 Dhriti Sundar Das
Dhriti Sundar Das- Department of General Medicine, All India Institute of Medical Sciences, Bhubaneswar, India
Autoimmune diseases and sickle cell abnormalities have been reported to share some common pathogenic pathways. Here, we describe two cases of individuals with sickle cell trait (SCT) who presented with antibodies to antinuclear antigens and clinical features consistent with systemic lupus erythematosus (SLE). These patients were from the eastern state of India, i.e., Odisha. Both patients had documented sickle cell trait and tested positive for anti-double-stranded DNA antibodies. They presented with lupus nephritis without any obvious features of sickle cell hemoglobinopathies. Crosstalk between SLE and SCT may explain this association, which needs validation in future large-scale studies.
Introduction
Sickle cell hemoglobinopathies are common genetic disorders that are widely prevalent in certain parts of eastern India (1, 2). Systemic lupus erythematosus (SLE) is an autoimmune entity characterized by multisystem involvement and variable manifestations. Both diseases have certain clinical features in common, such as episodes of joint pain, renal manifestations, and acute chest syndrome, which can pose diagnostic challenges for lupus in patients with these hemoglobinopathies. These common features may represent some common intersecting pathogenic pathways in these diseases. Hence, early diagnosis may sometimes be delayed in such endemic areas (3, 4). This report describes two cases of sickle cell trait (SCT) with seropositivity for lupus-related autoantibodies. The coexistence of SLE and SCT remains a field of active research.
Case presentation
Case 1
A 17-year-old woman with known sickle cell trait presented with a low-grade fever, cough associated with hemoptysis for 2 weeks, chest discomfort, and dyspnea for 3 days. There was no history suggestive of rashes or joint pain. Her medical history included cervical lymphadenopathy. Lymph node biopsy showed only reactive lymphadenitis . Physical examination revealed a pulse rate of 112/min, blood pressure of 136/86 mmHg, respiratory rate of 30/min, and oxygen saturation of 90%. Respiratory examination showed bilateral crepitations and wheezing in all lung fields. Over time, her oxygen saturation dropped to 84%. She was treated with intravenous systemic antibiotics, nebulization, and intravenous fluids. Laboratory investigations revealed an erythrocyte sedimentation rate (ESR) of 151 mm with anemia. Urinalysis revealed 3+ proteinuria. A chest x-ray revealed bilateral patchy infiltrates (Figure 1). Further examination revealed rashes on both feet, along with bilateral cervical lymphadenopathy and an oral ulcer. A peripheral smear showed features suggestive of hemolysis with target cells. The direct Coombs test was positive, and serum ferritin was elevated. Antinuclear antibodies (ANA) and double-stranded DNA (dsDNA) were positive (3+; mixed pattern) with a titer of ≥1:80. Splenomegaly was noted in an ultrasound of the abdomen. A computed tomography scan of the thorax revealed ground glass opacities, bilateral pleural effusions on the right side, and mediastinal and axillary lymphadenopathy (Figure 2). Sputum culture, acid-fast bacilli, and cartridge-based nucleic acid amplification test (CBNAAT) were negative. The 24-h urine protein was 3,521 mg. perinuclear ANCA (p-ANCA) and cytoplasmic ANCA (c-ANCA), tests were negative. The patient received pulse methylprednisolone, followed by oral prednisolone and hydroxychloroquine. Her condition gradually improved, and she was discharged in stable clinical condition. A renal biopsy obtained after discharge showed class 4 lupus nephritis. The case fulfilled the ACR/EULAR 2019 criteria for the diagnosis of SLE (5) (Table 1).
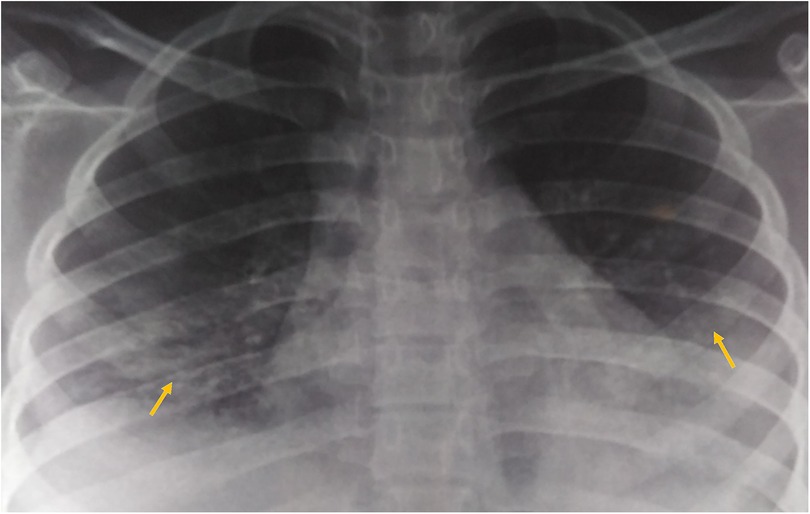
Figure 1. Chest x-ray (PA view) showing bilateral patchy infiltrates.
