 Romain Gastineau1*
Romain Gastineau1* Kamila Mianowicz2
Kamila Mianowicz2 Przemysław Dąbek1
Przemysław Dąbek1 Christian Otis3
Christian Otis3 Claude Lemieux4
Claude Lemieux4 Monique Turmel4
Monique Turmel4 Brygida Wawrzyniak-Wydrowska1
Brygida Wawrzyniak-Wydrowska1 Valcana Stoyanova2Artur Krawcewicz2Rafał J. Wróbel5Tomasz Abramowski2,6
Valcana Stoyanova2Artur Krawcewicz2Rafał J. Wróbel5Tomasz Abramowski2,6 Andrew J. Gooday7,8
Andrew J. Gooday7,8- 1Institute of Marine and Environmental Sciences, University of Szczecin, Szczecin, Poland
- 2Interoceanmetal Joint Organization, Szczecin, Poland
- 3Plateforme d’Analyse Génomique, Institut de Biologie Intégrative et des Systèmes, Université Laval, Québec, QC, Canada
- 4Institut de Biologie Intégrative et des Systèmes, Université Laval, Québec, QC, Canada
- 5West Pomeranian University of Technology, Szczecin, Poland
- 6Maritime University of Szczecin, Szczecin, Poland
- 7National Oceanography Centre, Southampton, United Kingdom
- 8Life Sciences Department, Natural History Museum, London, United Kingdom
Introduction: Xenophyophores are large benthic agglutinated Foraminifera that are a major component of the Clarion-Clipperton Zone megafauna.
Methods: Two xenophyophore specimens were obtained and submitted to genomic investigations.
Results: For both specimens, it was possible to obtain a ca. 25 kb circular xenophyophore-associated mitochondrial genome (XAM) showing similar gene contents with other Retaria, with which they are associated by a maximum likelihood multiprotein phylogeny. One of the specimens yielded a complete cluster of nuclear rRNA genes, the first to be obtained from a xenophyophore. Another full cluster of rRNA, likely belonging to Endomyxa parasites, was found within both specimens.
Discussion: Although the agglutinated nature of xenophyophores currently prevents a definitive conclusion, the mitogenomes obtained may represent the first to be obtained from those foraminifera. Deeper enquiries are required in order to properly ascribe these genomes to their host organism and to clarify the nature of the possibly parasitic Rhizaria associated with the xenophyophores.
Introduction
Xenophyophores are large, benthic protists in the supergroup Rhizaria that are confined to deep-sea habitats (Tendal, 1972, 1996). They have a test made of agglutinated material encapsulating a single branched, multinucleate cell enclosed within an organic tube (together forming the ‘granellare’ system) and large masses (the ‘stercomare’) of waste particles (stercomata) contained within an organic envelope (Tendal, 1972). The first species was described almost 150 years ago (Brady, 1883), but the taxonomic position of xenophyophores remained unresolved until the development of molecular methods, which revealed them to be giant agglutinated monothalamous foraminifera (monothalamids) (Pawlowski et al., 2003). In morphology-based foraminiferal classifications they are currently assigned to the monothalamid suborder Xenophyophoroidea Tendal, 1972 (Kaminski, 2014).
Xenophyophores are common at depths > 500 m in the bathyal, abyssal and hadal zones of the oceans (Tendal, 1996; Gallo et al., 2015). In some areas, such as the abyssal Clarion-Clipperton Zone (CCZ) in the Eastern Pacific, xenophyophores are particularly abundant and diverse (Gooday et al., 2017) and account for an important fraction of the megafauna (Ashford et al., 2014; Kamenskaya et al., 2013; Amon et al., 2016; Simon-Lledó et al., 2019a, 2019b; Durden et al., 2021; Uhlenkott et al., 2023). In the CCZ, they are often found associated with the polymetallic nodules that cover the seafloor (Gooday et al., 2020) and are of considerable potential commercial importance. The activities of contractors and their sponsoring countries interested in the possible future mining of nodules are regulated by the United Nations Convention on the Law of the Sea (UNCLOS) and the International Seabed Authority (ISA), the latter issuing detailed regulations and recommendations for exploration and prospecting activities. These include the sampling and identification of the benthic fauna and the acquisition of molecular barcodes (ISBA/25/LTC/6/Rev.2, 2022). As a result, a major international research effort has been devoted to the characterization of the benthic fauna in the CCZ (Rabone et al., 2023). Any attempt to collect massive amounts of nodules may impact xenophyophores, with additional consequences for associated organisms that rely on their often complex tests for multiple services, including the provision of food and shelter and breeding areas (Levin and Thomas, 1988; Levin and Gooday, 1992; Levin and Rouse, 2020).
The contractors licensed by ISA include the Interoceanmetal Joint Organization (IOM), a multinational consortium that has been assigned an exploration area in the eastern CCZ. Initially focusing mostly on geological-geochemical, engineering or environmental risk assessment studies (e.g., Abramowski and Nikończuk, 2019; Abramowski et al., 2021; Borkowski et al., 2022; Skowronek et al., 2021; Hikov et al., 2022; Milakovska et al., 2022; Radziejewska et al., 2022; Štyriaková et al., 2022; Wang et al., 2022), IOM has been expanding its field of expertise by successfully developing genomic studies of the CCZ megafauna (Gastineau et al., 2023a; 2025).
Generally speaking, the number of Rhizaria taxa whose mitochondrial genome has been sequenced is rather low when compared to other single-celled organisms (e.g., diatoms, chlorophyceae). This is especially true when taking into account the wide biodiversity of this supergroup. Several factors can account for this discrepancy, among them the difficulty of obtaining, identifying and cultivating Rhizaria. In the case of Foraminifera, it is only very recently that mitogenomes have been obtained from two planktonic species (Macher et al., 2023). Therefore, any additional taxa sequenced would be of interest, whatever their phylogenetic position within the Rhizaria.
Collections made by IOM during campaigns in the CCZ include some benthic megafaunal material, among which a few xenophyophores were present. Although most were in poor condition, two specimens were sufficiently well preserved to deserve investigation by next generation sequencing in order to obtain the molecular data required by ISA. The commonly used protocol of gene amplification by the polymerase chain reaction (PCR) followed by Sanger sequencing can be an issue in the case of Foraminifera. For example, the widely used gene coding for the small subunit of the nuclear ribosomal RNA (generally known as 18S or SSU) is characterized by its large and variable size in Retaria (Lecroq et al., 2009; Pawlowski et al., 2003, 2013). The extra length of the gene could create problems when trying to amplify it by PCR, needing an adaptation of the elongation time and a change of the polymerase used.
In the present study, the two xenophyophore specimens were considered as environmental samples rather than a monospecific isolate, with respect to their agglutinated nature, and we avoided any preconceived ideas regarding possible results. The sequencing reads were assembled with a stringent k-mer parameter and the resulting assemblies were data-mined for any sign of nuclear rRNA and mitochondrial genes. The contig files for both specimens yielded a circular mitochondrial genome with similar sizes and identical gene content. The current article refers to these as xenophyophore-associated mitogenomes (XAMs). This terminology reflects our careful approach to interpreting the results, prompted by the fact that we could not fully dissect the cytoplasm from the test. This would have raised a possibility of contamination of the sequenced DNA pool by organisms living on or inside the test. The gene content of the XAMs appeared to be close to that of planktonic foraminifera and clearly different from that of other Rhizaria. Data-mining the sequencing results for ribosomal RNA genes led to the discovery that another rhizarian, possibly parasitic, was present in both specimens. Finally, attempts were made to compare our results with the few genomic and transcriptomic data available for xenophyophores. Although these attempts did not invalidate our findings, they did prompt additional questions that will be discussed below.
Materials and methods
Sampling and characterization
Two xenophyophore specimens were obtained, together with the nodules they were attached to, by box-coring (a standard 0.25m2 box corer) during the 2014 IOM cruise to the CCZ (Figure 1) and stored in 90% ethanol. Every effort was made to keep the cold chain during sampling, transportation and storage, following the recommendations of ISA’s Legal and Technical Commission (LTC) s (ISBA, 2022). Specimen 2014_24 (Figure 2) was collected at station 3513 and specimen 2014_45 (Figure 3) at station 3535. Photographs of the specimen attached to polymetallic nodules were taken on board of the ship immediately after sampling, using a Nikon D700 camera equipped with an AF-S MICRO Nikkor 105mm 1:2.8G ED lens (Tokyo, Japan). Specimen 2014_24 was also imaged using a Nikon AZ100 stereo microscope with a Nikon DS-Fi2 camera (NIS Elements BR 4.13.05 software) (Tokyo, Japan). A portion of the test of specimen 2014_24 was dried on a SEM stubs without coating and observed in a SU8020 ultra-high resolution Hitachi scanning microscope (Tokyo, Japan) (Figure 4).

Figure 1. Location of the two sampling stations in the IOM claim area of the Clarion-Clipperton Zone (Pacific Ocean). The IOM claim area is represented in orange on the top left part. The block where the two samplings occurred is surrounded in red in the top right part. The two sampling stations are represented by red dots on the lower part of the figure.

Figure 2. Picture of the specimen of Spiculammina delicata IOM_2014_24 taken on its polymetallic nodule just after sampling (unscaled).

Figure 3. Picture of the specimen of Psammina aff. limbata IOM_2014_45 taken on its polymetallic nodule just after sampling (unscaled).
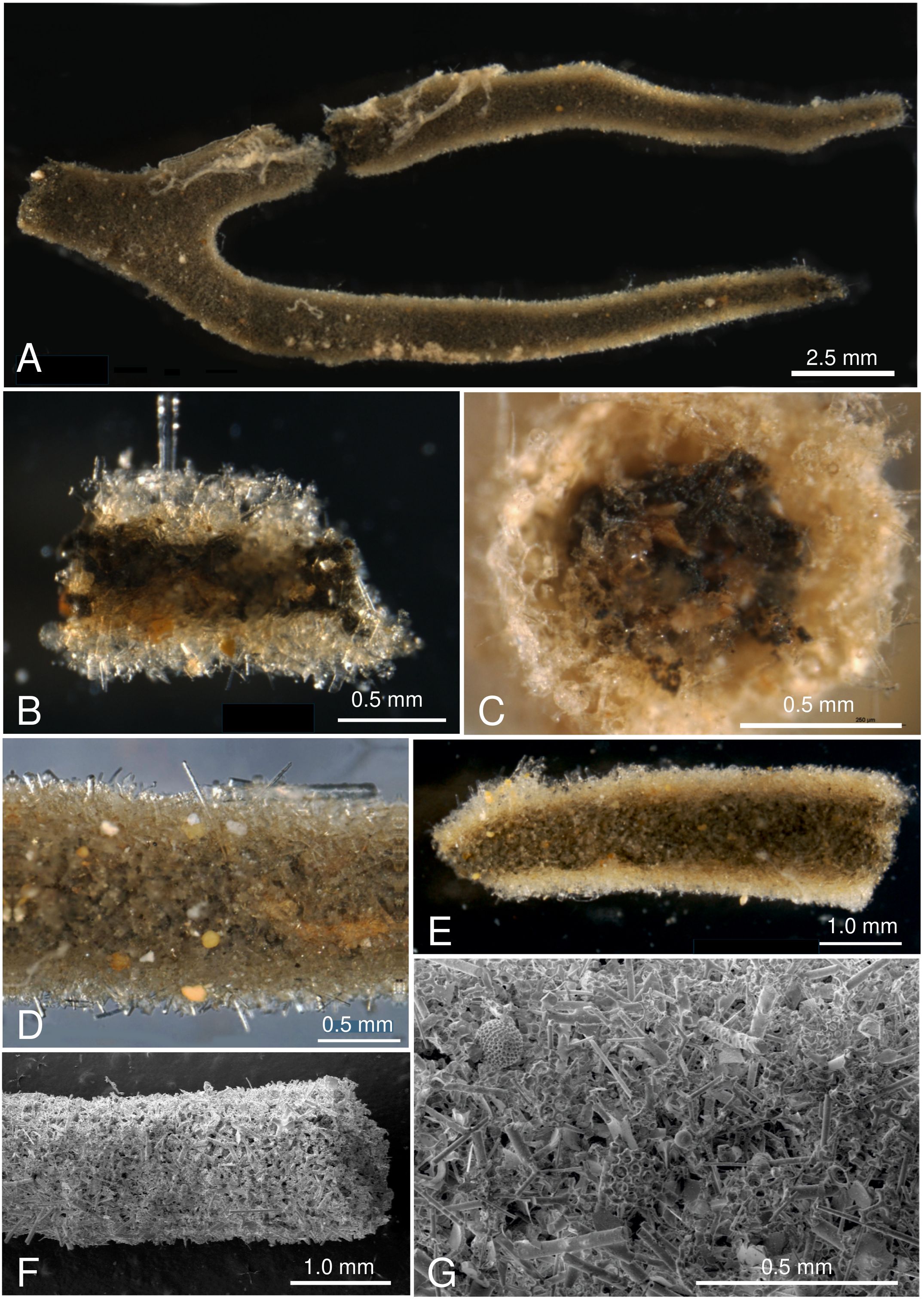
Figure 4. Pictures of test fragments of Spiculammina delicata IOM_2014_24. (A-E) Light photographs. (A). Overview of largest fragment. (B). Fragment broken longitudinally to show the thick test wall and interior with stercomare masses. (C). End of tubular fragment showing interior with stercomare masses and pale reddish granellare strands. (D) Exterior view of test with projecting sponge spicules and colored mineral grains. (E) Exterior view of test fragment immersed in glycerol to make it more transparent, revealing interior filled with dark stercomare masses. (F-G). SEM images of test wall composed of sponge spicules mixed with radiolarian fragments and mineral grains. Scales indicated on the pictures.
