 Charly A. Moras
Charly A. Moras Renaud Joannes-Boyau
Renaud Joannes-Boyau Lennart T. Bach
Lennart T. Bach Tyler Cyronak
Tyler Cyronak Kai G. Schulz
Kai G. Schulz- 1Faculty of Science and Engineering, Southern Cross University, Lismore, NSW, Australia
- 2Ecology and Biodiversity, Institute for Marine and Antarctic Studies, University of Tasmania, Hobart, TAS, Australia
- 3Institute for Coastal Plain Science, Georgia Southern University, Savannah, GA, United States
Ocean alkalinity enhancement (OAE) via the enhanced weathering of alkaline minerals is a promising carbon dioxide removal (CDR) technology. Theoretically, these includes iron and steel slags, although their dissolution kinetics in seawater are unknown. Here, we conducted lab-scale experiments to assess the alkalinity generation potential and dissolution kinetics of various slags in seawater. We show that the alkalinity generated per mass of iron slag was logarithmic, i.e., higher amounts of iron slag added had diminishing alkalinity returns. In contrast, the relatively quick dissolution of steel slags and their linear generation of alkalinity per mass of feedstock dissolved in seawater makes them better OAE candidates. Furthermore, despite the presence of potentially toxic metals in these feedstocks, their low to non-existent presence as dissolution products suggests that harmful concentrations should not be reached, at least for the slag tested here. Finally, if all steel slag produced annually was used for OAE, between 10 and 22 gigatonnes of CO2 could be captured cumulatively by 2,100, highlighting significant CDR potential by slags.
1 Introduction
Human activities are increasing atmospheric carbon dioxide (CO2) concentrations, the major cause of global warming. To date, global mean temperatures have already surpassed pre-industrial levels by 1.1°C and continue to rise. The Intergovernmental Panel on Climate Change (IPCC) predicts that atmospheric carbon dioxide removal (CDR) is necessary to reach the climate goals set by the Paris Agreement (IPCC, 2022), regardless of the shared socioeconomic pathway. Ocean alkalinity enhancement (OAE) is a promising CDR approach due to its potential scalability, durability of stored carbon, and relatively low energy demands (Keller et al., 2014; Feng et al., 2017; National Academies of Sciences, Engineering, and Medicine, 2022; Foteinis et al., 2023). One OAE strategy is to grind up alkaline minerals to speed up the natural weathering process that draws down atmospheric CO2 on millennial timescales.
Mineral based OAE increases seawater total alkalinity (TA) by dissolving alkaline minerals, such as olivine, lime, or brucite (Renforth and Henderson, 2017). Upon dissolution, seawater pH is increased, shifting carbonate chemistry speciation (Kheshgi, 1995). As a result, dissolved [CO2] is decreased, subsequently enhancing atmospheric CO2 uptake, or limiting CO2 degassing from CO2-rich seawater (Renforth and Henderson, 2017). Slag, a highly alkaline by-product of iron and steel manufacturing, has been suggested as a potentially effective alkaline feedstock for OAE (Reddy et al., 2019; Renforth, 2019). The annual global production of slag is approximately 500 megatonnes (1Mt = 106 tonnes), with most used in the construction industry, and significant amounts stockpiled on land as waste (Renforth, 2019). Using slags for OAE could be a cost-effective solution as only grinding and transport are required (Renforth et al., 2013).
The composition of slag varies depending on the iron and steel making processes. Blast furnace slag (BFS) is the first by-product in the conversion of iron ore to pig iron (Reddy et al., 2019). It contains between 30 and 36% of silicon dioxide (SiO2) and between 32 and 45% of calcium oxide (CaO). Basic oxygen furnace slag (BOFS) is obtained by converting molten iron into molten steel (Reddy et al., 2006). This slag contains between 30 and 35% of CaO, and between 10 and 35% of iron oxide (Fe2O3). Similar to BOFS, electric arc furnace slag (EAFS) is produced by converting steel scraps into molten steel. This slag contains between 22 to 60% of CaO and about 10 to 40% of Fe2O3. All slags contain aluminium oxides at varying concentrations (1 to 35%), and various trace elements such as chromium, copper, lead, nickel, or zinc (Luxán et al., 2000; Tossavainen et al., 2007; Yildirim and Prezzi, 2011). Since these elements may dissolve in seawater, a potential risk for marine ecosystems exists. Guo et al. (2024) investigated the potential risks of trace elements leaching during slag-based OAE in a microcosms study, and reported that steel slag presented a lower risk for plankton communities when compared to olivine. However, they also emphasized on the importance of studying the elemental composition of various slags, since this can affect the carbon removal efficiency as well as the amount of trace elements leaching during slag dissolution.
This study presents lab-scale experiments where dissolution kinetics of three slags were investigated in MilliQ and seawater, and a TA generation potential for each slag was determined. This research explores whether slags are suitable for OAE, by (1) describing the elemental composition of various slags, (2) determining the efficiency of each slag to produce alkalinity, and (3) assessing which trace elements leach into seawater during dissolution. Using water quality guidelines for Australia and New Zealand, we also provide insights into whether dissolving slags might present a risk for organisms and the environment.
2 Materials and methods
2.1 Slag information and slag dissolution
In this study, an iron blast furnace slag (BFS) and a basic oxygen furnace slag (BOFS) were obtained from Liberty Steelworks in Whyalla, Australia, and an electric arc furnace slag (EAFS) was obtained from BlueScope in Wollongong, Australia. Each slag was crushed using a Retsch BB50 jaw crusher and ground using a Pulverizer Laboratory ring mill, resulting in grain sizes averaging between 50 and 200 μm (Supplementary material). The elemental composition of each slag was determined using an inductively coupled plasma mass spectrometer (ICP-MS). The slag dissolution experiments were conducted using MilliQ (18.2 MΩ) and natural seawater (NSW). NSW was collected and treated as described in Moras et al. (2023).
A series of dissolution experiments were conducted in MilliQ to estimate the potential for alkalinity formation per mg of slag dissolving. Each slag was added in concentrations of 10, 25 and 50 mg kg−1 of MilliQ in high-quality borosilicate 3.3 1 L Schott bottles. The nine bottles had initially little headspace to minimize CO2 gas exchange and hence being able to attribute measured changes in DIC upon slag addition to carbonate dissolution or precipitation. They were kept at room temperature (air conditioning set at 21°C) and placed on a roller table at about 6 rotations per minute to keep the added slag particles in suspension. The pH was recorded daily to monitor the dissolution kinetics (calibrated using Metrohm buffers). Once a stable value was reached, TA samples were collected. Another set of TA samples was taken two weeks later to confirm that a steady state had been reached. The released TA was considered stable when the changes over two weeks was less than 5 μmol kg−1.
