 Heather A. Noriega
Heather A. Noriega Xiang Simon Wang
Xiang Simon Wang- Artificial Intelligence and Drug Discovery (AIDD) Core Laboratory for District of Columbia Center for AIDS Research (DC CFAR), Department of Pharmaceutical Sciences, College of Pharmacy, Howard University, Washington, DC, United States
Antibody-drug conjugates (ADCs) represent a mechanistically defined class of targeted therapeutics that combine monoclonal antibodies with cytotoxic payloads to achieve selective delivery to antigen-expressing carcinoma cells. Conventional ADC development has primarily relied on empirical screening and structure-based design, often limited by incomplete structural information, non-systematic linker–payload selection, and constraints in experimental throughput. Computational methods, including artificial intelligence and machine learning (AI/ML) are increasingly being integrated into ADC discovery and optimization workflows (i.e., AI-driven ADC Design) to address these limitations. This review is organized into six sections: (1) the progression from traditional modeling approaches to AI-driven design of individual ADC components; (2) the application of deep learning (DL) to antibody structure prediction and identification of optimal conjugation sites; (3) the use of AI/ML models for forecasting pharmacokinetic properties and toxicity profiles; (4) emerging generative algorithms for antibody sequence diversification and affinity optimization; (5) case studies demonstrating the integration of computational tools with experimental pipelines, including systems that link in silico predictions to high-throughput validation; and (6) persistent challenges, including data sparsity, model interpretability, validation complexity, and regulatory considerations. The review concludes with a discussion of future directions, emphasizing the role of multimodal data integration, reinforcement learning (RL), and closed-loop design frameworks to support iterative ADC development.
Introduction
Antibody-drug conjugates (ADCs) represent a rapidly expanding class of targeted cancer therapeutics that combine the specificity of monoclonal antibodies with the potent cytotoxicity of small-molecule drugs. This dual mechanism enables the selective elimination of cancer cells while minimizing off-target toxicity, offering an enhanced therapeutic indicator compared to traditional therapies. However, despite significant clinical advances, conventional ADC development has been slowed down by empirical approaches, incomplete structural information, and inefficient linker-payload selections, resulting in a time-consuming and costly discovery process (Kim et al., 2023).
Over the past 3 decades, computational methods have steadily evolved to address these limitations, as illustrated in Figure 1. In the early 2000s, in silico modeling and basic artificial intelligence/machine learning (AI/ML) algorithms were applied to predict antibody-antigen interactions based on physicochemical features. However, the methods used were limited by simple computational resources and the lack of extensive biological datasets. The progress accelerated in the 2010s with the emergence of DL. DL allowed for more complex modeling and high-dimensional relationships critical for predicting antibody structures, binding affinities, and developability parameters (Bai et al., 2023). Today, AI/ML play fundamental roles across the ADC discovery pipeline. ML models have demonstrated efficacy in predicting drug-to-antibody ratios (DAR) and conjugation site preferences with significantly improved accuracy over empirical methods (Angiolini et al., 2025). Another example is DL models capable of learning from three-dimensional structural data, which have enhanced antibody paratope prediction and rational affinity maturation (Dewalker et al., 2025).
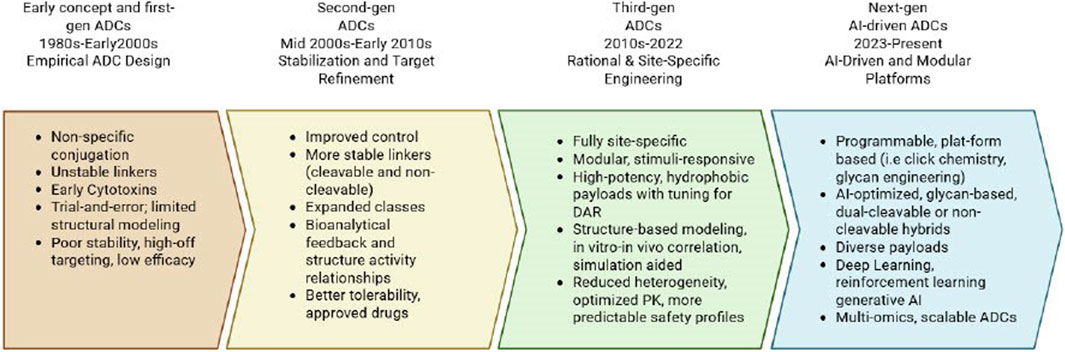
Figure 1. Timeline summarizing key developments in ADC design, from early empirical approaches to current AI-integrated and modular strategies.
The computational revolution has coincided with advances in molecular engineering. Glycan engineering is essential to ADC pharmacology, as it impacts antibody stability and immunogenicity and provides modifiable sites for site-specific conjugation. Recent computational advances have enabled the characterization of glycan microheterogeneity at the glycosylation sites, offering predictive tools to modulate glycan structures for optimal ADC properties (Moore et al., 2024; Cruz and Kayser, 2019). PEGylation, the covalent attachment of polyethylene glycol chains to antibodies, has been extensively used to improve ADC pharmacokinetics. AI-guided modeling optimizes PEG chain length, attachment sites, and structural shielding to prolong circulation while preserving target engagement (Smith, 2015). Small molecule payload optimization is another rapidly advancing area. Traditionally, this was selected via screening; small molecule payloads are now increasingly matched to antibodies using ML models that predict cytotoxic potency, stability, and intracellular trafficking properties (Debnath et al., 2022). ML has enabled the discovery of novel cytotoxic payloads, such as plitidepsin derivatives targeting eEF1A (Antunes et al., 2023). Furthermore, computational simulations of protein-protein interactions (PPIs) have allowed for engineering ADCs with minimized off-target binding and enhanced immune system modulation, including glycan-shielded or albumin-binding formats (Zhong and D'Antona, 2021).
Nanoparticle-enabled ADC systems represent another frontier for precision drug delivery. Antibody-nanoparticle conjugates (ANCs) integrate the targeting specificity of antibodies with the payload versatility and modifiable release kinetics of nanotechnology. Emerging studies demonstrate that ANC platforms can achieve improved pharmacokinetics, enhanced payloads, and better tumor penetration than conventional ADCs (Adhikari and Chen, 2025). AI models are being developed to optimize nanoparticle size, surface chemistry, and antibody orientation to maximize tumor accumulation and minimize off-target effects (Gholap et al., 2024; Chandrika et al., 2024). Most recent examples of antibody-conjugated nanoparticles targeting HEr2 positive breast cancer cells have demonstrated improved binding, enhanced drug release control, and superior therapeutic measures (Juan et al., 2020; Selepe et al., 2024).lipid nanoparticle (LNP) formulations decorated with antibodies are now used for siRNA and therapeutic protein delivery to lymphatic tissues (Sakurai et al., 2022).
Clinically, second-generation ADCs such as Polivy (Polatuzumab vedotin) and Enhertu (trastuzumab deruxtecan) showcase how structure-guided conjugation strategies, linker innovations, and computational payload optimizations are entering real-world practice (Shi and McHugh, 2023). Glycan and PEGylation modifications have refined the pharmacokinetics of investigational ADCs progressing through clinical pipelines (Sakhnini, 2019).
Despite these advances, several challenges remain. Among them are data scarcity for rare conjugation chemistries, interpretability of DL models, experimental validation burdens, and regulatory hurdles (Melo et al., 2018).
This review examines the application of computational approaches in designing and developing antibody-drug conjugates, spanning conventional modeling techniques through recent advances in AI/ML. The discussion is organized into six core areas: (1) the methodological shift from traditional design strategies to AI-enabled modeling of ADC components; (2) DL based approaches for antibody structure prediction and conjugation site identification; (3) ML frameworks for modeling pharmacokinetics and toxicity; (4) generative algorithms for antibody sequence diversification and affinity engineering; (5) the integration of computational tools with high-throughput experimental systems; and (6) unresolved challenges, validation requirements, and future directions for computational ADC research. Particular attention is given to how these approaches may enable the rational design of next-generation ADCs, including multimeric formats with enhanced modularity, combinatorial targeting capacity, and structural complexity. Together, these developments define a framework for advancing data-driven and mechanism-informed strategies in ADC engineering.
Traditional computational methods
Early ADC development relied on traditional structure-based design methods. X-ray crystallography provided atomic resolution structures of antibodies and target antigens, serving as templates for computational modeling. Structural determination through X-ray diffraction and small-angle X-ray scattering (SAXS) allowed for the initial mapping of epitope-paratope interactions essential for rational design (Chiu et al., 2019; Filntisi et al., 2014). Structural information from these methods informed initial assessments of conjugation sites and antigen binding compatibility. However, structural data were often incomplete. In the Protein Data Bank (PDB) and Electron Microscopy Data Bank (EMDB), only antibody fragments, primarily antigen-binding fragments (Fabs), single-chain variable fragments (scFvs), or isolated VH/VL domains, were typically available. Full-length immunoglobulin G (IgG) structures, encompassing flexible hinge regions and Fc domains, remained scarce due to the inherent difficulty in crystallizing or imaging large, flexible biomolecules (Chaves et al., 2024). This incomplete structural coverage constrained early ADC design efforts, especially in modeling linker attachment sites, steric hindrance, and glycosylation effects. Flexible regions such as the hinge domain, which is important for internalization and payload delivery, were poorly represented in available templates, which resulted in speculation modeling.
Molecular docking emerged as a primary computational tool, which allowed virtual predictions of antibody-antigen poses and assessed the potential effects of conjugation on antigen engagement. However, traditional docking algorithms were often optimized for small molecule ligands and struggled with large, flexible interface characteristics of antibody-antigen interactions. Scoring functions tended to oversimplify binding energetics, leading to overestimating affinities and frequent false positives (Garofalo et al., 2020; Siddiqui et al., 2025). To address these challenges, protein-protein docking platforms such as ClusPro and HADDOCK have been developed to model the flexibility and shape complementarity of larger biological complexes (Kozakov et al., 2017; Dominguez et al., 2003). These tools account for conformational changes and can incorporate experimental data to guide the docking process, making them better suited for simulating antibody-antigen and antibody-linker interactions. Incorporating such methods into ADC modeling has improved our ability to evaluate structural compatibility and binding site accessibility, though challenges still remain in modeling the full ADC assemblies at high resolution. Docking methods have been used to guide in silico affinity maturation and structural optimization of antibody variants during early-stage design.