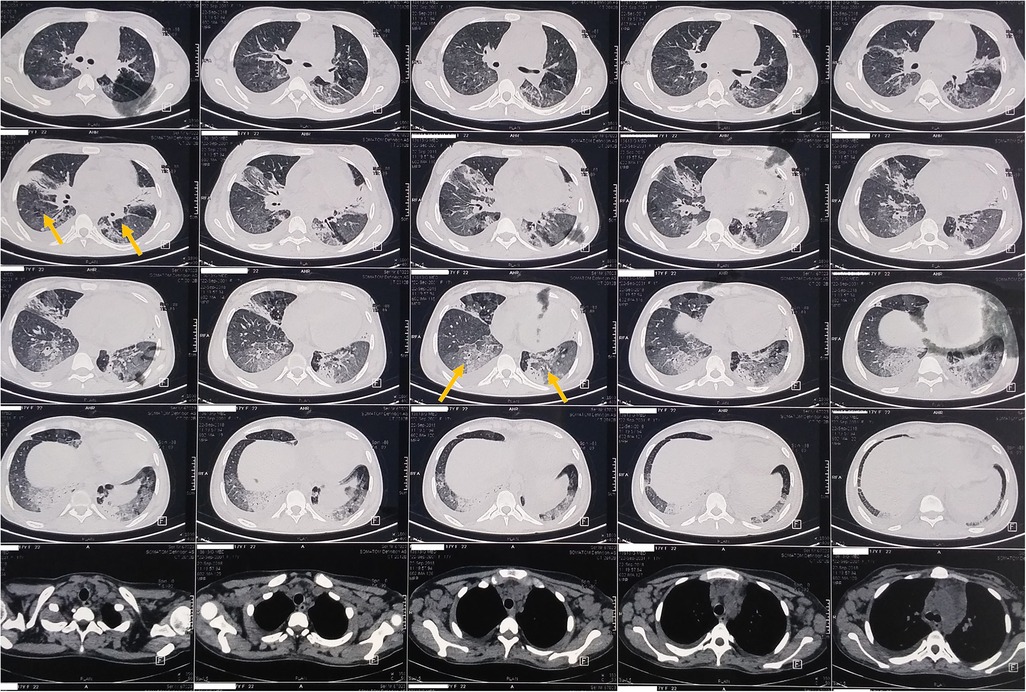
Figure 2. Thorax CT showing bilateral ground glass opacities.
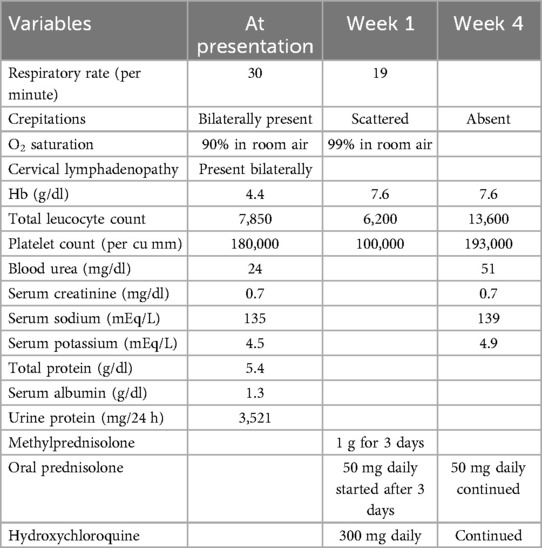
Table 1. Progress chart: case 1.
