 Fang Zhang
Fang Zhang Xiang Zhang
Xiang Zhang- Department of Cardiology, Chongzhou People’s Hospital, Chongzhou, Sichuan, China
Heart failure (HF) is a major global health problem associated with high illness rates, mortality, and healthcare costs. Although advances in diagnosis and therapy have improved outcomes for some patients, effective treatment—especially for HF with preserved ejection fraction (HFpEF)—remains limited. HF develops through complex interactions among neurohormonal activation, metabolic remodeling, mitochondrial dysfunction, inflammation, fibrosis, and microvascular impairment. Recent discoveries in these areas have revealed new molecular and cellular targets that may lead to more precise therapies. Novel pharmacological agents, metabolic modulators, device-based interventions, and regenerative approaches are reshaping the treatment landscape. In addition, personalized strategies such as multi-omics profiling, biomarker-guided management, and artificial intelligence–assisted diagnosis hold promise for better risk prediction and individualized care. However, translating mechanistic discoveries into clinical benefit remains a challenge. Future research integrating molecular insights with clinical phenotyping will be essential to achieve precision treatment and improved outcomes in patients with HF.
1 Introduction
Heart failure (HF) is a complex clinical syndrome characterized by impaired ventricular filling or ejection due to structural or functional cardiac abnormalities. It leads to symptoms such as dyspnea, fatigue, and edema, and is associated with poor quality of life and prognosis (1). Based on left ventricular ejection fraction (LVEF), HF is classified into HF with reduced ejection fraction (HFrEF, LVEF <40%), mildly reduced ejection fraction (HFmrEF, 40%–49%), and preserved ejection fraction (HFpEF, ≥50%) (2, 3). Notably, HFpEF now accounts for more than half of all HF cases and represents a major therapeutic challenge (4).
Despite significant progress in pharmacological and device-based therapies, the overall prognosis of HF remains poor, with high rates of hospitalization and mortality (5). While neurohormonal inhibition with ACE inhibitors, β-blockers, and mineralocorticoid receptor antagonists has greatly improved outcomes in HFrEF, similar success has not been achieved in HFpEF (6). No current treatment convincingly reduces mortality in HFpEF, largely because of its heterogeneous mechanisms involving metabolic abnormalities, inflammation, microvascular dysfunction (MVD), and systemic comorbidities (7). These limitations highlight an urgent unmet clinical need for new, mechanism-based and precision-oriented therapeutic strategies.
This review therefore summarizes recent advances in understanding the molecular and cellular mechanisms of HF and discusses how these insights can inform novel pharmacological, device-based, and personalized treatment approaches aimed at improving outcomes across diverse HF phenotypes.
2 Methodological framework
This narrative review was conducted to synthesize recent advances in the mechanistic understanding and emerging therapeutic strategies of HF. A structured literature search was performed across the PubMed, Scopus, and Web of Science databases for studies published between January 2010 and September 2025. The search combined the following keywords and Boolean operators: “heart failure” AND (“molecular mechanism” OR “pathophysiology” OR “metabolic remodeling” OR “mitochondrial dysfunction” OR “fibrosis” OR “epigenetic regulation” OR “precision medicine” OR “therapeutic strategies”). Inclusion criteria were: (1) peer-reviewed original articles or reviews; (2) studies involving human subjects, relevant animal models, or translational data; and (3) publications in English. Exclusion criteria included case reports, editorials, conference abstracts, and non-peer-reviewed materials (8).
Reference lists of relevant articles and recent high-impact reviews were also screened to ensure comprehensive coverage. The quality and relevance of studies were assessed based on methodological rigor and contribution to mechanistic or therapeutic understanding. While this review is primarily narrative in scope, it follows key principles of the PRISMA-ScR framework, ensuring transparent reporting of literature identification and selection. All included references were cross-verified for accuracy and represent the most recent and impactful studies available at the time of writing (9).
3 Novel insights into the pathophysiological mechanisms of HF
The pathophysiology of HF is highly complex, involving multiple mechanisms such as neuroendocrine activation, metabolic remodeling, inflammatory responses, mitochondrial dysfunction, and cellular senescence (10). These interacting processes, summarized in Figure 1, highlight the multi-layered nature of HF progression. In recent years, advances in experimental techniques have progressively elucidated more specific molecular mechanisms, providing a theoretical foundation for precision therapy.
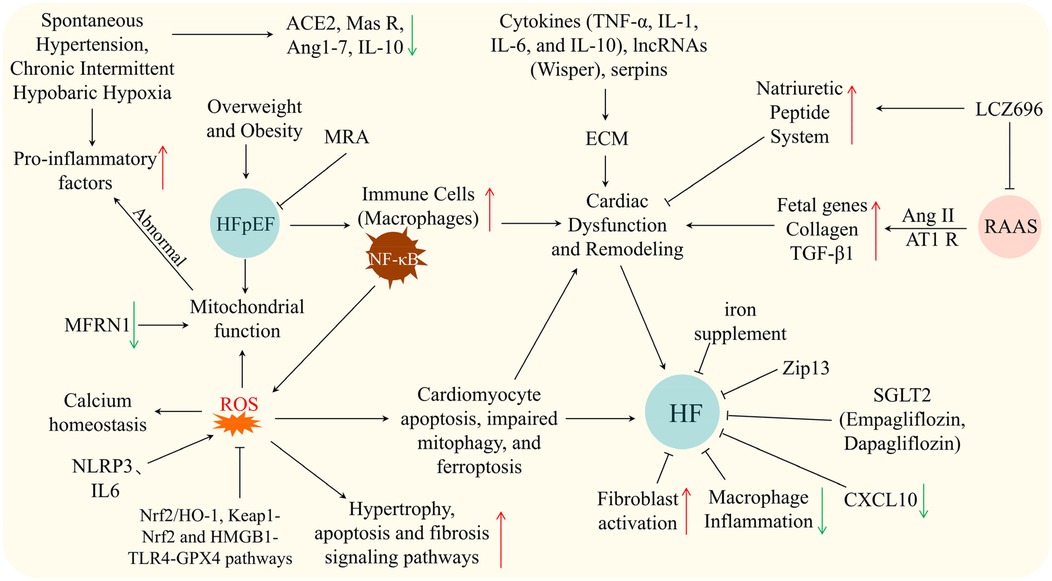
Figure 1. Schematic showing mechanisms and therapies in HF/HFpEF: triggers (hypertension, hypoxia, obesity), pathways (inflammation, ROS, RAAS, cytokines), cellular events, and interventions. Arrows: ↑ upregulation, ↓ downregulation, black for causal relationships, ⊥ for inhibition.
3.1 Experimental insights into RAAS-mediated neuroendocrine activation in HF
Overactivation of the renin–angiotensin–aldosterone system (RAAS) and the sympathetic nervous system (SNS) is a major driving factor in the onset and progression of HF (11, 12). Under pathological conditions such as myocardial injury and hypertension, the key RAAS effector molecule angiotensin II (Ang II) mediates signal transduction via the AT1A receptor, upregulating the expression of fetal genes, collagen, and TGF-β1 in non-infarcted regions, thereby promoting left ventricular dilation, fibrosis, and dysfunction, and accelerating ventricular remodeling and HF progression (13). Multiple animal studies have confirmed the cardioprotective effects of RAAS inhibition on cardiac structure and function. For instance, Woźniak et al. (14) demonstrated in a Tgaq*44 dilated cardiomyopathy mouse model that early combined administration of an ACE inhibitor (perindopril) and an aldosterone receptor antagonist (canrenone) preserved systolic function, whereas late intervention mainly attenuated ventricular dilation, indicating a stage-dependent therapeutic effect. Chen et al. (15) reported in spontaneously hypertensive rats that chronic intermittent hypobaric hypoxia downregulated ACE and AT1 receptor expression, upregulated ACE2 and Mas receptor expression, reduced Ang II and pro-inflammatory cytokines, and increased Ang1–7 and the anti-inflammatory cytokine IL-10, thereby improving vascular relaxation and remodeling. Hawlitschek et al. (16) found that captopril alone or in combination with nifedipine significantly reduced blood pressure, alleviated cardiac hypertrophy, and prevented myocardial fibrosis. Yi et al. (17) showed that time-restricted feeding suppressed the ACE–Ang II–AT1 axis, reducing Ang II-mediated cardiac remodeling and dysfunction, thereby lowering blood pressure and exerting cardioprotective effects. Clinically, McMurray et al. (18) demonstrated in the PARADIGM-HF trial that the angiotensin receptor–neprilysin inhibitor (ARNI) LCZ696 not only inhibited RAAS activity but also enhanced natriuretic peptide system function, synergistically improving cardiac load and neurohormonal imbalance, and producing greater reductions in cardiovascular mortality and HF rehospitalization compared with the ACE inhibitor enalapril. Collectively, these findings underscore the central pathological role of RAAS in cardiovascular disease progression, and indicate that RAAS inhibition yields significant improvements in cardiac structure and function in experimental models, with clear prognostic benefits in clinical practice.
3.2 Exercise and metabolic modulation in HF
Metabolic abnormalities are a key component of HF pathophysiology. Overweight and obesity, even in the absence of metabolic syndrome, significantly increase HF risk (by 37%–85%), with cardiovascular event risk rising proportionally with the number of metabolic syndrome components (19). In HFpEF, epicardial adipose tissue volume is markedly increased and correlates with cardiac dysfunction, metabolic abnormalities, and inflammatory markers (20).
Pharmacologically, mineralocorticoid receptor antagonists (MRAs) reduce HF hospitalization and cardiovascular mortality in HFrEF, with more modest benefits in HFmrEF/HFpEF and an increased risk of hyperkalemia (21). Non-steroidal MRA finerenone significantly reduces HF composite outcomes in patients with recent worsening HF (WHF) without increasing adverse events (22), while finerenone also lowers the risk of new-onset diabetes by 24% in HF patients without baseline diabetes (23). Among SGLT2 inhibitors, dapagliflozin achieves similar diuretic efficacy to metolazone but with less electrolyte disturbance and renal function deterioration (24), whereas empagliflozin shows no short-term improvement in myocardial energy metabolism (25). The mitochondrial uncoupler HU6 may enhance metabolic flexibility and potentially improve cardiac function in obesity-related HFpEF (26).
Exercise interventions show heterogeneous effects. In HFpEF, high-intensity interval training (HIIT) does not outperform moderate-intensity continuous training (MICT) in improving peak oxygen uptake (VO2peak) (27). However, in coronary artery disease and some chronic HF populations, HIIT significantly improves VO2peak, heart rate variability, and left ventricular function (28–30). Cardiac rehabilitation (CR) reduces HF and all-cause rehospitalization and improves exercise capacity and quality of life, with no mortality benefit (31), while hybrid comprehensive telerehabilitation (HCTR) yields short-term functional gains without long-term clinical benefit (32). Additionally, exercise oscillatory ventilation independently predicts mortality and heart transplantation in HFrEF (33). Other interventions, such as oral polysaccharide iron in iron-deficient HFrEF, do not improve exercise capacity or cardiac function (34). Collectively, these findings highlight the need for integrated metabolic control, optimized pharmacotherapy, and individualized exercise prescriptions to maximize benefits in HF management.
3.3 Mitochondrial dysfunction and iron metabolism in HF
Accumulating evidence from basic and clinical studies indicates that mitochondrial dysfunction is a central mechanism in the development and progression of HF. Impaired mitochondrial respiration in cardiomyocytes and peripheral blood mononuclear cells (PBMCs) exacerbates systemic inflammation. Zhou et al. (35) demonstrated that MitoDAMPs suppress complex I activity in PBMCs via IL-6 induction, whereas supplementation with the NAD+ precursor nicotinamide riboside (NR) enhances mitochondrial respiration and attenuates pro-inflammatory cytokine expression. Disruption of mitochondrial iron homeostasis also contributes critically to HF pathophysiology. Li et al. (36) reported that cardiac-specific Zip13 knockout mice exhibit severe contractile dysfunction, with decreased mitochondrial iron and elevated cytosolic iron; iron supplementation or overexpression of the mitochondrial iron transporter MFRN1 partially restores mitochondrial function, highlighting the essential role of ZIP13 in maintaining myocardial mitochondrial iron balance. Clinically, approximately 23% of HF patients present with left ventricular myocardial iron deficiency, which correlates with reduced activity of mitochondrial respiratory chain complexes and TCA cycle enzymes, as well as impaired oxidative stress defense (37). In vitro iron deprivation further confirms that impaired Fe–S cluster-dependent complex I–III activity significantly inhibits ATP production and cardiomyocyte contractility, which can be reversed by iron supplementation (38). Furthermore, frataxin deficiency or SLC25A3 loss perturbs NAD+ metabolism, mitochondrial biogenesis, and fusion/fission dynamics, leading to metabolic dysfunction, Ca2+ handling abnormalities, and cardiomyocyte hypertrophy (39, 40), while mitochondrial transplantation partially rescues these phenotypes. In endothelial cells, lipid droplet formation mitigates lipotoxicity, preserves mitochondrial function, and inhibits ferroptosis, thereby protecting cardiac microvascular integrity (41). Clinically, intravenous iron administration or SGLT2 inhibition with empagliflozin improves mitochondrial energy metabolism, enhances iron utilization and erythropoiesis, and subsequently improves cardiac and skeletal muscle function, leading to enhanced exercise capacity and left ventricular performance in HF patients (42–45). Collectively, mitochondrial dysfunction in HF involves deficits in energy metabolism as well as dysregulation of iron homeostasis and NAD+ levels, and targeting mitochondrial pathways represents a promising therapeutic strategy to improve cardiac function.
3.4 Inflammation in HF
A growing body of epidemiological and clinical evidence demonstrates that inflammation plays a central role in the onset, progression, and prognosis of HF, particularly HFpEF. Elevated systemic immune-inflammation index (SII) is positively associated with HF risk in multiple populations, including NHANES cohorts, smokers, and patients with diabetes (46, 47). Chronic immune-inflammatory conditions, such as rheumatoid arthritis, increase the risk of both ischemic and non-ischemic HF, with the highest risk observed in rheumatoid factor–positive individuals (48). HFpEF is frequently accompanied by metabolic abnormalities and activation of systemic inflammatory protein networks, in which specific mediators—TNFR1, UPAR, IGFBP7, and GDF-15—partially mediate structural and functional cardiac impairment (49), while distinct inflammatory patterns are associated with adverse outcomes, reduced exercise capacity, and impaired quality of life (50). These findings support the existence of a “comorbidity–inflammation–HF” pathological axis, in line with mechanistic insights that metabolic derangements and inflammatory burden interact to drive metabolic inflammation (metainflammation) (51), promoting ventricular remodeling through immune cell polarization, pro-inflammatory cytokine release, oxidative stress, and pathological fibrosis. Experimental studies further demonstrate that HF is associated with pro-inflammatory activation of immune cells (e.g., macrophages), mitochondrial dysfunction, and activation of key signaling pathways such as NF-κB; inhibiting macrophage inflammation, promoting M2 polarization, or restoring mitochondrial function can attenuate cardiac dysfunction and remodeling (35, 52). In terms of anti-inflammatory interventions, sodium-glucose cotransporter 2 (SGLT2) inhibitors such as empagliflozin and dapagliflozin exhibit significant cardioprotective effects in HF models and patients (53, 54), partly independent of SGLT2 itself, involving downregulation of CXCL10 (55), suppression of macrophage inflammation, and modulation of fibroblast activation. Additional approaches—including low-level transcutaneous vagus nerve stimulation (56, 57), selenium-enriched diets (58), NAD precursors (35), NF-κB pathway inhibitors, and certain herbal preparations such as processed Aconitum extracts (59)—have also demonstrated potential to improve cardiac function via inflammation attenuation. However, previous trials of anti-inflammatory agents such as TNF-α antagonists have shown limited or even harmful effects in HF (48), suggesting that non-specific or isolated anti-inflammatory strategies may be insufficient. Collectively, these findings underscore that although inflammation is an indispensable mechanistic link in HF, the efficacy of anti-inflammatory therapy depends on pathway selectivity, timing of intervention, and alignment with patient phenotype, rather than broad cytokine inhibition. Future therapies may benefit from biomarker-guided and phenotype-specific strategies that better match the underlying inflammatory pathways in HF.
