 Sabine Hahn
Sabine Hahn Romana Rashid
Romana Rashid Zaida G. Ramirez-Ortiz
Zaida G. Ramirez-Ortiz- Department of Medicine, Division of Infectious Disease and Immunology, University of Massachusetts Chan Medical School, Worcester, MA, United States
Scavenger receptors (SRs) play an important role in the innate immune response by recognizing and binding a variety of ligands to initiate the removal of both altered self- and non-self-antigens. Over the last two decades, SRs have become a forefront for their role influencing and contributing to inflammatory disease pathways. The findings discussed in this review show that the immunological role SRs play is (1) found in multiple organ systems and not limited to one disease or subset of symptoms; (2) part of both the innate and adaptive immune response in addition to influencing inflammatory signaling via non-immune cell subtypes; (3) both pro- or anti-inflammatory depending on which SR class or cell signaling pathway is being observed; (4) potentially useful for the development of therapeutics and diagnostic or prognostic biomarkers for autoimmune disease pathology. Understanding the role of SRs in the context of inflammation and autoimmunity will shed some light on the comprehension of heterogeneous diseases, such as Systemic Lupus Erythematosus.
1 Introduction
Systemic lupus erythematosus (SLE) is a complex and multifaceted autoimmune disease. It is represented by diverse clinical features impacting multiple organs, combined with a wide range of hematological and serological abnormalities, and disease comorbidities (1). The current classification criteria [2019 European League Against Rheumatism/American College of Rheumatology Immunologic Domains and Criteria for SLE (2019 EULAR/ACR)] requires patients to first test positive for antinuclear antibodies and is followed by an assessment of a combination of 10 symptoms or disorders, each weighted differently to calculate a final score (Figure 1) (2, 3). Many genetic deficiencies are associated with the development of SLE; however, most patients inherit the disease polygenically in conjunction with environmental, hormonal, or co-infectious triggers (1, 4, 5). The immunological defects leading to the development of SLE are a product of two stages: first, systemic autoimmunity that induces serum autoantibodies and, second, immunological events that induce organ damage (6). Immune dysregulation in both innate and adaptive immune systems can impact the activation and progression of SLE (1, 6). Nonetheless, diagnosis and subsequent treatment of SLE have been hampered due to heterogenic clinicopathological presentation of the disease (1, 4, 7).
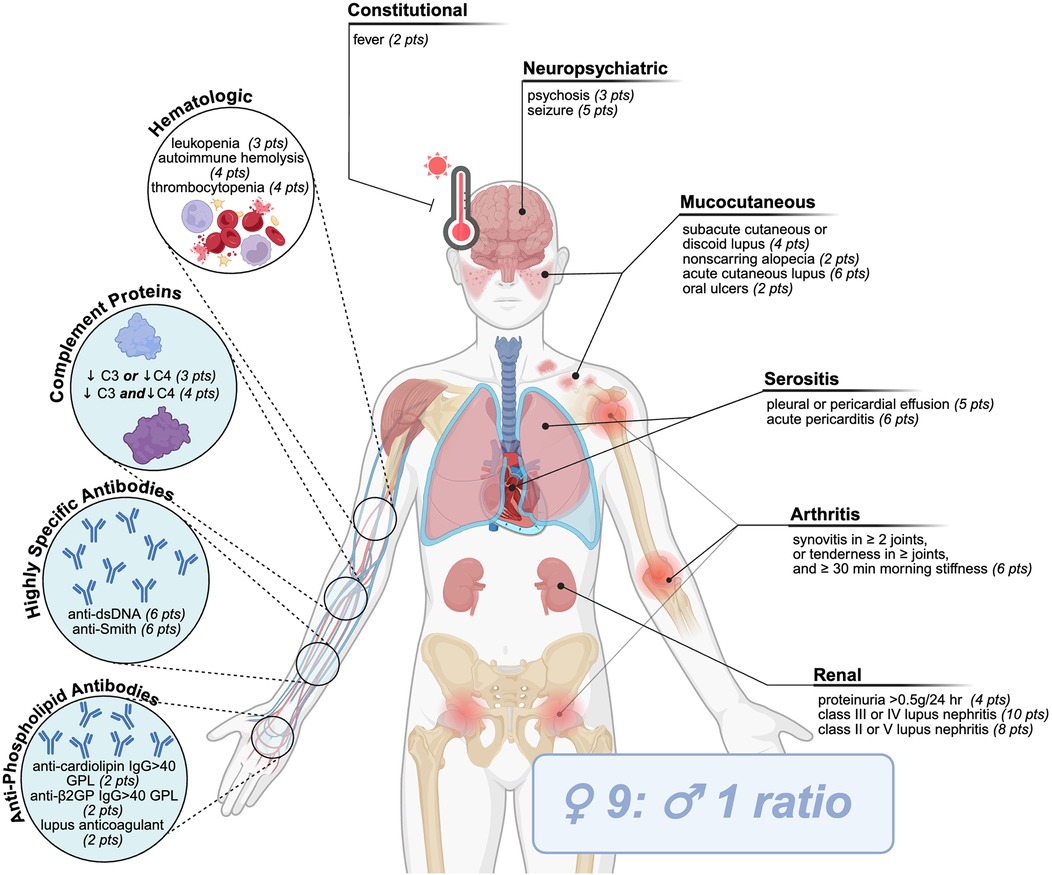
Figure 1. SLE classification criteria. Current criteria (2019 European League Against Rheumatism/American College of Rheumatology Immunologic Domains and Criteria for SLE) requires patients to first test positive for antinuclear antibodies, followed by an assessment of a combination of 10 clinical (uncolored) or immunological (blue circles) symptoms or disorders, each weighted differently to calculate a final score amounting to ≥10 points.
Immune homeostasis is vital for the balance of immune activation and suppression across tissues and organs to prevent damage due to excessive, malfunctioning, and/or self-targeting immune responses (8). The scavenger receptors (SRs) superfamily (Table 1) contributes to maintaining homeostasis through their roles- within cellular transport, nutrient exchange, and waste removal as well as their roles directly within immunity through the identification and presentation of antigens, regulation of inflammation, and adhesion of leukocytes (9, 10). SRs act as cell surface receptors, but can also be found intracellularly or as soluble forms within circulation where they can bind and promote the removal of an array of unwanted ligands—either self or non-self—via endocytosis, phagocytosis, and micropinocytosis (9, 11). Examples of these ligands include apoptotic cells, damage proteins, and heat shock proteins. Beyond scavenging for waste products to maintain homeostasis, SRs simultaneously help conserve the appropriate balance of endogenous molecules, identify damaged antigens, and elicit the proper immune responses (9). Over the past 40 years, numerous SRs have been identified. This superfamily of receptors has been divided into 10 distinct classes (12) based on their nucleotide sequence alignment and protein structure, with each class further divided into subclasses that share specific structural features (Supplementary Table 1) (11, 13, 14). However, as the field is evolving, some newly uncovered SRs remain unclassified while others are classified as noncanonical SRs, or proteins that simply exhibit scavenger receptor activity (12). Several SRs have been implicated in the progression of autoimmune diseases, including SLE, through mechanisms including inflammation, apoptosis, foam cell formation, T cell activation, and the complement pathway. This review will discuss the SRs identified to date that play a role in autoinflammation and autoimmunity, particularly in the context of SLE.
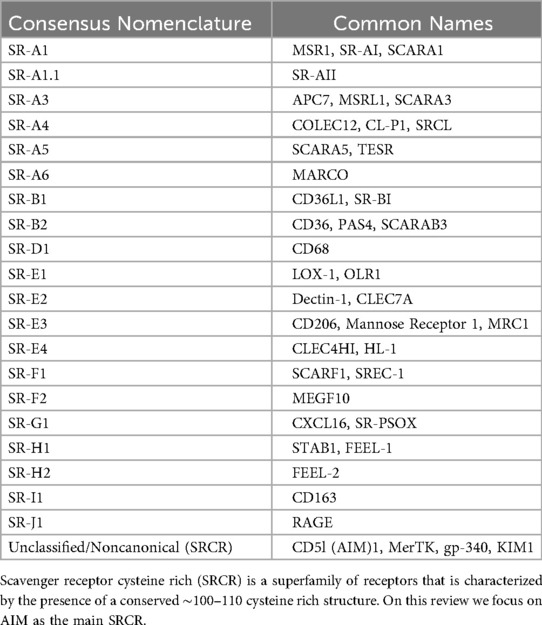
Table 1. Consensus nomenclature and common names of scavenger receptors.
2 Scavenger receptors in clinical hallmarks of SLE inflammation
Nearly twenty years ago an “immunological disease continuum model” was proposed by McGonagle and Dermott (15). The model suggests that noninfectious diseases lie on a spectrum from autoimmune to autoinflammatory, thereby classifying the roles of both the innate and adaptive immune responses in immunological disease, contextualizing noninfectious diseases (Figure 2). Ultimately, the model enhanced our understanding of the pathogenesis and treatment of immune reactivity against self, or as Paul Ehrlich himself called it, “horror autotoxicus” (15), the development of immune reactivity to self and loss of immune tolerance through aberrant dendritic, B, and T cell responses (15, 16). However, placing immunological diseases like SLE along this continuum provides an alternative perspective on the heterogeneity of clinical disease manifestation through the contribution of different inflammatory responses, without assuming that the adaptive immune response is central to disease pathogenesis. For the development of therapies, autoimmune diseases with autoinflammatory components could potentially benefit from innate immune pathway targeting (15, 16). Patients with SLE can experience inflammation in any organ (17). Local signs and symptoms of clinical inflammation were originally described by Celsus in the first century BC as rubor (redness), tumour (swelling), calor (heat), and dolor (pain) (Figure 3). A century later Galen added functio laesa (loss of function) and nearly two thousand years later, Mitchinson added a sixth, fluor (secretion) (18–21). These clinical hallmarks of inflammation may be inherently compromised or chronically unresolved in patients with SLE (18, 20).
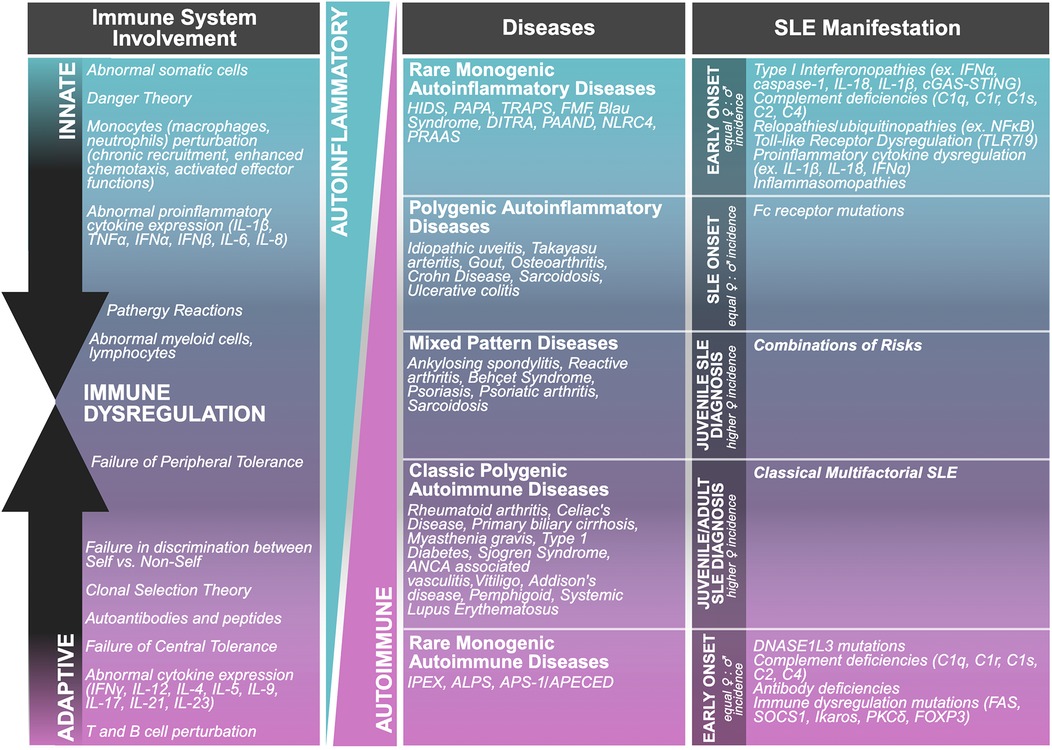
Figure 2. Immunological disease continuum model. This model proposes a spectrum of immune-mediated diseases that ranges from autoinflammatory to autoimmune condition.
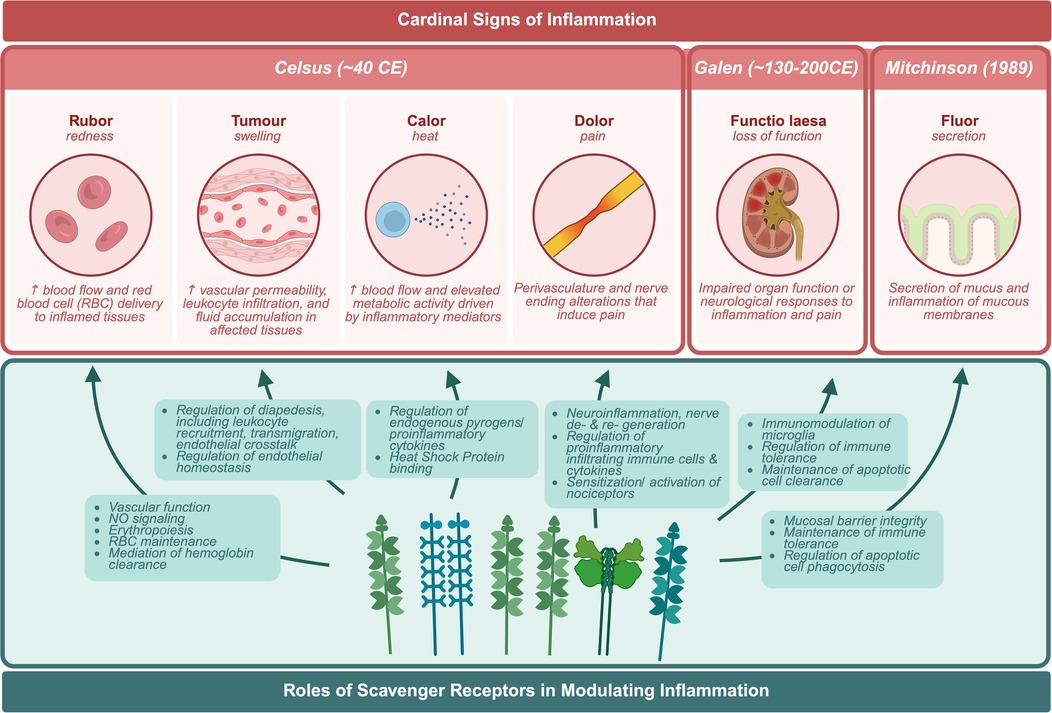
Figure 3. Cardinal signs of inflammation. Biological responses of the body to harmful stimuli in response to scavenger receptors.
2.1 Rubor
Rubor, or redness, is one of the classical signs of inflammation, arising from hyperemia—an increase in blood flow and red blood cell (RBC) delivery to inflamed tissues (22). SRs, initially characterized by their role in binding modified low- and high-density lipoproteins (LDL and HDL), have since been found to play multifaceted roles in vascular biology. They regulate endothelial integrity, vasodilation, red blood cell homeostasis, and immune responses, making them central to the inflammatory and vascular manifestations of autoimmune diseases such as SLE (23–25).
2.1.1 Rubor: vasculature & vasodilation
Scavenger receptor class B member 1 (SR-B1) is essential for lipid and cholesterol metabolism as well as vascular function. Expressed on endothelial cells, vascular smooth muscle cells, monocytes, and macrophages, SR-B1 facilitates HDL binding and mediates endothelium-dependent vasodilation, primarily through the synthesis and activity of nitric oxide (NO) (26–32). NO is a key signaling molecule in vascular homeostasis, responsible for modulating vasodilation, inhibiting leukocyte adhesion, and regulating platelet activity (33). In vivo studies demonstrate that SR-B1 deficiency leads to significant vascular dysfunction, including impaired NO signaling, dyslipidemia, increased platelet aggregation, and development of atherosclerotic lesions (26, 28–31) (34). For example, mice lacking SR-B1 exhibit thrombocytopenia, thrombomegaly, and heightened susceptibility to thrombotic events (34, 35). These symptoms are observed in 20%–40% of SLE patients. Moreover, SLE is associated with elevated systemic NO levels, suggesting that NO dysregulation may serve as a biomarker for disease activity (36, 37). Further, loss of SR-B1 in vivo contributes to coronary artery disease, myocardial infarction, ischemic cardiomyopathy, and heart failure—all of which are significantly more prevalent in SLE patients (5, 24–26, 38–41). Accelerated atherosclerosis remains a leading cause of morbidity and mortality in SLE patients (5, 24, 25, 40, 41). In Takayasu arteritis, a type of autoimmune vasculitis, autoantibodies against SR-B1 interfere with HDL uptake and suppress nitric oxide synthase (NOS) activity, promoting endothelial inflammation and vascular damage (42). Similarly, SR-B1 has been implicated in the pathogenesis of other autoimmune diseases (43) and is strongly linked to cardiovascular disease (CVD), a common comorbidity in SLE (44). SLE patients have a higher risk of developing cardiovascular disease (CVD) (23–25) as well as increased risk for a broad spectrum of cardiovascular complications, including aortic wall inflammation (45), development of atherosclerosis (46), peripheral arterial disease (47), dyslipidemia (48, 49), heart failure (50), angina pectoris (51), and myocardial infarction (51, 52).
2.1.2 Rubor: red blood cell development & maintenance
In addition to its vascular roles, SR-B1 also supports erythropoiesis and RBC maintenance (43). In SLE, RBCs frequently display abnormal morphology or size variability, which may impair oxygen delivery and contribute to common symptoms such as chronic fatigue, anemia, and cognitive dysfunction (53, 54). Autoimmune hemolytic anemia is another hematologic manifestation of SLE, leading to increased RBC destruction and free hemoglobin in circulation (55). SR-I1 or commonly known as CD163, is a hemoglobin-scavenging receptor primarily expressed on M2-polarized macrophages. It binds to haptoglobin–hemoglobin complexes to mediate hemoglobin clearance during intravascular hemolysis (55–58). In lupus nephritis (LN), kidney CD163+ macrophages infiltrate glomeruli, contributing to local inflammation and tissue damage (56, 59). Elevated levels of soluble SR-I1 are consistently found in the serum of patients with SLE, particularly those with macrophage activation syndrome, autoimmune hemolytic anemia, or immune thrombocytopenia (57, 60). Inflammatory conditions, including SLE, are characterized by increased expression of SR-I1, reflecting ongoing macrophage activation (55–57, 61).
Macrophage activation syndrome (MAS) is one of the most severe hyperinflammatory complications of lupus, characterized by uncontrolled macrophage and T cell activation, cytokine storm, and hemophagocytosis. While vascular and hematologic manifestations are well-established in SLE, defective SR pathways contributing to MAS susceptibility directly are limited (62, 63). In SLE, defective clearance of apoptotic debris [and release of nuclear danger/damage-associated molecular patterns (DAMPs) such as HMGB1] sustains TLR7/9 signaling and type I interferon production, which foster cytotoxic T-cell activation and excessive IFN- γ response—features characteristic of MAS (64–66). Under these inflammatory conditions, regulatory macrophage circuits such as the CD163-heme oxygenase-1 (HO-1) pathway, normally critical for heme detoxification and resolution of inflammation, may become functionally exhausted. Failure of this axis limits macrophage antioxidant and anti-inflammatory capacity and favors uncontrolled activation and hemophagocytosis, thereby lowering the threshold for MAS in SLE patients (67, 68). Serum soluble SR-I1 is consistently elevated in active MAS even under IL-6 blockade and closely reflects disease activity and the clinical relevance of SR dependent macrophage dysregulation (69, 70). Additional insights into SRs dysfunction and MAS pathogenesis emerge from related hyperinflammatory disorders. For example, in systemic juvenile idiopathic arthritis (sJIA), a disease with high MAS propensity, soluble SR-I1 correlates with MAS activity and TRIM8 upregulation in monocytes/macrophages augmenting IFN- γ responsiveness, providing a molecular mechanism for macrophage hyperactivation (70, 71).
Despite observed elevations in SR-I1, its precise role in SLE pathophysiology remains unclear. Two opposing hypotheses currently exist: the first suggests that SR-I1 is abnormally elevated, causing excessive phagocytosis of haptoglobin–hemoglobin complexes and downstream accumulation of iron, oxidative stress, and ferroptosis; the second proposes that SR-I1 is elevated as an adaptive response to prevent further tissue damage caused by ruptured red blood cells (55). Notably, soluble SR-I1 levels correlate with disease activity in rheumatoid arthritis (72) and serve as a biomarker for active renal involvement in SLE, including in the urine of patients with LN (73–75). Together, these findings underscore the dual vascular and hematologic roles of SRs such as SR-B1 and SR-I1 in the manifestation of rubor and broader SLE pathology. Their dysregulation contributes not only to visible signs of inflammation but also to systemic complications, making them potential biomarkers and therapeutic targets.
