 AC. Myo
AC. Myo R. Raju
R. Raju J. O. Piña
J. O. Piña P. Chattaraj
P. Chattaraj M. Furukawa
M. Furukawa- Section on Craniofacial Genetic Disorders, Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), National Institutes of Health (NIH), Bethesda, MD, United States
Cleft palate, a common congenital anomaly, is characterized by a failure of the palatal shelves to fuse during embryogenesis, resulting in an opening between the oral and nasal cavities. This malformation not only affects facial aesthetics but also significantly impacts speech, feeding, and hearing, necessitating multidisciplinary care from birth through adulthood. The etiology of cleft palate is complex, involving both genetic and environmental factors. Among the numerous genes implicated, Msx1 plays a pivotal role in palatal development. As a transcription factor, Msx1 regulates mesenchymal cell proliferation and epithelial-mesenchymal interactions, processes crucial for proper palatal shelf elevation and fusion. Disruptions in Msx1 expression or function have been directly linked to cleft palate through both animal and human studies, highlighting its significance in palatogenesis. This review focuses on the role of Msx1 in cleft palate, providing a comprehensive overview of its functions and the molecular mechanisms through which it influences palatal development. We examine recent research findings, including studies on Msx1 mutations, signaling pathways, and gene-environment interactions, to elucidate the complex relationship between Msx1 and cleft palate. Moreover, advancing research could establish Msx1 as a fundamental target in the creation of innovative therapeutic strategies for craniofacial disorders. By synthesizing current knowledge, this review aims to provide a deeper understanding of Msx1's role in cleft palate and pave the way for future research and clinical advancements.
Introduction
Cleft palate is a congenital malformation characterized by an opening between the oral and nasal cavities, resulting from the failure of the palatine processes to fuse during embryonic development (1). This condition can present in isolation or in conjunction with cleft lip, manifesting in various forms such as soft palate, hard palate, submucosal palate, and complete cleft lip and palate. As one of the most common congenital anomalies (2), cleft lip and palate necessitate multidisciplinary, long-term care from birth through adulthood. The condition significantly impacts aesthetics, pronunciation, feeding, swallowing, hearing, and psychology while also imposing a substantial economic burden. Affecting approximately 1 in 700–1,000 live births, cleft palate incidence varies across racial/ethnic groups, with a tendency for higher rates in Asian populations (3, 4).
Common complications include speech difficulties, such as nasal air emission and mispronunciation, feeding challenges leading to nasal regurgitation and nutritional deficiencies, and increased risk for otitis media and hearing loss. Cleft palate is a multifactorial disorder resulting from a complex interplay of genetic and environmental factors. Several genes, including MSX1, TGFB3, IRF6, TBX1, and PAX9 have been identified as contributors to cleft palate. Among them, Msx1 is particularly crucial as it encodes a transcription factor that regulates the growth and fusion of the palatine processes, making it one of the key determinants of palatal development.
The etiology of cleft palate involves both genetic predispositions and environmental influences, such as smoking, alcohol consumption, and nutritional deficiencies during critical periods of palatal development. This review will focus on recent research investigating the role of Msx1 in cleft palate, particularly its mutations and their involvement in relevant signaling pathways.
History of cleft palate research
Early studies on cleft palate date back to the 1840s (5, 6). Research expanded to animal models in the early 20th century, including dogs, horses, and chickens (7–9). Around 1950, cortisone-induced cleft palate in mice became a widely used experimental model (10–13). By the 1970s, researchers began identifying cleft palate as a component of various syndromes, such as Duane's Retraction Syndrome, adducted thumbs syndrome, and Pierre Robin sequence (14–16). Subsequent studies explored the role of etiological factors, including nutritional deficiencies, toxicological exposures, drugs, maternal health, and smoking (17–20). Notably, folic acid supplementation was identified as a protective factor against cleft lip and palate (21, 22). These studies collectively demonstrate that cleft palate development is a complex process influenced by multiple genetic and environmental factors.
Gene family, expression, and functions of MSX1
MSX1 is a homeobox transcription factor containing a highly conserved homeodomain that facilitates DNA binding and regulation of downstream target genes (23). In humans, the MSX gene family comprises MSX1 and MSX2, both of which are expressed in the embryonic craniofacial region and play crucial roles during development (24). MSX1 functions primarily as a transcriptional repressor, modulating key signaling pathways such as BMP4, Wnt, and Shh—pathways essential for craniofacial morphogenesis (25). During embryogenesis, Msx1/ msx1 exhibits strong expression in developing craniofacial and dental tissues (26, 27). In the dental mesenchyme, Msx1 regulates cell proliferation (28) and guides tooth morphogenesis (29). In mice, Msx1 deficiency results in arrested tooth development at the bud stage (30), while in humans, MSX1 mutations are linked to secondary cleft palate and dental agenesis (24, 30–32). Genome-wide association studies (GWAS) have further implicated MSX1 in the etiology of cleft palate (33). Functional studies across multiple model organisms, including Drosophila, chicken, and zebrafish, support an evolutionarily conserved role for MSX genes in craniofacial development (34–37).
