Abstract
The Portuguese oyster (Magallana angulata) is a valuable aquaculture species with sexual dimorphism in growth and nutritional traits, making it a promising candidate for sex-controlled breeding. However, the genetic mechanisms underlying its sex determination remain poorly understood. This study represents the first population-level investigation to identify sex-linked genetic markers and candidate genes in M. angulata using genome-wide association studies (GWAS) and transcriptomic analysis. A total of 1,613 sex-associated SNPs were identified, primarily located on Chr. 9 (7.99 Mb–10.51Mb). SNP annotation revealed 57 genes around these sex-associated SNPs. Transcriptomic analysis identified 8,635 differentially expressed genes (DEGs) between male and female gonads, of which 12 overlapped with GWAS-identified genes. Among these, four genes (5-HTR, ADPKD1-a, SCP, and SLC28A3) exhibited strong sex-specific genetic differentiation, with 5-HTR showing the most pronounced association. Further population genetic analysis revealed higher genetic diversity and heterozygosity of 5-HTR in males. Six sex-associated haplotypes based on SNPs within 5-HTR were constructed and validated in an independent population, although with reduced significance, suggesting both continuity and variation in sex determination mechanisms across populations. These findings provide a critical foundation for understanding the genetic basis of sex determination and advancing sex-controlled breeding strategies in M. angulata.
1 Introduction
Sex is a critical trait in aquatic animals, as sexual dimorphism significantly influences growth, size, shape, color, behavior, and ornamentation (Martínez et al., 2014; Horne et al., 2020; Li et al., 2022). These sex-specific differences have driven global efforts to develop sex-controlled breeding programs aimed at generating populations with a high proportion of either males or females, depending on production goals (Budd et al., 2015; Li et al., 2022). Achieving such goals necessitates a thorough understanding of the genetic architecture underlying sex determination, alongside the identification of reliable sex-specific or sex-linked genetic markers. Recent advances in molecular genetics and genome sequencing technologies have enabled the discovery of numerous sex-specific and sex-linked DNA markers in over 100 aquatic species, including fish, amphibians, crustaceans, echinoderms, and mollusks (Li et al., 2022). These markers have been successfully applied in molecular-assisted sex-controlled breeding programs for species such as Nile tilapia (Oreochromis niloticus), rainbow trout (Oncorhynchus mykiss), and yellow catfish (Pelteobagrus fulvidraco) (Sun et al., 2014; Guiguen et al., 2018; Mei and Gui, 2018).
Mollusks, the second-largest animal phylum after arthropods, exhibit extraordinary diverse and plastic sex determination mechanisms (Carlton, 1999; Gofas and Zenetos, 2003; Collin, 2013; Breton et al., 2018). Understanding these systems is critical not only for the development of sex-controlled breeding strategies in aquaculture but also for elucidating the broader evolutionary trajectories of sex chromosomes in invertebrates. Early studies on molluscan sex determination utilized karyotyping (Vitturi and Catalano, 1988; Rolan-Alvarez et al., 1996; Vitturi et al., 1998; Baršienė et al., 2000), sex ratio analysis in family lines, triploidy and gynogenesis (Allen et al., 1986; Guo and Allen, 1994) to infer underlying sex determination mechanisms and sex chromosomal configurations. Recent advances in genomic technology have provided deeper insights into molluscan sex determination. For example, studies in abalones have identified sex-linked markers across three species, revealing interspecies turnover of sex chromosome (Luo et al., 2021; Weng et al., 2022; Tang et al., 2025). Research on scallops has localized sex determination regions associated with transposons, indicating chromosomal transitions (Han et al., 2022). Collectively, these findings highlight the evolutionary diversity and genomic complexity of sex determination systems in mollusks, and emphasize the importance of integrative genomics for unraveling conserved and lineage-specific regulatory mechanisms.
Oysters, the world’s largest marine aquaculture group, are distributed across all continents except Antarctica (Forrest et al., 2009). They are predominantly dioecious, capable of bidirectional sex reversal, and occasionally exhibit hermaphroditism (Park et al., 2012). Since the 1970s, researchers have studied the genetic regulation of oyster sex determination. In Crassostrea virginica and M. gigas, paternal effects on sex ratios in half-sibling families were observed, leading to the proposal of a “three-locus” and “single-locus” models of sex determination (Haley, 1977, 1979; Guo et al., 1998; Hedrick and Hedgecock, 2010). More recently, microsatellite and GBS sequencing have identified major sex-linked QTLs in M. gigas, highlighting the role of genetic factors in sex determination (Guo et al., 2012; Han et al., 2021).
The Portuguese oyster (M. angulata), a warm-water subspecies of the Pacific oyster, is distributed along the southeastern coasts of China and Vietnam (Wang et al., 2010). It was inadvertently introduced to France in the late 19th century and later to other European countries for commercial aquaculture (Grade et al., 2016; Hsiao et al., 2016). Portuguese oysters exhibit sexual dimorphism in growth and nutritional traits, making them ideal candidates for sex-controlled breeding (Zeng et al., 2013; Qin et al., 2023; Xie et al., 2024). Furthermore, extreme sex ratios are often observed in some families, such as those highly biased toward males or females, suggesting that it is feasible to use marker-assisted selection to develop high-androgenic or high-femaleogenic lines (Vu et al., 2020). However, the genetic basis of sex determination in Portuguese oyster remains unexplored. In this study, we applied genome-wide association studies (GWAS), comparative transcriptomic analysis, and genetic structure analysis to identify sex-associated loci and genes in a cultured population of Portuguese oysters. The results provide a foundation for elucidating the genetic mechanisms underlying sex determination and advancing sex-controlled breeding strategies for this species.
2 Materials and methods
2.1 Sample collection, and whole-genome resequencing
Samples for GWAS analysis and transcriptomics were collected from a culture strain in Weitou Bay, Fujian Province, China, in November 2023. A total of 112 one-year-old oysters (57 females and 55 males), were included in the study. After collection, the samples were brought back to the laboratory, where the sex was identified and recorded through microscopic examination of gonadal tissue smears. Muscle and gonadal tissues were then sampled, rapidly frozen in liquid nitrogen, and stored at -80°C for subsequent DNA and RNA extraction.
The adductor muscle samples were sent to Annoroad Gene Technology Co., Ltd. (Beijing, China) for total DNA extraction and whole-genome resequencing at an average sequencing depth of 9×. Sequencing data were processed with Fastp v0.22.0 (Chen et al., 2018) to remove low-quality reads, resulting in clean reads. The clean reads were aligned to the Portuguese oyster genome (GenBank: GCA_025612915.2) using BWA v0.7.17 software (Li and Durbin, 2009). SNPs were identified with GATK v4.1.6.0 (Yang et al., 2011) and filtered using Plink v1.90 based on standard thresholds (MAF ≥ 0.05, missing genotype rate ≤ 5%, and Hardy-Weinberg equilibrium P-value ≥ 1e-3). Missing genotypes were imputed with Beagle v5.1 (Browning and Browning, 2016), and high-quality SNPs were used for further analysis.
2.2 Identification and validation of sex-associated SNPs and genes
GWAS was performed with Plink v1.90 (Purcell et al., 2007) using a logistic regression model. A Bonferroni correction was applied to set a significance threshold (0.05/total SNPs). SNPs surpassing this threshold were considered significantly associated with sex, and results were visualized as Manhattan plots using the Cmplot package (v4.5.1) in R.
To validate the GWAS results, sex-associated SNPs were used for principal component analysis (PCA) and phylogenetic tree analysis. PCA was conducted with Plink v1.90 (Purcell et al., 2007), and a maximum likelihood (ML) tree in NEWICK format was generated using IQ-TREE v2.2.0 (Minh et al., 2020). The tree was visualized on the iTOL platform (https://itol.embl.de/). SNPs that grouped individuals by sex in both PCA and phylogenetic analyses were deemed reliable.
Sex-associated SNPs were functionally annotated using the M. angulata genome (GenBank: GCA_025612915.2) and its annotation file (GCF_025612915.1-RS_2022_12) with the snpEff.jar package. Additionally, the 30 kb upstream and downstream regions of sex-associated SNPs were annotated to identify sex-associated genes.
2.3 Genome-wide analysis of intersex genetic differentiation and sex-biased SNP distribution
The FST value, a measure of genetic differentiation, was calculated for 20 kb genomic windows with a step size of 10 kb using VCFtools v0.1.16 (Danecek et al., 2011). Windows with FST values in the top 1% were identified as outliers, indicating potential involvement in sex differentiation. The genome-wide distribution of FST values was visualized using the Cmplot package in R (v4.5.1).
Sex-biased SNPs were identified based on genotype frequency differences between sexes. Specifically, SNPs where the reference allele homozygous genotype frequency (0|0) exceeded 85% in females and the alternate allele heterozygous or homozygous genotype frequency (0|1 or 1|1) exceeded 85% in males were classified as sex-biased (Luo et al., 2021). The genome-wide distribution of these sex-biased SNPs was visualized in 20 kb windows using the Cmplot package in R.
2.4 RNA extraction and transcriptome analysis
Gonadal tissues from four female and four male samples were randomly selected for RNA extraction, library construction, and sequencing, conducted by Annoroad Gene Technology Co., Ltd. Transcriptome sequencing was performed on the Illumina NovaSeq X Plus platform at a depth of 9×. After quality control with Fastp v0.22.0 (Chen et al., 2018), the clean reads were aligned to the M. angulata genome (GenBank: GCA_025612915.2) using HISAT2 v2.1.0 (Pertea et al., 2016). Alignment files were processed for sorting, indexing and format conversion using SAMtools v1.10 (Li et al., 2009). Gene expression levels were quantified as TPM (transcripts per million) using the run-featurecounts.R script (Liao et al., 2014) (https://github.com/mskcc/RNAseqDB/blob/master/run-featurecounts.R). Differentially expressed genes (DEGs) between sexes were identified with DESeq2 v1.40.2 (Love et al., 2014), with the thresholds |log2 FC| ≥ 1 and P < 0.05. Identified DEGs were further subjected to GO and KEGG enrichment analysis using the ClusterProfiler package (v4.8.2) (Yu et al., 2012).
2.5 Identification and variation analysis of sex-determining candidate genes
An integrative approach was applied to identify genes involved in sex determination by combining genetic variation, gene expression data, and clustering analysis. Sex-associated genes identified from GWAS were compared with significant DEGs from transcriptome analysis, and overlapping genes were visualized using the VennDiagram package (Chen and Boutros, 2011). The expression levels of candidate genes, measured as TPM, were displayed as heatmaps generated with the pheatmap package (Zhao et al., 2021).
For further analysis the genetic divergence pattern of the candidate genes, SNPs within the candidate gene regions were examined extracted and subjected to principal component analysis (PCA) using Plink v1.90. Genetic distance matrices were generated with VCF2Dis v1.47, and neighbor-joining (NJ) trees were constructed and visualized on the iTOL platform (https://itol.embl.de/). Genes that exhibited distinct sex-associated clustering in both PCA and NJ trees were identified as key sex-determining candidate genes.