3.5 Oxidative stress and cardiomyocyte injury in HF
In HF, the high metabolic activity of cardiomyocytes renders them a primary source of reactive oxygen and nitrogen species (ROS/RNS), while neurohormonal activation, adrenergic overstimulation, and excessive mechanical stress induce cellular stress that disrupts redox homeostasis and impairs mitochondrial function (60, 61). Major ROS sources include mitochondrial electron transport chain complexes I/III, NADPH oxidases (Nox2/Nox4), xanthine oxidoreductase (XOR), nitric oxide synthases (NOS), monoamine oxidases (MAO), and p66shc. Enhanced activity of these enzymes establishes a ROS-driven vicious cycle, damaging mitochondrial DNA, proteins, and lipids, perturbing calcium homeostasis, and activating hypertrophic, apoptotic, and fibrotic signaling pathways (62–64). Excess ROS can further induce cardiomyocyte apoptosis, impaired mitophagy, and ferroptosis through IGF2BP2-dynamin2, PDE4D-CREB-SIRT1-PINK1/Parkin, and Piezo1/Yap1 signaling axes, accelerating myocardial remodeling and HF progression (65–67). Activation of the NLRP3 inflammasome also exacerbates cardiomyocyte injury by promoting oxidative stress and pyroptosis (68). Under pathological conditions, excessive ROS generation combined with impaired antioxidant defense systems, including SOD, GSHPx, catalase, Trx/TrxR, and GSH, leads to cumulative oxidative damage, further compromising cardiomyocyte structure and function (62). Therapeutic strategies activating Nrf2/HO-1, Keap1-Nrf2, and HMGB1-TLR4-GPX4 pathways can suppress ROS production, restore mitochondrial function, and mitigate cardiomyocyte apoptosis, inflammation, and fibrosis, thereby improving pathological cardiac remodeling (69–71). Collectively, ROS generation in cardiomyocytes and its dynamic regulation constitute a central driver of myocardial remodeling and HF progression, providing a mechanistic basis for targeted interventions against cardiomyocyte oxidative stress (72).
3.6 Fibrosis and extracellular matrix remodeling in HF
Myocardial fibrosis is a central pathological process in the development and progression of HF, characterized by the activation of cardiac fibroblasts (CFs) and their differentiation into myofibroblasts (myoFbs), which mediate excessive deposition and crosslinking of extracellular matrix (ECM) proteins, ultimately leading to ventricular stiffening, myocardial remodeling, and functional impairment (73–75). Fibrosis can represent either a reparative response or maladaptive remodeling: acute myocardial injury, such as myocardial infarction, induces replacement fibrosis to preserve myocardial structural integrity, whereas chronic pressure overload triggers reactive fibrosis, resulting in diffuse interstitial and perivascular collagen accumulation and persistent disruption of left and right ventricular function (76, 77).
CFs sense mechanical stress and integrate signaling via integrin-FAK pathways, AngII downstream cascades, and TGF-β/Smad and non-canonical MAPK/PI3K/AKT pathways to regulate proliferation, migration, and phenotypic transformation, thereby driving excessive deposition of type I/III collagen and other matrix proteins (73, 74, 78, 79). Additionally, immune cell-mediated inflammation, cytokines (including TNF-α, IL-1, IL-6, IL-10), long noncoding RNAs such as Wisper, fibronectin, and serpins participate in myocardial ECM remodeling by modulating fibroblast activity, collagen crosslinking, and matrix degradation, further influencing fibrotic progression (80–83).
Myocardial fibrosis not only reduces diastolic compliance and impairs systolic function but also disrupts intercellular signaling and increases the risk of arrhythmias (84, 85). Interventions such as left ventricular assist device (LVAD) unloading, HDAC inhibitors, SGLT2 inhibitors, and direct fibroblast reprogramming have been shown to partially reverse fibrosis and structural-functional abnormalities, highlighting its potential as a therapeutic target in HF (75, 86–88). Collectively, myocardial fibrosis, through ECM remodeling, plays a pivotal role in HF pathogenesis, and the complex molecular regulatory networks and cellular heterogeneity underlying this process provide important directions for future precision therapies (89–91).
3.7 Autophagy and proteostasis dysregulation in HF
Autophagic dysregulation and protein homeostasis imbalance play central roles in the pathogenesis and progression of HF. Autophagy maintains cardiomyocyte protein quality control (PQC), clears misfolded and toxic proteins, and preserves mitochondrial function, with moderate activation conferring cardioprotection (92, 93). However, excessive or insufficient autophagy disrupts protein homeostasis, induces mitochondrial damage, and triggers apoptosis or autophagy-dependent cell death (autosis), exacerbating cardiomyopathy, atrial fibrillation, and HF progression (94, 95). PKG signaling and PQC dysfunction accelerate hypertrophy, remodeling, and cardiomyocyte loss, whereas pharmacological PKG activation restores PQC and improves cardiac function (96). BAG3 maintains protein homeostasis via chaperone-assisted selective autophagy (CASA), and its deficiency leads to age-dependent autophagic dysregulation and early-onset cardiomyopathy (97).
In HFpEF, diabetes promotes NF-κB/IL-6/NLRP3-mediated inflammation and oxidative stress, suppresses NO-sGC-cGMP-PKG and AKT/AMPK/mTOR-regulated autophagy, and impairs HSP27/HSP70-dependent PQC, increasing myocardial stiffness and diastolic dysfunction (98). Nrf2 regulates the ubiquitin–proteasome system and autophagy-related genes, and its dysfunction drives protein misfolding, hypertrophy, and HF development (99). Persistent mTORC1/4EBP1 activation under aging or metabolic–hypertensive stress disrupts protein homeostasis, accelerates toxic protein accumulation, and exacerbates HFpEF and cardiac aging (100, 101). Additional regulators, including ROR2, TRIM24, and Txlnb, modulate protein folding, ubiquitination, and calcium handling, further destabilizing protein homeostasis and impairing contractile function (102–105). Models of chronic kidney disease, premature aging, and rapid-growth broiler cardiomyopathy demonstrate that protein oxidation, glycation, and PQC disruption, combined with autophagic and mitochondrial dysfunction, accelerate pathological remodeling (106–108).
Recent studies have further delineated the crosstalk between autophagy, mitochondrial health, and proteostasis. Dysregulated mitophagy contributes to the accumulation of dysfunctional mitochondria and excessive ROS generation, amplifying cardiomyocyte injury and maladaptive remodeling (109). Crosstalk between mTOR, AMPK, and sirtuin signaling fine-tunes autophagic flux, linking nutrient status, metabolic stress, and cellular aging to HF progression (110). Moreover, key autophagy mediators such as Beclin-1, ATG5, and TFEB are increasingly recognized as potential therapeutic targets because restoration of balanced autophagy—neither excessive nor suppressed—attenuates hypertrophy, fibrosis, and contractile dysfunction in preclinical models (111). These findings reinforce the translational potential of modulating the AMPK–mTOR–TFEB axis or enhancing mitophagy to improve proteostasis and delay HF progression.
Therapeutically, interventions such as tanshinone IIA, rapamycin, ginsenosides, and components of Si-Miao-Yong-An decoction modulate AMPK–mTOR–Beclin1-dependent autophagy, inhibit mTORC1 and ER stress, or restore mitophagy, thereby reducing apoptosis, improving function, and attenuating remodeling (112–115). Nutritional status further influences autophagic activity, highlighting the interplay between autophagy and protein homeostasis as a potential therapeutic target in chronic HF (116). Collectively, autophagic dysregulation and protein homeostasis imbalance constitute central mechanisms driving cardiomyocyte injury, remodeling, and HF progression, providing avenues for precision therapy (117).
3.8 MVD and endothelial abnormalities in HF
Multiple studies have demonstrated that MVD and coronary microvascular endothelial dysfunction are highly prevalent and play critical pathogenic roles in HF, particularly in HFpEF. Even in HFpEF patients without obstructive coronary artery disease, approximately 81% exhibit coronary microvascular dysfunction (CMD), suggesting it as a potential therapeutic target (118). CMD can reduce coronary flow reserve and myocardial perfusion, thereby exacerbating myocardial ischemia, diastolic dysfunction, and oxidative stress, ultimately promoting HF progression and increasing cardiovascular risk (119). In HFpEF patients, coronary microvascular endothelial dysfunction involves both endothelium-dependent and endothelium-independent mechanisms, with the latter closely associated with impaired diastolic function and adverse outcomes (120, 121).
MVD impairs endothelial cell function, decreases nitric oxide (NO) bioavailability, and reduces coronary flow reserve, promoting left ventricular diastolic dysfunction and restrictive remodeling while compromising myocardial oxygen supply and preload reserve through multiple interacting pathways (122). Peripheral microvascular reactivity is also correlated with left ventricular structural and diastolic alterations, highlighting its role in early cardiac remodeling in HFpEF (123). Endothelial dysfunction in HF patients manifests as impaired vascular tone regulation, antioxidant capacity, and inflammatory modulation, which may be systemic or localized, and can serve as a prognostic marker while being partially modifiable by ACE inhibitors, statins, and regular exercise (124). Clinical studies further indicate that CMD restricts cardiac filling during exercise, reduces exercise capacity, and is closely linked to left ventricular diastolic dysfunction and myocardial ischemia (120). Additionally, HFpEF patients often present with arterial stiffening, impaired microvascular vasodilation, and abnormal venous capacitance, which collectively contribute to a complex pathophysiological network (125).
In summary, MVD and endothelial impairment constitute key pathogenic mechanisms in HF and HFpEF, promoting disease progression through myocardial ischemia, diastolic dysfunction, and adverse cardiac remodeling, while representing critical targets for early diagnosis, risk stratification, and individualized therapeutic intervention.
3.9 Epigenetic and transcriptomic regulation in HF
HF, particularly as a consequence of dilated cardiomyopathy (DCM), is intricately regulated by epigenetic and transcriptomic mechanisms. CFs, in response to injury, transition from a quiescent state to a highly collagen-secreting phenotype, thereby driving fibrosis and cardiac remodeling through excessive ECM deposition. This activation is precisely controlled by gene transcription, DNA methylation, histone modifications, chromatin remodeling, and intercellular signaling networks (126). Non-coding RNAs, including the lncRNA Wisper, interact with TIA1-related proteins to modulate pro-fibrotic enzyme expression, promoting fibrosis and adverse remodeling post-myocardial infarction, whereas antisense oligonucleotide-mediated silencing significantly alleviates fibrosis and cardiac dysfunction (83). Similarly, NAT10-mediated N4-acetylcytidine (ac4C) modification of mRNAs enhances the stability and translation efficiency of CD47 and ROCK2 transcripts, thereby facilitating cardiomyocyte hypertrophy, fibrosis, and inflammatory responses; inhibition of NAT10 or treatment with Remodelin effectively improves left ventricular structure and function (127). Epitranscriptomic enzymes and non-coding RNAs—including miRNAs, lncRNAs, and circRNAs—coordinate gene transcription and translation in cardiomyocytes and the cardiac immune microenvironment, influencing remodeling, diastolic dysfunction, inflammation, and fibrosis, and thus represent promising targets for diagnosis, prognosis, and therapeutic intervention in HF (128, 129).
Bromodomain and extra terminal domain (BET) family proteins, particularly BRD4, recognize histone acetylation marks and integrate super-enhancer and promoter regions of pro-fibrotic genes under cardiac stress, orchestrating NF-κB- and TGF-β-dependent fibrosis and contractile dysfunction while maintaining mitochondrial respiration and energy homeostasis under baseline conditions (130–132). Other epigenetic regulators, including DNA methyltransferases, protein methyltransferases, MLF1, HAND1, and Bmi1, modulate chromatin accessibility, enhancer-promoter looping, and mRNA splicing, thereby governing cardiomyocyte hypertrophy, fibrosis, calcium handling, and age-associated transcriptional programs (133–138). Environmental factors, such as bisphenol A and its analogs, can induce cardiomyocyte hypertrophy and alter DNA methylation patterns, illustrating the interplay between transcriptional and epigenetic mechanisms in cardiac toxicity (139). Multi-omics analyses reveal disease-specific DNA methylation and non-coding RNA expression patterns across HF patients of distinct etiologies, with epigenetic reprogramming and transcriptional dysregulation jointly dictating myocardial function, fibrosis, and remodeling (140, 141). Furthermore, stem cells and their derived cardiomyocytes, through optimized differentiation and maturation, can repair myocardial infarction-induced injury and improve cardiac function in HF patients (142).
Beyond individual epigenetic modifiers, emerging multi-omics studies have revealed coordinated epigenetic remodeling that regulates transcriptional plasticity in both cardiomyocytes and fibroblasts. Disease-specific DNA methylation signatures correlate with metabolic impairment and hypertrophy, whereas histone acetylation and m6A RNA modifications fine-tune gene networks that govern fibrosis, inflammation, and energy metabolism (143). Notably, pharmacological modulation of epigenetic pathways—such as inhibition of BRD4 or HDACs—has demonstrated the ability to reverse maladaptive gene expression, attenuate fibrosis, and improve ventricular function in preclinical models (144). These findings underscore epigenetic regulation as a mechanistically distinct yet clinically relevant avenue for precision therapy in both HFrEF and HFpEF (Table 1).
