 Christian Humpel
Christian Humpel- Laboratory of Psychiatry and Experimental Alzheimer’s Research, Medical University of Innsbruck, Innsbruck, Austria
Alzheimer’s disease (AD) is a severe neurodegenerative brain disorder molecularly characterized by extracellular β-amyloid plaques, intraneuronal tau neurofibrillary tangles, cholinergic neuron death, neuroinflammation, vascular damage, and astroglial and microglial activation. AD is a complex disorder, with >99% of all cases being sporadic and typically occuring around the age of 65. Due to this intricate nature of the disorder, in vitro experiments have limitations; however, three-dimensional organotypic brain slices may offer the best alternative for studying the mechanisms involved in the progression of AD. This review provides an overview of how to study the general aspects of AD ex vivo, focusing on (a) β-amyloid plaques in brain slices, (b) tau pathology induced by chemical drugs, (c) cell death of cholinergic neurons and protection by nerve growth factor, (d) activation of astrocytes and microglia, and (e) vascular pathologies, including the role of platelets. Furthermore, we investigated (f) how microcontact printing on brain slices can be used to study the spread of β-amyloid and tau, and (g) how brain slices can help identify novel human AD biomarkers.
Organotypic brain slices and Alzheimer’s disease
Alzheimer’s disease (AD) is a severe neurodegenerative brain disorder, characterized by two main pathologies, namely extracellular β-amyloid plaques and intraneuronal neurofibrillary tangles (NFTs) composed of phosphorylated Tau (Braak and del Tredici, 2015; Swerdlow, 2012). However, AD is a complex disease with many other pathologies such as neuroinflammation, silent strokes, activation of astrocytes and microglia, and dysfunction of microglia (Irvine et al., 2008; Obulesu and Jhansilakshmi, 2014; Glass and Arnold, 2012). There is also severe vascular damage, with the breakdown of the blood–brain barrier (BBB), vascular damage, angiogenesis, and activation of platelets, as well as deposition of β-amyloid in vessels, known as cerebral amyloid angiopathy (CAA) (Perlmutter, 1994; Greenberg et al., 2020). Finally, cholinergic neurons degenerate in the brain, and the loss of acetylcholine results in severe memory defects (Berry and Harrison, 2023; Dunnett and Fibiger, 1993).
Therefore, studying this complex disorder is challenging. AD is a disease of old age (>65 years) and is a non-genetic sporadic disease (Braak and del Tredici, 2015; Glass and Arnold, 2012). The exact cause for the emergence of AD is not yet fully understood. However, many risk factors are known, such as lifestyle factors, that affect vascular brain support. Sporadic AD is the most common form of AD (>99%), and only a few families have a genetic form of AD (1% of cases) (Braak and del Tredici, 2015; Glass and Arnold, 2012). As mentioned, the analysis of human brains would be the best strategy to study AD using brain imaging [positron emission tomography (PET) or magnetic resonance tomography (MRT)]; however, these techniques do not provide high resolution. Post-mortem brain studies are an alternative, but the complexity of the disease makes it difficult to study, and post-mortem delay results in false-positive results. The human Nun Study (Lemonick and Park, 2001) aimed to overcome this problem, as this is a well-characterized homogenous population. The Nun Study of aging and AD is a continuing longitudinal study, begun in 1986, with the aim to examine the onset and progression of AD in a well-controlled human population (same lifestyle, education, nutrition) with 678 nuns aged 75–106 years. The main outcome was that about one-third of the nuns showed β-amyloid plaques in the brain without any symptoms and with a normal cognitive function, which challenged the β-amyloid-cascade hypothesis (see below) and suggested that lifelong learning might partly counteract the neurodegenerative process.
Animal studies are complex and have primarily focused on transgenic mice (Humpel, 2019). However, transgenic mice do not reflect complex disorders and can only provide information on the progression of plaques or NFTs, but not on the development of sporadic disease. Thus, transgenic mice are useful for studying genetic but not sporadic AD (Foidl and Humpel, 2019a). It would be highly interesting to establish a mouse model of sporadic AD by treating mice for months with risk factors such as drugs that increase blood pressure, glucose, or cholesterol (Korde and Humpel, 2024; Foidl and Humpel, 2019b). Using mouse models is limited, as they live for only 24–28 months. To date, no mouse model of sporadic AD has been established, and we can only study single pathologies using cell culture models. To the best of my knowledge, the organotypic brain slice model is the most suitable for studying different aspects of dementia as it contains all brain cells in a complex three-dimensional architecture (Humpel, 2015). The use of organotypic brain slices began in the 1980s by Gähwiler (1981), improved using membrane inserts by Stoppini’s research (Stoppini et al., 1991), and further developed in my lab using whole coronal vibratome slices (Humpel, 2018) (Figure 1). This review summarizes and discusses how brain slices may be useful for studying different aspects of AD.

Figure 1. Organotypic brain slices are prepared from a postnatal day 8–10 mouse brain, sectioned with a vibratome 150 μm; (A) and then placed on a semipermeable membrane insert and cultured in 6-wells with medium (B) for several weeks at 37°C and 5% CO2. (B) shows a coronal brain slice at the hippocampal level. Scale bar = 2.2 cm (A); 0.5 cm (B).
β-amyloid plaques in brain slices
Aβ is a small peptide of 40 or 42 amino acids. The larger 42 amino acid form is more toxic and tends to form aggressive aggregates, leading to the development of extracellular β-amyloid plaques in the brain, as described in the so-called “β-amyloid cascade hypothesis” (Hardy and Higgins, 1992; Ittner and Götz, 2011; Swerdlow, 2012). The smaller form is deposited in vessels and is found at higher levels in the blood. β-amyloid is formed from a large transmembrane protein, the amyloid precursor protein (APP), which is processed by secretases (LaFerla et al., 2007). Many studies have used transgenic mice with mutated APP that develop β-amyloid plaques (Price et al., 1998; Lannfelt et al., 1993). In our experiments, we used the APP_SweDI mouse model, in which β-amyloid plaques develop in 6-month-old mice (Humpel, 2019), and we aimed to prepare adult brain slices to study the progression of β-amyloid plaques (Humpel, 2019). Unfortunately, a main disadvantage of adult brain slices is that brain cells do not survive well (Humpel, 2019). Usually, slices from postnatal mice flatten and become transparent, which is a good sign for healthy slices. Adult brain slices (and slices taken from mice older than 14 days) do not flatten, and therefore, we cultured thinner (110 μm) slices. Thus, as plaques develop in older mice and not in postnatal mice, it was not possible to study the progression of the plaques, but we could study plaques at different ages of the mice (Figure 2). In another experiment, we showed that neprilysin can degrade these plaques (Humpel, 2022), and slices are useful to study plaque elimination ex vivo.