Using the data from the MilliQ experiments, another set of experiments was conducted to assess the dissolution of slag in NSW. Five slag additions were performed, aiming to increase TA by 50, 100, 200, 350, and 500 μmol kg−1. This corresponded to 5.04, 8.53, 24.84, 122.73, and 606.61 mg kg−1 for BFS, 3.87, 7.80, 15.73, 27.58, and 38.44 mg kg−1 for BOFS, and 4.48, 8.98, 17.95, 31.26, and 44.73 mg kg−1 for EAFS. These additions were selected to cover a wide range of TA increase with a maximum total alkalinity target of +500 μmol kg−1, identified as the limit beyond which secondary CaCO3 precipitation could occur (Moras et al., 2022). The high-quality borosilicate 3.3 2 L Schott bottles were constantly rotated on a roller table for four weeks at about 6 rotations per minute and at room temperature (air conditioning set at 21°C). TA and DIC samples were collected before slag addition, and on days 1, 4, 7, 14, 21, and 28 days after slag addition. Similarly, samples for ICP-MS analysis were taken before slag addition for elemental analysis and 7, 14, 21, and 28 days after (see below for sampling procedures).
2.2 Carbonate chemistry measurements and ICP-MS analyses
Samples for TA and DIC were collected and analyzed as per Moras et al. (2022). TA was analyzed in duplicate and DIC in triplicate. The TA and DIC values were corrected against secondary in-house reference materials since certified ones from Prof. Andrew G. Dickson’s laboratory could not be supplied during the COVID-19 pandemic (Moras et al., 2023). The TA and DIC measurement uncertainties were calculated by error propagation. The DIC measurement uncertainty was estimated at ±1.1 μmol kg−1, and the TA measurement uncertainty at ±1.8 μmol kg−1. Remaining carbonate chemistry parameters such as CaCO3 saturation state of aragonite (ΩA) or pH on the total scale were estimated from TA, DIC, salinity and temperature data (see Moras et al., 2022 for details) using the CO2SYS script for MATLAB (Sharp et al., 2021).
Elemental composition of slag was determined on an Agilent 7,700 ICP-MS, coupled to a laser ablation unit (NWR213, Electro Scientific Industries, Inc.). Powdered slags were embedded in resin for solid ICP-MS analysis. The samples were then polished and measured, before being corrected against standard reference materials, obtained from the National Institute of Standards and Technology (NIST, batch #610 and #612). For liquid elemental ICP-MS analysis, 12 mL of water were taken with a 60 mL luer lock sterile syringe and filtered through a Nylon Captiva EconoFilter (0.45 μm) into 15 mL Falcon tubes. Samples were diluted 10 times with 3%v/v nitric acid. Measurements were carried out on the same ICP-MS as for the solid analyses and measurements were corrected against a seawater reference material from the National Research Council of Canada NASS-6.
Additionally, to estimate the carbon content in powdered slags, ~100 mg of each slag was weighed and wrapped in tin capsules in triplicates, before being pressed into small balls of about 5 mm diameter. The samples were then analyzed using a Thermo Fisher Flash Elemental Analyzer, coupled to a Delta V Plus isotope ratio mass spectrometer.
3 Results
3.1 Grain size and slag composition
The grain size distribution for individual slags was very similar, ranging mostly between 0.5 and 350 μm in diameter, with peaks around 50–200 μm (Appendix Figure A1). Concerning elemental composition, all three slags were rich in calcium, varying from 227 ± 51 mg g−1 for BFS, to 387 ± 144 and 338 ± 95 mg g−1 for BOFS and EAFS, respectively. Silicon and magnesium were also present in all slags, varying between ~64 and ~ 115 mg g−1 for silicon, and ~ 43 and ~ 81 mg g−1 for magnesium. With 61 ± 13 mg g−1 of aluminium, BFS was nearly four times richer than BOFS and EAFS. In contrast, BFS contained hardly any iron, while concentrations in BOFS and EAFS were 134 ± 58 and 310 ± 35 mg g−1, respectively. Some potentially toxic elements were also found, with 17–61 mg g−1 of aluminium in all slags, up to 4.3 mg g−1 of chromium in EAFS as well as 52.5 mg g−1 of manganese. Finally, BFS contained ~1.5 ± 0.3 mg of carbon per gram of slag, BOFS ~7.6 ± 0.4 mg g−1 and EAFS ~4.2 ± 0.1 mg g−1 (Appendix Table A1).
3.2 Alkalinity production potential from MilliQ experiments
The alkalinity generation potential of the three slags was tested in MilliQ water, at concentrations of 10, 25 and 50 mg of slag per kg of MilliQ. For BFS additions, stable conditions were reached after three weeks, while for BOFS and EAFS additions stable conditions were already observed after five days of experiment, meaning that complete alkalinity generation was about 4 times faster than for BFS. The alkalinity generation for BFS was not linear (Figure 1). In fact, with increasing amounts of BFS added, less alkalinity was generated. In contrast, over the range tested, alkalinity generation in BOFS and EAFS was linear. Also, about 10% more alkalinity was leached per mg of BOFS compared to EAFS.
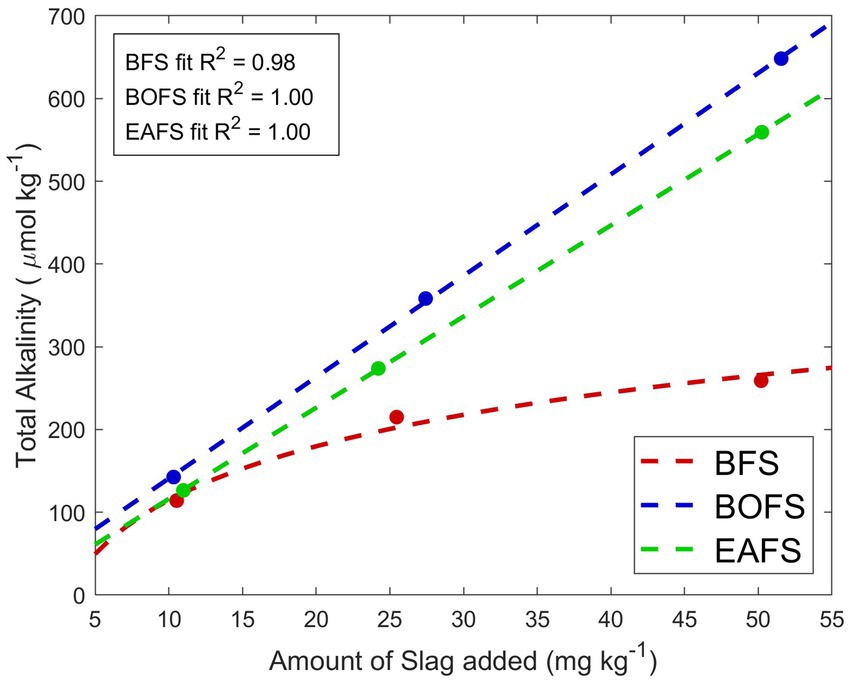
Figure 1. Amount of alkalinity leached per mg of slag added to one kg of MilliQ, averaged from two TA measurements, one once a stable pHF was observed and one measurement two weeks later. The coefficient of determination R2 for BFS (red logarithmic fit), BOFS (blue linear fit) and EAFS (green linear fit) is also reported. See Materials and Methods for details.