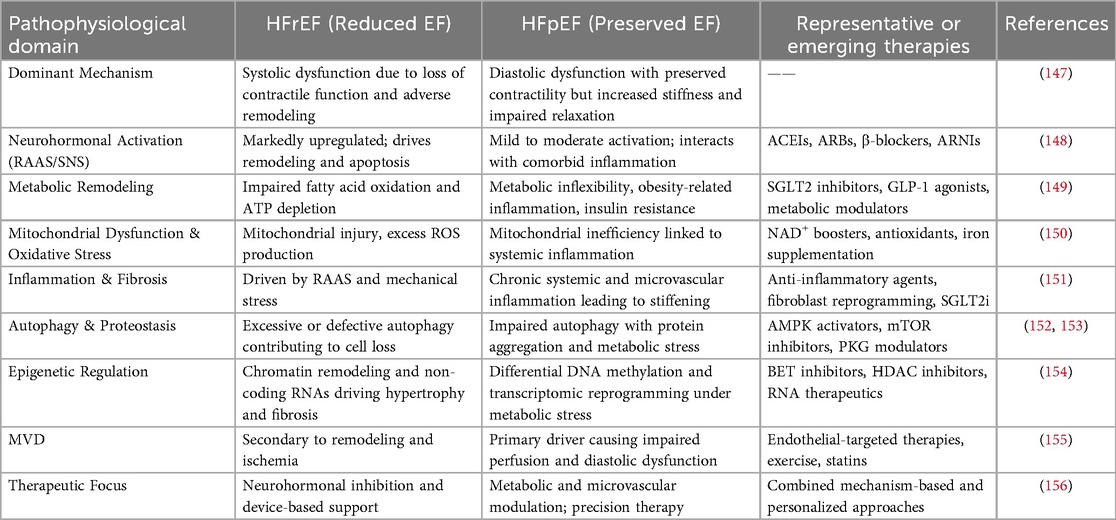
Table 1. Comparative summary of key mechanisms and therapeutic targets in HFrEF vs. HFpEF.
Collectively, HF pathogenesis is orchestrated by multilayered networks encompassing non-coding RNA regulation, epigenetic enzymes, chromatin remodeling, and epitranscriptomic modifications, providing a comprehensive molecular framework for mechanistic understanding and potential precision therapeutic strategies (145, 146).
3.10 Evidence quality and limitations of current data
The strength of available evidence across HF mechanisms and therapies varies substantially. Mechanistic insights are often derived from preclinical or single-center translational studies, which provide valuable biological understanding but are limited by small sample sizes, short durations, and lack of clinical endpoints (157). Conversely, large RCTs—such as those evaluating RAAS inhibition, β-blockers, and SGLT2 inhibitors—offer high-level evidence but typically target broad HF populations without molecular stratification (158). Conflicting findings among studies frequently reflect differences in experimental design, patient phenotype, and outcome measures. Future research should aim to integrate mechanistic precision with rigorous clinical trial methodology to close this translational gap.
4 Therapeutic advances in HF
HF results from complex interactions among neurohormonal activation, metabolic dysregulation, inflammation, fibrosis, and vascular dysfunction. Understanding these mechanisms has guided the development of targeted therapies. The following sections summarize current and emerging strategies in HF, including pharmacological, device-based, and regenerative approaches.
4.1 Pharmacological and metabolic therapies in HF
HF is a complex cardiovascular syndrome with multifactorial pathophysiology, in which neurohormonal dysregulation plays a pivotal role. In HFrEF, chronic activation of the SNS and the RAAS initially maintains hemodynamic stability but ultimately exacerbates cardiac workload, promotes myocardial remodeling, and accelerates disease progression (159, 160). RAAS and SNS hyperactivity are also implicated in the pathogenesis of cardiorenal syndrome and pulmonary arterial hypertension-related right ventricular failure, highlighting the potential clinical value of neurohormonal inhibition (161, 162).
Evidence-based pharmacotherapy for HFrEF includes ACE inhibitors (ACEIs), angiotensin II receptor blockers (ARBs), β-adrenergic blockers, and MRAs. ACEIs and ARBs improve cardiovascular and renal outcomes by suppressing RAAS activity, whereas β-blockers attenuate β1-adrenergic overstimulation, reducing apoptosis, inflammation, and adverse remodeling; their effects on myocyte enhancer factor 2 (MEF2) signaling and downstream gene networks further contribute to improved left ventricular function and survival (163–165). A comprehensive overview of the mechanisms, clinical evidence, and limitations of these pharmacological strategies is provided in Table 2. Combination therapy strategies, such as ACEI/ARB plus β-blocker and MRA, or the use of ARNIs, which simultaneously block RAAS and enhance natriuretic peptide (NP) signaling, have demonstrated significant reductions in cardiovascular mortality and HF hospitalization, with favorable tolerability in clinical practice (166–170). Nevertheless, real-world utilization of these guideline-directed therapies remains suboptimal, and polypharmacy may increase the risk of renal impairment and hyperkalemia, underscoring the need for individualized dosing and combination strategies (171, 172).
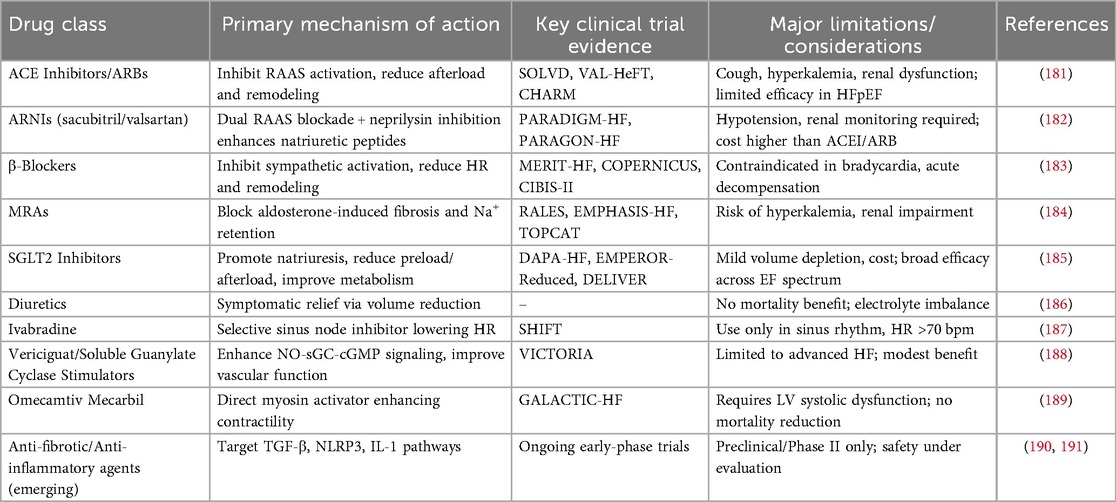
Table 2. Pharmacologic therapies in HF: mechanisms, evidence, and limitations.
For HFpEF and HFmrEF, conventional RAAS inhibitors and β-blockers show limited efficacy, and no definitive treatment exists (173–175). Recently, metabolic-targeted therapies, including sodium–glucose cotransporter 2 inhibitors (SGLT2i) and GLP-1 receptor agonists, have shown promise. SGLT2i exert cardiovascular benefits not only through renal glucose and sodium transport inhibition but also via modulation of cardiomyocyte ionic homeostasis, attenuation of inflammation and oxidative stress, thereby improving outcomes in both HFrEF and HFpEF patients (25, 176–178). GLP-1 receptor agonists ameliorate cardiac metabolic dysfunction, reduce myocardial hypertrophy and fibrosis, and improve cardiac function (179). Moreover, SGLT2i confer substantial cardiovascular benefits in elderly patients with type 2 diabetes mellitus, and metabolic interventions may also mitigate oxidative stress, neuroinflammation, and mitochondrial dysfunction, potentially improving cognitive decline, though clinical evidence remains limited (180).
Overall, HF pharmacotherapy is transitioning from simple suppression of maladaptive neurohormonal activation toward restoration of neuroendocrine balance and multi-targeted interventions. Future HF management may integrate conventional RAAS and β-blocker therapy with novel agents such as ARNIs, SGLT2i, and GLP-1 receptor agonists, along with metabolic interventions, to achieve cardiac protection, hemodynamic optimization, and metabolic homeostasis, providing a foundation for precision and individualized therapy (159).
4.2 Device-based and advanced interventional therapies in HF
HF patients continue to experience substantial residual risk despite guideline-directed medical therapy, including persistent symptoms, high rates of hospitalization, and mortality (192–194). These limitations have driven the rapid development of device-based interventions, which provide individualized therapies according to HF phenotype and severity. Key device therapies include cardiac resynchronization therapy (CRT), implantable cardioverter-defibrillators (ICD), mechanical circulatory support (MCS), and heart transplantation. An overview of these device-based and interventional approaches, including their indications, underlying mechanisms, and clinical limitations, is summarized in Table 3. CRT improves ventricular mechanical synchrony, enhancing left ventricular systolic function, reducing mitral regurgitation, promoting reverse remodeling, improving NYHA functional class and exercise capacity, and decreasing hospitalization and mortality (195–197). ICDs are primarily indicated for patients with moderate to severe HF or those at high risk, effectively preventing sudden cardiac death and ventricular arrhythmias, with outcomes influenced by NYHA class and cardiac function (198). For end-stage HF, MCS and heart transplantation provide definitive interventions, improving survival and quality of life, although device-related complications and limited availability remain challenges.
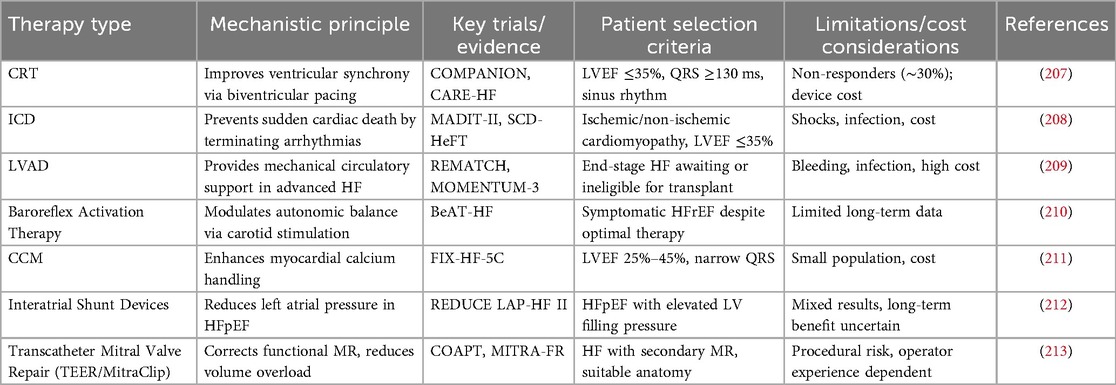
Table 3. Device-based and interventional therapies in HF.
Clinical studies indicate that approximately one-third of CRT recipients are non-responders, with key contributing factors including suboptimal atrioventricular (AV) and interventricular (VV) timing, non-left bundle branch block (non-LBBB), and electromechanical dyssynchrony (199, 200). To address this, emerging device-based sensor technologies and automated optimization algorithms have shown promise in improving long-term clinical outcomes and reducing HF-related rehospitalization, with some evidence suggesting superiority over conventional echocardiography-guided optimization (201, 202). Moreover, concomitant atrial fibrillation (AF) may attenuate CRT-mediated improvements in left ventricular function, and AF burden should be considered in therapeutic strategy and device selection (203). Cardiac contractility modulation (CCM) improves left ventricular systolic function and promotes reverse remodeling in patients with mild QRS prolongation, achieving effects comparable to CRT in this subgroup, though CRT remains more effective in patients with severe QRS prolongation (204).
In elderly HF patients (≥75 years), CRT-D therapy has been shown to reduce HF progression, mortality, and the risk of ventricular arrhythmias, with device reintervention rates comparable to younger populations. However, older patients are at higher risk of device-related complications, and current clinical trials provide limited evidence regarding quality of life and end-of-life care considerations in this population (205, 206). Real-world data also indicate that HF functional class significantly impacts device outcomes: in patients receiving ICD alone, NYHA class III/IV patients exhibit higher risk of HF-related events or death compared with class I/II, whereas CRT-D attenuates this disparity; additionally, patients with milder HF are more prone to ventricular arrhythmias (198).
In summary, device-based therapies play a pivotal role in HF management by improving ventricular synchrony, preventing sudden cardiac death, and providing advanced circulatory support. Integration of individualized indications, outcome evaluation, and emerging device technologies—including automated optimization algorithms, minimally invasive lead placement, and sensor-based monitoring—offers precise and personalized interventions, highlighting the potential of these strategies to reduce residual risk and optimize clinical outcomes across diverse HF populations.
4.3 Emerging biological and personalized therapies in HF
HF is a complex syndrome involving interactions among cardiomyocytes, fibroblasts, immune cells, and vascular endothelial cells. Myocardial fibrosis plays a central role in HF progression, and its extent and pattern are closely associated with disease development and prognosis, which can be assessed via imaging and circulating biomarkers (214). Traditional biomarkers such as BNP and NT-proBNP remain essential indicators of myocardial stress and volume overload, whereas multi-marker strategies integrating ST2, Galectin-3, and Copeptin provide deeper insight into HF pathophysiology and potential therapeutic targets (215–219).
Intervention strategies have expanded beyond conventional therapy. SGLT2 inhibitors have demonstrated reductions in HF hospitalization and mortality, potentially through modulation of myocardial inflammation and the STAT1-STING-mediated cellular senescence pathway (220, 221). Lifestyle interventions, including adherence to a Mediterranean diet, can improve metabolic profiles and reduce systemic inflammation (222). Cutting-edge biological approaches, such as targeting activated CFs, gene- and cell-based therapies, and recombinant human ACE2 administration, aim to repair or mitigate pathological cardiac remodeling (223, 224). Integrating multi-omics, circulating biomarkers, and clinical variables facilitates precision management, particularly in patients with HFpEF, renal comorbidities, or metabolic disturbances (225–228).
Clinical studies indicate that rapid initiation and titration of guideline-directed therapies in acute HF reduces short-term mortality and rehospitalization, whereas strict sodium restriction in chronic HF has not shown significant outcome improvement (229, 230). Mechanism-targeted interventions, including IL-1/IL-6 blockade, corticosteroids, and colchicine, show potential in acute HF, while triacylglycerol supplementation demonstrates efficacy in triglyceride deposit cardiomyopathy (231, 232). Combining genetic markers, advanced imaging, and artificial intelligence-assisted diagnostics can optimize patient stratification and individualized therapeutic strategies (233). Collectively, emerging biological and personalized therapies provide multidimensional approaches to improve outcomes and slow disease progression in HF patients.
5 Future perspectives and conclusions
Although significant advances have been made in delineating the pathophysiological mechanisms and therapeutic strategies for HF, several challenges persist. Current treatments predominantly target neurohormonal pathways (e.g., RAAS and SNS), yet they only partially mitigate maladaptive cardiac remodeling and fail to adequately address the phenotypic heterogeneity observed in HFpEF patients (160, 234). Furthermore, limited mechanistic understanding of metabolic dysregulation, inflammation, and MVD constrains the development of fully effective interventions. Emerging strategies integrating mechanistic insights and advanced technologies may overcome these limitations. Machine learning–based models have demonstrated superior predictive accuracy for hospitalization and mortality by combining clinical parameters with biomarker and imaging data (235). Multi-omics approaches, including transcriptomic, proteomic, and metabolomic profiling, have revealed novel pathways involved in cardiomyocyte energy metabolism, iron handling, and ECM remodeling, offering potential targets for personalized therapy (74, 236).