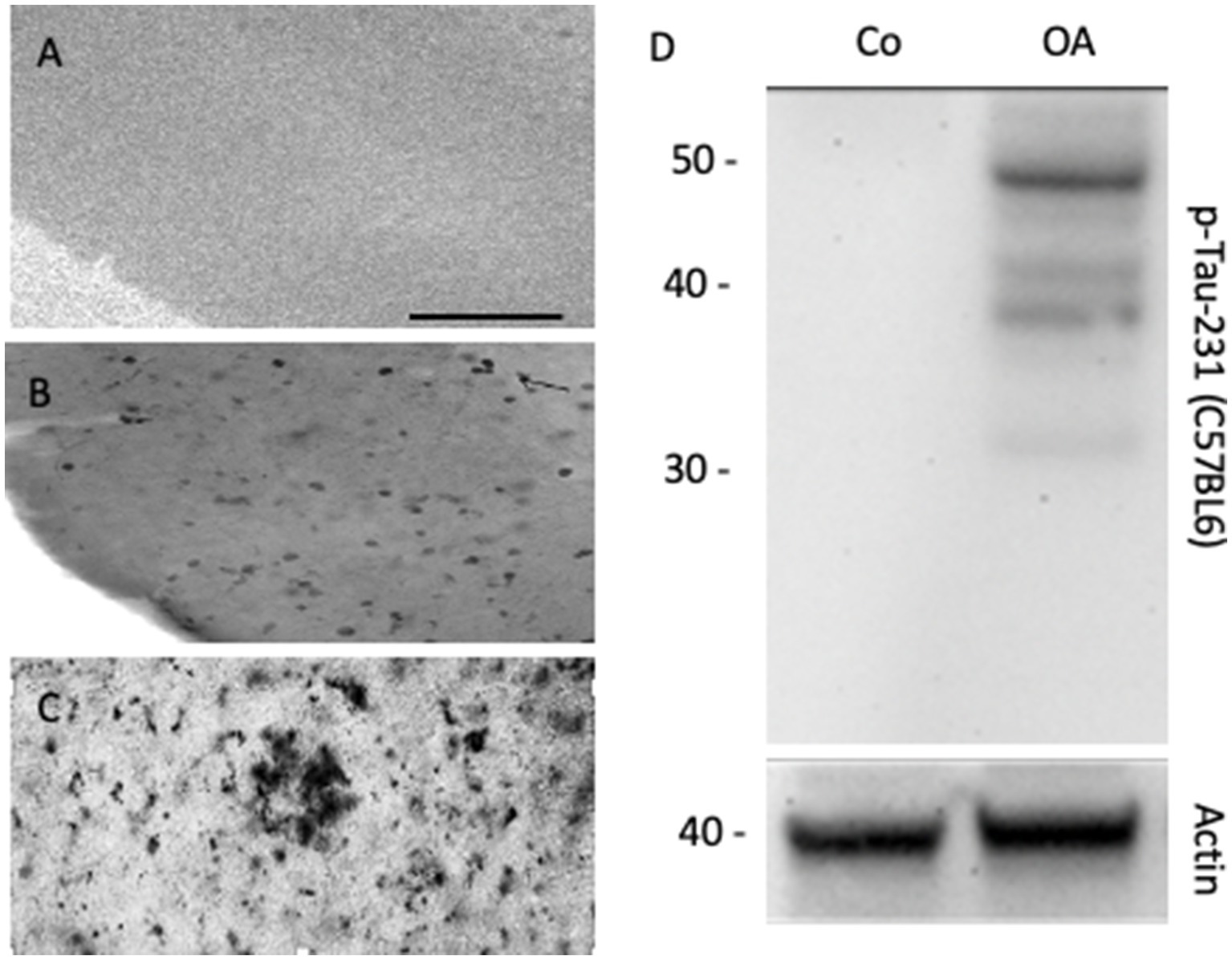
Figure 2. Organotypic brain slices can be cultured from adult APP_SweDI transgenic mice where β-amyloid plaques progressively develop, when incubated from 4-month-old brains (A), 5-month-old brains (B), or 6-month-old brains (C), where β-amyloid plaques are visualized. In organotypic mouse brain slices, the phosphorylation of Tau (e.g., phospho-Tau-231) can be studied when stimulated with okadaic acid (OA) versus a control (Co). (D) shows a representative Western Blot, where Tau is seen as a 50 kDa phosphorylated protein after treatment of slices with OA. Actin (40 kDa) serves as a control. Scale bar = 220 μm (A–C).
Tau pathology in brain slices
Tau is a large 60 kDa microtubule-associated protein that plays a role in axonal transport (Gallardo and Holtzman, 2019; Busche and Hyman, 2020). Tau is highly regulated by phosphorylation at about 40 sites. In AD, tau becomes hyperphosphorylated, and this process leads to a defect in axonal transport and thus the neurons die (“Tau-hypothesis of AD”) (Ittner and Götz, 2011; Swerdlow, 2012; Braak and del Tredici, 2012). The reasons for this hyperphosphorylation are unknown; however, enhanced protein kinase activity or decreased phosphatase activity may cause this effect (Bakota and Brandt, 2016; Busche and Hyman, 2020). Furthermore, it is not clear how β-amyloid and tau interact. The slice model may allow the study of transgenic mice with defects in tau protein, leading to NFTs. More importantly, we explored the mechanisms by which tau is phosphorylated and enters the toxicity pathway. Pharmacological treatments should be tested to block hyperphosphorylation, especially of different protein phosphatases. In a previous report, we showed that okadaic acid enhances tau phosphorylation in brain slices (Foidl and Humpel, 2018) (Figure 2D). Okadaic acid is a cytotoxin and was originally isolated from the black sponge Halichondria okadai. Amongst others, it particularly leads to the activation of GSK-3β, neuroinflammation, and oxidative stress.
Cholinergic neurons in brain slices
Acetylcholine is a major neurotransmitter in the brain that is responsible for memory (Berry and Harrison, 2023; Dunnett and Fibiger, 1993). Cholinergic neurons are located in the basal nucleus of Meynert, where they project to the cortex and are responsible for long-term memory. These neurons are also located in the septum and diagonal band of Broca, project to the hippocampus, and are responsible for short-term memory. Interestingly, cholinergic neurons degenerate in AD, although the reason is unknown (“cholinergic hypothesis of AD”) (Berry and Harrison, 2023; Dunnett and Fibiger, 1993). It is well known that the classical growth factor nerve growth factor (NGF) supports the survival of cholinergic neurons (Humpel, 2021). We have a long experience in culturing cholinergic neurons of the basal nucleus of Meynert (nBM) and have shown that NGF supports the survival of these neurons (Lapchak, 1993; Hefti and Will, 1987) (Figure 3). Interestingly, these neurons require long-term support for survival. Thus, the brain slice model may be suitable for studying how growth factors support survival and how pharmaceutical drugs can be developed. Generation of BBB-permeable NGF-like drugs may delay the degeneration of these neurons in the brain.
