 Sundeep Chaitanya Vedithi1*†
Sundeep Chaitanya Vedithi1*† Sony Malhotra2†
Sony Malhotra2† Marta Acebrón-García-de-Eulate1
Marta Acebrón-García-de-Eulate1 Modestas Matusevicius1
Modestas Matusevicius1 Pedro Henrique Monteiro Torres3
Pedro Henrique Monteiro Torres3 Tom L. Blundell1*
Tom L. Blundell1*- 1Department of Biochemistry, University of Cambridge, Cambridge, United Kingdom
- 2Rutherford Appleton Laboratory, Science and Technology Facilities Council, Oxon, United Kingdom
- 3Laboratório de Modelagem e Dinâmica Molecular, Instituto de Biofísica Carlos Chagas Filho, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
Leprosy, caused by Mycobacterium leprae (M. leprae), is treated with a multidrug regimen comprising Dapsone, Rifampicin, and Clofazimine. These drugs exhibit bacteriostatic, bactericidal and anti-inflammatory properties, respectively, and control the dissemination of infection in the host. However, the current treatment is not cost-effective, does not favor patient compliance due to its long duration (12 months) and does not protect against the incumbent nerve damage, which is a severe leprosy complication. The chronic infectious peripheral neuropathy associated with the disease is primarily due to the bacterial components infiltrating the Schwann cells that protect neuronal axons, thereby inducing a demyelinating phenotype. There is a need to discover novel/repurposed drugs that can act as short duration and effective alternatives to the existing treatment regimens, preventing nerve damage and consequent disability associated with the disease. Mycobacterium leprae is an obligate pathogen resulting in experimental intractability to cultivate the bacillus in vitro and limiting drug discovery efforts to repositioning screens in mouse footpad models. The dearth of knowledge related to structural proteomics of M. leprae, coupled with emerging antimicrobial resistance to all the three drugs in the multidrug therapy, poses a need for concerted novel drug discovery efforts. A comprehensive understanding of the proteomic landscape of M. leprae is indispensable to unravel druggable targets that are essential for bacterial survival and predilection of human neuronal Schwann cells. Of the 1,614 protein-coding genes in the genome of M. leprae, only 17 protein structures are available in the Protein Data Bank. In this review, we discussed efforts made to model the proteome of M. leprae using a suite of software for protein modeling that has been developed in the Blundell laboratory. Precise template selection by employing sequence-structure homology recognition software, multi-template modeling of the monomeric models and accurate quality assessment are the hallmarks of the modeling process. Tools that map interfaces and enable building of homo-oligomers are discussed in the context of interface stability. Other software is described to determine the druggable proteome by using information related to the chokepoint analysis of the metabolic pathways, gene essentiality, homology to human proteins, functional sites, druggable pockets and fragment hotspot maps.
Introduction
Mycobacterium leprae causes leprosy in about 200,000 people each year globally. Leprosy is a dermato-neurological infectious disease with varied clinical manifestations, often resulting in peripheral sensorimotor/demyelinating neuropathy leading to permanent nerve damage and disability. The World Health Organization currently recommends a combinatorial therapy [multidrug therapy (MDT)] with Dapsone, Rifampicin (Rifampin) and Clofazimine to treat leprosy (Manglani and Arif, 2006). MDT has proven effective in reducing the prevalence and controlling the incidence from about 5 million new cases in the 1990s to ∼200,000 new cases from the year 2005 (after India declared the elimination of leprosy). However, with the emergence of single and multidrug-resistant strains of M. leprae, novel therapies are essential to curb ongoing transmission of the disease. Also, the current therapy duration with MDT is 1 year leading to reduced treatment compliance and increased defaulter rates globally (Cambau et al., 2018).
Mycobacterium leprae is phylogenetically the closest bacterial species to Mycobacterium tuberculosis (M. tuberculosis). However, the M. leprae has a reduced genome of 3.2 Mbp, compared to 4.4 Mbp in M. tuberculosis, and survive with only 1,614 protein coding genes which are largely annotated based the features of their homologues in M. tuberculosis and other mycobacterial species (Cole et al., 2001). Dapsone interacts with bacterial dihydropteroate synthase, an enzyme essential for folic acid biosynthesis in bacteria. It is absent in humans (Cambau et al., 2006). Rifampin interacts with RNA polymerase, an enzyme critical for DNA dependent RNA synthesis (transcription) in M. leprae. These drugs are either bacteriostatic or bactericidal. However, they do not interfere with predilection of M. leprae for human nerve cells, which is a significant cause for demyelinating neuropathy in leprosy (Lockwood and Saunderson, 2012). Newer antibacterial agents that can effectively treat the disease within a short duration of time and prevent nerve damage are essential to reduce morbidity associated with the disease (Rao and Jain, 2013). Currently known drugs for leprosy, their drug target proteins and references related to their mechanisms of action are listed in Table 1.
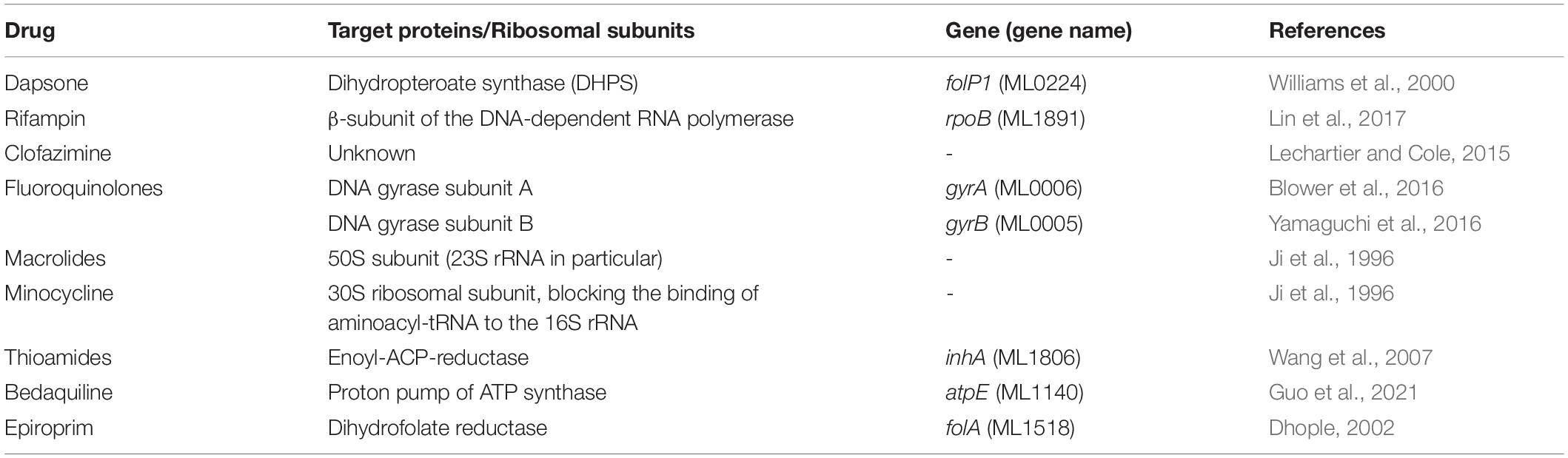
Table 1. Drugs and their corresponding target proteins in M. leprae.
Knowledge of the structural components of the proteome of M. leprae is critical for identifying drug target proteins and deciphering their essential roles in the survival of the pathogen. Key enzymes that catalyze chokepoint reactions can act as potential drug targets for antimycobacterial discovery. However, information related to 3D structures of these proteins is scarce for M. leprae, necessitating a more focussed structural genomics effort to unravel the druggable proteomic landscape of this bacillus long known to humankind.
Software tools that predict stability and affinity changes in drug-target proteins due to substitution mutations are discussed in the context of antimicrobial resistance. While the emphasis is on deciphering the druggable proteome, we provide a brief overview of the structure-guided virtual screening tools and methods that facilitate the chemical expansion of fragment-like small molecules to lead-like or drug-like compounds in the active or allosteric sites of the target protein.
