelial dysfunction, decreased PAECs proliferation but did not affecte apoptosis, and reducted nitric oxide likely through activating the ERK1/2 pathway (46). The difference between the two mechanisms may be due to the cellar heterogeneity and experimental conditions. Further studies investigating the role of ANO1 in endothelial cells are warranted.
3.3 The role of ANO1 in portal hypertension
Portal hypertension is a group of syndromes caused by persistent elevation of portal pressure, the vast majority of which are caused by cirrhosis of the liver. ANO1 contributed to spontaneous contractions, a significant stimulus of blood flow from the mesenteric vascular bed to the liver (61). Recently, several investigators found that ANO1 was downregulated in portal smooth muscle cells (PVSMC) from bile duct ligation mice with cirrhotic portal hypertension via increased AngII, followed by a reduction in spontaneous contractions (61). ANO1 is also involved in PVSMC proliferation and hyperexpression of ANO1 facilitates PVSMC proliferation in vitro but inhibits PVSMC proliferation from portal hypertensive rats in vivo, suggesting that ANO1 may also be a negative modulator of PVSMC proliferation under the influence of multiple in vivo factors (62). More research is necessary to establish whether ANO1 is a viable candidate for the treatment of portal hypertension.
4 The role of ANO1 in stroke
Stroke, a cerebrovascular accident, comprises a group of disorders resulting from a sudden blocked or ruptured blood vessel in the brain, causing damage to brain tissue. It is widely recognised that vascular remodelling of cerebral arterioles caused by hypertension is a major factor contributing to the increased incidence of ischemic stroke due to the narrowing atrial and reduced blood flow (63). ANO1 participates in the regulation of myogenic tone via non-selective cation channel-generated local Ca2+ signalling in cerebral arteries when membrane distension, including cell swelling and pressure-induced membrane stretch (64). Activated ANO1 enhanced AngII-induced phosphorylation of MYPT1 and MLC through the RhoA/ROCK signalling pathway, which triggered basilar vasoconstriction in male Sprague-Dawley rats (65). To improve our understanding of new approaches to control stroke development and ameliorate blood-brain barrier (BBB) damage, it's necessary to elucidate the pathological role of ANO1 in cerebrovascular remodelling during the development of hypertension and BBB injury after stroke (as shown in Figure 3).
Researchers found that ANO1 is expressed in brain capillary endothelial cells (BCECs) and is upregulated after ischaemic stroke caused by middle cerebral artery occlusion or under hypoxia (54, 66). A recent study demonstrated that the lncRNA ENST00000530525 may affect ANO1 expression by recruiting transcription factors or enhancers after ischaemic stroke (67). Inhibiting or silencing ANO1 alimerate ischemic-induced BBB injury via downregulating intracellular adhesion molecule-1 and neutrophil accumulation in a nuclear factor-kappaB-dependent manner, indicating that downregulation of ANO1 protects BBB disruption after ischemia stroke (66). In addition, the function of ANO1 regulates the proliferation and migration of BCECs, thus ensuring the maintenance of the barrier function of the BBB (68). This team further studied that the upregulated expression of ANO1 stimulated the proliferation and transendothelial permeability of BCECs, which may lead to BBB dysfunction under hypoxic stress (54). The administration of the ANO1 inhibitor T16Ainh-A01 or siRNA attenuated these effects. Contractile pericytes, a vital component of the BBB, serve as principal regulators of capillary diameter and cerebral blood flow (69). Following ischemic events, these pericytes augment capillary constriction and diminish cerebral blood flow through ANO1-mediated Ca2+-activated chloride efflux. Targeting ANO1 with Ani9 or deletion promotes pericyte relaxation and slows pericyte loss, attenuating the microvascular blood flow and BBB function (70).
Cerebrovascular remodelling is accompanied by increased proliferation and migration of VSMCs, deposition of ECM, and inhibition of VSMC apoptosis. Numerous studies concluded that ANO1 was negatively associated with cerebrovascular remodelling during hypertension. ANO1 expression and activity are decreased in basilar artery smooth muscle cells (BASMCs) from 2-kidney, 2-clip(2k2c) renohypertensive rats (71) and AngII-infused hypertensive mice (72), which are negatively regulated by the enhanced CaMKIIγ activity via phosphorylation of serine 727 within ANO1 in basilar arteries. One study revealed that AngII suppressed the expression of ANO1 in BASMCs from 2k2c renohypertensive rats and subsequently accelerated vascular remodelling, with the underlying mechanism being that AngII increased endophilin A2 and subsequently enhanced ubiquitin-mediated degradation of the ANO1 protein (73). The reduction of ANO1 promotes BASMC proliferation and cerebral vascular remodelling by promoting the G1/S transition via increasing cyclin D1 and cyclin E expression without affecting the cell cycle negative regulators p21 and p27 (71, 72).