Syndromic manifestations of MSX1 mutations
MSX1/Msx1 mutations have been identified as a significant genetic contributor to cleft palate, with functional studies confirming its role in palate, tooth, and craniofacial development (24, 30). These mutations are typically loss-of-function variants, including nonsense, frameshift, and missense mutations that result in reduced or abolished transcriptional activity. Many of these pathogenic variants are located within the highly conserved homeodomain, which is essential for DNA binding and gene regulation (38). Notably, phenotypic variability has been observed depending on the specific domain affected by the mutation, with mutations outside the homeodomain sometimes resulting in milder or distinct phenotypes. Zhao et al. found five new MSX1 variations in Chinese families exhibiting autosomal-dominant nonsyndromic oligodontia (38). These comprised three missense variants (Q221P, R224C, S270l), one nonsense variant (G122*), and one frameshift variant (A93Rfs*67). Significantly, 75% of missense variations and 33% of frameshift variants were situated within the highly conserved homeodomain (HD), indicating that missense variants are more prone to arise in evolutionarily limited locations. Conversely, frameshift variations exhibited no such trend, suggesting that their position is unrelated to amino acid conservation (38).
Mutations in MSX1 have been linked not only to isolated cleft palate but also to syndromic conditions characterized by craniofacial and dental anomalies. For example, Wolf-Hirschhorn syndrome (39) and Witkop syndrome (40, 41) exhibit orofacial clefting and tooth development abnormalities, highlighting the broader impact of MSX1 dysfunction in craniofacial biology. Furthermore, population-based studies suggest that certain ethnic groups may carry MSX1 variants that confer increased susceptibility to cleft palate (42), suggesting a role for MSX1 in future personalized risk assessments (Table 1).
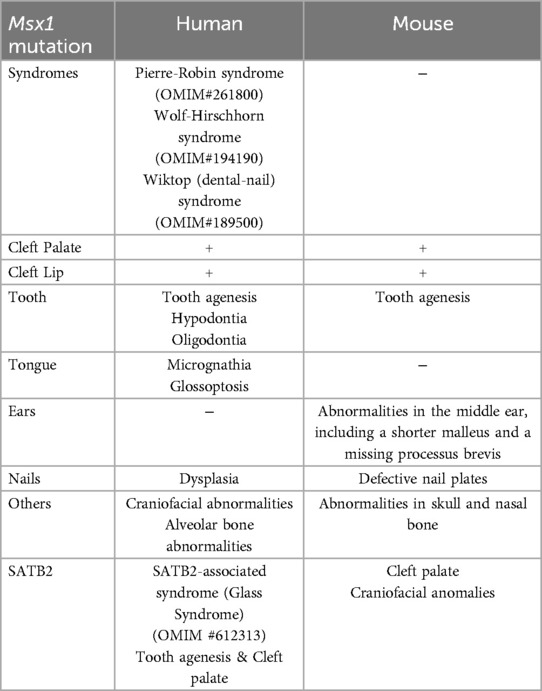
Table 1. A summary of Msx1 mutation related syndromes and phenotypes of human and mice.
Recent insights into Msx1-linked cleft palate: current research findings
Msx1 promoter activity is regulated by SATB2
SATB2 gene plays a major role in various organ and tissue developments such as brain, dental, and jaw (43). SATB2-associated syndrome is characterized by severe intellectual disability, neurodevelopmental disorders, cleft palate, and dental abnormalities (43, 44). In Satb2−/− mice, reduced Msx1 expressions were observed at the base of the developing palate (45). Luciferase reporter assay was performed to explore the effects of SATB2 on the Msx1 promoter using wild-type (WT) and variant proteins of SATB2 (A383P, R389C, G392Q, R399H, and F662fs*9). Results showed that SATB2 in WT elevated the activity of the Msx1 promoter, which plays a role in palatogenesis and odontogenesis. However, variants in the CUT1 domain of SATB2 disrupted this transcriptional activation. In immunolocalization studies variants in CUT2 domain of SATB2 localized to cytoplasm instead of nucleus suggesting that the nuclear localization signal of SATB2 resides in the CUT2 domain and that Msx1 promoter due to SATB2 variants may contribute to cleft palate and tooth agenesis in SATB2-associated syndrome (46) (Figure 1).
