Abstract
Danon Disease (DD) is a rare X-linked inherited disorder caused by severe deficiency of lysosome-associated membrane protein-2 (LAMP-2), encoded by the LAMP2 gene. Characteristic clinical features include a triad of cardiomyopathy, skeletal myopathy and cognitive impairment in males. Females usually exhibit milder, cardiac-predominant manifestations later in life. In this case, we report a 30-year-old woman with a novel suspected pathogenic LAMP2 mutation (c.1A > T, initiation codon mutation). She developed Wolff-Parkinson-White (WPW) syndrome in her twenties, acute heart failure post cesarean section at age 29, and persistent left ventricular hypertrophy. Positive result of Technetium-99 m pyrophosphate (99mTc-PYP) scintigraphy strongly indicated transthyretin amyloid cardiomyopathy (ATTR-CM). However, whole-exome sequencing (WES) identified the novel A to T transition in initiation codon (c.1A > T) of LAMP2 gene, establishing the diagnosis of DD and revealing the false-positive result of PYP scintigraphy in DD.
Introduction
Danon Disease is a rare X-linked dominant disorder characterized by severe cardiomyopathy, skeletal myopathy, and cognitive impairment, often accompanied by retinopathy and multi-system (neurologic, hepatic, gastrointestinal, pulmonary) involvement (1). The disease stems from deficiency of lysosome-associated membrane protein-2 (LAMP-2), encoded by the X-chromosomal LAMP2 gene (Xq24). LAMP-2, essential for autophagosome maturation via fusion with lysosomes and endosomes, is predominantly localized to these organelles' membranes (2, 3). Deficiency of its cardiac/skeletal muscle-enriched isoform LAMP-2B impairs autophagy, causing autophagosome accumulation, cellular hypertrophy, and ultimately myocyte death with fibrosis (1, 2).
Due to its X-linked dominant inheritance, Danon Disease demonstrates significant gender differences in phenotypic expression (4). Male patients typically develop cardiac symptoms in their mid-twenties, often progressing to severe heart failure requiring transplantation; they also frequently exhibit the classic triad and retinopathy. In contrast, cardiomyopathy in female patients usually manifests during adulthood, typically as left ventricular hypertrophy and conduction abnormalities; extracardiac manifestations are often absent or mild (4).
Here, we describe a 30-year-old woman presenting with mild left ventricular hypertrophy and conduction abnormalities, Whole-exome sequencing (WES) identified a novel initiation codon mutation in the LAMP2 gene, strongly suggesting the diagnosis.
Case presentation
A 30-year-old woman was referred to our hospital for cardiac assessment. A summarized timeline of key clinical events is presented in Table 1.
Table 1
| Time/Age | Manifestations/Events | Management | Therapy |
|---|---|---|---|
| Early 20s | Diagnosed with WPW Syndrome | Received a catheter ablation procedure | No pharmacotherapy |
| 2019/02/20, aged 28 | ECG showed no abnormality during pregnancy | ||
| 2019/08/23, aged 28 | Fever, cough, acute respiratory and cardiac failure after cesarean section, ECG showed reduced LVEF of 48%, with 13mm-thick interventricular septum and 12mm-thick posterior wall | Intensive Care Unit admission | |
| 2019/10/05, aged 28 | Mildly improved LVEF of 52%, with 13mm-thick interventricular septum and 12mm-thick posterior wall | Metoprolol 12.5 mg twice daily, Perindopril 4 mg once daily | |
| 2019/11/20, aged 28 | LVEF improved to 60%, with 12mm-thick interventricular septum and 12mm-thick posterior wall | As above | |
| 2020/04/02, aged 29 | LVEF 59%, with 13mm-thick interventricular septum and 13mm-thick posterior wall | As above | |
| 2020/09/17, aged 29 | LVEF 55%, with 13mm-thick interventricular septum and 13mm-thick posterior wall |
Timeline of the course of disease.