DNA extraction and sequencing
Specimen 2014_24 was in a better overall condition than specimen 2014_45, and contained larger quantities of cytoplasm. However, we were unable to fully isolate the cytoplasm and some test material remained prior to DNA extraction. In the case of 2014_45, dissection was not attempted and large parts of the test remained during extraction. DNA was extracted using a DNeasy Blood & Tissue Kit from Qiagen (Hilden, Germany), with a protocol adapted to include a 12h maceration in proteinase K at 65°C. DNA samples were sent to the Beijing Genomics Institute in Shenzhen. Sequencing took place on a DNBSEQ platform, with insert sizes of 400 bp. The sequencing returned distinct quantities of 150 bp paired-end reads depending on the sample, as explained in the Results part.
Assembly, data-mining, annotation and analyses of the gene content
Reads were assembled using SPAdes 3.15.5 (Bankevich et al., 2012) with a k-mer of 125. Basic assembly statistics were obtained using QUAST (Gurevich et al., 2013). Contigs of interest were identified by data-mining using blastn and blastx (Camacho et al., 2009). The blastn query for the rRNA used the Syringammina corbicula 18S partial sequence (GenBank: EU672993). The blastx query used the conserved proteins of Lotharella oceanica S.Ota, 2009 (KT806043) and Rhizaria sp. (MN082144), with the option query_gencode 4 and a e-value filter of 1e-20. We used orffinder (https://www.ncbi.nlm.nih.gov/orffinder/) with genetic code 4 to extract open-reading frames, and performed blastp analyses on each of them. The boundaries of all rRNA were found by using the Rfam portal (https://rfam.xfam.org/) (Kalvari et al., 2021). Arwen 1.2.3 (Laslett and Canbäck, 2008) was employed to find tRNA, and the secondary structure of the tRNA-Met was folded using Forna (Kerpedjiev et al., 2015). The non-conserved ORF were analyzed using InterPro (Paysan-Lafosse et al., 2023). The map of the XAM were drawn with OGDRAW (Lohse et al., 2013). Tandem repeats were searched using Tandem Repeat Finder online (Benson, 1999). Sequence logos were drawn with WebLogo 3.0 (Crooks et al., 2004) after multiple-sequence alignment with either Clustal Omega (Sievers and Higgins, 2014) or MUSCLE (Edgar, 2004).
Maximum likelihood multiprotein phylogeny of the XAMs
A set of protein sequences was selected from the ten available mitogenomes of Rhizaria and the two XAMs based on the following criteria: identified among all taxa, not split (e.g. ATP1 in Paracercomonas marina Cavalier-Smith & D.Bass, 2006), not fused (eg. Cox1/Cox2 in P. marina). The following proteins were selected: Cox3, ND1, ND2, ND3, ND4, ND4L, ND5, ND7, Cob, ATP6, ATP9. Each selected protein sequence was aligned separately using MAFFT 7 (Katoh and Standley, 2013) with the -auto option before being trimmed with trimAl (Capella-Gutiérrez et al., 2009) with the -automated1 option. The best model of evolution was evaluated on each alignment by Model-Test-NG (Darriba et al., 2020) using default parameters. Alignments were then concatenated by Phyutility 2.7.1 (Smith and Dunn, 2008) and formatted using the standalone version of ALTER (Glez-Peña et al., 2010). A partition file was created using the best model of evolution according to the Bayesian information criterion. Maximum-Likelihood phylogenetic analysis was conducted based on the alignment and the partition file using IQ-TREE v.2.2.0 (Minh et al., 2020) with 1000 ultra-fast bootstrap replicates. The tree was rooted with the Chlorarachniophyte L. oceanica and Bigelowiella natans Moestrup, 2001. Alignment and partition file can be obtained as Supplementary Data Sheets 1 and 2 respectively.
Two-dimensional folding of the xenophyophore 18S gene
In foraminifera, the gene sequence encoding the 18S contains several variable regions and unique nucleotide insertions, causing difficulties when trying to align it with other eukaryotic 18S sequences. Identifying the 5’ and 3’ ends of this gene, which have highly variable sequences, is no exception. To accurately define the position of the gene sequence, we have manually produced the secondary structure of the 18S rRNA of xenophyophores and compared it with the one proposed by Xie et al. (2011).
Maximum likelihood phylogeny of the contaminant rRNA
The contaminant 18S rRNA genes were aligned with the first 100 best megablast results filtered with a query cover of 70% and above, plus the 18S rRNA gene of L. oceanica and B. natans to root the tree. The software used were identical to those employed for the multiprotein phylogeny. The best model of evolution returned by Model-Test-NG was TIM2+I+G4 and 10,000 ultrafast bootstrap replicates were performed.
Data-mining the available genomic and transcriptomic data
The 310 bp sequence ascribed to the cox1 gene of Psammina limbata (OM719650) Kamenskaya et al., 2015 was used as a database, against which blastn queries of the contigs files from 2014_24 and 2014_45 were performed (no evalue filter applied).
Transcriptomic data of a specimen of Psammina sp (Sierra et al., 2022). were downloaded from SRA (SRX8544917), converted with SRA Toolkit and assembled using rnaSPAdes 3.15.5 (Bushmanova et al., 2019) with default parameters, and its completeness was evaluated with BUSCO 5.8.0 (Simão et al., 2015). The transcript file was data-mined by standalone blastn queries with the complete XAM of specimen 2014_45 and the fragment ascribed to the cox1 gene of Ps. limbata mentioned above. It was also submitted to blastx queries using the different protein-coding genes (PCGs) and ORFs of this XAM, with code 4 query code and an evalue filter of 1e-10.
Results
Species identifications
Shipboard photographs of the intact, freshly-collected specimens show that both xenophyophores were originally attached to nodules. Specimen 2014_24 had an arborescent morphology with a short basal trunk giving rise to a system of branches that divide dichotomously and taper towards pale extremities. The test wall was composed of a meshwork of sponge spicules. It was confidently identified as Spiculammina delicata Kamenskaya, 2005, a species described from the Russian exploration contract area in the central CCZ (Kamenskaya, 2005) and subsequently reported by Kamenskaya et al. (2015, 2017) from the same area where it appears to be the most common species. A single fragment was also found further to the east in the French area (Gooday and Wawrzyniak-Wydrowska, 2023).
Specimen 2014_45 had a flat, semicircular, fan-shaped test with vague concentric wrinkles that was attached to the nodule surface at 2–3 points on its lower margin. A root-like structure extended across the surface from one of these attachment points, and a similar branched root-like structure arose from the unattached part of the lower margin. Originally, this branched ‘root’ may have been attached to a second nodule. The specimen was confidently identified as Psammina aff. limbata Form 1, sensu Gooday et al. (2018), a species that is quite common in the eastern CCZ.
These two specimens are hereafter referred to as Spiculammina and Psammina, respectively.
Xenophyophora-associated mitogenomes
The sequencing of Spiculammina returned overall better results, with ca. 70M clean paired-end reads obtained. From the sequencing of Psammina, it was only possible to derive ca. 20M paired-end reads totally. After assembly, for Spiculammina, the total number of contigs was 372,064, the largest being 71,092 bp long with a N50 of 856 bp. For Psammina, these values were 36,960 bp, 66,642 bp and 1,341 bp respectively (complete analyzes available as Supplementary Data Sheet 3 for Spiculammina and Supplementary Data Sheet 4 for Psammina). Complete mitochondrial genome sequences with redundant endings were retrieved from the contig file of both specimens. The contig recovered from Spiculammina was 26,100 bp long with a coverage of 113.27X, while that of Psammina was 26,725 bp long with a coverage of 139.60X. For both contigs, the redundancy at the endings was 125 bp long. Later examination, following annotation, showed that these redundancies occurred within conserved genes, namely cob for Spiculammina and rnL for Psammina. After circularization and trimming, the genomes are 25,975 bp and 26,600 bp long for Spiculammina (Figure 5) (GenBank: PV138223) and Psammina (Figure 6) (GenBank: PV138224), respectively. Tandem Repeat Finder did not return any significant results. The sizes of these mitogenomes are smaller than those of other Rhizaria (Table 1), which range from 33,862 bp for Polymyxa betae Keskin, 1964 to 114,663 bp for Plasmodiophora brassicae Woronin, 1877, both being parasites of terrestrial plants. The Spiculammina and Psammina mitochondrial genomes are also half the size of those reported for the planktonic foraminifera Calcarina hispida Brady, 1876 and Neorotalia gaimardi (d’Orbigny in Fornasini, 1908) (Table 1). Excluded from analyses are the radiolarian mitogenomes described as fragmented (Macher et al., 2023) and not available in GenBank.
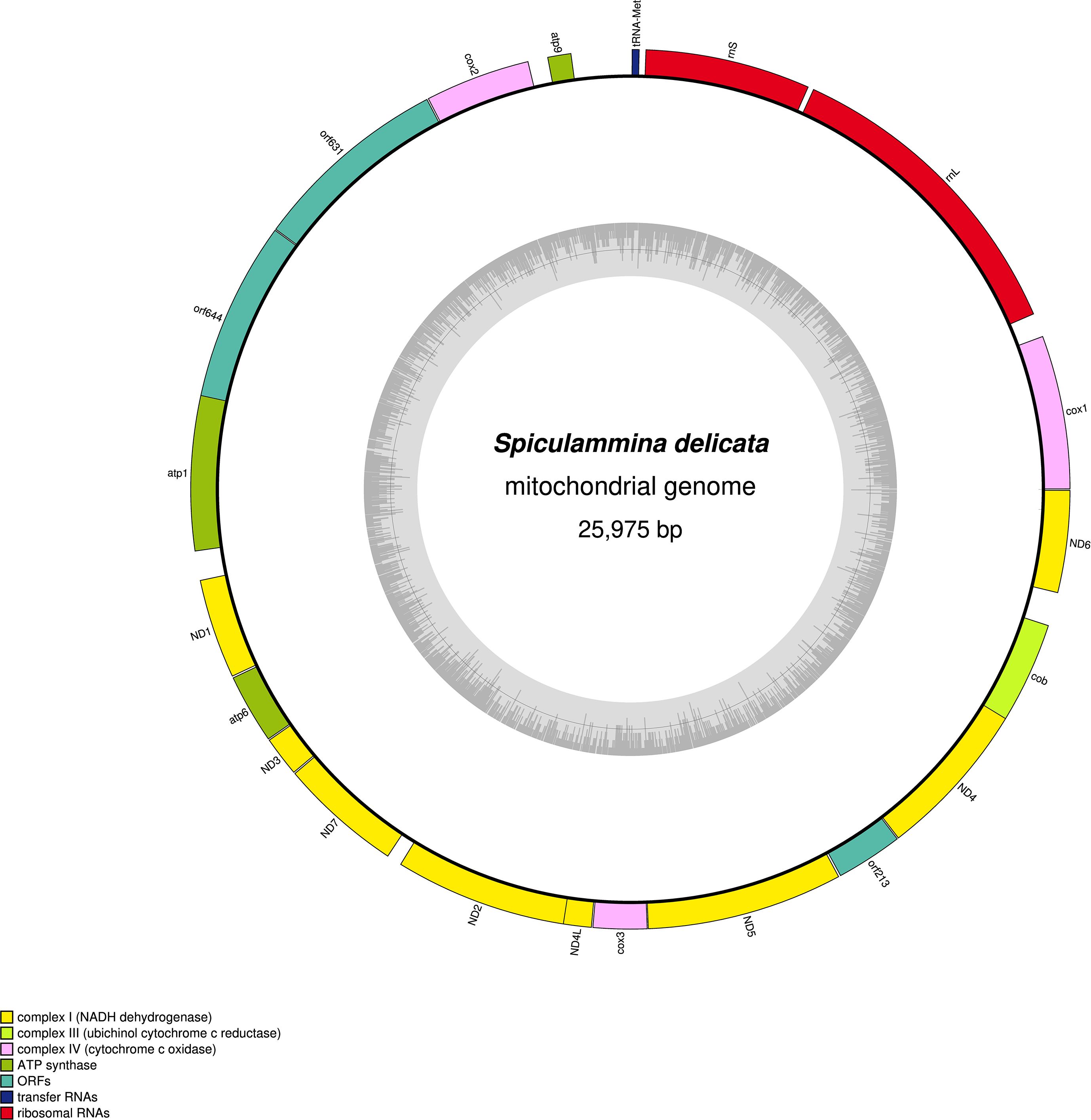
Figure 5. Map of the mitochondrial genome of the Spiculammina delicata-associated mitogenome.