Building on these developments, future research should prioritize integrating omics-derived biomarkers with machine learning and systems biology tools to refine risk prediction models and uncover mechanistically distinct HF subgroups. Such integrative frameworks may enable dynamic, individualized monitoring and more accurate identification of patients who are likely to respond to specific therapeutic modalities (236). In addition, developing personalized therapeutic approaches for distinct HF phenotypes—including HFpEF subgroups defined by metabolic dysfunction, systemic inflammation, or microvascular disease—will be crucial. Aligning therapeutic selection with molecular signatures and comorbidity profiles may help overcome the historical limitations of “one-size-fits-all” treatment paradigms (237). Finally, future clinical trial designs should better incorporate real-world patient complexity, including frailty, multimorbidity, and sex-specific differences that influence treatment response and tolerability. Adaptive and phenotype-stratified trial frameworks may enhance the evaluation of both established and emerging therapies, while digital health tools and patient-reported outcomes can improve longitudinal assessment of functional status and quality of life (238).
Together, these directions underscore the importance of integrating high-resolution mechanistic data with patient-specific phenotyping to enable precision-guided interventions. Translational studies combining experimental models, omics-based biomarker discovery, and clinical validation are expected to refine risk stratification, optimize therapeutic selection, and ultimately improve functional outcomes and survival in HF.
Author contributions
FZ: Writing – original draft, Data curation, Investigation, Conceptualization, Writing – review & editing. XZ: Writing – review & editing, Data curation. JJ: Writing – review & editing, Data curation. XZ: Writing – review & editing, Investigation. CZ: Investigation, Writing – review & editing. YZ: Investigation, Writing – review & editing. JG: Writing – review & editing, Supervision, Formal analysis.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that this work was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bozkurt B, Coats A, Tsutsui H. Universal definition and classification of heart failure. J Card Fail. (2021) 27(4):387–413. doi: 10.1016/j.cardfail.2021.01.022
2. Heidenreich PA, Bozkurt B, Aguilar D, Allen LA, Byun JJ, Colvin MM, et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure: a report of the American college of cardiology/American heart association joint committee on clinical practice guidelines. Circulation. (2022) 145:e895–e1032. doi: 10.1161/CIR.0000000000001063
3. Maryam , Varghese TP, Tazneem B. Unraveling the complex pathophysiology of heart failure: insights into the role of renin-angiotensin-aldosterone system (RAAS) and sympathetic nervous system (SNS). Curr Probl Cardiol. (2024) 49:102411. doi: 10.1016/j.cpcardiol.2024.102411
4. Abdin A, Bohm M, Shahim B, Karlstrom P, Kulenthiran S, Skouri H, et al. Heart failure with preserved ejection fraction epidemiology, pathophysiology, diagnosis and treatment strategies. Int J Cardiol. (2024) 412:132304. doi: 10.1016/j.ijcard.2024.132304
5. Laborante R, Restivo A, Mele D, Di Francesco M, Ferreira JP, Vasques-Novoa F, et al. Device-based strategies for monitoring congestion and guideline-directed therapy in heart failure: the who, when and how of personalised care. Card Fail Rev. (2025) 11:e11. doi: 10.15420/cfr.2025.01
6. Castiglione V, Gentile F, Ghionzoli N, Chiriaco M, Panichella G, Aimo A, et al. Pathophysiological rationale and clinical evidence for neurohormonal modulation in heart failure with preserved ejection fraction. Card Fail Rev. (2023) 9:e09. doi: 10.15420/cfr.2022.23
7. Juillière Y, Venner C, Filippetti L, Popovic B, Huttin O, Selton-Suty C. Heart failure with preserved ejection fraction: a systemic disease linked to multiple comorbidities, targeting new therapeutic options. Arch Cardiovasc Dis. (2018) 111:766–81. doi: 10.1016/j.acvd.2018.04.007
8. Palaparthi EC, Padala T, Singamaneni R, Manaswini R, Kantula A, Aditya Reddy P, et al. Emerging therapeutic strategies for heart failure: a comprehensive review of novel pharmacological and molecular targets. Cureus. (2025) 17:e81573. doi: 10.7759/cureus.81573
9. Koontalay A, Botti M, Hutchinson A. Narrative synthesis of the effectiveness and characteristics of heart failure disease self-management support programmes. ESC Heart Fail. (2024) 11:1329–40. doi: 10.1002/ehf2.14701
10. Schwinger RHG. Pathophysiology of heart failure. Cardiovasc Diagn Ther. (2021) 11:263–76. doi: 10.21037/cdt-20-302
11. Jia G, Aroor AR, Hill MA, Sowers JR. Role of renin-angiotensin-aldosterone system activation in promoting cardiovascular fibrosis and stiffness. Hypertension. (2018) 72:537–48. doi: 10.1161/HYPERTENSIONAHA.118.11065
12. Volpe M, Tocci G, Pagannone E. Activation of the renin-angiotensin-aldosterone system in heart failure. Ital Heart J. (2005) 6(1):16S–23. Available online at: https://pubmed.ncbi.nlm.nih.gov/15945296/15945296
13. Harada K, Sugaya T, Murakami K, Yazaki Y, Komuro I. Angiotensin II type 1A receptor knockout mice display less left ventricular remodeling and improved survival after myocardial infarction. Circulation. (1999) 100:2093–9. doi: 10.1161/01.cir.100.20.2093
14. Wozniak M, Tyrankiewicz U, Drelicharz L, Skorka T, Jablonska M, Heinze-Paluchowska S, et al. The effect of the renin-angiotensin-aldosterone system inhibition on myocardial function in early and late phases of dilated cardiomyopathy in Tgaq*44 mice. Kardiol Pol. (2013) 71:730–7. doi: 10.5603/KP.2013.0161
15. Chen H, Yu B, Guo X, Hua H, Cui F, Guan Y, et al. Chronic intermittent hypobaric hypoxia decreases high blood pressure by stabilizing the vascular renin-angiotensin system in spontaneously hypertensive rats. Front Physiol. (2021) 12:639454. doi: 10.3389/fphys.2021.639454
16. Hawlitschek C, Brendel J, Gabriel P, Schierle K, Salameh A, Zimmer HG, et al. Antihypertensive and cardioprotective effects of different monotherapies and combination therapies in young spontaneously hypertensive rats—a pilot study. Saudi J Biol Sci. (2022) 29:339–45. doi: 10.1016/j.sjbs.2021.08.093
17. Yi X, Abas R, Raja Muhammad Rooshdi RAW, Yan J, Liu C, Yang C, et al. Time-restricted feeding reduced blood pressure and improved cardiac structure and function by regulating both circulating and local renin-angiotensin systems in spontaneously hypertensive rat model. PLoS One. (2025) 20:e0321078. doi: 10.1371/journal.pone.0321078
18. McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, et al. Dual angiotensin receptor and neprilysin inhibition as an alternative to angiotensin-converting enzyme inhibition in patients with chronic systolic heart failure: rationale for and design of the prospective comparison of ARNI with ACEI to determine impact on global mortality and morbidity in heart failure trial (PARADIGM-HF). Eur J Heart Fail. (2013) 15:1062–73. doi: 10.1093/eurjhf/hft052
19. Lind L, Riserus U, Elmstahl S, Arnlov J, Michaelsson K, Titova OE. Combinations of BMI and metabolic syndrome and the risk of myocardial infarction, stroke, and heart failure. Nutr Metab Cardiovasc Dis. (2025) 35:104102. doi: 10.1016/j.numecd.2025.104102
20. Menghoum N, Badii MC, Leroy M, Parra M, Roy C, Lejeune S, et al. Exploring the impact of metabolic comorbidities on epicardial adipose tissue in heart failure with preserved ejection fraction. Cardiovasc Diabetol. (2025) 24:134. doi: 10.1186/s12933-025-02688-7
21. Jhund PS, Talebi A, Henderson AD, Claggett BL, Vaduganathan M, Desai AS, et al. Mineralocorticoid receptor antagonists in heart failure: an individual patient level meta-analysis. Lancet. (2024) 404:1119–31. doi: 10.1016/S0140-6736(24)01733-1
22. Desai AS, Vaduganathan M, Claggett BL, Kulac IJ, Jhund PS, Cunningham J, et al. Finerenone in patients with a recent worsening heart failure event: the FINEARTS-HF trial. J Am Coll Cardiol. (2025) 85:106–16. doi: 10.1016/j.jacc.2024.09.004
23. Butt JH, Jhund PS, Henderson AD, Claggett BL, Desai AS, Viswanathan P, et al. Finerenone and new-onset diabetes in heart failure: a prespecified analysis of the FINEARTS-HF trial. Lancet Diabetes Endocrinol. (2025) 13:107–18. doi: 10.1016/S2213-8587(24)00309-7
24. Yeoh SE, Osmanska J, Petrie MC, Brooksbank KJM, Clark AL, Docherty KF, et al. Dapagliflozin vs. metolazone in heart failure resistant to loop diuretics. Eur Heart J. (2023) 44:2966–77. doi: 10.1093/eurheartj/ehad341
25. Hundertmark MJ, Adler A, Antoniades C, Coleman R, Griffin JL, Holman RR, et al. Assessment of cardiac energy metabolism, function, and physiology in patients with heart failure taking empagliflozin: the randomized, controlled EMPA-VISION trial. Circulation. (2023) 147:1654–69. doi: 10.1161/CIRCULATIONAHA.122.062021
26. Kitzman DW, Lewis GD, Pandey A, Borlaug BA, Sauer AJ, Litwin SE, et al. A novel controlled metabolic accelerator for the treatment of obesity-related heart failure with preserved ejection fraction: rationale and design of the Phase 2a HuMAIN trial. Eur J Heart Fail. (2024) 26:2013–24. doi: 10.1002/ejhf.3305
27. Mueller S, Winzer EB, Duvinage A, Gevaert AB, Edelmann F, Haller B, et al. Effect of high-intensity interval training, moderate continuous training, or guideline-based physical activity advice on peak oxygen consumption in patients with heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. (2021) 325:542–51. doi: 10.1001/jama.2020.26812
28. Besnier F, Labrunee M, Richard L, Faggianelli F, Kerros H, Soukarie L, et al. Short-term effects of a 3-week interval training program on heart rate variability in chronic heart failure. A randomised controlled trial. Ann Phys Rehabil Med. (2019) 62:321–8. doi: 10.1016/j.rehab.2019.06.013
29. Wang C, Xing J, Zhao B, Wang Y, Zhang L, Wang Y, et al. The effects of high-intensity interval training on exercise capacity and prognosis in heart failure and coronary artery disease: a systematic review and meta-analysis. Cardiovasc Ther. (2022) 2022:4273809. doi: 10.1155/2022/4273809
30. Wisloff U, Stoylen A, Loennechen JP, Bruvold M, Rognmo O, Haram PM, et al. Superior cardiovascular effect of aerobic interval training versus moderate continuous training in heart failure patients: a randomized study. Circulation. (2007) 115:3086–94. doi: 10.1161/CIRCULATIONAHA.106.675041
31. Yamamoto S, Okamura M, Akashi YJ, Tanaka S, Shimizu M, Tsuchikawa Y, et al. Impact of long-term exercise-based cardiac rehabilitation in patients with chronic heart failure- A systematic review and meta-analysis. Circ J. (2024) 88:1360–71. doi: 10.1253/circj.CJ-23-0820
32. Piotrowicz E, Pencina MJ, Opolski G, Zareba W, Banach M, Kowalik I, et al. Effects of a 9-week hybrid comprehensive telerehabilitation program on long-term outcomes in patients with heart failure: the telerehabilitation in heart failure patients (TELEREH-HF) randomized clinical trial. JAMA Cardiol. (2020) 5:300–8. doi: 10.1001/jamacardio.2019.5006
33. Gama F, Rocha B, Aguiar C, Strong C, Freitas P, Brizido C, et al. Exercise oscillatory ventilation improves heart failure prognostic scores. Heart Lung Circ. (2023) 32:949–57. doi: 10.1016/j.hlc.2023.04.291
34. Lewis GD, Malhotra R, Hernandez AF, McNulty SE, Smith A, Felker GM, et al. Effect of oral iron repletion on exercise capacity in patients with heart failure with reduced ejection fraction and iron deficiency: the IRONOUT HF randomized clinical trial. JAMA. (2017) 317:1958–66. doi: 10.1001/jama.2017.5427
35. Zhou B, Wang DD, Qiu Y, Airhart S, Liu Y, Stempien-Otero A, et al. Boosting NAD level suppresses inflammatory activation of PBMCs in heart failure. J Clin Invest. (2020) 130:6054–63. doi: 10.1172/JCI138538
36. Li H, Wang X, Zhang Y, Yang Y, Zhang JZ, Zhou B. SLC39A13 Regulates heart function via mitochondrial iron homeostasis maintenance. Circ Res. (2025) 137(6):e144–56. doi: 10.1161/CIRCRESAHA.125.326201
37. Zhang H, Jamieson KL, Grenier J, Nikhanj A, Tang Z, Wang F, et al. Myocardial iron deficiency and mitochondrial dysfunction in advanced heart failure in humans. J Am Heart Assoc. (2022) 11:e022853. doi: 10.1161/JAHA.121.022853
38. Hoes MF, Grote Beverborg N, Kijlstra JD, Kuipers J, Swinkels DW, Giepmans BNG, et al. Iron deficiency impairs contractility of human cardiomyocytes through decreased mitochondrial function. Eur J Heart Fail. (2018) 20:910–9. doi: 10.1002/ejhf.1154