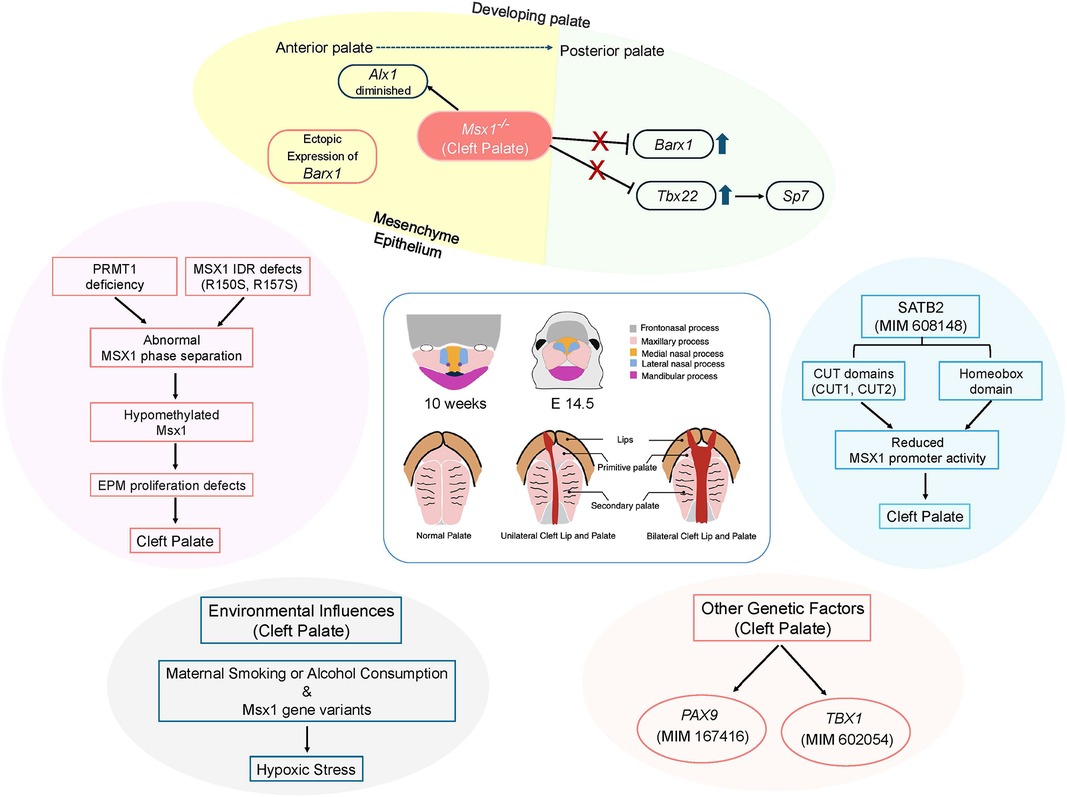
Figure 1. A schematic diagram showing different factors that can lead to cleft palate of human and mice. IDR, intrinsically disordered region; EPM, embryonic palate mesenchyme. Created with BioRender.com.
MSX1 phase separation leads to the development of cleft palate
Phase separation is a key phenomenon in various biological processes that regulate genetic expressions, signaling, and biochemical reactions (47, 48). This process is often driven by intrinsically disordered regions (IDRs) in proteins (47, 49). In addition to IDRs, posttranslational modifications (PTMs) such as phosphorylation and methylation also play significant roles in regulating phase separation (50) (Figure 1).
The repression domain containing the N-terminal region in MSX1 is a largely unfolded IDR. Protein analysis revealed that under physiological conditions, endogenous MSX1-formed circular droplet-like condensates uniformly distributed in the nucleus in human HEK293 T cells, human embryonic palate mesenchyme (EPM) (HEPM), and mouse EPM (MEPM). During osteogenic and chondrogenic differentiation in cultured HEPM, and in mouse palate tissues from gestational day E13.5 to postnatal day P1, MSX1 similarly formed droplet-like condensates in the nucleus indicating its important role in the spatiotemporal regulation of palatal fusion in mouse embryos. It was identified that only ΔIDR mutant, which lacks the IDR, exhibited significant defects in nuclear condensate formation, indicating that the IDR is essential for MSX1's droplet-like condensation in cells. This suggests that MSX1 likely undergoes phase separation via its IDR.