Molecular dynamics (MD) simulations provided a deeper understanding of antibody flexibility, linker behavior, and payload exposure under dynamic biological conditions. MD simulations enabled the prediction of domain motions, solvent accessibility, and aggregation-prone regions (Codina et al., 2019). These simulations continue to support ADC design efforts by offering atomic-level insight into conformational variability, linker strain, and local solvation effects that influence stability and binding. However, MD was constrained by limitations in simulation timescales, force field inaccuracies, and high computational costs, restricting simulations to relatively short times and small system sizes. Even when used with docking, MD refinements often failed to fully account for flexible loops, glycan motions, or hinge dynamics (Dixit, 2015). Ongoing advances in GPU acceleration, enhanced sampling techniques, and hybrid modeling approaches have improved the feasibility of MD in larger systems, enabling their continued integration alongside AI-driven workflows.
Developability assessments were another primary focus of traditional workflows. The early model evaluated candidate antibodies and ADCs for aggregation susceptibility, chemical stability, and solubility properties. These methods relied on sequence-based descriptors such as hydrophobic patches, charged residues, and coarse-grained structure-based features (Khetan et al., 2022). For example, Evers et al. demonstrated the structure-based in silico prediction of aggregation hotspots in biparatopic ADCs targeting c-MET, highlighting the need for early aggregation control to improve manufacturability (Evers et al., 2024). These tools were effective in screening candidates prior to experimental validation and were integrated into early-stage selection protocols to reduce downstream formulation issues.
However, early developability models faced limitations due to small training datasets, narrow antibody diversity coverage, and lack of generalization across different payload-linker combinations. The absence of high-quality 3D structures further compounded these challenges, often forcing researchers to project predictions from fragmentary or homology-modeled structures from software like SWISS-MODEL (Waterhouse et al., 2018). Despite these limitations, sequence-based models provided actionable insights into charge distribution, surface hydrophobicity, and isoelectric point, which remain relevant parameters in manufacturability risk assessment. These tools served as a decision support layer that complemented experimental assays and informed downstream engineering strategies. These traditional computational techniques, covering structure determination, docking, MD, and developability prediction, have established the foundation for modern AI-driven frameworks.
ADC’s structural prediction with AlphaFold series and other DL tools
The release of AlphaFold2 introduced a transformative approach to protein structure prediction using novel neural network DL architectures (Jumper et al., 2021; Yang et al., 2023; Skolnick et al., 2021; Marcu et al., 2022). AlphaFold2 achieves near-experimental accuracy for monomeric proteins, and early applications to antibody variable domains demonstrated reliable framework modeling. However, hypervariable complementarity-determining regions (CDRs), particularly CDR-H3 loops, remain challenging to predict accurately due to their intrinsic conformational flexibility (Yin and Pierce, 2023). Studies applying AlphaFold2 to antibodies revealed that while framework regions were modeled with RMSD <2 Å, long antigen-contacting CDR-H3 loops showed structural inaccuracies (Chen et al., 2024). Recognizing the need for complex modeling, DeepMind introduced AlphaFold-Multimer, expanding the system to co-fold protein-protein interactions (Evans et al., 2022). Applications to antibody-antigen complexes, such as CD20-targeted antibodies, demonstrated superior interface prediction compared to traditional docking, although induced-fit interactions remain challenging (Dabkowska et al., 2024; Boross and Leusen, 2012).
While AlphaFold2 and its multimer extension advanced structural modeling of individual proteins and some complexes, they were not designed to predict interactions with small molecules, glycans, or ions. AlphaFold2 primarily focused on monomeric folding and limited protein-protein assemblies, with no support for ligand or post-translational modification modeling. DeepMind recently introduced AlphaFold3 as a next-generation structure prediction system (Abramson et al., 2024; Krokidis et al., 2025; Desai et al., 2024). Compared to other structural modeling approaches such as RFdiffusion and ProteinMPNN, AlphaFold3 employs a diffusion-based generative framework, extending predictive capabilities to complexes involving proteins, nucleic acids, small molecules, ions, and glycans as shown in Figure 2 (Watson et al., 2022; Dauparas et al., 2022). For ADC design, AlphaFold3 offers the potential to predict glycosylated Fc domains, linker-payload interactions, and antigen-binding epitopes in the presence of cofactors (Roy and Al-Hashimi, 2024). However, despite improved static modeling of Fc glycans and payload-conjugated domains, AlphaFold3 still struggles to fully capture glycan microheterogeneity, dynamic shielding effects, and flexible linker behavior due to the limited availability of data. They also struggle with unusual DNA and RNA structures, such as single mutations (Bergonzo and Grishaev, 2025). Benchmark datasets have demonstrated that AlphaFold3 outperforms traditional methods such as AutoDock Vina as well as deep learning-based RoseTTAFold in analyzing protein-protein interactions, nucleic acid complexes, and glycosylated proteins (Abramson et al., 2024; Eberhardt et al., 2021; Baek et al., 2021). In ligand docking benchmarks, AlphaFold3 exhibited significantly higher success rates than conventional approaches. Evaluations from the 15th Critical Assessment of Structure Prediction (CASP15) further highlighted the advances achieved with AlphaFold3, demonstrating state-of-the-art performance in modeling multimeric protein complexes, protein-small molecule complexes, and protein–glycan assemblies. (Abramson et al., 2024) AlphaFold-Multimer previously achieved interface root-mean-square deviations (iRMSDs) often below 2.5 Å for antibody-antigen complexes (Liu et al., 2023), and AlphaFold3 extended these capabilities further, setting new benchmarks for backbone accuracy, ligand positioning, and covalent modification modeling. Although modeling glycan flexibility and microheterogeneity remains a limitation, AlphaFold3 excels at capturing static glycosylation states, antigenic surfaces, and linker-conjugated domains, making it highly valuable for ADC structural modeling workflows, as shown in Figure 3 using Pertuzumab and HER2 as an example.
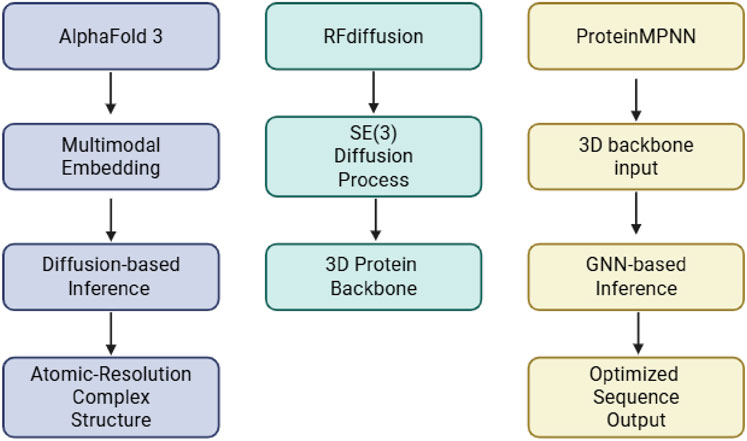
Figure 2. Comparison of structural modeling workflows. AlphaFold3 predicts complexes using multimodal diffusion, while RFdiffusion generates 3D protein backbones from structural constraints. ProteinMPNN designs sequences for fixed backbones via graph-based inference.
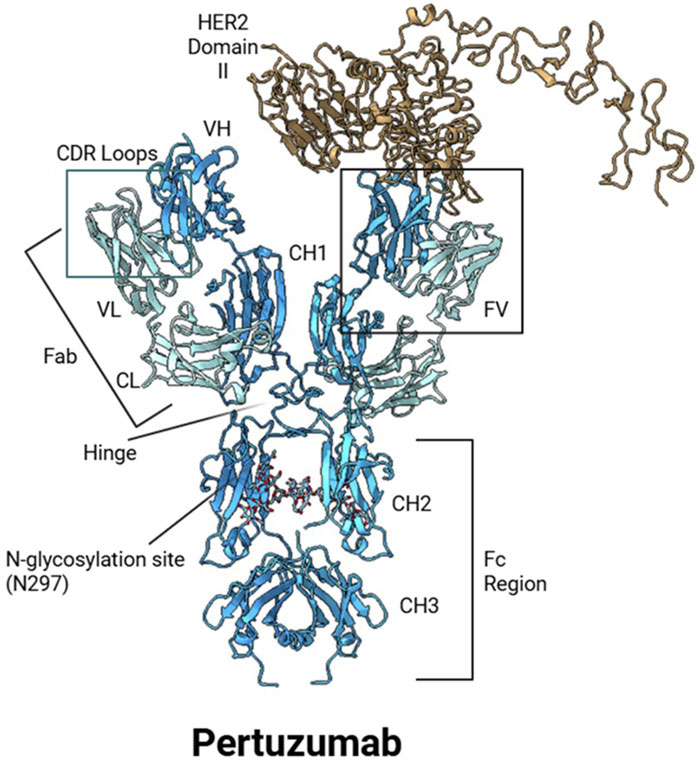
Figure 3. AlphaFold3-predicted structure of pertuzumab bound to HER2 Domain II, with modeled N-linked glycans. The model illustrates full antibody architecture, including variable (VH, VL) and constant (CH1, CH2, CH3, CL) domains. The antigen-binding fragment (Fab) regions contain the complementarity-determining regions (CDRs), which mediate interaction with HER2 Domain II. The Fc region includes a glycosylation site at Asn297, with glycans modeled by AlphaFold3. The HER2 epitope is positioned near the FV region, demonstrating structural alignment relevant to ADC targeting and conjugation strategies.