For the representative key gene, SNP variations were further analyzed. The upstream and downstream SNP distributions of these key genes were visualized using ggplot v2.2.1, while local GWAS, heterozygosity, and nucleotide diversity analyses were conducted using Plink v1.90 and VCFtools v0.1.16. Genotype patterns were presented as heatmaps generated with RectChr v1.37 (https://github.com/hewm2008/RectChr/), providing a detailed visualization of genetic variation around the candidate genes.
2.6 Construction and validation of sex-associated haplotypes
Single-marker association analysis was performed for all SNPs within the critical candidate gene region using GCTA v1.94.1 software (Yang et al., 2011), identifying SNPs significantly associated with sex (P < 0.05). Linkage disequilibrium (LD) blocks were then detected using Haploview v4.2 (Barrett, 2009), and haplotype associations with sex were analyzed using SHEsisPlus (http://shesisplus.bio-x.cn/).
To validate the reliability of sex-associated haplotypes, an independent validation population consisting of 50 one-year-old angulata individuals (25 males and 25 females) were collected from Weitou Bay in November 2022. Whole-genome resequencing was conducted, followed by SNP calling and haplotype association analysis using the same protocols as those applied to the discovery population. This approach ensured methodological consistency across datasets and allowed for a robust assessment of the reproducibility of sex-linked genetic signals.
3 Results
3.1 Whole-genome sequencing and variant calling
A total of 112 individuals (57 females and 55 males) were subjected to Illumina paired-end sequencing, generating 748.67 GB of raw data. After quality control, 713.71 GB of clean bases were retained (NCBI PRJNA1185961, Supplementary Table S1), with an average sequencing depth of 9×. From the clean data, 8,183,713 high-quality SNPs were identified through SNP calling and filtering, which were used for subsequent analyses.
3.2 Identification of sex-associated variants
A GWAS analysis using a logistic regression model identified 1,613 SNPs significantly associated with phenotypic sex (P < 6.31 × 10−9). Among these, 1,599 SNPs (99.13%) were concentrated within a 2.52-Mb region on Chr.9 (7.99–10.51 Mb), six SNPs (0.37%) were located on Chr.6, and the remaining eight SNPs were distributed across Chr.2, Chr.5, Chr.7, Chr.8, and Chr.10 (Figure 1A; Supplementary Table S2).
Figure 1
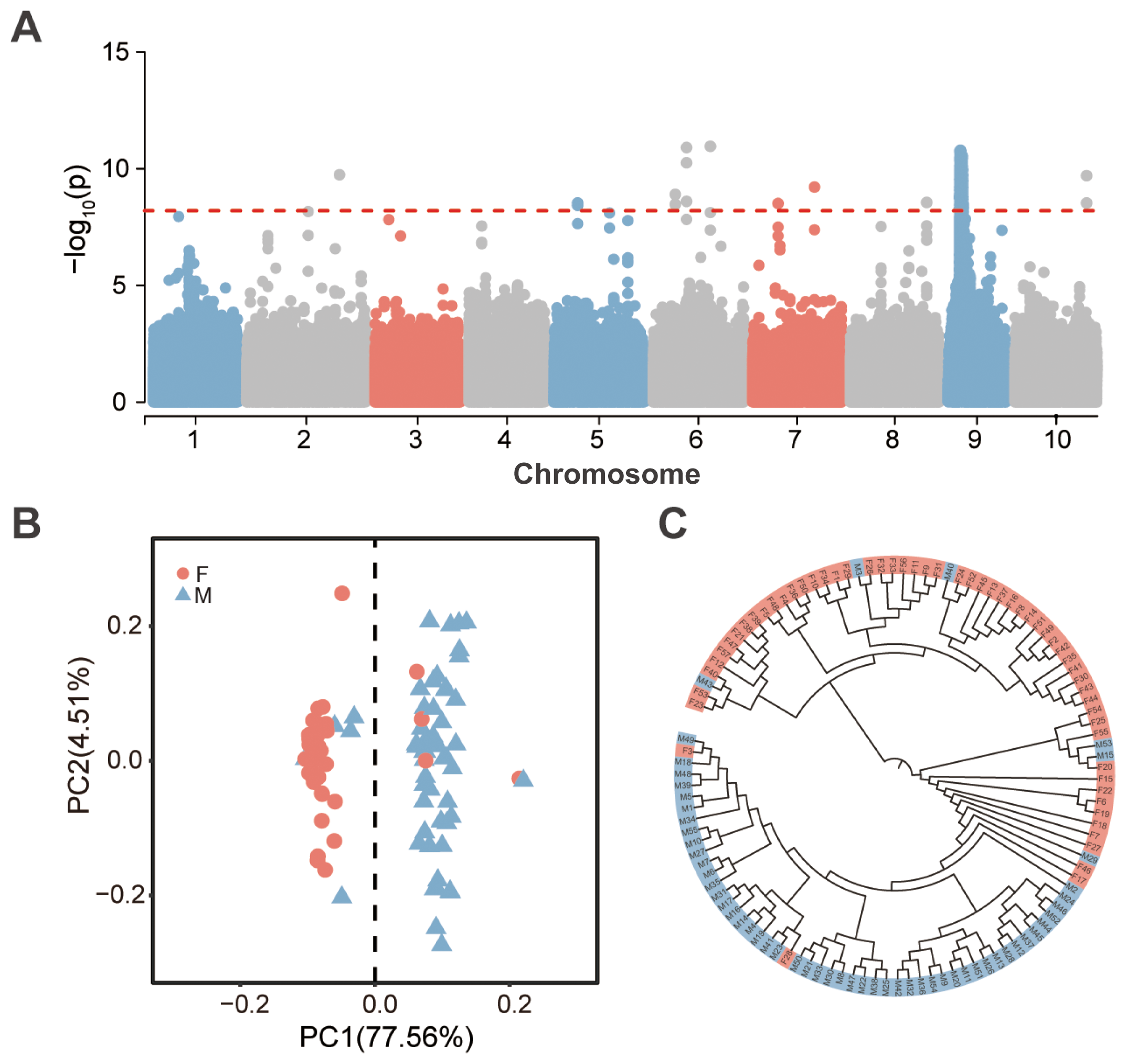
Identification of sex-associated loci in Portuguese oyster. (A) Manhattan plot of GWAS results for phenotypic sex, with the dashed line [-log10P = 8.20] indicating the significance threshold. (B) Principal component analysis (PCA) based on significantly sex-associated loci, with pink and gray-blue representing female and male individuals, respectively. (C) Maximum likelihood tree constructed based on significantly sex-associated loci, with pink and gray-blue representing female and male individuals, respectively.
Principal component analysis (PCA) based on the identified sex-associated SNPs revealed that PC1 accounted for 77.56% of the variance, effectively separating the samples into two distinct groups: a predominantly male group (92.59% male; 50 males and 4 females) and a predominantly female group (91.38% female; 53 females and 5 males) (Figure 1B). A similar pattern was observed when PCA was conducted using only the sex-associated SNPs on Chr.9, where PC1 explained 76.41% of the variance, segregating the samples into two groups with male proportions of 92.45% and 10.17%, respectively (Supplementary Figure S1B).
A maximum likelihood tree constructed using genome-wide sex-associated SNPs grouped the 112 individuals into two major branches (Figure 1C). One branch consisted predominantly of a female monophyletic group (designated FI), containing 42 females and 3 males (93.33% female). The other branch comprised a male-dominant monophyletic group (designated MI), including 49 males and 2 females (96.08% male), along with a polyphyletic group enriched for females (designated FII), which contained 13 females and 3 males (81.25% female). Sub branching within FI and FII indicated high genetic diversity at sex-associated loci. Additionally, a maximum likelihood tree based on sex-associated SNPs from Chr.9 revealed that females clustered near the base of the tree, while males were positioned in more derived branches.
To further investigate sex-associated genomic regions, we employed two complementary approaches: (1) identification of highly differentiated genomic windows between sexes, defined as those with intersex FST values in the top 1%; and (2) analysis of distribution of sex-biased SNPs, defined as loci where the homozygous reference genotype (0|0) frequency exceeded 85% in females, while the heterozygous or homozygous alternative genotype (0|1 or 1|1) frequency exceeded 85% in males. Using the FST approach, 564 highly differentiated windows were identified, with 369 windows (65.4%) located on Chr. 9. The remaining windows were distributed across the other nine chromosomes, ranging from 2 to 52 windows per chromosome (Figure 2A; Supplementary Table S3). For sex-biased SNPs, 82 loci were identified (Figure 2B; Supplementary Table S4), 80 of which (97.56%) were concentrated in the 8.19–11.24 Mb interval on Chr. 9. This interval overlapped with the distribution of sex-associated SNPs identified through GWAS (Figures 2B, 3E, F). Notably, no loci meeting the opposite criteria (0|0 in males and 0|1 or 1|1 in females) were detected. Moreover, a local zoomed-in view of the sex-linked region on Chr. 9 revealed a plateau-like distribution pattern of both sex-associated and highly differentiated SNPs (Figures 3A–D), suggesting the existence of a relatively broad and stable sex-linked genomic region.
Figure 2
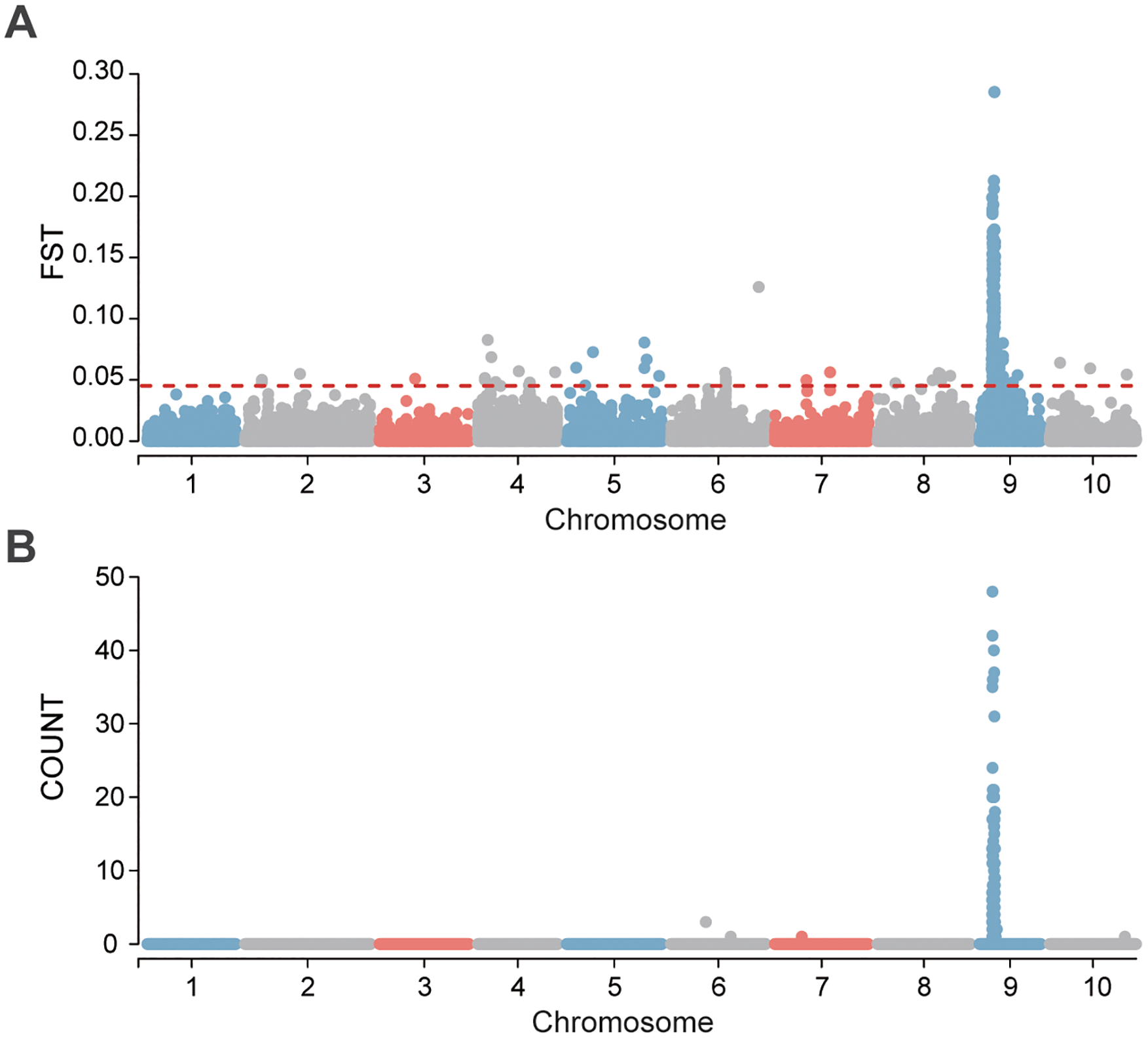
(A) Genetic differentiation (FST) between sexes, with a 20 kb window size. The black dashed lines indicate the significance threshold where FST values exceed the top 1%. (B) Distribution of sexually dimorphic genotype differentiation SNPs with a 20 kb window size.