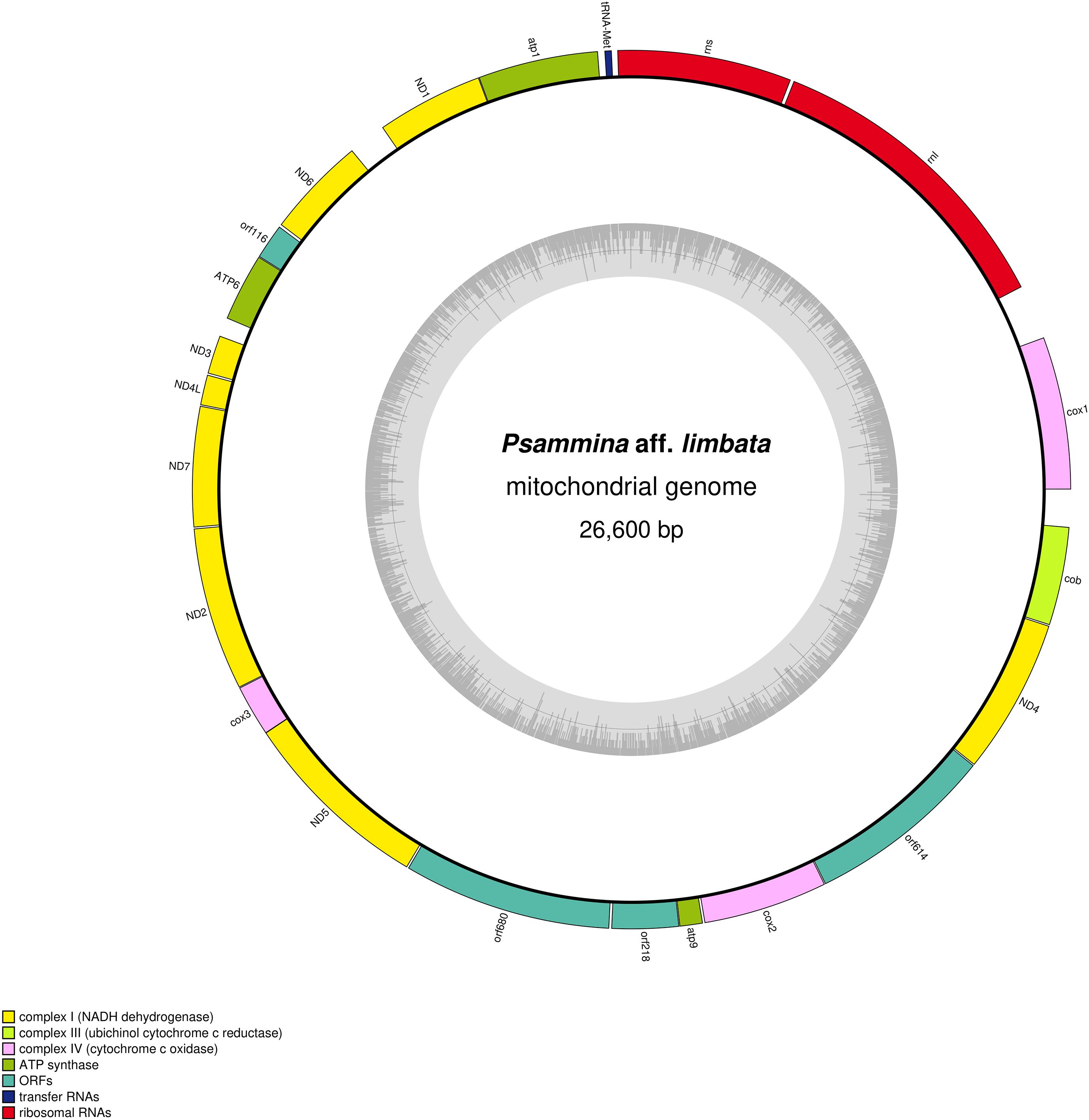
Figure 6. Map of the mitochondrial genome of the Psammina aff. limbata-associated mitogenome.
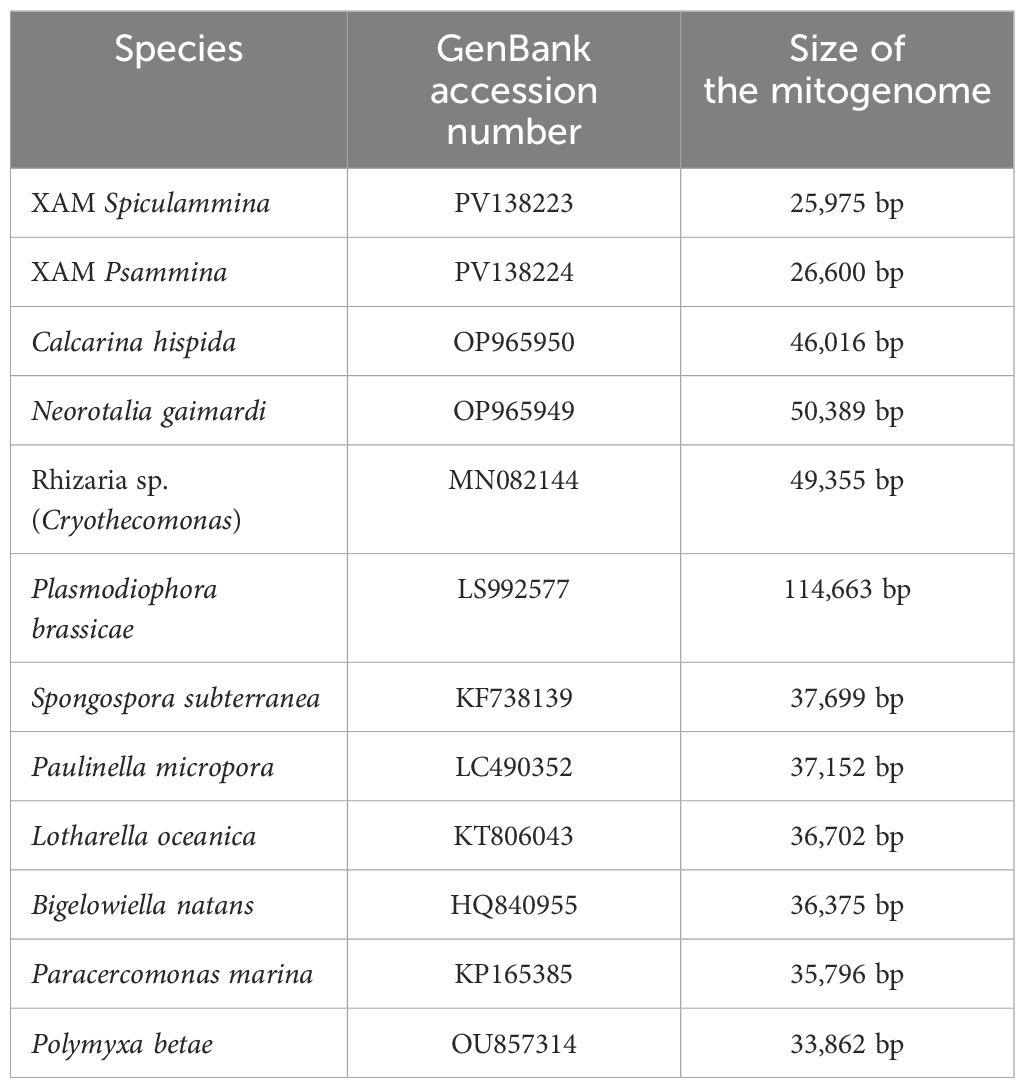
Table 1. Accession numbers and sizes of the available Rhizaria mitochondrial genomes.
No intron was detected in both the Spiculammina and Psammina mitochondrial genomes, and there is no gene duplication, such as that observed in L. oceanica (Tanifuji et al., 2016). The two genomes are not colinear with respect to gene order. They share a total of 16 conserved protein-coding genes that require NCBI genetic code 4 for translation. No atp8 or rps/rpl genes could be identified. These genes also appear to be missing in other Retaria (Macher et al., 2023), although they are sporadically found among Rhizaria. A total of 12 mitochondrial encoded rps/rpl genes are indeed present in the unidentified species of Rhizaria mentioned as Rhizaria sp. (MN082144) and Po. betae (OU857314). So far, atp8 has only been detected in B. natans, Po. betae and Pl. brassicae.
The sizes of the 16 conserved proteins of Spiculammina and Psammina are listed in Table 2, along those of others available for Retaria, and their percentages of identity are presented in Table 3. The Spiculammina and Psammina cox3 proteins are smaller than in other Rhizaria and they could not be extended because of the presence of stop codons. The cox1 and cox2 proteins are encoded in distinct ORFs and are missing conserved residues at their N termini, in contrast to their homologs in Paulinella micropora D. Lhee, E.C. Yang, J.I. Kim, R.A. Andersen & H.S. Yoon 2017 (Lhee et al., 2017) and Pa. marina (Valach et al., 2014) that are fused into single proteins. Also noteworthy is a 71 amino-acid difference in the middle of Cox2 that might correspond to an extension segment of the protein. The putative proteins encoded by the divergent ND6 display 10 and 9 transmembrane helix domains for Spiculammina and Psammina, respectively. The N- and C-terminal regions of these proteins successfully align with the corresponding portions of Rhizaria ND6 proteins, but there is a large and variable extension segment in the central portion of the predicted protein (Figure 7). Note that ND6 has not been found in planktonic foraminifera (Macher et al., 2023).
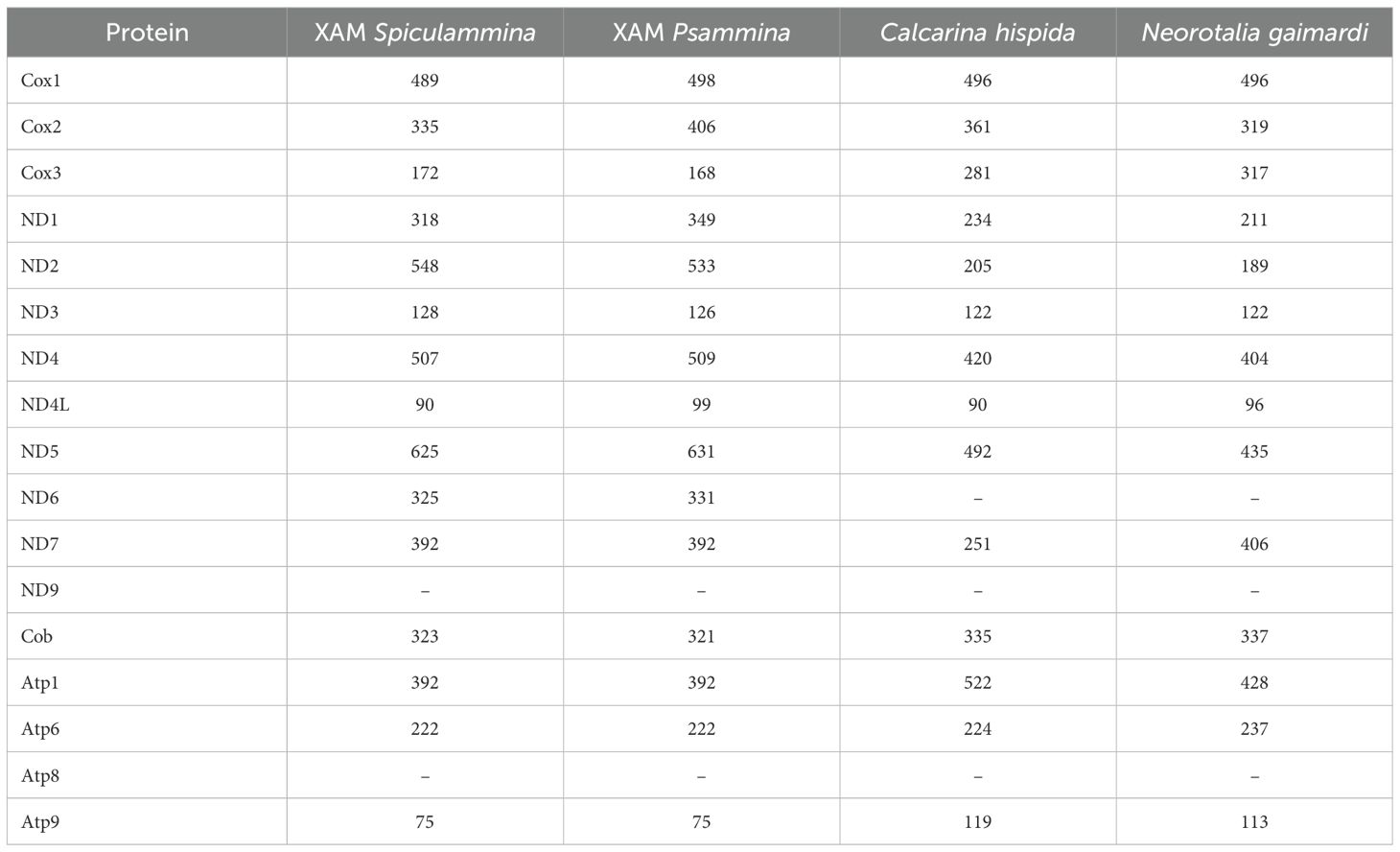
Table 2. Sizes of the putative mitochondrial proteins encoded by the XAM and the two available references for Retaria (in amino-acids).