Proteome Modeling in Mycobacterium leprae and Its Relevance to Structure-Guided Drug Discovery
Of the 1,614 annotated genes that are expressed in M. leprae, the structures of only 17 proteins are available (see Table 2) to date in the publicly available databases [Protein Data Bank (PDB) (Berman et al., 2000)], as opposed to around 1,277 entries for Mycobacterium tuberculosis. Solving the crystal/cryoEM structures of all the potential drug targets in M. leprae requires costly and labor intensive effort. Given the high sequence identity of many of the M. leprae proteins with their homologous counterparts in M. tuberculosis with solved structures in the PDB, employing computational tools to perform comparative modeling of proteins in M. leprae can be a robust alternative for acquiring a preliminary understanding of the functional sites and small molecule interactions.

Table 2. List of protein structures available for M. leprae in Protein Data Bank
Different groups have made several attempts to model the proteins of M. leprae. Table 3 lists two web-resources where such information is available. Computational protein structure modeling can reduce the sequence-structure gaps and enable genome-scale modeling of infectious pathogens (Bienert et al., 2017). Although the paucity of structural proteomics information for M. leprae in the publicly available databases is a challenge, the software developed in the Blundell laboratory will be useful in performing proteome scale modeling pipeline (Vivace) for proteomes of Mycobacterial pathogens and other bacterial species (Skwark et al., 2019). Vivace optimizes template selection, enables sequence-structure alignments, and constructs optimal quality models in both apo- and ligand-bound states. To facilitate multi-template modeling, protein structures from the entire PDB are initially organized in a structural profile database named TOCCATA (Ochoa-Montaño et al., 2015). Protein structures within each profile are classified based on domain annotations in CATH (Sillitoe et al., 2019) and SCOP (Andreeva et al., 2020) databases, pre-aligned and functionally annotated with information derived from UniProt (The UniProt Consortium, 2021) and PDB. The query protein sequence is aligned with representative structures from each cluster using a sequence-structure alignment tool named FUGUE (Shi et al., 2001). FUGUE recognizes distant homologues by sequence-structure comparison using environment-specific substitution tables and structure-dependent gap penalties. Alignments generated by FUGUE are fed into Modeler 9.24 (Webb and Sali, 2016) for model building. The ligands and other small molecules are modeled into corresponding protein structure models at sites recognized from the ligand-bound templates. Multiple models are generated based on the number of cluster hits, ranging from 3 to ∼1,000 models per query sequence in the M. leprae proteome. These models are of different states (ligand-bound and apomeric) and of varying quality based on the templates used in each profile.

Table 3. Web resources with models of M. leprae proteins (modelled using single templates).
Once modeled, each of the protein structure models undergoes a rigorous quality assessment by employing methods such as NDOPE, GA341 (Shen and Sali, 2006), GOAP (Zhou and Skolnick, 2011), SOAP (Webb and Sali, 2016), Molprobity (Chen et al., 2010) and secondary structure agreement score (Eramian et al., 2006). Models with extensive chain clashes, poorly resolved loops and improperly fitted ligands are ranked low in the consensus quality scoring process described in CHOPIN—a web resource for structural and functional proteome of Mycobacterium tuberculosis (Ochoa-Montaño et al., 2015).
Vivace is being used to model the proteome of M. leprae. Sequence and structural features at the genome-scale are being analyzed to identify essential enzymes that drive chokepoint metabolic reactions. Models in apomeric, ligand-bound and oligomeric (discussed in the later sections) states are being generated and analyzed for surface topology, cavities (Binkowski et al., 2003) and fragment hotspots (sites for potential small molecule binding) (Radoux et al., 2016). The schematic workflow shown in Figure 1 illustrates the modeling procedures adopted by our group to model proteomes of mycobacterial pathogens.
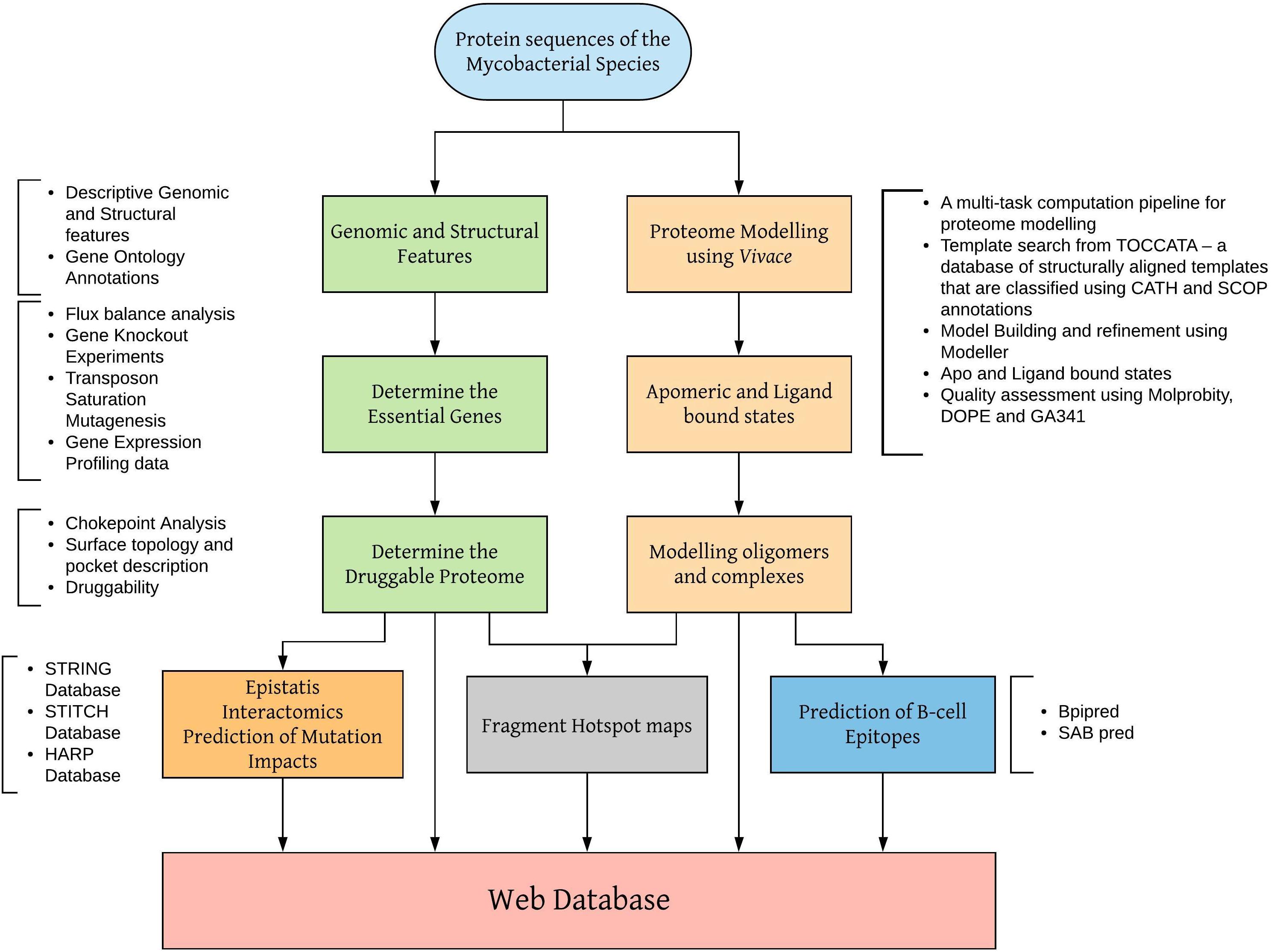
Figure 1. Workflow for modeling mycobacterial proteomes and developing web databases.
Approaches to Predict Homo/Hetero-Oligomeric Complexes
Protein-protein interactions (homo/hetero) govern a majority of the cellular processes. Structure determination of these complexes is crucial for understanding their functions. Usually, the experimental techniques used to unravel interacting protein partners are time consuming, challenging and expensive. There have been significant advances in the development of computational methods and tools to identify interacting pairs and predict the structures of protein-protein complexes (Das and Chakrabarti, 2021).
The computational tools for predicting protein-protein interactions developed over the years can be classified into the knowledge-based or de novo prediction methods. If the structures of the interacting partners are known, the interactions can be predicted using template-based, or template free and/or restraint-based docking. Template-based docking can provide the multi-component modeled complex but requires the presence of multi-component template structures (Ogmen et al., 2005; Mukherjee and Zhang, 2011). If the homologous multi-component template is unavailable, protein-protein docking approaches can be used to sample the conformational space and predict the docked complexes which are further scored using different schemes to discriminate native-like conformations from a pool of docked solutions. These different approaches for computational modeling of protein interactions were recently reviewed by Soni and Madhusudhan (2017).