In her early twenties, she was diagnosed with Wolff-Parkinson-White (WPW) Syndrome and underwent catheter ablation procedure at a local hospital. Despite persistent intermittent palpitations, she received no pharmacotherapy. In 2019, at 39 weeks' gestation, she developed a fever requiring intensive care unit admission for acute respiratory and cardiac failure following emergency cesarean section. Echocardiography (ECG) showed mild left ventricular hypertrophy with thickness of 13 mm in interventricular septum and 12 mm in posterior wall, and impaired systolic function with left ventricular ejection fraction (LVEF) of 48%. Diagnosed with peripartum cardiomyopathy, she was initiated on Metoprolol 12.5 mg twice daily and Perindopril 4 mg once daily. Follow-up echocardiography revealed improved left ventricular systolic function (LVEF 60%), although left ventricular wall thickness remained unchanged.
The patient presented to our clinic seeking evaluation regarding her suitability for another pregnancy. During the consultation, we elicited a concerning family history. She reported three miscarriages and the premature death of her firstborn son at age 1 due to undetermined cardiac causes. Her mother had suspected hypertrophic cardiomyopathy and died at age 30. The family pedigree (Supplementary Figure S1) is suggestive of an X-linked inheritance pattern for cardiomyopathy.
On admission, the patient's vital signs were within normal range, and she demonstrated no clinical sign of heart failure. Laboratory findings are detailed in Supplementary Table S1. Electrocardiogram (EKG) revealed a short PR interval of 106 ms, ventricular pre-excitation and sinus rhythm with