2.2 Calor
Calor, or heat, is a hallmark of inflammation and reflects increased blood flow and elevated metabolic activity driven by inflammatory mediators. During infection, fever (hyperthermia) is a protective physiological response that enhances immune function, promotes leukocyte trafficking, and inhibits pathogen replication (76–78). Historically, controlled elevation of body temperature—therapeutic hyperthermia—has even been employed to treat infectious disease by enhancing organ perfusion and activating immune pathways (76–78). However, excessive or sustained fever can lead to tissue damage and adverse outcomes, such as heat stroke (76).
In autoimmune diseases such as SLE, fever is a common manifestation—even in the absence of infection. Recurrent, unexplained fever is often a presenting symptom and is considered a clinical clue in early diagnosis (79–81). Studies report fever in 36%–86% of SLE patients, although the prevalence has declined due to routine use of nonsteroidal anti-inflammatory drugs (NSAIDs) (79, 81–86). Distinguishing SLE-related fever from infection remains clinically challenging, particularly in patients receiving immunosuppressive therapies that increase infection risk or due to inherent disease-associated immune perturbations (87, 88). In cases of Fever of Unknown Origin (FUO), infection must be rigorously ruled out before attributing symptoms to SLE (79). While infection is the most common cause of FUO, approximately 5% of cases are ultimately diagnosed as autoimmune conditions, including SLE and autoimmune thyroiditis (89, 90). Although SRs have not been directly linked to the development of fever, they influence proinflammatory signaling pathways, such as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and JAK/STAT, that regulate the febrile response (383).
2.2.1 Calor: endogenous pyrogens/cytokines
Fever is largely mediated by endogenous pyrogens—proinflammatory cytokines that are released by immune cells in response to infection, tissue damage, or autoimmune activity. Key endogenous pyrogens include tumor necrosis factor-α (TNF-α), interleukins (IL-1β, IL-6, IL-8), and interferons (IFN-β, IFN-γ), among others (91, 92). These cytokines circulate systemically and act on the hypothalamus to elevate body temperature. Many of these same cytokines are found at abnormally high levels in SLE patients, and their activity contributes to immune dysregulation, tissue damage, and systemic inflammation (93–100).
Due to this, they are also considered potential targets for SLE treatments (101). Scavenger receptor class E member 1 (SR-E1), also known as LOX-1, is a receptor for oxidized low-density lipoproteins (oxLDL) that is primarily expressed on vascular endothelial cells (102, 103), but also on vascular smooth muscle and lymphoid cells (104). SR-E1 mediates oxLDL endocytosis and promotes atherogenesis (104, 105) and has been shown to be a key player in the development of atherosclerosis, myocardial ischemia, hypertension, and inflammation (105, 106). SR-E1 deficiency in vivo protects mice from developing atherosclerosis (102), whereas SR-E1 overexpression promotes it (107). In SLE patients, SR-E1 expression is elevated even in early disease onset and low disease activity and correlates with high-sensitivity C-reactive protein (hsCRP), proinflammatory HDL, and oxLDL (108). Moreover, increased SR-E1 in SLE is associated with elevated IL-8 and reduced IFN-γ levels, while other cytokines such as IL-6, IL-10, and TNF-α remain unchanged (105). Interestingly, proinflammatory cytokines (e.g., IL-1β, TNF-α) can themselves induce SR-E1 expression, potentially creating a self-perpetuating inflammatory loop (108–110). Perturbing this feedback loop is a possible option to reduce inflammation-driven CVD in SLE (108, 111, 112).
2.2.2 Calor: prostaglandins & thermoregulatory neurons
Fever can also be triggered by non-immune cells. Pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α stimulate cyclooxygenase-2 (COX-2), which catalyzes the production of prostaglandin E2 (PGE2). PGE2, in turn, binds to PGE2 receptors stimulating the part of the brain where thermoregulatory neurons modulate body temperature and induce fever (113). Elevated PGE2 has been detected in the cerebrospinal fluid (CSF) of patients with neuropsychiatric SLE (NPSLE), along with increased levels of IL-6, IgG, and autoantibodies against calf thymus antigens (114). Among these, CSF IL-6 has shown the strongest correlation with NPSLE severity and may serve as a potential biomarker (115, 116). In pristane-induced lupus mouse models, PGE2 mediates the production of proinflammatory cytokines, such as IL-6, IL-10, and IFN-γ, and NO (117). While no direct link between PGE2 and SRs in SLE has yet been confirmed, emerging evidence suggests functional intersections. For example, SR-B2–mediated microglial phagocytosis of amyloid-β is regulated by PGE2 receptor signaling in in vivo Alzheimer's disease models (118). Additionally, celecoxib, a selective COX-2 inhibitor used in treating inflammation in SLE, has been shown to upregulate SR-B2 and downregulate SR-E1 in macrophages—indicating that prostaglandin pathways can modulate SR expression and potentially influence inflammatory outcomes (119).
2.2.3 Calor: heat shock proteins
Heat shock proteins (HSPs), molecular chaperones released from cells undergoing stress, help maintain protein homeostasis (120). Incubating murine skin explants at fever-range hyperthermia (40°C) upregulates HSP70 expression and leads to dendritic cell (DC) migration (121). At similar temperatures, expression of HSP70 is induced in lymphocytes (122, 123). HSPs can help immune cells withstand and react to inflammatory environments (121).
HSPs are typically expressed intracellularly; however, under conditions of cellular stress (e.g., cell damage, necrosis), HSPs may be released extracellularly, where they act as DAMPs. Extracellular HSPs can prime antigen-presenting cells, promote cytokine production (e.g., TNF-α, IL-6), and contribute to the generation of autoantibodies (124–132). This immune activation correlates with HSP levels: more cell damage triggers higher HSP expression and a more intense immune response (133–135). Elevated HSP70 levels, HSP gene polymorphisms, and anti-HSP autoantibodies have all been associated with SLE pathogenesis (136–141).
Several SRs have been identified as HSP-binding receptors, including SR-E1, SR-F1, SR-A, SR-H1, and SR-L1 (141–145). SR-E1 and SR-F1 bind strongly to HSP70, HSP90, Grp94, Hsp110, and Grp170 (146–149). SR-B2 deficiency in LDL receptor–deficient mice exposed to hyperthermic stress leads to HSP70 overexpression and enhanced atherosclerosis, highlighting the interplay between SRs, heat shock responses, and vascular inflammation (150–152). In lupus-prone mice, HSP70-based DNA vaccines have shown promise in suppressing anti-dsDNA antibody production, reducing proinflammatory responses, promoting tolerogenic immune responses, and prolonging survival (153). Other therapeutic approaches, such as epitope-based immunization with HSP70-derived peptides, have demonstrated TReg activation without inducing systemic immunosuppression (154). SLE is associated with increased auto-antibody productions of other HSP-like proteins and HSPs, namely grp94 and calreticulin (155, 156), which has been found to play a role in during physiological stresses like fever (157) and in the pathogenesis of SLE (144, 158). SR-L1, an HSP receptor, is particularly relevant in this context. Membrane-bound SR-L1 mediates antigen presentation, cytokine secretion, and T helper cell priming (126, 159). SR-L1 plays an immuno-protective role by suppressing the expression of inflammatory mediators (MCP-1/CCL2, TNF-α, and MMP-9) (160, 161). To suppress inflammation, SR-L1 sheds its ectodomain, generating soluble SR-L1 (162), which can be detected in the serum of patients with SLE and Rheumatoid Arthritis (RA) (163). Bruton's tyrosine kinase (Btk) also plays a key role by phosphorylating calreticulin on apoptotic cells (ACs), enabling membrane-bound SR-L1 to mediate the clearance of C1q-opsonized ACs. In the absence of Btk, SR-L1 cannot recognize calreticulin, resulting in the accumulation of apoptotic cell debris (164). Together, these findings suggest a multifaceted role for SRs in regulating immune responses to thermal and inflammatory stress. By binding HSPs and modulating cytokine production, SRs represent promising therapeutic targets and potential biomarkers for disease severity in SLE. Similarly, many HSPs themselves have been identified as targets for the treatment of autoimmunity, such as in RA, diabetes, multiple sclerosis (MS), and SLE (165, 166).
2.3 Dolor
In inflammation, dolor represents pain due to changes associated with perivasculature and nerve endings. Pain, especially chronic pain, is a hallmark of SLE and is often one of the first reported symptoms (167). Data shows that 85% of SLE patients report joint pain (168, 169) and 32%–66% report headaches (169). Up to 80% of SLE patients experiencing pain, fatigue, or joint pain rate these symptoms as moderate or severe (170). Pain in SLE can be both inflammatory and non-inflammatory in nature including musculoskeletal [arthritis, myositis, avascular necrosis, fracture, osteoarthritis (OA)], fibromyalgia), neuropsychological (headache, small fiber neuropathy), serositis (pericarditis, pleuritis, peritonitis), immunological disturbance, drug side effects, etc. as reviewed by Pisetsky et al. (171).
2.3.1 Dolor: neuropathic pain
SR-L1 (known as LRP-1 or CD91), plays a major role in neuroinflammation, nerve de- and re-generation, and neuropathic pain (172, 173). SR-L1 deficiency in Schwann cells is linked to mechanical allodynia—pain from normally non-painful stimuli such as light touch—and impaired motor function, both of which contribute to peripheral nerve injury and chronic pain (174, 175). SR-L1 deletion on macrophages induces an increase in NFκB pathway activation and inflammation (176). SR-L1 downregulates proinflammatory cytokines (IL-1β, TNF-α, and IL-6) released due to NFκB activation. Perturbed SR-L1 across several cell types results in increased secretion (172).
Similarly, SR-L1 has a role in neuronal cell survival by modulating c-Jun N-terminal kinase (JNK)-mediated apoptosis (172). Perturbing SR-L1 leads to JNK pathway activation and the release of several proinflammatory cytokines (e.g., TNF-α and IL-1ß) and chemokines such as chemokine (C-C motif) ligand (CCL) 2 (CCL2), CCL3, or CCL4 (177–179). Consequently, SR-L1 downregulation promotes synaptic and neuronal loss, leading to cognitive impairment (172, 180–184). In MS, autoantibodies against SR-L1 have been found to inhibit function and contribute to poor clinical outcomes (185). In response to inflammatory triggers, membrane-bound SR-L1 has been found to be anti-inflammatory (178, 186–189); however, increased levels of soluble SR-L1 correlate with inflammation in patients with SLE and RA (163). SR-L1, through its ligands and its biologically active soluble form, has been explored as a potential therapeutic target for neuropathic pain, based on studies of axonal injury and Alzheimer's disease (172, 177, 183). Although auto-antibodies to SR-L1 have not been attributed to SLE, auto-antibodies to scavenger receptor class L member 2 (SR-L2) have been found to be a major player in systemic autoimmune diseases (190). In a cohort of 147 patients, anti-LRP2 autoantibodies were detected in 87% with RA, 40% with SLE, 35% with systemic sclerosis, 15% with osteoarthritis, and 3% with Behçet's disease (190).
Beyond their role in systemic autoimmunity, immune cells such as macrophages are increasingly recognized for their involvement in the pathophysiology of chronic pain (191–193). CD68 (SR-D1) is commonly used as a biomarker to quantify inflammatory cell and macrophage infiltration (194). Often associated with RA (195), Morton's neuroma is an entrapment neuropathy characterized by compression of a plantar digital nerve in the foot, leading to neuropathic symptoms such as burning, paroxysmal pain, and paresthesia. In nerve samples from patients with Morton's neuroma, increased intraneural CD68+ macrophages have been positively correlated with burning pain, while higher expression of SR-A6 has been linked to paroxysmal pain, as measured by the Neuropathic Pain Symptom Inventory (196). Similarly, intervertebral disc tissue from patients with lower back pain shows elevated infiltration of TNF-α+ and CD68+ cells (197). Conversely, in post-amputation patients experiencing phantom pain, nerve biopsies revealed a lower presence of CD68+ macrophages. This finding led researchers to propose a potential protective role for these cells against the development of chronic pain in certain contexts (191). These observations suggest that the role of infiltrating CD68+ macrophages in pain may vary depending on the condition and anatomical site. For example, pain in chronic pancreatitis—whether alcoholic, biliary, hereditary, or autoimmune in origin—is typically classified as inflammatory rather than neuropathic, though these categories are not mutually exclusive. Notably, in cases of chronic pancreatitis, CD68+ macrophage levels did not correlate with pain severity (194).
2.3.2 Dolor: inflammatory pain
Building on the role of macrophage infiltration in various pain states, joint inflammation in diseases such as OA, RA, and SLE further illustrates the link between immune cell activity and pain. In these conditions, increased infiltration of immune cells and the production of pro-inflammatory mediators (often triggered by IgG immune complexes) sensitize and activate sensory nociceptors innervating joint tissues (198–201). Several SRs associated with macrophage activation have been implicated in inflammatory joint pain. For instance, elevated expression of CD163 or SF-I1 and the pro-inflammatory cytokine TNF-α correlates with higher resting pain scores in patients with hip OA (202). Similarly, IL-1β, another key pro-inflammatory cytokine, contributes to pain through its role in prostaglandin production (201, 203).
In RA murine models, macrophages expressing SR-D1, SR-I1, and SR-E3 are enriched in inflamed joints; however, a highly pathogenic and pro-inflammatory subset of macrophages co-expressing SR-I1 and SR-E3 has been identified in RA synovial tissue. These SR-E3+ SR-I1+ macrophages spontaneously secrete pro-inflammatory mediators including IL-6, IL-8, IL-1β, and TNF-α, and exhibit strong co-expression of CD40, a costimulatory activation marker known to drive chronic inflammation in RA (204–206). Remarkably, inhibition of CD40-TRAF6 signaling reversed the secretion of these mediators, suggesting that targeting this pathway may offer therapeutic benefit (207). Even more compelling is the observation that these pathogenic macrophages were present before the clinical onset of RA symptoms (207).
Extending beyond the joints, macrophage subsets expressing SR-E3 and SR-I1 have also been implicated in chronic pain development in the dorsal root ganglia (DRG). In mouse models genetically predisposed to chronic pain following peripheral injury, DRG-resident macrophages expressing SR-I1 alone (SR-E3⁻ SR-I1+) or both SR-E3 and SR-I1 (SR-E3+ SR-I1+) were found to promote chronic pain. Targeted depletion of these macrophage subsets effectively prevented the development of injury-induced chronic pain, highlighting their critical role in pain pathogenesis (208).
Relatively few SRs have been directly linked to pain in SLE or to pain mechanisms more broadly. However, further investigation into how SRs contribute to immune response pathways in SLE, particularly those leading to neuropathic and inflammatory pain, may reveal additional SRs involved in early immune dysregulation and the onset of autoimmunity. Uncovering these connections could offer novel insights into both SLE pathogenesis and pain modulation.
2.4 Tumor
The classical hallmark of tumor in inflammation, or swelling, is primarily caused by increased vascular permeability, leukocyte infiltration, and fluid accumulation in affected tissues (22). A key event in this process is diapedesis, or trans-endothelial migration, in which leukocytes exit the bloodstream and traverse the vascular and lymphatic endothelial barriers to reach sites of inflammation (209). This involves a tightly regulated sequence: leukocyte recruitment, adhesion to ECs, and eventual transmigration into surrounding tissues (210). Persistent vascular inflammation is a hallmark of several autoimmune conditions, including SLE (211, 212), where dysregulated immune cell infiltration exacerbates tissue injury and chronic inflammation.
2.4.1 Tumor: leukocyte recruitment & transmigration
Effective leukocyte recruitment is essential for host defense and tissue repair; however, in autoimmune diseases such as SLE, excessive or uncontrolled leukocyte accumulation leads to chronic inflammation and tissue damage (210).
Myeloid cells, particularly neutrophils and monocytes, play a central role in initiating and sustaining vascular inflammation in SLE (212–215). Neutrophils contribute to inflammation not only through phagocytosis but also by producing reactive oxygen species (ROS), releasing neutrophil extracellular traps (NETs), and modulating adaptive immunity via crosstalk with dendritic cells, macrophages, and lymphocytes (211, 216, 217). SRs, particularly SR-A, have been implicated in neutrophil activation, including via mitogen-activated protein kinase (MAPK) signaling pathways, leading to increased production of proinflammatory cytokines (i.e., IL-6, TNF-α) and NET formation (218).
SLE is also characterized by elevated Type I interferon levels, which drive monocyte chemotaxis through increased expression of MCP-1 and MIP-1α. This promotes the upregulation of SRs such as SR-A and SR-B2, further enhancing monocyte and neutrophil activation (219–223). These receptors facilitate the uptake of modified low-density lipoproteins (LDLs), linking innate immune activation with lipid metabolism and vascular inflammation. For example, SR-B2 supports macrophage spreading and migration (224), inflammasome activation (225), and has been implicated in lesional macrophage proliferation (223, 226).
2.4.2 Tumor: diapedesis & SR-mediated endothelial crosstalk
The transmigration of leukocytes across the endothelium is a tightly regulated, multi-step process involving changes in both leukocytes and endothelial cells. Inflammatory cytokines upregulate endothelial adhesion molecules, while chemokines activate leukocyte integrins to promote firm adhesion and extravasation (210). Leukocytes then alter their morphology to traverse the endothelial barrier and surrounding pericyte layer, ultimately infiltrating the inflamed interstitial tissue (210).
Among scavenger receptors, SR-G1 is unique in exhibiting both receptor and chemokine-like functions. In its membrane-bound form, SR-G1 binds oxLDLs and phosphatidylserine (PS), contributing to phagocytosis and waste clearance. In its soluble form, SR-G1 acts as a chemoattractant through its interaction with the CXCR6 receptor on bone marrow plasma cells and T cells (227). Soluble SR-G1 is particularly relevant to SLE: elevated serum levels correlate with disease severity, organ involvement, and prognosis in both adult (228) and juvenile SLE (229). Levels decrease with effective treatment, supporting its potential utility as a biomarker for SLE progression and therapeutic response (228).
2.4.3 Tumor: SRs in endothelial dysfunction
A major complication in SLE is the premature development of atherosclerotic cardiovascular disease (ASCVD). Early plaque formation is marked by endothelial dysfunction and the infiltration of pro-inflammatory leukocytes beneath the endothelial monolayer (226, 230). Monocyte-derived macrophages proliferate in response to hematopoietic growth factors such as macrophage colony-stimulating factor (M-CSF) and accumulate cholesteryl esters, forming foam cells (231–234). M-CSF also enhances SRs expression, particularly SR-A and SR-B2, promoting oxLDL uptake and foam cell formation (233, 235).
SR-B2 plays a central role in this process. Suppression of SR-B2 in murine models reduces aortic lesion size, suggesting that its function is non-redundant and critical in lesion development (236, 237). Importantly, SLE patient blood samples contain higher levels of oxLDL-containing immune complexes (238), which upregulate SR-B2 expression in healthy cells exposed to this SLE plasma (235). This mechanism likely contributes to accelerated foam cell formation and atherosclerosis in SLE, positioning SR-B2 as a potential therapeutic target (239, 240).
Additionally, Mer tyrosine kinase (MerTK), another SR, is essential for the clearance of apoptotic cells within plaques. Loss of MerTK impairs efferocytosis, promoting the formation of a lipid-rich necrotic core and driving plaque instability (241) as well as T cell–mediated β cell autoimmunity (242). Elevated levels of soluble MerTK in SLE patients correlate with disease activity (243), complement depletion, and anti-dsDNA titers, suggesting a role in both cardiovascular and autoimmune pathology (243, 244).
Another key player is SR-E1 (LOX-1), which mediates uptake of modified LDLs and contributes to foam cell formation. OxLDL exposure reduces DNA methylation of the SR-E1 promoter, creating a positive feedback loop that amplifies SR-E1 expression and promotes plaque progression (245). In SLE patients with ASCVD, serum levels of soluble SR-E1 correlate with inflammatory biomarkers such as high-sensitivity C-reactive protein (hsCRP), proinflammatory HDL, and oxLDL (108). Furthermore, higher sSR-E1 levels are associated with earlier age of SLE diagnosis (108), suggesting that SR-E1 could serve both as a biomarker and a therapeutic target in SLE-related vascular disease.
2.4.4 Tumor: endothelial cell scavenger receptors and tissue crosstalk
In SLE, the endothelium has been shown to be in a dysregulated state, even during low disease activity (246). SRs are expressed on endothelial cells, where they regulate vascular permeability, immune cell trafficking, and antigen presentation. These scavenger endothelial cells (SECs) are especially prominent in the liver, where hepatic sinusoidal endothelial cells (HSECs) act as filters for bloodborne antigens and macromolecules, given the liver's extensive exposure to gut-derived microbial products (247–251). HSECs express a range of SRs including SR-H1 (252–254), SR-H2 (255), SR-B2 (256), SR-B1 (257), SR-E3 (258, 259), and SR-F1 (250). SR-H1, for example, is induced by proinflammatory stimuli (260) and, in vitro, regulates lymphocyte trafficking to inflamed tissues (261). SR-H1 expression on monocytes is considered a predictive biomarker for increased cardiovascular-related disease risk (13). Both SR-H1 and SR-H2 bind to a variety of ligands including acLDL, advanced glycation end-products (AGEs), and both gram-positive and gram-negative bacteria (13, 262). SR-H1 also facilitates T and B cell trans-endothelial migration via interactions with adhesion molecules like ICAM-1 and VAP-1 (13, 253, 263–266).
