 Cécile Exertier
1*
Cécile Exertier
1*
 Lorenzo Antonelli
1
Lorenzo Antonelli
1
 Vittorio Brufani
2
Vittorio Brufani
2
 Gianni Colotti
1
Gianni Colotti
1
 Andrea Ilari
1
Andrea Ilari
1
 Annarita Fiorillo
1,2*
Annarita Fiorillo
1,2*- 1 Institute of Molecular Biology and Pathology, Italian National Research Council (IBPM-CNR), c/o Department Biochemical Sciences, Sapienza University of Rome, Rome, Italy
- 2 Department Biochemical Sciences, Sapienza University of Rome, Rome, Italy
Crystallographic fragment screening is a powerful methodology that enables the identification of low molecular weight ligands and has shown great promises in drug discovery. In this work we report the results of a fragment screening carried out in an effort to further map the cavities of trypanothione reductase from Trypanosoma brucei (TbTR), a critical target for drug design against human African trypanosomiases (HAT), for which efficient and non-toxic trypanocidal drugs are lacking. Moreover, the conservation of trypanothione reductase among trypanosomatids, including Leishmania, could facilitate the design of a wide-spectrum drug against many parasitic diseases. At the XCHEM facility (Diamond Light Sources, United Kingdom) we performed the soaking of TbTR monoclinic crystals with fragments from DSIpoised and EubOPEN DSIp libraries and we identified eight new hits binding to different cavities of TR including the trypanothione and the NADPH binding cavities. These fragments exhibited affinities ranging from submillimolar to millimolar, as determined by surface plasmon resonance (SPR). While the newly identified fragments did not significantly alter TbTR’s enzymatic activity—consistent with the nature of low-affinity ligands—they provide valuable insights into key interactions of fragments with TR and, together with prior fragment screening campaigns, pave the way towards follow-up chemical optimization into lead compounds.
1 Introduction
Trypanosoma brucei is the causative agent of Human African Trypanosomiasis (HAT), a neglected tropical disease also known as sleeping sickness, that is endemic mainly in low-income countries. The transmission of Trypanosoma brucei to the human host occurs upon the bloodmeal of the tsetse fly and causes fever, skin eruptions and pruritus. If the patient is not treated, the protozoan parasites reach the central nervous system inducing progressive neurological damage (Fall et al., 2022; Moreno et al., 2019). More precisely, the two sub-species, namely T. brucei gambiense and T. brucei rhodesiense lead to two different disease types: the first one is endemic in western and central Africa whereas the second one affects predominantly eastern and southern Africa. However, the main difference lies in the velocity at which the disease progresses towards its fatal outcome: the acute phase is reached over weeks to months or over years whether the host is infected by the rhodesiense or the gambiense strain (Lindner et al., 2025). Notably, the WHO 2025 guidelines regarding the treatment of T. brucei rhodesiense infections recommend the oral intake of fexinidazole, a 2-substituted 5-nitroimidazole whose trypanocidal activity has been established since the 1980s, as the primary treatment. However, it is not yet recommended for young patients (under 6 years old) and numerous side effects have been witnessed including nausea and vomiting, neutropenia, and neuropsychiatric adverse reactions including psychotic disorder and suicidal ideation (Barrett, 2025).
Considering the toxicity of such treatments and the ever-present risk of resistance development, it is of the utmost importance to keep on providing new and safer alternatives. Although an important effort has been put into drug repurposing and drug design based on SAR (structure-activity relationship) on pre-existing ligands and suboptimal inhibitors, it is mandatory to identify new potent lead-compounds.
Fragment-based screening (FBS) is an innovative and powerful technique to identify new ligand scaffolds (∼300 Da) binding to a protein using X-ray crystallography, a technique providing high resolution information and paving the way towards the development of tailor-made drug leads. X-ray crystallography represents a gold-standard methodology for the highly sensitive identification of weak binders, providing direct structural information for chemical follow-up. Technological developments, including crystallization and data collection automation, have made crystallography more efficient, and better suited for high-throughput applications, such as structure-based fragment screening (Thomas et al., 2019; Douangamath et al., 2021). As of now, FBS has been widely used to map out cavities of numerous proteins as shown by the ample number of so-called “PanDDA analysis group depositions” present in the Protein Data Bank (PDB). More precisely, PanDDA (Pan-Dataset Density Analysis) is a method that enhances the detection of weak or low-occupancy ligand binding in crystallographic fragment screening by statistically comparing electron density maps across multiple datasets to reveal subtle binding events that are otherwise hidden (Pearse et al., 2017). The “PanDDA analysis group depositions” entries regard the human proteome as well as proteins from pathogenic agents such as viruses (SARS Cov-2; (Gahbauer et al., 2023), Chikungunya, enterovirus, zika), bacteria (Bacillus subtilis, Pseudomonas aeruginosa, Porphyromonas gingivalis) and parasites (Trypanosoma cruzi, Trypanosoma brucei). Despite the great number of entries already deposited on the PDB, crystallographic fragment screening campaigns have largely remained undiscussed and have not been fully exploited by medicinal chemists in scientific publications (McIntyre et al., 2017; Bradshaw et al., 2024; Huang et al., 2024; Huang et al., 2025; Füsser et al., 2023; Wever et al., 2024; Exertier et al., 2024; Fiorillo et al., 2022; Ni et al., 2024; Gahbauer et al., 2023).
However in this context, we recently reported a first crystallographic fragment screening towards a protein from Trypanosoma brucei, namely the trypanothione reductase (TbTR) and we described the successful merging and elongation of the identified hits into potent leads, testifying of the potency of crystallography-based fragment screening (Fiorillo et al., 2022; Exertier et al., 2024).
Trypanothione reductase (TR) is a homodimeric enzyme crucial for trypanosomatids as it is essential to survive the extreme oxidative environment inside the host macrophages. In fact, TR carries out the NADPH-dependent reduction of the trypanothione, a peculiar molecule made of two glutathione linked by a spermidine, that is employed by trypanosomatids to maintain thiol homeostasis (Jaeger and Flohé, 2006).
Trypanothione reductase is a well-known target in drug discovery against trypanosomiases and leishmaniases. As a matter of fact, TR gathers several advantages: (i) it is well characterized and druggable (Baiocco et al., 2009b; Baiocco et al., 2013; Angiulli et al., 2015; Baiocco et al., 2009a), (ii) the trypanothione binding site significatively differs from that of its closest human homolog glutathione reductase (<40% overall sequence identity), therefore it enables the design of selective drugs, (iii) it is nearly impossible to generate viable Leishmania mutants devoid of TR catalytic activity (Krieger et al., 2002; Tovar et al., 1998a; Tovar et al., 1998b), (iv) the trypanothione binding site is highly conserved among all trypanosomatids and could therefore favor the design of a multi-parasite drug (Krieger et al., 2002; Tovar et al., 1998a; Battista et al., 2020).
In this study, we enrich the mapping of TbTR cavities through fragment screening, identifying novel scaffolds by leveraging the vast chemical space of the DSI-poised and EubOPEN libraries (Douangamath et al., 2021). Notably, the initial FBS campaign by Fiorillo et al. focused on soaking orthorhombic crystals with the first half of the DSI-poised library (Fiorillo et al., 2022). Here, we report the successful soaking of TbTR monoclinic crystals with fragments from the second half of the DSI-poised library, as well as the EubOPEN library, to further explore TR cavity mapping. The binding and inhibition activity of the most interesting hits were evaluated by surface plasmon resonance experiments and spectroscopic inhibition assays. The mapping of TR hotspots reported here contributes to the effort of providing new scaffolds amenable for chemical optimization into lead-like compounds.