Figure 7. LOGO representation of the alignment of ND6 proteins from available Rhizaria and the putative corresponding protein of the Xenophyophore-associated mitogenomes.
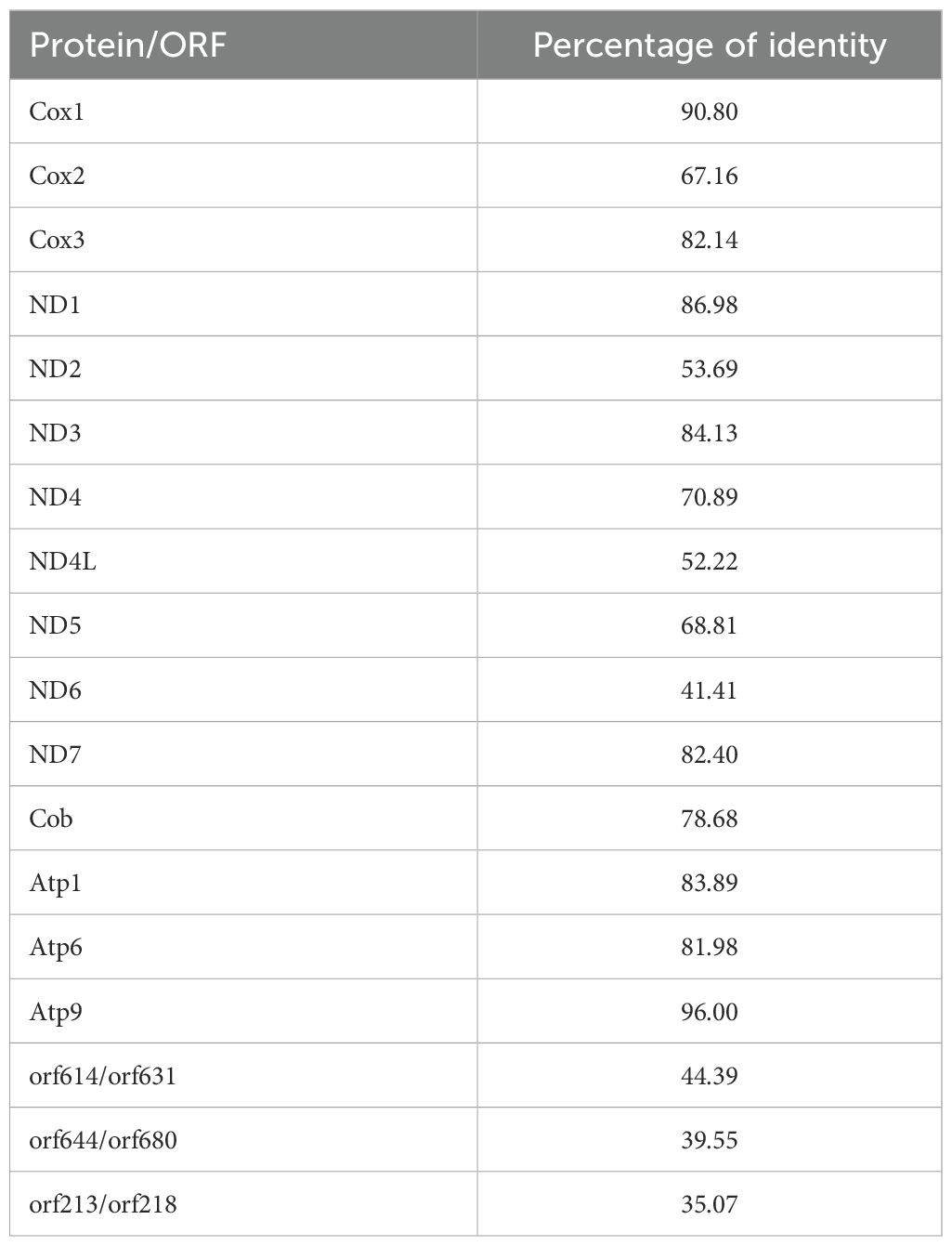
Table 3. Percentage of identities between the protein and non-identified ORF found in both XAMs.
Considering their sizes, orf213/orf218 might represent a divergent ND9, a gene that also appear to be missing in planktonic foraminifera (Macher et al., 2023). However, alignments of their putative proteins with reference sequences from the Conserved Domain Database (Wang et al., 2023) and other Rhizaria provide no support for this interpretation. InterPro failed to detect any conserved domain for the orf213/orf218 and orf614/orf631 pairs, but in the case of the orf644/orf680 pair, five domains of transmembrane helix were identified, four of which are conserved enough to be aligned (Figure 8).

Figure 8. LOGO representation of the alignment of the two putative protein encoded by orf644 and orf680. The four conserved transmembrane helix domains are highlighted by a yellow frame.
The Spiculammina and Psammina mitochondrial genomes encode two rRNA genes (rnS and rnL) and a single tRNA gene (tRNA-Met) (Table 4). These three genes form a conserved cluster with cox1 in both genomes (Figures 5, 6). Modelling of the tRNA-Met reveals that its D-loop is quite divergent although its anticodon and T-loops are conserved (Figure 9). Note that all our efforts to identify additional tRNA genes failed. The presence of other tRNA genes was initially suggested by Arwen, but these later proved to be artifactual because their positions were in conflict with rRNA or protein-coding genes.
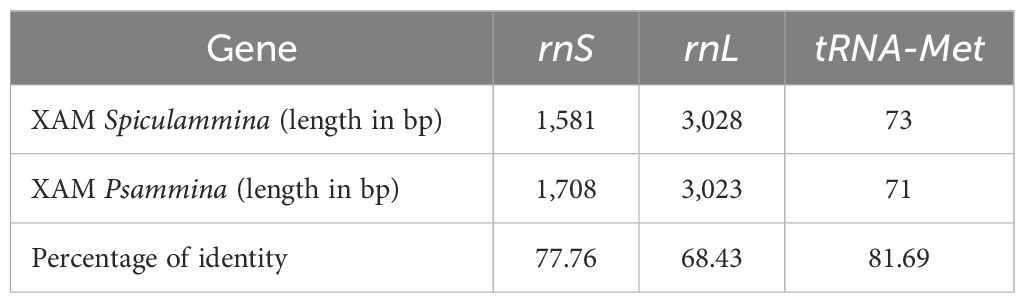
Table 4. Lengths and percentages of identity between the rRNA and tRNA identified in the two XAMs.
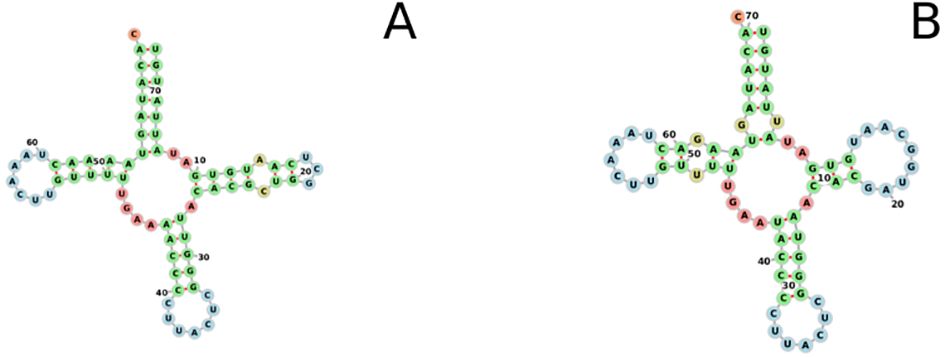
Figure 9. Two-dimensional folding of the single tRNA (tRNA-Met) found within the Xenophyophore-associated mitogenomes. Figure(A). Spiculammina delicata. Figure(B). Psammina aff. Limbata.
Multiprotein phylogeny
The maximum likelihood phylogenetic tree associated the two XAMs to planktonic Foraminifera with a strong support (Figure 10), forming a clade with very long branches. The distance between the two XAMs is greater than that between the planktonic foraminifera, and is comparable to the distance between the two species of Chlorarachniophyte used to root the tree. For the purpose of the following discussion, it should be underlined that XAMs are completely distinct from those of Endomyxa species (namely Po. betae, S. subterranea and Pl. brassicae).
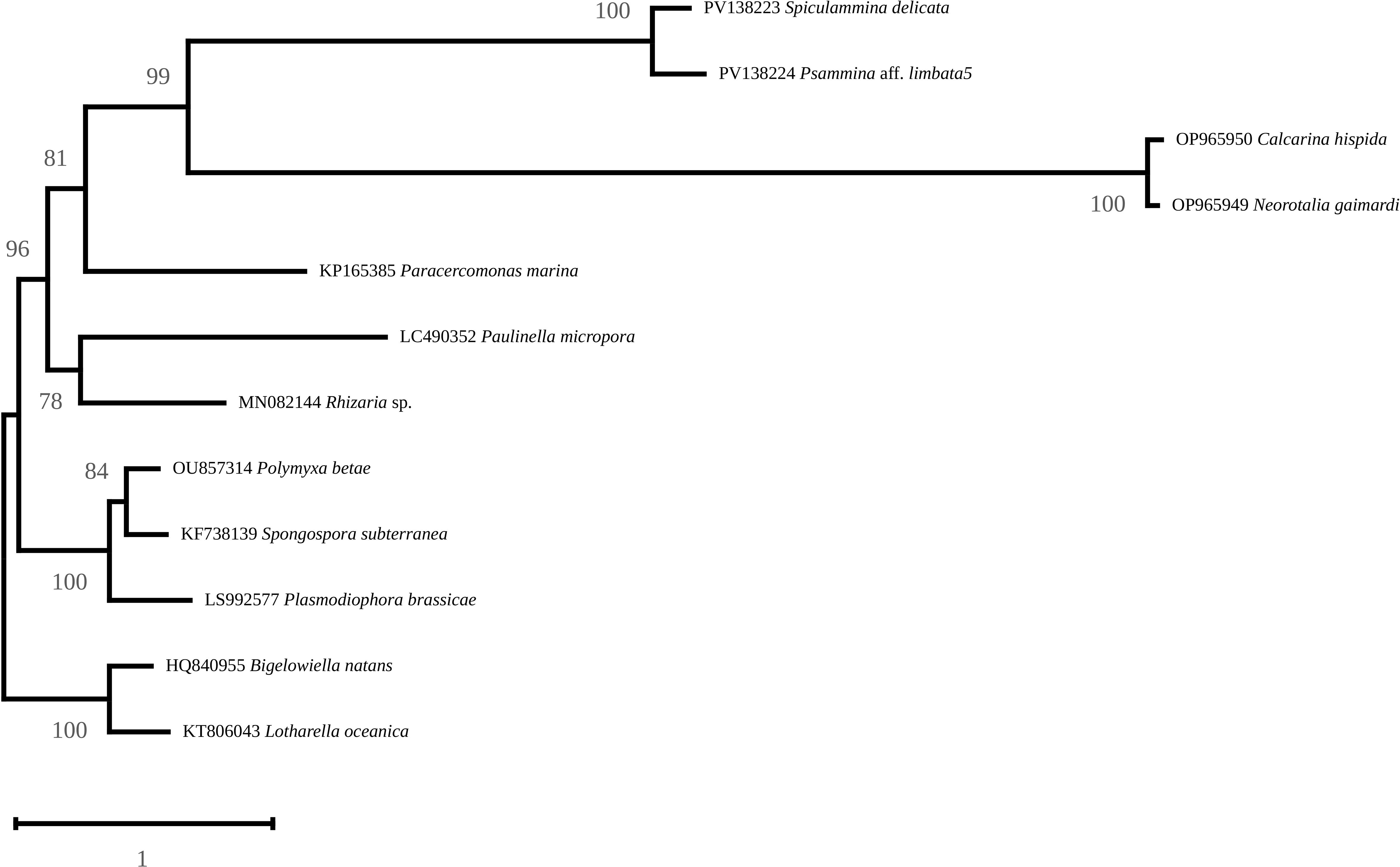
Figure 10. Maximum-Likelihood phylogenetic tree obtained from an alignment of nine mitochondrial protein sequences from 12 Rhizaria and rooted with Bigelowiella natans and Lotharella oceanica. Support is indicated at the nodes.