In addition to inhibiting abnormal BASMC proliferation, VSMC-specific transgenic expression of ANO1 also exerts a protective effect on BASMC apoptosis, migration, and extracellular matrix accumulation (74–76). Zeng et al. investigated that ANO1 contributed to hydrogen peroxide-induced mitochondria-dependent BASMC apoptosis from rats and mice via interaction with CypD, which could be alleviated by silencing ANO1 and applying T16Ainh-A01 (74). In addition, ANO1 suppressed WNK1, which in turn inhibited the RhoA/ROCK2/MLCP/MLC20 and integrinb3/FAK pathways, thereby antagonising AngII-induced BASMC migration and protecting the basilar artery from remodelling during hypertension (75). Further investigation showed that the ANO1 decreased the activation of WNK1 and subsequently reduced the levels of MMPs, which was followed by attenuation of the activity of the typical TGF-β1/Smad3, non-typical TGF-β1/ERK and JNK pathways, ultimately protecting against extracellular matrix deposition and antagonising AngII-induced cerebrovascular remodelling in mice in vivo and in vitro (76). It may be a potential target for treating ischemic stroke and ischemia-induced BBB damage, but this effect should be tested in the whole model, not just the cerebrovascular one. It would be interesting to see further studies aimed at validating ANO1 as a therapeutic target for brain stroke.
5 ANO1-targeted pharmacological modulation and therapy
Following the diverse functions of ANO1 mentioned above, applying ANO1 modulators should be cautious and specific in different cardiocerebral vascular diseases. A growing body of literature has investigated the application of ANO1 inhibitors in the cardiocerebral vascular system, especially anti-hypertension or anti-cardiac diseases (77). The inhibitors are divided into two categories: natural chemicals and synthetic agents.
Several compounds from natural daily foods have been shown to protect against cardiovascular disease, including vegetables, fruits, cereals, green tea, and red wine (78, 79). Plant lignan compounds, such as kobusin and eudesmin, show an antihypertensive effect in patients in clinical trials (80), which partially explains the therapeutic effect of ANO1 active inhibitors in vitro and in vivo (78). In addition, green tea and red wine had cardioprotective effects, underlying that tannic acid, related gallotannins, and polyphenols are potent and effective ANO1 inhibitors (79). However, the 50% inhibitory concentration (IC50) of these natural inhibitors was higher than that of chemical inhibitors, possibly due to the lack of purity of the ANO1 inhibitors.
The discovery of new therapeutic avenues from drugs already on the market is a viable strategy for obtaining safe and effective therapeutic drugs (81). Several clinical agents have been discovered that exhibited inhibitory effects on ANO1, particularly benzbromarone and nimodipine (as shown in Table 2) (36, 82). Benzbromarone, a gout drug, lowered right ventricular pressure, reduced elevated PAH, and attenuated vascular remodelling in established PAH in a dose-dependent manner. The application of benzbromarone in treating hypertension, especially PAH, is considered a priority in the clinic (36). Nimodipine, an LTCC blocker, inhibited ANO1 indirectly by decreasing intracellular Ca2+ concentration or directly in a dose-dependent manner, and interestingly, the effect on relaxation of aortic VSMC contraction was reversible since the inhibitory effect disappeared completely 8 min after removal of nimodipine (82).
Previous studies have shown that there is a new class of ANO1 antihypertensive blockers, such as T16Ainh-A01 (50), CaCCinh-A01 (83, 84), MONNA (83), TMinh-23 (37), Ani9 (85), Niclosamide (86) (as shown in Table 2). T16Ainh-A01 could reduce VSMC constriction and local BP elevation in SHRs (50) and alleviate Ang II-induced rat basilar arteries constriction (65). Additionally, T16Ainh-A01 could reverse PAH and ameliorate small pulmonary artery remodelling and right ventricular hypertrophy, possibly inhibiting ERK phosphorylation (60). It was demonstrated that TMinh-23 was one of the most potent inhibitors of ANO1 according to the minimum IC50, which was approximately close to 30 nM (87). It reduced vasoconstriction in vitro and lowered systolic and diastolic BP in SHR in vivo without a rebound effect after withdrawal but produced a transient increase in heart rate (37). Furthermore, MONNA, T16Ainh-A01, and Ani9 also impaired coronary artery constriction and increased coronary flow and may be potential development candidates for drug therapy for ischaemic heart disease and MI (16). Among them, T16Ainh-A01 and CaCCinh-A01 could suppress CFs proliferation, migration, and collagen secretion in a dose-dependent manner and may serve as novel anti-cardiac fibrotic therapeutics in the future (22).
Recent advances in potent and selective chemical probes and drug candidates targeting specific ANO1 will enable study designs that carefully incorporate targeted genetic approaches, including human induced pluripotent stem cells, condition/site-specific knockout in mice, or CRISPR gene editing in native cells (88). A new strategy may be to treat ANO1-related diseases by targeting small biological molecules. For example, the miR-144-3p/ANO1 axis may be a promising therapeutic target in myocardial ischaemia (26, 89). In addition, endophilin A2 and ET-1 have provided novel mechanistic insights and therapeutic targets for ANO1-induced vascular remodelling and BP elevation (73, 90). Moreover, alternative splicing of the primary ANO1 transcript could generate multiple variants that were altered in voltage-dependent and Ca2+ sensitivity and the rate of channel activation and deactivation, suggesting a putative therapeutic regimen (15).