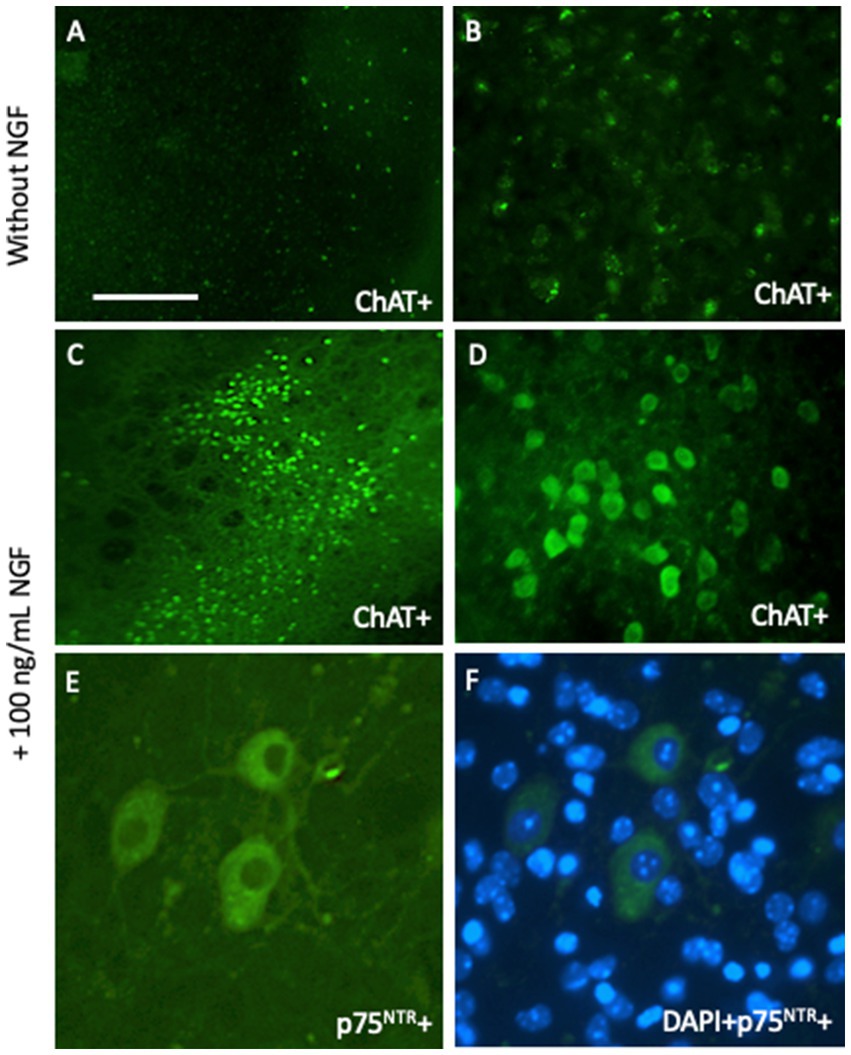
Figure 3. Cholinergic neurons from the basal nucleus of Meynert can be cultured for 2 weeks and degenerate in brain slices without any growth support (A,B), but survive when incubated with 100 ng/mL nerve growth factor (NGF) (C–F). Brain slices are immunohistochemically stained with antibodies against choline acetyltransferase (A–D) or the low-affinity NGF receptor p75 neurotrophin receptor (p75NTR) (E,F). Scale bar = 660 μm (A,C), 165 μm (B,D), 40 μm (E,F).
Cholinergic neurons offer a nice way to study cellular survival. The first issue is to judge if slices flatten and become transparent, as this is a good sign of healthy slices. Thick slices should be deleted. Second, a general blue fluorescent DAPI staining is suggested to evaluate the slice quality and the homogenous distribution of the cellular layer and the orientation of the slices. Third, a fast and simple propidium iodide staining is recommended to see cellular death, as visualized by red fluorescent nuclei. And finally, I suggest specific neuronal markers, like neurofilament or microtubule-associated protein-2, or in the case of cholinergic neurons, choline acetyltransferase. Such cell death assays can be performed with any kind of slices, not only cholinergic neurons. Cholinergic neurons offer a simple way to study cell death as neurons degenerate without NGF and survive with NGF (Humpel and Weis, 2002). In our hands, cholinergic neurons survive for 2–4 weeks with NGF, but we have also seen cholinergic neurons 50 weeks in culture with NGF (Marksteiner and Humpel, 2008).
Astrocytes and microglia in brain slices
Glial cells are important in the brain and play a role in stabilizing neurons, BBB function, and electrolytic balance (astroglia), formation of myelin sheets along axons, fast signaling (oligodendrocytes), and immune and phagocytic reactions (microglia) (Preeti et al., 2022; Hashioka et al., 2021; Kim et al., 2020; Kim et al., 2018; Schubert et al., 2001; Mrak and Griffin, 1996). These cells are present in cultured brain slices and can be stained for glial fibrillary acidic protein (GFAP, astroglia) (Daschil and Humpel, 2016), myelin oligodendrocyte protein (MOG, oligodendrocytes) (Yilmaz et al., 2024), Iba1, or CD11b (microglia) (Steiner et al., 2024) (Figure 4). In AD, astroglia is found around β-amyloid plaques in the halo and is activated to counteract plaque development and support plaque elimination (Daschil and Humpel, 2016). The main immune cells of the brain are microglia, which are present in different forms: round-amoeboid, ramified, and large macrophages, and their major aim is to eliminate brain debris and plaques (Steiner et al., 2024). Thus, it is of great interest to study microglia, as they become dysfunctional during AD progression. All of these cells can be studied in mouse organotypic brain slices and their respective pathological processes in AD.