Cluster of xenophyophore nuclear rRNA genes
For Spiculammina, we retrieved a 15,625 bp long fragment with a coverage of 140.48X that contains the complete cluster of nuclear ribosomal genes (GenBank: PV146256). The sizes of the 18S, 5.8S and 28S genes are 3,618 bp, 178 bp and 4,793 bp, respectively. The large size of the 18S is reminiscent to that observed for the freshwater foraminifera Haplomyxa saranae Dellinger, 2014, where the 18S gene (HE965431) is 4,863 bp long (Dellinger et al., 2014), or to that of the xenophyophores Shinkaiya lindsayi Lecroq et al., 2009 (EU649778) (Lecroq et al., 2009) and Syringammina corbicula Richardson, 2001 (EU672993) (Pawlowski et al., 2003) the sizes of which were 4,054 bp and 3,304 bp, respectively. Interestingly, an ORF lies at the 3’ end of the 28S gene. This ORF encodes a putative 496 AA transposase that blastp queries associate with piggyBac transposable element-derived protein. The proposed secondary structure is presented in Figure 11. The bases in red are identical to the structure proposed by Xie et al. (2011), while the bases and positions in blue represent variable regions that seem specific to Foraminifera. The nucleotide numbers of the additional sequences in Xenophyophores are indicated in the semicircles.
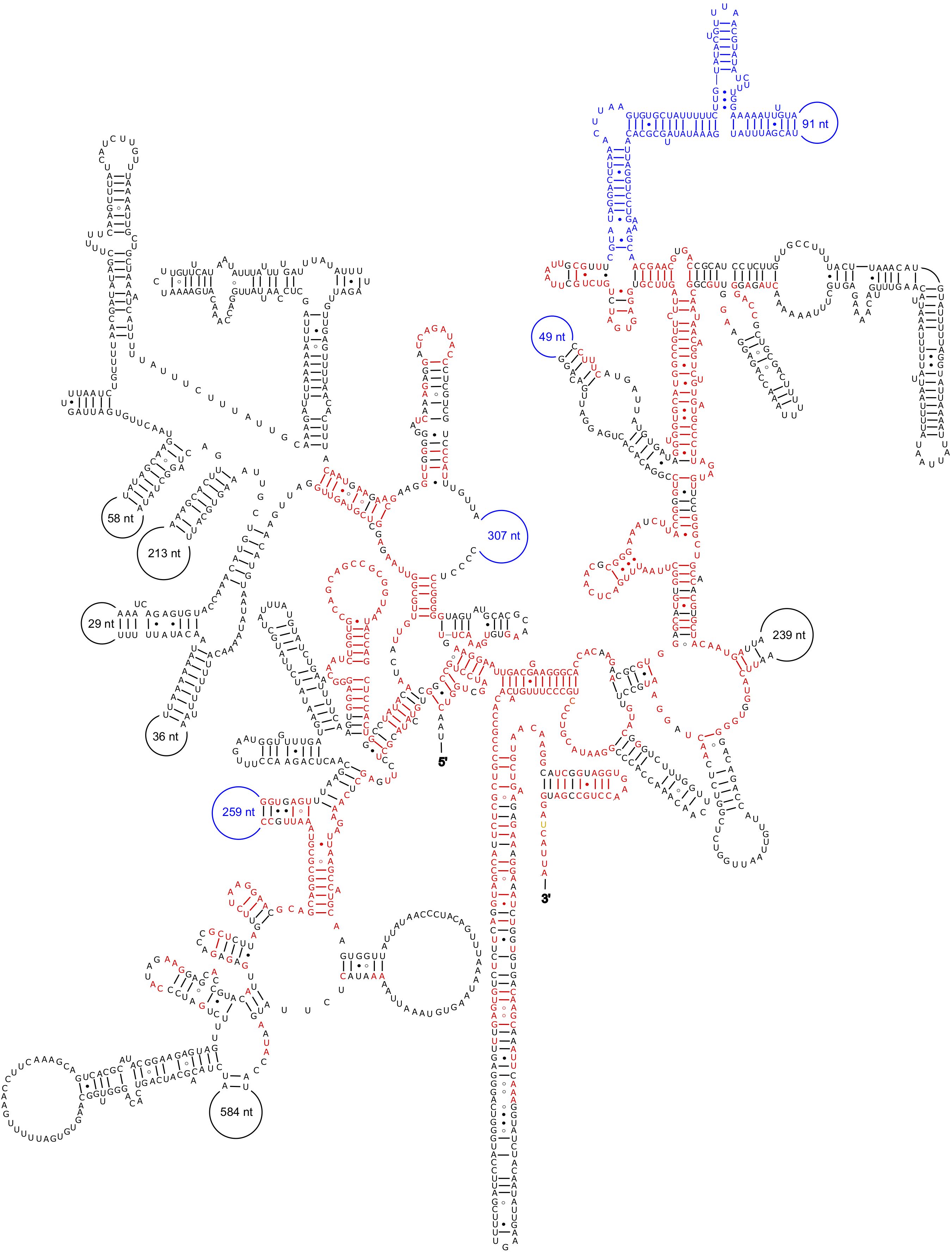
Figure 11. Secondary structure of the 18S rRNA of Spiculammin delicata. The bases in red are identical to the structure proposed by Xie et al. (2011). The bases and positions in blue represent the variable regions specific to Foraminifera. Standard pairing (A-U, G-C) is represented by dashes, the G○U pairs by dots, and the G○A pairs by an open circle. The nucleotide numbers of the sequences that seem specific to S. delicata are indicated in the semicircles.
For Psammina, we failed to assemble the complete cluster into a single contig. By lowering the kmer for assembly to 85, it was possible to extract several contigs that matched the rRNA queries. However, they came with a rather low coverage of about 6X. Our results suggest that there might be repeated portions within these genes, especially the 18S. It was possible to merge some of these contigs to obtain larger fragments, but this protocol was not considered to be reliable and the resulting contigs were therefore not retained. These contigs have not been submitted to GenBank but are available on a public repository as detailed in the data availability statement.
Unexpected guests: signals of unidentified Rhizaria associated with xenophyophores
An additional cluster of nuclear rRNA genes originating from unidentified Rhizaria was also found within each XAM specimen. The contigs were 13,003 bp long with a coverage of 45.94X for Spiculammina and 8,651 bp long with a coverage of 12.84X for Psammina. After trimming, the complete nuclear rRNA operons of Spiculammina (GenBank: PV134890) and Psammina (GenBank: PV134975) proved to be 6,557 bp and 6,545 bp long, respectively. Megablast queries returned partial 18S sequences ascribed mostly to uncultured eukaryotes/Rhizaria or species of the genus Gromia (Rothe et al., 2009). The inferred 18S maximum likelihood phylogeny is presented as a subtree (Figure 12) (complete available as Supplementary Data Sheet 5) that strictly associates the contaminant sequences of Spiculammina and Psammina with each other, together with a sequence ascribed to an unidentified haplosporidian from the hydrothermal vents of the Lost City (Mid-Atlantic Ridge) (López-García et al., 2007). These sequences are included in a larger clade that contains Endomyxa parasites of the shrimp Pandalus platyceros Brandt, 1851 (Reece et al., 2004) and species of the parasite genus Paradinium Chatton, formerly described as dinoflagellates but transferred to the Endomyxa based on, inter alia, molecular phylogeny (Skovgaard and Daugbjerg, 2008). All Gromia spp. associate into a different strongly supported clade. In Table 5, a comparison between the lengths of the different parts of the cluster is provided, and the conservation between both sequences indicated as a percentage of identity. 18S, 5.8S and 28S have identical lengths, and the differences between the internal transcribed spacers ranged only between five and 17 bp. At 95.87% of identity, 18S was the most polymorph, while 5.8S was the most conserved.
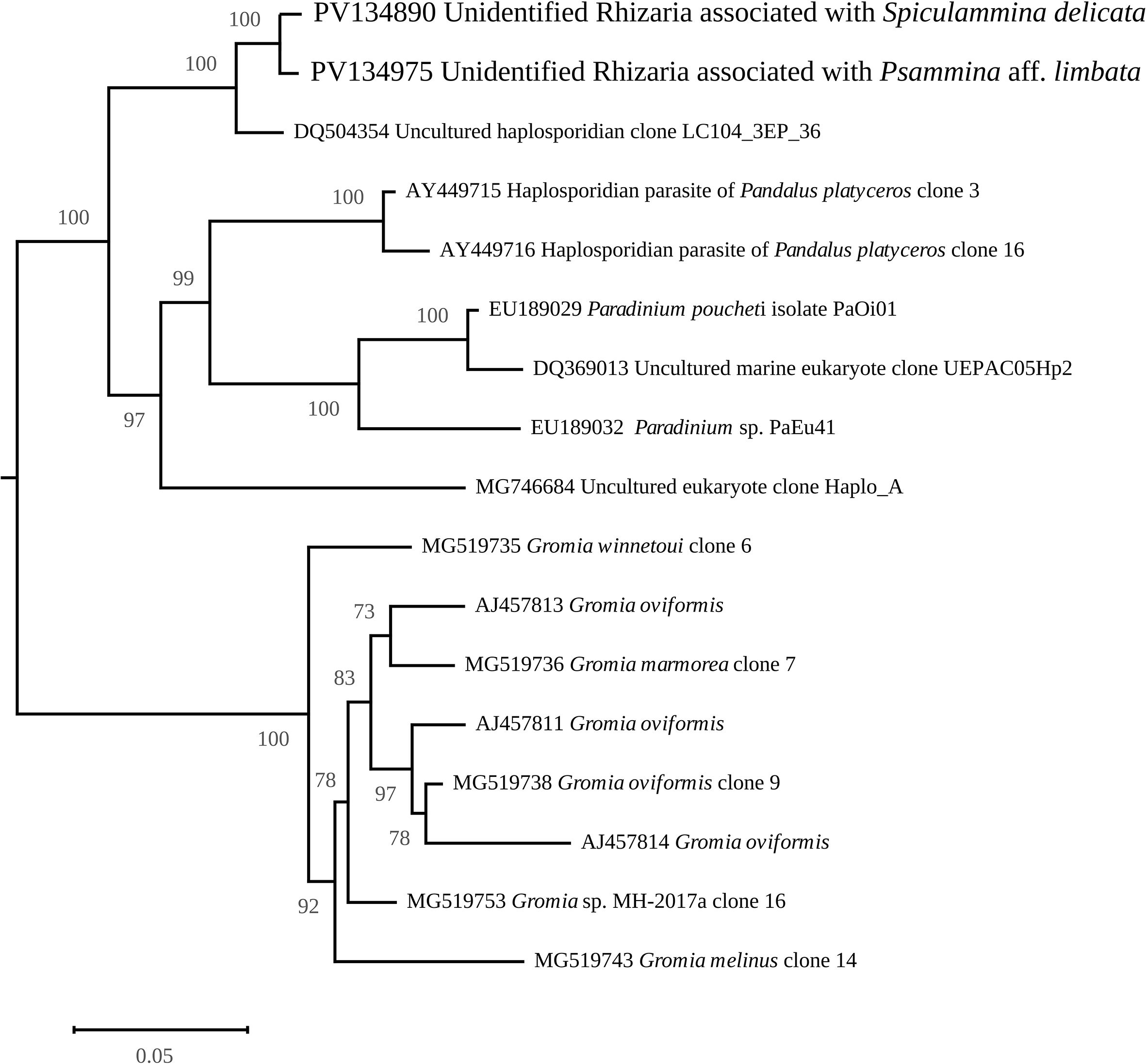
Figure 12. Maximum-Likelihood phylogenetic tree from an alignment of the 18S gene of the Rhizaria associated with xenophyophores with other various sequences obtained from GenBank. The figure shows the subtree containing the sequences of interest.