Case 2
A 20-year-old woman presented with a low-grade fever, multiple small joint pains of 1.5 months, and lower leg swelling, along with abdominal and facial puffiness of 1 month. The joint pain was additive in nature and associated with early morning stiffness lasting approximately 2–3 h. The swelling was not associated with any alteration in urine output or frothy urine. She also reported intermittent abdominal pain. Her medical history included repeated blood transfusions for anemia and a sickle cell abnormality, although no documentation was available. Physical examination revealed malar rashes, hyperpigmented rashes over the upper chest, and oral ulcers (Figures 3, 4). Her scalp hair was sparse, and she had pallor and pitting pedal edema. Laboratory evaluation showed a hemoglobin level of 6.7 g/dl, serum albumin of 3 g/dl, and a urine albumin-to-creatinine ratio of 100 mg/g creatinine. The direct Coombs test was 3+, and serum C3 was low. The 24-h urine protein was 840 mg. An abdominal ultrasound revealed no abnormalities. However, the anti-dsDNA test was positive with 2+ intensity. She developed a bloodstream infection with methicillin-resistant Staphylococcus aureus, for which intravenous antibiotics were administered. High-performance liquid chromatography revealed an HbA2 level of 3.4% and an HbF level of less than 0.8%, suggestive of sickle cell trait. Renal biopsy was deferred as the patient wished to get discharged and reviewed at follow-up. The case fulfilled the ACR/EULAR 2019 criteria for the diagnosis of SLE (5). The patient was discharged on oral corticosteroids. Her joint pain improved considerably, along with the resolution of pedal edema and facial puffiness (Tables 1, 2).
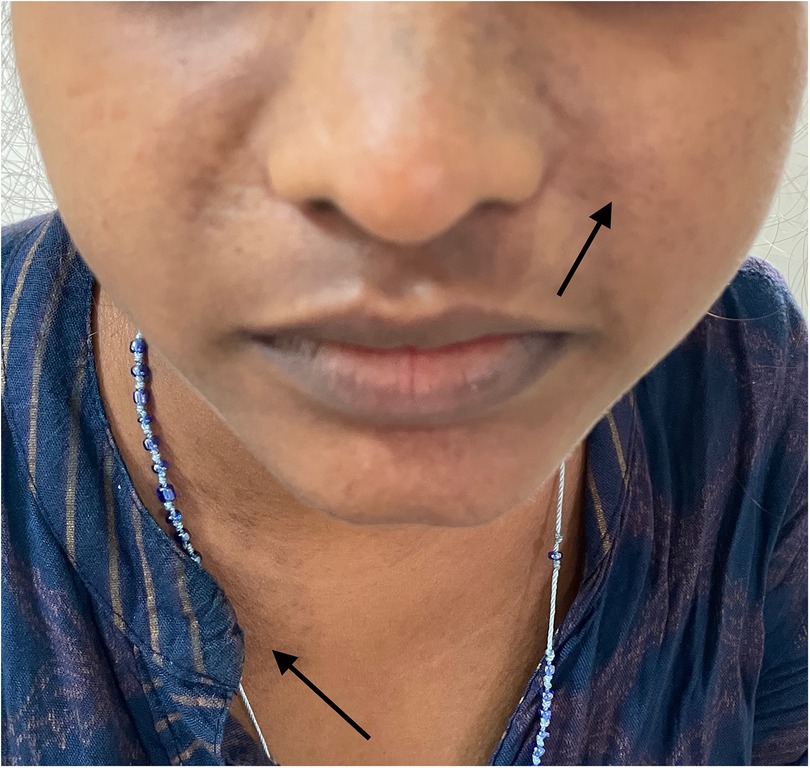
Figure 3. Rash on the face and upper chest.