PRMT1 is a crucial methyltransferase responsible for R residue methylation in proteins, acting as an upstream regulator of MSX1, thus playing a crucial role in craniofacial development (51). Inhibition of PRMT1 disrupted MSX1 phase separation. Co-localization and reciprocal coimmunoprecipitation of PRMT1 with MSX1 was observed in HEK293T and HEPM cell nuclei. Purified PRMT1 protein was able to directly methylate purified MSX1 protein. Unmethylated MSX1 protein exhibited altered phase separation, with a greater tendency to form condensates compared to PRMT1-methylated MSX1 proteins. The R150S and R157S mutants, which are potential dimethylation sites, showed reduced binding to PRMT1 and had lower methylation levels compared to MSX1-FL (full-length wild-type MSX1). The R150S and R157S mutants exhibited a significant reduction in PRMT1-catalyzed methylation in vitro. The abnormal phase separation behavior of R150S and R157S mutants resembled that of MSX1 phase separation when PRMT1 is inhibited. Methylation of the R150S and R157S proteins by PRMT1 significantly reduced their abnormal condensates, indicating that R150 and R157 in the MSX1 IDR, as PRMT1-targeted methylation sites, are crucial for MSX1 phase separation behavior.
The R150S and R157S MSX1 mutants exhibited a reduced proportion of S-phase cells and an increased proportion of G1-phase cells compared to MSX1-FL. These mutants also showed significantly lower expression of both the PCNA gene and protein compared to MSX1-FL, suggesting that the less dynamic MSX1 phase separation caused by R150S and R157S is linked to defects in EPM cell cycle progression and cell proliferation. The increased cell proliferation induced by MSX1-FL was diminished by co-transfection with siPRMT1. Mutations at the MSX1 methylation sites reduced the promoter activities of Tbx22 and Bmp4, important downstream targets of MSX1 in palate development (25). Furthermore, PRMT1-regulated MSX1 methylation and its effects on phase separation influenced the SHH, FGF, BMP, TGFβ, and WNT signaling pathways.
In prmt1 MO and msx1 MO zebrafish, there were significantly fewer egfp/phh3 double-positive cells compared to controls, indicating defects in EPM proliferation in vivo. These zebrafish also exhibited a higher incidence of cleft palate and defective gel-like condensates. Overexpression of PRMT1 only partially rescued the EPM proliferation defect and palate cleft in msx1 MO zebrafish. MSX1-FL mRNA rescued the reduced EPM cell proliferation and cleft palate defects in msx1 MO zebrafish, while MSX1 ΔIDR mRNA did not. In mouse models, the knockdown of prmt1 and msx1 at E10.5 resulted in a higher incidence of cleft palate compared to controls. Co-injection with adenovirus expressing MSX1-FL (AdFL) promoted palatal fusion and partially rescued the cleft palate caused by shMSX1, while R150S (AdR150S) and R157S (AdR157S) were less effective. This study demonstrates that PRMT1-regulated MSX1 phase separation is a key mechanism underlying cleft palate formation during craniofacial development, with MSX1 phase separation being triggered by its IDR and precisely modulated by PRMT1-mediated dimethylation of R150 and R157 in the MSX1 IDR.
Hypoxic stress causing cleft palate in Msx1 heterozygosity
Orofacial clefts (comprising cleft anomalies of the lips, palate, and/or facial primordia) can be either syndromic or nonsyndromic malformations with a multifactorial origin, involving both genetic factors and environmental influences (42). Interactions between MSX1 gene variants and maternal smoking or alcohol consumption have been linked to an increased risk of orofacial clefting (52, 53). Additionally, a connection between maternal smoking and MSX1 variants has been shown to elevate the risk for developmental limb malformations (54). Evidence indicates that embryonic hypoxia in the first trimester is associated with craniofacial malformations, including cleft lip and palate (55). While Msx1 deficiency causes growth defects in the medial nasal process in mouse embryos, it does not affect lip formation (30). To explore potential interactions between Msx1 deficiency and hypoxia, heterozygous Msx1 mutant mice were exposed to 10% O2 during early lip formation (E10.5 to E12.5). At E15.5, 72% of Msx1−/− developed cleft lips, either bilateral or unilateral. To model pharmacologically induced hypoxia, pregnant Msx1+/− females were exposed to varying doses of phenytoin from E10.5 to E11.5. This exposure significantly increased the incidence of cleft lip in Msx1−/− embryos, reaching 91.7% at the highest dose, with all affected mutants showing a bilateral cleft lip. While Msx1+/− embryos typically do not have cleft palates, phenytoin treatment caused a higher incidence of cleft palate in these embryos compared to wild-type controls (56). Phenytoin administration also decreased cell proliferation in the palatal processes of both wild-type and Msx1+/− embryos, and slightly reduced Bmp4 expression in the anterior palatal process of Msx1+/− embryos (57).