3.3 Alkalinity production in natural seawater
Using the TA generation potential data for MilliQ, five additions per slag were conducted in NSW, with ΔTA targets of +50, +100, +200, +350 and + 500 μmol kg−1. The maximum TA measured for the BFS experiments was much lower than the expected one, i.e., about half (Figure 2). While the TA remained stable in most treatments once reaching its maximum concentrations, the TA in the highest BFS addition decreased slightly after it had reached its maximum, i.e., ~230 μmol kg−1, between day 7 and 14 (Figure 2). DIC increased overtime, varying between +20.2 and + 32.9 μmol kg−1 over the 28 days of experiments, except for the highest BFS addition, where DIC increased by 56.3 μmol kg−1. ΩA followed the TA trends, with similar patterns for each addition except for the highest one. In the higher BFS experiment, ΩA increased over the first 7 days to up to 4.8, before decreasing to 3.8 and then remaining stable around 4.0. In the BOFS and EAFS experiments, similar DIC, TA and ΩA trends were observed (Figure 2). For both steel slags, TA and ΩA quickly increased during the first day, reaching a steady plateau about a week later. In contrast to TA, DIC increased steadily over time, ranging between 20 and 40 μmol kg−1, with lower concentrations for low and higher for high slag additions.
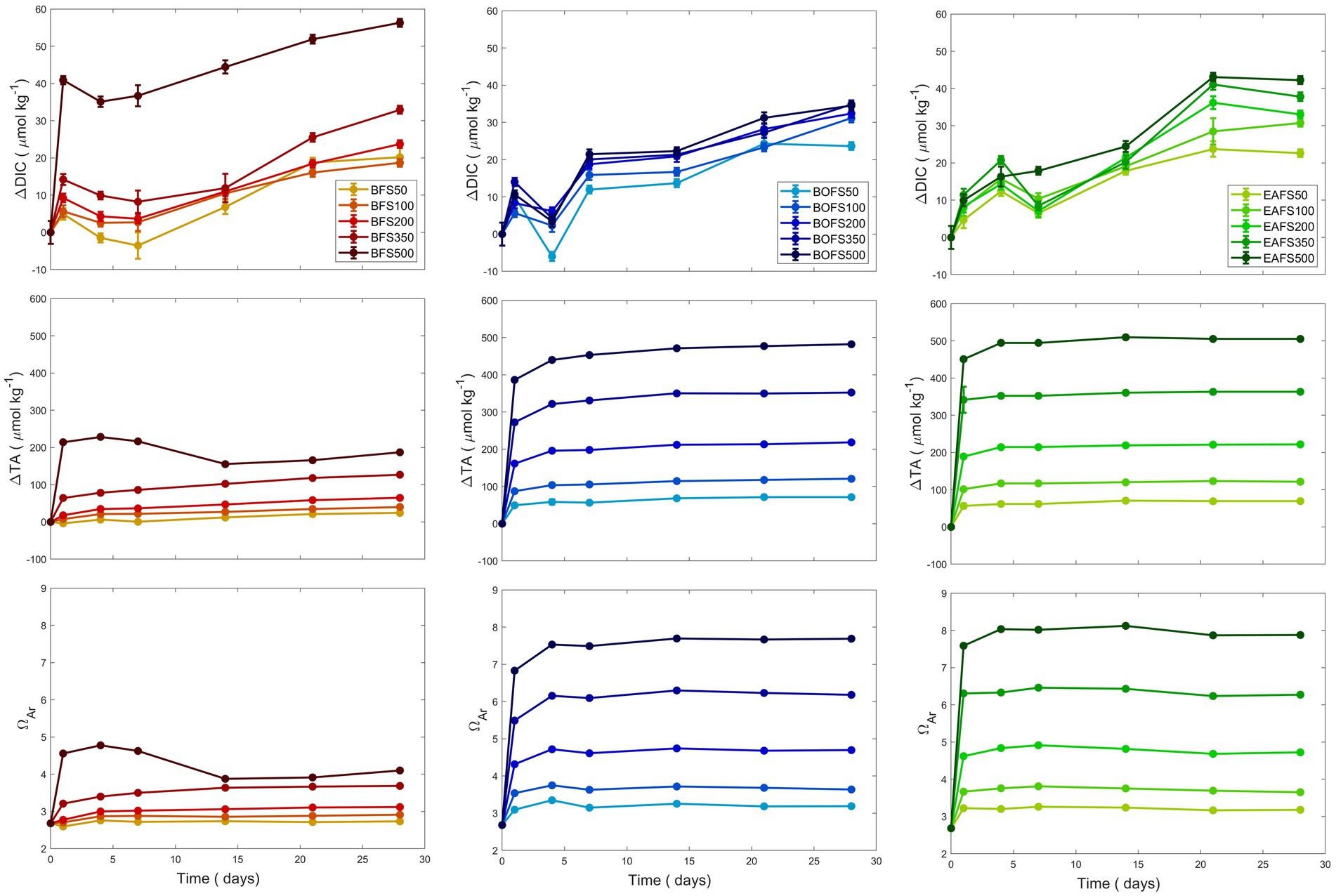
Figure 2. Changes in TA, DIC and ΩA over 28 days following five different BFS (red), BOFS (blue) and EAFS (green) additions, aiming for a TA increase by 50, 100, 200, 350, and 500 μmol kg−1.
During the dissolution of all slags, [Si], [Al], and [Mn] increased gradually over the four weeks of experiments with more pronounced increases when more slag had been added (Figure 3; Appendix Figures A3–A5). This pattern was particularly clear in the changes in [Al] and [Mn] from BOFS and EAFS. However, in some instances, stable concentrations were observed over 4 weeks, in particularly for [P] or [Ni], and some elements concentrations decreased over time, e.g., [Fe], [Cr], or [Sn] (Figure 3; Appendix Figures A3–A5).