a heart rate of 67 beats per minute (Supplementary Figure S2). ECG indicated mild left ventricular hypertrophy with thickness of 12 mm in interventricular septum and posterior wall, and impaired systolic function with an estimated LVEF of approximately 48% (Supplementary Figure S3). Further evaluation with cardiac magnetic resonance (CMR) imaging revealed diminished left ventricular wall contraction and late gadolinium enhancement (LGE) involving the subendocardium (indicated by triangles in Figure 1).
Figure 1
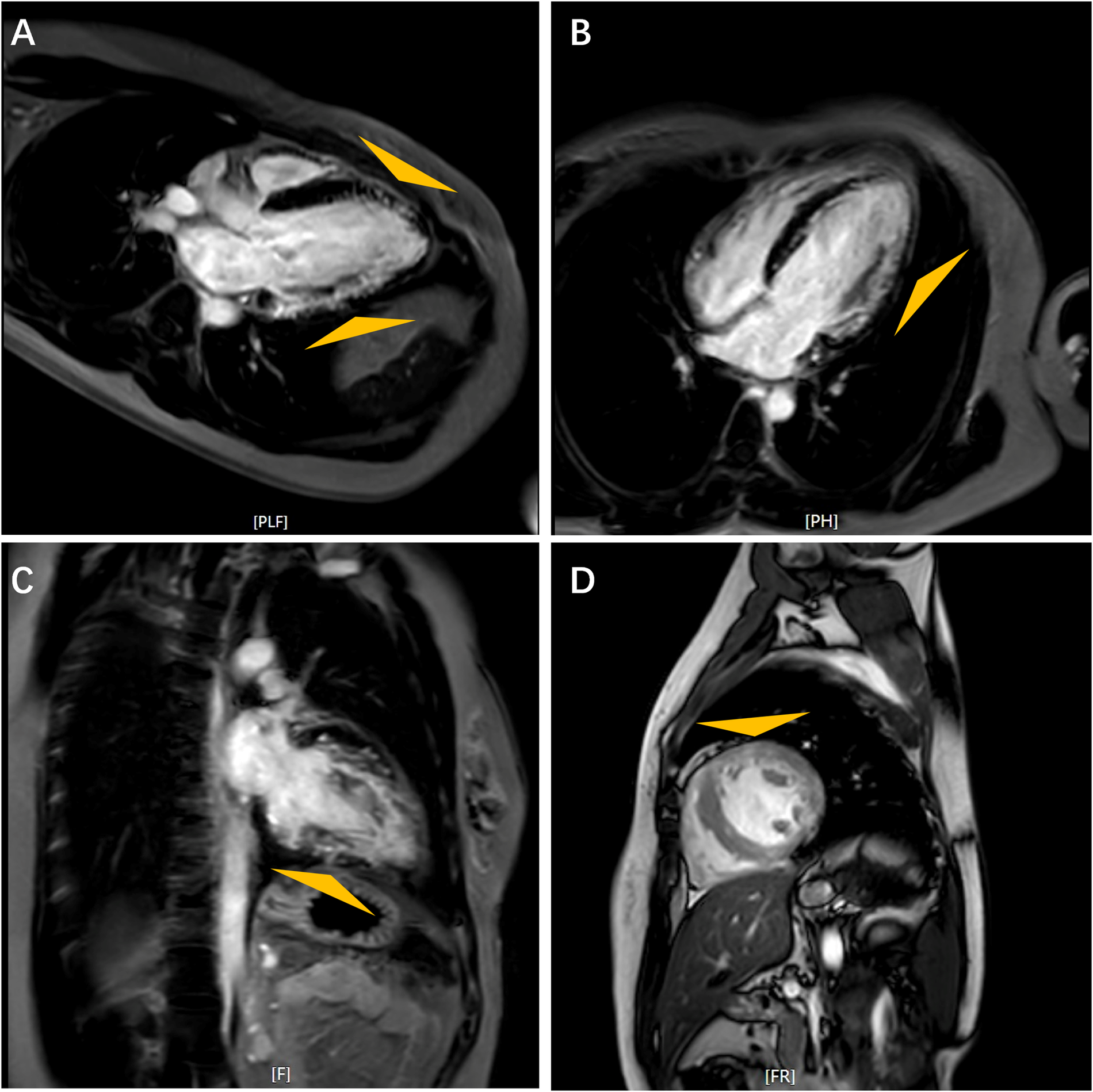
Cardiac magnetic resonance images. Feather-like delayed gadolinium enhancement in the left ventricular wall as indicated by triangles. Long Axis (A); Horizontal Long Axis (B); Four-Chamber View (C); Short Axis (D).
Given the subendocardial LGE pattern on CMR, which raised suspicion for infiltrative disease including cardiac amyloidosis, Technetium-99 m pyrophosphate (99mTc-PYP) scintigraphy was performed, which result demonstrated intense myocardial uptake [visual score Grade 3; heart-to-contralateral lung (H/CL) ratio: 1.82] (Figure 2). With immunofixation electrophoresis (IFE) of serum and urine revealing no presence of immunoglobulin monoclonal proteins, the positive result of 99mTc-PYP scintigraphy seemed to indicate a potential diagnosis of transthyretin amyloid cardiomyopathy (ATTR-CM). Genetic testing detected no pathogenic variant in the TTR gene, effectively excluding hereditary ATTR (ATTRv-CM) and supporting wild-type ATTR (ATTRwt-CM). However, ATTRwt-CM typically affects older individuals, and the clinical presentation was atypical for several reasons: the patient was a young woman of reproductive