Interestingly, SR-H1 promotes antigen presentation in a tolerogenic context by cross-presenting exogenous antigens on MHC-I and MHC-II molecules with high expression of inhibitory ligands (e.g., PD-L1), HSECs help induce regulatory T cells rather than proinflammatory responses (267–271). Inhibition of SR-H1 and SR-H2 has been shown to induce an anti-inflammatory plasma proteome and reduce monocyte-driven atherogenesis, pointing to their therapeutic potential (272).
SR-F1 (SCARF1, SREC-1), initially identified in human umbilical vein endothelial cells (264), is also upregulated in chronic liver diseases such as primary sclerosing cholangitis (PSC), primary biliary cholangitis (PBC), ALD, and non-alcoholic steatohepatitis (NASH) (250). SR-F1 activation by TNF-α or LPS enhances CD4+ T cell recruitment, working in concert with VCAM-1 to facilitate immune cell adhesion during liver inflammation (250). Other SRs, including SR-E1 (273, 274), SR-H1 (253, 254, 275), SR-H2 (255) also mediate leukocyte adhesion to endothelial cells, contributing to inflammatory crosstalk between the vasculature and the immune system.
2.4.5 Tumor: SRs in regulating neutrophil NETS and endothelial injury
Neutrophils recruited to the endothelium are activated by immune complexes (276) and perform several effector functions including phagocytosis, degranulation, and formation of neutrophil extracellular traps (NETs) (276, 277). While NETs are antimicrobial, excessive NET formation (NETosis) and elevated levels of circulating NETs is pathogenic in SLE, where it contributes to endothelial injury, immune complex formation, and the development of autoantibodies (277).
Increased NETs have been observed in patients with LN and in MRL-lpr mice (278). SR-J1 (RAGE) is an SR expressed on endothelial cells that plays a role in triggering NETosis (279) and, together with clathrin, mediates the uptake of NETs. However, endothelial phagocytic capacity is limited, and NET overload disrupts vascular integrity. Specifically, NET-associated elastase degrades VE-cadherin at intercellular junctions, increasing vascular permeability and leakage (278). This mechanism links NETosis and SR-mediated uptake to vascular damage and albumin extravasation, ultimately highlighting the role of NET clearance in preserving endothelial barrier function in SLE.
2.5 Functio laesa
Functio laesa, Latin for “loss of function,” can refer to either impaired organ function (280) or neurological responses to inflammation and pain (281). In the context of systemic inflammation, such as in SLE, functio laesa often reflects multi-organ dysfunction or outright failure (282). SLE can affect nearly every organ system, manifesting in complications such as lupus nephritis (kidneys), neuropsychiatric disease (central nervous system), cutaneous lupus (skin), lymphadenopathy (lymphatic system), and various cardiovascular conditions (283, 284). Organ damage occurs in at least 50% of SLE patients (285), though some studies report even higher rates. A Taiwanese cohort found that over 80% of SLE patients developed organ damage within 6 months of diagnosis (286), while U.S. studies report damage in 33%–50% of patients within the first five years (287). The most frequently affected systems vary by region: ocular, neuropsychiatric, and cardiovascular systems are common in the U.S. and Germany (287, 288), while renal, neuropsychiatric, pulmonary, gastrointestinal, and cutaneous systems are prominent in Taiwanese patients (286, 289).
2.5.1 Functio laesa: neuropsychiatric
Neuropsychiatric symptoms are among the most common manifestations of SLE, affecting 80%–90% of patients worldwide (290). These symptoms include cognitive impairment, motor dysfunction, sleep disruption, fatigue, mood disorders, and behavioral changes. Cognitive dysfunction often presents early—prior to the development of dementia or confusion—whereas symptoms like depression or headaches are more difficult to diagnose and frequently missed in early screenings (290).
Up to 40% of neuropsychiatric SLE (NP-SLE) symptoms arise before or at the time of SLE diagnosis, and 60% typically develop within a year (291, 292). Current research is focused on whether NP symptoms are driven by central nervous system (CNS) inflammation in chronic SLE, CNS dysfunction and damage, or treatments and medications (290, 293). While lesions are not always evident in NP-SLE patients, functional abnormalities have been identified, including cerebral hypoperfusion (292, 294–298), metabolic deficiencies (292, 299–302), and—most commonly—progressive neuronal atrophy (292, 298, 300, 303–310).
In SLE-prone mouse models, neuropsychiatric and behavioral symptoms precede systemic autoimmunity, immune cell infiltration, or vascular damage (311). Interestingly, microglia in SLE-prone mice exhibit neurodegenerative disease-associated signatures prior to systemic SLE manifestations. Microglia from SLE mouse models show upregulation of genes involved in SR activity and downregulation of genes involved in inflammation and chemotaxis, suggesting that microglia in SLE-prone mice may not be able to regulate inflammation appropriately (311). Makinde et al. found SRs, SR-G1 (CXCL16) and LGALS3BP, are upregulated in lupus-prone mouse microglia. In healthy conditions, microglial cells maintain tissue homeostasis in the brain by sensing changes in the environment and responding appropriately. Anti-inflammatory microglia release immunomodulatory factors to support tissue repair in the brain. In disease contexts, anti-inflammatory microglia may also reduce immune response and promote cell invasion and tumorigenesis (312). Outside of SLE, microglial SR-G1 has been proposed as a therapeutic target to reduce neuroinflammation (312), and LGALS3BP may be a therapeutic target for its roles in angiogenesis and tumor progression (313). In SLE, serum SR-G1 and platelet LGALS3BP levels also correlate with LN severity (229, 314), pointing to systemic involvement beyond the CNS.
2.5.2 Functio laesa: tissue homeostasis & clearance of apoptotic cells
Dysregulation of both innate and adaptive immune responses contribute to SLE (315). Adaptive immunity establishes a highly specific immunological memory to pathogens. B and T cells of the adaptive immune system are responsible for ensuring a specific, controlled spatiotemporal response to limit or prevent excessive tissue damage (316). The breakdown in immune tolerance is a hallmark of SLE development (315), as well as many other autoimmune disorders (316). Tissue homeostasis depends on the efficient clearance of apoptotic cells (ACs) by phagocytes in an immunologically silent process that prevents inflammation (317). Dying cells release “find-me” signals that attract phagocytes, while surface markers like phosphatidylserine (PS) serve as “eat-me” signals (317). Failure to clear ACs leads to their secondary necrosis, triggering inflammation, disruption of self-tolerance, and immune activation—a major contributor to SLE pathogenesis (318–321).
Elevated levels of uncleared ACs in SLE patients support the notion that defective clearance contributes to disease (319, 320). Scavenger receptors are critical in mediating efferocytosis, including SR-A, SR-F, and SR-H families (11, 262, 264, 322–325). SR-A1 and MARCO (SR-A6) are key receptors in this process. SR-A1 expressed on thymic macrophages was first shown to mediate apoptotic thymocyte clearance, and its blockade reduces phagocytosis by ∼50% (326). Mice lacking SR-A1 and MARCO develop higher levels of autoantibodies and lupus-like disease due to impaired clearance of apoptotic debris by marginal zone macrophages in the spleen (323). Additionally, SLE patients and murine models have been found to spontaneously produce autoantibodies against these SRs, impairing the apoptotic cell removal ability of these two SRs (323, 327).
SR-F1 (SCARF1) also plays a role in apoptotic cell clearance. Mice lacking SCARF1 develop lupus-like symptoms, including nephritis and dermatitis, driven by defective phagocytosis of PS- and C1q-labeled apoptotic cells (328). SCARF1 is a non-redundant efferocytosis receptor expressed on BDCA1+ dendritic cells, where its engagement promotes anti-inflammatory IL-10 production via STAT1/STAT3 signaling (329). Interestingly, while SCARF1 expression is not reduced in SLE patients, they exhibit anti-SCARF1 autoantibodies, which correlate with impaired efferocytosis (329). Additional work is needed to understand the function and action of the anti-SCARF1 autoantibodies, and whether these autoantibodies can be used as biomarkers for SLE.
2.5.3 Functio laesa: systemic organ damage
SLE-associated systemic inflammation leads to damage in organs such as the heart, lungs, and liver, particularly in patients with hematological symptoms like leukopenia or thrombocytopenia due to the circulation and accumulation of auto-antibodies and inflammatory mediators throughout the body (330). Scavenger receptors are again implicated—SR-A expression on peripheral blood mononuclear cells (PBMCs) correlates with systemic inflammatory response syndrome (SIRS) and multiple organ dysfunction syndrome (MODS), both indicators of poor survival outcomes (331). Further, mechanistic studies further demonstrate that MSR1 (SR-A) can physically synergize with Toll-like receptor 4 to amplify NF-kB signaling and pro-inflammatory cytokine release, providing a potential molecular bridge to the cytokine storm characteristic of MAS leading to systemic organ damage (332, 333). Similarly, soluble SR-I1 (CD163) levels correlate with MODS severity and prognosis in sepsis (334). Increased SR-I1 expression is found on macrophages in Macrophage Activation Syndrome patients who later develop SLE (65) and on PBMCs of prior diagnosed SLE patients (335). In LN and glomerulonephritis (GN), CD163+ macrophage infiltration is positively associated with disease severity and renal function decline (336, 337).
Effective SR-mediated clearance of dying cells is protective in acute injury, limiting the release of intracellular contents and preventing secondary necrosis and inflammation (338, 339). Conversely, impaired clearance promotes chronic inflammation and autoimmunity, as seen in SLE (340). The soluble scavenger receptor CD5l (also known as AIM, apoptosis inhibitor of macrophage) binds cellular debris and promotes phagocytic internalization of dead cells. AIM is primarily expressed by macrophages in the liver, lymphoid, and inflamed tissues (338, 341, 342). Elevated AIM levels are observed in both SLE patients (343) and lupus prone mice (344), correlating with SLE disease activity (343). AIM is most well-known for its role in Acute Kidney Injury (AKI) and chronic liver injury in which the accumulation of circulating AIM correlates with the progression of organ damage (345, 346). Two other SRs, SR-B2 and kidney injury molecule-1 (KIM-1), can recognize AIM when bound to cellular debris (338, 347). KIM-1, a scavenger receptor not typically expressed in healthy kidneys, is markedly elevated in injured renal tissue (348, 349). Elevated urinary KIM-1 levels in SLE patients suggest its utility as a non-invasive biomarker for renal involvement and progression of LN (350).
2.6 Fluor
Fluor refers to the secretion of mucus and inflammation of mucous membranes (19). Patients with autoimmune diseases, including SLE, exhibit an increased risk of developing chronic sinusitis (351, 352). Interestingly, chronic sinusitis may also serve as an early indicator of autoimmune disease onset (353). This relationship is thought to arise either from intrinsic immune dysregulation—leading to impaired mucosal defense and tolerance breakdown—or from extrinsic factors such as medications or infections that trigger or exacerbate autoimmunity (354).
One key player in mucosal immunity is glycoprotein-340 (gp-340), a member of the SR family (355, 356). Gp-340 is highly expressed in the sinonasal, ocular, and pulmonary mucosa, where it contributes to innate defense through pathogen recognition and clearance (355–357) and is found to be up-regulated in patients with chronic sinusitis (357).
Gp-340 functions in part through its interaction with Surfactant Protein D (SP-D), a collectin involved in pathogen recognition, modulation of inflammation, and phagocytosis (355, 358, 359). In SP-D-deficient mice, bacterial lung infections result in elevated inflammation, increased oxidative stress, and impaired macrophage function (360). Low circulating levels of SP-D have also been linked to the development of SLE (359). Although a direct mechanistic connection between gp-340 and SLE has not yet been established, its encoding gene, Deleted in Malignant Brain Tumor 1 (DMBT1), has been associated with several autoimmune and immune-related conditions, including SLE (361, 362).
A similar compromise in mucosal barrier integrity is seen in the gastrointestinal tract, which plays a vital role in immune homeostasis. The intestinal epithelium and its associated mucosal layer protect against microbial invasion while allowing for nutrient absorption (363, 364). Increased intestinal permeability, commonly referred to as “leaky gut syndrome”, permits the translocation of microbial products into the bloodstream, triggering both acute and systemic inflammation (363, 364). “Leaky Gut Syndrome” is increasingly implicated in the pathogenesis of SLE, as it has been in other autoimmune diseases such as RA, multiple sclerosis (MS), and type 1 diabetes (365).
In lupus, impaired gut barrier function allows pathogen-associated molecular patterns (PAMPs) and DAMPs to enter the bloodstream, activating dendritic cells, macrophages, and neutrophils (364). Bacterial components such as lipopolysaccharide (LPS), lipoteichoic acid (LTA), and β-glucans have been detected in the serum of SLE patients, reflecting microbial translocation and systemic immune activation (366). Scavenger receptor SR-E3 (CD206), also known as the mannose receptor, has been implicated in both gut dysbiosis and LN (367). In lupus-prone mouse models, colonization with Segmented Filamentous Bacteria (SFB) exacerbates glomerulonephritis, alters immune cell profiles, and disrupts gut barrier integrity (368). Notably, kidney-infiltrating CD206+ macrophages were observed in SFB-colonized mice, suggesting that gut-primed immune cells migrate to distal inflammatory sites, such as the kidney (368). In human SLE patients, the presence of CD206+ macrophages in the kidney correlates with disease severity (369).
The importance of mucosal SRs in maintaining immune tolerance is further supported by findings in Celiac disease, another autoimmune disorder affecting the gastrointestinal mucosa. Like SLE, Celiac disease is characterized by impaired clearance of ACs, which leads to the accumulation of cellular debris and chronic inflammation. Studies have shown that duodenal tissues from Celiac patients exhibit reduced expression of several scavenger receptors, including SR-B2, thrombospondin-1 (TSP-1), and CD61, alongside elevated levels of inflammatory cytokines such as IL-15, IL21, and IFN-γ (370). Furthermore, lamina propria mononuclear cells (LPMCs) isolated from Celiac patients display a diminished capacity to phagocytose ACs (370). Disruption in these localized immune defenses may initiate or amplify systemic inflammation and contribute to disease pathogenesis.
3 Scavenger receptors as biomarkers and targets to treat SLE
The complexity and heterogeneity of SLE—both in terms of clinical presentation and underlying immunological mechanisms—have long posed significant challenges in diagnosis, prognosis, and treatment. While serological markers such as anti-dsDNA and anti-Smith antibodies are commonly used, they lack sensitivity and specificity across all disease stages and patient populations. Therefore, there is growing interest in identifying novel, more reliable biomarkers that reflect disease activity, organ involvement, and therapeutic responsiveness.
Among the most promising candidates are SRs. SRs play a critical role in innate immunity through the recognition and clearance of endogenous and exogenous ligands, including apoptotic cells, modified lipids, and microbial products. Dysregulated expression or function of SRs has been implicated in the breakdown of self-tolerance and chronic inflammation, hallmarks of SLE pathogenesis. Notably, SRs also participate in intracellular signaling cascades such as the MAPK, NF-κB, and JAK-STAT pathways—many of which are known to be dysregulated in autoimmune conditions (316, 371–373). This positions SRs not only as mediators of immune response but also as potential biomarkers for disease progression and targets for immunomodulatory therapy.
Biomarkers are essential for the diagnosis, prognosis and the monitoring of SLE. Markers like dsDNA, complement levels and the presence of certain autoantibodies have been validated and are being used in clinical practice. However, it wasn't until 2007 that Wermeling et al. showed the presence of antibodies responsible for recognizing MARCO and SR-A (323). The discovery was then followed up by Chen et al., where the group identified an increase of anti-SR-A and anti-MARCO IgG in SLE patients when compared to controls (327, 374). In 2022, Jorge et al. showed an increase in anti-SCARF1 antibodies that correlate with increase dsDNA in the serum (329). This data was true for 26% of the patients suffering from SLE, however no additional correlation was found with other disease markers. Recently it was identified that the SR sCD163 (SR-I1) was elevated in patients suffering from LN, and it was suggested to be used as a prognostic biomarker (59). These discoveries are only in the initial stages and additional work is necessary to take this finding to the clinic. There is one thing all these discoveries had in common, the presence of these antibodies to SRs contribute to the breakdown of self-tolerance and increase autoimmune pathogenesis, making SRs the perfect candidate as disease progression biomarker.
Current SLE management strategies aim to (1) minimize disease activity, (2) prevent irreversible organ damage, (3) reduce the burden of comorbidities and treatment-related side effects, and (4) improve quality of life by alleviating pain and fatigue (5). However, due to the heterogeneous nature of the disease, a universal diagnostic or treatment protocol remains elusive. While glucocorticosteroids and antimalarial drugs have historically formed the backbone of treatment, their long-term use is associated with serious side effects and complications (Table 2). In recent years, the therapeutic landscape has shifted toward biologics, with only two FDA-approved options currently available—belimumab and anifrolumab (1, 375–377). Despite their promise, many patients exhibit incomplete or variable responses, necessitating adjunct or personalized therapies that account for unique immune signatures and organ involvement (375).
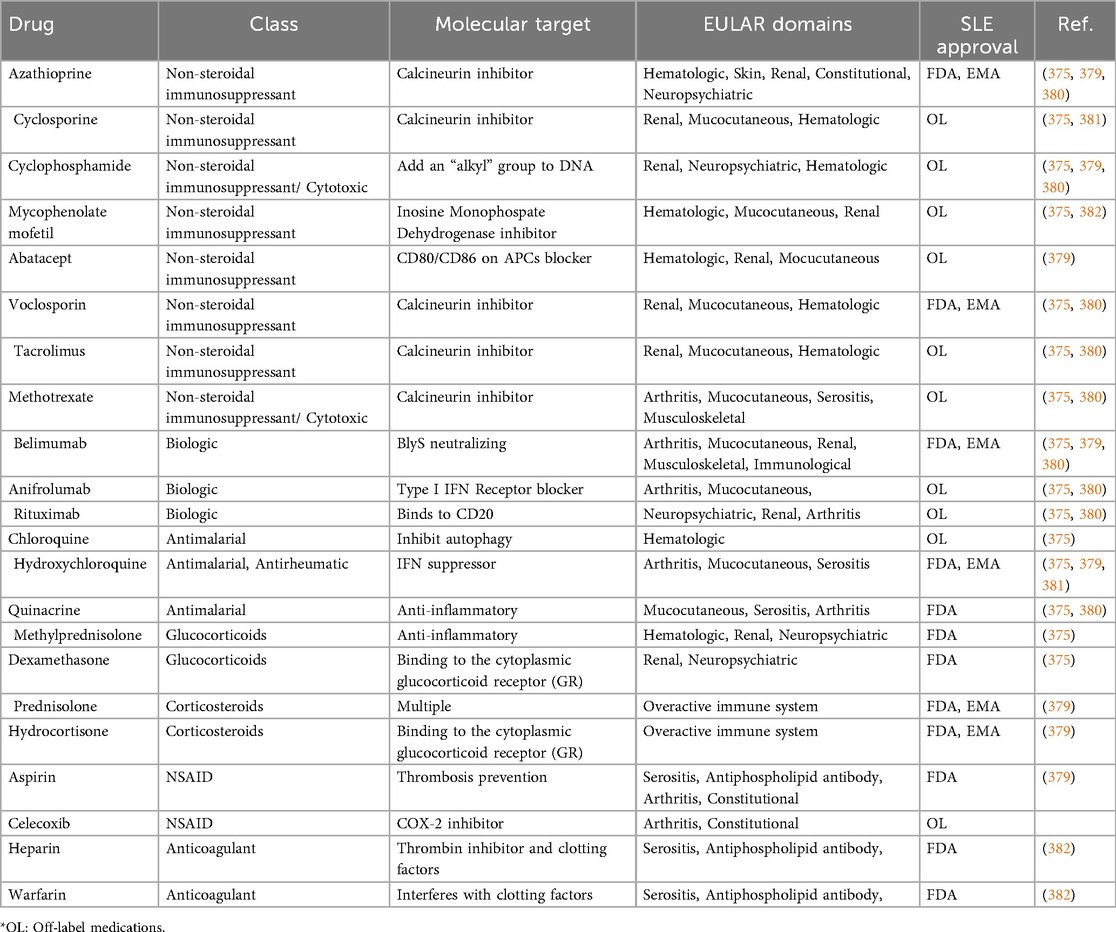
Table 2. Current SLE treatment options.