Specialized antibody-specific modeling tools have further enhanced precision and contributed to ADC development. DeepAb uses recurrent graph neural networks and recurrent architectures trained on curated antibody datasets to improve the CDR modeling, particularly emphasizing structural diversity in CDR-H3 loops (Ruffolo et al., 2020). DeepAb has performed better than Ablang, which was developed by utilizing antibody-specific language modeling to predict and complete missing regions in antibody sequences accurately (Olsen et al., 2022). ABlooper also uses mathematical neural networks, which enables rapid CDR modeling at scale but with reduced accuracy for long or kinked H3 loops (Abanades et al., 2022). SimpleDH3 provides a simple end-to-end DL framework for predicting CDR H3 loop structure. It directly outputs backbone atomic coordinates without relying on post-processing pipelines like Rosetta (Zenkova et al., 2021). Using ELMo embeddings and bi-directional LSTM architectures, SimpleDH3 achieves comparable RMSD performance to state-of-the-art methods such as DeepH3, offering a faster inference speed. It focuses on modeling only the highly variable CDR-H3 loops rather than full Fv domains, enabling efficient large-scale antibody screening for ADC design applications (Chungyoun and Gray, 2024).
While AlphaFold3 and related deep learning models represent significant progress in structural prediction, particularly for complex and multicomponent assemblies, they do not fully resolve the longstanding challenges associated with structure-based design. These model remain limited in their ability to capture dynamic behaviors, glycan heterogeneity, induced fit effects, and other context-dependent molecular phenomena critical to ADC functionality. As such, their outputs should be interpreted as static approximations within broader design frameworks that still require empirical testing, molecular simulation, and domain-specific validation. The increasing accuracy of predicted structures enhances the utility of in silico workflows but does not eliminate the need for mechanistic interpretation or experimental confirmation.
Integration of AI/ML in ADC ADMET prediction and linker design
AI/ML plays a significant role in designing linker architectures for ADCs, moving beyond static structure prediction to proactive optimization of linker flexibility, stability, and payload compatibility. Recent studies have demonstrated that DL models integrated with molecular simulations can efficiently propose linker sequences optimized for specific mechanical properties and conformational flexibility, influencing ADC internalization and payload release (Su and Zhang, 2021). By learning from structural ensembles, these AI-driven methods can anticipate steric clashes, predict linker degradation pathways, generate linker-payload combinations made to diverse intracellular environments, and enhance ADC efficacy and pharmacokinetics.
Transcending static structure optimization, ML, and DL models have become implementations in predicting key developability properties in early ADC discovery. Computational platforms now routinely evaluate solubility, aggregation propensity, chemical stability, and expression titer using ensemble ML methods trained on large antibody engineering datasets (Prihoda et al., 2022; Raybould et al., 2019). Aggregation-prone regions can be computationally mapped using support vector machine (SVM) classifiers and recurrent neural networks, while solubility predictors such as CamSol and SoluProt provide additional developability screening (Ghomi et al., 2020; Oeller et al., 2023). These approaches have reduced attrition rates and accelerated the selection of viable ADC lead candidates.
AI/ML have also been fundamental in linker and payload optimization, critical parameters that govern ADC stability, efficacy, and pharmacokinetics. Machine learning-based predictive models are trained to match linker properties to payload hydrophobicity, steric constraints, and chemical reactivity, ensuring optimal intracellular release profiles (Shen et al., 2023; Xiong et al., 2024). Novel informatics platforms now curate large libraries of ADC chemical structures and employ AI to identify linker–payload–antibody compatibility rulesets, streamlining rational ADC design (Shen et al., 2023).
To evaluate the outcomes of linker optimization, several metrics are employed. These include predictions of plasma stability, cleavage rate in lysosomal conditions, linker exposure under solvent-accessible surface area (SASA) analysis, and simulated steric compatibility with antibody and payload components. Additionally, docking scores, binding energy (ΔG), and RMSD across conformational ensembles are employed to assess spatial fit and flexibility. ML models often use predicted ADMET properties, such as half-life, cell permeability, and intracellular release kinetics as surrogate endpoints. For example, Su and Zhang demonstrated a model that predicted intracellular release profiles based on linker-payload hydrophobicity and steric load, validated against in vitro lysosomal degradation assays. (Su and Zhang, 2021) In glycan-based linker studies, the Woods Group used MD simulations to analyze hydrogen bond occupancy and glycosidic torsion angle variability, providing insight into stability and solvent exposure (Woods Group, 2025) More broadly, benchmarking efforts described by Bhatt and Shea have highlighted the use of hybrid ML-mechanistic evaluation pipelines, supported by publicly available ADC performance datasets (Bhatt and Shea, 2025).
Efficient, flexible linker modeling and design are further augmented by combining DL and MD simulations. Models trained on simulated ensembles can predict linker dynamics, steric hindrance, and degradation pathways, tailoring linker structures for optimal tumor penetration and cytotoxic payload release (Imrie et al., 2020). These advances allow ADC developers to model static conjugates and dynamic, physiologically relevant states critical for in vivo efficacy. Glycan-based linkers represent a modular and biocompatible strategy for next-generation ADCs. These linkers are engineered to incorporate site-specific conjugation, enzymatically cleavable motifs, or sterically protective elements that modulate payload exposure and tumor microenvironment responsiveness. Using MD simulations with the GLYCAM force field, glycan linkers can be modeled at atomic resolution to evaluate conformational flexibility, hydrogen bonding patterns, and solvent accessibility under physiologically relevant conditions. Such simulations enable the rational design of glycan linkers with controlled degradation profiles and enhanced plasma stability (Woods Group, 2025).
State-of-the-art AI pipelines built on NVIDIA GPU architecture have been increasingly adopted. These platforms offer scalable, parallelized computing that supports large-scale simulation and high-throughput docking workflows. Tools like DiffDock, a diffusion-based structure prediction and docking algorithm (Corso et al., 2022), can leverage these GPU-accelerated environments to model glycan–antibody and glycan–payload interactions with high spatial accuracy and computational efficiency. DiffDock’s ability to integrate conformational sampling and ligand flexibility makes it particularly suited for evaluating the dynamic behavior of glycan-containing conjugates. When deployed on NVIDIA’s optimized inference engines and CUDA-based infrastructure, these pipelines enable rapid screening and prioritization of glycan linker candidates across diverse structural and chemical configurations (St. John et al., 2024). This integration of DL-based docking, MD simulation, and high-performance computing provides a framework for the rational design of ADC–glycan conjugates that meet structural and functional constraints, supporting the development of next-generation modular linkers with tunable therapeutic properties.
Reinforcement learning (RL) methods represent an unexplored territory for de novo ADC design. Recent studies have demonstrated that RL frameworks can simulate iterative mutation and optimization cycles across antibody sequences, linker chemistries, and payload combinations. (Schneider, 2021). These algorithms allow the system to learn from each design iteration, refining candidates based on developability scores, predicted cytotoxicity, and pharmacokinetic parameters. Integrating RL into closed-loop experimental workflows offers the potential for fully autonomous ADC engineering platforms capable of continuous improvement and optimization.
Recent computational reviews have emphasized the growing sophistication of AI-driven ADC design strategies. For instance, Bhatt D and Shea J. summarized advances in computational lead optimization for antibody–linker–payload systems (Bhatt and Shea, 2025), while Lodge et al. described emerging technologies for quantifying antibody binding properties and their downstream implications for ADC design (Lodge et al., 2025). Together, these innovations show how AI/ML are no longer supplementary to ADC development but are becoming essential at every stage, from early antigen selection and antibody optimization to chemical linker matching and payload stability prediction. As datasets expand and DL algorithms grow, the integration of AI/ML holds great promise, and this is only the beginning.
Emerging generative AI models for ADCs
A notable advancement in this field is the emergence of generative AI frameworks specifically adapted for antibody engineering, which contributes to ADC development, as illustrated in Figure 4. Generative adversarial networks (GANs) and variational autoencoders (VAEs) have generated diversified CDR loop libraries with enhanced antigen-binding potential. For example, PALM-H3, a recent GAN-based generative framework, was created for SARS-CoV-2 antibody studies, which enables the design of the CDR-H3 sequences conditioned on antibody structural context by learning potential representations that preserve loop geometry and canonical structural motif while also introducing functional diversity for antigen recognition (He et al., 2024). Similarly, the Ig-VAE model efficiently learns potential representations of the antibody Fv domains, which allows the conditional generation of variant antibodies with user-friendly and user-defined properties (Eguchi et al., 2022).
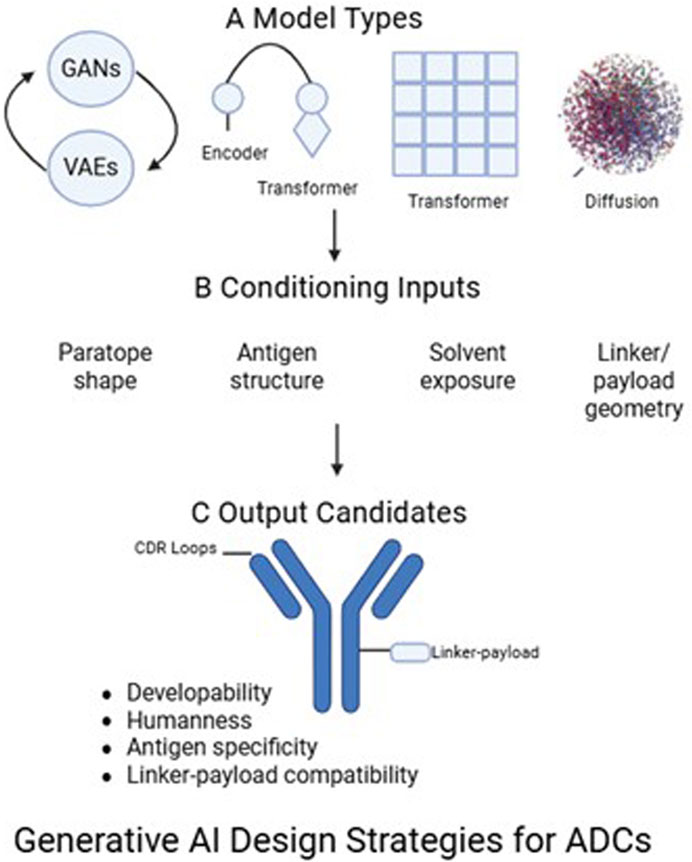
Figure 4. The diagram outlines three components of generative modeling for ADC design. (A) Model types (B) Conditioning inputs incorporate structural and functional constraints (C) Output candidates are antibody variants optimized for CDR loop structure, developability, humanness, antigen specificity, and linker-payload compatibility.