Figure 3
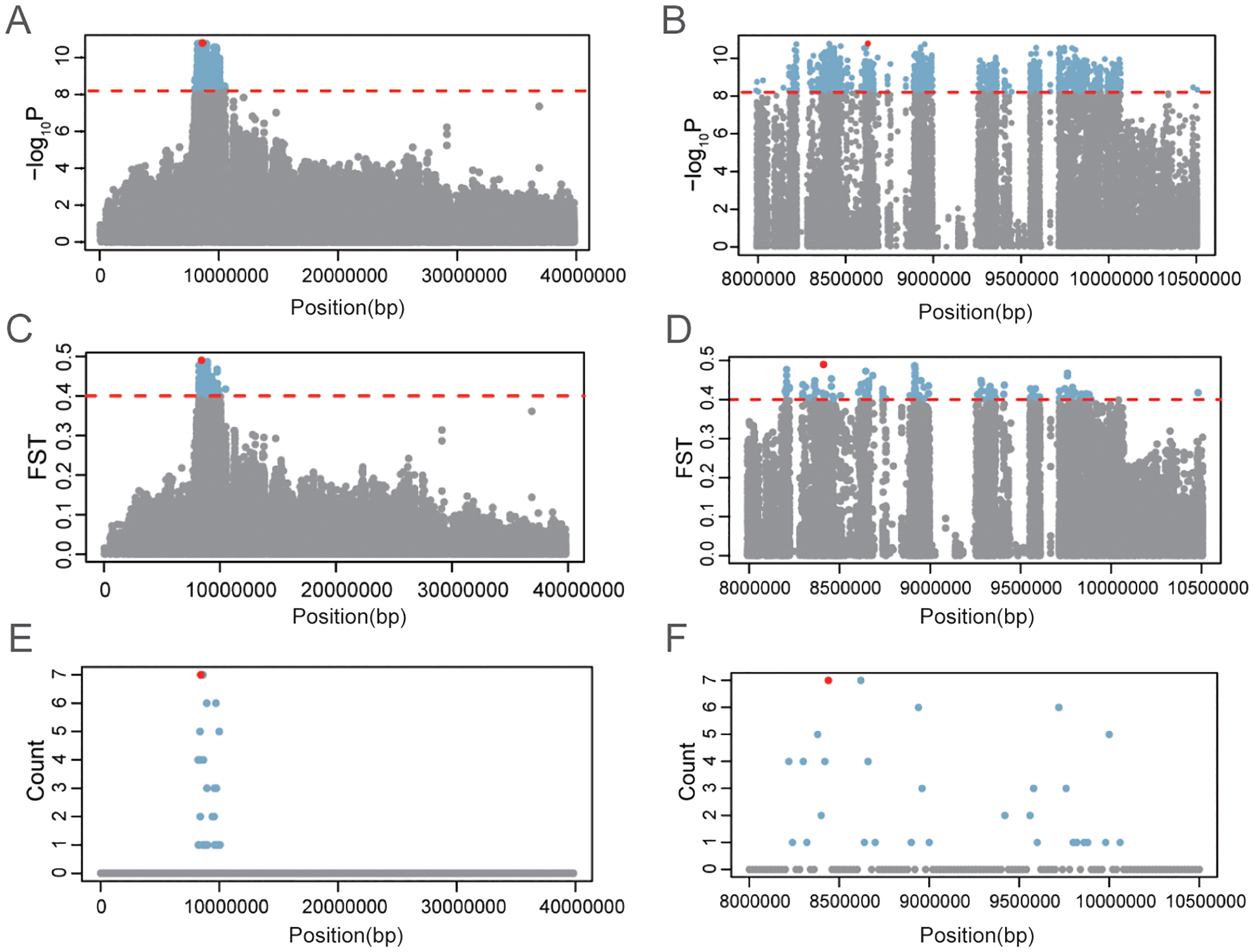
Sex-associated genomic region on Chr.9 in Portuguese oyster. Significance of sex association (A, B), intersex FST(C, D), and distribution of sexually dimorphic loci on Chr.9 (E, F).
3.3 Annotation of sex-associated variants
SNP annotation revealed that 1,484 of 1,613 sex-associated SNPs (92.00%) were located within genes, while 129 SNPs (8.00%) were in intergenic regions. Among the intragenic SNPs, 678 (45.69%) were in introns, 452 (30.46%) in upstream regions, 177 (11.93%) in exons, 80 (5.39%) in downstream regions, and 97 (6.54%) in untranslated regions (UTRs) (Figure 4). Gene annotation within 30 kb upstream and downstream of these SNPs identified 57 genes, including 54 on Chr.9 and 3 on Chr.5 (Supplementary Table S5). Of these, 20 genes contained missense mutations (Supplementary Table S6).
Figure 4
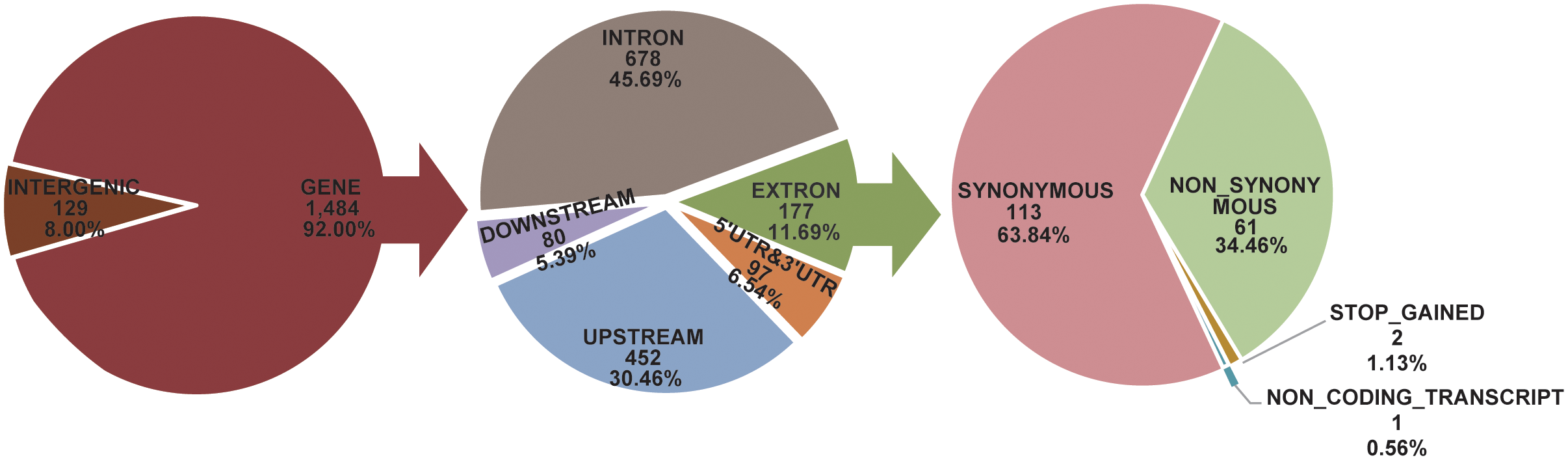
Annotation and prediction of effects for sex association SNPs.
3.4 Comparative transcriptomic analysis of male and female gonads
Transcriptomic analysis of gonadal tissues (testis and ovary) from four male and four female individuals generated a total of 51.64 Gb of raw sequencing data. After quality filtering, 50.82 Gb of clean data were retained, with Q30 scores exceeding 94.53% for all libraries, indicating high sequencing quality (Supplementary Table S7). Pearson correlation analysis revealed higher similarity within the same sex compared to between sexes, underscoring significant differences in gene expression patterns between testis and ovary (Supplementary Figure S2).
Differential expression analysis identified 8,635 differentially expressed genes (DEGs) between testis and ovary (|log2 FC| ≥ 1, P < 0.05), with 3,792 genes upregulated in the ovary and 4,843 genes upregulated in the testis (Figure 5). GO enrichment analysis showed that ovary-upregulated genes were primarily associated with chromosomal separation, chromatin binding, nuclear division, DNA replication, repair, and conformational changes (Supplementary Table S8; Supplementary Figure S3A). In contrast, testis-upregulated genes were enriched in processes related to G protein-coupled receptor signaling, transmembrane signaling, and cytoplasmic composition (Supplementary Table S9; Supplementary Figure S3B). KEGG pathway enrichment analysis revealed distinct functional roles for DEGs in two sexes. Ovary-upregulated genes were enriched in pathways related to DNA replication, repair, transcription, and the cell cycle (Supplementary Table S10; Supplementary Figure S3C). Testis-upregulated genes were associated with transport processes, transcription factors, ribosome biogenesis, and G protein-coupled receptor signaling pathways (Supplementary Table S11; Supplementary Figure S4D). Additionally, key sex differentiation pathways, such as Wnt/β-Catenin, MAPK, and cAMP/PKA signaling, were also found in KEGG enriched pathways (Supplementary Tables S10, S11), highlighting the potential roles of DEGs in regulating sex determination and differentiation.