age, and her EKG lacked low voltage. Furthermore, multiple reports describe false-positive results of 99mTc-PYP scintigraphy in patients with hypertrophic cardiomyopathy (HCM) phenotype (5–11). These discrepancies prompted reconsideration of the diagnosis and led to the recommendation for further genetic testing to evaluate alternative etiologies.
Figure 2
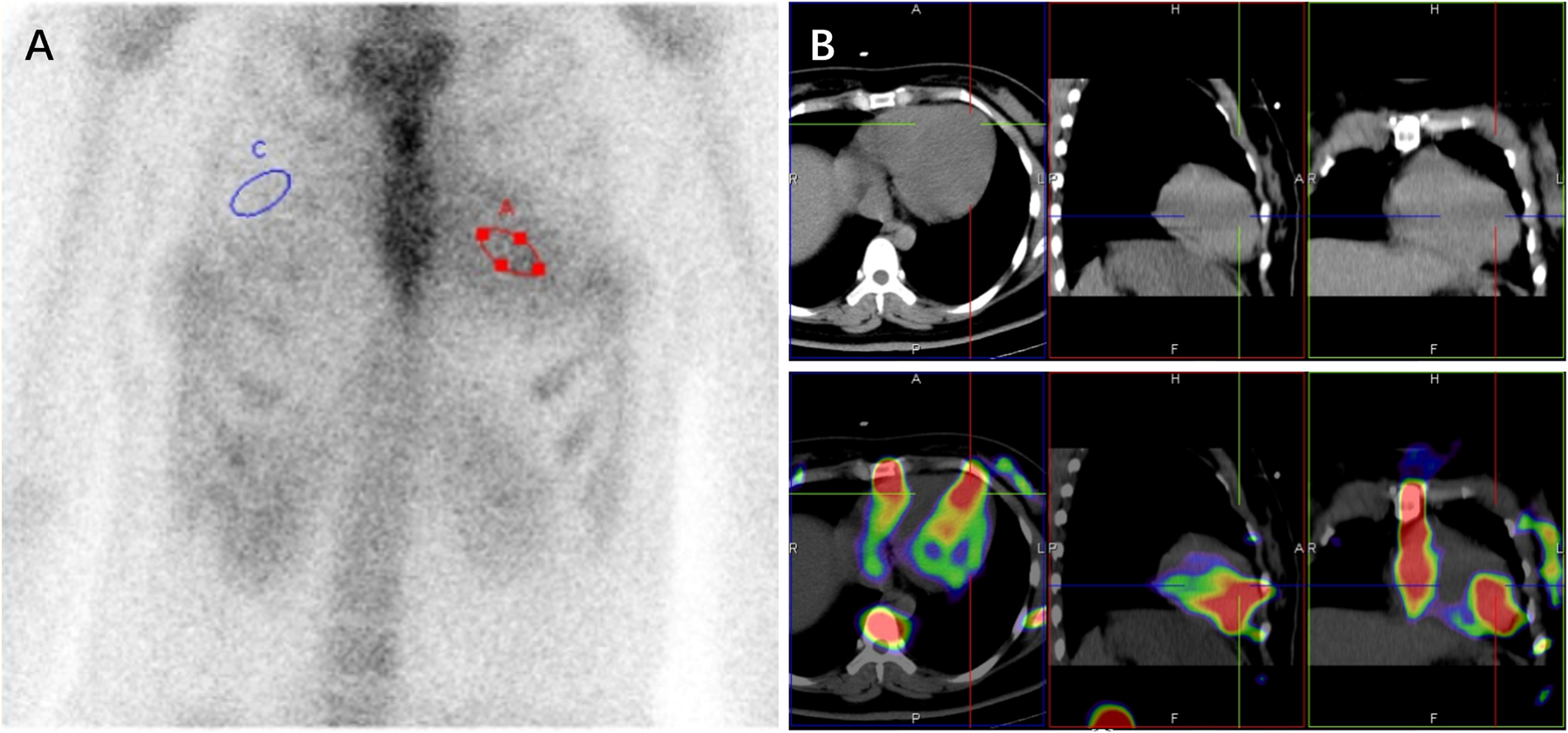
99mTc-PYP scintigraphy (A) and SPECT/CT images (B) revealed a grade 3 myocardial uptake with heart (red circle) to contralateral lung (blue circle) ratio of 1.82 at 3hr (A).
Ultimately, whole-exome sequencing (WES) identified that the patient carries a heterozygous variant with a novel mutation in the lysosome-associated membrane protein-2 (LAMP2) gene: an A-T transition (c.1A > T) at the initiation codon (ATG) in exon 1. This variant was classified as suspected pathogenic (PSV1 + PM2) according to ACMG guidelines (12). We utilized MutationTaster to predict the mutation's functional impact on the LAMP-2 protein. As mutations in the start codon typically preclude translation initiation at the native site, a downstream ATG at positions c.80–82 was predicted to become the new translation start site. This shift results in a frameshift mutation, generating a truncated protein of only 6 amino acids before encountering a premature stop codon. Structure prediction by AlphaFold shows that the mutant protein lacks nearly all functional domains and is unlikely to retain its original function (Figure 3). The combination of this genetic mutation with the patient's phenotype of ventricular hypertrophy and conduction abnormalities strongly suggested the diagnosis of Danon Disease.