2 Materials and methods
2.1 Expression and purification
TR from Trypanosoma brucei (TbTR) was expressed and purified as previously reported (Fiorillo et al., 2022; Exertier et al., 2024). In details, the gene of TbTR was subcloned in a pET15b vector. After transformation in E. coli BL21, a 10 mL starter culture was grown overnight to inoculate the next day 1 L of LB medium supplemented with antibiotics. The culture was incubated at 37°C, 180 rpm until the optical density reached 0.6. TbTR overexpression was induced upon addition of 1 mM IPTG. The cells were incubated at 37°C, 180 rpm for four more hours prior to harvest by centrifugation.
Cell pellets were resuspended in lysis buffer (50 mM Tris pH 8.0, 300 mM NaCl, 5 mM imidazole, 10 mM MgCl2, 0.1 mM PMSF and one tablet of Pierce antiprotease cocktail). Cells were lysed by sonication, cell debris were discarded by centrifugation and the soluble phase was loaded onto a HiTrap column for immobilized-metal affinity chromatography (IMAC). Elution of TbTR occurred upon application of an imidazole gradient. The His-tag was cleaved from TbTR construct using 1U of Thrombin per 0.1 mg of protein. Tag-free TbTR was further purified from the protease using a benzamidine resin and from the uncleaved protein by reverse-IMAC. Finally, TbTR buffer was exchanged into 20 mM HEPES pH 7.4 by dialysis.
2.2 Fragment screening experiment
Crystallization plates were partly set up at the XCHEM facility of Diamond Light Source using a Mosquito crystal robot from SPT Labtech and partly set up in our home laboratory using an Oryx4 robot from Douglas Instruments. In both cases, TbTR crystallizes at 17 mg/mL in 22% 2-Methyl-2,4-pentanediol (MPD), 14% PEG3350, imidazole 40 mM pH 8 and 50 mM NaBr. Surprisingly, the crystallization conditions were similar to those used in the previous screening, yet yielded crystals with a different symmetry (monoclinic vs. orthorhombic). This behavior, which we could not rationalize or control, is attributed to variations between sample batches. An ECHO acoustic liquid dispenser was used to dispense 503 fragments from second half of DSI Poised and the entire EubOPEN libraries onto the crystallization drops, resulting in a maximum concentration of 50 mM fragment and 10% DMSO (Collins et al., 2017). Soaked crystals were then incubated for 1 h at 20°C. Crystals were rapidly mounted on cryoloops thanks to the OLT shifter (Wright et al., 2021) and directly flash frozen in liquid nitrogen prior the diffraction experiments, which were carried out both at the I03 and I04-1 beamline. Data processing was carried out using the XCHEM Explorer (XCE) pipeline (Krojer et al., 2017). Hits were identified using Pan-Dataset Density Analysis (PANDDA) (Pearce et al., 2017), a first refinement was performed by Buster (Smart et al., 2012), implemented on XCE. The final refinement of the occupancy and geometry was performed using Phenix-1.21.2-5419 (Liebschner et al., 2019). Supplementary Table S1 reports the data collection and refinement statistics.
2.3 Surface plasmon resonance
Surface Plasmon Resonance (SPR) experiments were carried out to assess the thermodynamic parameters of the interaction between TbTR (ligand) and analytes (fragments) in a Sartorius Octet SF3 apparatus. The ligand was immobilized on an Octet CDL Sensor Chip, which consists of a carboxymethyl dextran 3D matrix which allows a highly stable covalent bond with the ligand. The ligand was immobilized with amine coupling chemistry. The amount of immobilized TbTR was detected by mass concentration-dependent changes in the refractive index on the sensorchip surface, and corresponded to about 3,800 resonance units (RU). Analytes were dissolved in 100% DMSO at a concentration of 50 mM, and subsequently diluted in sterile HEPES 20 mM, pH 7.4, NaCl 150 mM, 0.005% surfactant Tween20 to yield 2% DMSO final concentration (HSP-2%D buffer) and final analyte concentration: 1 mM. Further dilutions and all the experiments were carried out at 25°C in degassed HSP-2%D buffer. After ligand immobilization, analytes were injected at different concentrations, i.e., 33 μM, 100 μM, and 300 μM, into a moving stream of buffer (Valentini et al., 2022; Masciarelli et al., 2014). At higher concentrations, the signal traces became increasingly difficult to interpret, likely due to interference from non-specific binding and contributions from solubilized components in the solution. In all experiments, the increase in RU relative to baseline indicates complex formation between the immobilized TbTR ligand and the fragments (analytes). The plateau region, when present, represents the steady-state phase of the interaction. The decrease in RU after 200 s indicates analyte dissociation from the immobilized ligand after HSP-2%D buffer injection. As a negative control, sensor chips were treated as described above in the absence of immobilized TbTR. Values of the plateau signal at steady-state (Req) and full fittings with 1 and 2 sites were calculated from overall kinetic evaluation of the sensorgrams using the Octet SPR Analysis software with a one-site kinetic model.
2.4 Inhibition assays
Fragments were purchased from Enamine and were solubilized in DMSO to reach a stock concentration of 50 mM. Typically, TbTR enzymatic reaction at 25°C was followed in the presence of 50 nM TbTR, 100 μM fragments (2% DMSO final concentration), 150 μM trypanothione, 100 uM NADPH in 50 mM HEPES pH 7.4, 40 mM NaCl at 340 nm using a spectrophotometer (JASCO V650) equipped with a Peltier (JASCO EHC 716). Experiments were carried out only for the fragments identified in catalytic cavities (trypanothione and NADPH cavities). The reaction initiated upon NADPH addition was followed at 340 nm. Initial velocities were used to calculate the residual activity of TbTR compared to the activity in the absence of fragments. Each data point is the average of triplicate experiments. Data analysis was performed with Prism 9, and graphs were made with Labplot2.
3 Results
3.1 Fragment screening at XCHEM
Fragment screening has been performed on TbTR monoclinic crystals grown in 13% PEG3350, 24% MPD, 40 mM imidazole pH 8, 50 mM NaBr. It is worth mentioning that although the crystallization conditions are rather identical, the previously reported fragment screening experiment was performed on orthorhombic crystals of TbTR (Fiorillo et al., 2022). The monoclinic packing did not affect the quality of crystals, as these TbTR P21 crystals diffracted at high resolution (1.62–2.11Å) compared to the orthorombic ones (1.65–1.97Å, Fiorillo et al., 2022). Molecular replacement revealed the presence of two TbTR homodimers in the asymmetric unit instead of the single homodimer observed for the orthorhombic crystals, and the different packing arrangement did not significantly alter the solvent-accessible surface area (SASA) of the TbTR dimer (Supplementary Figures S1, S2).
The 1-h soaking of monoclinic TbTR crystals with 50 mM fragments from the second fraction of DSIpoised and the entire EubOPEN libraries, designed for favoring a follow-up chemical expansion (Cox et al., 2016) was performed at the XCHEM facility while X-ray diffraction was carried out at both Diamond Light Source I03 and I04-1 beamlines.