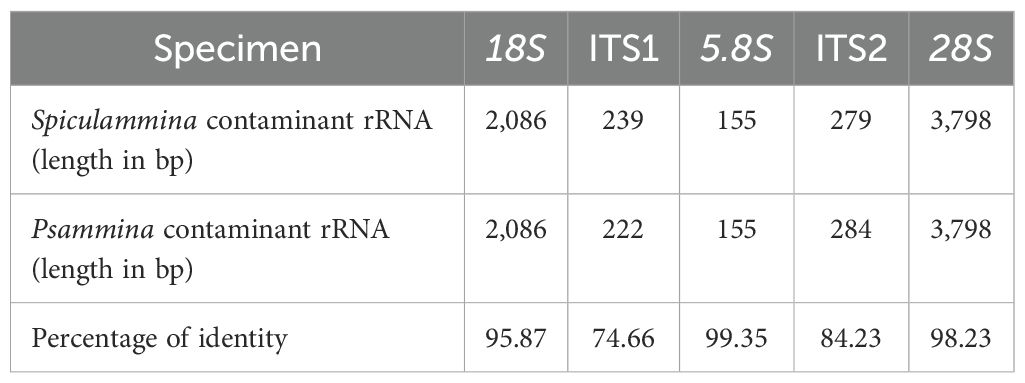
Table 5. Lengths and percentages of identity between the nuclear clusters of rRNA of the contaminant sequences evidenced among Spiculamminna and Psammina.
Comparisons between XAMs, published sequences and transcriptomic data
There were massive discrepancies between the XAMs and the only two available sequences ascribed to the cox1 gene of xenophyophores (Macher et al., 2022), and they did not even seem to belong to the XAMs. We data-mined the contigs files of both specimen with the 310 bp sequence ascribed to Psammina limbata Kamenskaya et al., 2015 (OM719650). It was possible in both cases to find contigs larger than this reference, with very high coverages, but none of them belonged to a larger, circular chromosome. In the case of Spiculammina, a 1,569 bp fragment with a very high coverage of 1600.05X was retrieved, displaying 95% identity with OM719650 (Supplementary Data Sheet 6). This contig contained a 431 AA long ORF (in code 4), that could be extended at its N terminal ending to a 467 AA long ORF starting with a TAA encoded Leucine without encountering a stop codon. Blastp queries showed strong similarities with the 496 AA long Cox1 proteins of C. hispida (evalue 0.0, identity 67.22%) and N. gaimardi (evalue 0.0, identity 67.22%), followed by a 419 AA long sequence ascribed to the Cox1 protein of another benthic foraminifera described as Globigerinidae sp. (PV558267), with evalue 2e-169 and identity 65.44%. The 16 first results for this query all belonged to foraminifera (mostly partial cox1 genes) but were followed by bacteria or stramenopiles. Using code 1 for translation, this ORF did not appear. Different attempts made to expand this 1,569 bp contig failed at returning a circular chromosome. The contig was used as a seed to perform an assembly with NOVOPlasty 4.3.3 (Dierckxsens et al., 2017) with a k-mer of 45. Instead of returning a single complete circular chromosome, NOVOplasty proposed 25 options to extend this seed, with lengths ranging from 3,174 bp to 5,673 bp (Supplementary Data Sheet 7). At its 5’ ending, the seed could never be extended beyond eight nucleotides (all identicals between the 25 options) while the 3’ extensions were very variable in length and sequence, as illustrated by the LOGO representation included as Supplementary Data Sheet 8. The largest contig derived from NOVOplasty (labelled ‘Contig3’ in Supplementary Data Sheet 7) was analyzed with orffinder, and three ORF of length above 100 AA were found. One corresponded to the aforementioned Cox1-like ORF, that could be extended to 479 AA. Close to it, a 186 AA ORF that could not be extended because of stop codons returned as best blastp result the 337 AA and 335 AA Cob proteins of N. gaimardi and C. hispida, but with high evalues (1e-40 and 4e-40 respectively) and low identities (53.90% for both). These matches were followed by sequences belonging to metazoa, mostly arthropoda. There was also a 593 AA long ORF, whose blastp query done on this sequence was surprising, as it returned two different proteins: Cox3 on its N-terminal part, ND1 on its C-terminal part. The two best results belonged to the Cox3 protein of N. gaimardi and C. hispida, followed by the ND1 protein of bacteria known to belong to the microbiome of benthic foraminifera (Woehle et al., 2022). When using the addSolexaReads.perl script included in the Consed package (Gordon and Green, 2013), the aim of which is to extend contigs by adding reads at their endings, the software suggested various possibilities for which each ending was supposed to display a single alternative extension. This could suggest that this element might be repeated many times but with variable neighboring. In the case of Psammina, the queries returned four different results, corresponding to contigs ranging from 204 bp to 608 bp. Only the 608 bp contig could align with the whole 310 bp of the reference, while the other contigs all only aligned with 125 bp. Identities ranged from 95% to 97%, but coverages varied from 158.35X to 276.64X.
The BUSCO analyze on the transcript file can be accessed as Supplementary Data Sheet 9. A total of 119 complete BUSCOs were found, 61 were fragmented and 75 were missing. The only match returned by the query with the cox1-ascribed fragment of Ps. limbata was a 2,032 bp fragment with a coverage of 457.98X (Supplementary Data Sheet 10). When translated using code 4, this transcript contained a 408 AA ORF, which returned as best blastp query the Cox1 protein of C. hispida, with evalue 0.0 and 68.80% identity. However, the Cox1 from C. hispida and N. gaimardi are both 496 AA long. The maximum extension that could have been reached on the N terminal part for this ORF would have started with a TAT-encoded Tyrosine residue, with a size of 483 AA. This ORF would also be split into several short fragments when using code 1 for translation.
Blastx queries returned nine matches, with lengths ranging from 140 to 235 bp, and coverage from 8.59X to 27.12X (Supplementary Data Sheet 11). E-values ranged between 1e-43 and 3e-24, with identities of 82% to 96%. These transcripts could be ascribed to the following genes with which they also aligned: ATP1, ATP6, cox1, cox2, cob, ND1 and ND7. The best match in terms of identity was also the most covered and corresponded to a 152 bp transcript ascribed to the 3’ part of the cox1 gene, aligning from positions 1,192 to 1,343 of the reference gene of XAM Psammina with no gap and an identity of 84.87%.
The blastn query done against the whole sequence of the XAM Psammina returned two interesting results, that could potentially be ascribed to mitochondrial pre-rRNA. One was 1,742 bp long with a coverage of 194.28X and could be ascribed to rnS, while the other was 1,463 bp long with a coverage of 58.01X and could be ascribed to a partial rnL at the 3’ part of the gene. Alignments between these transcripts and the putatively corresponding genes of the Spiculammina and Psammina XAMs are displayed in Figure 13 (for rnS) and Figure 14 (for rnL).

Figure 13. LOGO representation of the alignment of the rnS gene from both Xenophyophore-associated mitogenomes with the matching transcript from Psammina sp. biosample SAMN15195970.

Figure 14. LOGO representation of the alignment of the rnL gene from both Xenophyophore-associated mitogenomes with the matching transcript from Psammina sp. biosample SAMN15195970.
As a means to compare the coverages with a reference gene, the transcript file was data-mined for the actin-2 gene. The corresponding 375 AA long ORF was retrieved in a 1,312 bp long transcript (Supplementary Data Sheet 12) with a coverage of 802.64X.
Discussion
Ascribing XAMs as the functional mitogenomes of xenophyophores: the pros and the cons
As explained in the introduction, we avoid definitively assigning the mitogenomes to the two xenophyophore species and instead refer to them as xenophyophore-associated mitogenomes (XAMs). This conservative approach is supported by the suggested presence of rRNA from other Rhizaria associated with both xenophyophore specimens. No information is currently available about the organisms from which this rRNA is derived, whether they live on or inside the test and their degree of association, if any, with the xenophyophores. It cannot be ruled out, therefore, that the two XAMs belong to these contaminants. However, all our data suggest that these contaminants are members of the Endomyxa. On the other hand, the multiprotein phylogeny does not link the XAMs to the Endomyxa. Recent investigations also underlined the challenges involved in sequencing the mitochondrial genomes of the Endomyxa, which seem in some cases strongly reduced or possibly lost (Hiltunen Thorén et al., 2024).
There were massive discrepancies between the XAMs and the only two available sequences ascribed to the cox1 gene of xenophyophores (Macher et al., 2022). We cannot explain our inability to assemble larger mitogenomes that would contain the corresponding sequences from the two xenophyophore specimens. The fact that they could not be extended either by NOVOplasty or by Consed suggests that they might be surrounded by various environments. The fact that several contigs of various lengths and different degrees of coverage matched with the cox1 ascribed references is also difficult to understand. These features might all suggest that these elements are repeated many times but with variable neighboring sequences, in a manner that could be compared with repeated copies of mitochondrial genes transferred to the nuclear genome (aka NUMTs). The type of sequencing employed here (short reads) will not be sufficient to solve this question, which might require long-reads instead.
Data-mining the publicly available transcriptome of Ps. limbata failed to bring a definitive conclusion to our work. From the start, we did not expect it to yield complete mRNA corresponding to the mitochondrial protein coding genes. Indeed, with a few exceptions, polyadenylation of the mRNA in organelles serves as a signal for degradation (Slomovic et al., 2005). Thus, polyA mRNA are meant to be short lived, and a polyA enriched library, such as that prepared for transcriptomic analyses, may incorporate only little if any mitochondrial mRNA. It is possible, however, that reads representing mitochondrial DNA might end up being sequenced and later assembled, perhaps because of lighter DNAse treatment (e.g., Gastineau et al., 2023b). The results retrieved from data-mining the transcriptome of Ps. limbata, however, were not as decisive as for example in Gastineau et al. (2023b). Only short fragments could represent mRNA of the XAMs, and it would be premature and careless to assume that this is the case. Of greater interest is the presence of pre-rRNA that could be sequenced, possibly corresponding to rnS and rnL, although again, this result is inconclusive. Moreover, we do not know how to interpret the presence of a transcript corresponding to the cox1 reported by Macher et al. (2022) which had a coverage suggesting a rather high level of transcription comparable to that of nuclear genes.
Another important point is the protein coding gene content of the mitogenomes, which is comparable to that of planktonic Foraminifera and is rather distinct from other Rhizaria. The common loss of atp8 is noticeable, but has been found in many other Rhizarian. This gene is rather short (e.g., 141 bp in Pa. marina), highly variable, and reportedly lost among other phyla such as platyhelminthes (Le et al., 2002). The common loss of ND9 is more intriguing, considering the fact that all other Rhizaria have retained it, even as two diverging copies in L. oceanica. Finally, the complete loss of the rps/rpl gene is an important feature in common between XAMs and planktonic Foraminifera. The loss of rps/rpl is widely documented among metazoans; however, extreme differences sometimes occur within a same class of protists, for example in the Prasinophyceae (Chlorophyta) in which some species have a large set of rps/rpl genes in their mitogenome, while others have lost them altogether (Turmel et al., 1999, 2020).
In the case of the other coding genes, however, there are noticeable differences between XAMs and planktonic foraminifera. The rRNA genes do not appear fragmented in XAMs, but instead compact and rather conserved, both in length and sequence as well as in synteny with other genes. Also, at least one tRNA gene was found in the XAMs while none were described in planktonic foraminifera. When compared to Pa. marina, which appears to be the closest to Retaria based on the very limited sample of mitogenomes, the difference is rather obvious, as Pa. marina encodes a total of 17 tRNA genes. In a more general context, a reduced content in tRNA of mitogenomes has been observed previously. It is known for several protists (Gray et al., 1998), for example Chlamydomonas spp. (Chlorophyceae) (Denovan-Wright et al., 1998; Boer and Gray, 1988), and among metazoans it has been reported in Cnidaria (Haen et al., 2010). Also, parasites such as Plasmodium falciparum Welch, 1897 and Trypanosoma brucei Plimmer & Bradford, 1899 have no mitochondrial encoded tRNA at all (Gray et al., 1998).
Conclusion and future perspectives
This exploratory study has perhaps generated more questions than answers, particularly regarding whether or not the mitogenomes belong to the xenophyophores. If this can be shown to be the case, then the results will provide a robust background for further studies on population genetics and phylogeny. On the other hand, if the XAMs are derived from organisms living in association with the xenophyophores, then it will be necessary to characterize those organisms.