Figure 5
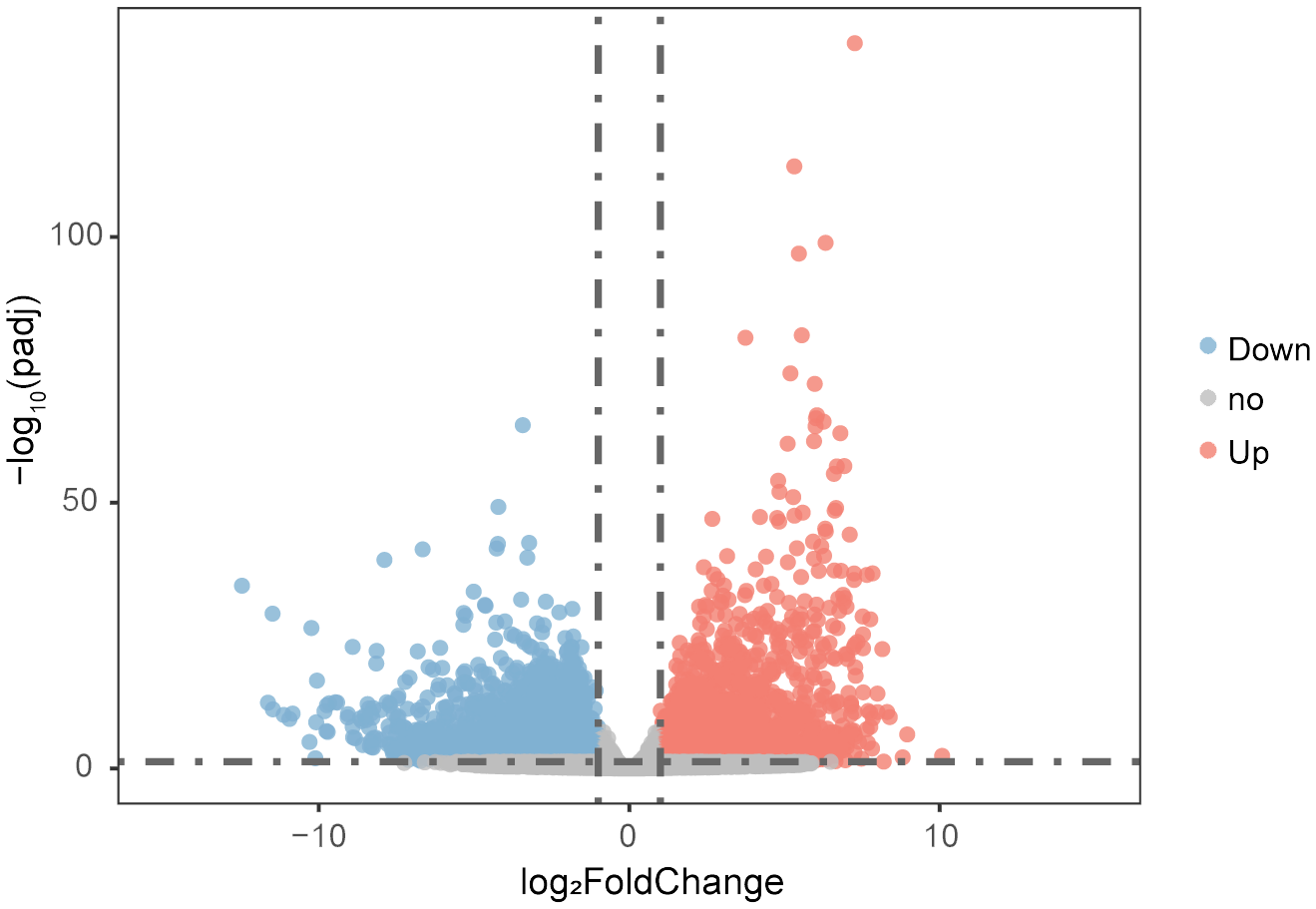
Volcano plot showing differentially expressed genes in testes and ovaries based on RNA-seq. Red and blue dots represent significantly upregulated and downregulated genes in ovary (|log2 FC| ≥ 1 and P < 0.05), respectively. Gray dots represent genes without significant differential expression.
3.5 Identification of candidate genes
Among the 57 sex-associated genes identified through GWAS analysis and SNP annotation, only 12 were differentially expressed between male and female gonads, including 10 male-upregulated genes and 2 female-upregulated genes (Figure 6; Supplementary Table S12). All 12 genes were located within the sexually-differentiated region on Chr. 9. Notably, ABCF2 and SPATA48 were consistently and highly expressed in the ovary, while SCP, 5-HTR, ADPK1-a, ADPK1-b, and CCKAR were stably and highly expressed in the testis (Figure 6B).
Figure 6
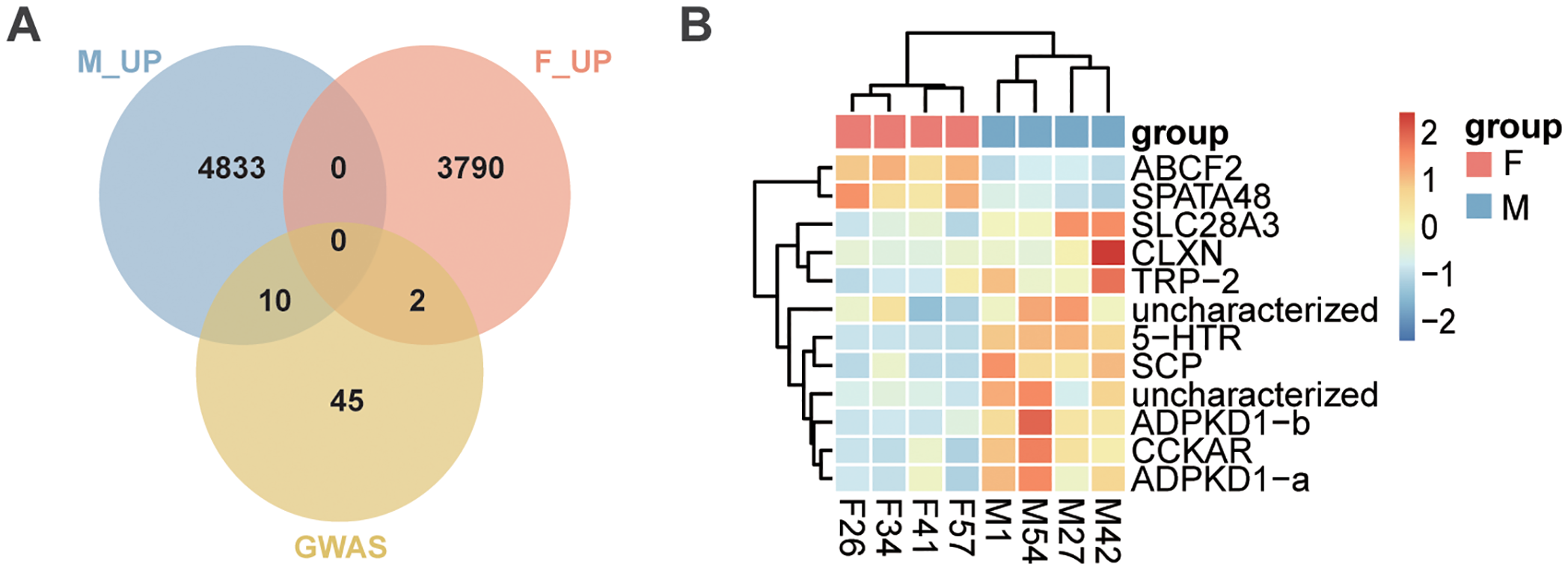
Intersection of sex-associated genes with DEGs between ovary and testis. (A) Venn diagram illustrating the overlap between sex-associated genes identified through GWAS and DEGs between testis and ovary. M_UP represents genes upregulated in testis, F_UP represents genes upregulated in ovary, and GWAS represents sex-associated genes. (B) Heatmap displaying the expression levels of 12 sex-determining candidate genes in testis and ovary.
To assess whether the 12 identified genes exhibit sex-associated genetic divergence pattern, NJ trees and PCA analyses were independently constructed based on SNPs within each gene. The results revealed that SNPs from 5-HTR, ADPKD1-a, SCP, and SLC28A3 demonstrated strong ability to distinguish individuals by sex in both NJ trees and PCA analyses, whereas SNPs from the other genes did not show similar discriminatory power (Figure 7; Supplementary Figures S4, S5). These findings suggest that these four genes may represent the earliest instances of genetic differentiation between sexes and may play a pivotal role in the sex determination process of Portuguese oysters. Consequently, 5-HTR, ADPKD1-a, SCP, and SLC28A3 are proposed as key candidate genes for future studies on the mechanisms of sex determination in this species (Table 1).
Figure 7
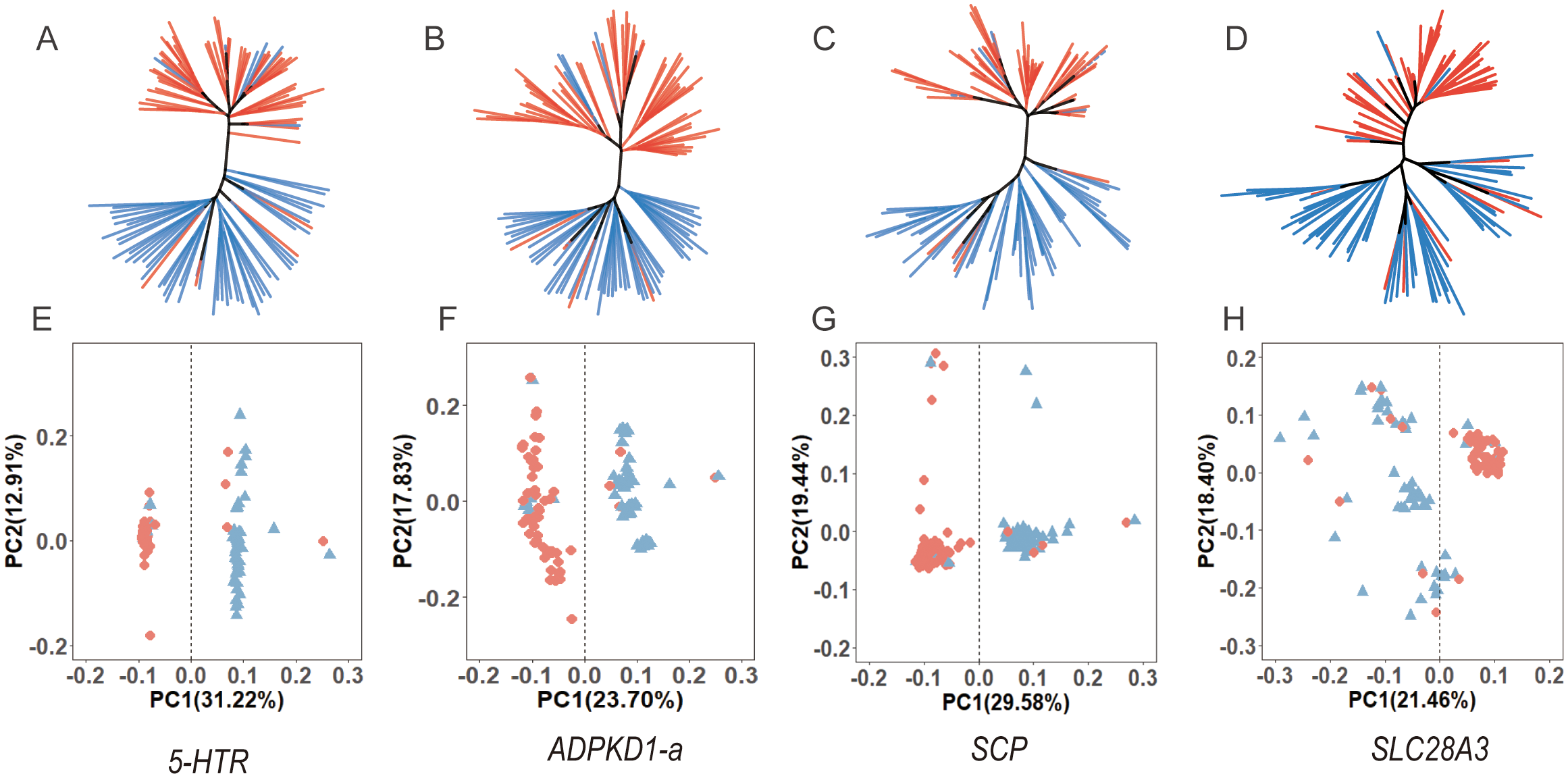
Neighbor-Joining (NJ) trees (A–D) and PCA plots (E–H) for four sex-associated genes, showing the correlation between genetic structure and sex.