Eight clear hits have been identified using the Pan-Dataset Density Analysis (PanDDA) implemented on the semi-automatic XCHEM-Explorer pipeline (Supplementary Table S2). These fragments were observed in five different sites with 0.6–1.0 occupancy (Figure 1; Table 1): (i) the most populated site is the trypanothione cavity, where compounds target several sub-pockets; (ii) the NADPH cavity, in the close vicinity of the doorstop pocket; (iii) three other sites accounting for pockets with no known enzymatic function. Notably fragment 79, a rather promiscuous compound, is found in three distinct cavities.
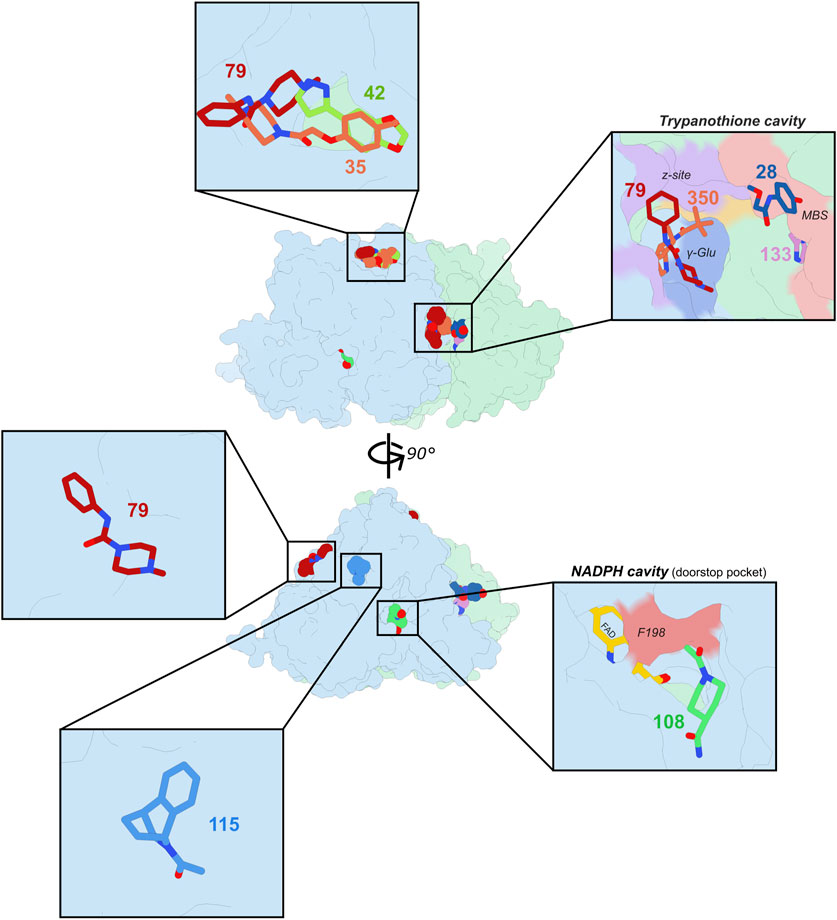
Figure 1. Fragment screening carried out on TbTR. Two 90°-related orientations of TbTR are shown at the center with the fragments identified by the PANDDA analysis. Inserts show more in details the pockets, cavities or surfaces with which fragments interact. Monomers from the TbTR homodimer are shown in light blue and light green and fragments are represented as colored spheres (center) or sticks (insets).
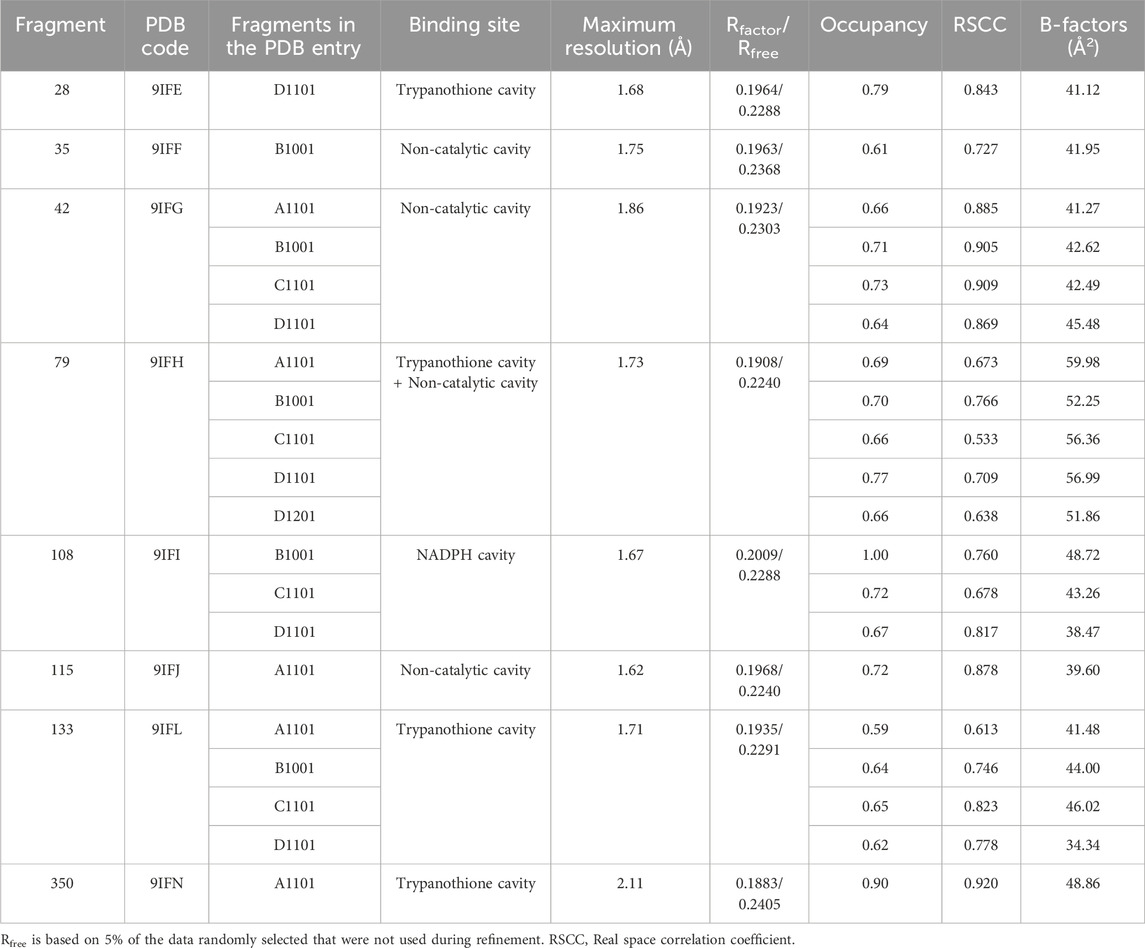
Table 1. Ligand details and overall quality.
3.2 The trypanothione binding cavity is the most populated site
Over the years, the trypanothione cavity has been the preferred target for the design of TR inhibitors (Exertier et al., 2024; Baiocco et al., 2013; Battista et al., 2020; Ilari et al., 2018; Battista et al., 2022; Turcano et al., 2018; Colotti et al., 2020; Patterson et al., 2011). This is primarily due to the larger size of the cavity and its distinct surface charge distribution compared to that of the glutathione cavity found in the closest human homolog, glutathione reductase (GR), which provide a significant advantage in the development of selective inhibitors through structure-activity relationship (SAR) studies. We counted four fragments accommodating in the trypanothione cavity. The “trypanothione cavity” inset of Figure 1 clearly shows that fragments may be divided in two groups: those that interact with the so-called mepacrine binding site (MBS), i.e., a hydrophobic patch localized at the entrance of the trypanothione cavity; and those binding to the z-site, a more profound hydrophobic alcove.