Transformer-based architecture has further expanded the landscape of antibody sequence generation for mutations. Pretrained models like AntiBERTa and AbLang influence antibody sequence databases to learn language-like patterns (Gao et al., 2024). These models enable the generation of synthetic antibody libraries, the prediction of structural features from sequences, and the scoring of sequences for humanness, which is essential to the ADC developmental pipeline. For example, Hu-mAB is designed using ML classifiers that can discriminate between human and non-human antibody variable domain sequences using the larger available repertoire data (Marks et al., 2021). As discussed earlier, recent extensions of these models incorporate paratope conditioning and allow for targeted CDR diversity, which is aimed at antigens and optimizes workflows (Peng and Yang, 2022).
In silico affinity maturation and humanness, scoring have also been transformed by integrating experimental deep mutational scanning (DMS) datasets with computational optimization strategies. Early work demonstrated that mutation libraries coupled with binding assays could map affinity and excel in this area (Barderas et al., 2008). Building on these computational frameworks, such as computational affinity maturation (CAM) models, has led to the development of models that predict point mutations that enhance antigen-binding affinity without compromising solubility or how it works (Koenig et al., 2015). These approaches allow ADC developers to simulate mutations in silico, guiding rational design for affinity and developability improvements.
Introducing diffusion-based generative models into antibody engineering has been a paradigm-shifting development. For example, EvoDiff, a diffusion-based model developed for protein generation, has recently been adapted for antibody design tasks, enabling the controllable generation of novel binders with desired paratope features (Alamdari et al., 2023). Similarly, DiffAb, a diffusion model trained specifically on antibody structural ensembles, allows users to sample structurally diverse yet functionally plausible antibody variants, providing new avenues for ADC diversification (Luo et al., 2022). These emerging models offer a path toward more integrated structure-function co-design, enabling the simultaneous optimization of sequence and structural properties. Traditional antibody modeling workflows often separate sequence optimization from structural validation. Diffusion models can generate sequences directly constrained by desired structural outcomes, such as loop angles, epitope curvature, or solvent accessibility parameters relevant to ADC linker engineering and payload accessibility.
Case applications and platforms
Case applications further showed the impact of AI-driven ADC design. In human epidermal growth factor receptor 2 (HER2) targeted ADCs, machine learning predicts optimal conjugation sites that maintain receptor affinity while maximizing internalization and endosomal escape efficiency. DL frameworks trained on antibody-antigen complex structures have guided the selection of paratope configurations that preserve epitope accessibility after conjugation, improving both binding and cytotoxicity (Sobhani et al., 2024; Dewalker et al., 2025). For the cluster of differentiation 30 (CD30) targeted ADCs, AI-based classification models have aided in predicting trafficking behavior and lysosomal delivery, leading to improved cytotoxic payload delivery in hematologic malignancies (Goeij et al., 2016). Another example is the epidermal growth factor receptor (EGFR) targeted ADCs, convolutional neural networks (CNNs) and graph-based models have been applied to forecast receptor expression heterogeneity across tumor subtypes, guiding rational selection and payload tuning to avoid off-target toxicity (Zhang et al., 2024).
Tools such as the ADCdb provide a curated repository of structural, functional, and clinical metadata for over 200 ADCs, which serves as a valuable foundation for multi-tasking learning models that integrate linker chemistry, payload class, and efficacy metrics (Shen et al., 2023).
Recent studies have demonstrated the efficacy of Neural Ordinary Differential Equations (Neural-ODEs) with an example of ADCnet, a deep neural network of the model, in capturing the dynamic behavior of ADCs, including intracellular trafficking, linker cleavage kinetics and payload release over time (Ahmed et al., 2022; Losada and Terranova, 2024; Bram et al., 2023).These continuous-time models are well suited for simulating the nonlinear kinetics observed in lysosomal degradation and drug activation. They provide valuable insights into structure-activity relationships relevant to payload potency and release (Jin et al., 2023), while IgFold, DeepAb, and DiffAb support rapid antibody structure prediction and paratope design specific for stable conjugation (Luo et al., 2022; Ruffolo et al., 2023; Ruffolo et al., 2022). Generative tools such as EvoDesign facilitate the structure-guided design of the antibody variant compatible with linkers and drug loading, while Mabtope aids in identifying conjugation-tolerant epitopes and evaluating developability and immunogenicity (Pearce et al., 2019; Bourquard et al., 2018; Tahir et al., 2021). Prihoda et al. (2022) This is to name a few, but we have provided Table 1 with a more in-depth explanation to others in the Antibody and ADC pipeline that are accessible as open source and through the industry.
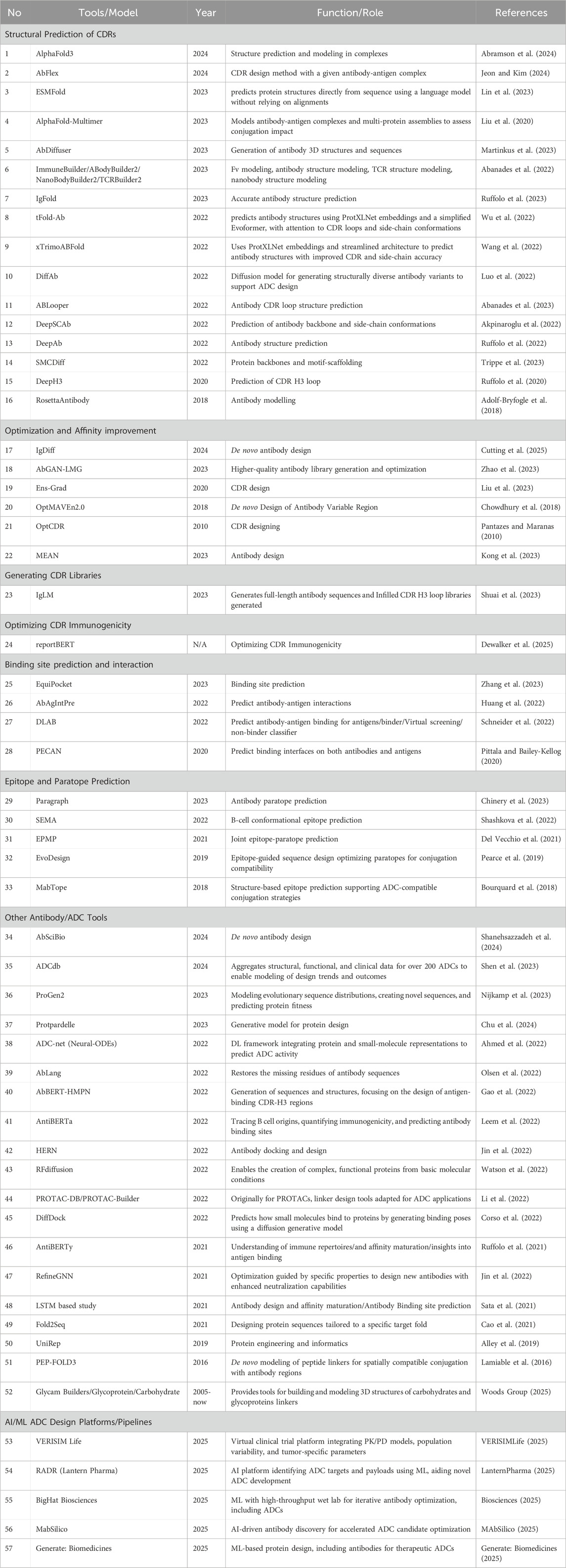
Table 1. Antibody and ADC design tools.
Challenges and future directions in AI-Driven ADC design
Integrating AI/ML into ADC design faces several challenges despite growing enthusiasm for their potential to accelerate discovery (Visan and Negut, 2024). One of the issues is the scarcity of high-quality, publicly available data. Much of the experimental information surrounding ADCs, such as conjugation chemistry, drug-to-antibody ratio (DAR), linker stability, pharmacokinetics, and toxicity, is either proprietary or inconsistently reported, limiting the development of generalizable models (Mckertish and Kayser, 2021). Even when data is available, the inherent complexity of ADCs poses unique modeling difficulties. Unlike traditional small molecules or monoclonal antibodies, ADCs comprise three interdependent components: the antibody, the chemical linker, and the cytotoxic payload, which can influence efficacy, stability, and safety in nonlinear and context-dependent ways (Dewalker et al., 2025). Current models often struggle to capture these multimodal interactions, especially when detailed 3D structural information of the complete ADC is lacking (Sapoval et al., 2022). Additionally, AI models developed for small molecule or biologic therapeutics frequently fail to translate to ADCs, as they inadequately predict developability factors such as aggregation, solubility, and clearance (Chen et al., 2023). Off-target effects and antigen heterogeneity complicate predictive modeling, particularly when simulating tumor selectivity or toxicity across diverse patient populations (Zhang and Liu, 2025). The reliance on black-box neural networks also introduces interpretability and regulatory acceptance issues, particularly in safety-critical environments (Hassija et al., 2023).
Future progress in AI-driven ADC development will rely on integrated, multi-scale modeling approaches that bridge protein structure prediction, systems pharmacology, and real-world clinical and omics data. Recent advancements such as AlphaFold3 enable structural inference of antibody–ligand complexes, including glycan and payload binding, which can support atomic-level modeling of full ADCs. (Abramson et al., 2024) These structural insights offer a path toward more predictive therapeutic design when coupled with system-level models incorporating tumor heterogeneity, immune microenvironment data, and patient-specific expression profiles. Generative models based on diffusion processes and variational inference are increasingly capable of proposing novel antibody scaffolds, conjugation sites, and payload chemistries that conform to structural and functional constraints (Corso et al., 2022). Transfer learning from related biologic modalities, including multimeric, bispecific antibodies, and nanoparticle conjugates, can further improve predictive accuracy when direct ADC training data is limited. Several methodological strategies have been introduced to address constraints related to the limited availability of training data and the narrow diversity of antibody sequences. Transfer learning allows models trained on large-scale protein datasets to be adapted to smaller, task-specific datasets through fine-tuning. Data augmentation techniques, including in silico mutagenesis and synthetic sequence generation, increase the number of training examples while preserving biologically grounded features. Self-supervised learning methods, such as masked language modeling and contrastive representation learning, are used to extract structural and sequence-level patterns without requiring labeled outputs. These approaches are designed to improve model performance under limited data conditions and reduce overfitting by enhancing feature extraction and sampling variability. Techniques such as federated learning enable the development of multi-institutional models without compromising data privacy, while active learning frameworks prioritize the experimental validation of high-uncertainty predictions to refine model performance iteratively. Interpretability methods, including attention-based attribution and saliency mapping, can help identify biologically relevant features driving model outputs, facilitating mechanistic understanding and regulatory transparency. Addressing these challenges systematically will be essential to establish AI-augmented workflows for the rational engineering multimeric and next-generation ADCs.