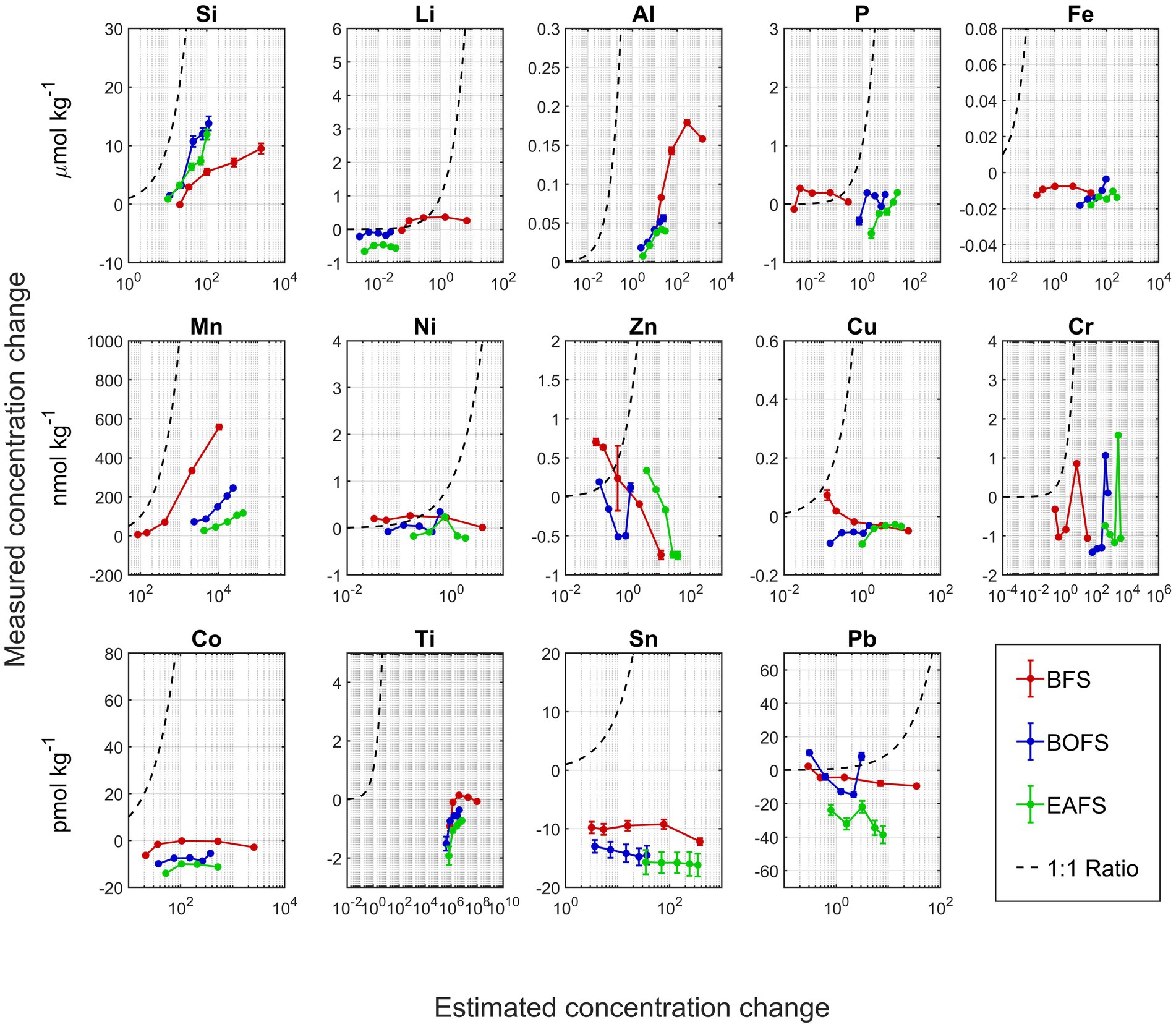
Figure 3. Comparison of measured concentration change of various elements upon slag addition in NSW in relation to the potential changes based on solid ICP-MS analyses (assuming full dissolution). Elemental concentration changes for BFS (red), BOFS (blue) and EAFS (green) are based on measurements from week 3 and week 4, i.e., when the TA was considered stable and dissolution complete. The 1: 1 line of measured versus potential concentration change is shown as a black dashed line.
4 Discussion
4.1 TA generation and CO2 carbon capture potential of various slag types
This study tested the dissolution of iron and steel slags in seawater, and their potential to generate alkalinity and leach trace elements in lab-scale experiments. Among the three slags sourced, one iron slag and two steel slags were used, with elemental compositions similar to those reported in the literature (Reddy et al., 2019). A first very interesting observation was that BFS dissolved much slower than BOFS and EAFS (Figures 1, 2). A logarithmic trend between BFS addition and TA generated was observed in MilliQ, indicative of reaching saturation, i.e., the more BFS is added, the less TA is generated per mass of feedstock. Furthermore, in all NSW treatments, BFS solubility was 46–48% lower than in MilliQ, further reducing its OAE suitability. The reason for the saturation of TA generation and lower solubility of BFS in seawater than in MilliQ is unclear at this stage. Based on the calcium and magnesium measurements in each slag, and assuming that all calcium and magnesium are present as CaO and MgO, respectively, all the TA generated by BOFS and EAFS (~12.72 mmol g−1 and ~ 11.18 mmol g−1, respectively) can be attributed to fully dissolving these compounds (Reddy et al., 2019). This is only the case for the lowest BFS addition in MilliQ, so it appears that calcium is present in a less soluble form than CaO. Which one is unknown, but it is not calcite or aragonite (insoluble in seawater) due to the low carbon content measured in all slags.
Nevertheless, an estimate of the carbon capture potential using slag can be derived. Ignoring BFS, due to the saturation behavior and further reduced solubility in seawater, on average 12.0 ± 1.1 mmol of alkalinity was generated per g of steel slag. With an average of ~0.8 mole of CO2 captured per mole of TA generated in oceanic surface waters (Moras et al., 2022; He and Tyka, 2023; Schulz et al., 2023), ~9.6 mmol of CO2 could be captured, corresponding to about 420 mg of CO2 per gram of steel slag. At a global annual steel production of nearly 1.9 gigatonnes (1 Gt = 109 tonnes) in 2021 (Worldsteel, 2022), and assuming that for every tonne of crude steel produced, ~0.125 tonnes of steel slag are generated (Tuck, 2023), using all available steel slag for OAE purposes could lead to about 100 Mt. of CO2 captured per year at the present day. Similarly, modeling global steel production into the future for various shared socioeconomic pathways (Renforth, 2019), up to 22 Gt of cumulative CO2 capture could be realized until 2,100 (Figure 4). Hence, the CO2 sequestration potential of steel slags is significant and may be economically viable as no extra investments are required for production (Renforth and Henderson, 2017; Renforth, 2019). The numbers presented here are at the upper boundary for steel slag as they assume full CO2 equilibration with the atmosphere after the alkalinity increase. However, they do not account for the CO2 emissions during steel production. Furthermore, the capture potential is highly dependent on the location of OAE implementation. For example, while model simulations suggest that a ratio of TA generated to CO2 captured close to 0.8 can be realized within years to decades, there are some places in which surface waters with increased TA will not stay in contact with the atmosphere for long enough to allow for full equilibration (He and Tyka, 2023).