Nevertheless, the pharmacological regulation and potential therapeutic strategies related to ANO1 remain limited. Firstly, ANO1 is present in different tissues and organs, and the structure of ANO1 is similar to other ion channels (ANO2, GFTR, ENaC, BEST1, etc.) (91), suggesting that the drugs acting on ANO1 may have off-target or non-specific effects. Secondly, although several ANO1 inhibitors have been discovered by high-throughput chemical library screening, the specific binding sites and interaction mechanisms are not addressed. The search for new potent and highly selective ANO1 inhibitors is still in its infancy, and most ANO1 inhibitors have only been tested in cell or animal experiments. Future studies are required to obtain insight into the precise mechanism of these molecules acting on ANO1 and to explore more organ- and tissue-specific compounds, which especially need to be validated in clinical trials.
6 Challenges for clinical use
There is still a long way to go to elucidate the mechanisms and clinically turn ANO1 into therapeutic targets for cardiocerebral vascular dieases. Firstly, there is a discrepancy in the expression of ANO1 in different models with different humoral factors. For all laboratory forms and models of hypertension, a complete investigation of the effects of blocking ANO1 to reduce BP is needed, as this may have unfavourable outcomes in cerebral and portal microcirculation. Secondly, the role of ANO1 in several pathophysiological cardiocerebral vascular progresses remains complex and contradictory. Especially in cardiac fibrosis under different experimental conditions, overexpression of ANO1 inhibits cardiac fibrosis by inhibiting the TGF-1 pathway, while another team verified that ANO1 promotes cardiac fibrosis via the AT1R-mediated MAPK signalling pathway. How ANO1 interacts with the MAPK pathway and the exact upstream and downstream relationships remain are unclear. Therefore, there is an unmet need for further cell and animal experiments to determine the precise signalling pathways involved in ANO1-associated cardiac fibrosis. Finally, there is an urgent need to systematically elaborate the crystal structure of ANO1 and its modulators, which will require much-needed effort and technological innovation to understand the structure-function relationship, physiological action, and pharmacological modulation of ANO1. The novel ANO1-targeted therapeutic approaches will be widely applied to cardiocerebral vascular disease in the future.
7 Conclusions
The ANO protein family is emerging as a fascinating class of ion channels, particularly because of their Ca2+ dependence and voltage sensitivity. The widespread distribution and involvement of ANO1 in multiple interferences in cardiocerebral vascular systems, in combination with the improvement of significant adverse effects of ANO1 knockout or ANO1 channel inhibition, render ANO1 an attractive therapeutic target. Herein, we present an overview of ANO1 and ANO1-related cardiac and cerebrovascular disease, emphasise that the disturbance of ANO1 is involved in the pathogenesis of the cardiac and cerebrovascular disease, and summarise existing and potential treatments targeting ANO1 for cardiac and cerebrovascular disease. Hopefully, this review will lay the groundwork for a deeper understanding of the cardiac and cerebrovascular mechanisms and treatments associated with ANO1 in the future.
Statements
Author contributions
J-qZ: Writing – review & editing, Writing – original draft. Z-jW: Writing – review & editing. X-hH: Writing – review & editing, Investigation, Supervision. Z-kQ: Conceptualization, Writing – review & editing, Supervision. Q-yY: Conceptualization, Writing – review & editing, Supervision. YQ: Validation, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the Natural Science Foundation of Shandong Province (ZR2023QH190), Shandong Province Natural Science Foundation (ZR2019MH110), and Bureau Science and Technology of Qingdao (19-6-1-19-nsh).
Acknowledgments
We thank Yuren 930 for allowing the authors to collaborate on this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ANO1, anoctamin-1; BASMC, basilar artery smooth muscle cell; BBB, blood brain barrier; BCEC, brain capillary endothelial cell; BP, blood pressure; CF, cardiac fibroblasts; EC, endothelial cell; MIRI, myocardial ischemia/reperfusion injury; PAEC, pulmonary artery endothelial cell; PAH, pulmonary arterial hypertension; PASMC, pulmonary artery smooth muscle cell; SHR, spontaneously hypertensive rat; VSMC, vascular smooth muscle cell.
References
1.
Pedemonte N Galietta LJ . Structure and function of TMEM16 proteins (anoctamins). Physiol Rev. (2014) 94(2):419–59. 10.1152/physrev.00039.2011
2.
Arreola J Pérez-Cornejo P Segura-Covarrubias G Corral-Fernández N León-Aparicio D Guzmán-Hernández ML . Function and regulation of the calcium-activated chloride channel anoctamin 1 (TMEM16A). Handb Exp Pharmacol. (2022) 283:101–51. 10.1007/164_2022_592
3.
Yang YD Cho H Koo JY Tak MH Cho Y Shim WS et al TMEM16A Confers receptor-activated calcium-dependent chloride conductance. Nature. (2008) 455(7217):1210–5. 10.1038/nature07313
4.
Schroeder BC Cheng T Jan YN Jan LY . Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell. (2008) 134(6):1019–29. 10.1016/j.cell.2008.09.003
5.
Caputo A Caci E Ferrera L Pedemonte N Barsanti C Sondo E et al TMEM16A, A membrane protein associated with calcium-dependent chloride channel activity. Science. (2008) 322(5901):590–4. 10.1126/science.1163518
6.
Brunner JD Lim NK Schenck S Duerst A Dutzler R . X-ray structure of a calcium-activated TMEM16 lipid scramblase. Nature. (2014) 516(7530):207–12. 10.1038/nature13984
7.