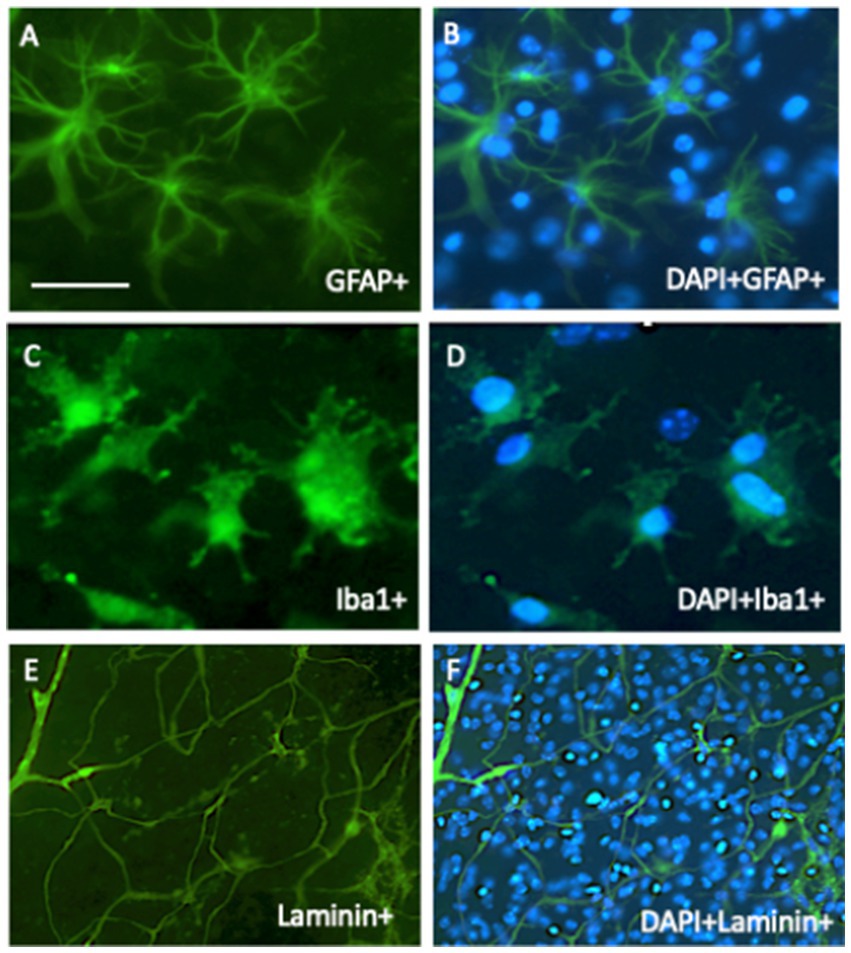
Figure 4. In organotypic brain slices, all brain cells survive and can be studied. In 2-week-old cultured slices, astroglia (A,B) survive and are stained with glial-fibrillary acidic protein (GFA), microglia (C,D) are stained with Iba1 antibody, and vessel/endothelial cells (E,F) with laminin. All cells were stained immunohistochemically with green fluorescent Alexa-488 secondary antibodies. (B,D,F) show the counterstaining with blue fluorescent nuclear DAPI. Scale bar = 60 μm (A–D); 260 μm (E,F).
Vascular pathologies and platelets in brain slices
Evidently, vascular structures are damaged in the AD brain, and the smaller β-amyloid (40) peptide is deposited in the brain vessels, causing CAA (Koemans and van Etten, 2025; van den Brink et al., 2024; Godrich et al., 2024; Vinters, 1987). This defect in blood vessels, together with the breakdown of the BBB, causes a loss of energy supply (glucose and O2) to the brain and an influx of toxic molecules from the blood. Defects in the vascular structure play a role in the progression of sporadic AD (Scheffer et al., 2021; de la Torre, 2018, 2010; Milionis et al., 2008; Lucas and Rifkind, 2013). It is suggested to maintain healthy vascular brain vessels in order to delay the progression of AD and 14 (vascular) risk factors should be considered: low education, hearing loss, high blood pressure, smoking, fatty food, depression, no sports, diabetes, alcohol, head trauma, air pollution, social isolation, high low-density lipoprotein cholesterol, and visual loss.
Organotypic brain vessels contain the complete vessel network, which can be easily stained with laminin (Figures 4E,F). We studied the role of the vessel system in our laboratory and found an intact vessel that was rearranged and connected in co-cultures. However, we could not prove that the vessels were intact, as they did not transport blood and were not tube-like; thus, we refer to them as vessel-like (Moser et al., 2003). In this context, it is worth mentioning that the major disadvantage of culturing brain slices is the lack of a BBB, and thus, influx and efflux cannot be studied (Humpel, 2015). Vascular structure degenerates in AD, and angiogenic factors can be easily studied, showing re-formation of vessels (Ucar et al., 2020). In addition, vessel repair, such as clotting, can be studied; in this context, the function of platelets is of great interest. Platelets contain much APP and secrete smaller β-amyloid (40) forms that may play a role in blood clotting (Humpel, 2017). It has also been hypothesized that platelets contribute to the pathology of AD. We and others have formed the “vascular hypothesis of AD,” that β-amyloid depositions (formed during CAA) in and around the vessel may spread over into the whole brain (Scheffer et al., 2021; de la Torre, 2018, 2010; Milionis et al., 2008; Lucas and Rifkind, 2013; Humpel, 2011). It has been suggested that damage to brain vessels causes the activation and migration of platelets to repair these vessels, and a dysfunctional cascade may activate the toxic β-amyloid cascade (Humpel, 2017). Such a process can be studied in brain slices: in a previous study, we (Kniewallner et al., 2018) isolated platelets from wildtype and transgenic AD mice, labelled them with a red fluorescent dye, and infused the labelled platelets back into anesthetized wildtype mice. From these mice, brain slices were prepared and cultured. Our data showed that platelets from AD mice severely damaged cortical vessels and entered the parenchyma, causing increased β-amyloid staining, inflammation, and activation of microglia around the lesions.
Spreading of “prion-like” proteins in slices
It is not clear how the AD pathology starts, but the “spreading hypothesis” (Walsh and Selkoe, 2016) suggests that toxic mutated prion-like proteins (such as alpha-synuclein or β-amyloid or mutated tau) are consumed, taken up by the gut, and transported along the vagus nerve into the brainstem (Wang et al., 2019). As AD develops over the decades, these aberrant/misfolded proteins spread throughout the brain in a prion-like manner (Walker, 2018). It is evident that plaques are already present in young individuals, develop over decades, spread from the brain stem to the entorhinal cortex and hippocampus, and then to the cortex. But how can we study spreading in slices? We have developed a method where proteins or peptides can be delivered onto brain slices loaded into collagen scaffolds (Ucar and Humpel, 2018). When we studied the survival of cholinergic neurons, growth factors (in this case, NGF) were added to the medium. This was never a problem, as long as the growth factors diffuse through the semipermeable 0.4 μm membranes and are absorbed into the brain slices. However, in some experiments, it was necessary to load the factors of interest directly onto the brain slices. The pipetting of a 1 μL solution directly onto the slice is not successful, as the fluid rapidly diffuses all over the brain slice. Thus, to apply the substance locally, we developed and optimized the collagen scaffold. Collagen is a natural, biocompatible, nontoxic molecule that can aggregate when mixed with 4-arm-poly (ethylene glycol) (PEG) succinimidyl succinate (PEG) (Ucar and Humpel, 2018). Very small collagen scaffolds with a diameter of 1 mm can be produced and loaded with any molecule of interest. These scaffolds can then be placed directly onto the brain slice in any area of interest (Ucar and Humpel, 2018). The collagen scaffold degrades over time and slowly releases the substance of interest. Using these collagen scaffolds, we applied β-amyloid or tau, or alpha-synuclein onto brain slices and studied their spreading in the brain slice (Korde and Humpel, 2022; Ucar et al., 2022; Moelgg et al., 2021).