In the z-site, event maps arising from the PanDDA analysis clearly indicated the presence of fragments 79 and 350 (Figures 2A,B; Supplementary Table S2). Importantly, 79 contains a piperazine ring linked to a phenyl group by an amide. The piperazine is stabilized due to interactions with E467; this interaction has also been observed previously with piperazine-containing fragments binding to TbTR (Supplementary Figure S3) (Fiorillo et al., 2022), although abolished upon merging and elongation of these fragments (Exertier et al., 2024).
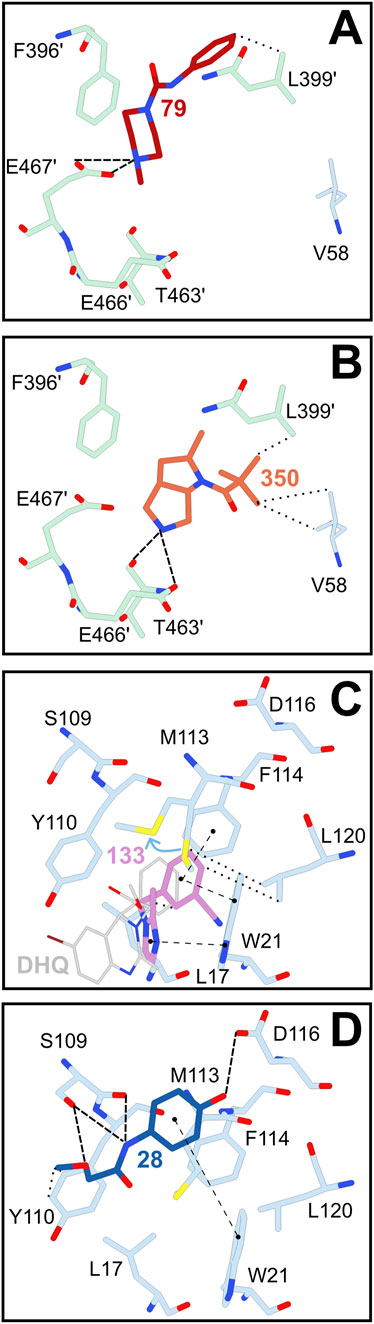
Figure 2. Hits identified in the trypanothione cavity of TbTR. Fragments 79 (A) and 350 (B) were identified close to the z-site. Fragments 133 (C) and 28 (D) were identified in the mepacrine binding site (MBS), a more external region of the trypanothione binding site. Notably, fragment 133 accomodates in the same sub-cavity formed upon displacement of M113 (indicated by a light blue arrow) as dihydroquinazolinic (DHQ in grey) compounds (pdb entry 2WP5 from Patterson et al. (2011)). Residues and fragments are displayed as sticks. Less than 3.2 Å-long interactions between residues and fragments are displayed as follows: π-π stackings are represented as dashed lines with round extremities, hydrophobic interactions as dotted lines and electrostatic interactions as simple dashed lines.
Other two fragments, namely 28 and 133, bind to the mepacrine binding site but in rather separate and mutually exclusive locations (Figures 2C,D). Interestingly, fragment 133 occupies a sub-cavity generated by the displacement of M113. A similar mode of binding has been observed for dihydroquinazoline compounds (DHQ), that account for some of the most potent TR inhibitors (Figure 2C; Patterson et al., 2011). Indeed, fragment 133 enters deeper into this sub-cavity compared to DHQs, although the moiety exposed towards the cavity is less bulky.
3.3 One fragment targets the doorstop pocket
We identified only one hit in the NADPH binding region, close to the so-called doorstop pocket (Ardini et al., 2024). F198 plays the role of a gatekeeper: in the absence of NADPH, F198 is locked in a perpendicular position with respect to the FAD. When NADPH enters the cavity however, F198 swings out from its position to allow the nicotinamide moiety to interact with the FAD isoallaxazine ring (Battista et al., 2020). Ligand binding within the doorstop pocket can restrict the movement of F198, consequently hindering NADPH binding and TR activity.
Fragment 108 is stabilized in this position mainly through hydrophobic interactions. In fact, it is surrounded by numerous apolar side chains, amongst them F198, F230, L332, L334 and V362. Nonetheless, the position of this fragment is also supported by H-bonds with water molecules (Figure 3).
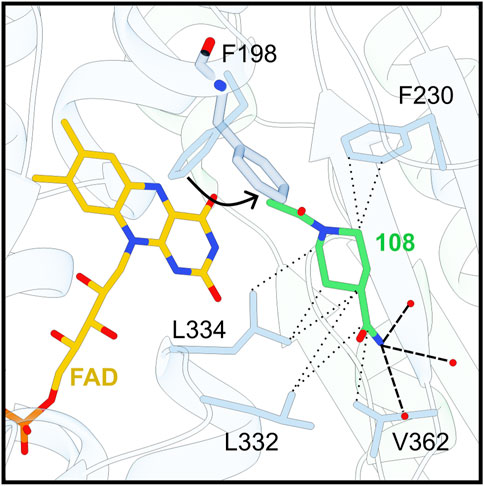
Figure 3. Fragment 108 accommodates in the doorstop pocket. F198 swing out movement is indicated by a black arrow. Secondary structures are displayed as ribbons, side chains and chemical compounds as sticks, and waters as red dots. Less than 3.2 Å-long interactions between residues and 108 are displayed as follows: Electrostatic interactions and hydrophobic/π-π interactions are shown respectively as simple dashed lines and dashed lines with dot extremities.
Fiorillo and colleagues previously reported the identification of several other fragments accommodating in the doorstop pocket, with the hypothesis that they could act as allosteric inhibitors. Amongst them, Fiorillo’s 68 (hereafter referred to as F-68) also contains two amide groups connected to a nitrogen heterocycle (piperidine instead of piperazine), and its position partially overlaps to that of fragment 108 (Supplementary Figure S4; Fiorillo et al., 2022).
3.4 Non-enzymatic cavities
Four fragments were found in other binding sites with no known enzymatic activity. One of these sites, containing the three fragments 35, 42 and 79 (Figure 4), was already detected in the previous fragment screening performed by Fiorillo and coworkers.
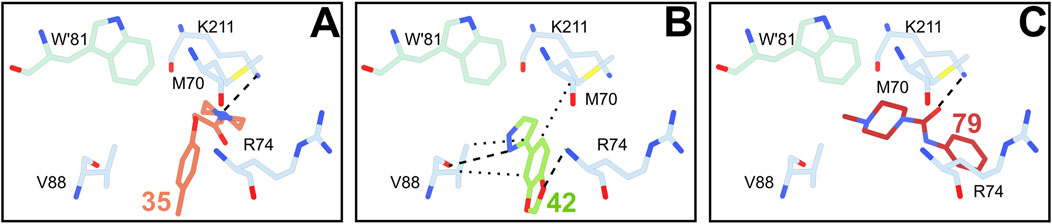
Figure 4. Fragments 35 (A), 42 (B) and 79 (C) binding to the main non-enzymatic site. Residue side chains and chemical compounds as represented as sticks, and waters as red dots. Less than 3.5 Å-long interactions between residues and fragments are displayed as follows: Electrostatic interactions and hydrophobic are shown respectively as simple dashed and dotted lines.