Dang S Feng S Tien J Peters CJ Bulkley D Lolicato M et al Cryo-EM structures of the TMEM16A calcium-activated chloride channel. Nature. (2017) 552(7685):426–9. 10.1038/nature25024
8.
Paulino C Kalienkova V Lam AKM Neldner Y Dutzler R . Activation mechanism of the calcium-activated chloride channel TMEM16A revealed by cryo-EM. Nature. (2017) 552(7685):421–5. 10.1038/nature24652
9.
Shi S Pang C Ren S Sun F Ma B Guo S et al Molecular dynamics simulation of TMEM16A channel: linking structure with gating. Biochim Biophys Acta Biomembr. (2022) 1864(1):183777. 10.1016/j.bbamem.2021.183777
10.
Schreiber R Ousingsawat J Kunzelmann K . The anoctamins: structure and function. Cell Calcium. (2024) 120:102885. 10.1016/j.ceca.2024.102885
11.
Xiao Q Yu K Perez-Cornejo P Cui Y Arreola J Hartzell HC . Voltage- and calcium-dependent gating of TMEM16A/Ano1 chloride channels are physically coupled by the first intracellular loop. Proc Natl Acad Sci U S A. (2011) 108(21):8891–6. 10.1073/pnas.1102147108
12.
Contreras-Vite JA Cruz-Rangel S De Jesús-Pérez JJ Figueroa IAA Rodríguez-Menchaca AA Pérez-Cornejo P et al Revealing the activation pathway for TMEM16A chloride channels from macroscopic currents and kinetic models. Pflugers Arch. (2016) 468(7):1241–57. 10.1007/s00424-016-1830-9
13.
Cabrita I Benedetto R Fonseca A Wanitchakool P Sirianant L Skryabin BV et al Differential effects of anoctamins on intracellular calcium signals. FASEB J. (2017) 31(5):2123–34. 10.1096/fj.201600797RR
14.
Tang ZZ Sharma S Zheng S Chawla G Nikolic J Black DL . Regulation of the mutually exclusive exons 8a and 8 in the CaV1.2 calcium channel transcript by polypyrimidine tract-binding protein. J Biol Chem. (2011) 286(12):10007–16. 10.1074/jbc.M110.208116
15.
Wu MM Lou J Song BL Gong YF Li YC Yu CJ et al Hypoxia augments the calcium-activated chloride current carried by anoctamin-1 in cardiac vascular endothelial cells of neonatal mice. Br J Pharmacol. (2014) 171(15):3680–92. 10.1111/bph.12730
16.
Askew Page HR Dalsgaard T Baldwin SN Jepps TA Povstyan O Olesen SP et al TMEM16A is implicated in the regulation of coronary flow and is altered in hypertension. Br J Pharmacol. (2019) 176(11):1635–48. 10.1111/bph.14598
17.
Ye Z Wu MM Wang CY Li YC Yu CJ Gong YF et al Characterization of cardiac anoctamin1 Ca2+-activated chloride channels and functional role in ischemia-induced arrhythmias. J Cell Physiol. (2015) 230(2):337–46. 10.1002/jcp.24709
18.
DeSimone CV McLeod CJ Gomez Pinilla PJ Beyder A Farrugia G Asirvatham SJ et al Telocytes express ANO-1-encoded chloride channels in canine ventricular myocardium. J Arrhythm. (2019) 35(3):515–21. 10.1002/joa3.12176
19.
El Chemaly A Norez C Magaud C Bescond J Chatelier A Fares N et al ANO1 contributes to angiotensin-II-activated Ca2+-dependent cl- current in human atrial fibroblasts. J Mol Cell Cardiol. (2014) 68:12–9. 10.1016/j.yjmcc.2013.12.027
20.
Gao Y Zhang YM Qian LJ Chu M Hong J Xu D . ANO1 inhibits cardiac fibrosis after myocardial infraction via TGF-β/smad3 pathway. Sci Rep. (2017) 7(1):2355. 10.1038/s41598-017-02585-4
21.
Kong JC Miao WQ Wang Y Zhou SF . ANO1 relieves pressure overload-induced myocardial fibrosis in mice by inhibiting TGF-β/Smad3 signaling pathway. Eur Rev Med Pharmacol Sci. (2020) 24(16):8493–501. 10.26355/eurrev_202008_22646
22.
Tian XQ Ma KT Wang XW Wang Y Guo ZK Si JQ . Effects of the calcium-activated chloride channel inhibitors T16Ainh-A01 and CaCCinh-A01 on cardiac fibroblast function. Cell Physiol Biochem. (2018) 49(2):706–16. 10.1159/000493036
23.
Wen ZJ Xin H Wang YC Liu HW Gao YY Zhang YF . Emerging roles of circRNAs in the pathological process of myocardial infarction. Mol Ther Nucleic Acids. (2021) 26:828–48. 10.1016/j.omtn.2021.10.002
24.
Wang Y Guo L Zhang Z Fu S Huang P Wang A et al A bibliometric analysis of myocardial ischemia/reperfusion injury from 2000 to 2023. Front Cardiovasc Med. (2023) 10:1180792. 10.3389/fcvm.2023.1180792
25.
Bellanti F . Hypoxia-inducible factor-1 in myocardial ischaemia/reperfusion injury. Acta Physiol (Oxf). (2017) 221(2):93–4. 10.1111/apha.12903
26.