Msx1/Tbx22/Sp7 axis in the regulation of palatogenesis
The expression of various signaling molecules and transcription factors is tightly regulated along the anteroposterior axis during palate development, with MSX1 playing a specific role in the anterior palate (58) (Figure 1). In a gain-of-function study using RosaMsx1Wnt1−Cre embryos, abnormal secondary palates were observed (59). These mice exhibited reduced cell proliferation and increased apoptosis in the mesenchyme of the palatal shelves. The size of the maxillary palatine process and palatine bone was notably smaller in RosaMsx1Wnt1−Cre mice with cleft palate, compared to control mice. The vomer bone was exposed due to the hypoplastic palatal bone, and the presphenoid was also deformed. The expression of RUNX2 was altered, and SP7 was significantly reduced in the palatine process of RosaMsx1Wnt1−Cre mice. In wild-type mice, Alx1 was specifically expressed in the mesenchyme of the anterior palatal shelves, but in Msx1−/− mice, anterior Alx1 expression was diminished, whereas it increased with Msx1 overexpression. Barx1, expressed in the mesenchyme of the medial and posterior palate in WT embryos, was ectopically expressed in the anterior palate of Msx1−/− palates, with increased expression in the medial palate. Conversely, Barx1 expression was significantly reduced in the medial palates of RosaMsx1Wnt1−Cre mice. These findings suggest that MSX1 is crucial for promoting the expression of anterior-specific genes and inhibiting the expansion of posterior genes into the anterior palate. Similarly, like RosaMsx1Wnt1−Cre mice, Tbx22−/− cleft palate mice also showed reduced bone formation in the posterior hard palate. Tbx22 expression was significantly increased in the medial and posterior palate regions of Msx1−/− mice but decreased in these regions in RosaMsx1Wnt1−Cre mice. MSX1 inhibited Tbx22 promoter activity in a dose-dependent manner. Both protein analysis and RNA-Seq data revealed a significant reduction in Sp7 in palates with Msx1 ectopic expression. Knockdown of Tbx22 expression led to a marked reduction in Sp7 expression in palatal mesenchymal cells. Tbx22 overexpression restored SP7 levels in Msx1-overexpressing palatal mesenchymal cells. This study shows that Sp7 is downstream of Tbx22 in palatal mesenchymal cells and that the Msx1/Tbx22/Sp7 axis plays a key role in regulating palate development.
Conclusion
Recent studies have revealed that Msx1 is not merely an associated factor in cleft palate development but a key transcriptional regulator of craniofacial morphogenesis. As a transcriptional repressor, Msx1 governs multiple developmental pathways, including cell proliferation, differentiation, and epithelial-mesenchymal interactions. Its mutations or dysregulated expression are linked not only to cleft palate but also to dental anomalies and mandibular underdevelopment. These findings reaffirm that cleft palate is a multifactorial disease rather than a consequence of a single genetic mutation.
However, Msx1 mutations alone do not account for all cleft palate cases, indicating that complex interactions with genetic background and environmental influences must be further investigated. While rescue studies have shown partial recovery, complete phenotypic correction remains elusive, likely due to Msx1's interplay with diverse signaling pathways.
Additionally, Msx1 expression must be finely tuned—both excessive and insufficient levels can disrupt normal craniofacial development. Future research should focus on elucidating the upstream and downstream molecular networks regulating Msx1 to uncover its precise role in craniofacial patterning. Defining how genetic diversity and environmental exposures modulate Msx1 function will be critical for developing targeted interventions.
Advances in gene-editing technologies, such as CRISPR-Cas9 and single-cell transcriptomics, offer promising avenues for dissecting Msx1-related disease mechanisms with unprecedented precision. Ultimately, a deeper understanding of Msx1 will not only advance basic science but also pave the way for innovative strategies in cleft palate prevention, early diagnosis, and personalized therapy. As research progresses, targeting Msx1 could become a cornerstone in the development of novel therapeutic approaches for craniofacial disorders.