Conclusion
AI/ML have begun to redefine the field of ADC design by introducing advanced capabilities for prediction, optimization, and iterative refinement across the development pipeline. Unlike traditional trial-and-error approaches, AI/ML models can extract subtle structure-activity relationships from complex, high-dimensional data and identify candidate designs that might be overlooked. These tools have already demonstrated utility in predicting antigen-antibody interactions, optimizing conjugation sites, forecasting pharmacokinetics and off-target liabilities, and selecting linker–payload combinations with improved stability and therapeutic index. As a result, researchers are shifting toward a rational, data-driven paradigm for ADC discovery, minimizing the need for resource-intensive empirical screening.
With the emergence of next-generation ADCs, including multimeric constructs, bispecific formats, and modular payload systems, there is an urgent need for more advanced computational frameworks capable of modeling their increased structural and functional complexity. Multimeric ADCs, which incorporate multiple antigen-binding domains or payload units, hold the potential for enhanced avidity, dual-target engagement, and improved tumor selectivity, particularly in heterogeneous or resistant cancers. However, the design of these molecules requires a detailed understanding of inter-domain spatial orientation, linker flexibility, steric constraints, and payload-release kinetics, all of which are challenging to assess experimentally. AI-driven platforms, particularly those employing graph neural networks, transformer-based architectures, and multimodal representation learning, are well-positioned to address these needs.
Recent breakthroughs such as AlphaFold3 offer unprecedented accuracy in predicting antibody structures and multicomponent protein complexes with bound ligands, glycans, and small molecules. This capability enables the structural modeling of full ADCs, including multimeric variants, at atomic resolution, facilitating in silico evaluation of conjugation strategies, epitope accessibility, and linker spatial compatibility. Complementing these efforts, diffusion-based generative models have emerged as powerful tools for the de novo design of antibody scaffolds, linkers, and payload-functional groups. These models operate by iteratively denoising latent molecular representations, allowing for the exploration of chemical and structural design spaces while maintaining biologically relevant constraints. Generative approaches, active learning, and experimental feedback transform the ADC design process from an enumeration-based approach to an intelligent, hypothesis-driven synthesis.
Continued investment is required in standardized data infrastructures, experimentally verified datasets, and regulatory frameworks that support algorithmic validation and model interpretability. However, increases in model complexity and dataset size do not resolve fundamental limitations in statistical learning. The performance of AI/ML models is influenced by data quality, including annotation accuracy, measurement consistency, and the completeness of molecular descriptors. Limitations in training set diversity, class imbalance, and non-independent sampling introduce bias, while improper separation of training and validation sets, as well as test sets, reduces reliability and inflate performance estimates. Without systematic evaluation protocols and benchmarking against external datasets, model generalizability remains constrained.
As AI/ML methods are incorporated into experimental and preclinical workflows, their utility depends on the alignment between the model structure, biological context, and the design of the training and validation processes. Establishing iterative feedback between in silico prediction and empirical testing, alongside coordination across computational, chemical, and clinical disciplines, become essential to enable the reliable application of these technologies. Ultimately, AI/ML approaches are positioned to transform the design and development of ADCs, including multimeric and next-generation formats, by enabling scalable and mechanistically grounded strategies. Approaches such as transfer learning, data augmentation, and representation learning can improve model performance in data-constrained settings and be incorporated into AI workflows without relying on large, labeled datasets. Their impact, however, will depend on the integration of these tools with high-quality datasets, validated model architecture, and coordinated experimental feedback to ensure biological relevance and translational applicability.
Author contributions
HN: Writing – original draft, Writing – review and editing. XW: Writing – review and editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. Generative AI tools were used during the early stages of manuscript preparation to assist in organizing the outline and drafting portions of the introduction. All generated content was reviewed, assessed, and rewritten in the authors’ own words to ensure accuracy, originality, and alignment with scholarly standards.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fddsv.2025.1628789/full#supplementary-material
References
Abanades, B., Georges, G., Bujotez, A., and Deane, C. M. (2022). “ABlooper: fast accurate antibody CDR loop structure prediction with accuracy estimation.” Bioinformatics 38.7. doi:10.1093/bioinformatics/btac016
Abanades, B., Wong, W. K., Boyles, F., Georges, G., Bujotzek, A., and Deane, C. M. (2023). “ImmuneBuilder: deep-Learning models for predicting the structures of immune proteins.” Nat. Commun. Biol. 6, 575. doi:10.1038/s42003-023-04927-7
Abramson, J., Adler, J., Dunger, J., Evans, R., Green, T., Pritzel, A., et al. (2024). “Accurate structure prediction of biomolecular interactions with AlphaFold 3.” Nature 630: 493–500. doi:10.1038/s41586-024-07487-w
Adhikari, A., and Chen, I. A. (2025). “Antibody-Nanoparticle Conjugates Ther. Comb. Best Two Worlds.” Wiley 21.15: 2409635. doi:10.1002/smll.202409635
Adolf-Bryfogle, J., Kalyuzhniy, O., Kubitz, M., Weitzner, B. D., Hu, X., Adachi, Y., et al. (2018). “RosettaAntibodyDesign (RAbD): a general framework for computational antibody design.” PLOS Comput. Biol. 14, e1006112. doi:10.1371/journal.pcbi.1006112
Ahmed, S., Le, D., Son, T., Adejumo, T., Ma, G., and Yao, X. (2022). “ADC-net: an open-source deep learning network for automated dispersion compensation in optical coherence tomography.” Front. Med. 9, 864879. doi:10.3389/fmed.2022.864879
Akpinaroglu, D., Ruffolo, J. A., Mahajan, S. P., and Gray, J. J. (2022). “Simultaneous prediction of antibody backbone and side-chain conformations with deep learning.” PLOS one 17, e0258173. doi:10.1371/journal.pone.0258173
Alamdari, S., Thakkar, N., van den Berg, R., Lu, A. X., Fusi, N., Amini, A. P., et al. (2023). “Protein generation with evolutionary diffusion: sequence is all you need.” bioRxiv. doi:10.1101/2023.09.11.556673
Alley, E. C., Khimulya, G., Biswas, S., alQuraishi, M., and Church, G. M. (2019). “Unified rational protein engineering with sequence-based deep representation learning.” Nat. Methods 16, 1315, 1322. doi:10.1038/s41592-019-0598-1
Angiolini, L., Manelli, F., Spiga, O. A., Tafi, A., Visibelli, A., Petricci, E., et al. (2025). “Machine learning for predicting the drug-to-antibody ratio (DAR) in the synthesis of antibody-drug conjugates (ADCs).” Am. Chem. J. Chem. Inf. Model. doi:10.1021/acs.jcim.5c00037
Antunes, E. M., Beukes, D. R., Caro-Diaz, E. J. E., Narchi, N. E., Tan, L. T., and Gerwick, W. H. (2023). “Chapter 5 - medicines from the sea,” in Oceans and human health 2nd edn. Editors L. E. Fleming, L. B. A. Creencia, W. H. Gerwick, H. C. Goh, M. O. Gribble, and B. Maycock, et al. (Cambridge, MA: Academic Press), 103–148. doi:10.1016/B978-0-323-95227-9.00009-9
Baek, M., Dinaio, F., Anishchenko, I., Dauparas, J., Ovchiinnikov, S., Lee, G. R., et al. (2021). “Accurate prediction of protein structures and interactions using a three-track neural network.” Science 373: 871–876. doi:10.1126/science.abj8754
Bai, G., Sun, C., Guo, Z., Wang, Y., Zeng, X., Su, Y., et al. (2023). “Accelerating antibody discovery and design with artificial intelligence: recent advances and prospects.” Seminars Cancer Biol. 95: 13–24. doi:10.1016/j.semcancer.2023.06.005
Barderas, R., Desmet, J., Timmerman, P., Casai, J. I., and Casal, J. I. (2008). “Affinity maturation of antibodies assisted by in silico modeling.” PNAS 105: 9029–9034. doi:10.1073/pnas.0801221105
Bergonzo, C., and Grishaev, A. (2025). “Critical assessment of RNA and DNA structure predictions via artificial intelligence: the imitation game.” J. Chem. Inf. Model. 65, 3544, 3554. doi:10.1021/acs.jcim.5c00245
Bhatt, D., and Shea, J. (2025). VERISIMLife. 9 1 2025. Available online at: https://www.verisimlife.com/publications-blog/knowledge-enhanced-ai-to-supercharge-adc-development-for-treatment-of-cancer#:∼:text=Given%20the%20combinatorial%20nature%20of,ADC%20that%20may%20be%20concerning.2842025.
Biosciences, B. H. (2025). Better biologics faster through ML-guided design. Available online at: https://www.bighatbio.com/May 1, 2025).
Boross, P., and Leusen, J. H. W. (2012). Mechanisms of action of CD20 antibodies. Am. J. Cancer Res. 2 (6), 676–90.
Bourquard, T., Musnier, A., Puard, V., Tahir, S., Ayoub, M. A., Jullian, Y., et al. (2018). “MAbTope: a method for improved epitope mapping.” J. Immunol. 201: 3096–3105. doi:10.4049/jimmunol.1701722
Bram, D. S., Nahum, U., Schropp, J., Pfister, M., and Koch, G. (2023). “Low-dimensional neural ODEs and their application in pharmacokinetics.” Springer: 123–140. doi:10.1007/s10928-023-09886-4
Cao, Y., Das, P., Chenthamarakshan, V., Chen, P. Y., Melnyk, I., and Shen, Y. (2021). Fold2Seq: a joint sequence(1D)-Fold(3D) embedding-based generative model for protein design. Proc. Mach. Learn Res. 139, 1261–1271.