Table 1
| Gene Symbol | Lead SNP | P-value of GWAS for lead SNP | Intersex FST | Description |
|---|---|---|---|---|
| 5-HTR | 9-8406434 | 1.69E-11 | 0.14 | 5-hydroxytryptamine receptor-like |
| ADPKD1-a | 9-8950441 | 1.79E-11 | 0.11 | polycystic kidney disease protein1-like 1 |
| SCP-a | 9-9596815 | 2.58E-10 | 0.10 | sarcoplasmic calcium-binding protein-like |
| SLC28A3 | 9-10480978 | 3.46E-09 | 0.07 | solute carrier family 28 member 3-like |
List of key candidate genes for sex determination in Portuguese oyster.
3.6 Haplotype construction and association analysis of the 5-HTR gene
Among the four key candidate genes, 5-HTR exhibited the strongest association with sex and contained a missense mutation, making it a representative gene for detailed SNP variation analysis (Table 1). This gene is located on Chr.9 between 8.37 and 8.43 Mb, comprising five exons and four introns (Figure 8A). Within this gene, 1,552 SNPs were identified, of which 239 (15.40%) were significantly associated with sex (Figure 8B). Local GWAS analysis revealed that sex-associated SNPs were distributed throughout the gene and its flanking genomic regions, with only 150 SNPs located in exon. Notably, a nonsynonymous mutation in exon 5 (Chr 9: 8,369,495 A>G) resulted in an amino acid substitution from isoleucine (Ile) to threonine (Thr) (Supplementary Table S6).
Figure 8
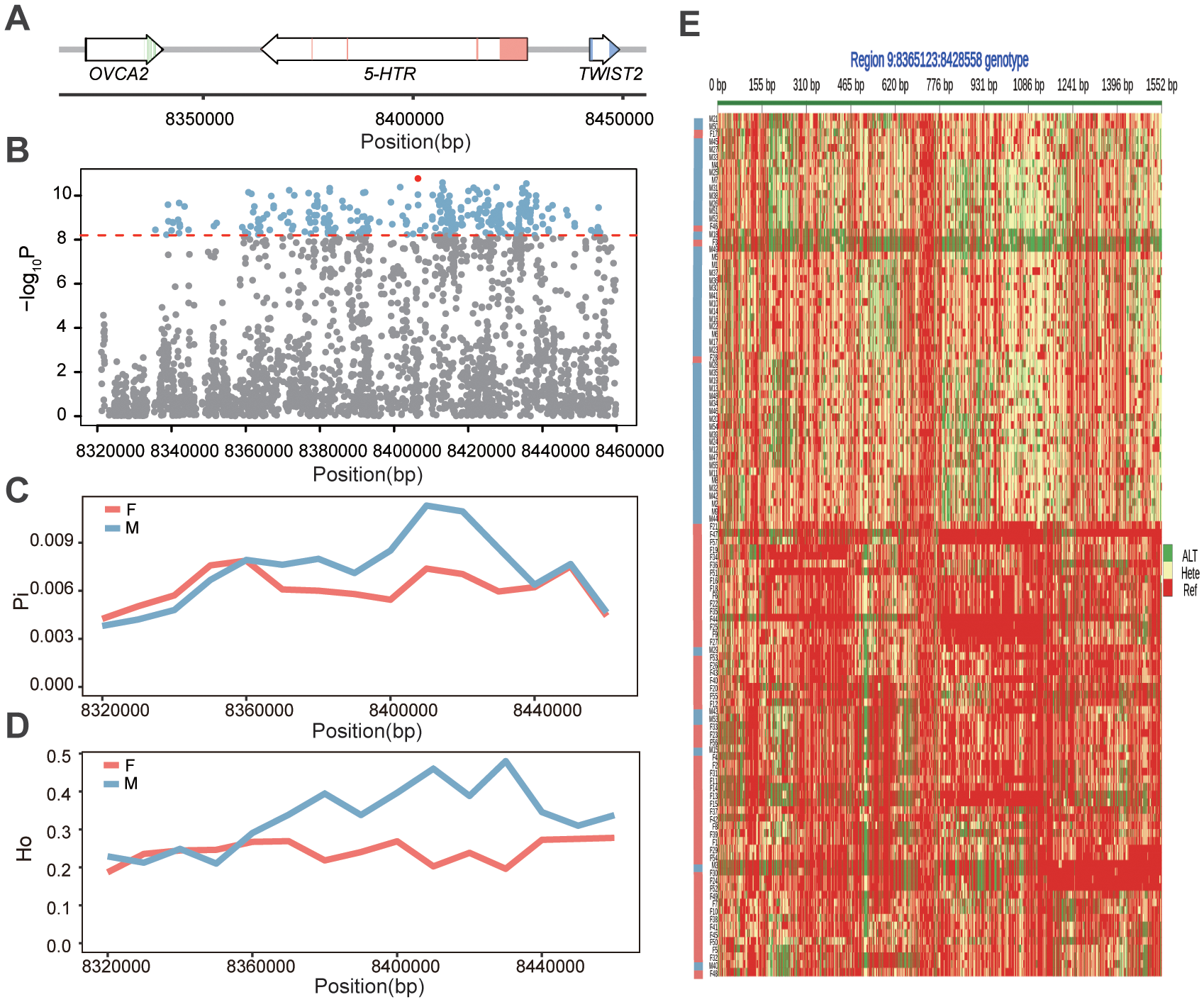
Genomic characterization of the 5-HTR locus in Portuguese oyster. (A) Genomic Structure of the 5-HTR gene and its upstream and downstream genes. (B) Local Manhattan plot showing the significance of SNP associations with phenotypic sex. (C) Nucleotide diversity (π) in male and female individuals. (D) Observed heterozygosity in females and males. (E) Genotype heatmap of SNPs within the 5-HTR region. Each row represents an individual sample, and each column represents a SNP. Red indicates homozygosity for the reference allele, green indicates homozygosity for the alternative allele, and yellow represents heterozygosity. Phenotypic sex is indicated by the colored bar on the left, with pink denoting females and gray-blue denoting males.
Interestingly, males exhibited higher genetic diversity (π) and observed heterozygosity (Ho) within the 5-HTR gene, particularly in the second intron region, suggesting the potential existence of a primitive male heterozygous pattern (Figures 8C–E). Genotype heatmaps further demonstrated that males had significantly more heterozygous locithan females across the four candidate genes (Figure 8E; Supplementary Figure S6). To validate the reliability of the resequencing data, Sanger sequencing of five female samples targeting the 5-HTR gene at Chr. 9: 8,369,495 A>G was performed, confirming the accuracy of the genotyping results (Supplementary Figures S7, S8).
Single-marker analysis identified 26 SNPs within 5-HTR that were significantly associated with sex. These SNPs formed six haplotypes, each containing sex-biased alleles, with association significance ranging from 9.41 × 10−³² to 6.92 × 10−¹³ (Table 2; Supplementary Figures S9, S10). Validation in an independent population confirmed that all six haplotypes were significantly associated with sex. However, differences in significance levels and allele frequencies between populations suggest that the genetic basis of sex determination exhibits both consistency and variability across populations (Figure 9).
Table 2
| Haplotype | Genotype | M_count | F_count | Sex ratio(M/F) | Beta | SE | P |
|---|---|---|---|---|---|---|---|
| Hap.1 | GA | 43 | 4 | 10.75 | -0.72 | 0.06 | 2.88E-20 |
| TT | 11 | 51 | 0.22 | 0.65 | 0.06 | 1.22E-17 | |
| Hap.2 | CTT | 47 | 6 | 7.83 | -0.56 | 0.06 | 1.77E-15 |
| TAG | 7 | 51 | 0.14 | 0.56 | 0.06 | 1.84E-16 | |
| Hap.3 | GAA | 45 | 4 | 11.25 | -0.72 | 0.06 | 6.75E-21 |
| ATT | 10 | 52 | 0.19 | 0.68 | 0.06 | 1.31E-20 | |
| Hap.4 | AG | 44 | 5 | 8.80 | -0.68 | 0.06 | 2.04E-19 |
| GT | 11 | 52 | 0.21 | 0.50 | 0.06 | 6.92E-13 | |
| Hap.5 | CTGG | 42 | 4 | 10.50 | -0.72 | 0.07 | 1.24E-19 |
| ACAA | 11 | 51 | 0.22 | 0.65 | 0.06 | 8.84E-19 | |
| Hap.6 | GT | 45 | 5 | 9.00 | -0.82 | 0.05 | 2.90E-28 |
| TC | 7 | 52 | 0.13 | 0.78 | 0.05 | 9.41E-32 |
Sex associated haplotype within the 5-HTR gene.
Beta, effect size of each haplotype; SE, standard error.