Figure 3
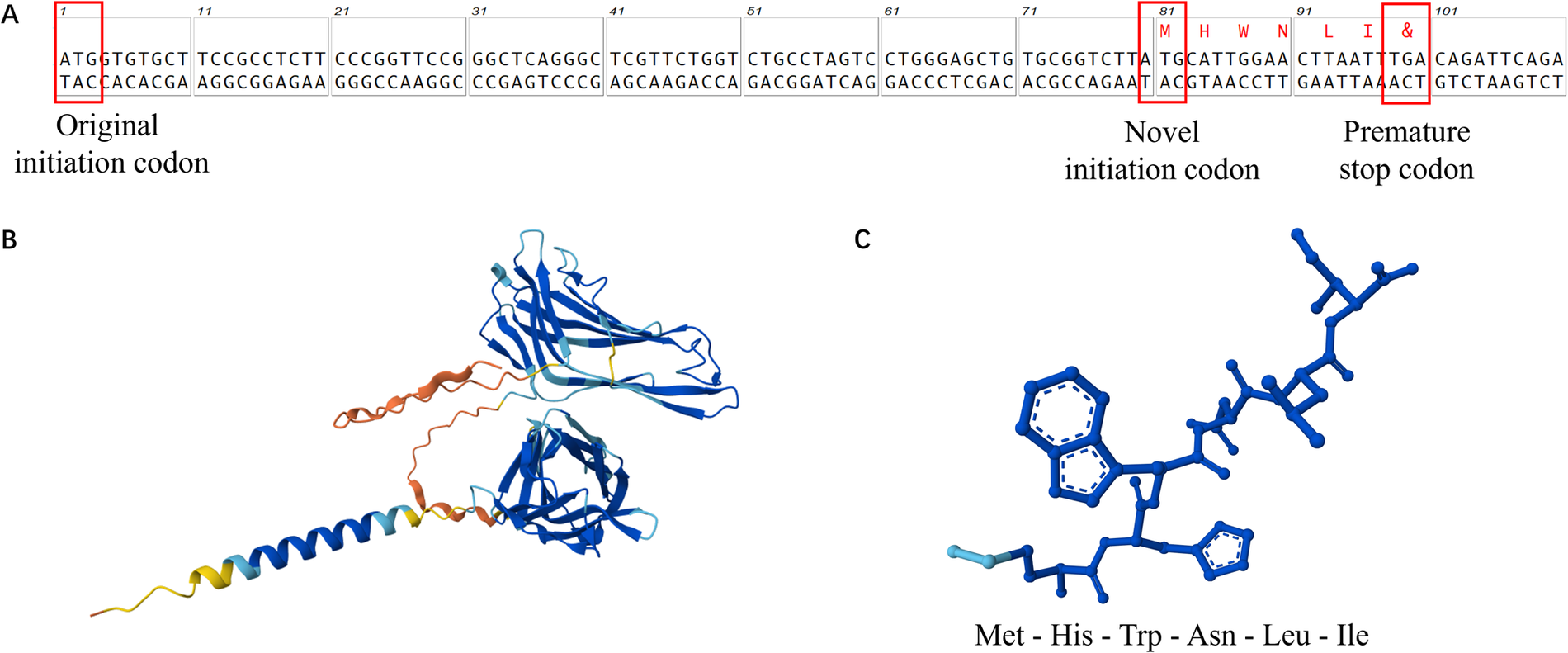
Schematic diagram of the new start codon (A) and the predicted structure of the LAMP-2 protein (B) compared with the mutant LAMP-2 protein (C).
The patient was administered with oral spironolactone (20 mg once daily), Sacubitril/Valsartan (50 mg twice daily), Metoprolol succinate (23.75 mg once daily), Dapagliflozin (10 mg once daily), and Amiodarone (400 mg once daily) for arrhythmia management. At the 2-year follow-up, echocardiography demonstrated significant improvement, with the left ventricular ejection fraction (LVEF) reaching 65%. The patient remained free from heart failure-related hospitalizations and adopted a child from a local welfare institution.
Discussion
Danon Disease is a rare, X-linked dominant inherited myopathy caused by defects in the LAMP2 gene, which encodes lysosomal-associated membrane protein-2. Severe deficiency of this protein leads to cellular autophagy, accumulation of autophagosomes, mitochondrial dysfunction, and myocyte death (1).
To date, 472 distinct molecular consequences associated with LAMP2 gene variants have been documented in the ClinVar Database, including 50 frameshifts, 248 missense, 38 nonsense, 22 splice sites and 114 untranslated regions. The majority of these mutations are pathogenic, with many definitively linked to Danon Disease. In this case, we identified a novel initiation codon mutation (c.1A > T) in LAMP2 gene by whole-exome sequencing (WES). Our findings strongly suggested its pathogenicity for Danon Disease. This mutation, located within the initiation codon of exon 1, is classified as a missense initiation codon variant in ClinVar Database. Consistent with ACMG guidelines (12), it is categorized as suspected pathogenic (PSV1 + PM2). Notably, this specific mutation is absent from databases such as Exome Aggregation Consortium (EXAC) or Human Gene Mutation Database (HGMD).