Chandrika, S., Rani, A. P., Meghana, K., Jyothsna, K., Yamini, S., and Kumar, V. K. (2024). AI: THE Revolutionary impact on drug delivery systems– a new era in medicine. World J. Pharm. Res., 109–130. doi:10.20959/wjpr202424-34871
Chaves, E. J. F., Coelho, D. F., Cruz, C. H. B., Moreira, E. G., Simoes, J. C. M., Nascimento-Filho, M. J., et al. (2024). “Structure-based computational design of antibody mimetics: challenges and perspectives.” FEBS Open Bio 15: 223–235. doi:10.1002/2211-5463.13855
Chen, H., Fan, X., Zhu, S., Pei, Y., Zhang, X., Zhang, X., et al. “Accurate prediction of CDR-H3 loop structures of antibodies with deep learning.” eLife 12(2024). doi:10.7554/eLife.91512
Chen, Z., Wang, X., Chen, X., Huang, J., Wnag, C., Wang, J., et al. (2023). Accelerating therapeutic protein design with computational approaches toward the clinical stage. Comput. Struct. Biotechnol. J. 21, 2909–2926. doi:10.1016/j.csbj.2023.04.027
Chinery, L., Wahome, N., Moal, I., and Deane, C. M. (2023). “Paragraph-antibody paratope prediction using graph neural networks with minimal feature vectors.” Bioinformatics 39, btac732. doi:10.1093/bioinformatics/btac732
Chiu, M. L., Goulet, D. R., Teplyakov, A., and Gilliland, G. L. (2019). “Antibody structure and function: the basis for engineering therapeutics.” MDPI 8.4: 55. doi:10.3390/antib8040055
Chowdhury, R., Allan, M. F., and Maranas, C. D. (2018). “OptMAVEn-2.0: de novo design of variable antibody regions against targeted antigen epitopes.” Antibodies 7, 23. doi:10.3390/antib7030023
Chu, A. E., Kim, J., Cheng, L., Nesr, G. E., Xui, M., Shuai, R. W., et al. (2024). “An all-atom protein generative model.” PNAS 121, e2311500121. doi:10.1073/pnas.2311500121
Chungyoun, M., and Gray, J. J. (2024). AI models for protein design are driving antibody engineering. Curr. Opin. Biomed. Eng. 28, 100473. doi:10.1016/j.cobme.2023.100473
Codina, N., Zhang, C., Chakroun, N., and Dalby, P. A. (2019). “Insights into the stability of a therapeutic antibody Fab fragment by molecular dynamics and its stabilization by computational design.” bioRxiv. doi:10.1101/644369
Corso, G., Stark, H., Jing, B., Barzilay, R., and Jaakkola, T. (2022). DiffDock: diffusion steps, twists, and turns for molecular docking. arXiv. doi:10.48550/arXiv.2210.01776
Cruz, E., and Kayser, V. (2019). Monoclonal antibody therapy of solid tumors: Clinical limitations and novel strategies to enhance treatment efficacy. Biol.: Targets Ther. 13, 33–51. doi:10.2147/BTT.S166310
Cutting, D., Dreyer, F. A., Errington, D., Schneider, C., and Deane, C. M. (2025). De novo antibody design with SE(3) diffusion. J. Comput. Biol. 32 (4). doi:10.1089/cmb.2024.0768
Dauparas, J., Anishchenko, I., Bennett, N., Bai, H., Ragotte, R. J., Milles, L. F., et al. (2022). Robust deep learning–based protein sequence design using ProteinMPNN. Science. 378 (6615), 49–56. doi:10.1126/science.add2187
Dabkowska, A., Domka, K., and Firczul, M. (2024). “Advancements in cancer immunotherapies targeting CD20: from pioneering monoclonal antibodies to chimeric antigen receptor-modified T cells.” Front. Immunol. 15 1363102. doi:10.3389/fimmu.2024.1363102
Debnath, U., Verma, S. J., Patra, J., and Mandal, S. K. (2022). “A review on recent synthetic routes and computational approaches for antibody drug conjugation developments used in anti-cancer therapy.” J. Mol. Struct. 1256: 132524. doi:10.1016/j.molstruc.2022.132524
Del Vecchio, A., Deac, A., Lio, P., and Velckovic, P. (2021). “Neural message passing for joint paratope-epitope prediction.” arXiv. doi:10.48550/arXiv.2106.00757
Desai, D., Kantliwala, S., Vybhavi, J., Ravi, R., Patel, H., and Patel, J. (2024). “Review of AlphaFold 3: transformative advances in drug design and therapeutics.” Cureus 16: e63646. doi:10.7759/cureus.63646
Dewalker, V., Morya, V. K., Kim, Y. H., Park, S. T., Kim, H. S., and Koh, Y. H. (2025). “Revolutionizing oncology: the role of Artificial Intelligence (AI) as an antibody design, and optimization tools.” Biomark. Res. doi:10.1186/s40364-025-00764-4
Dixit, S. B. (2015). “Computational methods in the optimization of biologic modalities,” in Developability of biotherapeutics: computational approaches, Editors S. K. Singh and S. Kumar. CRC Press, 35–54.
Dominguez, C., Boelens, R., and Bonvin, AMJJ (2003). HADDOCK: a protein–protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125 (7), 1731–1737. doi:10.1021/ja026939x
Eberhardt, J., Santos-Martins, D., Tillack, A. F., and Forli, S. (2021). “AutoDock Vina 1.2.0: new docking methods, expanded force field, and Python bindings.” J. Chem. Inf. Model. 61, 3891, 3898. doi:10.1021/acs.jcim.1c00203
Eguchi, R. R., Choe, C. A., and Huang, P. S. (2022). “Ig-VAE: generative modeling of protein structure by direct 3D coordinate generation.” PLOS Comput. Biol. 18.6: e1010271. doi:10.1371/journal.pcbi.1010271
Evans, R., O'Neill, M., Pritzel, A., Antropova, N., Senior, A., Green, T., et al. (2022). “Protein complex prediction with AlphaFold-Multimer.” bioRxiv. doi:10.1101/2021.10.04.463034
Evers, A., Krah, S., Demir, D., Gaa, R., Elter, D., Schroeter, C., et al. (2024). “Engineering hydrophobicity and manufacturability for optimized biparatopic antibody-drug conjugates targeting c-MET.” mABs 16: 2302386. doi:10.1080/19420862.2024.2302386
Filntisi, A., Vlachakis, D., Matsopoulos, G. K., and Kossida, S. (2014). “Computational construction of antibody–drug conjugates using surface lysines as the antibody conjugation site and a non-cleavable linker.” Cancer Inform. 13, 1–12. doi:10.4137/CIN.S19222
Gao, K., Wu, L., Zhu, J., Peng, T., Xia, Y., He, L., et al. (2022). “Incorporating pre-training paradigm for antibody sequence-structure Co-design.” arXiv. doi:10.48550/arXiv.2211.08406
Gao, X., Cao, C., He, C., and Lai, L. (2024). “Pre-training with a rational approach for antibody sequence representation.” Front. Immunol. 15 1468599. doi:10.3389/fimmu.2024.1468599
Garofalo, M., Grazioso, G., Cavalli, A., and Sgrignani, J. (2020). “How computational chemistry and drug delivery techniques can support the development of new anticancer drugs.” Molecules 25.7: 1756. doi:10.3390/molecules25071756
Generate: Biomedicines (2025). Gener. Biomed. Available online at: https://generatebiomedicines.com/ (May 1, 2025).