Yang G Tang X Tan L Nong D Yang P Ning H . Upregulation of miR-144-3p protects myocardial function from ischemia-reperfusion injury through inhibition of TMEM16A ca(2+)-activated chloride channel. Hum Cell. (2021) 34(2):360–71. 10.1007/s13577-020-00482-z
27.
Brugada J Katritsis DG Arbelo E Arribas F Bax JJ Blomström-Lundqvist C et al 2019 ESC guidelines for the management of patients with supraventricular tachycardia. The task force for the management of patients with supraventricular tachycardia of the European Society of Cardiology (ESC). Eur Heart J. (2020) 41(5):655–720. 10.1093/eurheartj/ehz467
28.
Venero JV Doyle M Shah M Rathi VK Yamrozik JA Williams RB et al Mid wall fibrosis on CMR with late gadolinium enhancement may predict prognosis for LVAD and transplantation risk in patients with newly diagnosed dilated cardiomyopathy-preliminary observations from a high-volume transplant centre. ESC Heart Fail. (2015) 2(4):150–9. 10.1002/ehf2.12041
29.
Ma Y Iyer RP Jung M Czubryt MP Lindsey ML . Cardiac fibroblast activation post-myocardial infarction: current knowledge gaps. Trends Pharmacol Sci. (2017) 38(5):448–58. 10.1016/j.tips.2017.03.001
30.
Yuan W Zhang X Fan X . The role of the Piezo1 mechanosensitive channel in heart failure. Curr Issues Mol Biol. (2023) 45(7):5830–48. 10.3390/cimb45070369
31.
Tian X Sun C Wang X Ma K Chang Y Guo Z et al ANO1 regulates cardiac fibrosis via ATI-mediated MAPK pathway. Cell Calcium. (2020) 92:102306. 10.1016/j.ceca.2020.102306
32.
Tian X Zhang Y Gong H Bai M Sun C Jia Y et al Knockdown of ANO1 decreases TGF-β- and IL-6-induced adhesion and migration of cardiac fibroblasts by inhibiting the expression of integrin and focal adhesion kinase. Exp Cell Res. (2024) 443(1):114321. 10.1016/j.yexcr.2024.114321
33.
Jin H Jung D . Gender-specific association of the ANO1 genetic variations with hypertension. Biomedical Science Letters. (2015) 21(3):144–51. 10.15616/BSL.2015.21.3.144
34.
Li Q Cai H . ANO1 taking center stage: blood pressure regulation in SHRs. J Mol Cell Cardiol. (2015) 82:216–7. 10.1016/j.yjmcc.2015.03.013
35.
Heinze C Seniuk A Sokolov MV Huebner AK Klementowicz AE Szijártó IA et al Disruption of vascular Ca2+-activated chloride currents lowers blood pressure. J Clin Invest. (2014) 124(2):675–86. 10.1172/JCI70025
36.
Papp R Nagaraj C Zabini D Nagy BM Lengyel M Skofic Maurer D et al Targeting TMEM16A to reverse vasoconstriction and remodelling in idiopathic pulmonary arterial hypertension. Eur Respir J. (2019) 53(6):1800965. 10.1183/13993003.00965-2018
37.
Cil O Chen X Askew Page HR Baldwin SN Jordan MC Myat Thwe P et al A small molecule inhibitor of the chloride channel TMEM16A blocks vascular smooth muscle contraction and lowers blood pressure in spontaneously hypertensive rats. Kidney Int. (2021) 100(2):311–20. 10.1016/j.kint.2021.03.025
38.
Ma MM Gao M Guo KM Wang M Li XY Zeng XL et al TMEM16A contributes to endothelial dysfunction by facilitating Nox2 NADPH oxidase-derived reactive oxygen species generation in hypertension. Hypertension. (2017) 69(5):892–901. 10.1161/HYPERTENSIONAHA.116.08874
39.
Bulley S Jaggar JH . Cl− channels in smooth muscle cells. Pflugers Arch. (2014) 466(5):861–72. 10.1007/s00424-013-1357-2
40.
Goto K Kitazono T . Chloride ions, vascular function and hypertension. Biomedicines. (2022) 10(9):2316. 10.3390/biomedicines10092316
41.
Leblanc N Forrest AS Ayon RJ Wiwchar M Angermann JE Pritchard HA et al Molecular and functional significance of ca(2+)-activated cl(-) channels in pulmonary arterial smooth muscle. Pulm Circ. (2015) 5(2):244–68. 10.1086/680189
42.
Jensen AB Joergensen HB Dam VS Kamaev D Boedtkjer D Füchtbauer EM et al Variable contribution of TMEM16A to tone in murine arterial vasculature. Basic Clin Pharmacol Toxicol. (2018) 123(1):30–41. 10.1111/bcpt.12984
43.
Wang Q Leo MD Narayanan D Kuruvilla KP Jaggar JH . Local coupling of TRPC6 to ANO1/TMEM16A channels in smooth muscle cells amplifies vasoconstriction in cerebral arteries. Am J Physiol Cell Physiol. (2016) 310(11):C1001–9. 10.1152/ajpcell.00092.2016
44.