Targeting key signaling pathways associated with SR activation—such as MAPK, NF-κB, and JAK-STAT—has been explored in clinical trials with mixed outcomes, largely due to SLE's intrinsic heterogeneity and overlapping immunopathogenic mechanisms (101, 316, 372, 373). These challenges underscore the need for refined biomarkers and therapeutic targets. As research advances, scavenger receptors may provide dual utility as both indicators of disease activity and modulators of inflammation, offering a promising avenue for stratified medicine in SLE care.
4 Discussion
Scavenger receptors play an essential role in the immune system and their immunomodulatory function through a variety of immune and non-immune cell types in the context of inflammation and several autoimmune diseases. The past two decades of research have shown that the role of SRs in inflammatory disease might have been underestimated, and the more recent body of published work demonstrates the many ways SRs influence, modulate, and directly contribute to inflammatory disease-related pathways.
Although great strides have been made to understand how SRs contribute to inflammation, many unanswered questions remain. As shown in this review, there is quite a bit of redundancy regarding SRs and the pathways that they participate in or regulate. Still, it appears that one class is not responsible for the immunomodulatory effects of SRs (10, 260). SRs participate in pro- and anti-inflammatory signaling cascades, which muddies their contribution to the development and prognosis of inflammatory disease as well. Furthermore, they interact with other pattern recognition receptors to enhance or suppress a response (10, 378).
There is convincing evidence that some SRs can act as potentially useful biomarkers for autoimmune disease diagnostics and prognostics, but this is not the case for every SR family. For example, autoantibodies for SR-AI are elevated in SLE patients (327) and autoantibodies to SCARF1 are linked to defects in efferocytosis and autoimmunity (329). Further research is necessary to identify SR with subset or symptoms of autoimmunity. However, designing SR-based therapeutic approaches may prove challenging since SR levels do not necessarily vary between healthy individuals and those with inflammatory disease. Downstream research is needed to finetune current findings and provide greater clarity on these knowledge gaps.
Despite the knowledge gaps that remain regarding SRs and their role in inflammation and autoimmunity, current research demonstrates that SRs wear many immunological hats and contribute to a variety of inflammatory signaling pathways. Therefore, it is crucial to continue investigating the role of SRs in inflammation to determine the therapeutic potential of targeting SRs in the context of inflammatory and autoimmune disease.
Author contributions
SH: Writing – original draft, Writing – review & editing. MC: Writing – review & editing. DS: Writing – review & editing. RR: Writing – review & editing. ZR: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was funded by Lupus Research Alliance Innovation Award 2022 and DoD-Impact Award (DoD W81XWH-21-1-0803).
Acknowledgments
We would thank Dr. Stuart Levitz and his lab for providing additional advice in the preparation of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/flupu.2025.1679564/full#supplementary-material
References
1. Kaul A, Gordon C, Crow MK, Touma Z, Urowitz MB, van Vollenhoven R, et al. Systemic lupus erythematosus. Nat Rev Dis Primers. (2016) 2:16039. doi: 10.1038/nrdp.2016.39
2. Vaillant AJ, Goyal A, Bansal P, Varacallo M. Systemic Lupus Erythematosus. Florida, USA: StatPearls Publishing (2023).
3. Lam NV, Brown JA, Sharma R. Systemic lupus erythematosus: diagnosis and treatment. Am Fam Physician. (2023) 107:383–95. PMID: 37054414
4. Sang A, Yin Y, Zheng YY, Morel L. Animal models of molecular pathology systemic lupus erythematosus. Prog Mol Biol Transl Sci. (2012) 105:321–70. doi: 10.1016/B978-0-12-394596-9.00010-X
5. Fava A, Petri M. Systemic lupus erythematosus: diagnosis and clinical management. J Autoimmun. (2019) 96:1–13. doi: 10.1016/j.jaut.2018.11.001
6. Pathak S, Mohan C. Cellular and molecular pathogenesis of systemic lupus erythematosus: lessons from animal models. Arthritis Res Ther. (2011) 13:241. doi: 10.1186/ar3465
7. Gachpazan M, Akhlaghipour I, Rahimi HR, Saburi E, Mojarrad M, Abbaszadegan MR, et al. Genetic and molecular biology of systemic lupus erythematosus among Iranian patients: an overview. Auto Immun Highlights. (2021) 12:2. doi: 10.1186/s13317-020-00144-y
8. da Gama Duarte J, Woods K, Andrews MC, Behren A. The good, the (not so) bad and the ugly of immune homeostasis in melanoma. Immunology & Cell Biology. (2018) 96:497–506. doi: 10.1111/imcb.12001
9. Patten DA, Wilkinson AL, O'Keeffe A, Shetty S. Scavenger receptors: novel roles in the pathogenesis of liver inflammation and cancer. Semin Liver Dis. (2022) 42:61–76. doi: 10.1055/s-0041-1733876
10. Canton J, Neculai D, Grinstein S. Scavenger receptors in homeostasis and immunity. Nat Rev Immunol. (2013) 13:621–34. doi: 10.1038/nri3515
11. PrabhuDas MR, Baldwin CL, Bollyky PL, Bowdish DME, Drickamer K, Febbraio M, et al. A consensus definitive classification of scavenger receptors and their roles in health and disease. J Immunol. (2017) 198:3775–89. doi: 10.4049/jimmunol.1700373
12. Prabhudas M, Bowdish D, Drickamer K, Febbraio M, Herz J, Kobzik L, et al. Standardizing scavenger receptor nomenclature. J Immunol. (2014) 192:1997–2006. doi: 10.4049/jimmunol.1490003
13. Zani IA, Stephen SL, Mughal NA, Russell D, Homer-Vanniasinkam S, Wheatcroft SB, et al. Scavenger receptor structure and function in health and disease. Cells. (2015) 4:178–201. doi: 10.3390/cells4020178
14. Krieger M. The other side of scavenger receptors: pattern recognition for host defense. Curr Opin Lipidol. (1997) 8:275–80. doi: 10.1097/00041433-199710000-00006
15. McGonagle D, McDermott MF. A proposed classification of the immunological diseases. PLoS Med. (2006) 3:e297. doi: 10.1371/journal.pmed.0030297
16. Marcuzzi A, Melloni E, Zauli G, Romani A, Secchiero P, Maximova N, et al. Autoinflammatory diseases and cytokine storms—imbalances of innate and adaptative immunity. Int J Mol Sci. (2021) 22:11241. doi: 10.3390/ijms222011241
17. Cervera R, Khamashta MA, Font J, Sebastiani GD, Gil A, Lavilla P, et al. Morbidity and mortality in systemic lupus erythematosus during a 10-year period: a comparison of early and late manifestations in a cohort of 1,000 patients. Medicine (Baltimore). (2003) 82:299–308. doi: 10.1097/01.md.0000091181.93122.55
18. Fayyaz A, Igoe A, Kurien BT, Danda D, James JA, Stafford HA, et al. Haematological manifestations of lupus. Lupus Sci Med. (2015) 2:e000078. doi: 10.1136/lupus-2014-000078
19. Mitchinson MJ. Fluor: another cardinal sign of inflammation. Lancet. (1989) 2:1520. doi: 10.1016/S0140-6736(89)92957-7
20. Punchard NA, Whelan CJ, Adcock I. The journal of inflammation. J Inflamm (Lond. (2004) 1:1. doi: 10.1186/1476-9255-1-1
22. Hannoodee S, Nasuruddin DN. Acute Inflammatory Response. Florida, USA: StatPearls Publishing (2024).
23. Stojan G, Petri M. Atherosclerosis in systemic lupus erythematosus. J Cardiovasc Pharmacol. (2013) 62:255–62. doi: 10.1097/FJC.0b013e31829dd857
24. Wilhelm AJ, Major AS. Accelerated atherosclerosis in SLE: mechanisms and prevention approaches. Int J Clin Rheumtol. (2012) 7:527–39. doi: 10.2217/ijr.12.46
25. van Leuven SI, Major AS. Accelerated atherosclerosis in systemic lupus erythematosus: don't Forget the devil we know!. Rheumatology. (2023) 63:3–5. doi: 10.1093/rheumatology/kead329
26. Pearson JT, Yoshimoto M, Chen YC, Sultani R, Edgley AJ, Nakaoka H, et al. Widespread coronary dysfunction in the absence of HDL receptor SR-B1 in an ischemic cardiomyopathy mouse model. Sci Rep. (2017) 7:18108. doi: 10.1038/s41598-017-18485-6
27. Staršíchová A. SR-B1-/-ApoE-R61h/h mice mimic human coronary heart disease. Cardiovasc Drugs Ther. (2023) 38(6):1123–37. doi: 10.1007/s10557-023-07475-8
28. Hein TW, Kuo L. LDLs impair vasomotor function of the coronary microcirculation: role of superoxide anions. Circ Res. (1998) 83:404–14. doi: 10.1161/01.RES.83.4.404
29. Hein TW, Liao JC, Kuo L. oxLDL specifically impairs endothelium-dependent, NO-mediated dilation of coronary arterioles. Am J Physiol Heart Circ Physiol. (2000) 278:H175–83. doi: 10.1152/ajpheart.2000.278.1.H175
30. Zeiher AM, Drexler H, Wollschläger H, Just H. Endothelial dysfunction of the coronary microvasculature is associated with coronary blood flow regulation in patients with early atherosclerosis. Circulation. (1991) 84:1984–92. doi: 10.1161/01.CIR.84.5.1984
31. Van Eck M, Hoekstra M, Hildebrand RB, Yaong Y, Stengel D, Kruijt JK, et al. Increased oxidative stress in scavenger receptor BI knockout mice with dysfunctional HDL. Arterioscler Thromb Vasc Biol. (2007) 27:2413–9. doi: 10.1161/ATVBAHA.107.145474
32. Gracia-Rubio I, Martín C, Civeira F, Cenarro A. SR-B1, a key receptor involved in the progression of cardiovascular disease: a perspective from mice and human genetic studies. Biomedicines. (2021) 9:1–18. doi: 10.3390/biomedicines9060612
33. Anderson TJ. Assessment and treatment of endothelial dysfunction in humans. J Am Coll Cardiol. (1999) 34:631–8. doi: 10.1016/S0735-1097(99)00259-4
34. Korporaal SJ, Meurs I, Hauer AD, Hildebrand RB, Hoekstra M, Cate HT, et al. Deletion of the high-density lipoprotein receptor scavenger receptor BI in mice modulates thrombosis susceptibility and indirectly affects platelet function by elevation of plasma free cholesterol. Arterioscler Thromb Vasc Biol. (2011) 31:34–42. doi: 10.1161/ATVBAHA.110.210252
35. Galanopoulos N, Christoforidou A, Bezirgiannidou Z. Lupus thrombocytopenia: pathogenesis and therapeutic implications. Mediterr J Rheumatol. (2017) 28:20–6. doi: 10.31138/mjr.28.1.20
36. Wanchu A, Khullar M, Deodhar SD, Bambery P, Sud A. Nitric oxide synthesis is increased in patients with systemic lupus erythematosus. Rheumatol Int. (1998) 18:41–3. doi: 10.1007/s002960050055
37. Al Gadban MM, German J, Truman J-P, Soodavar F, Riemer EC, Twal WO, et al. Lack of nitric oxide synthases increases lipoprotein immune complex deposition in the aorta and elevates plasma sphingolipid levels in lupus. Cell Immunol. (2012) 276:42–51. doi: 10.1016/j.cellimm.2012.03.007
38. Zhang S, Picard MH, Vasile E, Zhu Y, Raffai RL, Weisgraber KH, et al. Diet-induced occlusive coronary atherosclerosis, myocardial infarction, cardiac dysfunction, and premature death in scavenger receptor class B type I-deficient, hypomorphic apolipoprotein ER61 mice. Circulation. (2005) 111:3457–64. doi: 10.1161/CIRCULATIONAHA.104.523563
39. Nakaoka H, Nakagawa-Toyama Y, Nishida M, Okada T, Kawase R, Yamashita T, et al. Establishment of a novel murine model of ischemic cardiomyopathy with multiple diffuse coronary lesions. PLoS One. (2013) 8:e70755. doi: 10.1371/journal.pone.0070755
40. Nossent J, Cikes N, Kiss E, Marchesoni A, Nassonova V, Mosca M, et al. Current causes of death in systemic lupus erythematosus in Europe, 2000–2004: relation to disease activity and damage accrual. Lupus. (2007) 16:309–17. doi: 10.1177/0961203307077987
41. Bernatsky S, Boivin JF, Joseph L, Manzi S, Ginzler E, Gladman DD, et al. Mortality in systemic lupus erythematosus. Arthritis Rheum. (2006) 54:2550–7. doi: 10.1002/art.21955
42. Mutoh T, Shirai T, Ishii T, Shirota Y, Fujishima F, Takahashi F, et al. Identification of two major autoantigens negatively regulating endothelial activation in takayasu arteritis. Nat Commun. (2020) 11:1253. doi: 10.1038/s41467-020-15088-0
43. Feng H, Guo L, Wang D, Gao H, Hou G, Zheng Z, et al. Deficiency of scavenger receptor BI leads to impaired lymphocyte homeostasis and autoimmune disorders in mice. Arterioscler Thromb Vasc Biol. (2011) 31:2543–51. doi: 10.1161/ATVBAHA.111.234716
44. Porsch F, Binder CJ. Autoimmune diseases and atherosclerotic cardiovascular disease. Nat Rev Cardiol. (2024) 21:780–807. doi: 10.1038/s41569-024-01045-7
45. Carlucci PM, Purmalek MM, Dey AK, Temesgen-Oyelakin Y, Sakhardande S, Joshi AA, et al. Neutrophil subsets and their gene signature associate with vascular inflammation and coronary atherosclerosis in lupus. JCI Insight. (2018) 3(8):1–16. doi: 10.1172/jci.insight.99276
46. Smith CK, Vivekanandan-Giri A, Tang C, Knight JS, Mathew A, Padilla RL, et al. Neutrophil extracellular trap-derived enzymes oxidize high-density lipoprotein: an additional proatherogenic mechanism in systemic lupus erythematosus. Arthritis Rheumatol. (2014) 66:2532–44. doi: 10.1002/art.38703
47. Forte F, Buonaiuto A, Calcaterra I, Iannuzzo G, Ambrosino P, Di Minno MND. Association of systemic lupus erythematosus with peripheral arterial disease: a meta-analysis of literature studies. Rheumatology (Oxford). (2020) 59:3181–92. doi: 10.1093/rheumatology/keaa414
48. Purmalek MM, Carlucci PM, Dey AK, Sampson M, Temesgen-Oyelakin Y, Sakhardande S, et al. Association of lipoprotein subfractions and glycoprotein acetylation with coronary plaque burden in SLE. Lupus Sci Med. (2019) 6:e000332. doi: 10.1136/lupus-2019-000332
49. Szabó MZ, Szodoray P, Kiss E. Dyslipidemia in systemic lupus erythematosus. Immunol Res. (2017) 65:543–50. doi: 10.1007/s12026-016-8892-9
50. Yafasova A, Fosbøl EL, Schou M, Baslund B, Faurschou M, Docherty KF, et al. Long-term cardiovascular outcomes in systemic lupus erythematosus. J Am Coll Cardiol. (2021) 77:1717–27. doi: 10.1016/j.jacc.2021.02.029
51. Manzi S, Meilahn EN, Rairie JE, Conte CG, Medsger TA Jr., Jansen-McWilliams L, et al. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: comparison with the framingham study. Am J Epidemiol. (1997) 145:408–15. doi: 10.1093/oxfordjournals.aje.a009122
52. Restivo V, Candiloro S, Daidone M, Norrito R, Cataldi M, Minutolo G, et al. Systematic review and meta-analysis of cardiovascular risk in rheumatological disease: symptomatic and non-symptomatic events in rheumatoid arthritis and systemic lupus erythematosus. Autoimmun Rev. (2022) 21:102925. doi: 10.1016/j.autrev.2021.102925
53. Mohamed OSD, Azmy GJ, Elfadl EMA. Clinical significance of red blood cell distribution width in systemic lupus erythematosus patients. Egypt Rheumatol Rehab. (2020) 47:38. doi: 10.1186/s43166-020-00037-y
54. Ghiran IC, Zeidel ML, Shevkoplyas SS, Burns JM, Tsokos GC, Kyttaris VC. Systemic lupus erythematosus serum deposits C4d on red blood cells, decreases red blood cell membrane deformability, and promotes nitric oxide production. Arthritis Rheum. (2011) 63:503–12. doi: 10.1002/art.30143
55. Cheng Q, Mou L, Su W, Chen X, Zhang T, Xie Y, et al. Ferroptosis of CD163 + tissue-infiltrating macrophages and CD10+ PC+ epithelial cells in lupus nephritis. Front Immunol. (2023) 14:1–11. doi: 10.3389/fimmu.2023.1171318
56. Endo N, Tsuboi N, Furuhashi K, Shi Y, Du Q, Abe T, et al. Urinary soluble CD163 level reflects glomerular inflammation in human lupus nephritis. Nephrol Dial Transplant. (2016) 31:2023–33. doi: 10.1093/ndt/gfw214
57. Moestrup SK, Møller HJ. CD163: a regulated hemoglobin scavenger receptor with a role in the anti-inflammatory response. Ann Med. (2004) 36:347–54. doi: 10.1080/07853890410033171
58. Kristiansen M, Graversen JH, Jacobsen C, Sonne O, Hoffman HJ, Law SK, et al. Identification of the haemoglobin scavenger receptor. Nature. (2001) 409:198–201. doi: 10.1038/35051594
59. Liu Y, Li M, Zhang H, Yin Z, Wang X. Clinical significance of serum soluble scavenger receptor CD163 in patients with lupus nephritis. Lupus. (2024) 33:1279–88. doi: 10.1177/09612033241276033
60. Nishino A, Katsumata Y, Kawasumi H, Hirahara S, Kawaguchi Y, Yamanaka H. Usefulness of soluble CD163 as a biomarker for macrophage activation syndrome associated with systemic lupus erythematosus. Lupus. (2019) 28:986–94. doi: 10.1177/0961203319860201
61. Skytthe MK, Graversen JH, Moestrup SK. Targeting of CD163(+) macrophages in inflammatory and malignant diseases. Int J Mol Sci. (2020) 21:1–32. doi: 10.3390/ijms21155497
62. Liu AC, Yang Y, Li MT, Jia Y, Chen S, Ye S, et al. Macrophage activation syndrome in systemic lupus erythematosus: a multicenter, case-control study in China. Clin Rheumatol. (2018) 37:93–100. doi: 10.1007/s10067-017-3625-6
63. Aziz A, Castaneda EE, Ahmad N, Veerapalli H, Rockferry AG, Lankala CR, et al. Exploring macrophage activation syndrome secondary to systemic lupus erythematosus in adults: a systematic review of the literature. Cureus. (2021) 13:e18822. doi: 10.7759/cureus.18822
64. Liu T, Son M, Diamond B. HMGB1 in systemic lupus erythematosus. Front Immunol. (2020) 11:1057. doi: 10.3389/fimmu.2020.01057
65. Crayne CB, Albeituni S, Nichols KE, Cron RQ. The immunology of macrophage activation syndrome. Front Immunol. (2019) 10:119. doi: 10.3389/fimmu.2019.00119
66. Elkon KB, Stone VV. Type I interferon and systemic lupus erythematosus. J Interferon Cytokine Res. (2011) 31:803–12. doi: 10.1089/jir.2011.0045
67. Abraham NG, Drummond G. CD163-mediated hemoglobin-heme uptake activates macrophage HO-1, providing an antiinflammatory function. Circ Res. (2006) 99:911–4. doi: 10.1161/01.RES.0000249616.10603.d6
68. Thomsen JH, Etzerodt A, Svendsen P, Moestrup SK. The haptoglobin-CD163-heme oxygenase-1 pathway for hemoglobin scavenging. Oxid Med Cell Longev. (2013) 2013:523652. doi: 10.1155/2013/523652
69. Gao Z, Wang Y, Wang J, Zhang J, Wang Z. Soluble ST2 and CD163 as potential biomarkers to differentiate primary hemophagocytic lymphohistiocytosis from macrophage activation syndrome. Mediterr J Hematol Infect Dis. (2019) 11:e2019008. doi: 10.4084/mjhid.2019.008
70. Sakumura N, Shimizu M, Mizuta M, Inoue N, Nakagishi Y, Yachie A. Soluble CD163, a unique biomarker to evaluate the disease activity, exhibits macrophage activation in systemic juvenile idiopathic arthritis. Cytokine. (2018) 110:459–65. doi: 10.1016/j.cyto.2018.05.017
71. Schulert GS, Pickering AV, Do T, Dhakal S, Fall N, Schnell D, et al. Monocyte and bone marrow macrophage transcriptional phenotypes in systemic juvenile idiopathic arthritis reveal TRIM8 as a mediator of IFN-gamma hyper-responsiveness and risk for macrophage activation syndrome. Ann Rheum Dis. (2021) 80:617–25. doi: 10.1136/annrheumdis-2020-217470
72. Greisen SR, Moller HJ, Stengaard-Pedersen K, Hetland ML, Hørslev-Petersen K, Jørgensen A, et al. Soluble macrophage-derived CD163 is a marker of disease activity and progression in early rheumatoid arthritis. Clin Exp Rheumatol. (2011) 29:689–92. PMID: 21813065