Recently, tools have been developed which can make use of the wealth of sequence information available for protein sequences to predict/model interactions accurately. Machine learning approaches including deep learning have played a significant role in training models which can predict the interactions using the features derived from protein sequences alone (Huang Y.-A. et al., 2016; Du et al., 2017; Sun et al., 2017; Chen et al., 2019). The inspection of co-evolving sites in two protein partners can provide strong signals to elucidate interacting partners (Yu et al., 2016). A recent method CoFex (Hu and Chan, 2017) used co-evolutionary features to predict protein interactions. Ensemble based approaches which use multiple machine learning methods to vote for predictions have been reported to achieve high sensitivity and accuracy (Zhang et al., 2019; Li et al., 2020). Deep learning has also been employed to train a convolutional neural network (CNN) to predict the protein interacting pairs with high accuracy (Wang et al., 2019; Torrisi et al., 2020).
However, in-silico approaches can often give false positive or negative results as well, hence one also needs validation strategies to assess the quality of predicted interactions. Efforts in the community such as CASP (Critical Assessment of Structure Prediction) and CAPRI (Critical Assessment of Prediction of Interfaces) competitions, aim to assess the field and the state-of-the-art methods and their ability to “correctly” model protein structures and their interactions, respectively. They define and use multiple scores for assessing the quality of protein structure and interfaces in the modeled complexes. CASP14 is the present ongoing competition, where deep learning approach-AlphaFold2 has outperformed and were able to accurately predict the protein structures (AlQuraishi, 2019).
To illustrate the modeling process adopted by Vivace, Figure 2 depicts the models of three potential drug targets in M. leprae, the menB [1,4-dihydroxy-2-naphthoyl-CoA synthase (ML2263)], menD [2-succinyl-5-enolpyruvyl-6-hydroxy-3-cyclohexene-1-carboxylate synthase (ML2270)] and coaA [Pantothenate kinase (ML1954)].
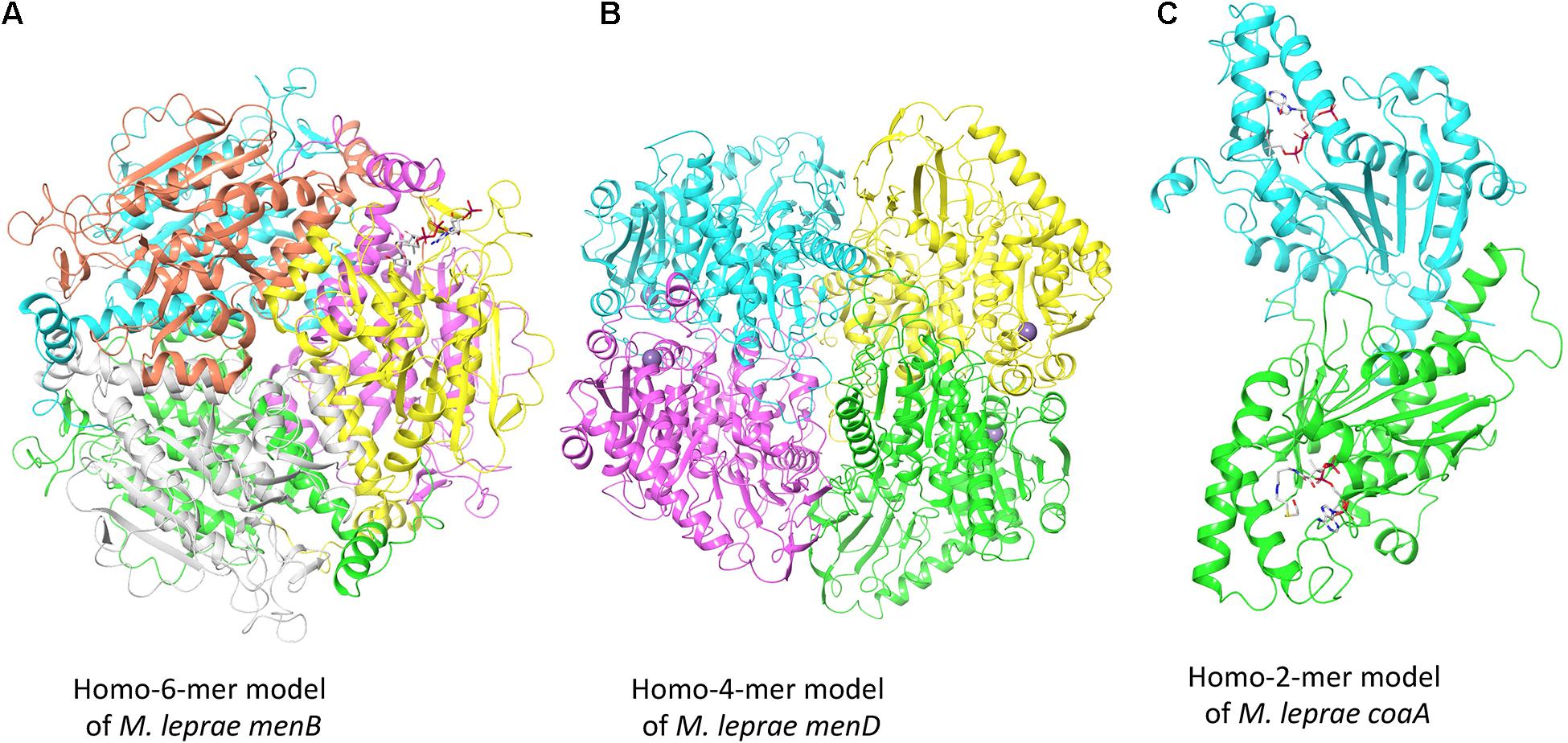
Figure 2. Oligomeric models of three potential drug targets in M. leprae. (A) The homohexameric model of M. leprae menB complexed with Salicylyl CoA. (B) The homotetrameric model of M. leprae menD bound to magnesium ions. (C) The homodimeric model of M. leprae coaA in complex with coenzyme A derivative.
The gene product of menB converts o-succinylbenzoyl-CoA (OSB-CoA) to 1,4-dihydroxy-2-naphthoyl-CoA (DHNA-CoA) and its homologue in M. tuberculosis (Rv0548c) is reported as essential (DeJesus et al., 2017). We built the model using the structure of its orthologous protein in M. tuberculosis (PDB Id: 4QII) as the template with sequence identity of 93% and sequence coverage of 100% (Figure 2A). The gene product of menD catalyzes the thiamine diphosphate-dependent decarboxylation of 2-oxoglutarate. Its homologue in M. tuberculosis (Rv0555) is noted to be essential for bacterial survival. We modeled menD of M. leprae using the structure of the M. tuberculosis orthlogue (PDB Id: 5ESD) as the template with the sequence identity of 86% and sequence coverage of 99% (Figure 2B). Finally, we modeled coaA which synthesizes coA from (R)—Pantothenate. CoaA has been recognized as a drug target in tuberculosis (Chiarelli et al., 2018). We modeled coaA using its orthologue in M. tuberculosis (PDB Id: 2GET) as the template with sequence identity of 93% and sequence coverage of 98% (Figure 2C).
Structural Implications of Substitution Mutations
Development of drug resistance is recognized as one of the major hurdles to disease management and control. For M. leprae, it is even more challenging as it relies on mouse footpad models (Vedithi et al., 2018). Antimicrobial resistance was noted in Dapsone, Rifampicin and Ofloxacin (a second-line drug). Treating and managing the disease is a big hurdle due to emerging drug resistance for these three drugs and lack of alternative effective treatments.
The drug-resistance mutations when mapped on to the three-dimensional structure of the drug target can provide crucial insights into effects on protein structure and function. There are several available software/web servers that can predict the impacts of mutation on protein stability and interactions with other proteins, ligands and nucleic acids. We have provided a list of some of the commonly used software tools for investigating the effects of mutations on protein structure and function (Table 4).