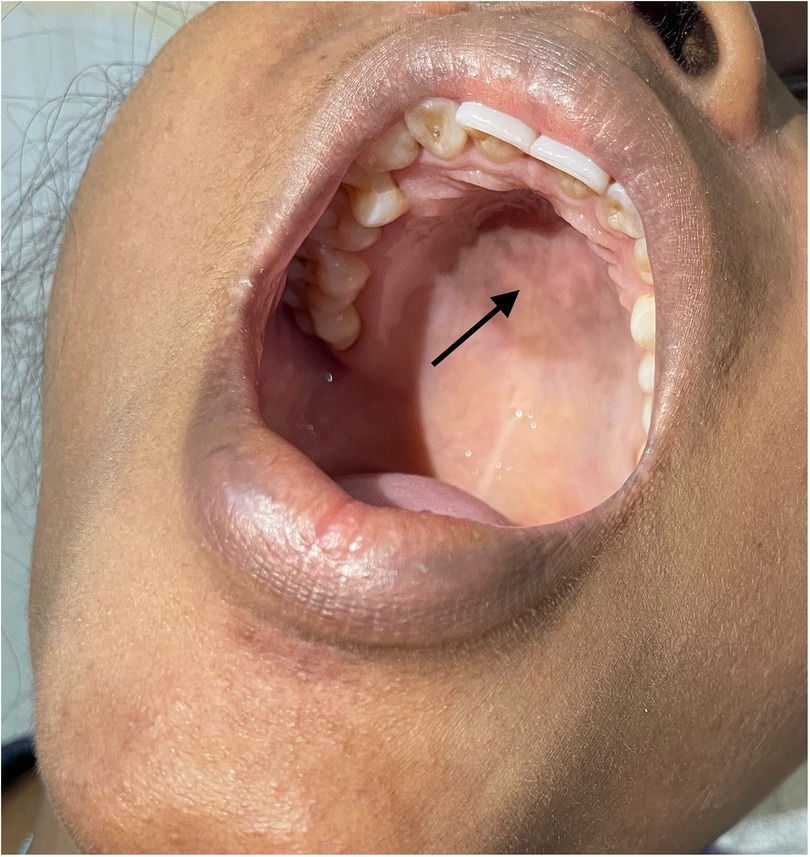
Figure 4. Ulcer of the hard palate.
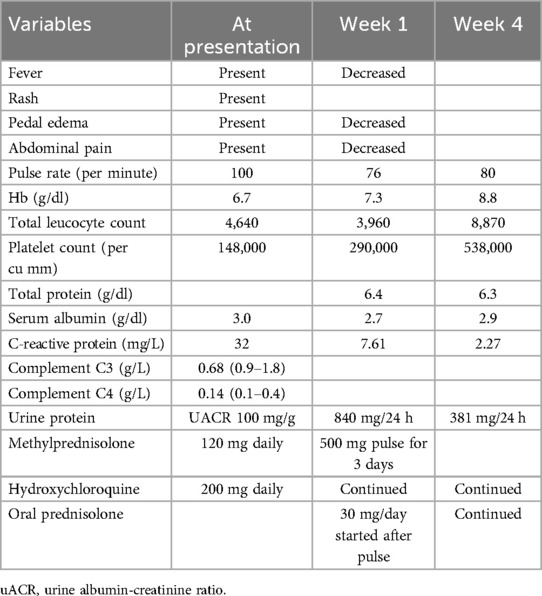
Table 2. Progress chart: case 2.
Discussion
We have presented two cases of sickle cell trait coexisting with SLE. Both of them have antibodies to ANA and anti-dsDNA. The coexistence of these conditions is of great importance, as autoimmune diseases are infrequently reported in the literature (6). Owing to the fact that sickle cell hemoglobinopathies are commonly reported in this part of the country, such cases highlight the importance of recognizing such associations. Usually, the presence of SCT is evident long before SLE is diagnosed (7). In these two cases, the diagnosis of sickle cell trait was made long before the onset of SLE. Both patients were young at the time of SLE diagnosis, as was reported in other literature work (7). In the first case, the patient presented with acute respiratory symptoms, which is in stark contrast to other reported cases where sickle cell patients presented with SLE symptoms like joint pain, swelling, and renal manifestations (3, 7). In contrast, the second case aligned with the more common presentation of SLE as reported elsewhere (8).
Articular manifestations were the most common presentation in such combinations of diseases, followed by serositis and renal involvement. Skin involvement was not frequently observed, which may be an uncommon observation compared to SLE alone (9).
Although the association of ANA positivity with sickle cell disease (SCD) has been described, very few cases have been reported with the SCT variant (3, 7). Patients with sickle cell disease have abnormalities of the alternate pathway of the complement system, which may make them susceptible to the development of autoimmune conditions (10).
Patients with sickle cell hemoglobinopathies and SLE present with hypocomplementemia, which leads to a failure in the activation of the alternate pathway (10). This results in reduced clearance of antigens, inciting autoimmunity. Sickle cell hemoglobinopathies have broad symptomatology, and ANA positivity alone cannot justify a diagnosis of SLE. Because ANA positivity has been found in approximately 50% of cases of sickle cell hemoglobinopathies, clinicians should not rely on this criterion alone (10). Therefore, when SLE is suspected, anti-dsDNA testing should be considered. Immune complexes are formed in such hemoglobinopathies, and this process can lead to the activation of autoimmunity.