Author contributions
ACM: Visualization, Writing – review & editing, Writing – original draft. RR: Writing – review & editing, Visualization, Writing – original draft. JOP: Writing – review & editing. PC: Writing – review & editing. MF: Writing – review & editing, Supervision, Writing – original draft.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Correction Note
A correction has been made to this article. Details can be found at: 10.3389/fdmed.2025.1662124.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Iwaya C, Suzuki A, Iwata J. MicroRNAs and gene regulatory networks related to cleft lip and palate. Int J Mol Sci. (2023) 24(4):3552. doi: 10.3390/ijms24043552
2. Saleem K, Zaib T, Sun W, Fu S. Assessment of candidate genes and genetic heterogeneity in human non syndromic orofacial clefts specifically non syndromic cleft lip with or without palate. Heliyon. (2019) 5(12):e03019. doi: 10.1016/j.heliyon.2019.e03019
3. Group IW. Prevalence at birth of cleft lip with or without cleft palate: data from the international perinatal database of typical oral clefts (IPDTOC). Cleft Palate Craniofac J. (2011) 48(1):66–81. doi: 10.1597/09-217
4. Leslie EJ, Marazita ML. Genetics of cleft lip and cleft palate. Am J Med Genet C Semin Med Genet. (2013) 163C(4):246–58. doi: 10.1002/ajmg.c.31381
7. Hobday F. Cleft palate in animals. Proc R Soc Med. (1922) 15(Pathol Sect):3–6. doi: 10.1177/003591572201501202
9. Juriloff DM, Roberts CW. Genetics of cleft palate in chickens and the relationship between the occurrence of the trait and maternal riboflavin deficiency. Poult Sci. (1975) 54(2):334–46. doi: 10.3382/ps.0540334
10. Baxter H, Fraser FC. The production of congenital defects in the offspring of female mice treated with cortisone. A preliminary report. McGill Med J. (1950) 19(4):245–9.14795899
11. Fraser FC, Fainstat TD. Production of congenital defects in the off-spring of pregnant mice treated with cortisone; progress report. Pediatrics. (1951) 8(4):527–33. doi: 10.1542/peds.8.4.527
12. Kalter H. Modification of teratogenic action of cortisone in mice by maternal age, maternal weight and litter size. Am J Physiol. (1956) 185(1):65–8. doi: 10.1152/ajplegacy.1956.185.1.65
13. Kalter H. Factors influencing the frequency of cortisone-induced cleft palate in mice. J Exp Zool. (1957) 134(3):449–67. doi: 10.1002/jez.1401340304
14. Kirkham TH. Duane’s retraction syndrome and cleft palate. Am J Ophthalmol. (1970) 70(2):209–12. doi: 10.1016/0002-9394(70)90004-8
15. Christian JC, Andrews PA, Conneally PM, Muller J. The adducted thumbs syndrome. An autosomal recessive disease with arthrogryposis, dysmyelination, craniostenosis, and cleft palate. Clin Genet. (1971) 2(2):95–103. doi: 10.1111/j.1399-0004.1971.tb00262.x
16. Bixler D, Christian JC. Pierre robin syndrome occurring in two related sibships. Birth Defects Orig Artic Ser. (1971) 7(7):67–71.5173245
17. Krapels IP, van Rooij IA, Ocke MC, van Cleef BA, Kuijpers-Jagtman AM, Steegers-Theunissen RP. Maternal dietary B vitamin intake, other than folate, and the association with orofacial cleft in the offspring. Eur J Nutr. (2004) 43(1):7–14. doi: 10.1007/s00394-004-0433-y
18. Alade A, Ismail W, Nair R, Schweizer M, Awotoye W, Oladayo A, et al. Periconceptional use of vitamin A and the risk of giving birth to a child with nonsyndromic orofacial clefts-A meta-analysis. Birth Defects Res. (2022) 114(10):467–77. doi: 10.1002/bdr2.2005
19. Dolk HM, Nau H, Hummler H, Barlow SM. Dietary vitamin A and teratogenic risk: european teratology society discussion paper. Eur J Obstet Gynecol Reprod Biol. (1999) 83(1):31–6. doi: 10.1016/S0301-2115(98)00228-0
20. Parker SE, Werler MM, Shaw GM, Anderka M, Yazdy MM. National birth defects prevention S. Dietary glycemic index and the risk of birth defects. Am J Epidemiol. (2012) 176(12):1110–20. doi: 10.1093/aje/kws201
21. Xu W, Yi L, Deng C, Zhao Z, Ran L, Ren Z, et al. Maternal periconceptional folic acid supplementation reduced risks of non-syndromic oral clefts in offspring. Sci Rep. (2021) 11(1):12316. doi: 10.1038/s41598-021-91825-9