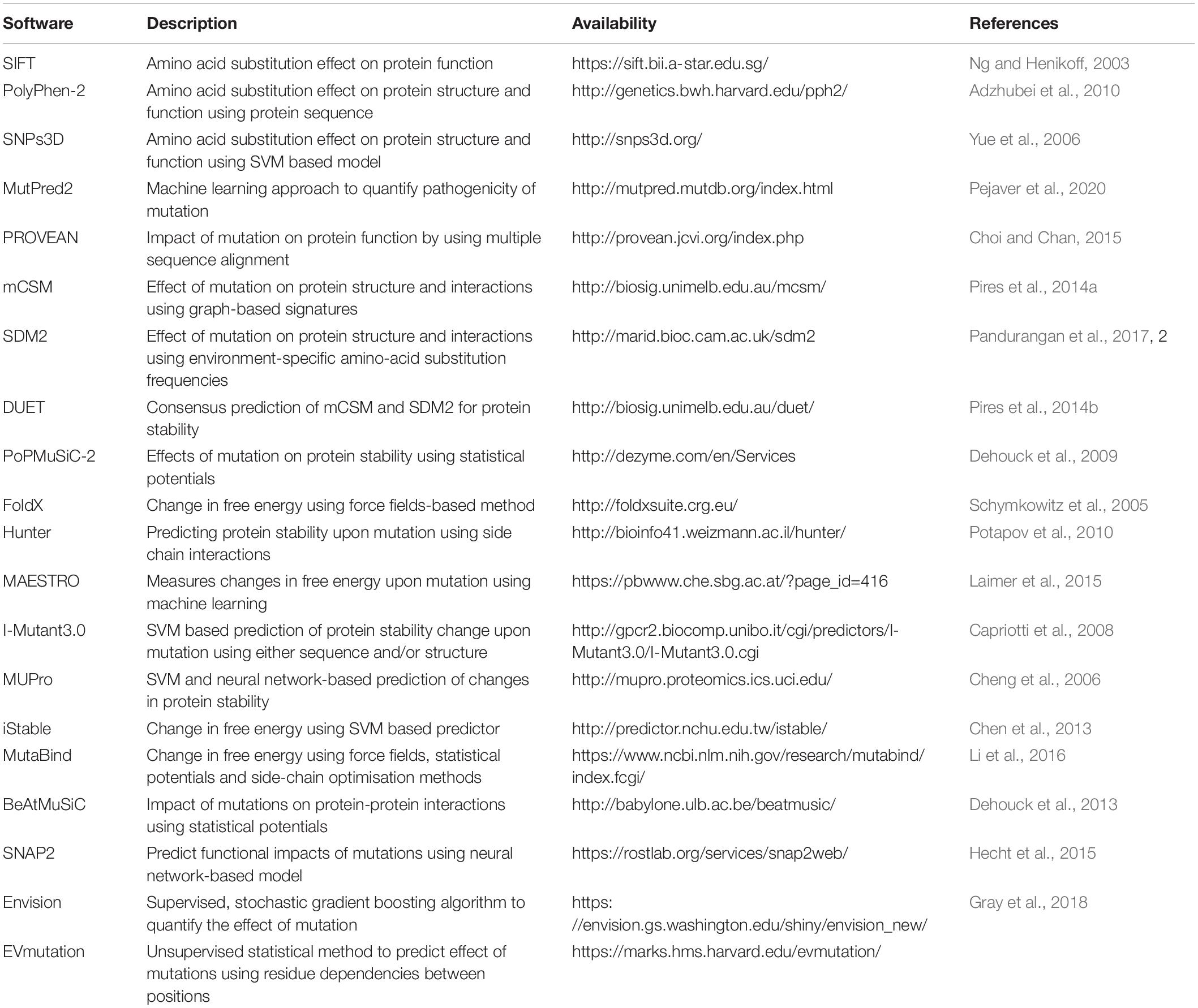
Table 4. Some of the commonly used tools for predicting the effect of mutations on protein structure and function.
Our own group have developed over the past decade the mCSM suite of computer programmes that use ML/AI approaches to predict the impacts of amino acid mutations not only on protomer stability (Pires et al., 2014a) but also on protein-protein, protein nucleic acid and protein-ligand interactions (Pires et al., 2016; Pires and Ascher, 2017). Recently, there have been further developments in the field where machine learning (ML)-based methods are gaining popularity. Many more recent ML methods also use features derived from protein structure and/or sequence to predict the effect of mutations (Hopf et al., 2017). A recent review, summarizes the performance of different ML methods and emphasizes the need for selecting reliable training dataset and informative features (Fang, 2020). Deep learning is an advanced training which is composed of multiple layers to learn different features from the input data and is proven to learn from the high-dimensional data. Recently, a method called DeepCLIP (Grønning et al., 2020) has been proposed which can predict protein binding to RNA using only sequence data. Another recently developed deep learning framework-MuPIPR (Zhou et al., 2020) (Mutation Effects in Protein–protein Interaction Prediction Using Contextualized Representations), can predict the effects of mutation on protein-protein interactions in terms of changes in buried surface area and binding affinity.
In-silico Saturation Mutagenesis
Using the tools described above, computational efforts exploiting recent growth in the availability of computing power can be immensely helpful to perform saturation mutagenesis, which can be used as a surveillance tool for drug resistance in leprosy. These mutational scanning exercises can provide crucial insights into the structure-function relationships by exploring all possible substitutions at a given site. This can provide a glimpse into the functional consequences of mutations in antimicrobial-resistance phenotypes. The extensive quantitative data from computational saturation mutagenesis experiments can guide experimental approaches and prove helpful for validation and/or engineering purposes. Recently published HARP (a database of Hansen’s Disease Antimicrobial Resistance Profiles) database (Vedithi et al., 2020) is a comprehensive repository of in-silico mutagenesis experiments for three important drug targets for M. leprae namely dihydropteroate synthase, RNA polymerase and DNA gyrase. A consensus impact for all the possible mutations on protein stability and function of these drug targets is provided in this database.
Druggability
Mycobacterium leprae genome is reduced to 3,268,203 bp preserving only 1,614 ORFs (Cole et al., 2001; Liu et al., 2004) of the Mycobacterial genus. The genome reduction is due to evolutionary adaptation of this intracellular obligate bacillus to Schwann and macrophages cells. Gene essentiality in M. leprae is deciphered based on essentiality of homologous genes, mainly in M. tuberculosis that are determined by experiments (Sassetti et al., 2003; DeJesus et al., 2017). Because of the evolutionary loss of non-essential genes by pseudogenization, only 65% of the existing total of M. leprae genes have been demonstrated to be essentials (Borah et al., 2020). In order to identify potential drug targets, a chokepoint reaction analysis helps to find proteins that are either consumers of unique substrates, or are unique producers of metabolites. It is predicted that the inhibition of chokepoint proteins produces an interruption of essential cell functions (Yeh et al., 2004). Determining the druggability of protein targets is important to avoid intractable targets. A druggable protein has the ability to bind with high affinity to a drug. In leprosy, the dihydropteroate synthase (DHPS), RNA polymerase (RNAP) and DNA gyrase (GYR) are known druggable proteins as they are the targets of Dapsone, Rifampicin and Ofloxacin, respectively. Nevertheless, protein druggability properties can be predicted by different bioinformatics tools based on the 3D structure/model of the protein. For example, the α-1,2-mannosyltransferase and mannosyltransferase proteins related to lipoarabinomannan pathway were identified as a possible drug targets using CASTp (Computer Altas of Surface Topography of proteins) (Gupta et al., 2020). CASTp determines the volume and the area of each cavity and pocket. Furthermore, for each pocket the solvent accessible surface and the molecular surface are calculated. Small-molecule virtual screening is another in-silico strategy used to ascertain druggability of the protein target. This approach provides an understanding of the physicochemical features of the binding sites and potential ligands that bind at these sites. In Mycobacterium tuberculosis, 2,809 proteins are identified as druggable using this in-silico approach (Anand and Chandra, 2014). Mammalian cell entry proteins of the class mce1A-E have been reported in M. leprae to facilitate bacterial entry into human nasal epithelial cells (Fadlitha et al., 2019). Mce1A has a significant role in the cell predilection and a comprehensive understanding of the structure and druggability of this protein can provide insights into host pathogen interactions and transmission in leprosy (Sato et al., 2007). In the case of ML2177c, this gene encodes for uridine phosphorylase and shows significantly high expression during leprosy infection. This is predicted as druggable using fragment-hotspot-map analysis (Malhotra et al., 2017). The fragment hotspots contain a juxtaposition of charge and lipophilicity that are essential for effective ligand binding through both enthalpic and entropic contributions. The hotspot map software uses different molecular probes (toluene, aniline and phenol) to calculate affinity maps that provide a visual guide of the pocket (Radoux et al., 2016). Figure 3 illustrates the recommended pathway to target prioritization in mycobacterial drug discovery.