In this case, this mutant LAMP2 variant manifested solely as cardiac involvement without extracardiac symptoms. Cardiac conductive abnormality presented early, with a diagnosis of Wolff-Parkinson-White (WPW) Syndrome in her twenties. Hypertrophic cardiomyopathy and heart failure emerged only under physiological stress, specifically, during late pregnancy culminating in cesarean section. Apart from mildly elevated AST and LDH levels, no evidence of skeletal myopathy was found. The patient reported no visual impairment and displayed no cognitive deficits. However, phenotypic expression of this mutation exhibits marked gender disparity. Her deceased son displayed signs of developmental delay and cardiac dilation at one month of age. According to next-generation sequencing (NGS) at the local hospital, the boy was also a carrier of this LAMP2 mutation and the clinical presentation strongly suggested he was succumbed to Danon Disease-related cardiomyopathy. Her mother's death around age 30 from suspected hypertrophic cardiomyopathy—likely attributable to this same LAMP2 variant—further underscores the mutation's poor prognosis. Heterogenous penetrance and expressivity observed in female carriers may stem from the gene's X-linked dominant inheritance pattern, skewed X-chromosome inactivation and functional mosaicism in LAMP-2 expression (1, 2, 4, 13, 14). Another initiation codon mutation in LAMP2 (c.2T > C) was recently reported by Wang et al. (15). Although both mutations (c.1A > T and c.2T > C) are predicted to disrupt the translation initiation codon, they are associated with strikingly divergent phenotypes. This observation suggests that additional factors may modulate phenotypic expression, warranting further mechanistic investigation.
False-positive results of 99mTc-PYP scintigraphy are documented in multiple conditions (7–11, 16–21). The most common mimic is cardiac amyloidosis, typically light-chain (AL) type (6, 8, 17, 18), though rare etiology like apolipoprotein A-IV amyloidosis (19) and primary hyperoxaluria (causing myocardial oxalate deposition) (20) also occur. Other recognized causes include hyperphosphatemia (9), myocardial injury [e.g., infarction (21), direct current cardioversion (22)], and extra-cardiac tracer uptake from thoracic/pulmonary lesions (10, 11, 23, 24), leading to misjudgment of semiquantitative measurement. Notably, Danon Disease has recently emerged as a novel cause of PYP false positivity (7). Antukh D, et al. first reported this phenomenon in a 23-year-old man in 2020, who presented the classical triad of cardiomyopathy, polyneuropathy and myopathy, highly suggestive of glycogen storage disorder. In our case, however, the atypical cardiac symptoms and absence of extracardiac manifestations created significant diagnostic challenges, initially masking the systemic nature of her disease. This experience underscores that even highly specific tests like PYP scintigraphy can yield misleading results. Critically, this diagnostic pitfall reinforces the essential role of genetic testing in achieving precise etiological classification for undetermined cardiomyopathy.
The limitation is that we were unable to conduct a comprehensive assessment, such as ophthalmic images, electromyography and intelligence assessment. Endomyocardial biopsy was not clinically indicated. The patient has not returned for scheduled follow-up. Furthermore, additional studies should be conducted to investigate the underlying mechanism linking this variant to LAMP-2 protein dysfunction and clinical phenotype.
Conclusion
In conclusion, this case study identified a novel initiation codon mutation in LAMP2 gene with strongly-suspected pathogenicity to Danon Disease. The first downstream ATG codon likely serves as an alternative translation start site, generating a truncated hexapeptide predicted to lack functional capacity in autophagy.