Gholap, A. D., Uddin, M. J., Faiyazuddin, M., Omri, A., Gowri, S., and Khalid, M. (2024). “Advances in artificial intelligence for drug delivery and development: a comprehensive review.” Comput. Biol. Med. 178: 108702. doi:10.1016/j.compbiomed.2024.108702
Ghomi, F. A., Kittila, T., and Welner, D. H. (2020). “A benchmark of protein solubility prediction methods on UDP-dependent glycosyltransferases.” bioRxiv. doi:10.1101/2020.02.28.962894
Goeij, B. F., Vink, T., Napel, H. T., Breij, E. C. W., Satijn, D., Wubbolts, R. W., et al. (2016). “Efficient payload delivery by a bispecific antibody-drug conjugate targeting HER2 and CD63.” Mol. Cancer Ther. 15, 2688, 2697. doi:10.1158/1535-7163.MCT-16-0364
Hassija, V., Chamola, V., Mahapatra, A., Singal, A., Goel, D., Huang, K., et al. (2023). “Interpreting black-box models: a review on explainable artificial intelligence.” Springer Nat. Link. 16: 45–74. doi:10.1007/s12559-023-10179-8
He, H., He, B., Guan, L., Zhao, Y., Jiang, F., Chen, G., et al. (2024). “De novo generation of SARS-CoV-2 antibody CDRH3 with a pre-trained generative large language model.” Nat. Commun. 15 6867. doi:10.1038/s41467-024-50903-y
Huang, Y., Zhang, Z., and Zhou, Y. (2022). “AbAgIntPre: a deep learning method for predicting antibody-antigen interactions based on sequence information.” Front. Immunol. 13, 1053617. doi:10.3389/fimmu.2022.1053617
Imrie, f, Bradley, A. R., Van der Schaar, M., and Deane, C. M. (2020). “Deep generative models for 3D linker design.” J. Chem. Inf. Model. 60, 1983, 1995. doi:10.1021/acs.jcim.9b01120
Jeon, W., and Kim, D. (2024). “AbFlex: designing antibody complementarity determining regions with flexible CDR definition.” Bioinformatics 40.btae122. doi:10.1093/bioinformatics/btae122
Jin, W., Barzilay, R., and Jaakkola, T. (2022). “Antibody-antigen docking and design via hierarchical equivariant refinement.” arXiv. doi:10.48550/arXiv.2207.06616
Jin, W., Wohlwend, J., Barzilay, R., and Jaakkola, T. (2022). “Iterative refinement graph neural network for antibody sequence-structure Co-design.” arXiv. doi:10.48550/arXiv.211
Jin, Y., Schladetsch, M. A., Huang, X., Balunas, M. J., and Wiemer, A. J. (2023). “Stepping forward in antibody-drug conjugate development.” Pharmacol. Ther. 229: 107917. doi:10.1016/j.pharmthera.2021.107917
Juan, A., Cimas, F. J., Bravo, I., Pandiella, A., Ocana, A., and Alonso-Moreno, C. (2020). “Antibody conjugation of nanoparticles as therapeutics for breast cancer treatment.” MDPI 21.6018. doi:10.3390/ijms21176018
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). “Highly accurate protein structure prediction with AlphaFold.” Nature 596: 583–589. doi:10.1038/s41586-021-03819-2
Khetan, R., Curtis, R., Deane, Cm, Hadsund, J. T., Kar, U., Krawczyk, K., et al. (2022). “Current advances in biopharmaceutical informatics: guidelines, impact and challenges in the computational developability assessment of antibody therapeutics.” mABs 14.1: 2020082. doi:10.1080/19420862.2021.2020082
Kim, J., McFee, M., Fang, Q., Abdin, O., and Kim, P. M. (2023). “Computational and artificial intelligence-based methods for antibody development.” Trends Pharmacol. Sci. 44.3: 175–189. doi:10.1016/j.tips.2022.12.005
Koenig, P., Lee, C. V., Sanowar, S., Wu, P., Stinson, J., Harris, S. F., et al. (2015). “Deep sequencing-guided design of a high affinity dual specificity antibody to target two angiogenic factors in neovascular age-related macular degeneration.” J. Biol. Chem. 290: 21773–21786. doi:10.1074/jbc.M115.662783
Kong, X., Huang, W., and Liu, Y. (2022). End-to-End full-atom antibody design. In Proceedings of the 40th International Conference on Machine Learning 202, 17526–17544. doi:10.48550/arXiv.2302.00203
Kozakov, D., Hall, D. R., Xia, B., Porter, K. A., Padhorny, D., Yueh, C., et al. (2017). The ClusPro web server for protein–protein docking. Nat. Protoc. 12 (2), 255–278. doi:10.1038/nprot.2016.169
Krokidis, M. G., Koumadorakis, D. E., Lazaros, K., Ivantsik, O., Exarchos, T., Vrahatis, A., et al. (2025). “AlphaFold3: an overview of applications and performance insights.” Mol. Sci. 26, 3671. doi:10.3390/ijms26083671
Lamiable, A., Thevenet, P., Rey, J., Vavrusa, M., Derreumaux, P., and Tuffery, P. (2016). PEP-FOLD3: faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 44, W449–W454. doi:10.1093/nar/gkw329
LanternPharma (2025). RADR® AI Platform: Transforming the pace and precision of oncology drug discovery. Lantern Pharma. Available online at: https://www.lanternpharma.com/ai-platform
Leem, J., Mitchell, L. S., Farmery, J. H. R., Barton, J., and Galson, J. D. (2022). “Deciphering the language of antibodies using self-supervised learning.” Patterns 3, 100513. doi:10.1016/j.patter.2022.100513
Li, F., Hu, O., Zhang, X., Liu, Z., Wu, S., Tian, S., et al. (2022). DeepPROTACs is a deep learning-based targeted degradation predictor for PROTACs. Nat. Commun. 13, 7133. doi:10.1038/s41467-022-34807-3
Lin, Z., Akin, H., Rao, R., Hie, B., Zhu, Z., Lu, W., et al. (2023). Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130. doi:10.1126/science.ade2574
Liu, G., Zeng, H., Mueller, J., Carter, B., Wang, Z., Schilz, J., et al. (2020). “Antibody complementarity determining region design using high-capacity machine learning.” Bioinformatics 36, 2126, 2133. doi:10.1093/bioinformatics/btz895
Liu, J., Guo, Z., Wu, T., Roy, R. S., Quadir, F., Chen, C., et al. (2023). “Enhancing alphafold-multimer-based protein complex structure prediction with MULTICOM in CASP15.” Nature 6, 1140. doi:10.1038/s42003-023-05525-3
Lodge, J., Kaitar, L., Duxbury, R., Hall, D., Burley, G. A., Cordy, J., et al. (2025). “Quantifying antibody binding: techniques and therapeutic implications.” mABs 17, 2459795. doi:10.1080/19420862.2025.2459795
Losada, I. B., and Terranova, N. (2024). “Bridging pharmacology and neural networks: a deep dive into neural ordinary differential equations.” CPT Pharmacometrics and Syst. Pharmacol. 13: 1289–1296. doi:10.1002/psp4.13149
Luo, S., Su, Y., Peng, X., Wang, S., Peng, J., and Ma, J. (2022). “Antigen-specific antibody design and optimization with diffusion-based generative models for protein structures.” bioRxiv. doi:10.1101/2022.07.10.499510
MAbSilico (2025). 21 days to design the best antibody candidates. Available online at: https://www.mabsilico.com/May 1, 2025).
Marcu, S. B., Tabirca, S., and Tangney, M. (2022). “An overview of alphafold's breakthrough.” Front. Artif. Intell. 5, 875587. doi:10.3389/frai.2022.875587
Marks, C., Hummer, A. M., Chin, M., and Deane, C. M. (2021). “Humanization of antibodies using a machine learning approach on large-scale repertoire data.” Bioinformatics 37.22: 4041–4047. doi:10.1093/bioinformatics/btab434
Martinkus, K., Ludwiczak, J., Cho, K., Liang, W. C., Lafrance-Vanasse, J., Hotzel, I., et al. (2023). AbDiffuser: full-atom generation of in vitro functioning antibodies. arXiv. doi:10.48550/arXiv.2308.05027
Mckertish, C. M., and Kayser, V. (2021). Advances and limitations of antibody drug conjugates for cancer. Biomedicines 9, 872. doi:10.3390/biomedicines9080872
Melo, R., Agostinho, L., Preto, A., Almeida, J. G., Correia, J. D. G., Ozge, S., et al. (2018). “Computational approaches in antibody-drug conjugate optimization for targeted cancer therapy.” Curr. Top. Med. Chem. 18: 1091–1109. doi:10.2174/1568026618666180731165222
Moore, E. J., Rice, M., Roy, G., Zhang, W., and Marelli, M. (2024). “Emerging conjugation strategies and protein engineering technologies aim to improve ADCs in the fight against cancer.” Xenobiotica 54. 469. 491. doi:10.1080/00498254.2024.2339993
Nijkamp, E., Ruffolo, J. A., Weinstein, E. N., Nikhil, N., Madani, A., Nijkamp, E., et al. (2023). ProGen2: exploring the boundaries of protein language models. Cell Syst. 14, 968–978.e3. doi:10.1016/j.cels.2023.10.002
Oeller, M., Kang, R., Bell, R., Ausserwoger, H., Sormanni, P., and Vendruscolo, M. (2023). “Sequence-based prediction of pH-dependent protein solubility using CamSol.” Brief. Bioinform. 24, bbad004. doi:10.1093/bib/bbad004
Olsen, T. H., Moal, I. H., and Deane, C. M. (2022). “AbLang: an antibody language model for completing antibody sequences.” Bioinform Adv. 2.1: vbac046. doi:10.1093/bioadv/vbac046
Pantazes, R. J., and Maranas, C. D. (2010). “OptCDR: a general computational method for the design of antibody complementarity determining regions for targeted epitope binding.” Protein Eng. Des. and Sel. 23, 849, 858. doi:10.1093/protein/gzq061
Pearce, R., Huang, X., Setiawan, D., and Zhang, Y. (2019). “EvoDesign: designing protein-protein binding interactions using evolutionary interface profiles in conjunction with an optimized physical energy function.” J. Mol. Biol. 431: 2467–2476. doi:10.1016/j.jmb.2019.02.028
Peng, H. P., and Yang, A. S. (2022). “Computational analysis of antibody paratopes for antibody sequences in antibody libraries.” Methods Mol. Biol. 2552. 437–445. doi:10.1007/978-1-0716-2609-2_24
Pittala, S., and Bailey-Kellog, C. (2020). “Learning context-aware structural representations to predict antigen and antibody binding interfaces.” Bioinformatics 36: 3996–4003. doi:10.1093/bioinformatics/btaa263
Prihoda, D., Maamary, J., Waight, A., Juan, V., Fayadat-Dilman, L., Svozil, D., et al. (2022). “BioPhi: a platform for antibody design, humanization, and humanness evaluation based on natural antibody repertoires and deep learning.” mABs 14, 2020203. doi:10.1080/19420862.2021.2020203
Raybould, M. J., Marks, C., Krawczyk, K., Deane, C. M., Nowak, J., Lewis, A. P., et al. (2019). “Five computational developability guidelines for therapeutic antibody profiling.” PNAS 116: 4025–4030. doi:10.1073/pnas.1810576116
Roy, R., and Al-Hashimi, H. M. (2024). AlphaFold3 takes a step toward decoding molecular behavior and biological computation. Nat. Struct. and Mol. Biol. 31, 997–1000. doi:10.1038/s41594-024-01350-2
Ruffolo, J. A., Chu, L. S., Mahajan, S. P., and Gray, J. J. (2023). “Fast, accurate antibody structure prediction from deep learning on massive set of natural antibodies.” Nat. Commun. 14: 2389. doi:10.1038/s41467-023-38063-x
Ruffolo, J. A., Gray, J. J., and Sulam, J. (2021). “Deciphering antibody affinity maturation with language models and weakly supervised learning.” arXiv. doi:10.48550/arXiv.2112.07782
Ruffolo, J. A., Guerra, C., Mahajan, S. P., Sulam, J., and Gray, J. J. (2020). “Geometric potentials from deep learning improve prediction of CDR H3 loop structures.” Bioinformatics 36: i268-i275. doi:10.1093/bioinformatics/btaa457
Ruffolo, J. A., Sulam, J., and Gray, J. J. (2022). “Antibody structure prediction using interpretable deep learning.” Patterns 3, 100406.2. doi:10.1016/j.patter.2021.100406
Sakhnini, L. I., Greisen, P. J., Wiberg, C., Bozoky, Z., Lund, S., Wolf Perez, A.-M., et al. (2019). Improving the developability of an antigen binding fragment by aspartate substitutions. Biochemistry 58 (24), 2750–2759. doi:10.1021/acs.biochem.9b00251
Sakurai, Y., Abe, N., Yoshikawa, K., Oyama, R., Ogasawara, S., Murata, T., et al. (2022). “Targeted delivery of lipid nanoparticle to lymphatic endothelial cells via anti-podoplanin antibody.” J. Control. Release 349: 379–387. doi:10.1016/j.jconrel.2022.06.052
Sapoval, N., Aghazadeh, A., Nute, M. G., Antunes, D. A., Balaji, A., Baraniuk, R., et al. (2022). Current progress and open challenges for applying deep learning across the biosciences. Nat. Commun. 13, 1728. doi:10.1038/s41467-022-29268-7
Sata, K., Kakuzaki, T., Metsugi, S., Kashiwagi, D., Yoshida, K., Wada, M., et al. (2021). “Antibody design using LSTM based deep generative model from phage display library for affinity maturation.” Nature. doi:10.1038/s41598-021-85274-7
Schneider, C. (2021). Deep learning algorithms for predicting association between antibody sequence, structure, and antibody properties (Doctoral dissertation). Ann Arbor, MI: ProQuest LLC. Available online at: https://www.proquest.com/openview/b38e647d7547fbd481d257a4291b5c0f/1?cbl=2026366&diss=y&pq-origsite=gscholar.