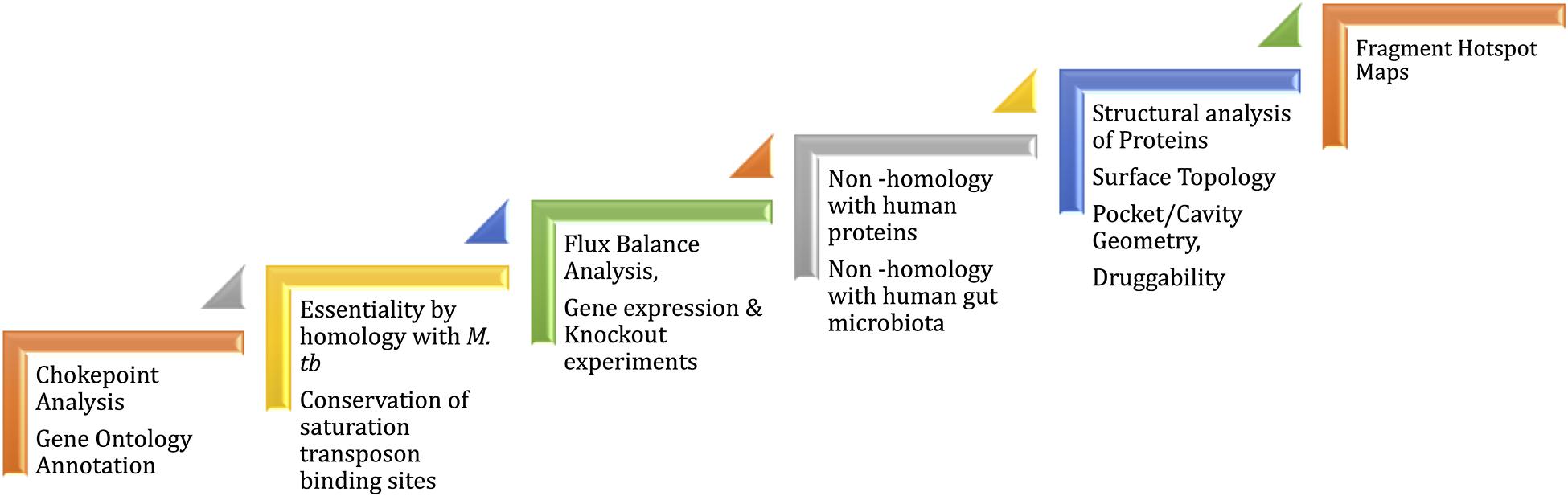
Figure 3. Workflow for drug target prioritization in M. leprae.
Structure-Guided Virtual Screening
Structure-guided virtual screening is a cost-effective computational tool for preliminary screening of proteins that are potential drug targets with chemical libraries ranging from small core fragments to large macrocyclic compounds in size and scaling from a few hundred molecules to billions (used in ultra-large-scale virtual screening campaigns). Since physical synthesis of drug molecules is not required, millions of virtual chemical entities can be swiftly docked into the active site of the protein structure/model and appropriate chemical scaffolds that fit with high scores and form relevant stabilizing interactions can be shortlisted for experimental validations. Virtual screening can be applied to novel drug discovery and also in drug repositioning experiments (screening with existing approved drugs to identify new target-protein interactions). A repurposing screen of LipU, a lipolytic protein that is conserved across mycobacterial species and noted to be essential for survival of M. leprae, revealed high docking scores for anti-viral drugs and anti-hypertensive (Kaur et al., 2019). Molecular docking software, such as Glide (Friesner et al., 2006), CCDC-GOLD (Jones et al., 1997), Autodock (Goodsell and Olson, 1990), Ledock (Wang et al., 2016), FlexX (Kramer et al., 1999), and SwissDock (Grosdidier et al., 2011) are used in virtual screening campaigns. Each algorithm has a unique scoring function to assess the fitness, number of stable interatomic interactions, and changes in energy landscape.
Discussion and Conclusion
Here, we have reviewed the tools and the advances made in protein structure prediction, modeling of genomes and impacts of amino acid replacements on protein structure and function. We have discussed these areas particularly focusing on the mycobacterial genomes, more specifically M. leprae. Given the paucity of information related to structural proteomic studies in leprosy, we discussed a multi-task protein modeling pipeline that enables proteome-scale template-based modeling of individual proteins encoded by various annotated genes in M. leprae. Homology-based structural and functional annotation of these protein models (Ochoa-Montaño et al., 2015; Skwark et al., 2019) using appropriate computational tools for modeling and druggability assessment can expedite characterization of the structural proteome of M. leprae and accelerate structure-guided novel drug discovery to combat nerve damage associated with leprosy.
Using the latest advancements in the field of protein structure bioinformatics, we describe our attempts to perform proteome scale modeling of mycobacterial genomes using in-house databases and pipelines. The modeled protein monomers or (homo/hetero) oligomers are subjected to multiple state-of-the-art validation scores. These models can be very helpful and provide useful insights to understand protein structure and function, identify drug targets and unravel their functional roles in the pathogen. The structures of selected drug targets can also help in experimental design and prioritizing the protein targets for validation.
The emergence of drug resistance to the multidrug therapy is a major challenge in treating mycobacterial infections especially leprosy where structural features of drug-target interactions are poorly understood. To complement the computational findings, our group has employed cryoEM methods to understand the impact of mutations on the structure of catalase peroxidase in M. tuberculosis (Munir et al., 2019, 2021). Protein sequences and structures can be used to model the impacts of drug resistance mutation on protein structure and function. We have described various software available for predicting the impacts of mutations using protein sequence or structure or both. In-silico saturation mutagenesis experiments can guide the experimental design and help in saving the time and labor required to conduct laboratory experiments on animal models. Structure-based drug design (Blundell et al., 2002; Blundell and Patel, 2004) is a way forward and is a promising approach to design new drugs and treatments.
Author Contributions
SV, SM, and MADGE conducted the review and written the manuscript. MM, PT, and TB reviewed the manuscript and provided necessary additions to the text. All authors contributed to the article and approved the submitted version.
Funding
SV, MADGE, and MM were supported by American Leprosy Missions Grant, United States of America (Grant No: G88726), SM was supported by the MRC DBT Grant, United Kingdom and India (RG78439), and TB was supported by the Wellcome Trust Programme Grant, United Kingdom (200814/Z/16/Z).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Authors would like to thank the rest of the computational biology team at the Department of Biochemistry, University of Cambridge, United Kingdom, for their overarching support and guidance in the review process.
References
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. doi: 10.1038/nmeth0410-248
AlQuraishi, M. (2019). AlphaFold at CASP13. Bioinformatics 35, 4862–4865. doi: 10.1093/bioinformatics/btz422
Anand, P., and Chandra, N. (2014). Characterizing the pocketome of Mycobacterium tuberculosis and application in rationalizing polypharmacological target selection. Sci. Rep. 4, 6356. doi: 10.1038/srep06356
Andreeva, A., Kulesha, E., Gough, J., and Murzin, A. G. (2020). The SCOP database in 2020: expanded classification of representative family and superfamily domains of known protein structures. Nucleic Acids Res. 48, D376–D382. doi: 10.1093/nar/gkz1064
Baugh, L., Phan, I., Begley, D. W., Clifton, M. C., Armour, B., Dranow, D. M., et al. (2015). Increasing the structural coverage of tuberculosis drug targets. Tuberculosis (Edinb) 95, 142–148. doi: 10.1016/j.tube.2014.12.003
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., et al. (2000). The protein data bank. Nucleic Acids Res. 28, 235–242.