73. Mejia-Vilet JM, Zhang XL, Cruz C, Cano-Verduzco ML, Shapiro JP, Nagaraja HN, et al. Urinary soluble CD163: a novel noninvasive biomarker of activity for lupus nephritis. J Am Soc Nephrol. (2020) 31:1335–47. doi: 10.1681/ASN.2019121285
74. Zhang T, Li H, Vanarsa K, Gidley G, Mok CC, Petri M, et al. Association of urine sCD163 with proliferative lupus nephritis, fibrinoid necrosis, cellular crescents and intrarenal M2 macrophages. Front Immunol. (2020) 11:671. doi: 10.3389/fimmu.2020.00671
75. Huang YJ, Lin CH, Yang HY, Luo SF, Kuo CF. Urine soluble CD163 is a promising biomarker for the diagnosis and evaluation of lupus nephritis. Front Immunol. (2022) 13:935700. doi: 10.3389/fimmu.2022.935700
76. Markota A, Kalamar Ž, Fluher J, Pirkmajer S. Therapeutic hyperthermia for the treatment of infection-a narrative review. Front Physiol. (2023) 14:1215686. doi: 10.3389/fphys.2023.1215686
77. Zhang H-G, Mehta K, Cohen P, Guha C. Hyperthermia on immune regulation: a temperature's Story. Cancer Lett. (2008) 271:191–204. doi: 10.1016/j.canlet.2008.05.026
78. Xinghua Gao HC. Hyperthermia on skin immune system and its application in the treatment of human papillomavirus-infected skin diseases. Front. Med. (2014) 8:1–5. doi: 10.1007/s11684-014-0309-3
79. Timlin H, Syed A, Haque U, Adler B, Law G, Machireddy K, et al. Fevers in adult lupus patients. Cureus. (2018) 10:e2098. doi: 10.7759/cureus.2098
80. Kallinich T, Gattorno M, Grattan CE, de Koning HD, Traidl-Hoffmann C, Feist E, et al. Unexplained recurrent fever: when is autoinflammation the explanation? Allergy. (2013) 68:285–96. doi: 10.1111/all.12084
81. Galloway J, Cope AP. The ying and yang of fever in rheumatic disease. Clinical Medicine. (2015) 15:288–91. doi: 10.7861/clinmedicine.15-3-288
82. Ryu S, Fu W, Petri MA. Associates and predictors of pleurisy or pericarditis in SLE. Lupus Sci Med. (2017) 4:e000221. doi: 10.1136/lupus-2017-000221
83. Font J, Cervera R, Ramos-Casals M, García-Carrasco M, Sents J, Herrero C, et al. Clusters of clinical and immunologic features in systemic lupus erythematosus: analysis of 600 patients from a single center. Semin Arthritis Rheum. (2004) 33:217–30. doi: 10.1053/S0049-0172(03)00133-1
84. Ambrose N, Morgan TA, Galloway J, Ionnoau Y, Beresford MW, Isenberg DA. Differences in disease phenotype and severity in SLE across age groups. Lupus. (2016) 25:1542–50. doi: 10.1177/0961203316644333
85. Swaak AJ, van den Brink HG, Smeenk RJ, Manger K, Kalden JR, Tosi S, et al. Systemic lupus erythematosus: clinical features in patients with a disease duration of over 10 years, first evaluation. Rheumatology (Oxford). (1999) 38:953–8. doi: 10.1093/rheumatology/38.10.953
86. Harvey AM, Shulman LE, Tumulty PA, Conley CL, Schoenrich EH. Systemic lupus erythematosus: review of the literature and clinical analysis of 138 cases. Medicine (Baltimore). (1954) 33:291–437. doi: 10.1097/00005792-195412000-00001
87. Barber MRW, Clarke AE. Systemic lupus erythematosus and risk of infection. Expert Rev Clin Immunol. (2020) 16:527–38. doi: 10.1080/1744666X.2020.1763793
88. Kunzler ALF, Tsokos GC. Infections in patients with systemic lupus erythematosus: the contribution of primary immune defects versus treatment-induced immunosuppression. Eur J Rheumatol. (2023) 10:148–58. doi: 10.5152/eurjrheum.2023.23068
89. Lunchenkov N, Filippov E, Prihodko O, Volchkova E. Classical fever of unknown origin: retrospective study in infectious clinical hospital №2. Open Forum Infect Dis. (2017) 4:S343–S343. doi: 10.1093/ofid/ofx163.818
90. Abdel-Aleem A, Zaki MS. Spinocerebellar ataxia type 2 (SCA2) in an Egyptian family presenting with polyphagia and marked CAG expansion in infancy. J Neurol. (2008) 255:413–9. doi: 10.1007/s00415-008-0690-4
91. Netea MG, Kullberg BJ, Van der Meer JWM. Circulating cytokines as mediators of fever. Clin Infect Dis. (2000) 31:S178–84. doi: 10.1086/317513
92. Zampronio AR, Soares DM, Souza GE. Central mediators involved in the febrile response: effects of antipyretic drugs. Temperature (Austin. (2015) 2:506–21. doi: 10.1080/23328940.2015.1102802
93. Damiati LA, Denetiu I, Bahlas S, Damiati S, Pushparaj PN. Immunoprofiling of cytokines, chemokines, and growth factors in female patients with systemic lupus erythematosus– a pilot study. BMC Immunol. (2023) 24:13. doi: 10.1186/s12865-023-00551-6
94. Weckerle CE, Mangale D, Franek BS, Kelly JA, Kumabe M, James JA, et al. Large-scale analysis of tumor necrosis factor α levels in systemic lupus erythematosus. Arthritis Rheum. (2012) 64:2947–52. doi: 10.1002/art.34483
95. Xiang N, Fang X, Sun XG, Zhou YB, Ma Y, Zhu C, et al. Expression profile of PU.1 in CD4(+)T cells from patients with systemic lupus erythematosus. Clin Exp Med. (2021) 21:621–32. doi: 10.1007/s10238-021-00717-9
96. La Cava A. Anticytokine therapies in systemic lupus erythematosus. Immunotherapy. (2010) 2:575–82. doi: 10.2217/imt.10.29
97. Park J, Jang W, Park HS, Park KH, Kwok SK, Park SH, et al. Cytokine clusters as potential diagnostic markers of disease activity and renal involvement in systemic lupus erythematosus. J Int Med Res. (2020) 48:300060520926882. doi: 10.1177/0300060520926882
98. Pacheco Y, Barahona-Correa J, Monsalve DM, Acosta-Ampudia Y, Rojas M, Rodríguez Y, et al. Cytokine and autoantibody clusters interaction in systemic lupus erythematosus. J Transl Med. (2017) 15:239. doi: 10.1186/s12967-017-1345-y
99. Reynolds JA, McCarthy EM, Haque S, Ngamjanyaporn P, Sergeant JC, Lee E, et al. Cytokine profiling in active and quiescent SLE reveals distinct patient subpopulations. Arthritis Res Ther. (2018) 20:173. doi: 10.1186/s13075-018-1666-0
100. Bahlas S, Damiati L, Dandachi N, Sait H, Alsefri M, Pushparaj PN. Rapid immunoprofiling of cytokines, chemokines and growth factors in patients with active rheumatoid arthritis using luminex multiple analyte profiling technology for precision medicine. Clin Exp Rheumatol. (2019) 37:112–9. PMID: 29998825
101. Allen ME, Rus V, Szeto GL. Leveraging heterogeneity in systemic lupus erythematosus for new therapies. Trends Mol Med. (2021) 27:152–71. doi: 10.1016/j.molmed.2020.09.009
102. Mehta JL, Sanada N, Hu CP, Chen J, Dandapat A, Sugawara F, et al. Deletion of LOX-1 reduces atherogenesis in LDLR knockout mice fed high cholesterol diet. Circ Res. (2007) 100:1634–42. doi: 10.1161/CIRCRESAHA.107.149724
103. Wang X, Phillips MI, Mehta JL. LOX-1 and angiotensin receptors, and their interplay. Cardiovasc Drugs Ther. (2011) 25:401–17. doi: 10.1007/s10557-011-6331-7
104. Twigg MW, Freestone K, Homer-Vanniasinkam S, Ponnambalam S. The LOX-1 scavenger receptor and its implications in the treatment of vascular disease. Cardiol Res Pract. (2012) 2012:632408. doi: 10.1155/2012/632408
105. Pyrpyris N, Dimitriadis K, Beneki E, Iliakis P, Soulaidopoulos S, Tsioufis P, et al. LOX-1 receptor: a diagnostic tool and therapeutic target in atherogenesis. Curr Probl Cardiol. (2024) 49:102117. doi: 10.1016/j.cpcardiol.2023.102117
106. Thakkar S, Wang X, Khaidakov M, Dai Y, Gokulan K, Mehta JL, et al. Structure-based design targeted at LOX-1, a receptor for oxidized low-density lipoprotein. Sci Rep. (2015) 5:16740. doi: 10.1038/srep16740
107. Akhmedov A, Rozenberg I, Paneni F, Camici GG, Shi Y, Doerries C, et al. Endothelial overexpression of LOX-1 increases plaque formation and promotes atherosclerosis in vivo. Eur Heart J. (2014) 35:2839–48. doi: 10.1093/eurheartj/eht532
108. Sagar D, Gaddipati R, Ongstad EL, Bhagroo N, An LL, Wang J, et al. LOX-1: a potential driver of cardiovascular risk in SLE patients. PLoS One. (2020) 15:e0229184. doi: 10.1371/journal.pone.0229184
109. Kume N, Moriwaki H, Kataoka H, Minami M, Murase T, Sawamura T, et al. Inducible expression of LOX-1, a novel receptor for oxidized LDL, in macrophages and vascular smooth muscle cells. Ann N Y Acad Sci. (2000) 902:323–7. doi: 10.1111/j.1749-6632.2000.tb06332.x
110. Hofnagel O, Luechtenborg B, Stolle K, Lorkowski S, Eschert H, Plenz G, et al. Proinflammatory cytokines regulate LOX-1 expression in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. (2004) 24:1789–95. doi: 10.1161/01.ATV.0000140061.89096.2b
111. Everett BM, Donath MY, Pradhan AD, Thuren T, Pais P, Nicolau JC, et al. Anti-Inflammatory therapy with canakinumab for the prevention and management of diabetes. J Am Coll Cardiol. (2018) 71:2392–401. doi: 10.1016/j.jacc.2018.03.002
112. Ridker PM, MacFadyen JG, Glynn RJ, Koenig W, Libby P, Everett BM, et al. Inhibition of interleukin-1β by canakinumab and cardiovascular outcomes in patients with chronic kidney disease. J Am Coll Cardiol. (2018) 71:2405–14. doi: 10.1016/j.jacc.2018.03.490
113. Santacroce L, Colella M, Charitos IA, Di Domenico M, Palmirotta R, Jirillo E. Microbial and host metabolites at the backstage of fever: current knowledge about the co-ordinate action of receptors and molecules underlying pathophysiology and clinical implications. Metabolites. (2023) 13:461. doi: 10.3390/metabo13030461
114. Tsai CY, Wu TH, Tsai ST, Chen KH, Thajeb P, Lin WM, et al. Cerebrospinal fluid lnterleukin-6, prostaglandin E2 and autoantibodies in patients with neuropsychiatric systemic lupus erythematosus and central nervous system infections. Scand J Rheumatol. (1994) 23:57–63. doi: 10.3109/03009749409103028
115. Okamoto H, Kobayashi A, Yamanaka H. Cytokines and chemokines in neuropsychiatric syndromes of systemic lupus erythematosus. BioMed Res Int. (2010) 2010:268436. doi: 10.1155/2010/268436
116. Hirohata S, Kikuchi H. Role of Serum IL-6 in neuropsychiatric systemic lupus erythematosus. ACR Open Rheumatol. (2021) 3:42–9. doi: 10.1002/acr2.11217
117. Chae BS, Shin TY, Kim DK, Eun JS, Leem JY, Yang JH. Prostaglandin E2-mediated dysregulation of proinflammatory cytokine production in pristane-induced lupus mice. Arch Pharm Res. (2008) 31:503–10. doi: 10.1007/s12272-001-1185-6
118. Li X, Melief E, Postupna N, Montine KS, Keene CD, Montine TJ. Prostaglandin E2 receptor subtype 2 regulation of scavenger receptor CD36 modulates microglial Aβ42 phagocytosis. Am J Pathol. (2015) 185:230–9. doi: 10.1016/j.ajpath.2014.09.016
119. Voloshyna I, Kasselman LJ, Carsons SE, Littlefield MJ, Gomolin IH, De Leon J, et al. Cox-2-dependent and independent effects of cox-2 inhibitors and nsaids on proatherogenic changes in human monocytes/macrophages. J Invest Med. (2017) 65:694–704. doi: 10.1136/jim-2016-000259
120. Hu C, Yang J, Qi Z, Wu H, Wang B, Zou F, et al. Heat shock proteins: biological functions, pathological roles, and therapeutic opportunities. MedComm. (2022) 3:e161. doi: 10.1002/mco2.161
121. Ostberg JR, Kabingu E, Repasky EA. Thermal regulation of dendritic cell activation and migration from skin explants. Int J Hyperthermia. (2003) 19:520–33. doi: 10.1080/02656730310001607986
122. Di YP, Repasky EA, Subjeck JR. Distribution of HSP70, protein kinase C, and spectrin is altered in lymphocytes during a fever-like hyperthermia exposure. J Cell Physiol. (1997) 172:44–54. doi: 10.1002/(SICI)1097-4652(199707)172:1%3C44::AID-JCP5%3E3.0.CO;2-D
123. Ostberg JR, Kaplan KC, Repasky EA. Induction of stress proteins in a panel of mouse tissues by fever-range whole body hyperthermia. Int J Hyperthermia. (2002) 18:552–62. doi: 10.1080/02656730210166168
124. Munoz LE, Janko C, Chaurio RA, Schett G, Gaipl US, Herrmann M. Igg opsonized nuclear remnants from dead cells cause systemic inflammation in SLE. Autoimmunity. (2010) 43:232–5. doi: 10.3109/08916930903510930
125. Tukaj S, Sitko K. Heat shock protein 90 (Hsp90) and Hsp70 as potential therapeutic targets in autoimmune skin diseases. Biomolecules. (2022) 12:1153. doi: 10.3390/biom12081153
126. Binder RJ, Srivastava PK. Essential role of CD91 in re-presentation of gp96-chaperoned peptides. Proc Natl Acad Sci USA. (2004) 101:6128–33. doi: 10.1073/pnas.0308180101
127. Vega VL, Rodríguez-Silva M, Frey T, Gehrmann M, Diaz JC, Steinem C, et al. Hsp70 translocates into the plasma membrane after stress and is released into the extracellular environment in a membrane-associated form that activates macrophages. J Immunol. (2008) 180:4299–307. doi: 10.4049/jimmunol.180.6.4299
128. Noessner E, Gastpar R, Milani V, Brandl A, Hutzler PJ, Kuppner MC, et al. Tumor-derived heat shock protein 70 peptide complexes are cross-presented by human dendritic cells. J Immunol. (2002) 169:5424–32. doi: 10.4049/jimmunol.169.10.5424
129. Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, et al. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. (2002) 277:15028–34. doi: 10.1074/jbc.M200497200
130. Lehner T, Bergmeier LA, Wang Y, Tao L, Sing M, Spallek R, et al. Heat shock proteins generate beta-chemokines which function as innate adjuvants enhancing adaptive immunity. Eur J Immunol. (2000) 30:594–603. doi: 10.1002/1521-4141(200002)30:2%3C594::AID-IMMU594%3E3.0.CO;2-1
131. Panjwani NN, Popova L, Srivastava PK. Heat shock proteins gp96 and hsp70 activate the release of nitric oxide by APCs. J Immunol. (2002) 168:2997–3003. doi: 10.4049/jimmunol.168.6.2997
132. Evans SS, Repasky EA, Fisher DT. Fever and the thermal regulation of immunity: the immune system feels the heat. Nat Rev Immunol. (2015) 15:335–49. doi: 10.1038/nri3843
133. Torigoe T, Tamura Y, Sato N. Heat shock proteins and immunity: application of hyperthermia for immunomodulation. Int J Hyperthermia. (2009) 25:610–6. doi: 10.3109/02656730903315831
134. Sugawara A, Torigoe T, Tamura Y, Kamiguchi K, Nemoto K, Oguro H, et al. Polyamine compound deoxyspergualin inhibits heat shock protein-induced activation of immature dendritic cells. Cell Stress Chaperones. (2009) 14:133–9. doi: 10.1007/s12192-008-0064-y
135. Chen T, Guo J, Han C, Yang M, Cao X. Heat shock protein 70, released from heat-stressed tumor cells, initiates antitumor immunity by inducing tumor cell chemokine production and activating dendritic cells via TLR4 pathway. J Immunol. (2009) 182:1449–59. doi: 10.4049/jimmunol.182.3.1449
136. Arora SK, Singh G, Sehgal S. Comparative evaluation of anti-heat shock protein antibodies in SLE and healthy controls. Scand J Rheumatol. (1995) 24:160–3. doi: 10.3109/03009749509099306
137. Ghoreishi M, Katayama I, Yokozeki H, Nishioka K. Analysis of 70 KD heat shock protein (HSP70) expression in the lesional skin of lupus erythematosus (LE) and LE related diseases. J Dermatol. (1993) 20:400–5. doi: 10.1111/j.1346-8138.1993.tb01307.x
138. Lovis C, Mach F, Donati YR, Bonventre JV, Polla BS. Heat shock proteins and the kidney. Ren Fail. (1994) 16:179–92. doi: 10.3109/08860229409044859
139. Fürnrohr BG, Wach S, Kelly JA, Haslbeck M, Weber CK, Stach CM, et al. Polymorphisms in the Hsp70 gene locus are genetically associated with systemic lupus erythematosus. Ann Rheum Dis. (2010) 69:1983–9. doi: 10.1136/ard.2009.122630
140. Minota S, Cameron B, Welch WJ, Winfield JB. Autoantibodies to the constitutive 73-kD member of the hsp70 family of heat shock proteins in systemic lupus erythematosus. J Exp Med. (1988) 168:1475–80. doi: 10.1084/jem.168.4.1475
141. Jacquemin C, Rambert J, Guillet S, Thiolat D, Boukhedouni N, Doutre M-S, et al. Heat shock protein 70 potentiates interferon alpha production by plasmacytoid dendritic cells: relevance for cutaneous lupus and vitiligo pathogenesis. Br J Dermatol. (2017) 177:1367–75. doi: 10.1111/bjd.15550
142. Thériault JR, Adachi H, Calderwood SK. Role of scavenger receptors in the binding and internalization of heat shock protein 70. J Immunol. (2006) 177:8604–11. doi: 10.4049/jimmunol.177.12.8604
143. Calderwood SK, Theriault J, Gray PJ, Gong J. Cell surface receptors for molecular chaperones. Methods. (2007) 43:199–206. doi: 10.1016/j.ymeth.2007.06.008
144. Berwin B, Delneste Y, Lovingood RV, Post SR, Pizzo SV. SREC-I, a type F scavenger receptor, is an endocytic receptor for calreticulin*. J Biol Chem. (2004) 279:51250–7. doi: 10.1074/jbc.M406202200
145. Facciponte JG, Wang XY, Subjeck JR. Hsp110 and Grp170, members of the Hsp70 superfamily, bind to scavenger receptor-A and scavenger receptor expressed by endothelial cells-I. Eur J Immunol. (2007) 37:2268–79. doi: 10.1002/eji.200737127
146. Murshid A, Gong J, Calderwood SK. Heat shock protein 90 mediates efficient antigen cross presentation through the scavenger receptor expressed by endothelial cells-I. J Immunol. (2010) 185:2903–17. doi: 10.4049/jimmunol.0903635
147. Greaves DR, Gough PJ, Gordon S. Recent progress in defining the role of scavenger receptors in lipid transport, atherosclerosis and host defence. Curr Opin Lipidol. (1998) 9:425–32. doi: 10.1097/00041433-199810000-00006
148. Manjili MH, Henderson R, Wang XY, Chen X, Li Y, Repasky E, et al. Development of a recombinant HSP110-HER-2/neu vaccine using the chaperoning properties of HSP110. Cancer Res. (2002) 62:1737–42. PMID: 11912148
149. Murshid A, Gong J, Calderwood SK. Hsp90-peptide complexes stimulate antigen presentation through the class II pathway after binding scavenger receptor SREC-I. Immunobiology. (2014) 219:924–31. doi: 10.1016/j.imbio.2014.08.001
150. Afek A, Keren G, Harats D, George J. Whole body hyperthermia accelerates atherogenesis in low-density lipoprotein receptor deficient mice. Exp Mol Pathol. (2001) 71:63–72. doi: 10.1006/exmp.2001.2379
151. Calderwood SK, Theriault JR, Gong J. How is the immune response affected by hyperthermia and heat shock proteins? Int J Hyperthermia. (2005) 21:713–6. doi: 10.1080/02656730500340794
152. Zhu X, Ng HP, Lai YC, Craigo JK, Nagilla PS, Raghani P, et al. Scavenger receptor function of mouse fcγ receptor III contributes to progression of atherosclerosis in apolipoprotein E hyperlipidemic mice. J Immunol. (2014) 193:2483–95. doi: 10.4049/jimmunol.1303075
153. Liu A, Ferretti C, Shi F-D, Cohen IR, Quintana FJ, La Cava A. DNA vaccination with Hsp70 protects against systemic lupus erythematosus in (NZB × NZW)F1 mice. Arthritis Rheumatol. (2020) 72:997–1002. doi: 10.1002/art.41202
154. van Herwijnen MJC, Wieten L, van der Zee R, van Kooten PJ, Wagenaar-Hilbers JP, Hoek A, et al. Regulatory T cells that recognize a ubiquitous stress-inducible self-antigen are long-lived suppressors of autoimmune arthritis. Proc Natl Acad Sci USA. (2012) 109:14134–9. doi: 10.1073/pnas.1206803109
155. Boehm J, Orth T, Van Nguyen P, Söling HD. Systemic lupus erythematosus is associated with increased auto-antibody titers against calreticulin and grp94, but calreticulin is not the ro/SS-A antigen. Eur J Clin Invest. (1994) 24:248–57. doi: 10.1111/j.1365-2362.1994.tb01082.x
156. Sontheimer RD, Racila D, Racila E, Eggleton P, Donnelly S. Calreticulin's role(s) in autoimmune disorders. In: Eggleton P, Michalak M, editors. Calreticulin. 2nd edn. Boston, MA: Springer US (2003). p. 180–92.