22. Zhou Y, Sinnathamby V, Yu Y, Sikora L, Johnson CY, Mossey P, et al. Folate intake, markers of folate status and oral clefts: an updated set of systematic reviews and meta-analyses. Birth Defects Res. (2020) 112(19):1699–719. doi: 10.1002/bdr2.1827
23. Woloshin P, Song K, Degnin C, Killary AM, Goldhamer DJ, Sassoon D, et al. MSX1 inhibits myoD expression in fibroblast x 10T1/2 cell hybrids. Cell. (1995) 82(4):611–20. doi: 10.1016/0092-8674(95)90033-0
24. van den Boogaard MJ, Dorland M, Beemer FA, van Amstel HK. MSX1 Mutation is associated with orofacial clefting and tooth agenesis in humans. Nat Genet. (2000) 24(4):342–3. doi: 10.1038/74155
25. Zhang Z, Song Y, Zhao X, Zhang X, Fermin C, Chen Y. Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and shh signaling in the regulation of mammalian palatogenesis. Development. (2002) 129(17):4135–46. doi: 10.1242/dev.129.17.4135
26. Jowett AK, Vainio S, Ferguson MW, Sharpe PT, Thesleff I. Epithelial-mesenchymal interactions are required for msx 1 and msx 2 gene expression in the developing murine molar tooth. Development. (1993) 117(2):461–70. doi: 10.1242/dev.117.2.461
27. Alappat S, Zhang ZY, Chen YP. Msx homeobox gene family and craniofacial development. Cell Res. (2003) 13(6):429–42. doi: 10.1038/sj.cr.7290185
28. Feng XY, Zhao YM, Wang WJ, Ge LH. Msx1 regulates proliferation and differentiation of mouse dental mesenchymal cells in culture. Eur J Oral Sci. (2013) 121(5):412–20. doi: 10.1111/eos.12078
29. Chen Y, Bei M, Woo I, Satokata I, Maas R. Msx1 controls inductive signaling in mammalian tooth morphogenesis. Development. (1996) 122(10):3035–44. doi: 10.1242/dev.122.10.3035
30. Satokata I, Maas R. Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat Genet. (1994) 6(4):348–56. doi: 10.1038/ng0494-348
31. Vastardis H, Karimbux N, Guthua SW, Seidman JG, Seidman CE. A human MSX1 homeodomain missense mutation causes selective tooth agenesis. Nat Genet. (1996) 13(4):417–21. doi: 10.1038/ng0896-417
32. Houzelstein D, Cohen A, Buckingham ME, Robert B. Insertional mutation of the mouse Msx1 homeobox gene by an nlacZ reporter gene. Mech Dev. (1997) 65(1–2):123–33. doi: 10.1016/S0925-4773(97)00065-8
33. Dixon MJ, Marazita ML, Beaty TH, Murray JC. Cleft lip and palate: understanding genetic and environmental influences. Nat Rev Genet. (2011) 12(3):167–78. doi: 10.1038/nrg2933
34. Hill RE, Jones PF, Rees AR, Sime CM, Justice MJ, Copeland NG, et al. A new family of mouse homeo box-containing genes: molecular structure, chromosomal location, and developmental expression of hox-7.1. Genes Dev. (1989) 3(1):26–37. doi: 10.1101/gad.3.1.26
35. Shimomura T, Kawakami M, Okuda H, Tatsumi K, Morita S, Nochioka K, et al. Retinoic acid regulates Lhx8 expression via FGF-8b to the upper jaw development of chick embryo. J Biosci Bioeng. (2015) 119(3):260–6. doi: 10.1016/j.jbiosc.2014.08.010
36. Postlethwait JH. The zebrafish genome: a review and msx gene case study. Genome Dyn. (2006) 2:183–97. doi: 10.1159/000095104
37. Raterman ST, Metz JR, Wagener F, Von den Hoff JW. Zebrafish models of craniofacial malformations: interactions of environmental factors. Front Cell Dev Biol. (2020) 8:600926. doi: 10.3389/fcell.2020.600926
38. Liang J, Von den Hoff J, Lange J, Ren Y, Bian Z, Carels CE. MSX1 mutations and associated disease phenotypes: genotype-phenotype relations. Eur J Hum Genet. (2016) 24(12):1663–70. doi: 10.1038/ejhg.2016.78
39. Battaglia A, Carey JC. Wolf-Hirschhorn syndrome and the 4p-related syndromes. Am J Med Genet C Semin Med Genet. (2008) 148C(4):241–3. doi: 10.1002/ajmg.c.30189
40. Paradowska-Stolarz A. MSX1 gene in the etiology orofacial deformities. Postepy Hig Med Dosw (Online). (2015) 69:1499–504.27259221
41. Jumlongras D, Bei M, Stimson JM, Wang WF, DePalma SR, Seidman CE, et al. A nonsense mutation in MSX1 causes witkop syndrome. Am J Hum Genet. (2001) 69(1):67–74. doi: 10.1086/321271
42. Mossey PA, Little J, Munger RG, Dixon MJ, Shaw WC. Cleft lip and palate. Lancet. (2009) 374(9703):1773–85. doi: 10.1016/S0140-6736(09)60695-4