Bienert, S., Waterhouse, A., de Beer, T. A. P., Tauriello, G., Studer, G., Bordoli, L., et al. (2017). The SWISS-MODEL repository—new features and functionality. Nucleic Acids Res. 45, D313–D319. doi: 10.1093/nar/gkw1132
Binkowski, T. A., Naghibzadeh, S., and Liang, J. (2003). CASTp: computed Atlas of surface topography of proteins. Nucleic Acids Res. 31, 3352–3355. doi: 10.1093/nar/gkg512
Blower, T. R., Williamson, B. H., Kerns, R. J., and Berger, J. M. (2016). Crystal structure and stability of gyrase–fluoroquinolone cleaved complexes from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 113, 1706–1713. doi: 10.1073/pnas.1525047113
Blundell, T. L., Jhoti, H., and Abell, C. (2002). High-throughput crystallography for lead discovery in drug design. Nat. Rev. Drug Discov. 1, 45–54. doi: 10.1038/nrd706
Blundell, T. L., and Patel, S. (2004). High-throughput X-ray crystallography for drug discovery. Curr. Opin. Pharmacol. 4, 490–496. doi: 10.1016/j.coph.2004.04.007
Borah, K., Kearney, J.-L., Banerjee, R., Vats, P., Wu, H., Dahale, S., et al. (2020). GSMN-ML- a genome scale metabolic network reconstruction of the obligate human pathogen Mycobacterium leprae. PLoS Negl. Trop. Dis. 14:e0007871. doi: 10.1371/journal.pntd.0007871
Cambau, E., Carthagena, L., Chauffour, A., Ji, B., and Jarlier, V. (2006). Dihydropteroate synthase mutations in the Folp1 gene predict dapsone resistance in relapsed cases of leprosy. Clin. Infect. Dis. 42, 238–241. doi: 10.1086/498506
Cambau, E., Saunderson, P., Matsuoka, M., Cole, S., Kai, M., Suffys, P., et al. (2018). Antimicrobial resistance in leprosy: results of the first prospective open survey conducted by a WHO surveillance network for the period 2009–15. Clin. Microbiol. Infect. 24, 1305–1310.
Capriotti, E., Fariselli, P., Rossi, I., and Casadio, R. (2008). A three-state prediction of single point mutations on protein stability changes. BMC Bioinformatics 9:S6. doi: 10.1186/1471-2105-9-S2-S6
Chen, C.-W., Lin, J., and Chu, Y.-W. (2013). iStable: off-the-shelf predictor integration for predicting protein stability changes. BMC Bioinformatics 14:S5. doi: 10.1186/1471-2105-14-S2-S5
Chen, K.-H., Wang, T.-F., and Hu, Y.-J. (2019). Protein-protein interaction prediction using a hybrid feature representation and a stacked generalization scheme. BMC Bioinformatics 20:308. doi: 10.1186/s12859-019-2907-1
Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., et al. (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21. doi: 10.1107/S0907444909042073
Cheng, J., Randall, A., and Baldi, P. (2006). Prediction of protein stability changes for single-site mutations using support vector machines. Proteins 62, 1125–1132. doi: 10.1002/prot.20810
Chiarelli, L. R., Mori, G., Orena, B. S., Esposito, M., Lane, T., de Jesus Lopes Ribeiro, A. L., et al. (2018). A multitarget approach to drug discovery inhibiting Mycobacterium tuberculosis PyrG and PanK. Sci. Rep. 8:3187. doi: 10.1038/s41598-018-21614-4
Choi, Y., and Chan, A. P. (2015). PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31, 2745–2747. doi: 10.1093/bioinformatics/btv195
Cole, S. T., Eiglmeier, K., Parkhill, J., James, K. D., Thomson, N. R., Wheeler, P. R., et al. (2001). Massive gene decay in the leprosy bacillus. Nature 409, 1007–1011. doi: 10.1038/35059006
Das, S., and Chakrabarti, S. (2021). Classification and prediction of protein–protein interaction interface using machine learning algorithm. Sci. Rep. 11:1761. doi: 10.1038/s41598-020-80900-2
Dehouck, Y., Grosfils, A., Folch, B., Gilis, D., Bogaerts, P., and Rooman, M. (2009). Fast and accurate predictions of protein stability changes upon mutations using statistical potentials and neural networks: PoPMuSiC-2.0. Bioinformatics 25, 2537–2543. doi: 10.1093/bioinformatics/btp445
Dehouck, Y., Kwasigroch, J. M., Rooman, M., and Gilis, D. (2013). BeAtMuSiC: prediction of changes in protein–protein binding affinity on mutations. Nucleic Acids Res. 41, W333–W339. doi: 10.1093/nar/gkt450
DeJesus, M. A., Gerrick, E. R., Xu, W., Park, S. W., Long, J. E., Boutte, C. C., et al. (2017). Comprehensive essentiality analysis of the Mycobacterium tuberculosis genome via saturating transposon mutagenesis. mBio 8:e02133–16. doi: 10.1128/mBio.02133-16
Dhople, A. M. (2002). In vivo activity of epiroprim, a dihydrofolate reductase inhibitor, singly and in combination with dapsone, against Mycobacterium leprae. Int. J. Antimicrob. Agents 19, 71–74. doi: 10.1016/s0924-8579(01)00470-8
Du, X., Sun, S., Hu, C., Yao, Y., Yan, Y., and Zhang, Y. (2017). DeepPPI: boosting prediction of protein–protein interactions with deep neural networks. J. Chem. Inf. Model. 57, 1499–1510. doi: 10.1021/acs.jcim.7b00028
Eramian, D., Shen, M., Devos, D., Melo, F., Sali, A., and Marti-Renom, M. A. (2006). A composite score for predicting errors in protein structure models. Protein Sci. 15, 1653–1666. doi: 10.1110/ps.062095806
Fadlitha, V. B., Yamamoto, F., Idris, I., Dahlan, H., Sato, N., Aftitah, V. B., et al. (2019). The unique tropism of Mycobacterium leprae to the nasal epithelial cells can be explained by the mammalian cell entry protein 1A. PLoS Negl. Trop. Dis. 13:e0006704. doi: 10.1371/journal.pntd.0006704
Fang, J. (2020). A critical review of five machine learning-based algorithms for predicting protein stability changes upon mutation. Brief Bioinform 21, 1285–1292. doi: 10.1093/bib/bbz071
Friesner, R. A., Murphy, R. B., Repasky, M. P., Frye, L. L., Greenwood, J. R., Halgren, T. A., et al. (2006). Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 49, 6177–6196. doi: 10.1021/jm051256o
Goodsell, D. S., and Olson, A. J. (1990). Automated docking of substrates to proteins by simulated annealing. Proteins 8, 195–202. doi: 10.1002/prot.340080302
Graña, M., Haouz, A., Buschiazzo, A., Miras, I., Wehenkel, A., Bondet, V., et al. (2007). The crystal structure of M. leprae ML2640c defines a large family of putative S-adenosylmethionine-dependent methyltransferases in mycobacteria. Protein Sci. 16, 1896–1904. doi: 10.1110/ps.072982707
Gray, V. E., Hause, R. J., Luebeck, J., Shendure, J., and Fowler, D. M. (2018). Quantitative missense variant effect prediction using large-scale mutagenesis data. Cell Syst. 6, 116–124.e3. doi: 10.1016/j.cels.2017.11.003
Grønning, A. G. B., Doktor, T. K., Larsen, S. J., Petersen, U. S. S., Holm, L. L., Bruun, G. H., et al. (2020). DeepCLIP: predicting the effect of mutations on protein–RNA binding with deep learning. Nucleic Acids Res. 48, 7099–7118. doi: 10.1093/nar/gkaa530