This site is rather exposed to solvent and close to the dimerization interface, involving both TR monomers through the protrusion of W61’ in the bottom of the cavity lined by M70, R74, V88 and K211. It must be noted that the binding of fragment 79 in this site is likely an artifact due to crystal packing. Indeed, although the binding pose of 79 is stabilized by a meaningful polar interaction between the amide oxygen and the side chain of K211, its benzene ring extends out of the cleft apparently towards the solvent but actually toward adjacent protein subunit. In contrast, 35 and 42 fully adhere to the cleft and share a common benzene ring whose position is conserved. Notably, other ligands have been previously detected in the same site, namely the fragment F-60 (Fiorillo et al., 2022) and a drug-like inhibitor binding this cleft as a secondary site (Turcano et al., 2020).
The two other non-enzymatic binding sites contain either fragment 79 or 115 (Supplementary Figure S5). These two sites are mostly superficial grooves that however show a certain affinity, although probably low, for these two fragments. While Fiorillo and colleagues already observed fragments binding to 115 groove, 79 binds to a shallow site that has never been identified before. In these last three binding sites, the occupancy of each fragment is relatively moderate (0.6–0.7) and interactions with the peptidic chains are rather scarce.
3.5 Fragment binding by SPR
The eight fragments identified here plus seven selected from the previous screen were tested by SPR to assess their affinity for TbTR. As previously stated, fragments are characterized by low mass (150–300 Da) and low affinity (μM-mM), making the characterization of their binding through SPR a challenging task that requires extensive experimental effort for optimization (Tiwari et al., 2021). However, in our study, we were more interested in obtaining a rough estimation of binding affinity rather than an exact measurement of the dissociation constant (KD) to rank the fragments for their binding properties. Thus, by testing three concentrations (33, 100, 300 μM), we could detect binding for 12 out of 15 fragments with TbTR, albeit with KD values in the submillimolar-millimolar range (Table 2; Supplementary Figure S6). The compounds endowed with the highest affinities were 28 and 350 in the trypanothione cavity, 108 and F-117 in the NADPH binding site, and 42 and F-60 in the non-enzymatic cavity. Although none of these ligands emerges as a particularly strong binder, their detectable binding indicates that some may potentially exert inhibitory effects on TbTR.
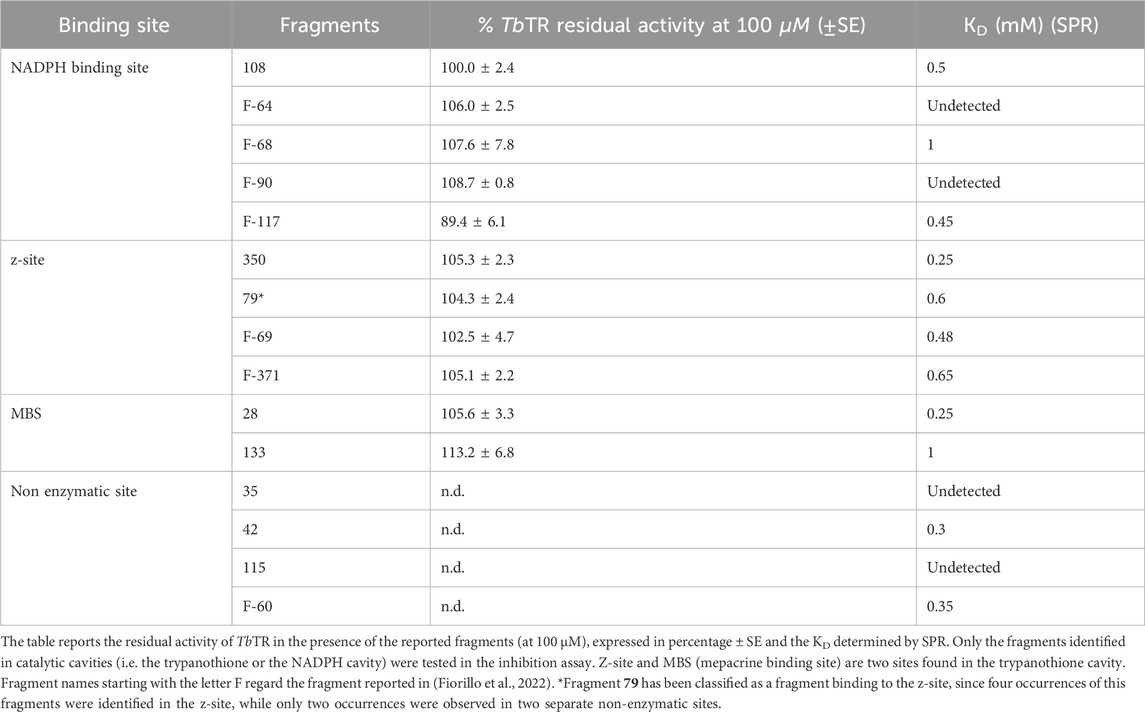
Table 2. Inhibition assay and binding by surface plasmon resonance.
3.6 Inhibition assays
The fragments found in the substrate cavities, namely the trypanothione and NADPH cavities, during the fragment screening reported in this work and in Fiorillo et al., 2022 were tested against TbTR to evaluate their inhibition activity (Table 2). Most of these fragments do not significantly alter the activity of TbTR even at a concentration of 100 μM. This is not unexpected as fragments are usually small low-affinity ligands and, in this context, their binding may not be sufficient to impede the entry and positioning of the substrates. Consequently, these fragments do not substantially hinder TbTR’s ability to perform its full and effective reduction of trypanothione.
Unlike hypothesized above, 108 does not affect TbTR activity, suggesting that the fragment does not sterically hinder the swinging movement of the gatekeeper F198.
However, it is worth mentioning that fragment F-117 at least inhibits TbTR reduction of trypanothione of ∼11% (Residual TbTR activity = 89.4%). F-117 has been identified in the doorstop pocket and appears to be among the fragments that protrude the most towards the FAD gatekeeper F198 (Supplementary Figure S4). This suggests that F-117 is capable to obstruct the movement of F198 side chain, thereby preventing NADPH nicotinamide moiety from positioning above the FAD isoalloxazine ring, disturbing the transfer of electrons from NADPH to the FAD. Thus, F-117 emerges as a good candidate for elongation and merging and a promising starting point for the design of a potent inhibitors.
Notably, a similar a strategy has been successfully applied for the design of noncovalent inhibitors targeting the doorstop pocket of the thioredoxin glutathione reductase from Schistosoma mansoni (Petukhova et al., 2023).
4 Discussion
This manuscript reports the findings from the second crystallographic fragment screening conducted on trypanothione reductase (TbTR) from Trypanosoma brucei, aimed at expanding the chemical space of binding fragments and enhancing the mapping of the protein surface. The screening, performed on monoclinic crystals, identified eight novel fragments distributed across five binding sites, complementing the 12 fragments previously characterized in orthorhombic crystals. Notably, all identified sites, except for one weakly populated and superficially engaged site, had been previously highlighted in earlier screening. While this experiment does not further extend the overall mapping of binding cavities of TbTR, it reinforces the classification of these sites as true hot spots that appear to be unaffected by crystal packing artifacts.