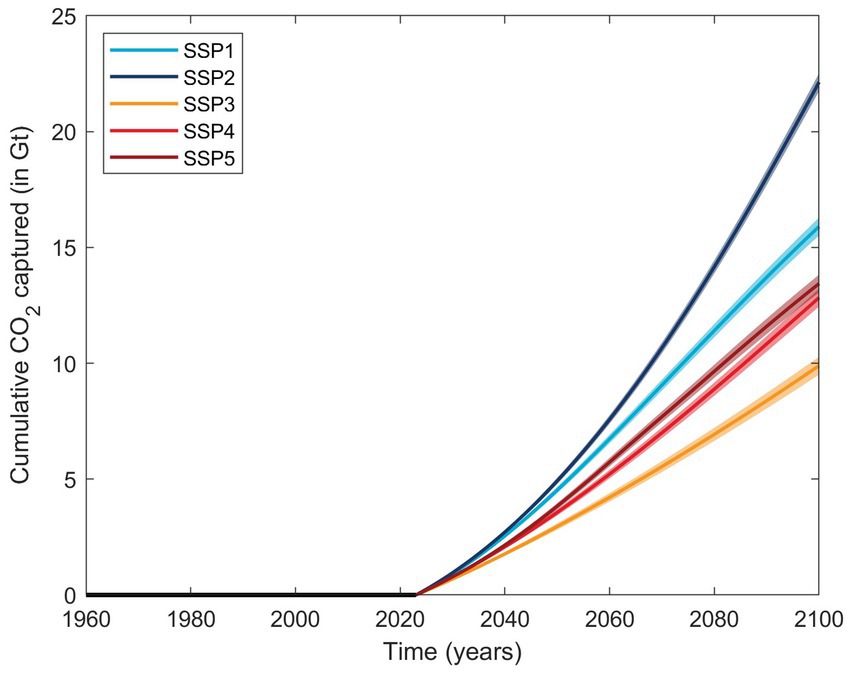
Figure 4. Estimated cumulative CO2 captured in Gt based on five shared socioeconomic pathways (SSPs) and assuming a 100% use of steel slag for OAE if started in 2023 (Renforth, 2019). The average CO2 potential is represented by the solid lines and the respective standard deviation range as transparent areas, but do not account for CO2 emissions during slag production.
4.2 Increase in DIC and absence of CaCO3 precipitation
For all three slags in all treatments, an immediate increase in DIC was measured, which cannot be explained by CO2 ingassing as there was no headspace initially. Although, for BOFS and EAFS, the increase was relatively small and a measured decrease at the second and third sampling point, respectively, suggests that there might have been sampling/measurement issues, which however has no implications for any conclusions drawn here. For BOFS and EAFS, there was a general trend of a steady DIC increase over time, consistent with CO2 ingassing, as expected. Altogether, this suggests that BOFS and EAFS do not release significant amounts of DIC during dissolution, which is important as it would reduce the CO2 uptake potential. One can also come to the same conclusion by assuming that all the carbon contained in the slags would be carbonates that would dissolve in seawater (either soluble calcium or magnesium carbonates). At 7.6 and 4.3 mg of CO32− per gram of BOFS and EAFS, respectively, only a ~ 5 and ~ 3 μmol kg−1 increase in DIC at the highest TA addition would be expected.
In contrast, for BFS, DIC could have increased via the dissolution of carbonates by up to 15 μmol kg−1 in the highest TA treatment. However, the measured DIC increase was much higher, and at this stage it is not clear what could explain this observation. In any case, this does not affect any of the conclusions drawn previously, and BFS has already been flagged as a suboptimal OAE feedstock. Finally, the apparent decrease in TA between the first and second week in the highest TA treatments with BFS is most likely a measurement artifact from imperfect sample filtration. CaCO3 precipitation is unlikely as it should have continued in a runaway fashion as described previously, and a drop in DIC should also have been observed (Moras et al., 2022). Instead, it could result from the dissolution of not yet dissolved slag during TA titrations, even though no visual observations of particles were made (note that BFS dissolution kinetics were much slower than for BOFS and EAFS).
An important finding was the general stability of TA observed after the increase at the start of the experiments. Recent studies have highlighted the issue of potential secondary CaCO3 precipitation following an increase in TA for OAE purposes (Fuhr et al., 2022; Moras et al., 2022; Hartmann et al., 2023). For instance, Moras et al. (2022) reported that at an ΩA ~ 7.0, so called “runaway CaCO3 precipitation” can occur, ultimately removing more TA than had been added. Surprisingly in our experiments, despite ΩA levels above 7.0 in two of the steel slag experiments, no drop in TA was observed for almost a month. In contrast, in experiments with Mg(OH)2, Ca(OH)2 and CaO, CaCO3 precipitation was observed within a few days at these conditions (Moras et al., 2022, Hartmann et al., 2023). The mechanisms behind CaCO3 precipitation are complex but several factors are known to enhance or inhibit precipitation. The presence of mineral particles in suspension have been found as a main trigger for runaway CaCO3 precipitation (Morse et al., 2003; Lioliou et al., 2007; Bustos-Serrano et al., 2009; Tang et al., 2020). Observed both in nature and in laboratory settings, these particles act as precipitation nuclei for CaCO3. The reason for not observing any CaCO3 precipitation here could be that steel slag has a lower lattice compatibility for CaCO3 than pure CaO, Ca(OH)2 and Mg(OH)2, although how that would work is not clear, as it is CaO in those slags that is mainly dissolving and generating TA. Also, rather-unlikely is that an increase in dissolved Mg concentrations during slag dissolution could act as a natural inhibitor of CaCO3 precipitation (Berner, 1975; Rushdi et al., 1992; Morse et al., 1997; Pan et al., 2021). Even if all Mg present in slags would be MgO, with Mg content varying between 4 and 8%, a maximum increase of about 90 μmol kg−1 could be explained at the highest TA treatment. This is relatively small, considering a natural Mg background of ~52.8 mmol kg−1 in natural seawater at a salinity of 35 (Dickson et al., 2007; Riebesell et al., 2011), and rather unlikely to significantly inhibit CaCO3 precipitation.
The most striking difference between steel slags and the calcium and magnesium minerals investigated previously are their dissolution kinetics. While CaO and MgO fully dissolved within hours, steel slag dissolution was on the order of days. A lower dissolution rate would suggest a lower pH in the diffusive boundary layer around particles suspended in the water column, given all other factors being equal (particles size, mixing, etc.). Despite similar bulk seawater ΩA, a lower pH at the particle surface could then indeed explain why no CaCO3 precipitation was found here. It has to be kept in mind, though, that dissolution rates measured here were for 21°C, meaning that minerals contained in slag with retrograde solubility such as Ca(OH)2 would dissolve faster at lower temperatures, which could lead to higher pH at particle surfaces. If that would lower the ΩA threshold at which secondary CaCO3 precipitation is actually observed, however, is not clear as at the same time, precipitation kinetics are slowed down at lower temperatures (Zhong and Mucci, 1989). Hence, slag dissolution experiments at different temperature are required.