Grosdidier, A., Zoete, V., and Michielin, O. (2011). SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 39, W270–W277. doi: 10.1093/nar/gkr366
Guo, H., Courbon, G. M., Bueler, S. A., Mai, J., Liu, J., and Rubinstein, J. L. (2021). Structure of mycobacterial ATP synthase bound to the tuberculosis drug bedaquiline. Nature 589, 143–147. doi: 10.1038/s41586-020-3004-3
Gupta, E., Gupta, S. R. R., and Niraj, R. R. K. (2020). Identification of drug and vaccine target in Mycobacterium leprae: a reverse vaccinology approach. Int. J. Pept. Res. Ther. 26, 1313–1326. doi: 10.1007/s10989-019-09936-x
Hatzopoulos, G. N., and Mueller-Dieckmann, J. (2010). Structure of translation initiation factor 1 from Mycobacterium tuberculosis and inferred binding to the 30S ribosomal subunit. FEBS Lett. 584, 1011–1015. doi: 10.1016/j.febslet.2010.01.051
Hecht, M., Bromberg, Y., and Rost, B. (2015). Better prediction of functional effects for sequence variants. BMC Genomics 16:S1. doi: 10.1186/1471-2164-16-S8-S1
Hentschel, J., Burnside, C., Mignot, I., Leibundgut, M., Boehringer, D., and Ban, N. (2017). The complete structure of the Mycobacterium smegmatis 70S ribosome. Cell Rep. 20, 149–160. doi: 10.1016/j.celrep.2017.06.029
Hopf, T. A., Ingraham, J. B., Poelwijk, F. J., Schärfe, C. P. I., Springer, M., Sander, C., et al. (2017). Mutation effects predicted from sequence co-variation. Nat. Biotechnol. 35, 128–135. doi: 10.1038/nbt.3769
Hu, L., and Chan, K. C. C. (2017). Extracting coevolutionary features from protein sequences for predicting protein-protein interactions. IEEE ACM Trans. Comput. Biol. Bioinform. 14, 155–166. doi: 10.1109/TCBB.2016.2520923
Huang, B., Fu, J., Guo, C., Wu, X., Lin, D., and Liao, X. (2016). (1)H, (15)N, (13)C resonance assignments for pyrazinoic acid binding domain of ribosomal protein S1 from Mycobacterium tuberculosis. Biomol. NMR Assign. 10, 321–324. doi: 10.1007/s12104-016-9692-9
Huang, Y.-A., You, Z.-H., Chen, X., Chan, K., and Luo, X. (2016). Sequence-based prediction of protein-protein interactions using weighted sparse representation model combined with global encoding. BMC Bioinformatics 17:184. doi: 10.1186/s12859-016-1035-4
Ji, B., Jamet, P., Perani, E. G., Sow, S., Lienhardt, C., Petinon, C., et al. (1996). Bactericidal activity of single dose of clarithromycin plus minocycline, with or without ofloxacin, against Mycobacterium leprae in patients. Antimicrob. Agents Chemother. 40, 2137–2141. doi: 10.1128/aac.40.9.2137
Jones, G., Willett, P., Glen, R. C., Leach, A. R., and Taylor, R. (1997). Development and validation of a genetic algorithm for flexible docking11Edited by F. E. Cohen. J. Mol. Biol. 267, 727–748. doi: 10.1006/jmbi.1996.0897
Kaur, G., Pandey, B., Kumar, A., Garewal, N., Grover, A., and Kaur, J. (2019). Drug targeted virtual screening and molecular dynamics of LipU protein of Mycobacterium tuberculosis and Mycobacterium leprae. J. Biomol. Struct. Dyn. 37, 1254–1269. doi: 10.1080/07391102.2018.1454852
Kaushal, P. S., Singh, P., Sharma, A., Muniyappa, K., and Vijayan, M. (2010). X-ray and molecular-dynamics studies on Mycobacterium leprae single-stranded DNA-binding protein and comparison with other eubacterial SSB structures. Acta Crystallogr. D Biol. Crystallogr. 66, 1048–1058. doi: 10.1107/S0907444910032208
Kramer, B., Rarey, M., and Lengauer, T. (1999). Evaluation of the FLEXX incremental construction algorithm for protein–ligand docking. Proteins 37, 228–241. doi: 10.1002/(sici)1097-0134(19991101)37:2<228::aid-prot8>3.0.co;2-8
Laimer, J., Hofer, H., Fritz, M., Wegenkittl, S., and Lackner, P. (2015). MAESTRO – multi agent stability prediction upon point mutations. BMC Bioinformatics 16:116. doi: 10.1186/s12859-015-0548-6
Lechartier, B., and Cole, S. T. (2015). Mode of Action of Clofazimine and Combination Therapy with Benzothiazinones against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 59, 4457–4463. doi: 10.1128/AAC.00395-15
Li, M., Simonetti, F. L., Goncearenco, A., and Panchenko, A. R. (2016). MutaBind estimates and interprets the effects of sequence variants on protein–protein interactions. Nucleic Acids Res. 44, W494–W501. doi: 10.1093/nar/gkw374
Li, Y., Golding, G. B., and Ilie, L. (2020). DELPHI: accurate deep ensemble model for protein interaction sites prediction. Bioinformatics btaa750. doi: 10.1093/bioinformatics/btaa750 [Epub ahead of print].
Lin, W., Mandal, S., Degen, D., Liu, Y., Ebright, Y. W., Li, S., et al. (2017). Structural basis of Mycobacterium tuberculosis transcription and transcription inhibition. Mol. Cell 66, 169–179.e8. doi: 10.1016/j.molcel.2017.03.001
Liu, Y., Harrison, P. M., Kunin, V., and Gerstein, M. (2004). Comprehensive analysis of pseudogenes in prokaryotes: widespread gene decay and failure of putative horizontally transferred genes. Genome Biol. 5:R64. doi: 10.1186/gb-2004-5-9-r64
Lockwood, D. N., and Saunderson, P. R. (2012). Nerve damage in leprosy: a continuing challenge to scientists, clinicians and service providers. Int. Health 4, 77–85. doi: 10.1016/j.inhe.2011.09.006
Malhotra, S., Vedithi, S. C., and Blundell, T. L. (2017). Decoding the similarities and differences among mycobacterial species. PLoS Negl. Trop. Dis. 11:e0005883. doi: 10.1371/journal.pntd.0005883
Mande, S. C., Mehra, V., Bloom, B. R., and Hol, W. G. (1996). Structure of the heat shock protein chaperonin-10 of Mycobacterium leprae. Science 271, 203–207. doi: 10.1126/science.271.5246.203
Manglani, P. R., and Arif, M. A. (2006). Multidrug therapy in leprosy. J. Indian Med. Assoc. 104, 686–688.
Mukherjee, S., and Zhang, Y. (2011). Protein-protein complex structure predictions by multimeric threading and template recombination. Structure 19, 955–966. doi: 10.1016/j.str.2011.04.006
Munir, A., Kumar, N., Ramalingam, S. B., Tamilzhalagan, S., Shanmugam, S. K., Palaniappan, A. N., et al. (2019). Identification and characterization of genetic determinants of isoniazid and rifampicin resistance in Mycobacterium tuberculosis in Southern India. Sci. Rep. 9:10283. doi: 10.1038/s41598-019-46756-x
Munir, A., Wilson, M. T., Hardwick, S. W., Chirgadze, D. Y., Worrall, J. A. R., Blundell, T. L., et al. (2021). Using cryo-EM to understand antimycobacterial resistance in the catalase-peroxidase (KatG) from Mycobacterium tuberculosis. Structure doi: 10.1016/j.str.2020.12.008 [Epub ahead of print].