Figure 9
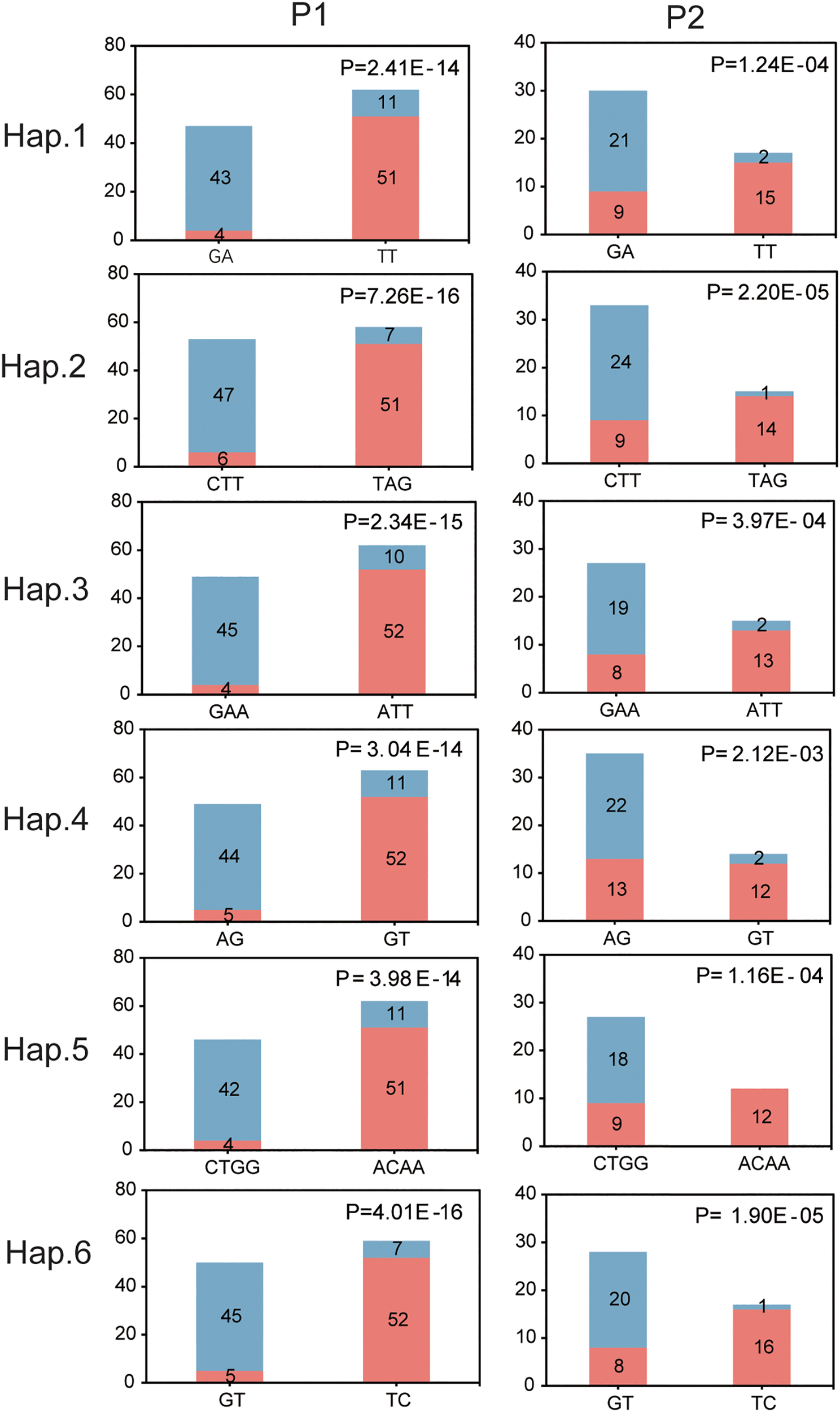
Six sex-associated haplotypes based on SNPs within 5-HTR across two independent populations. P1 denotes the GWAS discovery population, and P2 denotes the validation population. The x-axis represents haplotype categories, while the y-axis indicates the number of individuals carrying each haplotype. Pink and gray-blue bars correspond to female and male individuals, respectively. Statistical significance of the haplotype–sex association was assessed using chi-square tests, with P-values indicated.
4 Discussion
Oysters are dioecious, with bidirectional sex reversal potential across breeding seasons. In 1977, Haley identified paternal effect in half-sib families of C. virginica and proposed a “three-locus model”, in which oyster sex is determined by the additive effects of three loci (Haley, 1977, 1979). Subsequently, Guo et al. also observed paternal effect on sex ratios in half-sib families of M. gigas and suggested a “single-locus model”, positing that sex is controlled by one pair of alleles where genotype MF denotes males and FF may denote either males or females (Guo et al., 1998). More recently, two independent studies identified a major QTL linked to sex determination in M. gigas, using microsatellite markers and genotyping-by-sequencing (GBS), respectively (Guo et al., 2012; Han et al., 2021). Consistent with these findings, our study identified a major sex-associated genomic region on Chr.9 in M. angulata, supporting the hypothesis that sex determination in oysters involves a key genetic locus, particularly in certain families or populations.
Our study, along with the findings of Han et al., identified a major sex-determining locus on Chr.9 in Portuguese oysters and Pacific oysters, respectively (Han et al., 2021). However, the previously identified sex-linked genes PSMB3 and GPER1 were not located on Chr. 9 in Portuguese oysters, suggesting that the primary sex-linked loci on Chr. 9 may differ between these two species. This observation aligns with the well-documented variability in sex-determination systems among closely related species and even among populations within the same species, as observed in plants (Liston et al., 2014; VanBuren et al., 2015; Massonnet et al., 2020), bony fish (Lee et al., 2003; Franchini et al., 2018), amphibians (Jeffries et al., 2018), and shellfish (Han et al., 2022; Tang et al., 2025). Additionally, reduced significance of the 5-HTR gene in our validation population compared to the GWAS population suggests population-specific differences in genetic factors influencing sex determination. Such differences align with the frequently observed variation in sex ratios among families that were established in breeding programs (Guo et al., 1998; Vu et al., 2020), and highlight the complex and dynamic nature of sex determination mechanisms in oysters. Further studies warranted to comprehensively explorethese inter- and intra- species variations more comprehensively.
Chromosomal rearrangements are known to play a critical role in the origin and evolution of sex chromosomes. Examples include fragment duplication on LG9 in Oryzias latipes (Kondo et al., 2006), transpositions in salmonids (Bachtrog et al., 2008), and Y chromosome inversions in Drosophila miranda that restricted recombination with the X chromosome and accelerated differentiation (Bachtrog et al., 2008). In our study, the sex-linked region on Chr.9 exhibited distinct boundaries, elevated FST values, and a high density of sex-dimorphic loci over a 2.5 Mb interval, suggesting that one or more chromosomal rearrangements may have contributed to the formation of this sex-linked genomic region. We also observed multiple low-coverage gaps within this region, which is potentially due to the limitations of Illumina sequencing in high GC content, centromeric, or heterochromatic regions (Henikoff et al., 2001; Treangen and Salzberg, 2012; Ross et al., 2013). These findings highlight the need for further investigation into the structural genomic mechanisms underlying sex determination in oysters.
Sex-determining genes typically exhibit two critical characteristics: (1) the presence of sex-associated genetic variations, such as SNPs exhibiting significant differences between sexes; and (2) differential expression in male and female gonads, reflecting their functional role in sex differentiation. These dual features have been validated in well-known sex-determining genes, such as dmy in medaka (Matsuda et al., 2002), hsd17b1 in Japanese sea bream (Koyama et al., 2019), and fshr in flathead grey mullet (Ferraresso et al., 2021). By integrating GWAS and transcriptomic data, we identified 12 candidate genes that exhibit both sex-associated SNPs and differential expression. In addition, sex-determining genes are often located in the earliest evolutionary strata of sex chromosomes, representing the initial and strongest differentiation between sexes (Ferguson-Smith, 2006). Consequently, we further refined our candidate gene analysis by examining the association of their genetic structure with sex, narrowing down the 12 candidates to 4 key genes: 5-HTR, ADPKD1-a, SCP, and SLC28A3.
Among the four candidate genes identified, 5-HTR, ADPKD1-a, and SCP-a appear to participate in a regulatory framework involving neuroendocrine signaling and calcium dynamics. 5-HTR encodes a serotonin receptor, which regulates intracellular Ca²+ levels, activates the cAMP/PKA signaling cascade, and influences downstream transcriptional programs (Nichols and Nichols, 2008). In invertebrates such as pearl oysters and giant clams, serotonin (5-HT) signaling promotes oocyte maturation and gamete release (Wang and He, 2014; Yang et al., 2024), while in mosquitoes, it regulates ovarian development (Ling and Raikhel, 2018). In vertebrates, particularly fish, 5-HT has also been shown to modulate GnRH neuron activity and gonadotropin secretion, indicating a conserved role in the neuroendocrine regulation of reproduction (Prasad et al., 2015). ADPKD1-a belongs to the polycystin family and encodes a component of mechanosensitive calcium channels (e.g., Polycystin-1/2) that mediate Ca²+ influx in response to hydrodynamic or osmotic stimuli, thereby influencing cell fate decisions and gene expression (Wang et al., 2019; Lemos and Ehrlich, 2018). SCP-a, which encodes a sarcoplasmic calcium-binding protein, contributes to intracellular calcium buffering and signal precision, maintaining the spatial-temporal integrity of Ca²+ signaling (Asunción-Alvarez et al., 2024). Together, these genes may form a calcium-centered regulatory module that transduces environmental cues into developmental responses. Supporting this, studies in alligators and fish have shown that calcium signals activated by environmental changes regulate sex-determining genes such as Dmrt1 and Cyp19a1a via MAPK, CREB, and redox-sensitive pathways (Castelli et al., 2020; Heck and Woodward, 2025). In parallel, SLC28A3-like, a sodium-dependent nucleoside transporter, may link reproductive metabolism to sex-specific cell proliferation, particularly under high biosynthetic demand during gonadal development (Gray et al., 2004). Collectively, these findings suggest that 5-HTR, ADPKD1-a, and SCP-a may constitute a neuroendocrine–calcium signaling module that integrates environmental and genetic signals to orchestrate sex determination, gonadal development, and sexual phenotype maintenance in M. angulata.
In the present study, most of the identified sex-associated SNPs (92.0%) were located in non-coding regions, primarily in introns and flanking regulatory areas, suggesting a regulatory role in gene expression rather than direct protein modification. Non-coding SNPs may regulate gene expression through mechanisms such as cis-regulatory elements, splicing regulation, and epigenetic modifications (Pagani et al., 2000; Pastinen and Hudson, 2004; McVicker et al., 2013). In addition, higher heterozygosity observed in males within the 5-HTR region supports a male heterogametic-like segregation pattern, consistent with evolutionary trade-offs in sexual function optimization (Lesaffre et al., 2024).
Studies have demonstrated that oyster sex determination is influenced not only by genetic factors but also by environmental conditions. For instance, high temperatures have been shown to accelerate gonadal development in M. gigas, resulting in increased expression of male-biased genes and decreased expression of female-biased genes (Santerre et al., 2013). Similarly, starvation stress delays gonadal development in M. gigas, skews the sex ratio toward males, and alters both the methylation levels and expression of key sex-related genes (Sun et al., 2023). In this study, neither genome-wide sex-associated SNPs nor SNPs within key candidate genes were able to fully differentiate the sexes of the studied individuals. This finding suggests that environmental factors, in addition to genetic factors, may also contribute to sex determination in the studied population. These results highlight the complexity of oyster sex determination, which likely involves polygenic regulation and intricate gene-environment interactions.
6 Conclusion and perspectives
This study provides the first population-level genomic insight into sex determination in the Portuguese oyster (M. angulata). Through GWAS and transcriptomic integration, we identified a major sex-linked region on Chr.9 and four candidate genes (5-HTR, ADPKD1-a, SCP, and SLC28A3) with sex-associated SNPs and differential expression, suggesting their involvement in sex regulation. Among them, 5-HTR showed strong male-specific heterozygosity and haplotype signals. However, a key limitation is that all transcriptomic samples were derived from mature gonads, which may have missed early-acting sex-determining genes. We also observed reduced validation significance in an independent population, indicating possible population-specific variation. Future studies should focus on early gonadal stages and functional validation using gene editing and expression profiling. Together, our findings lay the groundwork for understanding sex determination in oysters and support the development of sex-controlled breeding strategies in aquaculture.
Statements
Data availability statement
Publicly available datasets were analyzed in this study. This data can be found here: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1185961.
Ethics statement
The animal study was reviewed and approved by the Animal Care and Use Committee of the Fisheries College of Jimei University.