In the attempt to prioritize the fragments identified, we conducted binding studies using surface plasmon resonance (SPR) alongside enzyme inhibition assays, including both the newly identified fragments as well as selected fragments from prior studies. Overall, most of the fragments demonstrated varying degrees of binding affinity, although none achieved strong binding characteristics, with dissociation constants ranging from sub-millimolar to millimolar levels, and only one exhibited significant inhibitory activity at a concentration of 100 µM. This finding is neither surprising nor unexpected considering the nature of small ligands; numerous studies have demonstrated that crystallographic fragment screening is a powerful tool for detecting weak binders that may elude other biophysical techniques (Douangamath et al., 2021; Patel et al., 2014; Schiebel et al., 2016; Dubianok, 2021). Nevertheless, the presence of weak binding interactions should not lead to the dismissal of these fragments. Rather, various works have shown that structure-based optimization of weak fragments can result in the development of potent inhibitors (Gahbauer et al., 2023; Exertier et al., 2024; Wever et al., 2024; Ni et al., 2024).
Therefore, only a thorough structural analysis of the interactions, complemented by relevant activity data, can truly reveal the potential of each fragment and each binding site.
Fragment 108 binds to the region known as the doorstop pocket, located near the NADPH binding site, joining four previously identified fragments: F-64, F-68, F-90, and F-117. A closer examination (Supplementary Figure S4) reveals that only F-64, F-68, and F-117 can effectively prevent the swing of Phe198 necessary for NADPH binding, whereas fragments 108 and F-90, which bind more distantly, do not have direct interactions with the gatekeeper residue. On the other hand, fragments 108 and F-117 exhibit the highest affinities, as determined by surface plasmon resonance (SPR). Taken together, these observations provide a rationale for the fact that only F-117 demonstrates inhibitory activity and suggest that any follow up for fragments binding this site should focus on creating steric hindrance as close as possible to Phe198. Moreover, it is important to highlight that the activity of F-117 provides the first significant validation of the doorstop pocket as an effective target for inhibition of TbTR.
Regarding the trypanothione cavity, none of the fragments exhibited inhibitory activity, although several displayed detectable affinities. This finding is somewhat unexpected, as other fragments that bind to the z-site were able to reduce TR activity by approximately 15% (Fiorillo et al., 2022). However, it can be speculated that these fragments had a higher affinity compared to those tested here. Notably, fragment 133, despite its low affinity and lack of inhibitory activity—indeed, it appears to enhance TR activity—exhibits a particularly appealing mode of binding. As previously described, fragment 133 anchors itself in a subpocket it creates in the MBS, similarly to dihydrochinazolinic (DHQ) compounds, but penetrates deeper into the subpocket (Figure 2C), although it protrudes slightly into the trypanothione cavity and therefore does not provide inhibition. DHQ derivatives demonstrate potent activity against TR (submicromolar IC50), yet they have been sidelined due to toxicity concerns (Patterson et al., 2011). Fragment 133 offers a unique opportunity to utilize the same binding mode as DHQ while allowing for modifications to the chemical scaffold that could potentially mitigate toxicity concerns. Notably, the superimposition of fragment 133 and compound 29a, a derivative of DHQ, indicates that replacing the 4-methylphenyl group with a bulkier condensed aromatic group may enhance the affinity and selectivity of DHQ compounds when targeting TR proteins.
Finally, noteworthy observations can also be drawn from fragments binding to the non-enzymatic sites: while ligands such as 35 and 42 may initially appear insignificant due to their binding to a site not recognized for altering activity, they could hold considerable relevance for the development of non-inhibitory ligands, such as PROTACs and molecular glue degraders.
Fragments are a valuable starting point for the development of drug leads, but significant effort is required to progress them into potent ligands. Recent advances in artificial intelligence (AI) and computational methods are increasingly complementing fragment-based drug discovery workflows. These tools can effectively aid fragment elaboration, from the prioritization of fragments to the design and synthesis of derivatives, thereby accelerating the entire fragment-to-lead process (Yoo et al., 2025). However, they still face challenges in accurately identifying weak small binders and defining binding poses, particularly in cryptic pockets, which underscores the continued pivotal role of experimental fragment screening.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
CE: Writing – review and editing, Validation, Investigation, Methodology, Supervision, Visualization, Formal Analysis, Software, Writing – original draft, Data curation. LA: Writing – review and editing, Formal Analysis, Methodology, Investigation. VB: Investigation, Writing – review and editing, Formal Analysis, Methodology. GC: Conceptualization, Visualization, Formal Analysis, Validation, Data curation, Methodology, Writing – review and editing, Software, Writing – original draft, Investigation. AI: Validation, Investigation, Resources, Conceptualization, Project administration, Writing – review and editing, Supervision, Funding acquisition, Writing – original draft. AF: Writing – review and editing, Software, Writing – original draft, Resources, Visualization, Investigation, Validation, Methodology, Funding acquisition, Data curation, Formal Analysis, Supervision, Project administration, Conceptualization.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was supported by MIUR-FISR2019, Project n°FISR2019_03796 “Proteolysis targeting chimeras (PROTACs) to treat leishmaniasis”-PROLEISH, by CNCCS (Collezione Nazionale di Composti Chimici e Centro di Screening) FOE 2021 (awarded to AI). This work benefited from access to the XChem facility at Diamond Light Source, an Instruct-ERIC centre, financial support was provided by iNEXT Discovery proposal 22027 (awarded to AF). Sapienza University of Rome is gratefully acknowledged for grant RM123188F763A9D5, RP12117A8675EBE7 to AF. GC and AI received financial support from the project PRIN MUR 2022HYF8KS.