Identify novel human AD biomarkers using microcontact printing
To decrease the amount of collagen in brain slices, we developed a microcontact printing (μCP) method (Steiner and Humpel, 2023). Using a master plate with respective stamps, small lines (30 μm width) can be printed directly onto the semipermeable membrane with any factor of interest (Figure 5). In our study, we loaded human plasma onto these lanes, coupled them to mouse brain slices, and visualized the migration of cells outside the slices (Steiner and Humpel, 2023). We hypothesized that the plasma of patients with AD contains factors that inhibit or activate mouse-derived cells compared to plasma from healthy controls.

Figure 5. Microcontact prints of human plasma are generated and checked with a red fluorescent Alexa-546 antibody, where several 30 μm lanes are seen (A,B). These microcontact prints are then connected to (half)brain slices at the hippocampal level and cultured for 2–4 weeks (C,D). Iba1-positive microglia migrate out from the brain slices along the lanes and can be quantified (E,F). Plasma from healthy controls and plasma from Alzheimer’s patients have different effects on the migration pattern of round-amoeboid microglia, and their protein pattern can be differentiated with differential mass spectrometry. This approach may allow the identification of new putative biomarkers. Scale bar = 260 μm (A); 870 μm (D); 150 μm (F).
Microcontact printing allows the transfer of proteins or peptides onto membranes in a sub-μm resolution. For printing, polydimethylsiloxane (PDMS) stamps were fabricated from a master mold with the desired lane pattern. For exploration of novel biomarkers, 30 μm lanes were printed onto cell culture membranes loaded with collagen. Collagen hydrogel-based microcontact prints allowed proteins to be gradually released into organotypic brain slices. Collagen also supports cell migration, attachment, differentiation, proliferation, and survival. To search for plasma biomarkers, plasma from patients with AD can be μCP together with collagen onto membranes as 30 μm lanes. Briefly, 100 μL of plasma is lyophilized and resuspended in the collagen-PEG solution and then printed as 30 μm lanes onto the membrane and ultraviolet (UV) sterilized. We believe that a short exposure (10 min) to UV light does not affect the proteins loaded in collagen, although we do not have data or evidence to support this claim, and further studies are needed to prove it. The next day, brain slices were placed onto these plasma prints in the culture inserts. After 2–4 weeks of culture, the slice-derived cells that migrated along the lanes or formed vessels along the lanes were quantified.
Using this method with subsequent differential mass spectrometry, we identified new putative biomarkers in AD plasma (Yilmaz et al., 2024; Steiner et al., 2024). First, we showed that the plasma of patients with AD inhibits the migration of round-amoeboid Iba1 positive microglia (Steiner et al., 2024). This process could be mediated via mannose-binding protein C, macrophage receptor MARCO, complement factor H-related protein-3, and C-reactive protein. Second, we showed that plasma of patients with AD enhances the formation of myelin oligodendrocyte glycoprotein+ dots along nerve fibers, possibly involving aldehyde-dehydrogenase 1A1, alpha-synuclein, and protein S100-A4 (Yilmaz et al., 2024). Finally, we showed that plasma of patients with AD increases migration of laminin+/lectin+ endothelial cells, possibly via modulation of C-reactive protein, basigin, and trem-like transcript 1 protein (Yilmaz et al., 2025).
Experimental applications using organotypic brain slices
In this review, I discuss how distinct pathologies of AD can be studied in organotypic brain slices: manipulation of β-amyloid plaques, phosphorylation of tau, inflammation and reactive gliosis, cell death and protection of cholinergic neurons, and immune responses, as well as to find novel biomarkers. Table 1 provides a summary of how ex vivo brain slices can help understand AD pathologies.
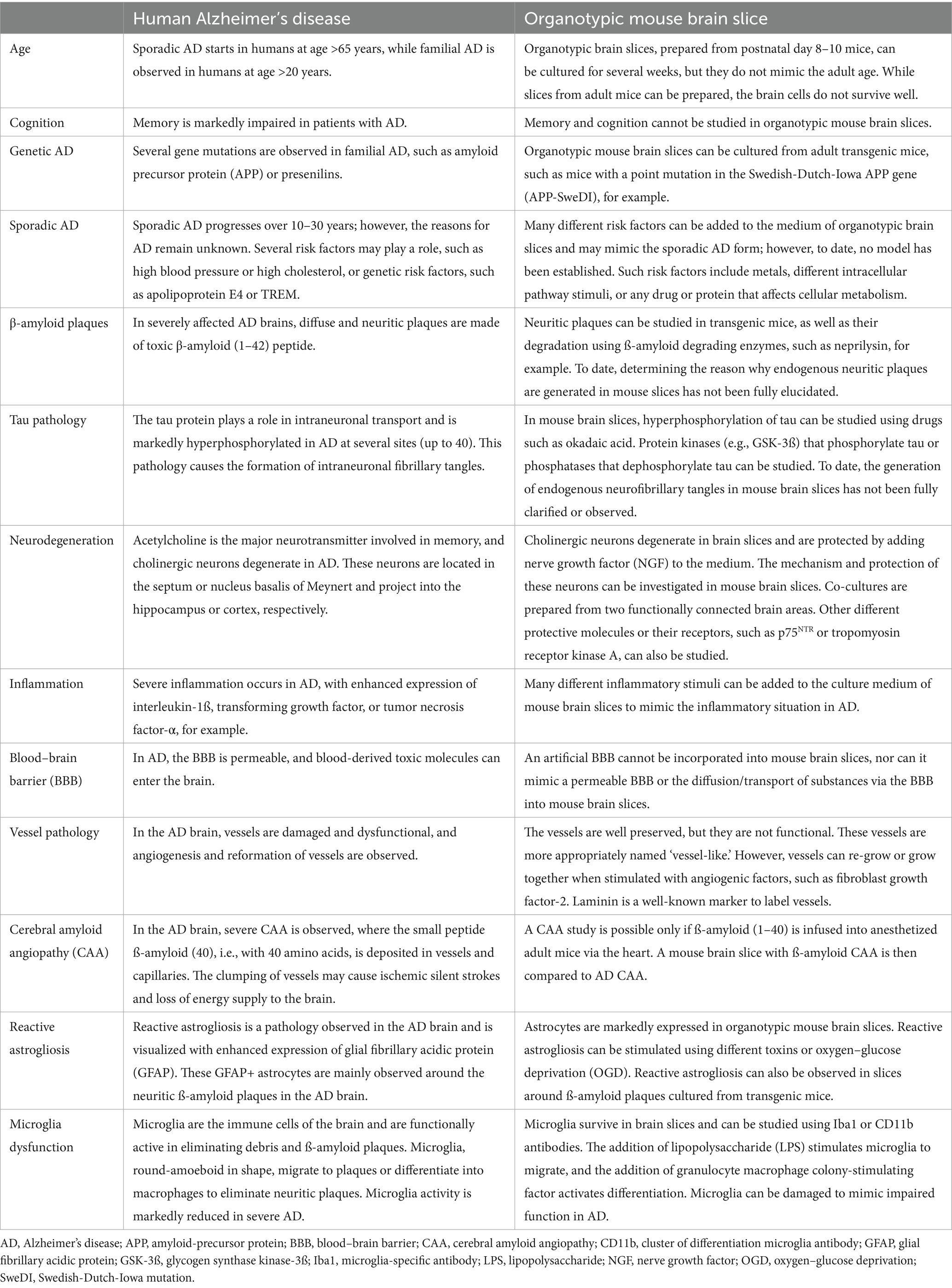
Table 1. Comparison between human Alzheimer’s disease (AD) and organotypic mouse brain slices.