Matchkov VV Black Joergensen H Kamaev D Hoegh Jensen A Beck HC Skryabin BV et al A paradoxical increase of force development in saphenous and tail arteries from heterozygous ANO1 knockout mice. Physiol Rep. (2020) 8(22):e14645. 10.14814/phy2.14645
45.
Allawzi AM Vang A Clements RT Jhun BS Kue NR Mancini TJ et al Activation of anoctamin-1 limits pulmonary endothelial cell proliferation via p38-mitogen-activated protein kinase-dependent apoptosis. Am J Respir Cell Mol Biol. (2018) 58(5):658–67. 10.1165/rcmb.2016-0344OC
46.
Skofic Maurer D Zabini D Nagaraj C Sharma N Lengyel M Nagy BM et al Endothelial dysfunction following enhanced TMEM16A activity in human pulmonary arteries. Cells. (2020) 9(9):1984. 10.3390/cells9091984
47.
Turner T Chen X Zahner M Opsahl A DeMarco G Boucher M et al FGF21 increases water intake, urine output and blood pressure in rats. PLoS One. (2018) 13(8):e0202182. 10.1371/journal.pone.0202182
48.
Mallappallil M Sabu J Friedman EA Salifu M . What do we know about opioids and the kidney?Int J Mol Sci. (2017) 18(1):223. 10.3390/ijms18010223
49.
Yip KP Balasubramanian L Kan C Wang L Liu R Ribeiro-Silva L et al Intraluminal pressure triggers myogenic response via activation of calcium spark and calcium-activated chloride channel in rat renal afferent arteriole. Am J Physiol Renal Physiol. (2018) 315(6):F1592–600. 10.1152/ajprenal.00239.2018
50.
Wang B Li C Huai R Qu Z . Overexpression of ANO1/TMEM16A, an arterial Ca2+-activated cl- channel, contributes to spontaneous hypertension. J Mol Cell Cardiol. (2015) 82:22–32. 10.1016/j.yjmcc.2015.02.020
51.
Zhang XH Zheng B Yang Z He M Yue LY Zhang RN et al TMEM16A And myocardin form a positive feedback loop that is disrupted by KLF5 during ang II-induced vascular remodeling. Hypertension. (2015) 66(2):412–21. 10.1161/HYPERTENSIONAHA.115.05280
52.
Lv XF Zhang YJ Liu X Zheng HQ Liu CZ Zeng XL et al TMEM16A ameliorates vascular remodeling by suppressing autophagy via inhibiting bcl-2-p62 complex formation. Theranostics. (2020) 10(9):3980–93. 10.7150/thno.41028
53.
Zhang Y Ye L Duan DD Yang H Ma T . TMEM16A plays an insignificant role in myocardium remodeling but may promote angiogenesis of heart during pressure-overload. Front Physiol. (2022) 13:897619. 10.3389/fphys.2022.897619
54.
Suzuki T Suzuki Y Asai K Imaizumi Y Yamamura H . Hypoxia increases the proliferation of brain capillary endothelial cells via upregulation of TMEM16A ca(2+)-activated cl(-) channels. J Pharmacol Sci. (2021) 146(1):65–9. 10.1016/j.jphs.2021.03.002
55.
Ma K Liu S Liang H Wang G Wang T Luo S et al Ca(2+)-activated cl(-) channel TMEM16A inhibition by cholesterol promotes angiogenesis in endothelial cells. J Adv Res. (2021) 29:23–32. 10.1016/j.jare.2020.09.003
56.
Cai H Fan S Cai L Zhu L Zhao Z Li Y et al Dihydroartemisinin attenuates hypoxia-induced pulmonary hypertension through the ELAVL2/miR-503/PI3K/AKT axis. J Cardiovasc Pharmacol. (2022) 80(1):95–109. 10.1097/FJC.0000000000001271
57.
Forrest AS Joyce TC Huebner ML Ayon RJ Wiwchar M Joyce J et al Increased TMEM16A-encoded calcium-activated chloride channel activity is associated with pulmonary hypertension. Am J Physiol Cell Physiol. (2012) 303(12):C1229–43. 10.1152/ajpcell.00044.2012
58.
Sun H Xia Y Paudel O Yang XR Sham JS . Chronic hypoxia-induced upregulation of Ca2+-activated cl- channel in pulmonary arterial myocytes: a mechanism contributing to enhanced vasoreactivity. J Physiol. (2012) 590(15):3507–21. 10.1113/jphysiol.2012.232520
59.
Liu D Wang K Su D Huang Y Shang L Zhao Y et al TMEM16A Regulates pulmonary arterial smooth muscle cells proliferation via p38MAPK/ERK pathway in high pulmonary blood flow-induced pulmonary arterial hypertension. J Vasc Res. (2020) 58(1):27–37. 10.1159/000511267
60.
Xie JY Liu WY Lv WJ Han XH Kong QN Wu YH et al Transmembrane protein 16A/anoctamin 1 inhibitor T16A(inh)-A01 reversed monocrotaline-induced rat pulmonary arterial hypertension. Pulm Circ. (2020) 10(4):1–11. 10.1177/2045894020946670
61.