Acknowledgments
The authors would like to acknowledge the Diamond Light Source for access to the fragment screening facility XChem, for usage of DSi-Poised library and for beamtime on beamlines I03 and I04-1 under proposal LB30489. The authors acknowledge the Biocrystal Facility and the support from the project “Potentiating the Italian Capacity for Structural Biology Services in Instruct Eric (ITACA.SB)” (Project No. IR0000009) within the call MUR 3264/2021 PNRR M4/C2/L3.1.1, funded by the European Union NextGenerationEU. CE and AI are members of the SBN@CNR network.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchbi.2025.1605579/full#supplementary-material
References
Angiulli, G., Lantella, A., Forte, E., Angelucci, F., Colotti, G., Ilari, A., et al. (2015). Leishmania infantum trypanothione reductase is a promiscuous enzyme carrying an NADPH:O2 oxidoreductase activity shared by glutathione reductase. Biochimica Biophysica Acta (BBA) - General Subj. 1850 (9), 1891–1897. doi:10.1016/j.bbagen.2015.05.022
Ardini, M., Aboagye, S. Y., Petukhova, V. Z., Kastrati, I., Ippoliti, R., Thatcher, G. R. J., et al. (2024). The “doorstop pocket” in thioredoxin Reductases─An unexpected druggable regulator of the catalytic machinery. J. Med. Chem. 67 (18), 15947–15967. doi:10.1021/acs.jmedchem.4c00669
Baiocco, P., Colotti, G., Franceschini, S., and Ilari, A. (2009b). Molecular basis of antimony treatment in leishmaniasis. J. Med. Chem. 52 (8), 2603–2612. doi:10.1021/jm900185q
Baiocco, P., Franceschini, S., Ilari, A., and Colotti, G. (2009a). Trypanothione reductase from Leishmania infantum: cloning, expression, purification, crystallization and preliminary X-ray data analysis. Protein Peptide Lett. 16 (2), 196–200. doi:10.2174/092986609787316306
Baiocco, P., Poce, G., Alfonso, S., Cocozza, M., Porretta, G. C., Colotti, G., et al. (2013). Inhibition of Leishmania infantum trypanothione reductase by azole-based compounds: a comparative analysis with its physiological substrate by X-ray crystallography. ChemMedChem 8 (7), 1175–1183. doi:10.1002/cmdc.201300176
Barrett, M. P. (2025). Transforming the chemotherapy of human African trypanosomiasis. Clin. Microbiol. Rev. 38, e0015323. doi:10.1128/cmr.00153-23
Battista, T., Colotti, G., Ilari, A., and Fiorillo, A. (2020). Targeting trypanothione reductase, a key enzyme in the redox trypanosomatid metabolism, to develop new drugs against leishmaniasis and trypanosomiases. Molecules 25 (8), 1924. doi:10.3390/molecules25081924
Battista, T., Federico, S., Brogi, S., Pozzetti, L., Khan, T., Butini, S., et al. (2022). Optimization of potent and specific trypanothione reductase inhibitors: a structure-based drug discovery approach. ACS Infect. Dis. 8 (8), 1687–1699. doi:10.1021/acsinfecdis.2c00325
Bradshaw, W. J., Kennedy, E. C., Moreira, T., Smith, L. A., Chalk, R., Katis, V. L., et al. (2024). Regulation of inositol 5-phosphatase activity by the C2 domain of SHIP1 and SHIP2. Structure 32 (4), 453–466.e6. doi:10.1016/j.str.2024.01.005
Collins, P. M., Ng, J. T., Talon, R., Nekrosiute, K., Krojer, T., Douangamath, A., et al. (2017). Gentle, fast and effective crystal soaking by acoustic dispensing. Acta Crystallogr. D. Struct. Biol. 73 (3), 246–255. doi:10.1107/s205979831700331x
Colotti, G., Saccoliti, F., Gramiccia, M., Di Muccio, T., Prakash, J., Yadav, S., et al. (2020). Structure-guided approach to identify a novel class of anti-leishmaniasis diaryl sulfide compounds targeting the trypanothione metabolism. Amino Acids 52 (2), 247–259. doi:10.1007/s00726-019-02731-4
Cox, O. B., Krojer, T., Collins, P., Monteiro, O., Talon, R., Bradley, A., et al. (2016). A poised fragment library enables rapid synthetic expansion yielding the first reported inhibitors of PHIP(2), an atypical bromodomain. Chem. Sci. 7 (3), 2322–2330. doi:10.1039/c5sc03115j
Douangamath, A., Powell, A., Fearon, D., Collins, P. M., Talon, R., Krojer, T., et al. (2021). Achieving efficient fragment screening at XChem facility at Diamond light Source. J. Vis. Exp. JoVE (171). doi:10.3791/62414
Dubianok, Y. (2021). Too weak for biophysics: prioritising and progressing fragment hits from X-ray crystallographic screening. University of Oxford. [PhD thesis]. Available online at: https://ora.ox.ac.uk/objects/uuid:7664169a-7965-4819-97df-fc463d795208
Exertier, C., Salerno, A., Antonelli, L., Fiorillo, A., Ocello, R., Seghetti, F., et al. (2024). Fragment merging, growing, and linking identify new trypanothione reductase inhibitors for leishmaniasis. J. Med. Chem. 67 (1), 402–419. doi:10.1021/acs.jmedchem.3c01439
Fall, F., Mamede, L., Schioppa, L., Ledoux, A., De Tullio, P., Michels, P., et al. (2022). Trypanosoma brucei: metabolomics for analysis of cellular metabolism and drug discovery. Metabolomics 18 (4), 20. doi:10.1007/s11306-022-01880-0
Fiorillo, A., Colotti, G., Exertier, C., Liuzzi, A., Seghetti, F., Salerno, A., et al. (2022). Innovative approach for a classic target: fragment screening on trypanothione reductase reveals new opportunities for drug design. Front. Mol. Biosci. 9, 900882. doi:10.3389/fmolb.2022.900882
Füsser, F. T., Wollenhaupt, J., Weiss, M. S., Kümmel, D., and Koch, O. (2023). Novel starting points for fragment-based drug design against mycobacterial thioredoxin reductase identified using crystallographic fragment screening. Acta Crystallogr. D. Struct. Biol. 79 (9), 857–865. doi:10.1107/s2059798323005223
Gahbauer, S., Correy, G. J., Schuller, M., Ferla, M. P., Doruk, Y. U., Rachman, M., et al. (2023). Iterative computational design and crystallographic screening identifies potent inhibitors targeting the Nsp3 macrodomain of SARS-CoV-2. Proc. Natl. Acad. Sci. U. S. A. 120 (2), e2212931120. doi:10.1073/pnas.2212931120
Huang, C. Y., Metz, A., Lange, R., Artico, N., Potot, C., Hazemann, J., et al. (2024). Fragment-based screening targeting an open form of the SARS-CoV-2 main protease binding pocket. Acta Crystallogr. D. Struct. Biol. 80 (2), 123–136. doi:10.1107/s2059798324000329
Huang, L., Wang, W., Zhu, Z., Li, Q., Li, M., Zhou, H., et al. (2025). Novel starting points for fragment-based drug design against human heat-shock protein 90 identified using crystallographic fragment screening. IUCrJ 12 (2), 177–187. doi:10.1107/s2052252524012247
Ilari, A., Genovese, I., Fiorillo, F., Battista, T., De Ionna, I., Fiorillo, A., et al. (2018). Toward a drug against all kinetoplastids: from LeishBox to specific and potent trypanothione reductase inhibitors. Mol. Pharm. 15 (8), 3069–3078. doi:10.1021/acs.molpharmaceut.8b00185
Jaeger, T., and Flohé, L. (2006). The thiol-based redox networks of pathogens: unexploited targets in the search for new drugs. BioFactors 27 (1–4), 109–120. doi:10.1002/biof.5520270110
Krieger, S., Schwarz, W., Ariyanayagam, M. R., Fairlamb, A. H., Krauth Siegel, R. L., and Clayton, C. (2002). Trypanosomes lacking trypanothione reductase are avirulent and show increased sensitivity to oxidative stress. Mol. Microbiol. 35 (3), 542–552. doi:10.1046/j.1365-2958.2000.01721.x
Krojer, T., Talon, R., Pearce, N., Collins, P., Douangamath, A., Brandao-Neto, J., et al. (2017). The XChemExplorer graphical workflow tool for routine or large-scale protein–ligand structure determination. Acta Crystallogr. D. Struct. Biol. 73 (3), 267–278. doi:10.1107/s2059798316020234
Liebschner, D., Afonine, P. V., Baker, M. L., Bunkóczi, G., Chen, V. B., Croll, T. I., et al. (2019). Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D. Struct. Biol. 75 (10), 861–877. doi:10.1107/s2059798319011471
Lindner, A. K., Lejon, V., Barrett, M. P., Blumberg, L., Bukachi, S. A., Chancey, R. J., et al. (2025). New WHO guidelines for treating rhodesiense human African trypanosomiasis: expanded indications for fexinidazole and pentamidine. Lancet Infect. Dis. 25 (2), e77–e85. doi:10.1016/s1473-3099(24)00581-4
Masciarelli, S., Quaranta, R., Iosue, I., Colotti, G., Padula, F., Varchi, G., et al. (2014). A small-molecule targeting the MicroRNA binding domain of argonaute 2 improves the retinoic acid differentiation response of the acute promyelocytic leukemia cell line NB4. ACS Chem. Biol. 9 (8), 1674–1679. doi:10.1021/cb500286b
McIntyre, P. J., Collins, P. M., Vrzal, L., Birchall, K., Arnold, L. H., Mpamhanga, C., et al. (2017). Characterization of three druggable hot-spots in the aurora-A/TPX2 interaction using biochemical, biophysical, and fragment-based approaches. ACS Chem. Biol. 12 (11), 2906–2914. doi:10.1021/acschembio.7b00537
Moreno, C. J. G., Temporão, A., Torres, T., and Sousa Silva, M. (2019). Trypanosoma brucei interaction with host: mechanism of VSG release as target for drug discovery for African trypanosomiasis. Int. J. Mol. Sci. 20 (6), 1484. doi:10.3390/ijms20061484
Ni, X., Richardson, R. B., Godoy, A. S., Ferla, M. P., Kikawa, C., Scheen, J., et al. (2024). Crystallographic fragment screening and deep mutational scanning of Zika virus NS2B-NS3 protease enable development of resistance-resilient inhibitors.