Limitations of brain slices
Organotypic brain slices have several limitations. It is a disadvantage that the complex brain pathology of AD cannot be studied in slices at the same time. To date, we have not been able to stimulate slices to produce β-amyloid plaques or to test the hyperphosphorylation of tau and its complex sporadic cascade of events. We do not yet have a brain slice culture system for modeling all pathologies of AD and their time-dependent progression. We also lack a functional BBB connected to brain slices to study vascular-related pathologies and influx and efflux processes in the blood and cerebrospinal fluid. Furthermore, postnatal slices do not mimic complex age-related diseases, as sporadic AD is seen in humans aged > 65 years, although the pathology begins 30 years prior. While there are limitations regarding AD pathology, the slice technology also has technical limitations. It is very difficult to establish and culture brain slices and requires at least 2–3 months of experience to learn and handle this technique. The main challenge is producing slices that flatten during culture and display high survival rates in all brain cells. The addition of serum and growth factors may selectively enhance the survival of specific cells. However, the major disadvantage is that we and others are unable to culture adult slices with healthy neurons or other brain cells. Further study is necessary to optimize the long-term survival (>2 weeks) of brain cells in adult slices, especially when considering adult human tissues.
Outlook and translation to humans
In summary, organotypic brain slices offer an excellent ex vivo model for mimicking different aspects of neurodegenerative diseases, including AD, Parkinson’s disease, and other brain disorders. Much research has been conducted to improve this model.
1. The culturing of brain slices on Millipore inserts is expensive, and thus far, Merck inserts are the best choice. We have already reduced costs by reusing undamaged inserts and using an extra membrane on top of the inserts. Pre-cut extra membranes are also cost-effective. Alternatively, we cut the membrane inserts from a roll, which markedly reduces cost. To date, we have developed novel “ring-inserts,” where we glued a membrane on plastic rings and culture slices. This is the cheapest and most reproducible method, and we have demonstrated the survival of cholinergic and dopaminergic neurons (Gern et al., submitted 2025).
2. These “ring inserts” were tested for live-cell imaging of brain slices. A disadvantage of slices is that they grow on membrane inserts and cannot be investigated without cutting the membranes. However, the use of additional membranes can overcome this problem. Using our novel “ring-inserts,” we can investigate organotypic brain slices directly under the microscope for hours or weeks. A recent report from our lab showed that astrocytes can be fluorescently labelled with GFAP and vessels with laminin using microcontact printing, and can be investigated over weeks using live cell imaging techniques (Humpel, 2025).
3. The major challenge is culturing brain slices from adult mice or rats. This will revolutionize experimental culture models, as such models are more likely to mimic the adult situation of AD or other brain disorders. To date, the survival of adult slice cells has been very low, and the conditions under which adult slices show high viability must be developed. Interestingly, slices from brains taken from mice older than 12–14 days do not flatten well, and brain cells do not survive well in such slices. It is unclear why such slices show a lower viability at this age, and we recommend using brains from postnatal day 8–10 for optimal culturing.
4. Finally, it would be extremely interesting to prepare and culture brain tissue from human brains and translate the findings to a real human AD situation. Definitely, the pioneering study of Mendes et al. (2018) may offer such a novel way to culture human brain slices at least for a short period of time. Brain slices should offer a way to culture brain sections after biopsy or from postmortem brains and culture slices over weeks, and to investigate their pathologies. Modern computer-assisted techniques and artificial intelligence may help optimize this technique in the future.
Author contributions
CH: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
Special thanks go to Anna Draxl and Mohadeseh Ragerdikashani for their technical assistance. Finally, the MS has been corrected by Editage (VIDXV_2 and VIDXV_3), and I thank them for their excellent corrections.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Bakota, L., and Brandt, R. (2016). Tau biology and tau-directed therapies for Alzheimer's disease. Drugs 76, 301–313. doi: 10.1007/s40265-015-0529-0
Berry, A. S., and Harrison, T. M. (2023). New perspectives on the basal forebrain cholinergic system in Alzheimer's disease. Neurosci. Biobehav. Rev. 150:105192. doi: 10.1016/j.neubiorev.2023.105192
Braak, H., and Del Tredici, K. (2012). Where, when, and in what form does sporadic Alzheimer's disease begin? Curr. Opin. Neurol. 25, 708–714. doi: 10.1097/WCO.0b013e32835a3432
Braak, H., and Del Tredici, K. (2015). Neuroanatomy and pathology of sporadic Alzheimer's disease. Adv. Anat. Embryol. Cell Biol. 215, 1–162.
Busche, M. A., and Hyman, B. T. (2020). Synergy between amyloid-beta and tau in Alzheimer's disease. Nat. Neurosci. 23, 1183–1193. doi: 10.1038/s41593-020-0687-6
Daschil, N., and Humpel, C. (2016). Green-fluorescent protein+ astrocytes attach to beta-amyloid plaques in an Alzheimer mouse model and GFP+ astrocytes are sensitive for clasmatodendrosis. Front. Aging Neurosci. 8:75. doi: 10.3389/fnagi.2016.00075
de la Torre, J. C. (2010). The vascular hypothesis of Alzheimer's disease: bench to bedside and beyond. Neurodegener Dis 7, 116–121. doi: 10.1159/000285520
de la Torre, J. (2018). The vascular hypothesis of Alzheimer's disease: a key to preclinical prediction of dementia using neuroimaging. J Alzheimer's Dis 63, 35–52. doi: 10.3233/JAD-180004
Dunnett, S. B., and Fibiger, H. C. (1993). Role of forebrain cholinergic systems in learning and memory: Relevance to the cognitive deficits of aging and Alzheimer's dementia. Prog Brain Res 98, 413–420. doi: 10.1016/s0079-6123(08)62425-5
Foidl, M., and Humpel, C. (2018). Differential hyperphosphorylation of tau-S199, −T231 and -S396 in organotypic brain slices of Alzheimer mice. A model to study early tau hyperphosphorylation using Okadaic acid. Front. Aging Neurosci. 10:113. doi: 10.3389/fnagi.2018.00113
Foidl, B., and Humpel, C. (2019a). Can mouse models mimic sporadic Alzheimer’s disease? Neural Regen. Res. 15, 401–406. doi: 10.4103/1673-5374.266046
Foidl, B. M., and Humpel, C. (2019b). Chronic treatment with five vascular risk factors causes cerebral amyloid angiopathy but no Alzheimer pathology in C57BL6 mice. Brain Behav. Immun. 78, 52–64. doi: 10.1016/j.bbi.2019.01.009
Gähwiler, B. H. (1981). Organotypic monolayer cultures of nervous tissue. J. Neurosci. Methods 4, 329–342. doi: 10.1016/0165-0270(81)90003-0
Gallardo, G., and Holtzman, D. M. (2019). Amyloid-beta and tau at the crossroads of Alzheimer's disease. Adv. Exp. Med. Biol. 1184, 187–203. doi: 10.1007/978-981-32-9358-8_16
Glass, D. J., and Arnold, S. E. (2012). Some evolutionary perspectives on Alzheimer's disease pathogenesis and pathology. Alzheimers Dement. 8, 343–351. doi: 10.1016/j.jalz.2011.05.2408
Godrich, D., Pasteris, J., Martin, E. R., Rundek, T., Schellenberg, G., Foroud, T., et al. (2024). Cerebral amyloid angiopathy impacts neurofibrillary tangle burden and cognition. Brain Commun. 6:fcae369. doi: 10.1093/braincomms/fcae369
Greenberg, S. M., Bacskaim, B. J., Hernandez-Guillamon, M., Pruzin, J., Sperling, R., and van Veluw, S. J. (2020). Cerebral amyloid angiopathy and Alzheimer disease - one peptide, two pathways. Nat. Rev. Neurol. 16, 30–42. doi: 10.1038/s41582-019-0281-2
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer's disease: the amyloid cascade hypothesis. Science 256, 184–185. doi: 10.1126/science.1566067
Hashioka, S., Wu, Z., and Klegeris, A. (2021). Glia-driven Neuroinflammation and systemic inflammation in Alzheimer's disease. Curr. Neuropharmacol. 19, 908–924. doi: 10.2174/1570159X18666201111104509
Hefti, F., and Will, B. (1987). Nerve growth factor is a neurotrophic factor for forebrain cholinergic neurons; implications for Alzheimer's disease. J. Neural Transm. Suppl. 24, 309–315.