Since xenophyophores are exclusively deep-sea organisms, they can only be collected during expeditions with large, ocean-going research vessels. They are therefore difficult and expensive to obtain. Their delicate tests are prone to fragmentation during collection, a particular problem in the case of older material collected using towed devices. Moreover, in a significant proportion of cases they are dead when collected (e.g., Hughes and Gooday, 2004). On the other hand, xenophyophores are part of the benthic megafauna and therefore much larger than other Retaria (apart from some other large foraminifera). They are also multinucleate and, in this regard, more comparable to metazoans than to many protists. Where abundant, xenophyophores are often obtained by chance in good condition in box cores, while targeted sampling can be carried out by remote operated vehicles (ROVs), autonomous underwater vehicles (AUVs) or manned submersibles. Thus, there are now numerous possibilities to obtain relatively pristine material. For genetic studies, it is important that the cytoplasm will be carefully fully dissected in order to separate it from the test material and, as much as possible, of the stercomare (waste material). By doing so, the risk of DNA contamination by other organisms will be minimized. The DNA extracted from cytoplasm prepared in this way could be used for long-read sequencing, thus providing opportunities for revealing a lot of information on the structure of the different genomes of xenophyophores.
Data availability statement
All the genomic sequences (XAM, rRNA clusters) have been deposited on GenBank and are also availabe on Zenodo following this link: https://doi.org/10.5281/zenodo.14918269. Reads have been submitted to the Sequence Read Archive and are available under project number PRJNA1223329.
Author contributions
RG: Investigation, Writing – original draft. KM: Investigation, Writing – review & editing. PD: Investigation, Writing – review & editing. CO: Investigation, Writing – review & editing. CL: Investigation, Writing – review & editing. MT: Investigation, Writing – review & editing. BW-W: Investigation, Writing – review & editing. VS: Investigation, Writing – review & editing. AK: Investigation, Writing – review & editing. RW: Investigation, Writing – review & editing. TA: Investigation, Writing – review & editing. AG: Investigation, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was co-funded by the Minister of Science under the “Regional Excellence Initiative” Programme for 2024-2027 (RID/SP/0045/2024/01). Claude Lemieux and Monique Turmel were supported by grant RGPIN-2017-04506 from the Natural Sciences and Engineering Research Council of Canada (NSERC). Christian Otis was supported by the “Programme d’appui aux plateformes technologiques stratégiques” from the “Ministère de l’Économie, de l’Innovation et de l’Énergie, Québec”.
Acknowledgments
We are greatly thankful to Pr. Teresa Radziejewska for her help and comments with the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2025.1582660/full#supplementary-material
References
Abramowski T. and Nikończuk P. (2019). “Preliminary balance of the cold accumulated in polymetallic nodules stored on the mining ship,” in Sustainable design and manufacturing 2019, vol. 155 . Eds. Ball P., Huaccho Huatuco L., Howlett R. J., and Setchi R. (Springer Singapore, Singapore), 545–552, ISBN: 9789811392702. doi: 10.1007/978-981-13-9271-9_45
Abramowski T., Urbanek M., and Baláž P. (2021). Structural economic assessment of polymetallic nodules mining project with updates to present market conditions. Minerals 11, 311. doi: 10.3390/min11030311
Amon D. J., Ziegler A. F., Dahlgren T. G., Glover A. G., Goineau A., Gooday A. J., et al. (2016). Insights into the abundance and diversity of abyssal megafauna in a polymetallic-nodule region in the eastern clarion-clipperton zone. Sci. Rep. 6, 30492. doi: 10.1038/srep30492
Ashford O. S., Davies A. J., and Jones D. O. B. (2014). Deep-sea benthic megafaunal habitat suitability modelling: A global-scale maximum entropy model for xenophyophores. Deep Sea Res. Part I: Oceanographic Res. Papers 94, 31–44. doi: 10.1016/j.dsr.2014.07.012
Bankevich A., Nurk S., Antipov D., Gurevich A. A., Dvorkin M., Kulikov A. S., et al. (2012). SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Benson G. (1999). Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Boer P. H. and Gray M. W. (1988). Transfer RNA genes and the genetic code in chlamydomonas reinhardtii mitochondria. Curr. Genet. 14, 583–590. doi: 10.1007/BF00434084
Borkowski P. J., Abramowski T., Szada-Borzyszkowska M., and Szada-Borzyszkowski W. (2022). Comminution of polymetallic nodules with a high-pressure water jet. Materials 15, 8228. doi: 10.3390/ma15228228
Brady H. B. (1883). Note on syringammina, a new type of arenaceous rhizopoda. Proc. R. Soc. London 35, 155–161. doi: 10.1098/rspl.1883.0031
Bushmanova E., Antipov D., Lapidus A., and Prjibelski A. D. (2019). rnaSPAdes: A de novo transcriptome assembler and its application to RNA-seq data. GigaScience 8, giz100. doi: 10.1093/gigascience/giz100
Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., et al. (2009). BLAST+: architecture and applications. BMC Bioinf. 10, 421. doi: 10.1186/1471-2105-10-421
Capella-Gutiérrez S., Silla-Martínez J. M., and Gabaldón T. (2009). trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Crooks G. E., Hon G., Chandonia J.-M., and Brenner S. E. (2004). WebLogo: A sequence logo generator: figure 1. Genome Res. 14, 1188–1190. doi: 10.1101/gr.849004
Darriba D., Posada D., Kozlov A. M., Stamatakis A., Morel B., and Flouri T. (2020). ModelTest-NG: A new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 37, 291–294. doi: 10.1093/molbev/msz189
Dellinger M., Labat A., Perrouault L., and Grellier P. (2014). Haplomyxa saranae gen. Nov. et sp. Nov., a new naked freshwater foraminifer. Protist 165, 317–329. doi: 10.1016/j.protis.2014.03.007
Denovan-Wright E. M., Nedelcu A. M., and Lee R. W. (1998). Complete sequence of the mitochondrial DNA of Chlamydomonas eugametos. Plant Mol. Biol. 36, 285–295. doi: 10.1023/A:1005995718091
Dierckxsens N., Mardulyn P., and Smits G. (2017). NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res, 45, e18. doi: 10.1093/nar/gkw955
Durden J. M., Putts M., Bingo S., Leitner A. B., Drazen J. C., Gooday A. J., et al. (2021). Megafaunal ecology of the western clarion clipperton zone. Front. Mar. Sci. 8. doi: 10.3389/fmars.2021.671062
Edgar R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Gallo N. D., Cameron J., Hardy K., Fryer P., Bartlett D. H., and Lisa A. (2015). Levin submersible- and lander-observed community patterns in the mariana and new britain trenches: influence of productivity and depth on epibenthic and scavenging communities. Deep Sea Res. Part I: Oceanographic Res. Papers 99, 119–133. doi: 10.1016/j.dsr.2014.12.012
Gastineau R., Dąbek P., Mianowicz K., Stoyanova V., Krawcewicz A., and Abramowski T. (2023a). Complete mitochondrial genome of the abyssal coral abyssoprimnoa gemina cairns 2015 (Octocorallia, primnoidae) from the clarion-clipperton zone, pacific ocean. ZK 1183, 81–98. doi: 10.3897/zookeys.1183.109000
Gastineau R., Harðardóttir S., Guilmette C., Lemieux C., Turmel M., Otis C., et al. (2023b). Mitochondrial genome sequence of the protist ancyromonas sigmoides kent 1881 (Ancyromonadida) from the sugluk inlet, hudson strait, nunavik, québec. Front. Microbiol. 14. doi: 10.3389/fmicb.2023.1275665
Gastineau R., Mianowicz K., Dąbek P., Otis C., Stoyanova V., Krawcewicz A., et al. (2025). Genomic investigation of benthic invertebrates from the clarion-clipperton fields of polymetallic nodules. ZK 1231, 11–44. doi: 10.3897/zookeys.1231.135347
Glez-Peña D., Gómez-Blanco D., Reboiro-Jato M., Fdez-Riverola F., and Posada D. (2010). ALTER: program-oriented conversion of DNA and protein alignments. Nucleic Acids Res. 38, W14–W18. doi: 10.1093/nar/gkq321
Gooday A. J., Durden J. M., and Smith C. R. (2020). Giant, highly diverse protists in the abyssal pacific: vulnerability to impacts from seabed mining and potential for recovery. Communicative Integr. Biol. 13, 189–197. doi: 10.1080/19420889.2020.1843818
Gooday A. J., Holzmann M., Caulle C., Goineau A., Kamenskaya O., Weber A. A.-T., et al. (2017). Giant protists (Xenophyophores, foraminifera) are exceptionally diverse in parts of the abyssal eastern pacific licensed for polymetallic nodule exploration. Biol. Conserv. 207, 106–116. doi: 10.1016/j.biocon.2017.01.006
Gooday A. J., Holzmann M., Goineau A., Kamenskaya O., Melnik V. F., Pearce R. B., et al. (2018). Xenophyophores (Rhizaria, foraminifera) from the eastern clarion-clipperton zone (Equatorial pacific): the genus psammina. Protist 169, 926–957. doi: 10.1016/j.protis.2018.09.003
Gooday A. J. and Wawrzyniak-Wydrowska B. (2023). Macrofauna-sized foraminifera in epibenthic sledge samples from five areas in the eastern clarion-clipperton zone (Equatorial pacific). Front. Mar. Sci. 9. doi: 10.3389/fmars.2022.1059616
Gordon D. and Green P. (2013). Consed: A graphical editor for next-generation sequencing. Bioinformatics 29, 2936–2937. doi: 10.1093/bioinformatics/btt515
Gray M. W., Lang B. F., Cedergren R., Golding G. B., Lemieux C., Sankoff D., et al (1998). Genome structure and gene content in protist mitochondrial DNAs. Nucleic Acids Res. 26, 865–878. doi: 10.1093/nar/26.4.865
Gurevich A., Saveliev V., Vyahhi N., and Tesler G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Haen K. M., Pett W., and Lavrov D. V. (2010). Parallel Loss of Nuclear-Encoded Mitochondrial Aminoacyl-tRNA Synthetases and mtDNA-Encoded tRNAs in Cnidaria. Mol. Biol. Evol. 27, 2216–2219. doi: 10.1093/molbev/msq112
Hikov A., Milakovska Z., Stoyanova V., Stefanova E., Abramowski T., Chavdarova S., et al. (2022). REE and trace elements distribution in the deep-sea sediments from the interoceanmetal (IOM) polymetallic nodule exploration area in the clarion-clipperton fractures zone, NE pacific. C. R. Acad. Bulg. Sci. 75, 1018–1027. doi: 10.7546/CRABS.2022.07.10
Hiltunen Thorén M., Onut-Brännström I., Alfjorden A., Pecková H., Swords F., Hooper C., et al. (2024). Comparative genomics of ascetosporea gives new insight into the evolutionary basis for animal parasitism in rhizaria. BMC Biol. 22, 103. doi: 10.1186/s12915-024-01898-x
Hughes J. A. and Gooday A. J. (2004). Associations between living benthic foraminifera and dead tests of syringammina fragilissima (Xenophyophorea) in the darwin mounds region (NE atlantic). Deep Sea Res. Part I: Oceanographic Res. Papers 51, 1741–1758. doi: 10.1016/j.dsr.2004.06.004
ISBA/25/LTC/6/Rev.2 (2022). Recommendations for the guidance of contractors on the content, format and structure of annual reports (Kingston, Jamaica: International Seabed Authority). Available online at: https://www.isa.org.jm/wp-content/uploads/2022/04/ISBA_25_LTC_6_Rev.2-2211076E.pdf.