Complement activation is intrinsic to sickle cell hemoglobinopathies, even in steady-state conditions, due to the exposure of phosphatidylserine and free heme released during intravascular hemolysis (10). The role of B regulatory cells may be key in the pathogenesis of the possible crosstalk between SLE and SCT. There is both a decrease and functional impairment of B regulatory cells in sickle cell syndromes with SLE, resulting in an imbalanced immune response (11).
Published literature suggests that SCT may not be a completely benign state or an actual disease condition but may posit itself as a risk factor for worse outcomes that herald from the interplay between the two (12–14). The sickle gene herein is envisaged to confer an increased susceptibility to infections like bloodstream infections, autoimmune hemolytic anemia, and lung infections.
Evidence suggests that sickle cell trait may potentially increase the risk of hypercoagulability. In addition, the literature supports that SLE itself is an independent risk factor for both arterial and venous thromboembolism (13). In situ, thrombus or hypercoagulability is known to promote microbial adhesion and colonization and thus is a potent risk factor for infection (15). Hence, SLE patients with SCT may be at higher risk for adverse clinical outcomes.
To date, only a few cases of SLE in individuals with sickle cell trait have been reported in the scientific literature (6). However, it is important to mention that there is no established evidence of a cause–and-effect relationship between sickle cell trait and SLE. Although the limitation of generalizability remains, a heightened awareness of sickle-associated complications may lead to a reduction of complications in such populations within the evolving SLE landscape.
Certain common genes have been implicated in sickle cell syndrome and SLE. The bone morphogenetic protein( BMP) pathway gene is involved in sickle vasculopathy and renal complications, like albuminuria and glomerular hyperfiltration, in sickle cell syndrome. Similarly, BMP single-nucleotide polymorphisms (SNPs) are associated with susceptibility to SLE and its pathogenesis. It has also been found that certain other genes, namely, human leukocyte antigen (HLA) and genes located on chromosomes 2 and 6, are involved in the pathogenesis of both SLE and sickle cell disease (16–19).
Although there are case series and case reports documenting lupus nephritis in patients with sickle cell syndrome, large epidemiological studies are lacking to convincingly state that lupus nephritis has a high prevalence in patients with SCT and SLE (20).
This report highlights that sickle cell trait may be one of the crucial risk factors for SLE. The decline in kidney function in individuals with sickle cell syndromes (SCD and SCT) is greater than in healthy individuals (21). Clinicians should be aware of this knowledge of the association for appropriate management. Awareness of such associations is important for the prognosis of the cases. In areas with a high prevalence of sickle cell syndromes, the diagnosis of SLE needs to be carefully considered based on clinical signs and symptoms.
Conclusion
This report highlights the importance of the appropriate use of diagnostic tests in the setting of sickle cell syndrome and SLE. Early intervention is the key to favorable outcomes. Large clinicoepidemiological studies are required to further explore the relationship between autoimmune diseases and sickle cell syndromes.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors without undue reservation.
Ethics statement
Ethical approval was not required for the studies involving humans because formal ethical approval is not required for case reports as per general institutional policy understanding. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
DD: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author declares that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
The author acknowledges all the doctors and staff at the Department of General Medicine, AIIMS Bhubaneswar, who were involved in patient care.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Balgir RS. Spectrum of hemoglobinopathies in the state of Orissa, India: a ten years cohort study. J Assoc Physicians India. (2005) 53:1021–6.16572956
2. Chhotray GP, Dash BP, Ranjit M. Spectrum of hemoglobinopathies in Orissa, India. Hemoglobin. (2004) 28(2):117–22. doi: 10.1081/HEM-120034244