Humpel, C. (2011). Chronic mild cerebrovascular dysfunction as a cause for Alzheimer's disease? Review. Exp. Gerontol. 46, 225–232. doi: 10.1016/j.exger.2010.11.032
Humpel, C. (2015). Organotypic brain slice cultures - review. Neuroscience 305, 86–98. doi: 10.1016/j.neuroscience.2015.07.086
Humpel, C. (2017). Platelets: their potential contribution to the generation of beta-amyloid plaques in Alzheimer’s disease. Curr. Neurovasc. Res. 14, 290–298. doi: 10.2174/1567202614666170705150535
Humpel, C. (2018). Organotypic brain slice cultures. Curr. Protoc. Immunol. 123:e59. doi: 10.1002/cpim.59
Humpel, C. (2019). Organotypic brain slices of adult transgenic mice: a tool to study Alzheimer’s disease. Curr. Alzheimer Res. 16, 172–181. doi: 10.2174/1567205016666181212153138
Humpel, C. (2021). NGF released from blood cells or collagen hydrogels as a therapeutic target in Alzheimer’s disease? Adv Exp Med Biol 1331, 193–202. doi: 10.1007/978-3-030-74046-7_12
Humpel, C. (2022). Intranasal neprilysin rapidly eliminates beta-amyloid plaques, but causes plaque compensations: the explanation why the beta-amyloid cascade may fail? Neural Regeneration Review 17, 1881–1884. doi: 10.4103/1673-5374.335138
Humpel, C. (2025). Long-term live-cell imaging of GFAP+ astroglia and laminin+ vessels in organotypic mouse brain slices using microcontact printing. Front. Cell. Neurosci. 19:1540150. doi: 10.3389/fncel.2025.1540150
Humpel, C., and Weis, C. (2002). Nerve growth factor and cholinergic CNS neurons studied in organotypic brain slices: implications in Alzheimer’s disease? J Neural Transmission Supplement 62, 253–263. doi: 10.1007/978-3-7091-6139-5_23
Irvine, G. B., El-Agnaf, O. M., Shankar, G. M., and Walsh, D. M. (2008). Protein aggregation in the brain: the molecular basis for Alzheimer's and Parkinson's diseases. Mol. Med. 14, 451–464. doi: 10.2119/2007-00100.Irvine
Ittner, L. M., and Götz, J. (2011). Amyloid-β and tau--a toxic pas de deux in Alzheimer's disease. Nat. Rev. Neurosci. 12, 65–72. doi: 10.1038/nrn2967
Kim, Y. S., Jung, H. M., and Yoon, B. E. (2018). Exploring glia to better understand Alzheimer's disease. Anim Cells Syst 22, 213–218. doi: 10.1080/19768354.2018.1508498
Kim, E., Otgontenger, U., Jamsranjav, A., and Kim, S. S. (2020). Deleterious alteration of glia in the brain of Alzheimer's disease. Int. J. Mol. Sci. 21:6676. doi: 10.3390/ijms21186676
Kniewallner, K., Foidl, B. M., and Humpel, C. (2018). Platelets isolated from an Alzheimer mouse damage healthy cortical vessels and cause inflammation in an organotypic ex vivo brain slice model. Sci. Rep. 8:15483. doi: 10.1038/s41598-018-33768-2
Koemans, E. A., and van Etten, E. S. (2025). Cerebral amyloid angiopathy: one single entity? Curr. Opin. Neurol. 38, 29–34. doi: 10.1097/WCO.0000000000001330
Korde, D. S., and Humpel, C. (2022). Spreading of P301S aggregated tau investigated in organotypic mouse brain slice cultures. Biomol. Ther. 12:1164. doi: 10.3390/biom12091164
Korde, D. S., and Humpel, C. (2024). A combination of heavy metals and intracellular pathway modulators induces Alzheimer disease-like pathologies in organotypic brain slices. Biomol. Ther. 14:165. doi: 10.3390/biom14020165.38397402
LaFerla, F. M., Green, K. N., and Oddo, S. (2007). Intracellular amyloid-beta in Alzheimer's disease. Nat. Rev. Neurosci. 8, 499–509. doi: 10.1038/nrn2168
Lannfelt, L., Folkesson, R., Mohammed, A. H., Winblad, B., Hellgren, D., Duff, K., et al. (1993). Alzheimer's disease: molecular genetics and transgenic animal models. Behav. Brain Res. 57, 207–213. doi: 10.1016/0166-4328(93)90137-f
Lapchak, P. A. (1993). Nerve growth factor pharmacology: application to the treatment of cholinergicneurodegeneration in Alzheimer's disease. Exp. Neurol. 124, 16–20. doi: 10.1006/exnr.1993.1168
Lemonick, M. D., and Park, A. (2001). The nun study. How one scientist and 678 sisters are helping unlock the secrets of Alzheimer's. Time 157, 54–59.