4.3 Potential environmental effects of slag dissolution
If alkaline materials such as slags with various metal impurities would be used for OAE, an open question is the potential release of bioactive elements during dissolution. Using the slag composition data and the known amounts added to NSW, the maximum increase in potentially bioactive element concentrations can be calculated. For example, about 100 μmol kg−1 of Ti could leach in the highest BFS addition and about 3.7 μmol kg−1 of Cr in the highest EAFS addition. When comparing the potential changes in NSW elemental composition from solid ICP-MS analysis to the actual measured changes in NSW elemental composition, in most cases, the measured concentration was much lower than the potential ones, i.e., the plotted data being positioned to the right of the 1:1 line (Figure 3). In some instances, such as for Li or Ni, the measured concentration was close to the potential one, at least for the lower addition. A few data points were above the 1:1 line, most likely the result of measurement uncertainties. In some instances, such as for Si, Al and Mg, seawater concentrations clearly increased toward higher slag additions. Finally, for elements such as Li, Ni and Sn, seawater concentrations hardly changed indicating that some elements do not dissolve in seawater, at least not within the experiment timeframe. These elements may be locked up in minerals that have a low solubility in seawater, such as for instance Fe(III).
Using the Australian and New Zealand guidelines for fresh and marine water quality, nine potentially toxic elements for which harmful concentrations are reported were identified in the slags (Commonwealth of Australia, 2023). These were aluminium, chromium, cobalt, copper, lead, manganese, nickel and zinc. In all of our experiments, potentially toxic element concentrations were all lower than the safe levels reported in the Australian and New Zealand guidelines for fresh and marine water quality (Table 1). For BFS, Al would be the first element to reach toxic levels, followed by Mn. The same would be the case for EAFS, while for BOFS, the element order would be reversed. This means that up to 509.9 mg kg−1 of BFS, 149.4 mg kg−1 of BOFS and 332.7 mg kg−1 of EAFS can be dissolved before potentially reaching toxic levels for an element. Such additions would correspond to an alkalinity increase of 483.7, 1899.8 and 3718.9 μmol kg−1, respectively. For the two steel slags, this is most likely much higher than one would aim for, as of potentially negative effects of increased pHT on marine biota (between 9.5 and 10.6 at 21°C, assuming open ocean conditions, i.e., initial TA = 2,300 μmol kg−1, DIC = 2,100 μmol kg−1 and salinity of 35), as well as pseudo-homogeneous CaCO3 precipitation onto naturally occurring colloids and organic matter at ΩA values above 12.3 (ΩA would increase between 24.6 and 31.8, respectively). In essence, dissolution of trace elements, particularly in steel slags, appears unlikely to exceed potentially toxic levels, at least for the type of slags tested here. Similarly, the quick dilution naturally occurring following slag dissolution, especially in an open-ocean setting, would further reduce the concentration of potentially toxic elements. Such dilution would also allow for more slag to be dissolved.
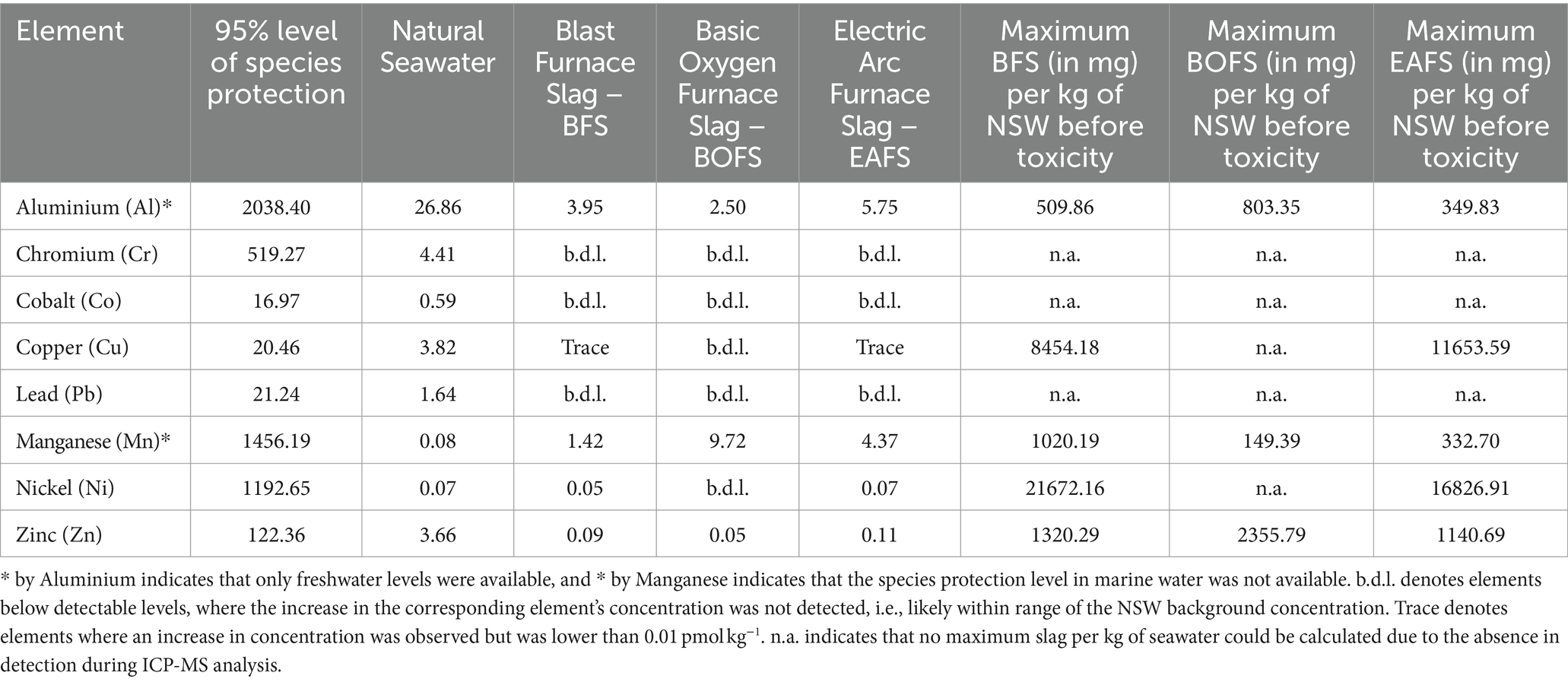
Table 1. Potentially toxic elements leached in pmol L−1 per mg of slag added per liter of natural seawater, and the concentration for the 95% species protection level, reported by the Australian and New Zealand Guidelines for Fresh and Marine Water Quality (Commonwealth of Australia, 2023).