Statements
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by Ruijin Hospital Ethics committee, Shanghai Jiao Tong University School of Medicine. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
QL: Writing – original draft, Data curation, Formal analysis, Visualization. ZS: Formal analysis, Writing – original draft. ZQ: Supervision, Writing – review & editing, Funding acquisition. YC: Supervision, Funding acquisition, Writing – review & editing, Conceptualization. WJ: Writing – review & editing, Funding acquisition, Supervision, Conceptualization.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was supported by grants from the National Natural Science Foundation of China (82270404 to JW, 82300421 to QZ, 82300422 to WZ, 82400451 to HS, 82400445 to CY), Shanghai Sailing Program of Science and Technology Commission (23YF14360000), Huangpu District Key Medical Discipline Program (2025ZDXK01) and Huangpu Special Studio Program (2023MY01).
Acknowledgments
We express gratitude to the patient and the personnel involved in this treatment.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2025.1699732/full#supplementary-material
References
1.
Hong KN Eshraghian EA Arad M Argirò A Brambatti M Bui Q et al International consensus on differential diagnosis and management of patients with danon disease. J Am Coll Cardiol. (2023) 82(16):1628–47. 10.1016/j.jacc.2023.08.014
2.
Endo Y Furuta A Nishino I . Danon disease: a phenotypic expression of LAMP-2 deficiency. Acta Neuropathol. (2015) 129(3):391–8. 10.1007/s00401-015-1385-4
3.
Huynh KK Eskelinen EL Scott CC Malevanets A Saftig P Grinstein S . LAMP Proteins are required for fusion of lysosomes with phagosomes. EMBO J. (2007) 26(2):313–24. 10.1038/sj.emboj.7601511
4.
Brambatti M Caspi O Maolo A Koshi E Greenberg B Taylor MRG et al Danon disease: gender differences in presentation and outcomes. Int J Cardiol. (2019) 286:92–8. 10.1016/j.ijcard.2019.01.020
5.
Hanna M Ruberg FL Maurer MS Dispenzieri A Dorbala S Falk RH et al Cardiac scintigraphy with technetium-99m-labeled bone-seeking tracers for suspected amyloidosis. J Am Coll Cardiol. (2020) 75(22):2851–62. 10.1016/j.jacc.2020.04.022
6.
Poterucha TJ Elias P Bokhari S Einstein AJ DeLuca A Kinkhabwala M et al Diagnosing transthyretin cardiac amyloidosis by technetium Tc 99 m pyrophosphate. JACC: Cardiovascular Imaging. (2021) 14(6):1221–31. 10.1016/j.jcmg.2020.08.027
7.
Antukh D Shchekochikhin D Rosina T Mershina E Larina O Pasha S et al Scintigraphy false-positive results for cardiac amyloidosis in a patient with danon disease. Clin Case Rep. (2021) 9(8):e04652. 10.1002/ccr3.4652
8.
Zeng Y Poterucha TJ Einstein AJ Zhang Q Chen Y Xie H et al False positive technetium-99 m pyrophosphate scintigraphy in a patient with cardiac amyloidosis light chain: case report. Case Report. Medicine. (2021) 100(17):e25582. 10.1097/MD.0000000000025582
9.
Hu LH Kuo Y Chang FP Wang WT Yang BH Huang WS et al Hyperphosphatemia-Related false-positive 99mTc-pyrophosphate myocardial scan: a case report with endomyocardial biopsy result. Clin Nucl Med. (2023) 48(11):e544–6. 10.1097/RLU.0000000000004869
10.
Nunes ARP Alves VM . Mitral annular calcification as a potential false-positive for cardiac amyloidosis in 99mTc-DPD scintigraphy accurately identified by SPECT/CT. Clin Nucl Med. (2024) 49(4):e179–81. 10.1097/RLU.0000000000005086
11.
Harford W Weinberg MN Buja LM Parkey RW Bonte FJ Willerson JT . Positive99m tc-stannous pyrophosphate myocardial image in a patient with carcinoma of the lung. Radiology. (1977) 122(3):747–8. 10.1148/122.3.747
12.
Richards S Aziz N Bale S Bick D Das S Gastier-Foster J et al Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17(5):405–24. 10.1038/gim.2015.30
13.
Cenacchi G Papa V Pegoraro V Marozzo R Fanin M Angelini C . Review: danon disease: review of natural history and recent advances. Neuropathology Appl Neurobio. (2020) 46(4):303–22. 10.1111/nan.12587
14.