Kondo R Furukawa N Deguchi A Kawata N Suzuki Y Imaizumi Y et al Downregulation of ca(2+)-activated cl(-) channel TMEM16A mediated by angiotensin II in cirrhotic portal hypertensive mice. Front Pharmacol. (2022) 13:831311. 10.3389/fphar.2022.831311
62.
Zeng X Huang P Chen M Liu S Wu N Wang F et al TMEM16A regulates portal vein smooth muscle cell proliferation in portal hypertension. Exp Ther Med. (2018) 15(1):1062–8. 10.3892/etm.2017.5466
63.
Cornwell WK 3rd Ambardekar AV Tran T Pal JD Cava L Lawley J et al Stroke incidence and impact of continuous-flow left ventricular assist devices on cerebrovascular physiology. Stroke. (2019) 50(2):542–8. 10.1161/STROKEAHA.118.022967
64.
Bulley S Neeb ZP Burris SK Bannister JP Thomas-Gatewood CM Jangsangthong W et al TMEM16A/ANO1 channels contribute to the myogenic response in cerebral arteries. Circ Res. (2012) 111(8):1027–36. 10.1161/CIRCRESAHA.112.277145
65.
Li RS Wang Y Chen HS Jiang FY Tu Q Li WJ et al TMEM16A contributes to angiotensin II-induced cerebral vasoconstriction via the RhoA/ROCK signaling pathway. Mol Med Rep. (2016) 13(4):3691–9. 10.3892/mmr.2016.4979
66.
Liu PY Zhang Z Liu Y Tang XL Shu S Bao XY et al TMEM16A inhibition preserves blood-brain barrier integrity after ischemic stroke. Front Cell Neurosci. (2019) 13:360. 10.3389/fncel.2019.00360
67.
Jiang W Li J Cai Y Liu W Chen M Xu X et al The novel lncRNA ENST00000530525 affects ANO1, contributing to blood-brain barrier injury in cultured hCMEC/D3 cells under OGD/R conditions. Front Genet. (2022) 13:873230. 10.3389/fgene.2022.873230
68.
Suzuki T Yasumoto M Suzuki Y Asai K Imaizumi Y Yamamura H . TMEM16A Ca(2+)-activated cl(-) channel regulates the proliferation and migration of brain capillary endothelial cells. Mol Pharmacol. (2020) 98(1):61–71. 10.1124/mol.119.118844
69.
Al-Hosni R Agostinelli E Ilkan Z Scofano L Kaye R Dinsdale RL et al Pharmacological profiling of small molecule modulators of the TMEM16A channel and their implications for the control of artery and capillary function. Br J Pharmacol. (2025) 182(8):1719–40. 10.1111/bph.17383
70.
Korte N Ilkan Z Pearson CL Pfeiffer T Singhal P Rock JR et al The Ca2+-gated channel TMEM16A amplifies capillary pericyte contraction and reduces cerebral blood flow after ischemia. J Clin Invest. (2022) 132(9):e154118. 10.1172/JCI154118
71.
Wang M Yang H Zheng LY Zhang Z Tang YB Wang GL et al Downregulation of TMEM16A calcium-activated chloride channel contributes to cerebrovascular remodeling during hypertension by promoting basilar smooth muscle cell proliferation. Circulation. (2012) 125(5):697–707. 10.1161/CIRCULATIONAHA.111.041806
72.
Lin CX Lv XF Yuan F Li XY Ma MM Liu CZ et al Ca(2+)/calmodulin-dependent protein kinase II γ-dependent Serine727 phosphorylation is required for TMEM16A ca(2+)-activated cl(-) channel regulation in cerebrovascular cells. Circ J. (2018) 82(3):903–13. 10.1253/circj.CJ-17-0585
73.
Liu CZ Li FY Lv XF Ma MM Li XY Lin CX et al Endophilin A2 regulates calcium-activated chloride channel activity via selective autophagy-mediated TMEM16A degradation. Acta Pharmacol Sin. (2020) 41(2):208–17. 10.1038/s41401-019-0298-5
74.
Zeng JW Chen BY Lv XF Sun L Zeng XL Zheng HQ et al Transmembrane member 16A participates in hydrogen peroxide-induced apoptosis by facilitating mitochondria-dependent pathway in vascular smooth muscle cells. Br J Pharmacol. (2018) 175(18):3669–84. 10.1111/bph.14432
75.
Zheng H Li X Zeng X Huang C Ma M Lv X et al TMEM16A inhibits angiotensin II-induced basilar artery smooth muscle cell migration in a WNK1-dependent manner. Acta Pharm Sin B. (2021) 11(12):3994–4007. 10.1016/j.apsb.2021.04.013
76.
Zeng XL Sun L Zheng HQ Wang GL Du YH Lv XF et al Smooth muscle-specific TMEM16A expression protects against angiotensin II-induced cerebrovascular remodeling via suppressing extracellular matrix deposition. J Mol Cell Cardiol. (2019) 134:131–43. 10.1016/j.yjmcc.2019.07.002
77.
Genovese M Galietta LJV . Anoctamin pharmacology. Cell Calcium. (2024) 121:102905. 10.1016/j.ceca.2024.102905
78.
Jiang Y Yu B Fang F Cao H Ma T Yang H . Modulation of chloride channel functions by the plant lignan compounds kobusin and eudesmin. Front Plant Sci. (2015) 6:1041. 10.3389/fpls.2015.01041
79.