5 Conclusion
The use of slag as alkaline mineral for OAE has been recently proposed, and their dissolution in seawater was tested here. Iron slag showed a saturation behavior where the amount of alkalinity generated per mg of slag decreased the more slag was added. In contrast, BOFS and EAFS had high carbon capture potentials, with about 0.4 g of CO2 per g of steel slag dissolved. This would correspond to about 100 Mt. of CO2 that could be captured annually at present day if all steel slag would be used for OAE. Extrapolating such estimates into the future for various shared socioeconomic pathways, a total of up to 22 Gt of CO2 could be captured until 2,100. Finally, the issue of metals reaching potentially toxic concentrations during slag dissolution appears to be limited.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material, and the data are available at: https://doi.org/10.5281/zenodo.10020699.
Author contributions
CM: Visualization, Validation, Resources, Methodology, Investigation, Funding acquisition, Formal analysis, Data curation, Conceptualization, Writing – review & editing, Writing – original draft. RJ-B: Writing – review & editing, Validation, Supervision, Software, Resources, Methodology, Investigation, Funding acquisition, Formal analysis, Data curation, Conceptualization. LB: Writing – review & editing, Visualization, Validation, Supervision, Resources, Methodology, Conceptualization. TC: Writing – review & editing, Visualization, Validation, Supervision, Methodology, Conceptualization. KS: Writing – review & editing, Writing – original draft, Visualization, Validation, Supervision, Resources, Methodology, Investigation, Funding acquisition, Formal analysis, Conceptualization.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The study was funded by the Cat. 5 – SCU Grad School scholarship granted to CM as part of his PhD project. ICP-MS analyses were made possible by Australian Research Council grants awarded to RJ-B and KS (Grant No. LE200100022) and to RJ-B (grant no. LE120100201).
Acknowledgments
The authors would like to express their appreciation to Liberty Steelworks in Whyalla and BlueScope in Wollongong, Australia, for providing the various slags. The authors also thank the Southern Cross Analytical Research Services of the Southern Cross University, Lismore, Australia, and particularly Nick Ward for their help with grinding and grain size analyses of the slags.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fclim.2024.1396487/full#supplementary-material
SUPPLEMENTARY FIGURE A1 | Grain size distribution of BFS (red), BOFS (blue), and EAFS (green), measured between 0.01 to 3000 µm.
SUPPLEMENTARY FIGURE A2 | pHF and TA changes during BFS (red), BOFS (blue) and EAFS (green) dissolution in MilliQ over 50, 25, and 25 days, respectively, at various concentrations, i.e., ~10, ~25, and ~50 mg kg-1. The changes in pHF are plotted on the left axis, and the TA changes in μmol kg-1 are plotted on the right axis.
SUPPLEMENTARY FIGURE A3 | Changes in NSW elemental composition (Δ Concentration) from before slag addition and during slag dissolution, following five different BFS additions aiming for a ΔTA of +50, +100, +200, +350, and +500 µmol kg-1, expressed in µmol or nmol kg-1. Standard deviations have been calculated from continuous 60 seconds solution measurements for each element.
SUPPLEMENTARY FIGURE A4 | Changes in NSW elemental composition (Δ Concentration) from before slag addition and during slag dissolution, following five different BOFS additions aiming for a ΔTA of +50, +100, +200, +350, and +500 µmol kg-1, expressed in µmol or nmol kg-1. Standard deviations have been calculated from continuous 60 seconds solution measurements for each element.
SUPPLEMENTARY FIGURE A5 | Changes in NSW elemental composition (Δ Concentration) from before slag addition and during slag dissolution, following five different EAFS additions aiming for a ΔTA of +50, +100, +200, +350, and +500 µmol kg-1, expressed in µmol or nmol kg-1. Standard deviations have been calculated from continuous 60 seconds solution measurements for each element.
References
Berner, R. A. (1975). The role of magnesium in the crystal growth of calcite and aragonite from sea water. Geochim. Cosmochim. Acta 39, 489–504. doi: 10.1016/0016-7037(75)90102-7
Bustos-serrano, H., Morse, J. W., and Millero, F. J. (2009). The formation of whitings on the little Bahama Bank. Mar. Chem. 113, 1–8. doi: 10.1016/j.marchem.2008.10.006
Commonwealth of Australia. (2023). Australian & New Zealand Guidelines for Fresh & Marine Water Quality. Available at: https://www.waterquality.gov.au/anz-guidelines/guideline-values/default/water-quality-toxicants/search (Accessed May 18, 2023)
Dickson, A. G., Sabine, C. L., and Christian, J. (2007). R: Guide to best practices for ocean CO2 measurements, PICES special publication 3; IOCCP report 8. Sidney, British Columbia: North Pacific Marine Science Organization, 191.
Feng, E. Y., Koeve, W., Keller, D. P., and Oschlies, A. (2017). Model-based assessment of the CO2 sequestration potential of coastal ocean alkalinization. Earth's Future 5, 1252–1266. doi: 10.1002/2017EF000659
Foteinis, S., Campbell, J. S., and Renforth, P. (2023). Life cycle assessment of coastal enhanced weathering for carbon dioxide removal from air. Environ. Sci. Technol. 57, 6169–6178. doi: 10.1021/acs.est.2c08633
Fuhr, M., Geilert, S., Schmidt, M., Liebetrau, V., Vogt, C., Ledwig, B., et al. (2022). Kinetics of olivine weathering in seawater: an experimental study. Front. Clim. 4:831587. doi: 10.3389/fclim.2022.831587
Guo, J. A., Strzepek, R. F., Swadling, K. M., Townsend, A. T., and Bach, L. T. (2024). Influence of ocean alkalinity enhancement with olivine or steel slag on a coastal plankton community in Tasmania. Biogeosciences 21, 2335–2354. doi: 10.5194/bg-21-2335-2024
Hartmann, J., Suitner, N., Lim, C., Schneider, J., Marín-Samper, L., Arístegui, J., et al. (2023). Stability of alkalinity in ocean alkalinity enhancement (OAE) approaches – consequences for durability of CO2 storage. Biogeosciences 20, 781–802. doi: 10.5194/Bg-20-781-2023
He, J., and Tyka, M. D. (2023). Limits and CO2 equilibration of near-coast alkalinity enhancement. Biogeosciences 20, 27–43. doi: 10.5194/bg-20-27-2023
IPCC. (2022). Summary for policymakers. In: Climate change 2022: Mitigation of climate change. Contribution of working group III to the sixth assessment report of the intergovernmental panel on climate change. P. R. Shukla, J. Skea, R. Slade, A. KhourdajieAl, R. Diemenvan, and D. McCollum, Cambridge University Press, Cambridge, pp. 3–33.