D'souza RS Levandowski C Slavov D Graw SL Allen LA Adler E et al Danon disease: clinical features, evaluation, and management. Circulation Heart Failure. (2014) 7(5):843–9. 10.1161/CIRCHEARTFAILURE.114.001105
15.
Wang Y Bai M Zhang P Peng Y Chen Z He Z et al Identification and functional analysis of a novel de novo missense mutation located in the initiation codon of LAMP2 associated with early onset female Danon disease. Molec Gen & Gen Med. (2023) 11(9):e2216. 10.1002/mgg3.2216
16.
Wu Z Xia S Ma D Li L . Negative methylene diphosphonate scintigraphy in biopsy-confirmed hereditary transthyretin Ala117Ser cardiac amyloidosis: a case report. Cardiology Plus. (2023) 8(3):206–10. 10.1097/CP9.0000000000000058
17.
Hsu TJ Tseng CT Kuo L Yang CY Lin YP Yu WC et al AHL Amyloidosis mimicking transthyretin amyloidosis on cardiac tc-99 m pyrophosphate scan: a diagnostic challenge. J Nucl Cardiol. (2025) 47:102147. 10.1016/j.nuclcard.2025.102147
18.
Arslan DF Inal E Isik EG Sanli Y . Extending amyloidosis diagnosis: a rare case of peritoneal AL amyloidosis revealed by 18F-FDG PET/CT and 99mTc-PYP SPECT/CT. Clin Nucl Med. (2025) 50(3):e192–3. 10.1097/RLU.0000000000005603
19.
Basel Allaw M Sinha A Ghafourian K Avery R Weinberg RL Lomasney JW et al Don’t judge a book by its cover: a case report of apolipoprotein A-IV cardiac amyloidosis. Eur Heart J Case Rep. (2023) 7(8):ytad341. 10.1093/ehjcr/ytad341
20.
Wang YW Xie YT Sun XX . Hot hearts on bone scintigraphy are not all amyloidosis: hyperoxaluria-associated cardiomyopathy. Clin Nucl Med. (2024) 49(12):1120–1. 10.1097/RLU.0000000000005411
21.
Codini MA Turner DA Battle WE Hassan P Ali A Messer JV . Value and limitations of technetium-99 m stannous pyrophosphate in the detection of acute myocardial infarction. Am Heart J. (1979) 98(6):752–62. 10.1016/0002-8703(79)90474-5
22.
Pugh BR Buja LM Parkey RW Poliner LR Stokely EM Bonte FJ et al Cardioversion and “false positive” technetium-99 m stannous pyrophosphate myocardial scintigrams. Circulation. (1976) 54(3):399–403. 10.1161/01.CIR.54.3.399
23.
Zhou Q Zuo J Xiao L Jiang L Zhao Z . Bilateral multiple fractures obstruct quantitation of 99mTc-pyrophosphate imaging for cardiac amyloidosis. Clin Nucl Med. (2024) 49(7):e362–3. 10.1097/RLU.0000000000005230
24.
Khatri W Polydefkis M Vaishnav J Saad E Solnes LB Rowe SP . Large pleural effusion: a pitfall in the quantitation of 99mTc-PYP imaging for ATTR cardiac amyloidosis. Clin Nucl Med. (2022) 47(9):e594–5. 10.1097/RLU.0000000000004168
Summary
Keywords
Danon Disease, LAMP2 gene, initiation codon mutation, false-positive scintigraphy, genetic test, gender differences, case report
Citation
Li Q, Sun Z, Qiu Z, Chen Y and Jin W (2025) A novel LAMP2 initiation codon mutation causes Danon Disease: a case report. Front. Cardiovasc. Med. 12:1699732. doi: 10.3389/fcvm.2025.1699732
Received
05 September 2025
Accepted
23 September 2025
Published
08 October 2025
Volume
12 - 2025
Edited by
Zhihua Wang, Chinese Academy of Medical Sciences and Peking Union Medical College, China
Reviewed by
Xujie Liu, Chinese Academy of Medical Sciences and Peking Union Medical College, China
Haocheng Lu, Southern University of Science and Technology, China
Updates
Copyright
© 2025 Li, Sun, Qiu, Chen and Jin.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
* Correspondence: Yanjia Chen cyj193009@163.com Wei Jin jinwei@shsmu.edu.cn
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.