157. Conway EM, Liu L, Nowakowski B, Steiner-Mosonyi M, Ribeiro SP, Michalak M. Heat shock-sensitive expression of calreticulin. in vitro and in vivo up-regulation. J Biol Chem. (1995) 270:17011–6. doi: 10.1074/jbc.270.28.17011
158. Eggleton P, Llewellyn DH. Pathophysiological roles of calreticulin in autoimmune disease. Scand J Immunol. (1999) 49:466–73. doi: 10.1046/j.1365-3083.1999.00542.x
159. Binder RJ, Zhou YJ, Messmer MN, Pawaria S. CD91-dependent modulation of immune responses by heat shock proteins: a role in autoimmunity. Autoimmune Dis. (2012) 2012:863041. doi: 10.1155/2012/863041
160. Overton CD, Yancey PG, Major AS, Linton MF, Fazio S. Deletion of macrophage LDL receptor-related protein increases atherogenesis in the mouse. Circ Res. (2007) 100:670–7. doi: 10.1161/01.RES.0000260204.40510.aa
161. Gaultier A, Arandjelovic S, Niessen S, Overton CD, Linton MF, Fazio S, et al. Regulation of tumor necrosis factor receptor-1 and the IKK-NF-kappaB pathway by LDL receptor-related protein explains the antiinflammatory activity of this receptor. Blood. (2008) 111:5316–25. doi: 10.1182/blood-2007-12-127613
162. Zurhove K, Nakajima C, Herz J, Bock HH, May P. Gamma-secretase limits the inflammatory response through the processing of LRP1. Sci Signal. (2008) 1:ra15. doi: 10.1126/scisignal.1164263
163. Gorovoy M, Gaultier A, Campana WM, Firestein GS, Gonias SL. Inflammatory mediators promote production of shed LRP1/CD91, which regulates cell signaling and cytokine expression by macrophages. J Leukoc Biol. (2010) 88:769–78. doi: 10.1189/jlb.0410220
164. Byrne JC, Ni Gabhann J, Stacey KB, Coffey BM, McCarthy E, Thomas W, et al. Bruton's tyrosine kinase is required for apoptotic cell uptake via regulating the phosphorylation and localization of calreticulin. J Immunol. (2013) 190:5207–15. doi: 10.4049/jimmunol.1300057
165. Rajaiah R, Moudgil KD. Heat-shock proteins can promote as well as regulate autoimmunity. Autoimmun Rev. (2009) 8:388–93. doi: 10.1016/j.autrev.2008.12.004
166. Schett G, Redlich K, Xu Q, Bizan P, Gröger M, Tohidast-Akrad M, et al. Enhanced expression of heat shock protein 70 (hsp70) and heat shock factor 1 (HSF1) activation in rheumatoid arthritis synovial tissue. Differential regulation of hsp70 expression and hsf1 activation in synovial fibroblasts by proinflammatory cytokines, shear stress, and antiinflammatory drugs. J Clin Invest. (1998) 102:302–11. doi: 10.1172/JCI2465
167. Waldheim E, Ajeganova S, Bergman S, Frostegård J, Welin E. Variation in pain related to systemic lupus erythematosus (SLE): a 7-year follow-up study. Clin Rheumatol. (2018) 37:1825–34. doi: 10.1007/s10067-018-4079-1
168. Gladman D, Urowitz M, Klippel J. Primer on the Rheumatic Diseases. New York, NY: Springer Science+Business Media (1997).
169. Greco CM, Rudy TE, Manzi S. Adaptation to chronic pain in systemic lupus erythematosus: applicability of the multidimensional pain inventory. Pain Medicine. (2003) 4:39–50. doi: 10.1046/j.1526-4637.2003.03001.x
170. Nyman E, Vaughan T, Desta B, Wang X, Barut V, Emmas C. Characteristics and symptom severity of patients reporting systemic lupus erythematosus in the PatientsLikeMe online health community: a retrospective observational study. Rheumatol Ther. (2020) 7:201–13. doi: 10.1007/s40744-020-00195-7
171. Pisetsky DS, Eudy AM, Clowse MEB, Rogers JL. The categorization of pain in systemic lupus erythematosus. Rheum Dis Clin North Am. (2021) 47:215–28. doi: 10.1016/j.rdc.2020.12.004
172. García-Fernández P, Üçeyler N, Sommer C. From the low-density lipoprotein receptor-related protein 1 to neuropathic pain: a potentially novel target. Pain Rep. (2021) 6:e898. doi: 10.1097/PR9.0000000000000898
173. Brifault C, Kwon H, Campana WM, Gonias SL. LRP1 deficiency in microglia blocks neuro-inflammation in the spinal dorsal horn and neuropathic pain processing. Glia. (2019) 67:1210–24. doi: 10.1002/glia.23599
174. Orita S, Henry K, Mantuano E, Yamauchi K, De Corato A, Ishikawa T, et al. Schwann cell LRP1 regulates remak bundle ultrastructure and axonal interactions to prevent neuropathic pain. J Neurosci. (2013) 33:5590–602. doi: 10.1523/JNEUROSCI.3342-12.2013
175. Poplawski G, Ishikawa T, Brifault C, Lee-Kubli C, Regestam R, Henry KW, et al. Schwann cells regulate sensory neuron gene expression before and after peripheral nerve injury. Glia. (2018) 66:1577–90. doi: 10.1002/glia.23325
176. Yancey PG, Blakemore J, Ding L, Fan D, Overton CD, Zhang Y, et al. Macrophage LRP-1 controls plaque cellularity by regulating efferocytosis and akt activation. Arterioscler Thromb Vasc Biol. (2010) 30:787–95. doi: 10.1161/ATVBAHA.109.202051
177. Gaultier A, Arandjelovic S, Li X, Janes J, Dragojlovic N, Zhou GP, et al. A shed form of LDL receptor-related protein-1 regulates peripheral nerve injury and neuropathic pain in rodents. J Clin Invest. (2008) 118:161–72. doi: 10.1172/JCI32371
178. Mantuano E, Brifault C, Lam MS, Azmoon P, Gilder AS, Gonias SL. LDL receptor-related protein-1 regulates NFκB and microRNA-155 in macrophages to control the inflammatory response. Proc Natl Acad Sci U S A. (2016) 113:1369–74. doi: 10.1073/pnas.1515480113
179. Yang L, Liu CC, Zheng H, Kanekiyo T, Atagi Y, Jia L, et al. LRP1 modulates the microglial immune response via regulation of JNK and NF-κB signaling pathways. J Neuroinflammation. (2016) 13:304. doi: 10.1186/s12974-016-0772-7
180. He Y, Ruganzu JB, Zheng Q, Wu X, Jin H, Peng X, et al. Silencing of LRP1 exacerbates inflammatory response via TLR4/NF-κB/MAPKs signaling pathways in APP/PS1 transgenic mice. Mol Neurobiol. (2020) 57:3727–43. doi: 10.1007/s12035-020-01982-7
181. Liu CC, Hu J, Zhao N, Wang J, Wang N, Cirrito JR, et al. Astrocytic LRP1 mediates brain aβ clearance and impacts amyloid deposition. J Neurosci. (2017) 37:4023–31. doi: 10.1523/JNEUROSCI.3442-16.2017
182. Ma LY, Fei YL, Wang XY, Wu SD, Du JH, Zhu M, et al. The research on the relationship of RAGE, LRP-1, and aβ accumulation in the hippocampus, prefrontal lobe, and amygdala of STZ-induced diabetic rats. J Mol Neurosci. (2017) 62:1–10. doi: 10.1007/s12031-017-0892-2
183. Patel P, Shah J. Role of vitamin D in amyloid clearance via LRP-1 upregulation in Alzheimer's disease: a potential therapeutic target? J Chem Neuroanat. (2017) 85:36–42. doi: 10.1016/j.jchemneu.2017.06.007
184. Zhang H, Chen W, Tan Z, Zhang L, Dong Z, Cui W, et al. A role of low-density lipoprotein receptor-related protein 4 (LRP4) in astrocytic aβ clearance. J Neurosci. (2020) 40:5347–61. doi: 10.1523/JNEUROSCI.0250-20.2020
185. Pazhouhandeh M, Sahraian MA, Siadat SD, Fateh A, Vaziri F, Tabrizi F, et al. A systems medicine approach reveals disordered immune system and lipid metabolism in multiple sclerosis patients. Clin Exp Immunol. (2018) 192:18–32. doi: 10.1111/cei.13087
186. Pocivavsek A, Mikhailenko I, Strickland DK, Rebeck GW. Microglial low-density lipoprotein receptor-related protein 1 modulates c-jun N-terminal kinase activation. J Neuroimmunol. (2009) 214:25–32. doi: 10.1016/j.jneuroim.2009.06.010
187. Pocivavsek A, Burns MP, Rebeck GW. Low-density lipoprotein receptors regulate microglial inflammation through c-jun N-terminal kinase. Glia. (2009) 57:444–53. doi: 10.1002/glia.20772
188. Staudt ND, Jo M, Hu J, Bristow JM, Pizzo DP, Gaultier A, et al. Myeloid cell receptor LRP1/CD91 regulates monocyte recruitment and angiogenesis in tumors. Cancer Res. (2013) 73:3902–12. doi: 10.1158/0008-5472.CAN-12-4233
189. Chuang TY, Guo Y, Seki SM, Rosen AM, Johanson DM, Mandell JW, et al. LRP1 expression in microglia is protective during CNS autoimmunity. Acta Neuropathol Commun. (2016) 4:68. doi: 10.1186/s40478-016-0343-2
190. Ooka S, Matsui T, Nishioka K, Kato T. Autoantibodies to low-density-lipoprotein-receptor-related protein 2 (LRP2) in systemic autoimmune diseases. Arthritis Res Ther. (2003) 5:R174–80. doi: 10.1186/ar754
191. Stremmel C, Horn C, Eder S, Dimmler A, Lang W. The impact of immunological parameters on the development of phantom pain after major amputation. Eur J Vasc Endovasc Surg. (2005) 30:79–82. doi: 10.1016/j.ejvs.2005.02.050
192. Wagner R, Heckman HM, Myers RR. Wallerian degeneration and hyperalgesia after peripheral nerve injury are glutathione-dependent. Pain. (1998) 77:173–9. doi: 10.1016/S0304-3959(98)00091-8
193. Wagner R, Janjigian M, Myers RR. Anti-inflammatory interleukin-10 therapy in CCI neuropathy decreases thermal hyperalgesia, macrophage recruitment, and endoneurial TNF-alpha expression. Pain. (1998) 74:35–42. doi: 10.1016/S0304-3959(97)00148-6
194. Ceyhan GO, Deucker S, Demir IE, Erkan M, Schmelz M, Bergmann F, et al. Neural fractalkine expression is closely linked to pain and pancreatic neuritis in human chronic pancreatitis. Lab Invest. (2009) 89:347–61. doi: 10.1038/labinvest.2008.170
196. Sandy-Hindmarch OP, Chang P-S, Scheuren PS, De Schoenmacker I, Hubli M, Loizou C, et al. The local molecular signature of human peripheral neuropathic pain. PAIN. (2024) 166(5):1143–56. doi: 10.1097/j.pain.0000000000003472
197. Dongfeng R, Hou S, Wu W, Wang H, Shang W, Tang J, et al. The expression of tumor necrosis factor-α and CD68 in high-intensity zone of lumbar intervertebral disc on magnetic resonance image in the patients with low back pain. Spine. (2011) 36:E429–33. doi: 10.1097/BRS.0b013e3181dfce9e
198. Wijesinghe SN, Ditchfield C, Flynn S, Agrawal J, Davis ET, Dajas-Bailador F, et al. Immunomodulation and fibroblast dynamics driving nociceptive joint pain within inflammatory synovium: unravelling mechanisms for therapeutic advancements in osteoarthritis. Osteoarthritis Cartilage. (2024) 32:1358–70. doi: 10.1016/j.joca.2024.06.011
199. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. (2016) 388:2023–38. doi: 10.1016/S0140-6736(16)30173-8
200. Ceccarelli F, Perricone C, Cipriano E, Massaro L, Natalucci F, Capalbo G, et al. Joint involvement in systemic lupus erythematosus: from pathogenesis to clinical assessment. Semin Arthritis Rheum. (2017) 47:53–64. doi: 10.1016/j.semarthrit.2017.03.022
201. Hasegawa T, Lee CYC, Hotchen AJ, Fleming A, Singh R, Suzuki K, et al. Macrophages and nociceptor neurons form a sentinel unit around fenestrated capillaries to defend the synovium from circulating immune challenge. Nat Immunol. (2024) 25:2270–83. doi: 10.1038/s41590-024-02011-8
202. Ohashi Y, Uchida K, Fukushima K, Satoh M, Koyama T, Tsuchiya M, et al. Correlation between CD163 expression and resting pain in patients with hip osteoarthritis: possible contribution of CD163 + monocytes/macrophages to pain pathogenesis. J Orthop Res. (2022) 40:1365–74. doi: 10.1002/jor.25157
203. Cunha TM, Verri WA, Silva JS, Poole S, Cunha FQ, Ferreira SH. A cascade of cytokines mediates mechanical inflammatory hypernociception in mice. Proc Natl Acad Sci U S A. (2005) 102:1755–60. doi: 10.1073/pnas.0409225102
204. Román-Fernández IV, García-Chagollán M, Cerpa-Cruz S, Jave-Suárez LF, Palafox-Sánchez CA, García-Arellano S, et al. Assessment of CD40 and CD40l expression in rheumatoid arthritis patients, association with clinical features and DAS28. Clin Exp Med. (2019) 19:427–37. doi: 10.1007/s10238-019-00568-5
205. Kawai T, Andrews D, Colvin RB, Sachs DH, Cosimi AB. Thromboembolic complications after treatment with monoclonal antibody against CD40 ligand. Nat Med. (2000) 6:114–114. doi: 10.1038/72162
206. Suttles J, Stout RD. Macrophage CD40 signaling: a pivotal regulator of disease protection and pathogenesis. Semin Immunol. (2009) 21:257–64. doi: 10.1016/j.smim.2009.05.011
207. Hanlon MM, Smith CM, Canavan M, Neto NGB, Song Q, Lewis MJ, et al. Loss of synovial tissue macrophage homeostasis precedes rheumatoid arthritis clinical onset. Sci Adv. (2024) 10:eadj1252. doi: 10.1126/sciadv.adj1252
208. Reynders A, Anissa Jhumka Z, Gaillard S, Mantilleri A, Malapert P, Magalon K, et al. Gut microbiota promotes pain chronicity in Myosin1A deficient male mice. Brain Behav Immun. (2024) 119:750–66. doi: 10.1016/j.bbi.2024.05.010
209. Muller WA. Getting leukocytes to the site of inflammation. Vet Pathol. (2013) 50:7–22. doi: 10.1177/0300985812469883
210. Filippi MD. Mechanism of diapedesis: importance of the transcellular route. Adv Immunol. (2016) 129:25–53. doi: 10.1016/bs.ai.2015.09.001
211. Ramirez GA, Rovere-Querini P, Sabbadini MG, Manfredi AA. Parietal and intravascular innate mechanisms of vascular inflammation. Arthritis Res Ther. (2015) 17:16. doi: 10.1186/s13075-015-0528-2
212. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol. (2011) 187:538–52. doi: 10.4049/jimmunol.1100450
213. Hakkim A, Fürnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A. (2010) 107:9813–8. doi: 10.1073/pnas.0909927107
214. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med. (2011) 3:73ra19. doi: 10.1126/scitranslmed.3001180
215. Leffler J, Martin M, Gullstrand B, Tydén H, Lood C, Truedsson L, et al. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol. (2012) 188:3522–31. doi: 10.4049/jimmunol.1102404
216. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. (2011) 11:519–31. doi: 10.1038/nri3024
217. Perdomo J, Leung HHL, Ahmadi Z, Yan F, Chong JJH, Passam FH, et al. Neutrophil activation and NETosis are the major drivers of thrombosis in heparin-induced thrombocytopenia. Nat Commun. (2019) 10:1322. doi: 10.1038/s41467-019-09160-7
218. Zhu Y, Yang Y, Li F, Fan S, Chen X, Lu Y, et al. Stimulation of the class-A scavenger receptor induces neutrophil extracellular traps (NETs) by ERK dependent NOX2 and ROMO1 activation. Biochem Biophys Res Commun. (2019) 511:847–54. doi: 10.1016/j.bbrc.2019.02.142
219. Manchanda AS, Kwan AC, Ishimori M, Thomson LEJ, Li D, Berman DS, et al. Coronary microvascular dysfunction in patients with systemic lupus erythematosus and chest pain. Front Cardiovasc Med. (2022) 9:867155. doi: 10.3389/fcvm.2022.867155
220. McCarthy EM, Smith S, Lee RZ, Cunnane G, Doran MF, Donnelly S, et al. The association of cytokines with disease activity and damage scores in systemic lupus erythematosus patients. Rheumatology (Oxford). (2014) 53:1586–94. doi: 10.1093/rheumatology/ket428
221. Živković V, Cvetković T, Mitić B, Stamenković B, Stojanović S, Radovanović-Dinić B, et al. Monocyte chemoattractant protein-1 as a marker of systemic lupus erythematosus: an observational study. Rheumatol Int. (2018) 38:1003–8. doi: 10.1007/s00296-017-3888-x
222. Lee YH, Song GG. Urinary MCP-1 as a biomarker for lupus nephritis: a meta-analysis. Z Rheumatol. (2017) 76:357–63. doi: 10.1007/s00393-016-0109-z
223. Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res. (2016) 118:653–67. doi: 10.1161/CIRCRESAHA.115.306256
224. Park YM, Febbraio M, Silverstein RL. CD36 modulates migration of mouse and human macrophages in response to oxidized LDL and may contribute to macrophage trapping in the arterial intima. J Clin Invest. (2009) 119:136–45. doi: 10.1172/JCI35535
225. Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol. (2013) 14:812–20. doi: 10.1038/ni.2639
226. Robbins CS, Hilgendorf I, Weber GF, Theurl I, Iwamoto Y, Figueiredo JL, et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med. (2013) 19:1166–72. doi: 10.1038/nm.3258
227. Alquraini A, El Khoury J. Scavenger receptors. Curr Biol. (2020) 30:R790–5. doi: 10.1016/j.cub.2020.05.051
228. Qin M, Guo Y, Jiang L, Wang X. Elevated levels of serum sCXCL16 in systemic lupus erythematosus; potential involvement in cutaneous and renal manifestations. Clin Rheumatol. (2014) 33:1595–601. doi: 10.1007/s10067-014-2741-9
229. Hassan AM, Farghal NMA, Hegab DS, Mohamed WS, Abd-Elnabi HH. Serum-soluble CXCL16 in juvenile systemic lupus erythematosus: a promising predictor of disease severity and lupus nephritis. Clin Rheumatol. (2018) 37:3025–32. doi: 10.1007/s10067-018-4203-2
230. Libby P, Nahrendorf M, Swirski FK. Leukocytes link local and systemic inflammation in ischemic cardiovascular disease: an expanded “cardiovascular Continuum”. J Am Coll Cardiol. (2016) 67:1091–103. doi: 10.1016/j.jacc.2015.12.048
231. Hilgendorf I, Theurl I, Gerhardt LMS, Robbins CS, Weber GF, Gonen A, et al. Innate response activator B cells aggravate atherosclerosis by stimulating T helper-1 adaptive immunity. Circulation. (2014) 129:1677–87. doi: 10.1161/CIRCULATIONAHA.113.006381
232. Swirski FK, Hilgendorf I, Robbins CS. From proliferation to proliferation: monocyte lineage comes full circle. Semin Immunopathol. (2014) 36:137–48. doi: 10.1007/s00281-013-0409-1
233. Clinton SK, Underwood R, Hayes L, Sherman ML, Kufe DW, Libby P. Macrophage colony-stimulating factor gene expression in vascular cells and in experimental and human atherosclerosis. Am J Pathol. (1992) 140:301–16. PMID: 1739124
234. Sugiyama S, Okada Y, Sukhova GK, Virmani R, Heinecke JW, Libby P. Macrophage myeloperoxidase regulation by granulocyte macrophage colony-stimulating factor in human atherosclerosis and implications in acute coronary syndromes. Am J Pathol. (2001) 158:879–91. doi: 10.1016/S0002-9440(10)64036-9