39. Chiang S, Braidy N, Maleki S, Lal S, Richardson DR, Huang ML. Mechanisms of impaired mitochondrial homeostasis and NAD(+) metabolism in a model of mitochondrial heart disease exhibiting redox active iron accumulation. Redox Biol. (2021) 46:102038. doi: 10.1016/j.redox.2021.102038
40. Li S, Zhang J, Fu W, Cao J, Li Z, Tian X, et al. Mitochondrial transplantation rescues Ca(2+) homeostasis imbalance and myocardial hypertrophy in SLC25A3-related hypertrophic cardiomyopathy. Cell Rep. (2024) 43:115065. doi: 10.1016/j.celrep.2024.115065
41. Wang YT, Moura AK, Zuo R, Wang Z, Roudbari K, Hu JZ, et al. Defective lipid droplet biogenesis exacerbates oleic acid-induced cellular homeostasis disruption and ferroptosis in mouse cardiac endothelial cells. Cell Death Discov. (2025) 11:374. doi: 10.1038/s41420-025-02669-5
42. Angermann CE, Sehner S, Gerhardt LMS, Santos-Gallego CG, Requena-Ibanez JA, Zeller T, et al. Anaemia predicts iron homoeostasis dysregulation and modulates the response to empagliflozin in heart failure with reduced ejection fraction: the EMPATROPISM-FE trial. Eur Heart J. (2025) 46:1507–23. doi: 10.1093/eurheartj/ehae917
43. Charles-Edwards G, Amaral N, Sleigh A, Ayis S, Catibog N, McDonagh T, et al. Effect of iron isomaltoside on skeletal muscle energetics in patients with chronic heart failure and iron deficiency. Circulation. (2019) 139:2386–98. doi: 10.1161/CIRCULATIONAHA.118.038516
44. Packer M, Ferreira JP, Butler J, Filippatos G, Januzzi JL Jr., Gonzalez Maldonado S, et al. Reaffirmation of mechanistic proteomic signatures accompanying SGLT2 inhibition in patients with heart failure: a validation cohort of the EMPEROR program. J Am Coll Cardiol. (2024) 84:1979–94. doi: 10.1016/j.jacc.2024.07.013
45. van der Meer P, van der Wal HH, Melenovsky V. Mitochondrial function, skeletal muscle metabolism, and iron deficiency in heart failure. Circulation. (2019) 139:2399–402. doi: 10.1161/CIRCULATIONAHA.119.040134
46. He Z, Gao B, Deng Y, Wu J, Hu X, Qin Z. Associations between systemic immune-inflammation index and heart failure: a cross-sectional study. Medicine. (2024) 103:e40096. doi: 10.1097/MD.0000000000040096
47. Zheng H, Yin Z, Luo X, Zhou Y, Zhang F, Guo Z. Associations between systemic immunity-inflammation index and heart failure: evidence from the NHANES 1999–2018. Int J Cardiol. (2024) 395:131400. doi: 10.1016/j.ijcard.2023.131400
48. Heidenreich P. Inflammation and heart failure: therapeutic or diagnostic opportunity? J Am Coll Cardiol. (2017) 69:1286–7. doi: 10.1016/j.jacc.2017.01.013
49. Sanders-van Wijk S, Tromp J, Beussink-Nelson L, Hage C, Svedlund S, Saraste A, et al. Proteomic evaluation of the comorbidity-inflammation paradigm in heart failure with preserved ejection fraction: results from the PROMIS-HFpEF study. Circulation. (2020) 142:2029–44. doi: 10.1161/CIRCULATIONAHA.120.045810
50. de Boer RA. Myeloperoxidase in heart failure with preserved ejection fraction: a target against inflammation? JACC Heart Fail. (2023) 11:788–90. doi: 10.1016/j.jchf.2023.04.009
51. Schiattarella GG, Rodolico D, Hill JA. Metabolic inflammation in heart failure with preserved ejection fraction. Cardiovasc Res. (2021) 117:423–34. doi: 10.1093/cvr/cvaa217
52. Dong X, Jiang J, Lin Z, Wen R, Zou L, Luo T, et al. Nuanxinkang protects against ischemia/reperfusion-induced heart failure through regulating IKKbeta/IkappaBalpha/NF-kappaB-mediated macrophage polarization. Phytomedicine. (2022) 101:154093. doi: 10.1016/j.phymed.2022.154093
53. Benedetti R, Chianese U, Papulino C, Scisciola L, Cortese M, Formisano P, et al. Unlocking the power of empagliflozin: rescuing inflammation in hyperglycaemia-exposed human cardiomyocytes through comprehensive multi-level analysis. Eur J Heart Fail. (2025) 27:844–56. doi: 10.1002/ejhf.3566
54. Wu Q, Yao Q, Hu T, Yu J, Jiang K, Wan Y, et al. Dapagliflozin protects against chronic heart failure in mice by inhibiting macrophage-mediated inflammation, independent of SGLT2. Cell Rep Med. (2023) 4:101334. doi: 10.1016/j.xcrm.2023.101334
55. Guo W, Zhao L, Huang W, Chen J, Zhong T, Yan S, et al. Sodium-glucose cotransporter 2 inhibitors, inflammation, and heart failure: a two-sample Mendelian randomization study. Cardiovasc Diabetol. (2024) 23:118. doi: 10.1186/s12933-024-02210-5
56. Kittipibul V, Fudim M. Tackling inflammation in heart failure with preserved ejection fraction: resurrection of vagus nerve stimulation? J Am Heart Assoc. (2022) 11:e024481. doi: 10.1161/JAHA.121.024481
57. Stavrakis S, Elkholey K, Morris L, Niewiadomska M, Asad ZUA, Humphrey MB. Neuromodulation of inflammation to treat heart failure with preserved ejection fraction: a pilot randomized clinical trial. J Am Heart Assoc. (2022) 11:e023582. doi: 10.1161/JAHA.121.023582
58. Bhattarai U, Xu R, He X, Pan L, Niu Z, Wang D, et al. High selenium diet attenuates pressure overload-induced cardiopulmonary oxidative stress, inflammation, and heart failure. Redox Biol. (2024) 76:103325. doi: 10.1016/j.redox.2024.103325
59. Xing Z, Chen J, Yu T, Li X, Dong W, Peng C, et al. Aconitum carmichaelii Debx. Attenuates heart failure through inhibiting inflammation and abnormal vascular remodeling. Int J Mol Sci. (2023) 24:5838. doi: 10.3390/ijms24065838
60. Aimo A, Borrelli C, Vergaro G, Piepoli MF, Caterina AR, Mirizzi G, et al. Targeting mitochondrial dysfunction in chronic heart failure: current evidence and potential approaches. Curr Pharm Des. (2016) 22:4807–22. doi: 10.2174/1381612822666160701075027
61. Aimo A, Castiglione V, Borrelli C, Saccaro LF, Franzini M, Masi S, et al. Oxidative stress and inflammation in the evolution of heart failure: from pathophysiology to therapeutic strategies. Eur J Prev Cardiol. (2020) 27:494–510. doi: 10.1177/2047487319870344
62. D'Oria R, Schipani R, Leonardini A, Natalicchio A, Perrini S, Cignarelli A, et al. The role of oxidative stress in cardiac disease: from physiological response to injury factor. Oxid Med Cell Longev. (2020) 2020:5732956. doi: 10.1155/2020/5732956
63. Li T, Wang N, Yi D, Xiao Y, Li X, Shao B, et al. ROS-mediated ferroptosis and pyroptosis in cardiomyocytes: an update. Life Sci. (2025) 370:123565. doi: 10.1016/j.lfs.2025.123565
64. Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol. (2011) 301:H2181–2190. doi: 10.1152/ajpheart.00554.2011
65. Fu J, Su C, Ge Y, Ao Z, Xia L, Chen Y, et al. PDE4D Inhibition ameliorates cardiac hypertrophy and heart failure by activating mitophagy. Redox Biol. (2025) 81:103563. doi: 10.1016/j.redox.2025.103563
66. Ren H, Hu W, Jiang T, Yao Q, Qi Y, Huang K. Mechanical stress induced mitochondrial dysfunction in cardiovascular diseases: novel mechanisms and therapeutic targets. Biomed Pharmacother. (2024) 174:116545. doi: 10.1016/j.biopha.2024.116545
67. Wang J, Li S, Yu H, Gao D. Oxidative stress regulates cardiomyocyte energy metabolism through the IGF2BP2-dynamin2 signaling pathway. Biochem Biophys Res Commun. (2022) 624:134–40. doi: 10.1016/j.bbrc.2022.07.089
68. Ye X, Lin ZJ, Hong GH, Wang ZM, Dou RT, Lin JY, et al. Pyroptosis inhibitors MCC950 and VX-765 mitigate myocardial injury by alleviating oxidative stress, inflammation, and apoptosis in acute myocardial hypoxia. Exp Cell Res. (2024) 438:114061. doi: 10.1016/j.yexcr.2024.114061
69. Hou X, Hu G, Wang H, Yang Y, Sun Q, Bai X. Acot1 overexpression alleviates heart failure by inhibiting oxidative stress and cardiomyocyte apoptosis through the Keap1-Nrf2 pathway. Exp Anim. (2025). doi: 10.1538/expanim.24-0129
70. Wang Z, Zhang Y, Wang L, Yang C, Yang H. NBP relieves cardiac injury and reduce oxidative stress and cell apoptosis in heart failure mice by activating Nrf2/HO-1/Ca(2+)-SERCA2a axis. Evid Based Complement Alternat Med. (2022) 2022:7464893. doi: 10.1155/2022/7464893
71. Zhu K, Fan R, Cao Y, Yang W, Zhang Z, Zhou Q, et al. Glycyrrhizin attenuates myocardial ischemia reperfusion injury by suppressing inflammation, oxidative stress, and ferroptosis via the HMGB1-TLR4-GPX4 pathway. Exp Cell Res. (2024) 435:113912. doi: 10.1016/j.yexcr.2024.113912
72. Peoples JN, Saraf A, Ghazal N, Pham TT, Kwong JQ. Mitochondrial dysfunction and oxidative stress in heart disease. Exp Mol Med. (2019) 51:1–13. doi: 10.1038/s12276-019-0355-7
73. Czubryt MP, Hale TM. Cardiac fibrosis: pathobiology and therapeutic targets. Cell Signal. (2021) 85:110066. doi: 10.1016/j.cellsig.2021.110066
74. Ghazal R, Wang M, Liu D, Tschumperlin DJ, Pereira NL. Cardiac fibrosis in the multi-omics era: implications for heart failure. Circ Res. (2025) 136:773–802. doi: 10.1161/CIRCRESAHA.124.325402
75. Liu M, Lopez de Juan Abad B, Cheng K. Cardiac fibrosis: myofibroblast-mediated pathological regulation and drug delivery strategies. Adv Drug Deliv Rev. (2021) 173:504–19. doi: 10.1016/j.addr.2021.03.021
76. Andersen S, Nielsen-Kudsk JE, Vonk Noordegraaf A, de Man FS. Right ventricular fibrosis. Circulation. (2019) 139:269–85. doi: 10.1161/CIRCULATIONAHA.118.035326
77. Schimmel K, Ichimura K, Reddy S, Haddad F, Spiekerkoetter E. Cardiac fibrosis in the pressure overloaded left and right ventricle as a therapeutic target. Front Cardiovasc Med. (2022) 9:886553. doi: 10.3389/fcvm.2022.886553
78. Li R, Frangogiannis NG. Integrins in cardiac fibrosis. J Mol Cell Cardiol. (2022) 172:1–13. doi: 10.1016/j.yjmcc.2022.07.006
79. Yao Y, Hu C, Song Q, Li Y, Da X, Yu Y, et al. ADAMTS16 Activates latent TGF-beta, accentuating fibrosis and dysfunction of the pressure-overloaded heart. Cardiovasc Res. (2020) 116:956–69. doi: 10.1093/cvr/cvz187
80. Andenaes K, Lunde IG, Mohammadzadeh N, Dahl CP, Aronsen JM, Strand ME, et al. The extracellular matrix proteoglycan fibromodulin is upregulated in clinical and experimental heart failure and affects cardiac remodeling. PLoS One. (2018) 13:e0201422. doi: 10.1371/journal.pone.0201422
81. Deng B, Zhang Y, Zhu C, Wang Y, Weatherford E, Xu B, et al. Divergent actions of myofibroblast and myocyte beta(2)-Adrenoceptor in heart failure and fibrotic remodeling. Circ Res. (2023) 132:106–8. doi: 10.1161/CIRCRESAHA.122.321816
82. Frangogiannis NG. Cardiac fibrosis: cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol Aspects Med. (2019) 65:70–99. doi: 10.1016/j.mam.2018.07.001
83. Micheletti R, Plaisance I, Abraham BJ, Sarre A, Ting CC, Alexanian M, et al. The long noncoding RNA Wisper controls cardiac fibrosis and remodeling. Sci Transl Med. (2017) 9:eaai9118. doi: 10.1126/scitranslmed.aai9118
84. Li L, Zhao Q, Kong W. Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol. (2018) 68–69:490–506. doi: 10.1016/j.matbio.2018.01.013
85. Segura AM, Frazier OH, Buja LM. Fibrosis and heart failure. Heart Fail Rev. (2014) 19:173–85. doi: 10.1007/s10741-012-9365-4
86. Farris SD, Don C, Helterline D, Costa C, Plummer T, Steffes S, et al. Cell-specific pathways supporting persistent fibrosis in heart failure. J Am Coll Cardiol. (2017) 70:344–54. doi: 10.1016/j.jacc.2017.05.040
87. Pandey AK, Bhatt DL, Pandey A, Marx N, Cosentino F, Pandey A, et al. Mechanisms of benefits of sodium-glucose cotransporter 2 inhibitors in heart failure with preserved ejection fraction. Eur Heart J. (2023) 44:3640–51. doi: 10.1093/eurheartj/ehad389
88. Travers JG, Wennersten SA, Pena B, Bagchi RA, Smith HE, Hirsch RA, et al. HDAC inhibition reverses preexisting diastolic dysfunction and blocks covert extracellular matrix remodeling. Circulation. (2021) 143:1874–90. doi: 10.1161/CIRCULATIONAHA.120.046462
89. Frangogiannis NG. Cardiac fibrosis. Cardiovasc Res. (2021) 117:1450–88. doi: 10.1093/cvr/cvaa324
90. Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac fibrosis: the fibroblast awakens. Circ Res. (2016) 118:1021–40. doi: 10.1161/CIRCRESAHA.115.306565
91. Valiente-Alandi I, Schafer AE, Blaxall BC. Extracellular matrix-mediated cellular communication in the heart. J Mol Cell Cardiol. (2016) 91:228–37. doi: 10.1016/j.yjmcc.2016.01.011
92. Bielawska M, Warszynska M, Stefanska M, Blyszczuk P. Autophagy in heart failure: insights into mechanisms and therapeutic implications. J Cardiovasc Dev Dis. (2023) 10:352. doi: 10.3390/jcdd10080352
93. Li J, Zhang D, Wiersma M, Brundel B. Role of autophagy in proteostasis: friend and foe in cardiac diseases. Cells. (2018) 7:279. doi: 10.3390/cells7120279
94. Nah J, Zablocki D, Sadoshima J. The role of autophagic cell death in cardiac disease. J Mol Cell Cardiol. (2022) 173:16–24. doi: 10.1016/j.yjmcc.2022.08.362
95. Tang Y, Xu W, Liu Y, Zhou J, Cui K, Chen Y. Autophagy protects mitochondrial health in heart failure. Heart Fail Rev. (2024) 29:113–23. doi: 10.1007/s10741-023-10354-x
96. Oeing CU, Mishra S, Dunkerly-Eyring BL, Ranek MJ. Targeting protein kinase G to treat cardiac proteotoxicity. Front Physiol. (2020) 11:858. doi: 10.3389/fphys.2020.00858