3. Appenzeller S, Fattori A, Saad ST, Costallat LT. Systemic lupus erythematosus in patients with sickle cell disease. Clin Rheumatol. (2008) 27(3):359–64. doi: 10.1007/s10067-007-0779-7
4. Elficki Y, Rawas A, Bossei AA, Bdawod A, Zabani R, Shams B. Coexistence of lupus nephritis and sickle cell trait, an electron microscopic assessment of renal glomerular damage: case report of a rare association. Electron Physician. (2017) 9(9):5298–302. doi: 10.19082/5298
5. Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, Ramsey-Goldman R, et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Arthritis Rheumatol. (2019) 71(9):1400–12. doi: 10.1002/art.40930
6. Das DS, Sahoo D. Coevality of systemic lupus erythematosus with sickle cell trait: a not so uncommon entity. Cureus. (2020) 12(8):e10119. doi: 10.7759/cureus.10119
7. Maamar M, Tazi-Mezalek Z, Harmouche H, Mounfaloti W, Adnaoui M, Aouni M. Systemic lupus erythematosus associated with sickle-cell disease: a case report and literature review. J Med Case Rep. (2012) 6:366. doi: 10.1186/1752-1947-6-366
8. Cojocaru M, Cojocaru IM, Silosi I, Vrabie CD. Manifestations of systemic lupus erythematosus. Maedica (Bucur). (2011) 6(4):330–6.22879850
9. Stull C, Sprow G, Werth VP. Cutaneous involvement in systemic lupus erythematosus: a review for the rheumatologist. J Rheumatol. (2023) 50(1):27–35. doi: 10.3899/jrheum.220089
10. Piccin A, O'Connor-Byrne N, Daves M, Lynch K, Farshbaf AD, Martin-Loeches I. Autoimmune disease and sickle cell anaemia: “intersecting pathways and differential diagnosis”. Br J Haematol. (2022) 197(5):518–28. doi: 10.1111/bjh.18109
11. Boulassel MR, Al-Naamani A, Al-Zubaidi A, Al-Qarni Z, Khan H, Oukil A, et al. Coexistence of sickle cell disease and systemic lupus erythematosus is associated with quantitative and qualitative impairments in circulating regulatory B cells. Hum Immunol. (2022) 83(12):818–25. doi: 10.1016/j.humimm.2022.09.005
12. Tsaras G, Owusu-Ansah A, Boateng FO, Amoateng-Adjepong Y. Complications associated with sickle cell trait: a brief narrative review. Am J Med. (2009) 122(6):507–12. doi: 10.1016/j.amjmed.2008.12.020
13. Key NS, Derebail VK. Sickle-cell trait: novel clinical significance. Hematol Am Soc Hematol Educ Program. (2010) 2010:418–22. doi: 10.1182/asheducation-2010.1.418
14. Naik RP, Smith-Whitley K, Hassell KL, Umeh NI, de Montalembert M, Sahota P, et al. Clinical outcomes associated with sickle cell trait: a systematic review. Ann Intern Med. (2018) 169(9):619–27. doi: 10.7326/M18-1161
15. Gigler CR, Oertel LB. Understanding hypercoagulopathies. Nursing. (2010) 40(8):52–6. doi: 10.1097/01.NURSE.0000383901.80719.b8
16. Steinberg MH, Sebastiani P. Genetic modifiers of sickle cell disease. Am J Hematol. (2012) 87(8):795–803. doi: 10.1002/ajh.23232
17. Ramos PS, Shedlock AM, Langefeld CD. Genetics of autoimmune diseases: insights from population genetics. J Hum Genet. (2015) 60(11):657–64. doi: 10.1038/jhg.2015.94
18. Nam SW, Lee KS, Yang JW, Ko Y, Eisenhut M, Lee KH, et al. Understanding the genetics of systemic lupus erythematosus using Bayesian statistics and gene network analysis. Clin Exp Pediatr. (2021) 64(5):208–22. doi: 10.3345/cep.2020.00633
19. Mo JS, Chae SC. Bone morphogenetic protein 6 polymorphisms are associated with systemic lupus erythematosus susceptibility in the Korean population. Arch Rheumatol. (2018) 33(4):424–30. doi: 10.5606/ArchRheumatol.2018.6644
20. Saxena VR, Mina R, Moallem HJ, Rao SP, Miller ST. Systemic lupus erythematosus in children with sickle cell disease. J Pediatr Hematol Oncol. (2003) 25(8):668–71. doi: 10.1097/00043426-200308000-00019
Keywords: sickle cell trait, systemic lupus erythematosus, autoimmune disease, lupus nephritis, crosstalk
Citation: Das DS (2024) Case Report: Crosstalk between systemic lupus erythematosus and sickle cell syndromes—two cases from eastern India. Front. Lupus 2:1484352. doi: 10.3389/flupu.2024.1484352
Received: 21 August 2024; Accepted: 10 October 2024;
Published: 6 November 2024.
Edited by:
Hiroyuki Wakiguchi, Oita University, JapanReviewed by:
Yusuke Miyazaki, University of Occupational and Environmental Health Japan, JapanKunio Hashimoto, Nagasaki University, Japan
Copyright: © 2024 Das. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dhriti Sundar Das, Z2VubWVkX2Rocml0aUBhaWltc2JodWJhbmVzd2FyLmVkdS5pbg==