Keller, D. P., Feng, E. Y., and Oschlies, A. (2014). Potential climate engineering effectiveness and side effects during a high carbon dioxide-emission scenario. Nat. Commun. 5:3304. doi: 10.1038/ncomms4304
Kheshgi, H. S. (1995). Sequestering atmospheric carbon dioxide by increasing ocean alkalinity. Energy 20, 915–922. doi: 10.1016/0360-5442(95)00035-F
Lioliou, M. G., Paraskeva, C. A., Koutsoukos, P. G., and Payatakes, A. C. (2007). Heterogeneous nucleation and growth of calcium carbonate on calcite and quartz. J. Colloid Interface Sci. 308, 421–428. doi: 10.1016/j.jcis.2006.12.045
Luxán, M. P., Sotolongo, R., Dorrego, F., and Herrero, E. (2000). Characteristics of the slags produced in the fusion of scrap steel by electric arc furnace. Cem. Concr. Res. 30, 517–519. doi: 10.1016/S0008-8846(99)00253-7
Moras, C. A., Bach, L. T., Cyronak, T., Joannes-Boyau, R., and Schulz, K. G. (2022). Ocean alkalinity enhancement – avoiding runaway CaCO3 precipitation during quick and hydrated lime dissolution. Biogeosciences 19, 3537–3557. doi: 10.5194/bg-19-3537-2022
Moras, C. A., Bach, L. T., Cyronak, T., Joannes-Boyau, R., and Schulz, K. G. (2023). Preparation and quality control of in-house reference materials for marine dissolved inorganic carbon and total alkalinity measurements. Limnol. Oceanogr. Methods 21, 637–644. doi: 10.1002/lom3.10570
Morse, J. W., Gledhill, D. K., and Millero, F. J. (2003). CaCO3 precipitation kinetics in waters from the great Bahama bank: implications for the relationship between bank hydrochemistry and whitings. Geochim. Cosmochim. Acta 67, 2819–2826. doi: 10.1016/S0016-7037(03)00103-0
Morse, J. W., Wang, Q., and Tsio, M. Y. (1997). Influences of temperature and mg:ca ratio on CaCO3 precipitates from seawater. Geology 25, 85–87. doi: 10.1130/0091-7613(1997)025<0085:Iotamc>2.3.Co;2
National Academies of Sciences, Engineering, and Medicine (2022). A research strategy for ocean-based carbon dioxide removal and sequestration. Washington, DC: The National Academies Press, 323.
Pan, Y., Li, Y., Ma, Q., He, H., Wang, S., Sun, Z., et al. (2021). The role of Mg2+ in inhibiting CaCO3 precipitation from seawater. Mar. Chem. 237:104036. doi: 10.1016/j.marchem.2021.104036
Reddy, K. R., Gopakumar, A., and Chetri, J. K. (2019). Critical review of applications of iron and steel slags for carbon sequestration and environmental remediation. Rev. Environ. Sci. Biotechnol. 18, 127–152. doi: 10.1007/s11157-018-09490-w
Reddy, A. S., Pradhan, R. K., and Chandra, S. (2006). Utilization of basic oxygen furnace (BOF) slag in the production of a hydraulic cement binder. Int. J. Miner. Process. 79, 98–105. doi: 10.1016/j.minpro.2006.01.001
Renforth, P. (2019). The negative emission potential of alkaline materials. Nat. Commun. 10:1401. doi: 10.1038/s41467-019-09475-5
Renforth, P., and Henderson, G. (2017). Assessing Ocean alkalinity for carbon sequestration. Rev. Geophys. 55, 636–674. doi: 10.1002/2016RG000533
Renforth, P., Jenkins, B. G., and Kruger, T. (2013). Engineering challenges of ocean liming. Energy 60, 442–452. doi: 10.1016/j.energy.2013.08.006
Riebesell, U., Fabry, V., Hansson, L., and Gattuso, J. (2011). Guide to best practices for ocean acidification research and data reporting. Luxembourg: Publications Office of the European Union, 260.
Rushdi, A. I., Pytkowicz, R. M., Suess, E., and Chen, C. T. (1992). The effects of magnesium-to-calcium ratios in artificial seawater, at different ionic products, upon the induction time, and the mineralogy of calcium carbonate: a laboratory study. Geol. Rundsch. 81, 571–578. doi: 10.1007/BF01828616
Schulz, K. G., Bach, L. T., and Dickson, A. G. (2023). Seawater carbonate system considerations for ocean alkalinity enhancement research. State Planet Discuss. 2023, 1–24. doi: 10.5194/sp-2023-12
Sharp, J., Pierrot, D., Humphreys, M., Epitalon, J., Orr, J., Lewis, E., et al. (2021). CO2SYSv3 for MATLAB. Zenodo 10:3950562. doi: 10.5281/zenodo.3950562
Tang, H., Wu, X., Xian, H., Zhu, J., Wei, J., Liu, H., et al. (2020). Heterogeneous nucleation and growth of CaCO3 on calcite (104) and aragonite (110) surfaces: implications for the formation of abiogenic carbonate cements in the ocean. Fortschr. Mineral. 10:294. doi: 10.3390/min10040294
Tossavainen, M., Engstrom, F., Yang, Q., Menad, N., Lidstrom Larsson, M., and Bjorkman, B. (2007). Characteristics of steel slag under different cooling conditions. Waste Manag. 27, 1335–1344. doi: 10.1016/j.wasman.2006.08.002
Tuck, C. C. (2023). Iron and steel slag, U.S. Geological Survey, mineral commodity summaries. Available at: https://worldsteel.org/steel-topics/statistics/world-steel-in-figures-2022/ (Accessed September 19, 2023)
Worldsteel. (2022). World steel association, world steel in figures 2022. Available at: https://worldsteel.org/steel-topics/statistics/world-steel-in-figures-2022/ (Accessed September 19, 2023)
Yildirim, I. Z., and Prezzi, M. (2011). Chemical, mineralogical, and morphological properties of steel slag. Adv. Civil Eng. 2011:463638, 1–13. doi: 10.1155/2011/463638
Keywords: ocean alkalinity enhancement, carbon capture potential, iron slag, steel slag, carbonate chemistry
Citation: Moras CA, Joannes-Boyau R, Bach LT, Cyronak T and Schulz KG (2024) Carbon dioxide removal efficiency of iron and steel slag in seawater via ocean alkalinity enhancement. Front. Clim. 6:1396487. doi: 10.3389/fclim.2024.1396487
Edited by:
Phil Renforth, Heriot-Watt University, United KingdomReviewed by:
Christopher Robert Pearce, University of Southampton, United KingdomSpyros Foteinis, Heriot-Watt University, United Kingdom
Copyright © 2024 Moras, Joannes-Boyau, Bach, Cyronak and Schulz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Charly A. Moras, Yy5tb3Jhcy4xMEBzdHVkZW50LnNjdS5lZHUuYXU=