97. Maroli G, Schanzer A, Gunther S, Garcia-Gonzalez C, Rupp S, Schlierbach H, et al. Inhibition of autophagy prevents cardiac dysfunction at early stages of cardiomyopathy in Bag3-deficient hearts. J Mol Cell Cardiol. (2024) 193:53–66. doi: 10.1016/j.yjmcc.2024.06.001
98. Delalat S, Sultana I, Osman H, Sieme M, Zhazykbayeva S, Herwig M, et al. Dysregulated inflammation, oxidative stress, and protein quality control in diabetic HFpEF: unraveling mechanisms and therapeutic targets. Cardiovasc Diabetol. (2025) 24:211. doi: 10.1186/s12933-025-02734-4
99. Cui T, Lai Y, Janicki JS, Wang X. Nuclear factor erythroid-2 related factor 2 (Nrf2)-mediated protein quality control in cardiomyocytes. Front Biosci. (2016) 21:192–202. doi: 10.2741/4384
100. Kobak KA, Zarzycka W, King CJ, Borowik AK, Peelor FF 3rd, Baehr LM, et al. Proteostatic imbalance drives the pathogenesis and age-related exacerbation of heart failure with preserved ejection fraction. JACC Basic Transl Sci. (2025) 10:475–97. doi: 10.1016/j.jacbts.2024.11.006
101. Zarzycka W, Kobak KA, King CJ, Peelor FF, Miller BF, Chiao YA. Hyperactive mTORC1/4EBP1 signaling dysregulates proteostasis and accelerates cardiac aging. bioRxiv. (2024). doi: 10.1101/2024.05.13.594044
102. Hartman H, Uy G, Uchida K, Scarborough EA, Yang Y, Barr E, et al. ROR2 drives right ventricular heart failure via disruption of proteostasis. bioRxiv. (2025). doi: 10.1101/2025.02.01.635961
103. McLendon JM, Zhang X, Stein CS, Baehr LM, Bodine SC, Boudreau RL. A specialized centrosome-proteasome axis mediates proteostasis and influences cardiac stress through Txlnb. bioRxiv. (2024). doi: 10.1101/2024.02.12.580020
104. McLendon JM, Zhang X, Stein CS, Baehr LM, Bodine SC, Boudreau RL. Gain and loss of the centrosomal protein taxilin-beta influences cardiac proteostasis and stress. J Mol Cell Cardiol. (2025) 201:56–69. doi: 10.1016/j.yjmcc.2025.02.008
105. Neu M, Deshpande A, Borlepawar A, Hammer E, Alameldeen A, Vocking P, et al. TRIM24 Regulates chromatin remodeling and calcium dynamics in cardiomyocytes. Cell Commun Signal. (2025) 23:312. doi: 10.1186/s12964-025-02323-8
106. Fanjul V, Jorge I, Camafeita E, Macias A, Gonzalez-Gomez C, Barettino A, et al. Identification of common cardiometabolic alterations and deregulated pathways in mouse and pig models of aging. Aging Cell. (2020) 19:e13203. doi: 10.1111/acel.13203
107. Narayanan G, Halim A, Hu A, Avin KG, Lu T, Zehnder D, et al. Molecular phenotyping and mechanisms of myocardial fibrosis in advanced chronic kidney disease. Kidney360. (2023) 4:1562–79. doi: 10.34067/KID.0000000000000276
108. Olkowski AA, Wojnarowicz C, Laarveld B. Pathophysiology and pathological remodelling associated with dilated cardiomyopathy in broiler chickens predisposed to heart pump failure. Avian Pathol. (2020) 49:428–39. doi: 10.1080/03079457.2020.1757620
109. Li A, Gao M, Liu B, Qin Y, Chen L, Liu H, et al. Mitochondrial autophagy: molecular mechanisms and implications for cardiovascular disease. Cell Death Dis. (2022) 13:444. doi: 10.1038/s41419-022-04906-6
110. Cetrullo S, D'Adamo S, Tantini B, Borzi RM, Flamigni F. mTOR, AMPK, and Sirt1: key players in metabolic stress management. Crit Rev Eukaryot Gene Expr. (2015) 25:59–75. doi: 10.1615/critreveukaryotgeneexpr.2015012975
111. Maejima Y, Titus AS, Zablocki D, Sadoshima J. Recent progress regarding the role of autophagy in cardiac disease. Cardiovasc Res. (2025):cvaf203. [Online ahead of print]. doi: 10.1093/cvr/cvaf203
112. Liao M, Xie Q, Zhao Y, Yang C, Lin C, Wang G, et al. Main active components of Si-Miao-Yong-An decoction (SMYAD) attenuate autophagy and apoptosis via the PDE5A-AKT and TLR4-NOX4 pathways in isoproterenol (ISO)-induced heart failure models. Pharmacol Res. (2022) 176:106077. doi: 10.1016/j.phrs.2022.106077
113. Wang D, Lv L, Xu Y, Jiang K, Chen F, Qian J, et al. Cardioprotection of Panax Notoginseng saponins against acute myocardial infarction and heart failure through inducing autophagy. Biomed Pharmacother. (2021) 136:111287. doi: 10.1016/j.biopha.2021.111287
114. Zhang X, Wang Q, Wang X, Chen X, Shao M, Zhang Q, et al. Tanshinone IIA protects against heart failure post-myocardial infarction via AMPKs/mTOR-dependent autophagy pathway. Biomed Pharmacother. (2019) 112:108599. doi: 10.1016/j.biopha.2019.108599
115. Gao G, Chen W, Yan M, Liu J, Luo H, Wang C, et al. Rapamycin regulates the balance between cardiomyocyte apoptosis and autophagy in chronic heart failure by inhibiting mTOR signaling. Int J Mol Med. (2020) 45:195–209. doi: 10.3892/ijmm.2019.4407
116. Corsetti G, Pasini E, Romano C, Chen-Scarabelli C, Scarabelli TM, Flati V, et al. How can malnutrition affect autophagy in chronic heart failure? Focus and perspectives. Int J Mol Sci. (2021) 22:3332. doi: 10.3390/ijms22073332
117. Hu L, Gao D, Lv H, Lian L, Wang M, Wang Y, et al. Finding new targets for the treatment of heart failure: endoplasmic reticulum stress and autophagy. J Cardiovasc Transl Res. (2023) 16:1349–56. doi: 10.1007/s12265-023-10410-9
118. Rush CJ, Berry C, Oldroyd KG, Rocchiccioli JP, Lindsay MM, Touyz RM, et al. Prevalence of coronary artery disease and coronary microvascular dysfunction in patients with heart failure with preserved ejection fraction. JAMA Cardiol. (2021) 6:1130–43. doi: 10.1001/jamacardio.2021.1825
119. Singh A, Ashraf S, Irfan H, Venjhraj F, Verma A, Shaukat A, et al. Heart failure and microvascular dysfunction: an in-depth review of mechanisms, diagnostic strategies, and innovative therapies. Ann Med Surg. (2025) 87:616–26. doi: 10.1097/MS9.0000000000002971
120. Ahmad A, Corban MT, Toya T, Verbrugge FH, Sara JD, Lerman LO, et al. Coronary microvascular dysfunction is associated with exertional haemodynamic abnormalities in patients with heart failure with preserved ejection fraction. Eur J Heart Fail. (2021) 23:765–72. doi: 10.1002/ejhf.2010
121. Yang JH, Obokata M, Reddy YNV, Redfield MM, Lerman A, Borlaug BA. Endothelium-dependent and independent coronary microvascular dysfunction in patients with heart failure with preserved ejection fraction. Eur J Heart Fail. (2020) 22:432–41. doi: 10.1002/ejhf.1671
122. Gamrat A, Surdacki MA, Chyrchel B, Surdacki A. Endothelial dysfunction: a contributor to adverse cardiovascular remodeling and heart failure development in type 2 diabetes beyond accelerated atherogenesis. J Clin Med. (2020) 9:2090. doi: 10.3390/jcm9072090
123. Cauwenberghs N, Godderis S, Sabovcik F, Cornelissen V, Kuznetsova T. Subclinical heart remodeling and dysfunction in relation to peripheral endothelial dysfunction: a general population study. Microcirculation. (2021) 28:e12731. doi: 10.1111/micc.12731
124. Shantsila E, Wrigley BJ, Blann AD, Gill PS, Lip GY. A contemporary view on endothelial function in heart failure. Eur J Heart Fail. (2012) 14:873–81. doi: 10.1093/eurjhf/hfs066
125. Balmain S, Padmanabhan N, Ferrell WR, Morton JJ, McMurray JJ. Differences in arterial compliance, microvascular function and venous capacitance between patients with heart failure and either preserved or reduced left ventricular systolic function. Eur J Heart Fail. (2007) 9:865–71. doi: 10.1016/j.ejheart.2007.06.003
126. Aguado-Alvaro LP, Garitano N, Pelacho B. Fibroblast diversity and epigenetic regulation in cardiac fibrosis. Int J Mol Sci. (2024) 25:6004. doi: 10.3390/ijms25116004
127. Shi J, Yang C, Zhang J, Zhao K, Li P, Kong C, et al. NAT10 is involved in cardiac remodeling through ac4C-mediated transcriptomic regulation. Circ Res. (2023) 133:989–1002. doi: 10.1161/CIRCRESAHA.122.322244
128. Gomes CPC, Schroen B, Kuster GM, Robinson EL, Ford K, Squire IB, et al. Regulatory RNAs in heart failure. Circulation. (2020) 141:313–28. doi: 10.1161/CIRCULATIONAHA.119.042474
129. Yang YL, Li XW, Chen HB, Tang QD, Li YH, Xu JY, et al. Single-cell transcriptomics reveals writers of RNA modification-mediated immune microenvironment and cardiac resident Macro-MYL2 macrophages in heart failure. BMC Cardiovasc Disord. (2024) 24:432. doi: 10.1186/s12872-024-04080-x
130. Ijaz T, Burke MA. BET protein-mediated transcriptional regulation in heart failure. Int J Mol Sci. (2021) 22:6059. doi: 10.3390/ijms22116059
131. Kim SY, Zhang X, Schiattarella GG, Altamirano F, Ramos TAR, French KM, et al. Epigenetic reader BRD4 (bromodomain-containing protein 4) governs nucleus-encoded mitochondrial transcriptome to regulate cardiac function. Circulation. (2020) 142:2356–70. doi: 10.1161/CIRCULATIONAHA.120.047239
132. Stratton MS, Bagchi RA, Felisbino MB, Hirsch RA, Smith HE, Riching AS, et al. Dynamic chromatin targeting of BRD4 stimulates cardiac fibroblast activation. Circ Res. (2019) 125:662–77. doi: 10.1161/CIRCRESAHA.119.315125
133. Feng Y, Cai L, Hong W, Zhang C, Tan N, Wang M, et al. Rewiring of 3D chromatin topology orchestrates transcriptional reprogramming and the development of human dilated cardiomyopathy. Circulation. (2022) 145:1663–83. doi: 10.1161/CIRCULATIONAHA.121.055781
134. Gi WT, Haas J, Sedaghat-Hamedani F, Kayvanpour E, Tappu R, Lehmann DH, et al. Epigenetic regulation of alternative mRNA splicing in dilated cardiomyopathy. J Clin Med. (2020) 9:1499. doi: 10.3390/jcm9051499
135. Gonzalez-Valdes I, Hidalgo I, Bujarrabal A, Lara-Pezzi E, Padron-Barthe L, Garcia-Pavia P, et al. Bmi1 limits dilated cardiomyopathy and heart failure by inhibiting cardiac senescence. Nat Commun. (2015) 6:6473. doi: 10.1038/ncomms7473
136. Lv J, Chen Q, Wang J, Guo N, Fang Y, Guo Q, et al. Downregulation of MLF1 safeguards cardiomyocytes against senescence-associated chromatin opening. Nucleic Acids Res. (2025) 53:gkae1176. doi: 10.1093/nar/gkae1176
137. Marques FZ, Chu PY, Ziemann M, Kaspi A, Kiriazis H, Du XJ, et al. Age-related differential structural and transcriptomic responses in the hypertensive heart. Front Physiol. (2018) 9:817. doi: 10.3389/fphys.2018.00817
138. Szulik MW, Davis K, Bakhtina A, Azarcon P, Bia R, Horiuchi E, et al. Transcriptional regulation by methyltransferases and their role in the heart: highlighting novel emerging functionality. Am J Physiol Heart Circ Physiol. (2020) 319:H847–65. doi: 10.1152/ajpheart.00382.2020
139. Cheng MD, Li CL, Pei XY, Zhang YF, Jia DD, Zuo YB, et al. Integrative analysis of DNA methylome and transcriptome reveals epigenetic regulation of bisphenols-induced cardiomyocyte hypertrophy. Ecotoxicol Environ Saf. (2023) 263:115391. doi: 10.1016/j.ecoenv.2023.115391
140. Glezeva N, Moran B, Collier P, Moravec CS, Phelan D, Donnellan E, et al. Targeted DNA methylation profiling of human cardiac tissue reveals novel epigenetic traits and gene deregulation across different heart failure patient subtypes. Circ Heart Fail. (2019) 12:e005765. doi: 10.1161/CIRCHEARTFAILURE.118.005765
141. Meder B, Haas J, Sedaghat-Hamedani F, Kayvanpour E, Frese K, Lai A, et al. Epigenome-wide association study identifies cardiac gene patterning and a novel class of biomarkers for heart failure. Circulation. (2017) 136:1528–44. doi: 10.1161/CIRCULATIONAHA.117.027355
142. Jackson AO, Rahman GA, Yin K, Long S. Enhancing matured stem-cardiac cell generation and transplantation: a novel strategy for heart failure therapy. J Cardiovasc Transl Res. (2021) 14:556–72. doi: 10.1007/s12265-020-10085-6
143. Junxi L, Mocun Y, Boda Z. Epigenetic and epitranscriptomic regulation of cardiac metabolism in aging and disease. J Cardiovasc Aging. (2025) 5:15. doi: 10.20517/jca.2025.06
144. Brand CS, Lighthouse JK, Trembley MA. Protective transcriptional mechanisms in cardiomyocytes and cardiac fibroblasts. J Mol Cell Cardiol. (2019) 132:1–12. doi: 10.1016/j.yjmcc.2019.04.023
145. Shao X, Zhang X, Yang L, Zhang R, Zhu R, Feng R. Integrated analysis of mRNA and microRNA expression profiles reveals differential transcriptome signature in ischaemic and dilated cardiomyopathy induced heart failure. Epigenetics. (2021) 16:917–32. doi: 10.1080/15592294.2020.1827721
146. Piran S, Liu P, Morales A, Hershberger RE. Where genome meets phenome: rationale for integrating genetic and protein biomarkers in the diagnosis and management of dilated cardiomyopathy and heart failure. J Am Coll Cardiol. (2012) 60:283–9. doi: 10.1016/j.jacc.2012.05.005
147. Maharaj R. Diastolic dysfunction and heart failure with a preserved ejection fraction: relevance in critical illness and anaesthesia. J Saudi Heart Assoc. (2012) 24:99–121. doi: 10.1016/j.jsha.2012.01.004
148. Manolis AA, Manolis TA, Manolis AS. Neurohumoral activation in heart failure. Int J Mol Sci. (2023) 24:15472. doi: 10.3390/ijms242015472
149. Checa-Ros A, Okojie OJ, D'Marco L. SGLT2 inhibitors: multifaceted therapeutic agents in cardiometabolic and renal diseases. Metabolites. (2025) 15:536. doi: 10.3390/metabo15080536