Kalvari I., Nawrocki E. P., Ontiveros-Palacios N., Argasinska J., Lamkiewicz K., Marz M., et al. (2021). Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 49, D192–D200. doi: 10.1093/nar/gkaa1047
Kamenskaya O. E. (2005). Spiculammina delicata gen. et sp. n., a new xenophyophore from the eastern Pacific (Psamminidae). Invertebrate zoology. 2, 23–27. doi: 10.15298/invertzool.02.1.03
Kamenskaya O. E., Melnik V. F., Gooday A. J., et al (2013). Giant protists (xenophyophores and komokiaceans) from the Clarion-Clipperton ferromanganese nodule field (eastern Pacific). Biol Bull Rev 3, 388–398. doi: 10.1134/S2079086413050046
Kamenskaya O. E., Gooday A. J., Tendal O. S., and Melnik V. F. (2015). Xenophyophores (Protista, foraminifera) from the clarion-clipperton fracture zone with description of three new species. Mar. Biodiv 45, 581–593. doi: 10.1007/s12526-015-0330-z
Kamenskaya O. E., Gooday A. J., Tendal O. S., and Melnik V. F. (2017). Xenophyophores (Rhizaria, foraminifera) from the Russian license area of the clarion-clipperton zone (Eastern equatorial pacific), with the description of three new species. Mar. Biodiv 47, 299–306. doi: 10.1007/s12526-016-0595-x
Kaminski M. A. (2014). The year 2010 classification of the agglutinated foraminifera. Micropaleontology 61, 89–108. doi: 10.47894/mpal.60.1.09
Katoh K. and Standley D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kerpedjiev P., Hammer S., and Hofacker I. L. (2015). Forna (Force-directed RNA): simple and effective online RNA secondary structure diagrams. Bioinformatics 31, 3377–3379. doi: 10.1093/bioinformatics/btv372
Laslett D. and Canbäck B. (2008). ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 24, 172–175. doi: 10.1093/bioinformatics/btm573
Le T. H., Blair D., and McManus D. P. (2002). Mitochondrial genomes of parasitic flatworms. Trends Parasitol. 18, 206–213. doi: 10.1016/S1471-4922(02)02252-3
Lecroq B., Gooday A. J., Tsuchiya M., and Pawlowski J. A. (2009). New genus of xenophyophores (Foraminifera) from Japan trench: morphological description, molecular phylogeny and elemental analysis. Zoological J. Linn. Soc. 156, 455–464. doi: 10.1111/j.1096-3642.2008.00493.x
Levin L. A. and Gooday A. J. (1992). "Possible Roles for Xenophyophores in Deep-Sea Carbon Cycling". in Deep-Sea Food Chains and the Global Carbon Cycle, eds. G. T. Rowe, V. Pariente. (Dordrecht: Springer Netherlands), pp. 93–104.
Levin L. A. and Rouse G. W. (2020). Giant protists (Xenophyophores) function as fish nurseries. Ecology 101, e02933. doi: 10.1002/ecy.2933
Levin L. A. and Thomas C. L. (1988). The ecology of xenophyophores (Protista) on eastern pacific seamounts. Deep Sea Res. Part A. Oceanographic Res. Papers 35, 2003–2027. doi: 10.1016/0198-0149(88)90122-7
Lhee D., Yang E. C., Kim J. I., Nakayama T., Zuccarello G., Andersen R. A., et al. (2017). Diversity of the photosynthetic paulinella species, with the description of paulinella micropora sp. Nov. and the chromatophore genome sequence for strain KR01. Protist 168, 155–170. doi: 10.1016/j.protis.2017.01.003
Lohse M., Drechsel O., Kahlau S., and Bock R. (2013). OrganellarGenomeDRAW—a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41, W575–W581. doi: 10.1093/nar/gkt289
López-García P., Vereshchaka A., and Moreira D. (2007). Eukaryotic diversity associated with carbonates and fluid–seawater interface in lost city hydrothermal field. Environ. Microbiol. 9, 546–554. doi: 10.1111/j.1462-2920.2006.01158.x
Macher J.-N., Bloska D. M., Holzmann M., Girard E. B., Pawlowski J., and Renema W. (2022). Mitochondrial cytochrome c oxidase subunit I (COI) metabarcoding of foraminifera communities using taxon-specific primers. PeerJ 10, e13952. doi: 10.7717/peerj.13952
Macher J.-N., Coots N. L., Poh Y.-P., Girard E. B., Langerak A., Muñoz-Gómez S. A., et al. (2023). Single-cell genomics reveals the divergent mitochondrial genomes of retaria (Foraminifera and radiolaria). mBio 14, e00302–e00323. doi: 10.1128/mbio.00302-23
Milakovska Z., Hikov A., Stoyanova V., Peytcheva I., Lyubomirova V., and Abramowski T. (2022). REY in pore waters of sediments hosting fe-mn nodules of the interoceanmetal exploration area in the clarion-clipperton fracture zone, NE pacific. Geologica Balcanica 51, 27–35. doi: 10.52321/GeolBalc.51.2.27
Minh B. Q., Schmidt H. A., Chernomor O., Schrempf D., Woodhams M. D., Von Haeseler A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Pawlowski J., Holzmann M., Fahrni J., and Richardson S. L. (2003). Small subunit ribosomal DNA suggests that the xenophyophorean syringammina corbicula is a foraminiferan. J. Eukaryotic Microbiol. 50, 483–487. doi: 10.1111/j.1550-7408.2003.tb00275.x
Pawlowski J., Holzmann M., and Tyszka J. (2013). New supraordinal classification of foraminifera: molecules meet morphology. Mar. Micropaleontology 100, 1–10. doi: 10.1016/j.marmicro.2013.04.002
Paysan-Lafosse T., Blum M., Chuguransky S., Grego T., Pinto B. L., Salazar G. A., et al. (2023). InterPro in 2022. Nucleic Acids Res. 51, D418–D427. doi: 10.1093/nar/gkac993
Rabone M., Wiethase J. H., Simon-Lledó E., Emery A. M., Jones D. O. B., Dahlgren T. G., et al. (2023). How many metazoan species live in the world’s largest mineral exploration region? Curr. Biol. 33, 2383–2396.e5. doi: 10.1016/j.cub.2023.04.052
Radziejewska T., Mianowicz K., and Abramowski T. (2022). “Natural variability versus anthropogenic impacts on deep-sea ecosystems of importance for deep-sea mining,” in Perspectives on deep-sea mining. Ed. Sharma R. (Springer International Publishing, Cham), 281–311, ISBN: 9783030879815.
Reece K. S., Siddall M. E., Stokes N. A., and Burreson E. M. (2004). Molecular phylogeny of the Haplosporidia based on two independent gene sequences. J. Parasitol. 90, 1111–1122. doi: 10.1645/GE-102R
Rothe N., Gooday A. J., Cedhagen T., Fahrni J., Hughes J. A., Page A., et al. (2009). Three new species of deep-sea gromia (Protista, rhizaria) from the bathyal and abyssal weddell sea, Antarctica: THREE NEW SPECIES OF GROMIA. Zoological J. Linn. Soc. 157, 451–469. doi: 10.1111/j.1096-3642.2009.00540.x
Sierra R., Mauffrey F., Cruz J., Holzmann M., Gooday A. J., Maurer-Alcalá X., et al. (2022). Taxon-rich transcriptomics supports higher-level phylogeny and major evolutionary trends in foraminifera. Mol. Phylogenet. Evol. 174, 107546. doi: 10.1016/j.ympev.2022.107546
Sievers F. and Higgins D. G. (2014). “Clustal omega, accurate alignment of very large numbers of sequences,” in Multiple sequence alignment methods, vol. 1079 . Ed. Russell D. J. (Humana Press, Totowa, NJ), 105–116, ISBN: 9781627036450.
Simão F. A., Waterhouse R. M., Ioannidis P., Kriventseva E. V., and Zdobnov E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Simon-Lledó E., Bett B. J., Huvenne V. A. I., Schoening T., Benoist N. M. A., Jeffreys R. M., et al. (2019b). Megafaunal variation in the abyssal landscape of the clarion clipperton zone. Prog. Oceanography 170, 119–133. doi: 10.1016/j.pocean.2018.11.003
Simon-Lledó E., Bett B. J., Huvenne V. A. I., Schoening T., Benoist N. M. A., and Jones D. O. B. (2019a). Ecology of a polymetallic nodule occurrence gradient: implications for deep-sea mining. Limnology Oceanography 64, 1883–1894. doi: 10.1002/lno.11157
Skovgaard A. and Daugbjerg N. (2008). Identity and systematic position of paradinium poucheti and other paradinium-like parasites of marine copepods based on morphology and nuclear-encoded SSU rDNA. Protist 159, 401–413. doi: 10.1016/j.protis.2008.02.003
Skowronek A., Maciąg Ł., Zawadzki D., Strzelecka A., Baláž P., Mianowicz K., et al. (2021). Chemostratigraphic and textural indicators of nucleation and growth of polymetallic nodules from the clarion-clipperton fracture zone (IOM claim area). Minerals 11, 868. doi: 10.3390/min11080868
Slomovic S., Laufer D., Geiger D., and Schuster G. (2005). Polyadenylation and degradation of human mitochondrial RNA: the prokaryotic past leaves its mark. Mol. Cell. Biol. 25, 6427–6435. c
Smith S. A. and Dunn C. W. (2008). Phyutility: A phyloinformatics tool for trees, alignments and molecular data. Bioinformatics 24, 715–716. doi: 10.1093/bioinformatics/btm619
Štyriaková D., Štyriaková I., Šuba J., Baláž P., and Abramowski T. (2022). Bioleaching test of polymetallic nodule samples from the IOM exploration area. Minerals 12, 1373. doi: 10.3390/min12111373
Tanifuji G., Archibald J. M., and Hashimoto T. (2016). Comparative genomics of mitochondria in chlorarachniophyte algae: endosymbiotic gene transfer and organellar genome dynamics. Sci. Rep. 6, 21016. doi: 10.1038/srep21016
Tendal O. S. (1972). A monograph of the xenophyophoria (Rhizopodea, protozoa). Galathea Rep. 12, 7–100.
Tendal O. S. (1996). Synoptic checklist and bibliography of the Xenophyophora (Protista), with a zoogeographical survey of the group. Galathea Rep. 17, 101.
Turmel M., Lemieux C., Burger G., Lang B. F., Otis C., Plante I., et al. (1999). The complete mitochondrial DNA sequences of nephroselmis olivacea and pedinomonas minor : two radically different evolutionary patterns within green algae. Plant Cell 11, 1717–1729. doi: 10.1105/tpc.11.9.1717
Turmel M., Otis C., and Lemieux C. (2020). Complete mitogenome of the chlorophyte green alga marsupiomonas sp. NIES 1824 (Pedinophyceae). Mitochondrial DNA Part B 5, 548–550. doi: 10.1080/23802359.2019.1710283
Uhlenkott K., Meyn K., Vink A., and Martínez Arbizu P. A. (2023). Review of megafauna diversity and abundance in an exploration area for polymetallic nodules in the eastern part of the clarion clipperton fracture zone (North east pacific), and implications for potential future deep-sea mining in this area. Mar. Biodivers. 53, 22. doi: 10.1007/s12526-022-01326-9
Valach M., Burger G., Gray M. W., and Lang B. F. (2014). Widespread occurrence of organelle genome-encoded 5S rRNAs including permuted molecules. Nucleic Acids Res. 42, 13764–13777. doi: 10.1093/nar/gku1266
Wang J., Chitsaz F., Derbyshire M. K., Gonzales N. R., Gwadz M., Lu S., et al. (2023). The conserved domain database in 2023. Nucleic Acids Res. 51, D384–D388. doi: 10.1093/nar/gkac1096
Wang R., Zhu Z., Su X., Mianowicz K., Jia H., and Wu K. (2022). Slurry pumps in deep-sea mining: A review of numerical and experimental studies. Ocean Eng. 251, 111150. doi: 10.1016/j.oceaneng.2022.111150
Woehle C., Roy A.-S., Glock N., Michels J., Wein T., Weissenbach J., et al. (2022). Denitrification in foraminifera has an ancient origin and is complemented by associated bacteria. Proc. Natl. Acad. Sci. U.S.A. 119, e2200198119. doi: 10.1073/pnas.2200198119
Keywords: deep sea, benthic foraminifera, Endomyxa, agglutinated protists, Retaria, mitochondrial, phylogeny, CCZ
Citation: Gastineau R, Mianowicz K, Dąbek P, Otis C, Lemieux C, Turmel M, Wawrzyniak-Wydrowska B, Stoyanova V, Krawcewicz A, Wróbel RJ, Abramowski T and Gooday AJ (2025) Xenophyophore-associated mitogenomes: genomic investigations of two specimens from the Clarion-Clipperton Zone. Front. Mar. Sci. 12:1582660. doi: 10.3389/fmars.2025.1582660
Received: 24 February 2025; Accepted: 24 June 2025;
Published: 15 July 2025.
Edited by:
Beata Szymczycha, Polish Academy of Sciences, PolandReviewed by:
Daniel Garcia-Souto, University of Vigo, SpainMichał Karlicki, Stanford University, United States
Copyright © 2025 Gastineau, Mianowicz, Dąbek, Otis, Lemieux, Turmel, Wawrzyniak-Wydrowska, Stoyanova, Krawcewicz, Wróbel, Abramowski and Gooday. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Romain Gastineau, cm9tYWluLmdhc3RpbmVhdUB1c3ouZWR1LnBs