235. Reiss AB, Wan DW, Anwar K, Merrill JT, Wirkowski PA, Shah N, et al. Enhanced CD36 scavenger receptor expression in THP-1 human monocytes in the presence of lupus plasma: linking autoimmunity and atherosclerosis. Exp Biol Med (Maywood). (2009) 234:354–60. doi: 10.3181/0806-BC-194
236. Febbraio M, Podrez EA, Smith JD, Hajjar DP, Hazen SL, Hoff HF, et al. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest. (2000) 105:1049–56. doi: 10.1172/JCI9259
237. Kuchibhotla S, Vanegas D, Kennedy DJ, Guy E, Nimako G, Morton RE, et al. Absence of CD36 protects against atherosclerosis in ApoE knock-out mice with no additional protection provided by absence of scavenger receptor A I/II. Cardiovasc Res. (2008) 78:185–96. doi: 10.1093/cvr/cvm093
238. Matsuura E, Kobayashi K, Hurley BL, Lopez LR. Atherogenic oxidized low-density lipoprotein/beta2-glycoprotein I (oxLDL/beta2GPI) complexes in patients with systemic lupus erythematosus and antiphospholipid syndrome. Lupus. (2006) 15:478–83. doi: 10.1191/0961203306lu2337oa
239. Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. (2010) 10:36–46. doi: 10.1038/nri2675
240. Tabas I, García-Cardeña G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol. (2015) 209:13–22. doi: 10.1083/jcb.201412052
241. Thorp E, Cui D, Schrijvers DM, Kuriakose G, Tabas I. Mertk receptor mutation reduces efferocytosis efficiency and promotes apoptotic cell accumulation and plaque necrosis in atherosclerotic lesions of apoe−/− mice. Arterioscler Thromb Vasc Biol. (2008) 28:1421–8. doi: 10.1161/ATVBAHA.108.167197
242. Wallet MA, Sen P, Flores RR, Wang Y, Yi Z, Huang Y, et al. MerTK is required for apoptotic cell–induced T cell tolerance. J Exp Med. (2008) 205:219–32. doi: 10.1084/jem.20062293
243. Wu J, Ekman C, Jönsen A, Sturfelt G, Bengtsson AA, Gottsäter A, et al. Increased plasma levels of the soluble mer tyrosine kinase receptor in systemic lupus erythematosus relate to disease activity and nephritis. Arthritis Res Ther. (2011) 13:R62. doi: 10.1186/ar3316
244. Zizzo G, Guerrieri J, Dittman LM, Merrill JT, Cohen PL. Circulating levels of soluble MER in lupus reflect M2c activation of monocytes/macrophages, autoantibody specificities and disease activity. Arthritis Res Ther. (2013) 15:R212. doi: 10.1186/ar4407
245. Kattoor AJ, Goel A, Mehta JL. LOX-1: regulation, signaling and its role in atherosclerosis. Antioxidants (Basel). (2019) 8(7):1–15. doi: 10.3390/antiox8070218
246. Bergkamp SC, Wahadat MJ, Salah A, Kuijpers TW, Smith V, Tas SW, et al. Dysregulated endothelial cell markers in systemic lupus erythematosus: a systematic review and meta-analysis. J Inflamm. (2023) 20:18. doi: 10.1186/s12950-023-00342-1
247. Seternes T, Sørensen K, Smedsrød B. Scavenger endothelial cells of vertebrates: a nonperipheral leukocyte system for high-capacity elimination of waste macromolecules. Proc Natl Acad Sci U S A. (2002) 99:7594–7. doi: 10.1073/pnas.102173299
248. Smedsrød B. Clearance function of scavenger endothelial cells. Comp Hepatol. (2004) 3(Suppl 1):S22. doi: 10.1186/1476-5926-2-S1-S22
249. Sørensen KK, McCourt P, Berg T, Crossley C, Couteur DL, Wake K, et al. The scavenger endothelial cell: a new player in homeostasis and immunity. Am J Physiol Regul Integr Comp Physiol. (2012) 303:R1217–30. doi: 10.1152/ajpregu.00686.2011
250. Patten DA, Kamarajah SK, Rose JM, Tickle J, Shepherd EL, Adams DH, et al. SCARF-1 promotes adhesion of CD4+ T cells to human hepatic sinusoidal endothelium under conditions of shear stress. Sci Rep. (2017) 7:17600. doi: 10.1038/s41598-017-17928-4
251. Amersfoort J, Eelen G, Carmeliet P. Immunomodulation by endothelial cells — partnering up with the immune system? Nat Rev Immunol. (2022) 22:576–88. doi: 10.1038/s41577-022-00694-4
252. Patten DA, Wilson GK, Bailey D, Shaw RK, Jalkanen S, Salmi M, et al. Human liver sinusoidal endothelial cells promote intracellular crawling of lymphocytes during recruitment: a new step in migration. Hepatology. (2017) 65:294–309. doi: 10.1002/hep.28879
253. Shetty S, Weston CJ, Oo YH, Westerlund N, Stamataki Z, Youster J, et al. Common lymphatic endothelial and vascular endothelial receptor-1 mediates the transmigration of regulatory T cells across human hepatic sinusoidal endothelium. J Immunol. (2011) 186:4147–55. doi: 10.4049/jimmunol.1002961
254. Shetty S, Bruns T, Weston CJ, Stamataki Z, Oo YH, Long HM, et al. Recruitment mechanisms of primary and malignant B cells to the human liver. Hepatology. (2012) 56:1521–31. doi: 10.1002/hep.25790
255. Jung M-Y, Park S-Y, Kim I-S. Stabilin-2 is involved in lymphocyte adhesion to the hepatic sinusoidal endothelium via the interaction with αMβ2 integrin. J Leukocyte Biol. (2007) 82:1156–65. doi: 10.1189/jlb.0107052
256. Duryee MJ, Freeman TL, Willis MS, Hunter CD, Hamilton BC 3rd, Suzuki H, et al. Thiele, scavenger receptors on sinusoidal liver endothelial cells are involved in the uptake of aldehyde-modified proteins. Mol Pharmacol. (2005) 68:1423–30. doi: 10.1124/mol.105.016121
257. Malerød L, Juvet K, Gjøen T, Berg T. The expression of scavenger receptor class B, type I (SR-BI) and caveolin-1 in parenchymal and nonparenchymal liver cells. Cell Tissue Res. (2002) 307:173–80. doi: 10.1007/s00441-001-0476-9
258. Elvevold K, Simon-Santamaria J, Hasvold H, McCourt P, Smedsrød B, Sørensen KK. Liver sinusoidal endothelial cells depend on mannose receptor-mediated recruitment of lysosomal enzymes for normal degradation capacity. Hepatology. (2008) 48:2007–15. doi: 10.1002/hep.22527
259. Malovic I, Sørensen KK, Elvevold KH, Nedredal GI, Paulsen S, Erofeev AV, et al. The mannose receptor on murine liver sinusoidal endothelial cells is the main denatured collagen clearance receptor. Hepatology. (2007) 45:1454–61. doi: 10.1002/hep.21639
260. Taban Q, Mumtaz PT, Masoodi KZ, Haq E, Ahmad SM. Scavenger receptors in host defense: from functional aspects to mode of action. Cell Commun Signal. (2022) 20:2. doi: 10.1186/s12964-021-00812-0
261. Irjala H, Elima K, Johansson EL, Merinen M, Kontula K, Alanen K, et al. The same endothelial receptor controls lymphocyte traffic both in vascular and lymphatic vessels. Eur J Immunol. (2003) 33:815–24. doi: 10.1002/eji.200323859
262. Ishii J, Adachi H, Aoki J, Koizumi H, Tomita S, Suzuki T, et al. SREC-II, a new member of the scavenger receptor type F family, trans-interacts with SREC-I through its extracellular domain. J Biol Chem. (2002) 277:39696–702. doi: 10.1074/jbc.M206140200
263. Karikoski M, Irjala H, Maksimow M, Miiluniemi M, Granfors K, Hernesniemi S, et al. Clever-1/stabilin-1 regulates lymphocyte migration within lymphatics and leukocyte entrance to sites of inflammation. Eur J Immunol. (2009) 39:3477–87. doi: 10.1002/eji.200939896
264. Adachi H, Tsujimoto M, Arai H, Inoue K. Expression cloning of a novel scavenger receptor from human endothelial cells. J Biol Chem. (1997) 272:31217–20. doi: 10.1074/jbc.272.50.31217
265. Bertolino P, Schrage A, Bowen DG, Klugewitz K, Ghani S, Eulenburg K, et al. Early intrahepatic antigen-specific retention of naïve CD8+ T cells is predominantly ICAM-1/LFA-1 dependent in mice. Hepatology. (2005) 42:1063–71. doi: 10.1002/hep.20885
266. John B, Crispe IN. Passive and active mechanisms trap activated CD8+ T cells in the liver. J Immunol. (2004) 172:5222–9. doi: 10.4049/jimmunol.172.9.5222
267. Limmer A, Ohl J, Wingender G, Berg M, Jüngerkes F, Schumak B, et al. Cross-presentation of oral antigens by liver sinusoidal endothelial cells leads to CD8T cell tolerance. Eur J Immunol. (2005) 35:2970–81. doi: 10.1002/eji.200526034
268. Limmer A, Ohl J, Kurts C, Ljunggren HG, Reiss Y, Groettrup M, et al. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat Med. (2000) 6:1348–54. doi: 10.1038/82161
269. Diehl L, Schurich A, Grochtmann R, Hegenbarth S, Chen L, Knolle PA. Tolerogenic maturation of liver sinusoidal endothelial cells promotes B7-homolog 1-dependent CD8+ T cell tolerance. Hepatology. (2008) 47:296–305. doi: 10.1002/hep.21965
270. Burgdorf S, Kautz A, Böhnert V, Knolle PA, Kurts C. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8T cell activation. Science. (2007) 316:612–6. doi: 10.1126/science.1137971
271. Carambia A, Freund B, Schwinge D, Heine M, Laschtowitz A, Huber S, et al. TGF-β-dependent induction of CD4+CD25+Foxp3+ tregs by liver sinusoidal endothelial cells. J Hepatol. (2014) 61:594–9. doi: 10.1016/j.jhep.2014.04.027
272. Manta CP, Leibing T, Friedrich M, Nolte H, Adrian M, Schledzewski K, et al. Targeting of scavenger receptors stabilin-1 and stabilin-2 ameliorates atherosclerosis by a plasma proteome switch mediating monocyte/macrophage suppression. Circulation. (2022) 146:1783–99. doi: 10.1161/CIRCULATIONAHA.121.058615
273. Hayashida K, Kume N, Minami M, Kita T. Lectin-like oxidized LDL receptor-1 (LOX-1) supports adhesion of mononuclear leukocytes and a monocyte-like cell line THP-1 cells under static and flow conditions. FEBS Lett. (2002) 511:133–8. doi: 10.1016/S0014-5793(01)03297-5
274. Honjo M, Nakamura K, Yamashiro K, Kiryu J, Tanihara H, McEvoy LM, et al. Lectin-like oxidized LDL receptor-1 is a cell-adhesion molecule involved in endotoxin-induced inflammation. Proc Natl Acad Sci U S A. (2003) 100:1274–9. doi: 10.1073/pnas.0337528100
275. Salmi M, Koskinen K, Henttinen T, Elima K, Jalkanen S. CLEVER-1 mediates lymphocyte transmigration through vascular and lymphatic endothelium. Blood. (2004) 104:3849–57. doi: 10.1182/blood-2004-01-0222
276. Wang L, Luqmani R, Udalova IA. The role of neutrophils in rheumatic disease-associated vascular inflammation. Nat Rev Rheumatol. (2022) 18:158–70. doi: 10.1038/s41584-021-00738-4
277. Moore S, Juo H-H, Nielsen CT, Tyden H, Bengtsson AA, Lood C. Role of neutrophil extracellular traps regarding patients at risk of increased disease activity and cardiovascular comorbidity in systemic lupus erythematosus. J Rheumatol. (2020) 47:1652–60. doi: 10.3899/jrheum.190875
278. Pieterse E, Rother N, Garsen M, Hofstra JM, Satchell SC, Hoffmann M, et al. Neutrophil extracellular traps drive endothelial-to-mesenchymal transition. Arterioscler Thromb Vasc Biol. (2017) 37:1371–9. doi: 10.1161/ATVBAHA.117.309002
279. Boone BA, Orlichenko L, Schapiro NE, Loughran P, Gianfrate GC, Ellis JT, et al. The receptor for advanced glycation end products (RAGE) enhances autophagy and neutrophil extracellular traps in pancreatic cancer. Cancer Gene Ther. (2015) 22:326–34. doi: 10.1038/cgt.2015.21
280. Megha KB, Joseph X, Akhil V, Mohanan PV. Cascade of immune mechanism and consequences of inflammatory disorders. Phytomedicine. (2021) 91:153712. doi: 10.1016/j.phymed.2021.153712
281. Bennett JM, Reeves G, Billman GE, Sturmberg JP. Inflammation–nature's way to efficiently respond to all types of challenges: implications for understanding and managing “the epidemic” of chronic diseases. Front Med (Lausanne). (2018) 5:1–30. doi: 10.3389/fmed.2018.00316
282. Marshall JC. SIRS and MODS: what is their relevance to the science and practice of intensive care? Shock. (2000) 14:586–9. doi: 10.1097/00024382-200014060-00002
283. Muhammad O, Jindal H, Sharath M, Khan AM, Choi S. Systemic lupus erythematosus with multi-organ involvement in a young female: lymphadenopathy, lupus cerebritis, lupus nephritis, and cardiac manifestations. Cureus. (2021) 13:e15517. doi: 10.7759/cureus.15517
284. Tsokos GC. Autoimmunity and organ damage in systemic lupus erythematosus. Nat Immunol. (2020) 21:605–14. doi: 10.1038/s41590-020-0677-6
285. Murimi-Worstell IB, Lin DH, Nab H, Kan HJ, Onasanya O, Tierce JC, et al. Association between organ damage and mortality in systemic lupus erythematosus: a systematic review and meta-analysis. BMJ Open. (2020) 10:e031850. doi: 10.1136/bmjopen-2019-031850
286. Chiu YM, Chuang MT, Lang HC. Medical costs incurred by organ damage caused by active disease, comorbidities and side effect of treatments in systemic lupus erythematosus patients: a Taiwan nationwide population-based study. Rheumatol Int. (2016) 36:1507–14. doi: 10.1007/s00296-016-3551-y
287. Bell CF, Ajmera MR, Meyers J. An evaluation of costs associated with overall organ damage in patients with systemic lupus erythematosus in the United States. Lupus. (2022) 31:202–11. doi: 10.1177/09612033211073670
288. Schultze M, Garal-Pantaler E, Pignot M, Levy RA, Carnarius H, Schneider M, et al. Clinical and economic burden of organ damage among patients with systemic lupus erythematosus in a real-world setting in Germany. BMC Rheumatology. (2024) 8:18. doi: 10.1186/s41927-024-00387-6
289. Kang SC, Hwang SJ, Chang YS, Chou CT, Tsai CY. Characteristics of comorbidities and costs among patients who died from systemic lupus erythematosus in Taiwan. Arch Med Sci. (2012) 8:690–6. doi: 10.5114/aoms.2012.30293
290. Gulinello M, Wen J, Putterman C. Neuropsychiatric symptoms in lupus. Psychiatr Ann. (2012) 42:322–8. doi: 10.3928/00485713-20120906-05
291. van Dam AP. Diagnosis and pathogenesis of CNS lupus. Rheumatol Int. (1991) 11:1–11. doi: 10.1007/BF00290244
292. Ballok DA. Neuroimmunopathology in a murine model of neuropsychiatric lupus. Brain Res Rev. (2007) 54:67–79. doi: 10.1016/j.brainresrev.2006.12.003
293. Antonchak MA, Saoudian M, Khan AR, Brunner HI, Luggen ME. Cognitive dysfunction in patients with systemic lupus erythematosus: a controlled study. J Rheumatol. (2011) 38:1020–5. doi: 10.3899/jrheum.100560
294. Colamussi P, Giganti M, Cittanti C, Dovigo L, Trotta F, Tola MR, et al. Brain single-photon emission tomography with 99mTc-HMPAO in neuropsychiatric systemic lupus erythematosus: relations with EEG and MRI findings and clinical manifestations. Eur J Nucl Med. (1995) 22:17–24. doi: 10.1007/BF00997243
295. Handa R, Sahota P, Kumar M, Jagannathan NR, Bal CS, Gulati M, et al. in vivo proton magnetic resonance spectroscopy (MRS) and single photon emission computerized tomography (SPECT) in systemic lupus erythematosus (SLE). Magn Reson Imaging. (2003) 21:1033–7. doi: 10.1016/S0730-725X(03)00200-5
296. Huang WS, Chiu PY, Tsai CH, Kao A, Lee CC. Objective evidence of abnormal regional cerebral blood flow in patients with systemic lupus erythematosus on tc-99 m ECD brain SPECT. Rheumatol Int. (2002) 22:178–81. doi: 10.1007/s00296-002-0224-9
297. López-Longo FJ, Carol N, Almoguera MI, Olazarán J, Alonso-Farto JC, Ortega A, et al. Cerebral hypoperfusion detected by SPECT in patients with systemic lupus erythematosus is related to clinical activity and cumulative tissue damage. Lupus. (2003) 12:813–9. doi: 10.1191/0961203303lu470oa
298. Luyendijk J, Steens SC, Ouwendijk WJ, Steup-Beekman GM, Bollen EL, van der Grond J, et al. Neuropsychiatric systemic lupus erythematosus: lessons learned from magnetic resonance imaging. Arthritis Rheum. (2011) 63:722–32. doi: 10.1002/art.30157
299. Komatsu N, Kodama K, Yamanouchi N, Okada S, Noda S, Nawata Y, et al. Decreased regional cerebral metabolic rate for glucose in systemic lupus erythematosus patients with psychiatric symptoms. Eur Neurol. (1999) 42:41–8. doi: 10.1159/000008067
300. Sibbitt WL Jr., Sibbitt RR. Magnetic resonance spectroscopy and positron emission tomography scanning in neuropsychiatric systemic lupus erythematosus. Rheum Dis Clin North Am. (1993) 19:851–68. doi: 10.1016/S0889-857X(21)00210-6
301. Brooks WM, Sabet A, Sibbitt WL Jr., Barker PB, van Zijl PC, Duyn JH, et al. Neurochemistry of brain lesions determined by spectroscopic imaging in systemic lupus erythematosus. J Rheumatol. (1997) 24:2323–9. PMID: 9415636
302. Volkow ND, Warner N, McIntyre R, Valentine A, Kulkarni M, Mullani N, et al. Cerebral involvement in systemic lupus erythematosus. Am J Physiol Imaging. (1988) 3:91–8. PMID: 3260788
303. Sibbitt WL Jr., Haseler LJ, Griffey RH, Hart BL, Sibbitt RR, Matwiyoff NA. Analysis of cerebral structural changes in systemic lupus erythematosus by proton MR spectroscopy. AJNR Am J Neuroradiol. (1994) 15:923–8. PMID: 8059662
304. Gonzalez-Scarano F, Lisak RP, Bilaniuk LT, Zimmerman RA, Atkins PC, Zweiman B. Cranial computed tomography in the diagnosis of systemic lupus erythematosus. Ann Neurol. (1979) 5:158–65. doi: 10.1002/ana.410050209
305. Kaell AT, Shetty M, Lee BC, Lockshin MD. The diversity of neurologic events in systemic lupus erythematosus. Prospective clinical and computed tomographic classification of 82 events in 71 patients. Arch Neurol. (1986) 43:273–6. doi: 10.1001/archneur.1986.00520030063016
306. Miguel EC, Pereira RM, Pereira CA, Baer L, Gomes RE, de Sá LC, et al. Psychiatric manifestations of systemic lupus erythematosus: clinical features, symptoms, and signs of central nervous system activity in 43 patients. Medicine (Baltimore). (1994) 73:224–32. doi: 10.1097/00005792-199407000-00005
307. Omdal R, Selseth B, Kløw NE, Husby G, Mellgren SI. Clinical neurological, electrophysiological, and cerebral CT scan findings in systemic lupus erythematosus. Scand J Rheumatol. (1989) 18:283–9. doi: 10.3109/03009748909095031