Namkung W Thiagarajah JR Phuan PW Verkman AS . Inhibition of Ca2+-activated cl- channels by gallotannins as a possible molecular basis for health benefits of red wine and green tea. FASEB J. (2010) 24(11):4178–86. 10.1096/fj.10-160648
80.
Ursoniu S Sahebkar A Andrica F Serban C Banach M . Effects of flaxseed supplements on blood pressure: a systematic review and meta-analysis of controlled clinical trial. Clin Nutr. (2016) 35(3):615–25. 10.1016/j.clnu.2015.05.012
81.
Ji Q Shi S Ma B Zhang W An H Guo S . Drug repurposing and molecular mechanisms of the antihypertensive drug candesartan as a TMEM16A channel inhibitor. Int J Biol Macromol. (2023) 235:123839. 10.1016/j.ijbiomac.2023.123839
82.
Wang H Ma D Zhu X Liu P Li S Yu B et al Nimodipine inhibits intestinal and aortic smooth muscle contraction by regulating ca(2+)-activated cl(-) channels. Toxicol Appl Pharmacol. (2021) 421:115543. 10.1016/j.taap.2021.115543
83.
Boedtkjer DM Kim S Jensen AB Matchkov VM Andersson KE . New selective inhibitors of calcium-activated chloride channels—t16A(inh) -A01, CaCC(inh) -A01 and MONNA—what do they inhibit?Br J Pharmacol. (2015) 172(16):4158–72. 10.1111/bph.13201
84.
Shi S Guo S Chen Y Sun F Pang C Ma B et al Molecular mechanism of CaCC(inh)-A01 inhibiting TMEM16A channel. Arch Biochem Biophys. (2020) 695:108650. 10.1016/j.abb.2020.108650
85.
Seo Y Lee HK Park J Jeon DK Jo S Jo M et al Ani9, A novel potent small-molecule ANO1 inhibitor with negligible effect on ANO2. PLoS One. (2016) 11(5):e0155771. 10.1371/journal.pone.0155771
86.
Kaye R Pearson C Babiker T Agostinelli E Al-Hosni R Tammaro P . Clinically relevant niclosamide concentrations modulate TMEM16A and ca(V)1.2 channels to control artery tone and capillary diameter. Br J Pharmacol. (2025) 182(19):4490–515. 10.1111/bph.70077
87.
Truong EC Phuan PW Reggi AL Ferrera L Galietta LJV Levy SE et al Substituted 2-acylaminocycloalkylthiophene-3-carboxylic acid arylamides as inhibitors of the calcium-activated chloride channel transmembrane protein 16A (TMEM16A). J Med Chem. (2017) 60(11):4626–35. 10.1021/acs.jmedchem.7b00020
88.
Jaboreck MC Lühmann JL Mielenz M Stanke F Göhring G Martin U et al Generation of two TMEM16A knockout iPSC clones each from a healthy human iPSC line, from a cystic fibrosis patient specific line with p.Phe508del mutation and from the gene corrected iPSC line. Stem Cell Res. (2022) 64:102918. 10.1016/j.scr.2022.102918
89.
Zuo YB Zhang YF Zhang R Tian JW Lv XB Li R et al Ferroptosis in cancer progression: role of noncoding RNAs. Int J Biol Sci. (2022) 18(5):1829–43. 10.7150/ijbs.66917
90.
Hiram R Rizcallah E Sirois C Sirois M Morin C Fortin S et al Resolvin D1 reverses reactivity and Ca2+ sensitivity induced by ET-1, TNF-α, and IL-6 in the human pulmonary artery. Am J Physiol Heart Circ Physiol. (2014) 307(11):H1547–58. 10.1152/ajpheart.00452.2014
91.
Lee YH Yi GS . Prediction of novel Anoctamin1 (ANO1) inhibitors using 3D-QSAR pharmacophore modeling and molecular docking. Int J Mol Sci. (2018) 19(10):3204. 10.3390/ijms19103204
92.
Genovese M Buccirossi M Guidone D De Cegli R Sarnataro S di Bernardo D et al Analysis of inhibitors of the anoctamin-1 chloride channel (transmembrane member 16A, TMEM16A) reveals indirect mechanisms involving alterations in calcium signalling. Br J Pharmacol. (2023) 180(6):775–85. 10.1111/bph.15995
Summary
Keywords
hypertension, ANO1, stroke, vascular remodelling, endothelial dysfunction
Citation
Zhang J-q, Wen Z-j, Han X-h, Qiu Z-k, Yang Q-y and Qiu Y (2025) The emerging role of anoctamin-1 in cardiac and cerebrovascular diseases. Front. Cardiovasc. Med. 12:1691331. doi: 10.3389/fcvm.2025.1691331
Received
23 August 2025
Accepted
08 October 2025
Published
24 October 2025
Volume
12 - 2025
Edited by
Gaurav Kumar, Medical College of Wisconsin, United States
Reviewed by
Kenichi Goto, Kyushu University, Japan
Tingting Zhou, Jiangnan University, China
Updates
Copyright
© 2025 Zhang, Wen, Han, Qiu, Yang and Qiu.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
* Correspondence: Yue Qiu yayaqiuyue@sina.com
†These authors have contributed equally to this work and share first authorship
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.