Author contributions
XZ: Formal analysis, Investigation, Software, Visualization, Writing – original draft. YN: Conceptualization, Formal analysis, Funding acquisition, Resources, Writing – original draft. JX: Formal analysis, Writing – original draft. YH: Formal analysis, Writing – original draft. CT: Formal analysis, Writing – original draft. CS: Formal analysis, Writing – original draft. QWa: Investigation, Writing – original draft. QWu: Investigation, Writing – original draft. XG: Investigation, Writing – original draft. JQ: Investigation, Writing – original draft. YK: Resources, Writing – original draft. HG: Investigation, Writing – original draft. MC: Conceptualization, Project administration, Formal analysis, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was funded by the Agriculture Research System of China of MOF and MARA (CARS-49), National Key Research and Development Program of China (2018YFD0901400), and Special Fund Project of High-Quality Development of Marine Services and Fisheries in Fujian Province (FJHY-YYKJ-2024-1-6).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Correction note
A correction has been made to this article. Details can be found at: 10.3389/fmars.2025.1643904.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2025.1542815/full#supplementary-material
References
1
Allen S. K. Hidu H. Stanley J. G. (1986). Abnormal gametogenesis and sex ratio in triploid soft-shell clams (Mya arenaria). Biol. Bull.170, 198–210. doi: 10.2307/1541803
2
Asunción-Alvarez D. Palacios J. Ybañez-Julca R. O. Rodriguez-Silva C. N. Nwokocha C. Cifuentes F. et al . (2024). Calcium signaling in endothelial and vascular smooth muscle cells: Sex differences and the influence of estrogens and androgens. Am J Physiol Heart Circ Physiol326 (4), H950–H970. doi: 10.1152/ajpheart.00600.2023
3
Bachtrog D. Hom E. Wong K. M. Maside X. de Jong P. (2008). Genomic degradation of a young Y chromosome in Drosophila miranda. Genome Biol.9, R30. doi: 10.1186/gb-2008-9-2-r30
4
Barrett J. C. (2009). Haploview: Visualization and analysis of SNP genotype data. Cold Spring Harb. Protoc.2009, pdb.ip71. doi: 10.1101/pdb.ip71
5
Baršienė J. Ribi G. Barsyte D. (2000). Comparative karyological analysis of five species of viviparus (gastropoda: Prosobranchia). J. Molluscan Stud.66, 259–271. doi: 10.1093/MOLLUS%2F66.2.259
6
Breton S. Capt C. Guerra D. Stewart D. (2018). “Sex-determining mechanisms in bivalves,” in Transitions between sexual systems: Understanding the mechanisms of, and pathways between, dioecy, hermaphroditism and other sexual systems. Ed. LeonardJ. L. (Springer International Publishing, Cham), 165–192.
7
Browning B. L. Browning S. R. (2016). Genotype imputation with millions of reference samples. Am. J. Hum. Genet.98, 116–126. doi: 10.1016/j.ajhg.2015.11.020
8
Budd A. Banh Q. Domingos J. Jerry D. (2015). Sex control in fish: Approaches, challenges and opportunities for aquaculture. J. Mar. Sci. Eng.2015, 329–355. doi: 10.3390/jmse3020329
9
Carlton J. T. (1999). Molluscan invasions in marine and estuarine communities. Malacologia41, 439–454.
10
Castelli M. A. Whiteley S. L. Georges A. Holleley C. E. (2020). Cellular calcium and redox regulation: The mediator of vertebrate environmental sex determination?. Biol Rev Camb Philos Soc95 (3), 680–695. doi: 10.1111/brv.12582
11
Chen H. Boutros P. C. (2011). Venndiagram: A package for the generation of highly-customizable venn and euler diagrams in R. BMC Bioinf.12, 35. doi: 10.1186/1471-2105-12-35
12
Chen S. Zhou Y. Chen Y. Gu J. (2018). Fastp: An ultra-fast all-in-one fastq preprocessor. Bioinformatics34, i884–i890. doi: 10.1093/bioinformatics/bty560
13
Collin R. (2013). Phylogenetic patterns and phenotypic plasticity of molluscan sexual systems. Integr. Comp. Biol.53, 723–725. doi: 10.1093/icb/ict076
14
Danecek P. Auton A. Abecasis G. Albers C. A. Banks E. DePristo M. A. et al . (2011). The variant call format and VCFtools. Bioinformatics27, 2156–2158. doi: 10.1093/bioinformatics/btr330
15
Ferguson-Smith M. A. (2006). The evolution of sex chromosomes and sex determination in vertebrates and the key role of DMRT1. Sexual Dev.1, 2–11. doi: 10.1159/000096234
16
Ferraresso S. Bargelloni L. Babbucci M. Cannas R. Follesa M. C. Carugati L. et al . (2021). Fshr: A fish sex-determining locus shows variable incomplete penetrance across flathead grey mullet populations. iScience24, 101886. doi: 10.1016/j.isci.2020.101886
17
Forrest B. M. Keeley N. B. Hopkins G. A. Webb S. C. Clement D. M. (2009). Bivalve aquaculture in estuaries: Review and synthesis of oyster cultivation effects. Aquaculture298, 1–15. doi: 10.1016/j.aquaculture.2009.09.032
18
Franchini P. Jones J. C. Xiong P. Kneitz S. Gompert Z. Warren W. C. et al . (2018). Long-term experimental hybridisation results in the evolution of a new sex chromosome in swordtail fish. Nat. Commun.9, 5136. doi: 10.1038/s41467-018-07648-2
19
Gofas S. Zenetos A. (2003). “Exotic molluscs in the mediterranean basin: Current status and perspectives,” in Oceanography and marine biology (London: CRC Press), 237–277.
20
Grade A. Chairi H. Lallias D. Power D. Ruano F. Leitão A. et al . (2016). New insights about the introduction of the Portuguese oyster, Crassostrea angulata, into the North East Atlantic from Asia based on a highly polymorphic mitochondrial region. Aquat. Living Resour.29, 404. doi: 10.1051/alr/2016035
21
Gray J. H. Owen R. P. Giacomini K. M. (2004). The concentrative nucleoside transporter family, slc28. Pflügers Archiv447, 728–734. doi: 10.1007/s00424-003-1107-y
22
Guiguen Y. Bertho S. Herpin A. Fostier A. (2018). Sex determination and sex control in Salmonidae (Hoboken: John Wiley & Sons Ltd).
23
Guo X. Allen S. (1994). Sex determination and polyploid gigantism in the dwarf surfclam (Mulinia lateralis say). Genetics138, 1199–1206. doi: 10.1093/genetics/138.4.1199
24
Guo X. Hedgecock D. Hershberger W. K. Cooper K. XXXJ.R.X.X.X. S. K. A. (1998). Genetic determinants of protandric sex in the Pacific oyster, Crassostrea gigas thunberg. Evolution52, 394–402. doi: 10.1111/j.1558-5646.1998.tb01640.x
25
Guo X. Li Q. Wang Q. Z. Kong L. F. (2012). Genetic mapping and QTL analysis of growth-related traits in the Pacific oyster. Mar. Biotechnol.14, 218–226. doi: 10.1007/s10126-011-9405-4
26
Haley L. E. (1977). Sex determination in the American oyster. J. Hered.68, 114–116. doi: 10.1093/oxfordjournals.jhered.a108784
27
Haley L. (1979). Genetics of sex determination in the American oyster. Proc. Natl. Shellfisheries Assoc.69, 54–57.
28
Han Z. Li Q. Xu C. Liu S. Yu H. Kong L. (2021). QTL mapping for orange shell color and sex in the Pacific oyster (Crassostrea gigas). Aquaculture530, 735781. doi: 10.1016/j.aquaculture.2020.735781
29
Han W. Liu L. Wang J. Wei H. Li Y. Zhang L. et al . (2022). Ancient homomorphy of molluscan sex chromosomes sustained by reversible sex-biased genes and sex determiner translocation. Nat. Ecol. Evol.6, 1891–1906. doi: 10.1038/s41559-022-01898-6
30
Heck C. T. Woodward H. N. (2025). The consequences of calcium: Investigating intracortical reproductive signals in the American alligator for sex determination. Anat Rec (Hoboken)308 (2), 629–635. doi: 10.1002/ar.25533
31
Hedrick P. W. Hedgecock D. (2010). Sex determination: Genetic models for oysters. J. Hered.101, 602–611. doi: 10.1093/jhered/esq065
32
Henikoff S. Ahmad K. Malik H. S. (2001). The centromere paradox: Stable inheritance with rapidly evolving DNA. Science293, 1098–1102. doi: 10.1126/science.1062939
33
Horne C. R. Hirst A. G. Atkinson D. (2020). Selection for increased male size predicts variation in sexual size dimorphism among fish species. Proc. Biol. Sci.287, 20192640. doi: 10.1098/rspb.2019.2640
34
Hsiao S.-T. Chuang S.-C. Chen K.-S. Ho P.-H. Wu C.-L. Chen C. A. (2016). DNA barcoding reveals that the common cupped oyster in Taiwan is the Portuguese oyster Crassostrea angulata (ostreoida; ostreidae), not C. gigas. Sci. Rep.6, 34057. doi: 10.1038/srep34057
35
Jeffries D. L. Lavanchy G. Sermier R. Sredl M. J. Miura I. Borzée A. et al . (2018). A rapid rate of sex-chromosome turnover and non-random transitions in true frogs. Nat. Commun.9, 4088. doi: 10.1038/s41467-018-06517-2
36
Kondo M. Hornung U. Nanda I. Imai S. Sasaki T. Shimizu A. et al . (2006). Genomic organization of the sex-determining and adjacent regions of the sex chromosomes of medaka. Genome Res.16, 815–826. doi: 10.1101/gr.5016106
37
Koyama T. Nakamoto M. Morishima K. Yamashita R. Yamashita T. Sasaki K. et al . (2019). A SNP in a steroidogenic enzyme is associated with phenotypic sex in Seriola fishes. Curr. Biol.29, 1901–1909.e1908. doi: 10.1016/j.cub.2019.04.069
38
Lee B.-Y. Penman D. Kocher T. (2003). Identification of a sex-determining region in Nile tilapia (Oreochromis niloticus) using bulked segregant analysis. Anim. Genet.34, 379–383. doi: 10.1046/j.1365-2052.2003.01035.x
39
Lemos F. d. O. Ehrlich B. E. (2018). Polycystin and calcium signaling in cell death and survival. Cell Calcium69, 37–45. doi: 10.1016/j.ceca.2017.05.011
40
Lesaffre T. Pannell J. R. Mullon C. (2024). An explanation for the prevalence of XY over ZW sex determination in species derived from hermaphroditism. Proc. Natl. Acad. Sci. United States America121 40, e2406305121. doi: 10.1073/pnas.2406305121
41
Li H. Durbin R. (2009). Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics25, 1754–1760. doi: 10.1093/bioinformatics/btp324
42
Li H. Handsaker B. Wysoker A. Fennell T. Ruan J. Homer N. et al . (2009). The sequence alignment/map format and samtools. Bioinformatics25, 2078–2079. doi: 10.1093/bioinformatics/btp352
43
Li X.-Y. Mei J. Ge C.-T. Liu X.-L. Gui J.-F. (2022). Sex determination mechanisms and sex control approaches in aquaculture animals. Sci. China Life Sci.65, 1091–1122. doi: 10.1007/s11427-021-2075-x
44
Liao Y. Smyth G. K. Shi W. (2014). Featurecounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics30, 923–930. doi: 10.1093/bioinformatics/btt656
45
Ling L. Raikhel A. S. (2018). Serotonin signaling regulates insulin-like peptides for growth, reproduction, and metabolism in the disease vector aedes aEgypti. Proc. Natl. Acad. Sci. U. S. A115, E9822–e9831. doi: 10.1073/pnas.1808243115
46
Liston A. Cronn R. C. Ashman T. L. (2014). Fragaria: A genus with deep historical roots and ripe for evolutionary and ecological insights. Am. J. Bot.101 10, 1686–1699. doi: 10.3732/ajb.1400140
47
Love M. I. Huber W. Anders S. (2014). Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol.15, 550. doi: 10.1186/s13059-014-0550-8
48
Luo H. Xiao J. Jiang Y. Ke Y. Ke C. Cai M. (2021). Mapping and marker identification for sex-determining in the Pacific abalone, Haliotis discus hannai ino. Aquaculture530, 735810. doi: 10.1016/j.aquaculture.2020.735810
49
Martínez P. Viñas A. M. Sánchez L. Díaz N. Ribas L. Piferrer F. (2014). Genetic architecture of sex determination in fish: Applications to sex ratio control in aquaculture. Front. Genet.5. doi: 10.3389/fgene.2014.00340
50
Massonnet M. Cochetel N. Minio A. Vondras A. M. Lin J. Muyle A. et al . (2020). The genetic basis of sex determination in grapes. Nat. Commun.11, 2902. doi: 10.1038/s41467-020-16700-z
51
Matsuda M. Nagahama Y. Shinomiya A. Sato T. Matsuda C. Kobayashi T. et al . (2002). Dmy is a Y-specific dm-domain gene required for male development in the medaka fish. Nature417, 559–563. doi: 10.1038/nature751
52
McVicker G. van de Geijn B. Degner J. F. Cain C. E. Banovich N. E. Raj A. et al . (2013). Identification of genetic variants that affect histone modifications in human cells. Science342, 747–749. doi: 10.1126/science.1242429
53
Mei J. Gui J.-F. (2018). “Sexual size dimorphism, sex determination, and sex control in yellow catfish,” in Sex control in aquaculture (Hoboken: John Wiley & Sons Ltd), 495–507.