308. Ainiala H, Dastidar P, Loukkola J, Lehtimäki T, Korpela M, Peltola J, et al. Cerebral MRI abnormalities and their association with neuropsychiatric manifestations in SLE: a population-based study. Scand J Rheumatol. (2005) 34:376–82. doi: 10.1080/03009740510026643
309. Waterloo K, Omdal R, Jacobsen EA, Kløw NE, Husby G, Torbergsen T, et al. Cerebral computed tomography and electroencephalography compared with neuropsychological findings in systemic lupus erythematosus. J Neurol. (1999) 246:706–11. doi: 10.1007/s004150050436
310. Kozora E, Filley CM. Cognitive dysfunction and white matter abnormalities in systemic lupus erythematosus. J Int Neuropsychol Soc. (2011) 17:385–92. doi: 10.1017/S1355617711000191
311. Makinde HM, Winter DR, Procissi D, Mike EV, Stock AD, Kando MJ, et al. A novel microglia-specific transcriptional signature correlates with behavioral deficits in neuropsychiatric lupus. Front Immunol. (2020) 11:230. doi: 10.3389/fimmu.2020.00230
312. Lepore F, D'Alessandro G, Antonangeli F, Santoro A, Esposito V, Limatola C, et al. CXCL16/CXCR6 axis drives microglia/macrophages phenotype in physiological conditions and plays a crucial role in glioma. Front Immunol. (2018) 9:2750. doi: 10.3389/fimmu.2018.02750
313. Zhao C, Liu Y, Meng J, Wang X, Liu X, Li W, et al. LGALS3BP in microglia promotes retinal angiogenesis through PI3K/AKT pathway during hypoxia. Invest Ophthalmol Vis Sci. (2022) 63:25. doi: 10.1167/iovs.63.8.25
314. El Bannoudi H, Cornwell M, Luttrell-Williams E, Engel A, Rolling C, Barrett TJ, et al. Platelet LGALS3BP as a mediator of myeloid inflammation in systemic lupus erythematosus. Arthritis Rheumatol. (2023) 75:711–22. doi: 10.1002/art.42382
315. Pan L, Lu MP, Wang JH, Xu M, Yang SR. Immunological pathogenesis and treatment of systemic lupus erythematosus. World J Pediatr. (2020) 16:19–30. doi: 10.1007/s12519-019-00229-3
316. Hedrich CM. Shaping the spectrum — from autoinflammation to autoimmunity. Clin Immunol. (2016) 165:21–8. doi: 10.1016/j.clim.2016.03.002
317. Mistry P, Kaplan MJ. Cell death in the pathogenesis of systemic lupus erythematosus and lupus nephritis. Clin Immunol. (2017) 185:59–73. doi: 10.1016/j.clim.2016.08.010
318. Mahajan A, Herrmann M, Muñoz LE. Clearance deficiency and cell death pathways: a model for the pathogenesis of SLE. Front Immunol. (2016) 7:35. doi: 10.3389/fimmu.2016.00035
319. Sheriff A, Gaipl US, Voll RE, Kalden JR, Herrmann M. Apoptosis and systemic lupus erythematosus. Rheum Dis Clin North Am. (2004) 30:505–27. doi: 10.1016/j.rdc.2004.04.006
320. Gaipl US, Kuhn A, Sheriff A, Munoz LE, Franz S, Voll RE, et al. Clearance of apoptotic cells in human SLE. Curr Dir Autoimmun. (2006) 9:173–87. doi: 10.1159/000090781
321. Schiller M, Bekeredjian-Ding I, Heyder P, Blank N, Ho AD, Lorenz HM. Autoantigens are translocated into small apoptotic bodies during early stages of apoptosis. Cell Death Differ. (2008) 15:183–91. doi: 10.1038/sj.cdd.4402239
322. Jo HS, Kim HJ. Stabilin-2 mediated apoptotic cell phagocytosis induces interleukin-10 expression by p38 and Pbx1 signaling. Cell Biochem Biophys. (2024) 82:919–25. doi: 10.1007/s12013-024-01243-7
323. Wermeling F, Chen Y, Pikkarainen T, Scheynius A, Winqvist O, Izui S, et al. Class A scavenger receptors regulate tolerance against apoptotic cells, and autoantibodies against these receptors are predictive of systemic lupus. J Exp Med. (2007) 204:2259–65. doi: 10.1084/jem.20070600
324. Wicker-Planquart C, Tacnet-Delorme P, Preisser L, Dufour S, Delneste Y, Housset D, et al. Insights into the ligand binding specificity of SREC-II (scavenger receptor expressed by endothelial cells). FEBS Open Bio. (2021) 11:2693–704. doi: 10.1002/2211-5463.13260
325. Iram T, Ramirez-Ortiz Z, Byrne MH, Coleman UA, Kingery ND, Means TK, et al. Megf10 is a receptor for C1Q that mediates clearance of apoptotic cells by astrocytes. J Neurosci. (2016) 36:5185–92. doi: 10.1523/JNEUROSCI.3850-15.2016
326. Platt N, Suzuki H, Kurihara Y, Kodama T, Gordon S. Role for the class A macrophage scavenger receptor in the phagocytosis of apoptotic thymocytes in vitro. Proc Natl Acad Sci U S A. (1996) 93:12456–60. doi: 10.1073/pnas.93.22.12456
327. Chen XW, Shen Y, Sun CY, Wu FX, Chen Y, Yang CD. Anti-class a scavenger receptor autoantibodies from systemic lupus erythematosus patients impair phagocytic clearance of apoptotic cells by macrophages in vitro. Arthritis Res Ther. (2011) 13:R9. doi: 10.1186/ar3230
328. Ramirez-Ortiz ZG, Pendergraft WF 3rd, Prasad A, Byrne MH, Iram T, Blanchette CJ, et al. The scavenger receptor SCARF1 mediates the clearance of apoptotic cells and prevents autoimmunity. Nat Immunol. (2013) 14:917–26. doi: 10.1038/ni.2670
329. Jorge AM, Lao T, Kim R, Licciardi S, El Khoury J, Luster AD, et al. SCARF1-induced Efferocytosis plays an immunomodulatory role in humans, and autoantibodies targeting SCARF1 are produced in patients with systemic lupus erythematosus. J Immunol. (2022) 208:955–67. doi: 10.4049/jimmunol.2100532
330. Tan L, Zhao Y. Analysis of multiple organ damage and clinical immunological characteristics in systemic lupus erythematosus patients with hematologic involvement. Int J Med Sci. (2021) 18:2624–9. doi: 10.7150/ijms.48997
331. Emura I, Usuda H. Appearance of large scavenger receptor A-positive cells and increased small scavenger receptor A positive cells in peripheral blood is associated with mortality in systemic inflammatory response syndrome and multiple organ dysfunction syndrome. Pathol Int. (2011) 61:7–12. doi: 10.1111/j.1440-1827.2010.02607.x
332. Onyishi CU, Desanti GE, Wilkinson AL, Lara-Reyna S, Frickel EM, Fejer G, et al. Toll-like receptor 4 and macrophage scavenger receptor 1 crosstalk regulates phagocytosis of a fungal pathogen. Nat Commun. (2023) 14:4895. doi: 10.1038/s41467-023-40635-w
333. Yu H, Ha T, Liu L, Wang X, Gao M, Kelley J, et al. Scavenger receptor A (SR-A) is required for LPS-induced TLR4 mediated NF-kappaB activation in macrophages. Biochim Biophys Acta. (2012) 1823:1192–8. doi: 10.1016/j.bbamcr.2012.05.004
334. Feng L, Zhou X, Su L-X, Feng D, Jia Y-H, Xie L-X. Clinical significance of soluble hemoglobin scavenger receptor CD163 (sCD163) in sepsis, a prospective study. PLoS One. (2012) 7:e38400. doi: 10.1371/journal.pone.0038400
335. Mukherjee R, Kanti Barman P, Kumar Thatoi P, Tripathy R, Kumar Das B, Ravindran B. Non-classical monocytes display inflammatory features: validation in sepsis and systemic lupus erythematous. Sci Rep. (2015) 5:13886. doi: 10.1038/srep13886
336. Olmes G, Buttner-Herold M, Ferrazzi F, Distel L, Amann K, Daniel C. CD163+ M2c-like macrophages predominate in renal biopsies from patients with lupus nephritis. Arthritis Res Ther. (2016) 18:90. doi: 10.1186/s13075-016-0989-y
337. Li J, Liu CH, Xu DL, Gao B. Significance of CD163-positive macrophages in proliferative glomerulonephritis. Am J Med Sci. (2015) 350:387–92. doi: 10.1097/MAJ.0000000000000569
338. Arai S, Miyazaki T. A scavenging system against internal pathogens promoted by the circulating protein apoptosis inhibitor of macrophage (AIM). Semin Immunopathol. (2018) 40:567–75. doi: 10.1007/s00281-018-0717-6
339. Nakaya M, Watari K, Tajima M, Nakaya T, Matsuda S, Ohara H, et al. Cardiac myofibroblast engulfment of dead cells facilitates recovery after myocardial infarction. J Clin Invest. (2017) 127:383–401. doi: 10.1172/JCI83822
340. Shao WH, Cohen PL. Disturbances of apoptotic cell clearance in systemic lupus erythematosus. Arthritis Res Ther. (2011) 13:202. doi: 10.1186/ar3206
341. Chen Q, Ishii K, Mori H, Nishijima A, Arai S, Miyazaki T, et al. Cryo-EM reveals structural basis for human AIM/CD5l recognition of polymeric immunoglobulin M. Nat Commun. (2024) 15:9387. doi: 10.1038/s41467-024-53615-5
342. Sanjurjo L, Aran G, Roher N, Valledor AF, Sarrias M-R. AIM/CD5l: a key protein in the control of immune homeostasis and inflammatory disease. J Leukocyte Biol. (2015) 98:173–84. doi: 10.1189/jlb.3RU0215-074R
343. Lai X, Xiang Y, Zou L, Li Y, Zhang L. Elevation of serum CD5l concentration is correlated with disease activity in patients with systemic lupus erythematosus. Int Immunopharmacol. (2018) 63:311–6. doi: 10.1016/j.intimp.2018.07.022
344. Akama-Garren EH, Carroll MC. Lupus susceptibility loci predispose mice to clonal lymphocytic responses and myeloid expansion. J Immunol. (2022) 208:2403–24. doi: 10.4049/jimmunol.2200098
345. Sugisawa R, Komatsu G, Hiramoto E, Takeda N, Yamamura K-i, Arai S, et al. Independent modes of disease repair by AIM protein distinguished in AIM-felinized mice. Sci Rep. (2018) 8:13157. doi: 10.1038/s41598-018-31580-6
346. Yamazaki T, Mori M, Arai S, Tateishi R, Abe M, Ban M, et al. Circulating AIM as an indicator of liver damage and hepatocellular carcinoma in humans. PLoS One. (2014) 9:e109123. doi: 10.1371/journal.pone.0109123
347. Arai S, Kitada K, Yamazaki T, Takai R, Zhang X, Tsugawa Y, et al. Apoptosis inhibitor of macrophage protein enhances intraluminal debris clearance and ameliorates acute kidney injury in mice. Nat Med. (2016) 22:183–93. doi: 10.1038/nm.4012
348. Dieudé M, West LJ, Muruve DA, Gunaratman L, Mohanakumar T, Zorn E, et al. New answers to old conundrums: what antibodies. Exosomes and inflammasomes bring to the conversation. Canadian national transplant research program international summit report. Transplantation. (2018) 102:209–14. doi: 10.1097/TP.0000000000001872
349. Ichimura T, Bonventre JV, Bailly V, Wei H, Hession CA, Cate RL, et al. Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up-regulated in renal cells after injury. J Biol Chem. (1998) 273:4135–42. doi: 10.1074/jbc.273.7.4135
350. Nozaki Y, Shiga T, Ashida C, Tomita D, Itami T, Kishimoto K, et al. U-KIM-1 as a predictor of treatment response in lupus nephritis. Lupus. (2023) 32:54–62. doi: 10.1177/09612033221135871
351. Shih LC, Hsieh HH, Tsay GJ, Lee IT, Tsou YA, Lin CL, et al. Chronic rhinosinusitis and premorbid autoimmune diseases: a population-based case-control study. Sci Rep. (2020) 10:18635. doi: 10.1038/s41598-020-75815-x
352. Kronzer VL, Crowson CS, Sparks JA, Vassallo R, Davis JM 3rd. Investigating asthma, allergic disease, passive smoke exposure, and risk of rheumatoid arthritis. Arthritis Rheumatol. (2019) 71:1217–24. doi: 10.1002/art.40858
353. Elcioglu OC, Afsar B, Bakan A, Takir M, Ozkok A, Oral A, et al. Chronic rhinosinusitis, endothelial dysfunction, and atherosclerosis. Am J Rhinol Allergy. (2016) 30:58–61. doi: 10.2500/ajra.2016.30.4325
354. Kronzer VL, Davis JM, Hanson AC, Sparks JA, Myasoedova E, Duarte-Garcia A, et al. Association between sinusitis and incident rheumatic diseases: a population-based study. RMD Open. (2024) 10(1):1–9. doi: 10.1136/rmdopen-2023-003622
355. Bikker FJ, Ligtenberg AJM, End C, Renner M, Blaich S, Lyer S, et al. Bacteria binding by DMBT1/SAG/gp-340 is confined to the VEVLXXXXW motif in its scavenger receptor cysteine-rich domains*. J Biol Chem. (2004) 279:47699–703. doi: 10.1074/jbc.M406095200
356. Osei KA, Deivanayagam C, Nichols JJ. Glycoprotein 340 in mucosal immunity and ocular surface. Ocul Surf. (2018) 16:282–8. doi: 10.1016/j.jtos.2018.04.006
357. Kim TH, Lee SH, Lee HM, Jung HH, Lee SH, Cho WS, et al. Increased expression of glycoprotein 340 in the ethmoid Sinus Mucosa of patients with chronic sinusitis. Arch Otolaryngol Head Neck Surg. (2007) 133:1111–4. doi: 10.1001/archotol.133.11.1111
358. Sorensen GL. Surfactant protein D in respiratory and non-respiratory diseases. Front Med (Lausanne). (2018) 5:18. doi: 10.3389/fmed.2018.00018
359. Hoegh SV, Voss A, Sorensen GL, Høj A, Bendixen C, Junker P, et al. Circulating surfactant protein D is decreased in systemic lupus erythematosus. J Rheumatol. (2009) 36:2449–53. doi: 10.3899/jrheum.090069
360. LeVine AM, Whitsett JA, Gwozdz JA, Richardson TR, Fisher JH, Burhans MS, et al. Distinct effects of surfactant protein A or D deficiency during bacterial infection on the lung. J Immunol. (2000) 165:3934–40. doi: 10.4049/jimmunol.165.7.3934
361. Gan XX, Li YY, Li SJ, Mo SS, Feng JH, Shen F, et al. Significance of DMBT1 in papillary thyroid carcinoma concurrent with Hashimoto's Thyroiditis. Front Oncol. (2021) 11:680873. doi: 10.3389/fonc.2021.680873
362. Choudhury K, Gandhi M, Kumar U, Chatterjee S, Purty RS. Evaluation of the deleted in malignant brain tumor 1 protein expression and DNA methylation profile in rheumatoid arthritis patients. Indian J Rheumatol. (2023) 18:68–73. doi: 10.4103/injr.injr_181_21
363. Mu Q, Kirby J, Reilly CM, Luo XM. Leaky gut as a danger signal for autoimmune diseases. Front Immunol. (2017) 8:598. doi: 10.3389/fimmu.2017.00598
364. Charoensappakit A, Sae-khow K, Leelahavanichkul A. Gut barrier damage and gut translocation of pathogen molecules in lupus, an impact of innate immunity (macrophages and neutrophils) in autoimmune disease. Int J Mol Sci. (2022) 23:8223. doi: 10.3390/ijms23158223
365. Battaglia M, Garrett-Sinha LA. Bacterial infections in lupus: roles in promoting immune activation and in pathogenesis of the disease. J Transl Autoimmun. (2021) 4:100078. doi: 10.1016/j.jtauto.2020.100078
366. Nockher WA, Wigand R, Schoeppe W, Scherberich JE. Elevated levels of soluble CD14 in serum of patients with systemic lupus erythematosus. Clin Exp Immunol. (1994) 96:15–9. doi: 10.1111/j.1365-2249.1994.tb06222.x
367. Gordon S. Pattern recognition receptors: doubling up for the innate immune response. Cell. (2002) 111:927–30. doi: 10.1016/S0092-8674(02)01201-1
368. Valiente GR, Munir A, Hart ML, Blough P, Wada TT, Dalan EE, et al. Gut dysbiosis is associated with acceleration of lupus nephritis. Sci Rep. (2022) 12:152. doi: 10.1038/s41598-021-03886-5
369. Li J, Yu YF, Liu CH, Wang CM. Significance of M2 macrophages in glomerulonephritis with crescents. Pathol Res Pract. (2017) 213:1215–20. doi: 10.1016/j.prp.2017.04.011
370. Cupi ML, Sarra M, De Nitto D, Franzè E, Marafini I, Monteleone I, et al. Defective expression of scavenger receptors in celiac disease Mucosa. PLoS One. (2014) 9:e100980. doi: 10.1371/journal.pone.0100980
371. Merino LMM, Parra-Medina R, Sarria MA, Galvis JAD. Innate immune system. In: Shoenfeld Y, Anaya JM, Rojas-Villarraga A, editors. Autoimmunity: From Bench to Bedside. Bogota, Colombia: El Rosario University Press (2013).
372. Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. (2018) 9:7204–18. doi: 10.18632/oncotarget.23208
373. Nair A, Thankachen RU, Raj J, Gopi S. 1 - Inflammation, symptoms, benefits, reaction, and biochemistry. In: Gopi S, Amalraj A, Kunnumakkara A, Thomas S, editors. Inflammation and Natural Products. New York, NY: Academic Press (2021). p. 1–19.
374. Warde N. Class A scavenger receptors as novel autoantigens in SLE. Nat Rev Rheumatol. (2011) 7:195–195. doi: 10.1038/nrrheum.2011.30
375. Katarzyna PB, Wiktor S, Ewa D, Piotr L. Current treatment of systemic lupus erythematosus: a clinician's Perspective. Rheumatol Int. (2023) 43:1395–407. doi: 10.1007/s00296-023-05306-5
376. Blair HA, Duggan ST. Belimumab: a review in systemic lupus erythematosus. Drugs. (2018) 78:355–66. doi: 10.1007/s40265-018-0872-z
377. Deeks ED. Anifrolumab: first approval. Drugs. (2021) 81:1795–802. doi: 10.1007/s40265-021-01604-z
378. Areschoug T, Gordon S. Scavenger receptors: role in innate immunity and microbial pathogenesis. Cell Microbiol. (2009) 11:1160–9. doi: 10.1111/j.1462-5822.2009.01326.x
379. Nayroles G, Frybourg S, Gabriel S, Kornfeld Å, Antoñanzas-Villar F, Espín J, et al. Unlocking the potential of established products: toward new incentives rewarding innovation in Europe. J Mark Access Health Policy. (2017) 5:1298190. doi: 10.1080/20016689.2017.1298190
380. Ruiz-Irastorza G, Bertsias G. Treating systemic lupus erythematosus in the 21st century: new drugs and new perspectives on old drugs. Rheumatology. (2020) 59:v69–81. doi: 10.1093/rheumatology/keaa403
381. Athanassiou P, Athanassiou L. Current treatment approach, emerging therapies and new horizons in systemic lupus erythematosus. Life. (2023) 13:1496. doi: 10.3390/life13071496
382. Yuan W, Guan F. Thrombosis and anticoagulation therapy in systemic lupus erythematosus. Autoimmune Dis. (2022) 2022:3208037. doi: 10.1155/2022/320803735795725
Keywords: scavenger receptors (SRs), autoimmunity, systemic lupus erythematosus (SLE), inflammation, cardinal signs
Citation: Hahn S, Chitre M, Shepard D, Rashid R and Ramirez-Ortiz ZG (2025) Scavenger receptors: key players in the immunological puzzle of lupus. Front. Lupus 3:1679564. doi: 10.3389/flupu.2025.1679564
Received: 4 August 2025; Accepted: 30 September 2025;
Published: 29 October 2025.
Edited by:
Selcan Demir, Eskişehir Osmangazi University, TürkiyeReviewed by:
Ozge Basaran, Hacettepe University, TürkiyeVildan Güngörer, University of Health Sciences, Türkiye
Copyright: © 2025 Hahn, Chitre, Shepard, Rashid and Ramirez-Ortiz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zaida G. Ramirez-Ortiz, WmFpZGEuUmFtaXJlek9ydGl6QHVtYXNzbWVkLmVkdQ==