Ng, P. C., and Henikoff, S. (2003). SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814. doi: 10.1093/nar/gkg509
Ochoa-Montaño, B., Mohan, N., and Blundell, T. L. (2015). CHOPIN: a web resource for the structural and functional proteome of Mycobacterium tuberculosis. Database (Oxford) 2015:bav026. doi: 10.1093/database/bav026
Ogmen, U., Keskin, O., Aytuna, A. S., Nussinov, R., and Gursoy, A. (2005). PRISM: protein interactions by structural matching. Nucleic Acids Res. 33, W331–W336. doi: 10.1093/nar/gki585
Pandurangan, A. P., Ochoa-Montaño, B., Ascher, D. B., and Blundell, T. L. (2017). SDM: a server for predicting effects of mutations on protein stability. Nucleic Acids Res. 45, W229–W235. doi: 10.1093/nar/gkx439
Pejaver, V., Urresti, J., Lugo-Martinez, J., Pagel, K. A., Lin, G. N., Nam, H.-J., et al. (2020). Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 11, 1–13. doi: 10.1038/s41467-020-19669-x
Pieper, U., Webb, B. M., Barkan, D. T., Schneidman-Duhovny, D., Schlessinger, A., Braberg, H., et al. (2011). ModBase, a database of annotated comparative protein structure models, and associated resources. Nucleic Acids Res. 39, D465–D474. doi: 10.1093/nar/gkq1091
Pires, D. E. V., and Ascher, D. B. (2017). mCSM–NA: predicting the effects of mutations on protein–nucleic acids interactions. Nucleic Acids Res. 45, W241–W246. doi: 10.1093/nar/gkx236
Pires, D. E. V., Ascher, D. B., and Blundell, T. L. (2014a). DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 42, W314–W319. doi: 10.1093/nar/gku411
Pires, D. E. V., Ascher, D. B., and Blundell, T. L. (2014b). mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 30, 335–342. doi: 10.1093/bioinformatics/btt691
Pires, D. E. V., Blundell, T. L., and Ascher, D. B. (2016). mCSM-lig: quantifying the effects of mutations on protein-small molecule affinity in genetic disease and emergence of drug resistance. Sci. Rep. 6:29575. doi: 10.1038/srep29575
Potapov, V., Cohen, M., Inbar, Y., and Schreiber, G. (2010). Protein structure modelling and evaluation based on a 4-distance description of side-chain interactions. BMC Bioinformatics 11:374. doi: 10.1186/1471-2105-11-374
Radoux, C. J., Olsson, T. S. G., Pitt, W. R., Groom, C. R., and Blundell, T. L. (2016). Identifying interactions that determine fragment binding at protein hotspots. J. Med. Chem. 59, 4314–4325. doi: 10.1021/acs.jmedchem.5b01980
Rao, P. N., and Jain, S. (2013). Newer Management Options in Leprosy. Indian J. Dermatol. 58, 6–11. doi: 10.4103/0019-5154.105274
Roe, S. M., Barlow, T., Brown, T., Oram, M., Keeley, A., Tsaneva, I. R., et al. (1998). Crystal structure of an octameric RuvA-holliday junction complex. Mol. Cell 2, 361–372. doi: 10.1016/s1097-2765(00)80280-4
Sassetti, C. M., Boyd, D. H., and Rubin, E. J. (2003). Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48, 77–84. doi: 10.1046/j.1365-2958.2003.03425.x
Sato, N., Fujimura, T., Masuzawa, M., Yogi, Y., Matsuoka, M., Kanoh, M., et al. (2007). Recombinant Mycobacterium leprae protein associated with entry into mammalian cells of respiratory and skin components. J. Dermatol. Sci. 46, 101–110. doi: 10.1016/j.jdermsci.2007.01.006
Schymkowitz, J., Borg, J., Stricher, F., Nys, R., Rousseau, F., and Serrano, L. (2005). The FoldX web server: an online force field. Nucleic Acids Res. 33, W382–W388. doi: 10.1093/nar/gki387
Shen, M., and Sali, A. (2006). Statistical potential for assessment and prediction of protein structures. Protein Sci. 15, 2507–2524. doi: 10.1110/ps.062416606
Shi, J., Blundell, T. L., and Mizuguchi, K. (2001). FUGUE: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J. Mol. Biol. 310, 243–257. doi: 10.1006/jmbi.2001.4762
Sillitoe, I., Dawson, N., Lewis, T. E., Das, S., Lees, J. G., Ashford, P., et al. (2019). CATH: expanding the horizons of structure-based functional annotations for genome sequences. Nucleic Acids Res. 47, D280–D284. doi: 10.1093/nar/gky1097
Skwark, M. J., Torres, P. H. M., Copoiu, L., Bannerman, B., Floto, R. A., and Blundell, T. L. (2019). Mabellini: a genome-wide database for understanding the structural proteome and evaluating prospective antimicrobial targets of the emerging pathogen Mycobacterium abscessus. Database 2019, baz113. doi: 10.1093/database/baz113
Soni, N., and Madhusudhan, M. S. (2017). Computational modeling of protein assemblies. Curr. Opin. Struct. Biol. 44, 179–189. doi: 10.1016/j.sbi.2017.04.006
Sun, T., Zhou, B., Lai, L., and Pei, J. (2017). Sequence-based prediction of protein protein interaction using a deep-learning algorithm. BMC Bioinformatics 18:277. doi: 10.1186/s12859-017-1700-2
The UniProt Consortium (2021). UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489. doi: 10.1093/nar/gkaa1100
Torrisi, M., Pollastri, G., and Le, Q. (2020). Deep learning methods in protein structure prediction. Comput. Struct. Biotechnol. J. 18, 1301–1310. doi: 10.1016/j.csbj.2019.12.011
Vedithi, S. C., Malhotra, S., Das, M., Daniel, S., Kishore, N., George, A., et al. (2018). Structural implications of mutations conferring rifampin resistance in Mycobacterium leprae. Sci. Rep. 8:5016. doi: 10.1038/s41598-018-23423-1
Vedithi, S. C., Malhotra, S., Skwark, M. J., Munir, A., Acebrón-García-De-Eulate, M., Waman, V. P., et al. (2020). HARP: a database of structural impacts of systematic missense mutations in drug targets of Mycobacterium leprae. Comput. Struct. Biotechnol. J. 18, 3692–3704. doi: 10.1016/j.csbj.2020.11.013
Wang, F., Langley, R., Gulten, G., Dover, L. G., Besra, G. S., Jacobs, W. R., et al. (2007). Mechanism of thioamide drug action against tuberculosis and leprosy. J. Exp. Med. 204, 73–78. doi: 10.1084/jem.20062100
Wang, L., Wang, H.-F., Liu, S.-R., Yan, X., and Song, K.-J. (2019). Predicting protein-protein interactions from matrix-based protein sequence using convolution neural network and feature-selective rotation forest. Sci. Rep. 9, 1–12. doi: 10.1038/s41598-019-46369-4
Wang, Z., Sun, H., Yao, X., Li, D., Xu, L., Li, Y., et al. (2016). Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: the prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 18, 12964–12975. doi: 10.1039/C6CP01555G
Webb, B., and Sali, A. (2016). Comparative Protein structure modeling using MODELLER. Curr. Protoc. Bioinformatics 54, 5.6.1–5.6.37. doi: 10.1002/cpbi.3
Williams, D. L., Spring, L., Harris, E., Roche, P., and Gillis, T. P. (2000). Dihydropteroate synthase of Mycobacterium leprae and dapsone resistance. Antimicrob. Agents Chemother. 44, 1530–1537. doi: 10.1128/aac.44.6.1530-1537.2000
Yamaguchi, T., Yokoyama, K., Nakajima, C., and Suzuki, Y. (2016). DC-159a shows inhibitory activity against DNA gyrases of Mycobacterium leprae. PLoS Negl. Trop. Dis. 10:e0005013. doi: 10.1371/journal.pntd.0005013
Yeh, I., Hanekamp, T., Tsoka, S., Karp, P. D., and Altman, R. B. (2004). Computational analysis of Plasmodium falciparum metabolism: organizing genomic information to facilitate drug discovery. Genome Res. 14, 917–924. doi: 10.1101/gr.2050304
Yu, J., Vavrusa, M., Andreani, J., Rey, J., Tufféry, P., and Guerois, R. (2016). InterEvDock: a docking server to predict the structure of protein–protein interactions using evolutionary information. Nucleic Acids Res. 44, W542–W549. doi: 10.1093/nar/gkw340
Yue, P., Melamud, E., and Moult, J. (2006). SNPs3D: candidate gene and SNP selection for association studies. BMC Bioinformatics 7:166. doi: 10.1186/1471-2105-7-166
Zhang, L., Yu, G., Xia, D., and Wang, J. (2019). Protein–protein interactions prediction based on ensemble deep neural networks. Neurocomputing 324, 10–19. doi: 10.1016/j.neucom.2018.02.097
Zhou, G., Chen, M., Ju, C. J. T., Wang, Z., Jiang, J.-Y., and Wang, W. (2020). Mutation effect estimation on protein–protein interactions using deep contextualized representation learning. NAR Genom Bioinform. 2. doi: 10.1093/nargab/lqaa015
Keywords: Mycobacterium leprae, amino acid substitution, chokepoint reactions, drug binging sites, homology (comparative) modeling, protein interface
Citation: Vedithi SC, Malhotra S, Acebrón-García-de-Eulate M, Matusevicius M, Torres PHM and Blundell TL (2021) Structure-Guided Computational Approaches to Unravel Druggable Proteomic Landscape of Mycobacterium leprae. Front. Mol. Biosci. 8:663301. doi: 10.3389/fmolb.2021.663301
Received: 02 February 2021; Accepted: 12 April 2021;
Published: 07 May 2021.
Edited by:
Alexandre G. De Brevern, INSERM U1134 Biologie Intégrée du Globule Rouge, FranceReviewed by:
Stéphane Téletchéa, Université de Nantes, FranceJeremy Esque, Institut Biotechnologique de Toulouse (INSA), France
Copyright © 2021 Vedithi, Malhotra, Acebrón-García-de-Eulate, Matusevicius, Torres and Blundell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sundeep Chaitanya Vedithi, c2N2MjZAY2FtLmFjLnVr; Tom L. Blundell, dGxiMjBAY2FtLmFjLnVr
†These authors have contributed equally to this work