43. Zarate YA, Fish JL. SATB2-associated syndrome: mechanisms, phenotype, and practical recommendations. Am J Med Genet A. (2017) 173(2):327–37. doi: 10.1002/ajmg.a.38022
44. Zarate YA, Bosanko KA, Thomas MA, Miller DT, Cusmano-Ozog K, Martinez-Monseny A, et al. Growth, development, and phenotypic spectrum of individuals with deletions of 2q33.1 involving SATB2. Clin Genet. (2021) 99(4):547–57. doi: 10.1111/cge.13912
45. Britanova O, Depew MJ, Schwark M, Thomas BL, Miletich I, Sharpe P, et al. Satb2 haploinsufficiency phenocopies 2q32-q33 deletions, whereas loss suggests a fundamental role in the coordination of jaw development. Am J Hum Genet. (2006) 79(4):668–78. doi: 10.1086/508214
46. Ukita N, Ogawa T, Yamada M, Takeuchi C, Kosaki K, Moriyama K. Functional analyses of SATB2 variants reveal pathogenicity mechanisms linked with SATB2-associated syndrome. Am J Med Genet A. (2025) 197(6):e64005. doi: 10.1002/ajmg.a.64005
47. Alberti S, Gladfelter A, Mittag T. Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cell. (2019) 176(3):419–34. doi: 10.1016/j.cell.2018.12.035
48. Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol. (2017) 18(5):285–98. doi: 10.1038/nrm.2017.7
49. Uversky VN. The multifaceted roles of intrinsic disorder in protein complexes. FEBS Lett. (2015) 589(19 Pt A):2498–506. doi: 10.1016/j.febslet.2015.06.004
50. Luo YY, Wu JJ, Li YM. Regulation of liquid-liquid phase separation with focus on post-translational modifications. Chem Commun (Camb). (2021) 57(98):13275–87. doi: 10.1039/D1CC05266G
51. Gou Y, Li J, Wu J, Gupta R, Cho I, Ho TV, et al. Prmt1 regulates craniofacial bone formation upstream of Msx1. Mech Dev. (2018) 152:13–20. doi: 10.1016/j.mod.2018.05.001
52. van den Boogaard MJ, de Costa D, Krapels IP, Liu F, van Duijn C, Sinke RJ, et al. The MSX1 allele 4 homozygous child exposed to smoking at periconception is most sensitive in developing nonsyndromic orofacial clefts. Hum Genet. (2008) 124(5):525–34. doi: 10.1007/s00439-008-0569-6
53. Fallin MD, Hetmanski JB, Park J, Scott AF, Ingersoll R, Fuernkranz HA, et al. Family-based analysis of MSX1 haplotypes for association with oral clefts. Genet Epidemiol. (2003) 25(2):168–75. doi: 10.1002/gepi.10255
54. Hwang SJ, Beaty TH, McIntosh I, Hefferon T, Panny SR. Association between homeobox-containing gene MSX1 and the occurrence of limb deficiency. Am J Med Genet. (1998) 75(4):419–23. doi: 10.1002/(SICI)1096-8628(19980203)75:4%3C419::AID-AJMG14%3E3.0.CO;2-R
55. Webster WS, Abela D. The effect of hypoxia in development. Birth Defects Res C Embryo Today. (2007) 81(3):215–28. doi: 10.1002/bdrc.20102
56. Nakatomi M, Ludwig KU, Knapp M, Kist R, Lisgo S, Ohshima H, et al. Msx1 deficiency interacts with hypoxia and induces a morphogenetic regulation during mouse lip development. Development. (2020) 147(21):dev189175. doi: 10.1242/dev.189175
57. Park J, Nakatomi M, Sasaguri M, Habu M, Takahashi O, Yoshiga D, et al. Msx1 heterozygosity in mice enhances susceptibility to phenytoin-induced hypoxic stress causing cleft palate. Cleft Palate Craniofac J. (2021) 58(6):697–706. doi: 10.1177/1055665620962690
58. Potter AS, Potter SS. Molecular anatomy of palate development. PLoS One. (2015) 10(7):e0132662. doi: 10.1371/journal.pone.0132662
Keywords: cleft palate, MSX1, palatal development, recent insights, genetics causes, environmental causes, msx1 downstream signaling pathway
Citation: Myo AC, Raju R, Piña JO, Chattaraj P and Furukawa M (2025) Current insights on the genetics and mechanisms of MSX1-associated cleft palate. Front. Dent. Med. 6:1610223. doi: 10.3389/fdmed.2025.1610223
Received: 11 April 2025; Accepted: 20 June 2025;
Published: 7 July 2025;
Corrected: 25 July 2025.
Edited by:
Andrea Ballini, University of Foggia, ItalyReviewed by:
Emanuela Iovino, IRCCS University Hospital of Bologna Sant Orsola Polyclinic, ItalyCopyright: © 2025 Myo, Raju, Piña, Chattaraj and Furukawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: M. Furukawa, bWFzYWUueWFtYWRhQG5paC5nb3Y=