150. Xu X, Pang Y, Fan X. Mitochondria in oxidative stress, inflammation and aging: from mechanisms to therapeutic advances. Signal Transduct Target Ther. (2025) 10:190. doi: 10.1038/s41392-025-02253-4
151. Rolski F, Maczewski M. Cardiac fibrosis: mechanistic discoveries linked to SGLT2 inhibitors. Pharmaceuticals. (2025) 18:313. doi: 10.3390/ph18030313
152. Ma C, Liu Y, Fu Z. Implications of endoplasmic reticulum stress and autophagy in aging and cardiovascular diseases. Front Pharmacol. (2024) 15:1413853. doi: 10.3389/fphar.2024.1413853
153. Numata G, Takimoto E. Cyclic GMP and PKG signaling in heart failure. Front Pharmacol. (2022) 13:792798. doi: 10.3389/fphar.2022.792798
154. Di Salvo TG, Haldar SM. Epigenetic mechanisms in heart failure pathogenesis. Circ Heart Fail. (2014) 7:850–63. doi: 10.1161/CIRCHEARTFAILURE.114.001193
155. Aldujeli A, Tsai TY, Haq A, Tatarunas V, Knokneris A, Briedis K, et al. Impact of coronary microvascular dysfunction on functional left ventricular remodeling and diastolic dysfunction. J Am Heart Assoc. (2024) 13:e033596. doi: 10.1161/JAHA.123.033596
156. Ansari RA, Senapati SG, Ahluwalia V, Panjwani GAR, Kaur A, Yerrapragada G, et al. Artificial intelligence-guided neuromodulation in heart failure with preserved and reduced ejection fraction: mechanisms, evidence, and future directions. J Cardiovasc Dev Dis. (2025) 12:314. doi: 10.3390/jcdd12080314
157. Mann DL, Felker GM. Mechanisms and models in heart failure: a translational approach. Circ Res. (2021) 128:1435–50. doi: 10.1161/CIRCRESAHA.121.318158
158. Chow SL, Maisel AS, Anand I, Bozkurt B, de Boer RA, Felker GM, et al. Role of biomarkers for the prevention, assessment, and management of heart failure: a scientific statement from the American heart association. Circulation. (2017) 135:e1054–91. doi: 10.1161/CIR.0000000000000490
159. Fu S, Ping P, Wang F, Luo L. Synthesis, secretion, function, metabolism and application of natriuretic peptides in heart failure. J Biol Eng. (2018) 12:2. doi: 10.1186/s13036-017-0093-0
160. Hartupee J, Mann DL. Neurohormonal activation in heart failure with reduced ejection fraction. Nat Rev Cardiol. (2017) 14:30–8. doi: 10.1038/nrcardio.2016.163
161. Ameri P, Bertero E, Meliota G, Cheli M, Canepa M, Brunelli C, et al. Neurohormonal activation and pharmacological inhibition in pulmonary arterial hypertension and related right ventricular failure. Heart Fail Rev. (2016) 21:539–47. doi: 10.1007/s10741-016-9566-3
162. Onuigbo MA. RAAS Inhibition and cardiorenal syndrome. Curr Hypertens Rev. (2014) 10:107–11. doi: 10.2174/1573402111666141231144228
163. Cruickshank JM. Beta-blockers and heart failure. Indian Heart J. (2010) 62:101–10. Available online at: https://pubmed.ncbi.nlm.nih.gov/21180298/21180298
164. Metra M, Cas LD, di Lenarda A, Poole-Wilson P. Beta-blockers in heart failure: are pharmacological differences clinically important? Heart Fail Rev. (2004) 9:123–30. doi: 10.1023/B:HREV.0000046367.99002.a4
165. Tobin SW, Hashemi S, Dadson K, Turdi S, Ebrahimian K, Zhao J, et al. Heart failure and MEF2 transcriptome dynamics in response to beta-blockers. Sci Rep. (2017) 7:4476. doi: 10.1038/s41598-017-04762-x
166. Fu S, Chang Z, Luo L, Deng J. Therapeutic progress and knowledge basis on the natriuretic peptide system in heart failure. Curr Top Med Chem. (2019) 19:1850–66. doi: 10.2174/1568026619666190826163536
167. Khder Y, Shi V, McMurray JJV, Lefkowitz MP. Sacubitril/valsartan (LCZ696) in heart failure. Handb Exp Pharmacol. (2017) 243:133–65. doi: 10.1007/164_2016_77
168. Nielsen PM, Grimm D, Wehland M, Simonsen U, Kruger M. The combination of valsartan and sacubitril in the treatment of hypertension and heart failure—an update. Basic Clin Pharmacol Toxicol. (2018) 122:9–18. doi: 10.1111/bcpt.12912
169. Volterrani M, Iellamo F, Senni M, Piepoli MF. Therapeutic options of angiotensin receptor neprilysin inhibitors (ARNis) in chronic heart failure with reduced ejection fraction: beyond RAAS and sympathetic nervous system inhibition. Int J Cardiol. (2017) 226:132–5. doi: 10.1016/j.ijcard.2016.04.180
170. von Lueder TG, Atar D, Krum H. Current role of neprilysin inhibitors in hypertension and heart failure. Pharmacol Ther. (2014) 144:41–9. doi: 10.1016/j.pharmthera.2014.05.002
171. Rossignol P, Zannad F, Pitt B. Writing group of 10th global cardio vascular clinical trialist forum held on December 6th-7th in Paris F. Time to retrieve the best benefits from renin angiotensin aldosterone system (RAAS) inhibition in heart failure patients with reduced ejection fraction: lessons from randomized controlled trials and registries. Int J Cardiol. (2014) 177:731–3. doi: 10.1016/j.ijcard.2014.11.004
172. Vaduganathan M, Fonarow GC, Greene SJ, Devore AD, Albert NM, Duffy CI, et al. Treatment persistence of renin-angiotensin-aldosterone-system inhibitors over time in heart failure with reduced ejection fraction. J Card Fail. (2022) 28:191–201. doi: 10.1016/j.cardfail.2021.08.008
173. Mascolo A, di Mauro G, Cappetta D, De Angelis A, Torella D, Urbanek K, et al. Current and future therapeutic perspective in chronic heart failure. Pharmacol Res. (2022) 175:106035. doi: 10.1016/j.phrs.2021.106035
174. Nochioka K, Sakata Y, Shimokawa H. Combination therapy of renin angiotensin system inhibitors and beta-blockers in patients with heart failure. Adv Exp Med Biol. (2018) 1067:17–30. doi: 10.1007/5584_2018_179
175. Yamamoto K. beta-Blocker therapy in heart failure with preserved ejection fraction: importance of dose and duration. J Cardiol. (2015) 66:189–94. doi: 10.1016/j.jjcc.2015.02.004
176. Billing AM, Kim YC, Gullaksen S, Schrage B, Raabe J, Hutzfeldt A, et al. Metabolic communication by SGLT2 inhibition. Circulation. (2024) 149:860–84. doi: 10.1161/CIRCULATIONAHA.123.065517
177. Chen S, Coronel R, Hollmann MW, Weber NC, Zuurbier CJ. Direct cardiac effects of SGLT2 inhibitors. Cardiovasc Diabetol. (2022) 21:45. doi: 10.1186/s12933-022-01480-1
178. Scheen AJ, Bonnet F. Efficacy and safety profile of SGLT2 inhibitors in the elderly: how is the benefit/risk balance? Diabetes Metab. (2023) 49:101419. doi: 10.1016/j.diabet.2023.101419
179. Withaar C, Meems LMG, Markousis-Mavrogenis G, Boogerd CJ, Sillje HHW, Schouten EM, et al. The effects of liraglutide and dapagliflozin on cardiac function and structure in a multi-hit mouse model of heart failure with preserved ejection fraction. Cardiovasc Res. (2021) 117:2108–24. doi: 10.1093/cvr/cvaa256
180. Rizzo MR, Di Meo I, Polito R, Auriemma MC, Gambardella A, di Mauro G, et al. Cognitive impairment and type 2 diabetes mellitus: focus of SGLT2 inhibitors treatment. Pharmacol Res. (2022) 176:106062. doi: 10.1016/j.phrs.2022.106062
181. Nandave M. Role of ACE inhibitors and angiotensin receptor blockers in acute heart failure. In: Nandave M, editor. Angiotensin-converting Enzyme Inhibitors vs Angiotensin Receptor Blockers: A Critical Analysis of Antihypertensive Strategies: A Machine-Generated Literature Overview. Singapore: Springer Nature Singapore (2024). p. 277–327.
182. Greenberg B. Angiotensin receptor-neprilysin inhibition (ARNI) in heart failure. Int J Heart Fail. (2020) 2:73–90. doi: 10.36628/ijhf.2020.0002
183. de Oliveira MT Jr., Baptista R, Chavez-Leal SA, Bonatto MG. Heart failure management with beta-blockers: can we do better? Curr Med Res Opin. (2024) 40:43–54. doi: 10.1080/03007995.2024.2318002
184. João P F, Rossello X, Eschalier R, McMurray John JV, Pocock S, Girerd N, et al. MRAs in elderly HF patients. JACC Heart Fail. (2019) 7:1012–21. doi: 10.1016/j.jchf.2019.08.017
185. Crispino SP, Segreti A, Nafisio V, Valente D, Crisci F, Ferro A, et al. The role of SGLT2-inhibitors across all stages of heart failure and mechanisms of early clinical benefit: from prevention to advanced heart failure. Biomedicines. (2025) 13:608. doi: 10.3390/biomedicines13030608
186. Magdy JS, McVeigh J, Indraratna P. Diuretics in the management of chronic heart failure: when and how. Aust Prescr. (2022) 45:200–4. doi: 10.18773/austprescr.2022.069
187. Parakh N, Chaturvedi V, Kurian S, Tyagi S. Effect of ivabradine vs atenolol on heart rate and effort tolerance in patients with mild to moderate mitral stenosis and normal sinus rhythm. J Card Fail. (2012) 18:282–8. doi: 10.1016/j.cardfail.2012.01.001
188. Armstrong Paul W, Roessig L, Patel Mahesh J, Anstrom Kevin J, Butler J, Voors Adriaan A, et al. A multicenter, randomized, double-blind, placebo-controlled trial of the efficacy and safety of the oral soluble guanylate cyclase stimulator. JACC Heart Fail. (2018) 6:96–104. doi: 10.1016/j.jchf.2017.08.013
189. John R T, Diaz R, Felker GM, McMurray John JV, Metra M, Solomon Scott D, et al. Omecamtiv mecarbil in chronic heart failure with reduced ejection fraction. JACC Heart Fail. (2020) 8:329–40. doi: 10.1016/j.jchf.2019.12.001
190. Wang W, Gao Y, Chen Y, Cheng M, Sang Y, Wei L, et al. TGF-beta inhibitors: the future for prevention and treatment of liver fibrosis? Front Immunol. (2025) 16:1583616. doi: 10.3389/fimmu.2025.1583616
191. Jiang Y, Qiang Z, Liu Y, Zhu L, Xiao L, Du Z, et al. Diverse functions of NLRP3 inflammasome in PANoptosis and diseases. Cell Death Discov. (2025) 11:389. doi: 10.1038/s41420-025-02689-1
192. Estep JD, Salah HM, Kapadia SR, Burkhoff D, Lala A, Butler J, et al. HFSA scientific statement: update on device based therapies in heart failure. J Card Fail. (2024) 30:1472–88. doi: 10.1016/j.cardfail.2024.07.007
193. Nguyen AH, Hurwitz M, Abraham J, Blumer V, Flanagan MC, Garan AR, et al. Medical management and device-based therapies in chronic heart failure. J Soc Cardiovasc Angiogr Interv. (2023) 2:101206. doi: 10.1016/j.jscai.2023.101206
194. Salah HM, Fudim M, Burkhoff D. Device interventions for heart failure. JACC Heart Fail. (2023) 11:1039–54. doi: 10.1016/j.jchf.2023.07.002
195. Debska-Kozlowska A, Ksiazczyk M, Warchol I, Lubinski A. Clinical usefulness of N-terminal prohormone of brain natriuretic peptide and high sensitivity troponin T in patients with heart failure undergoing cardiac resynchronization therapy. Curr Pharm Des. (2019) 25:1671–8. doi: 10.2174/1381612825666190621155718
196. Philippon F. Cardiac resynchronization therapy: device-based medicine for heart failure. J Card Surg. (2004) 19:270–4. doi: 10.1111/j.0886-0440.2004.04081.x
197. Sun S, Joglar JA. Cardiac resynchronization therapy: prospect for long-lasting heart failure remission. J Investig Med. (2011) 59:887–92. doi: 10.2310/JIM.0b013e3182186320
198. Suleiman M, Goldenberg I, Samniah N, Rosso R, Marai I, Pekar A, et al. Outcome of patients with advanced heart failure who receive device-based therapy for primary prevention of sudden cardiac death: insights from the Israeli ICD registry. Pacing Clin Electrophysiol. (2015) 38:738–45. doi: 10.1111/pace.12627
199. Belkin MN, Upadhyay GA. Does cardiac resynchronization therapy benefit patients with non-left bundle branch block prolonged QRS patterns? Curr Cardiol Rep. (2017) 19:125. doi: 10.1007/s11886-017-0929-8
200. Rowe MK, Kaye GC. Advances in atrioventricular and interventricular optimization of cardiac resynchronization therapy—what’s the gold standard? Expert Rev Cardiovasc Ther. (2018) 16:183–96. doi: 10.1080/14779072.2018.1427582
201. Lunati M, Magenta G, Cattafi G, Moreo A, Falaschi G, Contardi D, et al. Clinical relevance of systematic CRT device optimization. J Atr Fibrillation. (2014) 7:1077. doi: 10.4022/jafib.1077
202. Ueda N, Noda T, Ishibashi K, Nakajima K, Kataoka N, Kamakura T, et al. Efficacy of a device-based continuous optimization algorithm for patients with cardiac resynchronization therapy. Circ J. (2019) 84:18–25. doi: 10.1253/circj.CJ-19-0691
203. Gasparini M, Regoli F, Galimberti P, Ceriotti C, Cappelleri A. Cardiac resynchronization therapy in heart failure patients with atrial fibrillation. Europace. (2009) 11(5):v82–86. doi: 10.1093/europace/eup273
204. Zhang Q, Chan YS, Liang YJ, Fang F, Lam YY, Chan CP, et al. Comparison of left ventricular reverse remodeling induced by cardiac contractility modulation and cardiac resynchronization therapy in heart failure patients with different QRS durations. Int J Cardiol. (2013) 167:889–93. doi: 10.1016/j.ijcard.2012.01.066
205. Kramer DB, Reynolds MR, Mitchell SL. Resynchronization: considering device-based cardiac therapy in older adults. J Am Geriatr Soc. (2013) 61:615–21. doi: 10.1111/jgs.12174
206. Suleiman M, Goldenberg I, Haim M, Schliamser JE, Boulos M, Ilan M, et al. Clinical characteristics and outcomes of elderly patients treated with an implantable cardioverter-defibrillator or cardiac resynchronization therapy in a real-world setting: data from the Israeli ICD registry. Heart Rhythm. (2014) 11:435–41. doi: 10.1016/j.hrthm.2013.12.003