Patel, D., Bauman, J. D., and Arnold, E. (2014). Advantages of crystallographic fragment screening: functional and mechanistic insights from a powerful platform for efficient drug discovery. Prog. Biophysics Mol. Biol. 116 (2–3), 92–100. doi:10.1016/j.pbiomolbio.2014.08.004
Patterson, S., Alphey, M. S., Jones, D. C., Shanks, E. J., Street, I. P., Frearson, J. A., et al. (2011). Dihydroquinazolines as a novel class of trypanosoma brucei trypanothione reductase inhibitors: discovery, synthesis, and characterization of their binding mode by protein crystallography. J. Med. Chem. 54 (19), 6514–6530. doi:10.1021/jm200312v
Pearce, N. M., Krojer, T., Bradley, A. R., Collins, P., Nowak, R. P., Talon, R., et al. (2017). A multi-crystal method for extracting obscured crystallographic states from conventionally uninterpretable electron density. Nat. Commun. 8 (1), 15123. doi:10.1038/ncomms15123
Petukhova, V. Z., Aboagye, S. Y., Ardini, M., Lullo, R. P., Fata, F., Byrne, M. E., et al. (2023). Non-covalent inhibitors of thioredoxin glutathione reductase with schistosomicidal activity in vivo. Nat. Commun. 14 (1), 3737. doi:10.1038/s41467-023-39444-y
Schiebel, J., Radeva, N., Krimmer, S. G., Wang, X., Stieler, M., Ehrmann, F. R., et al. (2016). Six biophysical screening methods miss a large proportion of crystallographically discovered fragment hits: a case study. ACS Chem. Biol. 11 (6), 1693–1701. doi:10.1021/acschembio.5b01034
Smart, O. S., Womack, T. O., Flensburg, C., Keller, P., Paciorek, W., Sharff, A., et al. (2012). Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr. D. Biol. Crystallogr. 68 (4), 368–380. doi:10.1107/s0907444911056058
Thomas, S. E., Collins, P., James, R. H., Mendes, V., Charoensutthivarakul, S., Radoux, C., et al. (2019). Structure-guided fragment-based drug discovery at the synchrotron: screening binding sites and correlations with hotspot mapping. Philosophical Trans. R. Soc. A Math. Phys. Eng. Sci. 377 (2147), 20180422. doi:10.1098/rsta.2018.0422
Tiwari, P. B., Bencheqroun, C., Lemus, M., Shaw, T., Kouassi-Brou, M., Alaoui, A., et al. (2021). SPRD: a surface plasmon resonance database of common factors for better experimental planning. BMC Mol. Cell Biol. 22 (1), 17. doi:10.1186/s12860-021-00354-w
Tovar, J., Cunningham, M. L., Smith, A. C., Croft, S. L., and Fairlamb, A. H. (1998a). Down-regulation of Leishmania donovani trypanothione reductase by heterologous expression of a trans-dominant mutant homologue: effect on parasite intracellular survival. Proc. Natl. Acad. Sci. 95 (9), 5311–5316. doi:10.1073/pnas.95.9.5311
Tovar, J., Wilkinson, S., Mottram, J. C., and Fairlamb, A. H. (1998b). Evidence that trypanothione reductase is an essential enzyme in Leishmania by targeted replacement of the tryA gene locus. Mol. Microbiol. 29 (2), 653–660. doi:10.1046/j.1365-2958.1998.00968.x
Turcano, L., Battista, T., De Haro, E. T., Missineo, A., Alli, C., Paonessa, G., et al. (2020). Spiro-containing derivatives show antiparasitic activity against Trypanosoma brucei through inhibition of the trypanothione reductase enzyme. PLoS Negl. Trop. Dis. 14 (5), e0008339. doi:10.1371/journal.pntd.0008339
Turcano, L., Torrente, E., Missineo, A., Andreini, M., Gramiccia, M., Di Muccio, T., et al. (2018). Identification and binding mode of a novel Leishmania Trypanothione reductase inhibitor from high throughput screening. PLoS Negl. Trop. Dis. 12 (11), e0006969. doi:10.1371/journal.pntd.0006969
Valentini, E., D’Aguanno, S., Di Martile, M., Montesano, C., Ferraresi, V., Patsilinakos, A., et al. (2022). Targeting the anti-apoptotic Bcl-2 family proteins: machine learning virtual screening and biological evaluation of new small molecules. Theranostics 12 (5), 2427–2444. doi:10.7150/thno.64233
Wever, M. J. A., Scommegna, F. R., Egea-Rodriguez, S., Dehghani-Tafti, S., Brandao-Neto, J., Poisson, J. F., et al. (2024). Structure-based discovery of first inhibitors targeting the helicase activity of human PIF1. Nucleic Acids Res. 52 (20), 12616–12632. doi:10.1093/nar/gkae897
Wright, N. D., Collins, P., Koekemoer, L., Krojer, T., Talon, R., Nelson, E., et al. (2021). The low-cost Shifter microscope stage transforms the speed and robustness of protein crystal harvesting. Acta Crystallogr. D. Struct. Biol. 77 (1), 62–74. doi:10.1107/s2059798320014114
Keywords: trypanothione reductase, fragment screening, protein crystallography, surface plasmon resonance, drug discovery
Citation: Exertier C, Antonelli L, Brufani V, Colotti G, Ilari A and Fiorillo A (2025) Expanding the molecular landscape of fragments binding to trypanothione reductase, a legitimate target for drug design against human African trypanosomiasis. Front. Chem. Biol. 4:1605579. doi: 10.3389/fchbi.2025.1605579
Received: 03 April 2025; Accepted: 30 April 2025;
Published: 14 May 2025.
Edited by:
Massimiliano Perduca, University of Verona, ItalyReviewed by:
Rocco Caliandro, National Research Council (CNR), ItalyAlejandro Giorgetti, University of Verona, Italy
Copyright © 2025 Exertier, Antonelli, Brufani, Colotti, Ilari and Fiorillo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Annarita Fiorillo, YW5uYXJpdGEuZmlvcmlsbG9AdW5pcm9tYTEuaXQ=; Cécile Exertier, Y2VjaWxlLmV4ZXJ0aWVyQGNuci5pdA==