Lucas, H. R., and Rifkind, J. M. (2013). Considering the vascular hypothesis of Alzheimer's disease: effect of copper associated amyloid on red blood cells. Adv. Exp. Med. Biol. 765, 131–138. doi: 10.1007/978-1-4614-4989-8_19
Marksteiner, J., and Humpel, C. (2008). Beta-amyloid expression, release and extracellular deposition in aged rat brain slices. Mol. Psychiatry 13, 939–952. doi: 10.1038/sj.mp.4002072
Mendes, N. D., Fernandes, A., Almeida, G. M., Santos, L. E., Selles, M. C., Lyra E Silva, N. M., et al. (2018). Free-floating adult human brain-derived slice cultures as a model to study the neuronal impact of Alzheimer's disease-associated aβ oligomers. J. Neurosci. Methods 307, 203–209. doi: 10.1016/j.jneumeth.2018.05.021
Milionis, H. J., Florentin, M., and Giannopoulos, S. (2008). Metabolic syndrome and Alzheimer's disease: a link to a vascular hypothesis? CNS Spectr. 13, 606–613. doi: 10.1017/s1092852900016886
Moelgg, K., Jummun, F., and Humpel, C. (2021). Spreading of beta-amyloid in organotypic mouse brain slices and microglial protection of cholinergic neurons. Biomol. Ther. 11:434. doi: 10.3390/biom11030434
Moser, K. V., Schmidt-Kastner, R., Hinterhuber, H., and Humpel, C. (2003). Brain capillaries and cholinergic neurons persist in organotypic brain slices in the absence of blood flow. Eur. J. Neurosci. 18, 85–94. doi: 10.1046/j.1460-9568.2003.02728.x
Mrak, R. E., and Griffin, W. S. (1996). Role of activated glia and of glial cytokines in Alzheimer's disease: a review. Eos 16, 80–84.
Obulesu, M., and Jhansilakshmi, M. (2014). Neuroinflammation in Alzheimer's disease: an understanding of physiology and pathology. Int. J. Neurosci. 124, 227–235. doi: 10.3109/00207454.2013.831852
Perlmutter, L. S. (1994). Microvascular pathology and vascular basement membrane components in Alzheimer's disease. Mol. Neurobiol. 9, 33–40. doi: 10.1007/BF02816103
Preeti, K., Sood, A., and Fernandes, V. (2022). Metabolic regulation of glia and their Neuroinflammatory role in Alzheimer's disease. Cell. Mol. Neurobiol. 42, 2527–2551. doi: 10.1007/s10571-021-01147-7
Price, D. L., Tanzi, R. E., Borchelt, D. R., and Sisodia, S. S. (1998). Alzheimer's disease: genetic studies and transgenic models. Annu. Rev. Genet. 32, 461–493. doi: 10.1146/annurev.genet.32.1.461
Scheffer, S., Hermkens, D. M. A., van der Weerd, L., de Vries, H. E., and Daemen, M. J. A. P. (2021). Vascular hypothesis of Alzheimer disease: topical review of mouse models. Arterioscler. Thromb. Vasc. Biol. 41, 1265–1283. doi: 10.1161/ATVBAHA.120.311911
Schubert, P., Ogata, T., Marchini, C., and Ferroni, S. (2001). Glia-related pathomechanisms in Alzheimer's disease: a therapeutic target? Mech. Ageing Dev. 123, 47–57. doi: 10.1016/s0047-6374(01)00343-8
Steiner, K., and Humpel, C. (2023). Long-term organotypic brain slices cultured on collagen-based microcontact prints: a perspective for a brain-on-a-chip. J. Neurosci. Methods 399:109979. doi: 10.1016/j.jneumeth.2023.109979
Steiner, K., Yilmaz, S. N., Gern, A., Marksteiner, J., Faserl, K., Villunger, M., et al. (2024). From Organotypic mouse brain slices to human Alzheimer plasma biomarkers: a focus on microglia. Biomol. Ther. 14:1109. doi: 10.3390/biom14091109
Stoppini, L., Buchs, P. A., and Muller, D. (1991). A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods 37, 173–182. doi: 10.1016/0165-0270(91)90128-m
Swerdlow, R. H. (2012). Alzheimer's disease pathologic cascades: who comes first, what drives what. Neurotox. Res. 22, 182–194. doi: 10.1007/s12640-011-9272-9
Ucar, B., and Humpel, C. (2018). Collagen for brain repair: therapeutic perspectives - a mini-review. Neural Regen. Res. 13, 595–598. doi: 10.4103/1673-5374.230273
Ucar, B., Stefanova, N., and Humpel, C. (2022). Spreading of aggregated α synuclein in sagittal organotypic mouse brain slices. Biomol. Ther. 12:163. doi: 10.3390/biom12020163
Ucar, B., Yusufogullari, S., and Humpel, C. (2020). Collagen hydrogels loaded with fibroblast growth factor-2 as a bridge to repair brain vessels in organotypic brain slices. Exp. Brain Res. 238, 2521–2529. doi: 10.1007/s00221-020-05907-7
van den Brink, H., Voigt, S., Kozberg, M., and van Etten, E. S. (2024). The role of neuroinflammation in cerebral amyloid angiopathy. EBioMedicine 110:105466. doi: 10.1016/j.ebiom.2024.105466
Vinters, H. V. (1987). Cerebral amyloid angiopathy. A critical review. Stroke 18, 311–324. doi: 10.1161/01.str.18.2.311
Walker, L. C. (2018). Prion-like mechanisms in Alzheimer disease. Handb. Clin. Neurol. 153, 303–319. doi: 10.1016/B978-0-444-63945-5.00016-7
Walsh, D. M., and Selkoe, D. J. (2016). A critical appraisal of the pathogenic protein spread hypothesis of neurodegeneration.Nat. Rev. Neurosci. 17, 251–260. doi: 10.1038/nrn.2016.13
Wang, X., Sun, G., Feng, T., Zhang, J., Huang, X., Wang, T., et al. (2019). Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 29, 787–803. doi: 10.1038/s41422-019-0216-x
Yilmaz, S. N., Steiner, K., Marksteiner, J., Faserl, K., Sarg, B., and Humpel, C. (2025). Novel Plasma Biomarkers for Alzheimer’s Disease: Insights from Organotypic Brain Slice and Microcontact Printing Techniques. Front. Biosci. (Landmark Ed). 30:36257. doi: 10.31083/FBL36257
Keywords: Alzheimer’s disease, organotypic 3D culture, brain slice, pathology, review
Citation: Humpel C (2025) Three-dimensional organotypic mouse brain slices to study Alzheimer’s disease pathologies: a review. Front. Dement. 4:1585124. doi: 10.3389/frdem.2025.1585124
Edited by:
Charbel Moussa, Georgetown University, United StatesReviewed by:
Karin Mente, Case Western Reserve University, United StatesIgnazio Cali, Kore University of Enna, Italy
Copyright © 2025 Humpel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian Humpel, Y2hyaXN0aWFuLmh1bXBlbEBpLW1lZC5hYy5hdA==