207. Lewis GF, Gold MR. Developments in cardiac resynchronisation therapy. Arrhythm Electrophysiol Rev. (2015) 4(2):122–8. doi: 10.15420/aer.2015.04.02.122
208. Goldberger Z, Lampert R. Implantable cardioverter-DefibrillatorsExpanding indications and technologies. JAMA. (2006) 295:809–18. doi: 10.1001/jama.295.7.809
209. Sanchez-Enrique C, Jorde UP, Gonzalez-Costello J. Heart transplant and mechanical circulatory support in patients with advanced heart failure. Rev Esp Cardiol (Engl Ed). (2017) 70:371–81. doi: 10.1016/j.rec.2016.12.036
210. Sharif ZI, Galand V, Hucker WJ, Singh JP. Evolving cardiac electrical therapies for advanced heart failure patients. Circ Arrhythm Electrophysiol. (2021) 14:e009668. doi: 10.1161/CIRCEP.120.009668
211. Bazoukis G, Saplaouras A, Efthymiou P, Yiannikourides A, Liu T, Letsas KP, et al. Cardiac contractility modulation in patients with heart failure—a review of the literature. Heart Fail Rev. (2024) 29:689–705. doi: 10.1007/s10741-024-10390-1
212. Berry N, Mauri L, Feldman T, Komtebedde J, van Veldhuisen DJ, Solomon SD, et al. Transcatheter InterAtrial shunt device for the treatment of heart failure: rationale and design of the pivotal randomized trial to REDUCE elevated left atrial pressure in patients with heart failure II (REDUCE LAP-HF II). Am Heart J. (2020) 226:222–31. doi: 10.1016/j.ahj.2019.10.015
213. Tom SK, Kalra K, Perdoncin E, Tully A, Devireddy CM, Inci E, et al. Transcatheter treatment options for functional mitral regurgitation: which device for which patients? Interv Cardiol. (2024) 19:e10. doi: 10.15420/icr.2021.29
214. Ravassa S, Lopez B, Treibel TA, San Jose G, Losada-Fuentenebro B, Tapia L, et al. Cardiac fibrosis in heart failure: focus on non-invasive diagnosis and emerging therapeutic strategies. Mol Aspects Med. (2023) 93:101194. doi: 10.1016/j.mam.2023.101194
215. Gallagher MJ, McCullough PA. The emerging role of natriuretic peptides in the diagnosis and treatment of decompensated heart failure. Curr Heart Fail Rep. (2004) 1:129–35. doi: 10.1007/s11897-004-0022-7
216. Moayedi Y, Ross HJ. Advances in heart failure: a review of biomarkers, emerging pharmacological therapies, durable mechanical support and telemonitoring. Clin Sci. (2017) 131:553–66. doi: 10.1042/CS20160196
217. Neuen BL, Vaduganathan M, Claggett BL, Beldhuis I, Myhre P, Desai AS, et al. Natriuretic peptides, kidney function, and clinical outcomes in heart failure with preserved ejection fraction. JACC Heart Fail. (2025) 13:28–39. doi: 10.1016/j.jchf.2024.08.009
218. Salah K, Stienen S, Pinto YM, Eurlings LW, Metra M, Bayes-Genis A, et al. Prognosis and NT-proBNP in heart failure patients with preserved versus reduced ejection fraction. Heart. (2019) 105:1182–9. doi: 10.1136/heartjnl-2018-314173
219. Tsutsui H, Albert NM, Coats AJS, Anker SD, Bayes-Genis A, Butler J, et al. Natriuretic peptides: role in the diagnosis and management of heart failure: a scientific statement from the heart failure association of the European society of cardiology, heart failure society of America and Japanese heart failure society. J Card Fail. (2023) 29:787–804. doi: 10.1016/j.cardfail.2023.02.009
220. Packer M, Anker SD, Butler J, Filippatos G, Ferreira JP, Pocock SJ, et al. Effect of empagliflozin on the clinical stability of patients with heart failure and a reduced ejection fraction: the EMPEROR-reduced trial. Circulation. (2021) 143:326–36. doi: 10.1161/CIRCULATIONAHA.120.051783
221. Shi Y, Zhao L, Wang J, Liu X, Bai Y, Cong H, et al. Empagliflozin protects against heart failure with preserved ejection fraction partly by inhibiting the senescence-associated STAT1-STING axis. Cardiovasc Diabetol. (2024) 23:269. doi: 10.1186/s12933-024-02366-0
222. Tuttolomondo A, Simonetta I, Daidone M, Mogavero A, Ortello A, Pinto A. Metabolic and vascular effect of the Mediterranean diet. Int J Mol Sci. (2019) 20:4716. doi: 10.3390/ijms20194716
223. Basu R, Poglitsch M, Yogasundaram H, Thomas J, Rowe BH, Oudit GY. Roles of angiotensin peptides and recombinant human ACE2 in heart failure. J Am Coll Cardiol. (2017) 69:805–19. doi: 10.1016/j.jacc.2016.11.064
224. He X, Du T, Long T, Liao X, Dong Y, Huang ZP. Signaling cascades in the failing heart and emerging therapeutic strategies. Signal Transduct Target Ther. (2022) 7:134. doi: 10.1038/s41392-022-00972-6
225. deFilippi CR, Shah P, Shah SJ, Alemayehu W, Lam CSP, Butler J, et al. Proteomics identify clinical phenotypes and predict functional outcomes in heart failure with preserved ejection fraction: insights from VITALITY-HFpEF. Circ Heart Fail. (2024) 17:e011792. doi: 10.1161/CIRCHEARTFAILURE.124.011792
226. Georgiopoulou VV, Tang WHW, Giamouzis G, Li S, Deka A, Dunbar SB, et al. Renal biomarkers and outcomes in outpatients with heart failure: the Atlanta cardiomyopathy consortium. Int J Cardiol. (2016) 218:136–43. doi: 10.1016/j.ijcard.2016.05.041
227. Nagai T, Nakao M, Anzai T. Risk stratification towards precision medicine in heart failure—current progress and future perspectives. Circ J. (2021) 85:576–83. doi: 10.1253/circj.CJ-20-1299
228. Ni W, Jiang R, Xu D, Zhu J, Chen J, Lin Y, et al. Association between insulin resistance indices and outcomes in patients with heart failure with preserved ejection fraction. Cardiovasc Diabetol. (2025) 24:32. doi: 10.1186/s12933-025-02595-x
229. Ezekowitz JA, Colin-Ramirez E, Ross H, Escobedo J, Macdonald P, Troughton R, et al. Reduction of dietary sodium to less than 100 mmol in heart failure (SODIUM-HF): an international, open-label, randomised, controlled trial. Lancet. (2022) 399:1391–400. doi: 10.1016/S0140-6736(22)00369-5
230. Kimmoun A, Cotter G, Davison B, Takagi K, Addad F, Celutkiene J, et al. Safety, tolerability and efficacy of rapid optimization, helped by NT-proBNP and GDF-15, of heart failure therapies (STRONG-HF): rationale and design for a multicentre, randomized, parallel-group study. Eur J Heart Fail. (2019) 21:1459–67. doi: 10.1002/ejhf.1575
231. Davison BA, Abbate A, Cotter G, Pascual-Figal D, Van Tassell B, Villota JN, et al. Effects of anti-inflammatory therapy in acute heart failure: a systematic review and meta-analysis. Heart Fail Rev. (2025) 30:575–87. doi: 10.1007/s10741-025-10491-5
232. Hirano KI, Okamura S, Sugimura K, Miyauchi H, Nakano Y, Nochioka K, et al. Long-term survival and durable recovery of heart failure in patients with triglyceride deposit cardiomyovasculopathy treated with tricaprin. Nat Cardiovasc Res. (2025) 4:266–74. doi: 10.1038/s44161-025-00611-7
233. Figueiral M, Paldino A, Fazzini L, Pereira NL. Genetic biomarkers in heart failure: from gene panels to polygenic risk scores. Curr Heart Fail Rep. (2024) 21:554–69. doi: 10.1007/s11897-024-00687-5
234. von Lueder TG, Kotecha D, Atar D, Hopper I. Neurohormonal blockade in heart failure. Card Fail Rev. (2017) 3:19–24. doi: 10.15420/cfr.2016:22:2
235. Puniya BL. Artificial-intelligence-driven innovations in mechanistic computational modeling and digital twins for biomedical applications. J Mol Biol. (2025) 437:169181. doi: 10.1016/j.jmb.2025.169181
236. Lin M, Guo J, Gu Z, Tang W, Tao H, You S, et al. Machine learning and multi-omics integration: advancing cardiovascular translational research and clinical practice. J Transl Med. (2025) 23:388. doi: 10.1186/s12967-025-06425-2
237. Bastos JM, Colaço B, Baptista R, Gavina C, Vitorino R. Innovations in heart failure management: the role of cutting-edge biomarkers and multi-omics integration. J Mol Cell Cardiol Plus. (2025) 11:100290. doi: 10.1016/j.jmccpl.2025.100290
238. Ben-Eltriki M, Rafiq A, Paul A, Prabhu D, Afolabi MOS, Baslhaw R, et al. Adaptive designs in clinical trials: a systematic review-part I. BMC Med Res Methodol. (2024) 24:229. doi: 10.1186/s12874-024-02272-9
Glossary
4EBP1 eukaryotic translation initiation factor 4E binding protein 1
ac4C N4-acetylcytidine
ACE angiotensin-converting enzyme
ACE2 angiotensin-converting enzyme 2
AF atrial fibrillation
AKT protein kinase B
AMPK AMP-activated protein kinase
Ang1–7 angiotensin 1–7
ARBs angiotensin II receptor blockers
ARNIs angiotensin receptor–neprilysin inhibitors
AT1 angiotensin II type 1 receptor
AT1A angiotensin II type 1A receptor
ATG5 autophagy related 5
AV atrioventricular
BAG3 BCL2-associated athanogene 3
BET bromodomain and extra-terminal proteins
Bmi1 B-cell-specific moloney murine leukemia virus integration site 1
BRD4 bromodomain-containing protein 4
CASA chaperone-assisted selective autophagy
CCM cardiac contractility modulation
CD47 cluster of differentiation 47
CFs cardiac fibroblasts
circRNA circular RNA
CMD coronary microvascular dysfunction
CR cardiac rehabilitation
CRT cardiac resynchronization therapy
CXCL10 C-X-C motif chemokine ligand 10
DCM dilated cardiomyopathy
ECM extracellular matrix
Fe–S iron–sulfur cluster
GDF-15 growth differentiation factor 15
GLP-1 glucagon-like peptide-1
GPX4 glutathione peroxidase 4
GSH glutathione
GSHPx glutathione peroxidase
HAND1 heart and neural crest derivatives expressed 1
HCTR hybrid comprehensive telerehabilitation
HDAC histone deacetylase
HF heart failure
HFmrEF heart failure with mildly reduced ejection fraction
HFpEF heart failure with preserved ejection fraction
HFrEF heart failure with reduced ejection fraction
HIIT high-intensity interval training
HMGB1 high-mobility group box 1
HO-1 heme oxygenase-1
HSP27/HSP70 heat shock protein 27/70
ICD implantable cardioverter-defibrillator
IGFBP7 insulin-like growth factor binding protein 7
IL-1 interleukin-1
IL-10 interleukin-10
IL-6 interleukin-6
Keap1 kelch-like ECH-associated protein 1
LBBB left bundle branch block
LCZ696 sacubitril/valsartan
lncRNA long non-coding RNA
LVAD left ventricular assist device
LVEF left ventricular ejection fraction
MAO monoamine oxidases
MAPK mitogen-activated protein kinase
Mas R mas receptor
MCS mechanical circulatory support
MEF2 myocyte enhancer factor 2
MFRN1 mitoferrin 1
MICT moderate-intensity continuous training
miRNA microRNA
MitoDAMPs mitochondrial damage-associated molecular patterns
MLF1 myeloid leukemia factor 1
MR mitral regurgitation
MRA mineralocorticoid receptor antagonist
mTOR mammalian target of rapamycin
MVD microvascular dysfunction
myoFbs myofibroblasts
NAD+ nicotinamide adenine dinucleotide
NAT10 N-acetyltransferase 10
NF-κB nuclear factor-kappa B
NHANES national health and nutrition examination survey
NLRP3 NOD-like receptor pyrin domain-containing protein 3
NO nitric oxide
NOS nitric oxide synthases
Nox2/Nox4 NADPH oxidase 2/4
NP natriuretic peptide
NR nicotinamide riboside
Nrf2 NF-E2-related factor 2
NYHA New York heart association
PBMCs peripheral blood mononuclear cells
PI3K phosphatidylinositol 3-kinase
PKG protein kinase G
PQC protein quality control
RAAS renin–angiotensin–aldosterone system
RNS reactive nitrogen species
ROCK2 rho-associated protein kinase 2
ROR2 receptor tyrosine kinase-like orphan receptor 2
ROS reactive oxygen species
SGLT2 sodium–glucose cotransporter 2
SGLT2i sodium–glucose cotransporter 2 inhibitor
SII systemic immune-inflammation index
SLC25A3 solute carrier family 25 member 3
Smad Sma and mad related proteins
SNS sympathetic nervous system
SOD superoxide dismutase
ST2 suppression of tumorigenicity 2
STAT1 signal transducer and activator of transcription 1
STING stimulator of interferon genes
TCA tricarboxylic acid cycle
TGF-β transforming growth factor-β
TLR4 toll-like receptor 4
TNFR1 tumor necrosis factor receptor 1
TNF-α tumor necrosis factor-α
TRIM24 tripartite motif containing 24
Trx/TrxR thioredoxin/thioredoxin reductase
Txlnb taxilin beta
UPAR urokinase-type plasminogen activator receptor
VO2peak peak oxygen uptake
VV interventricular
WHF worsening heart failure
Wisper wound and scar-associated lncRNA
XOR xanthine oxidoreductase
Zip13 zinc transporter 13
Keywords: heart failure, molecular mechanisms, fibrosis, mitochondrial dysfunction, inflammation, precision medicine
Citation: Zhang F, Zhang X, Jian J, Zeng X, Zheng C, Zhang Y and Gao J (2025) Heart failure: mechanistic insights and precision therapeutic strategies. Front. Cardiovasc. Med. 12:1696793. doi: 10.3389/fcvm.2025.1696793
Received: 12 September 2025; Revised: 21 November 2025;
Accepted: 24 November 2025;
Published: 5 December 2025.
Edited by:
Alexander E. Berezin, Paracelsus Medical University, AustriaReviewed by:
Syed Nurul Hasan, New York University, United StatesPrashanth Kumar Patnaik, Osmania Medical College, India
Copyright: © 2025 Zhang, Zhang, Jian, Zeng, Zheng, Zhang and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fang Zhang, ZnpoYW5nMTEwM0AxNjMuY29t