Schneider, C., Buchanan, A., Taddese, B., and Deane, C. M. (2022). “DLAB: deep learning methods for structure-based virtual screening of antibodies.” Bioinformatics 38, 377, 383. doi:10.1093/bioinformatics/btab660
Selepe, C. T., Dhlamini, K. S., Tshweu, L., Moralo, M., Kwezi, L., Ray, S. S., et al. (2024). “Trastuzumab-based nanomedicines for breast cancer therapy: recent advances and future opportunities.” Wiley 5.5: 2300191. doi:10.1002/nano.202300191
Shanehsazzadeh, A., McPartlon, M., Kasun, G., Steiger, A. K., Sutton, J. M., Yassine, E., et al. (2024). “Unlocking de novo antibody design with generative artificial intelligence.” bioRxiv. doi:10.1101/2023.01.08.523187
Shashkova, T. I., Umerenkov, D., Salnikov, M., Strashnov, P. V., Konstantinova, A. V., Lebed, I., et al. (2022). “SEMA: antigen B-cell conformational epitope prediction using deep transfer learning.” Front. Immunol. 13, 960985. doi:10.3389/fimmu.2022.960985
Shen, L., Sun, X., Chen, Z., Guo, Y., Shen, Z., Song, Y., et al. (2023). “ADCdb: the database of antibody-drug conjugates.” Nucleic Acid Res. 52: D1097–D1109. doi:10.1093/nar/gkad831
Shi, M., and McHugh, K. J. (2023). “Strategies for overcoming protein and peptide instability in biodegradable drug delivery systems.” Adv. Drug Deliv. Rev. 199: 114904. doi:10.1016/j.addr.2023.114904
Shuai, R. W., Ruffolo, J. A., and Gray, J. J. (2023). “IgLM: infilling language modeling for antibody sequence design.” Cell Syst. 14, 979, 989.e4. doi:10.1016/j.cels.2023.10.001
Siddiqui, B., Yadav, C. H., Akil, M., Faiyyaz, M., Khan, A. R., Ahmad, N., et al. (2025). “Artificial intelligence in computer-aided drug design (CADD) tools for the finding of potent biologically active small molecules: traditional to modern approach.” Combinatorial chemistry and high throughput screening. doi:10.2174/0113862073334062241015043343
Skolnick, J., Gao, M., Zhou, H., and Singh, S. (2021). “AlphaFold 2: why it works and its implications for understanding the relationships of protein sequence, structure, and function.” J. Chem. Inf. Model 61: 4827–4831. doi:10.1021/acs.jcim.1c01114
Smith, A. J. (2015). “New horizons in therapeutic antibody discovery: opportunities and challenges versus small-molecule therapeutics.” SLAS Discov. 20.4: 437–453. doi:10.1177/1087057114562544
Sobhani, N., D'Angelo, A., Pittacolo, M., Mondani, G., and Generali, D. (2024). “Future AI will Most likely predict antibody-drug conjugate response in oncology: a review and expert opinion.” Cancers (Baseline) 16: 3089. doi:10.3390/cancers16173089
St. John, P., Lin, D., Binder, P., Greaves, M., Shah, V., St.John, J., et al. (2024). “BioNeMo framework: a modular, high-performance library for AI model development in drug discovery. arXiv. Available online at: https://arxiv.org/abs/2411.10548.
Su, D., and Zhang, D. (2021). “Linker design impacts antibody-drug conjugate pharmacokinetics and efficacy via modulating the stability and payload release efficiency.” Front. Pharmacol. 12: 687926. doi:10.3389/fphar.2021.687926
Tahir, S., Bourquard, T., Musiner, A., Jullian, Y., Corde, Y., Omahdi, Z., et al. (2021). “Accurate determination of epitope for antibodies with unknown 3D structures.” MAbs 13: 1961349. doi:10.1080/19420862.2021.1961349
Trippe, B. L., Yim, J., Tischer, D., Baker, D., Broderick, T., Barzilay, R., et al. (2023). “Diffusion probabilistic modeling of protein backbones in 3D for the motif-scaffolding problem.” Biomolecules. doi:10.48550/arXiv.2206.04119
VERISIMLife (2025). Available online at: https://www.verisimlife.com/May 1, 2025).
Visan, A. I., and Negut, I. (2024). Integrating artificial intelligence for drug discovery in the context of revolutionizing drug delivery. Life 14, 233. doi:10.3390/life14020233
Wang, Y., Gong, X., Li, S., Yang, B., Sun, Y., Shi, C., et al. (2022). xTrimoABFold: de novo antibody structure prediction without MSA. arXiv. doi:10.48550/arXiv.2212.00735
Waterhouse, A., Bertoni, M., Bienert, S., Struder, G., Tauriello, G., Gumienny, R., et al. (2018). “SWISS-MODEL: homology modelling of protein structures and complexes.” Nucleic Acid Res. 46: W296–W303. doi:10.1093/nar/gky427
Watson, J. L., Juergens, D., Bennett, N. R., Trippe, B. L., Yim, J., Eisenach, H. E., et al. (2022). “Broadly applicable and accurate protein design by integrating structure prediction networks and diffusion generative models.” bioRxiv. doi:10.1101/2022.12.09.519842
Woods Group (2025). “GLYCAM web,” in Carbohydrate research center. Athens, GA: University of Georgia. Available online at: http://glycam.org.
Wu, J., Wu, F., Jiang, B., Liu, W., and Zhao, P. (2022). tFold-ab: fast and accurate antibody structure prediction without sequence homologs. bioRxiv. doi:10.1101/2022.11.10.515918
Xiong, Z., Liu, K., Liu, S., Feng, J., Wang, J., Feng, Z., et al. (2024). “Precision HER2: a comprehensive AI system for accurate and consistent evaluation of HER2 expression in invasive breast Cancer.” BMC Cancer 24, 1204. doi:10.1186/s12885-024-12980-6
Yang, Z., Zeng, X., Zhao, Y., and Chen, R. (2023). “AlphaFold2 and its applications in the fields of biology and medicine.” Signal Transduct. Target. Ther. 8, 115. doi:10.1038/s41392-023-01381-z
Yin, R., and Pierce, B. G. (2023). Evaluation of AlphaFold antibody-antigen modeling with implications for improving predictive accuracy. bioRxiv, 2023.07.05.547832. doi:10.1101/2023.07.05.547832
Zenkova, N., Sedykh, E., Shugaeva, T., Strashko, V., Ermak, T., and Shpilman, A. (2021). Simple end-to-end deep learning model for CDR-H3Loop structure prediction. Mach. Learn. arXiv. doi:10.48550/arXiv.2111.10656
Zhang, C., and Liu, H. (2025). Advancements and future directions of dual-target chimeric antigen receptor T-cell therapy in preclinical and clinical studies. J. Immunol. Res. 2025, 5845167. doi:10.1155/jimr/5845167
Zhang, G., Shang, L., Cao, Y., Zhang, J., Li, S., Qian, R., et al. (2024). “Prediction of epidermal growth factor receptor (EGFR) mutation status in lung adenocarcinoma patients on computed tomography (CT) images using 3-dimensional (3D) convolutional neural network.” Quant. Imaging Med. Surg. 14.8: 6048–6059. doi:10.21037/qims-24-33
Zhang, Y., Huang, W., Wei, Z., Yuan, Y., and Ding, Z. (2023). “EquiPocket: an E(3)-equivariant geometric graph neural network for ligand binding site prediction.” arXiv. doi:10.48550/arXiv.2302.12177
Zhao, W., Luo, X., Tong, F., Zheng, X., Li, J., Zhao, G., et al. (2023). “Improving antibody optimization ability of generative adversarial network through large language model.” Comput. Struct. Biotechnol. J. 21, 5839, 5850. doi:10.1016/j.csbj.2023.11.041
Keywords: AI/ML (artificial intelligence/machine learning), antibody-drug conjugate (ADC), AlphaFold 3, neural ODEs, generative and algorithmic design
Citation: Noriega HA and Wang XS (2025) AI-driven innovation in antibody-drug conjugate design. Front. Drug Discov. 5:1628789. doi: 10.3389/fddsv.2025.1628789
Received: 15 May 2025; Accepted: 02 June 2025;
Published: 26 June 2025.
Edited by:
Gabriel Navarrete-Vazquez, Autonomous University of the State of Morelos, MexicoReviewed by:
Marco A. Loza-Mejía, Universidad La Salle, MexicoFernando Prieto-Martínez, National Autonomous University of Mexico, Mexico
Copyright © 2025 Noriega and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiang Simon Wang, eC5zaW1vbi53YW5nQGdtYWlsLmNvbQ==