54
Minh B. Q. Schmidt H. A. Chernomor O. Schrempf D. Woodhams M. D. von Haeseler A. et al . (2020). Iq-tree 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol.37, 1530–1534. doi: 10.1093/molbev/msaa015
55
Nichols D. E. Nichols C. D. (2008). Serotonin receptors. Chem. Rev.108, 1614–1641. doi: 10.1021/cr078224o
56
Pagani F. Buratti E. Stuani C. Romano M. Zuccato E. Niksic M. et al . (2000). Splicing factors induce cystic fibrosis transmembrane regulator exon 9 skipping through a nonevolutionary conserved intronic element. J. Biol. Chem.275, 21041–21047. doi: 10.1074/jbc.M910165199
57
Park J. J. Kim H. Kang S. W. An C. M. Lee S.-H. Gye M. C. et al . (2012). Sex ratio and sex reversal in two-year-old class of oyster, Crassostrea gigas (bivalvia: Ostreidae). Dev. Reprod.16, 385–388. doi: 10.12717/dr.2012.16.4.385
58
Pastinen T. Hudson T. J. (2004). Cis-acting regulatory variation in the human genome. Science306, 647–650. doi: 10.1126/science.1101659
59
Pertea M. Kim D. Pertea G. M. Leek J. T. Salzberg S. L. (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nature Protocols11 (9), 1650–1667. doi: 10.1038/nprot.2016.095
60
Prasad P. Ogawa S. Parhar I. S. (2015). Role of serotonin in fish reproduction. Front. Neurosci.9. doi: 10.3389/fnins.2015.00195
61
Purcell S. Neale B. Todd-Brown K. Thomas L. Ferreira M. A. R. Bender D. et al . (2007). Plink: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet.81, 559–575. doi: 10.1086/519795
62
Qin Y. Li R. Liao Q. Shi G. Zhou Y. Wan W. et al . (2023). Comparison of biochemical composition, nutritional quality, and metals concentrations between males and females of three different Crassostrea sp. Food Chem.398, 133868. doi: 10.1016/j.foodchem.2022.133868
63
Rolan-Alvarez E. Buño I. Gosalvez J. (1996). Sex is determined by sex chromosomes in littorina saxatilis (olivi) (gastropoda, prosobranchia). Hereditas124, 261–268. doi: 10.1111/j.1601-5223.1996.00261.x
64
Ross M. G. Russ C. Costello M. Hollinger A. Lennon N. J. Hegarty R. et al . (2013). Characterizing and measuring bias in sequence data. Genome Biol.14, R51. doi: 10.1186/gb-2013-14-5-r51
65
Santerre C. Sourdaine P. Marc N. Mingant C. Robert R. Martinez A.-S. (2013). Oyster sex determination is influenced by temperature — first clues in spat during first gonadic differentiation and gametogenesis. Comp. Biochem. Physiol. Part A.: Mol. Integr. Physiol.165, 61–69. doi: 10.1016/j.cbpa.2013.02.007
66
Sun Y.-L. Jiang D.-N. Zeng S. Hu C.-J. Ye K. Yang C. et al . (2014). Screening and characterization of sex-linked DNA markers and marker-assisted selection in the Nile tilapia (Oreochromis niloticus). Aquaculture433, 19–27. doi: 10.1016/j.aquaculture.2014.05.035
67
Sun D. Yu H. Li Q. (2023). Starvation-induced changes in sex ratio involve alterations in sex-related gene expression and methylation in Pacific oyster Crassostrea gigas. Comp. Biochem. Physiol. Part B: Biochem. Mol. Biol.267, 110863. doi: 10.1016/j.cbpb.2023.110863
68
Tang C. Luo H. Luo X. You W. Ke Y. Ren P. et al . (2025). Identification and mapping of sex-linked markers in giant abalone (haliotis gigantea gmelin). Aquaculture595, 741529. doi: 10.1016/j.aquaculture.2024.741529
69
Treangen T. J. Salzberg S. L. (2012). Repetitive DNA and next-generation sequencing: Computational challenges and solutions. Nat. Rev. Genet.13, 36–46. doi: 10.1038/nrg3117
70
VanBuren R. Zeng F. Chen C. Zhang J. Wai C. M. Han J. O. et al . (2015). Origin and domestication of papaya yh chromosome. Genome Res.25, 524–533. doi: 10.1101/gr.183905.114
71
Vitturi R. Catalano E. (1988). A male XO sex-determining mechanism in Theodoxus meridionalis (Neritidae) (prosobranchia, Archaeogastropoda). Cytologia53, 131–138. doi: 10.1508/cytologia.53.131
72
Vitturi R. Colomba M. Caputo V. Pandolfo A. (1998). Brief communication. XY chromosome sex systems in the neogastropods Fasciolaria lignaria and Pisania striata (mollusca: Prosobranchia). J. Hered.89, 538–543. doi: 10.1093/jhered/89.6.538
73
Vu S. V. Knibb W. Nguyen N. T. H. Vu I. V. O’Connor W. Dove M. et al . (2020). First breeding program of the Portuguese oyster Crassostrea angulata demonstrated significant selection response in traits of economic importance. Aquaculture518, 734664. doi: 10.1016/j.aquaculture.2019.734664
74
Wang Q. He M. (2014). Molecular characterization and analysis of a putative 5-ht receptor involved in reproduction process of the pearl oyster pinctada fucata. Gen. Comp. Endocrinol.204, 71–79. doi: 10.1016/j.ygcen.2014.05.010
75
Wang H. Qian L. Liu X. Zhang G. Guo X. (2010). Classification of a common cupped oyster from Southern China. J. Shellfish Res.29, 857–866. doi: 10.2983/035.029.0420
76
Wang Z. Ng C. Liu X. Wang Y. Li B. Kashyap P. et al . (2019). The ion channel function of polycystin-1 in the polycystin-1/polycystin-2 complex. EMBO Reports20 (11), e48336. doi: 10.15252/embr.201948336
77
Weng X. Xu Y. Dong X. Luo X. You W. Ke C. et al . (2022). Sex-specific markers developed by next-generation sequencing confirmed a male heterogametic sex determination in small abalone, Haliotis diversicolor. Aquaculture555, 738256. doi: 10.1016/j.aquaculture.2022.738256
78
Xie J. Ning Y. Han Y. Su C. Zhou X. Wu Q. et al . (2024). Identification of snps and candidate genes associated with growth using gwas and transcriptome analysis in portuguese oyster (Magallana angulata). Fishes9, 471. doi: 10.3390/fishes9120471
79
Yang J. Lee S. H. Goddard M. E. Visscher P. M. (2011). GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet.88, 76–82. doi: 10.1016/j.ajhg.2010.11.011
80
Yang X. Noor Z. Guo S. Zhao Z. Cai B. Huang G. et al . (2024). Analysis of molecular identity and function of putative serotonin receptors in the giant clam (tridacna crocea) and the potential role of 5-ht1d-like receptor in reproduction. Aquaculture593, 741247. doi: 10.1016/j.aquaculture.2024.741247
81
Yu G. Wang L.-G. Han Y. He Q.-Y. (2012). Clusterprofiler: An R package for comparing biological themes among gene clusters. OMICS: A J. Integr. Biol.16, 284–287. doi: 10.1089/omi.2011.0118
82
Zeng Z. Ni J. Ke C. (2013). Expression of glycogen synthase (GYS) and glycogen synthase kinase 3β (GSK3β) of the Fujian oyster, Crassostrea angulata, in relation to glycogen content in gonad development. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol.166 3-4, 203–214. doi: 10.1016/j.cbpb.2013.09.003
83
Zhao Y. Li M. C. Konaté M. M. Chen L. Das B. Karlovich C. et al . (2021). TPM, FPKM, or normalized counts? A comparative study of quantification measures for the analysis of rna-seq data from the NCI patient-derived models repository. J. Transl. Med.19, 269. doi: 10.1186/s12967-021-02936-w
Summary
Keywords
Magallana angulata , sex-linked markers, genome-wide association analysis, transcriptomic analysis, sex determination
Citation
Zhou X, Ning Y, Xie J, Han Y, Tang C, Su C, Wan Q, Wu Q, Guo X, Qi J, Ke Y, Ge H and Cai M (2025) Identification of sex-linked markers and genes in Portuguese oyster (Magallana angulata). Front. Mar. Sci. 12:1542815. doi: 10.3389/fmars.2025.1542815
Received
10 December 2024
Accepted
28 April 2025
Published
06 June 2025
Corrected
26 June 2025
Volume
12 - 2025
Edited by
Ivana Veneza, Federal University of Western Pará, Brazil
Reviewed by
Hongyu Ma, Shantou University, China
Dong-Neng Jiang, Guangdong Ocean University, China
Updates
Copyright
© 2025 Zhou, Ning, Xie, Han, Tang, Su, Wan, Wu, Guo, Qi, Ke, Ge and Cai.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mingyi Cai, myicai@jmu.edu.cn; Hui Ge, gehuizlj@163.com
†These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.