 Gwen B. Effgen1
Gwen B. Effgen1 Christopher D. Hue1
Christopher D. Hue1 Edward Vogel III1
Edward Vogel III1 Matthew B. Panzer2
Matthew B. Panzer2 David F. Meaney3
David F. Meaney3 Cameron R. Bass2
Cameron R. Bass2 Barclay Morrison III1*
Barclay Morrison III1*- 1 Department of Biomedical Engineering, Columbia University, New York, NY, USA
- 2 Department of Biomedical Engineering, Duke University, Durham, NC, USA
- 3 Department of Bioengineering, University of Pennsylvania, Philadelphia, PA, USA
Due to the prominent role of improvised explosive devices (IEDs) in wounding patterns of U.S. war-fighters in Iraq and Afghanistan, blast injury has risen to a new level of importance and is recognized to be a major cause of injuries to the brain. However, an injury risk-function for microscopic, macroscopic, behavioral, and neurological deficits has yet to be defined. While operational blast injuries can be very complex and thus difficult to analyze, a simplified blast injury model would facilitate studies correlating biological outcomes with blast biomechanics to define tolerance criteria. Blast-induced traumatic brain injury (bTBI) results from the translation of a shock wave in-air, such as that produced by an IED, into a pressure wave within the skull–brain complex. Our blast injury methodology recapitulates this phenomenon in vitro, allowing for control of the injury biomechanics via a compressed-gas shock tube used in conjunction with a custom-designed, fluid-filled receiver that contains the living culture. The receiver converts the air shock wave into a fast-rising pressure transient with minimal reflections, mimicking the intracranial pressure history in blast. We have developed an organotypic hippocampal slice culture model that exhibits cell death when exposed to a 530 ± 17.7-kPa peak overpressure with a 1.026 ± 0.017-ms duration and 190 ± 10.7 kPa-ms impulse in-air. We have also injured a simplified in vitro model of the blood–brain barrier, which exhibits disrupted integrity immediately following exposure to 581 ± 10.0 kPa peak overpressure with a 1.067 ± 0.006-ms duration and 222 ± 6.9 kPa-ms impulse in-air. To better prevent and treat bTBI, both the initiating biomechanics and the ensuing pathobiology must be understood in greater detail. A well-characterized, in vitro model of bTBI, in conjunction with animal models, will be a powerful tool for developing strategies to mitigate the risks of bTBI.
Introduction
The prevalence of injuries to service men and women resulting from improvised explosive devices (IEDs) in recent U.S. military conflicts has highlighted the knowledge-deficit that exists concerning the acute and long-term human health threat of blast-induced traumatic brain injury (bTBI). Since 2001, nearly 80% of all U.S. service member casualties in Iraq and Afghanistan have been the result of IEDs, and the incidence of bTBI has increased sharply (Owens et al., 2008; Tanielian and Jaycox, 2008; Livingston and O’hanlon, 2011; O’Hanlon and Livingston, 2011). The total number of service members reported to be affected by traumatic brain injury (TBI) from 2000 to 2011 is over 220,000 (Defense and Veterans Brain Injury Center, 2012). Experimental studies have shown that exposure to blasts below the threshold for pulmonary injury can result in acute and delayed behavioral deficits and neurodegeneration (Kaur et al., 1997; Cernak et al., 2001b; Risling et al., 2002; Saljo et al., 2002a; Moochhala et al., 2004; Rafaels et al., 2010). In light of these studies, there is an urgent need to develop an injury risk-function for bTBI and to improve our understanding of the mechanisms that lead to acute and long-term deficits resulting from bTBI.
Real-world blast loading can be exceedingly complex; therefore, simplified experimental models are necessary to begin developing blast injury risk-functions for brain tissue before investigating more complex conditions (Hooker, 1924). Peak overpressure, duration, and impulse of the positive pressure phase have been shown to be key parameters influencing the severity of bTBI (Bass et al., 2008; Chen et al., 2009; Rafaels et al., 2010, 2011; Panzer et al., 2011). In our system, a compressed-gas shock tube was used to reproduce a free-field blast wave (i.e., a Friedlander wave) characterized by a fast-rising overpressure with an exponential decay into a negative pressure phase. This idealized blast injury model has been used extensively to study the effects of primary blast on the body (Cassen et al., 1950; Celander et al., 1955; Richmond et al., 1961; Dodd et al., 1997; Elsayed, 1997; Cernak et al., 2001a,b; Gorbunov et al., 2005; Bass et al., 2008; Garman et al., 2009, 2011; Long et al., 2009; Rafaels et al., 2010, 2011; Panzer et al., 2011).
In vitro biological models offer several advantages including ease of accessibility, allowing the sample to be viewed directly, and functional or biochemical measures to be taken before and after injury at multiple time points. Serial sampling helps to determine the evolution of the pathology for identification of not only therapeutic targets but also critical delivery time points to treat bTBI. In vitro injury models have the added advantage of precise control over the injury biomechanics. Taken together, these advantages make it possible to correlate biological outcome measures to injury parameters for defining tissue-level tolerance criteria. This is an advantage over using computational models to deduce the tissue-level injury criteria, since computational models can be highly sensitive to assumptions, including material properties, loading conditions, and interface parameters. In vitro blast injury lays a strong foundation for the development of bTBI models with increased complexity, facilitating a correlation between microscopic physiological damage witnessed in vitro to macroscopic damage and behavioral deficits in vivo.
In vitro models have increased our mechanistic understanding of TBI caused by blunt trauma or inertial mechanisms (i.e., motor vehicle accidents, falls, assaults) for both the initiating biomechanics and the ensuing pathobiology (Morrison et al., 1998, 2011). The development of in vitro blast injury models that recapitulate intracranial blast physics could accelerate future bTBI research in a similar manner. For this reason, we have developed an in vitro blast injury model that provides precise control of the overpressure biomechanics for correlating loading parameters to the living biological response (Panzer et al., 2012). To reproduce the intracranial milieu where the shock wave is translated to a fast-rising pressure wave, tissue cultures were submerged in a fluid-filled reservoir to simulate the surrounding brain. The resulting effect is an increase in the duration of the external pressure input that replicated pressure histories measured within the brain in experimental studies (Clemedson and Pettersson, 1956; Romba et al., 1961; Chavko et al., 2007; Saljo et al., 2008). Of particular significance, the blast injury methodology described here is compatible with many in vitro biological preparations with only minor adjustments (Panzer et al., 2012); we present data utilizing models of the brain parenchyma (organotypic hippocampal slice culture, OHSC) and the blood–brain barrier (BBB). This work is in conjunction with complementary characterization of test devices for in vivo and in vitro blast injury and methodology for their implementation with in vivo models.
Materials and Methods
Shock Tube
Blast overpressures were generated with a 76-mm diameter aluminum shock tube with an adjustable-length driver section (25 mm used for the current studies) pressurized with helium and a 1240-mm long driven section (Panzer et al., 2012). The diaphragm was composed of polyethylene terephthalate (PET) membranes, and the thickness of the diaphragm was varied to control the burst pressure. Three piezoresistive pressure transducers (Endevco, San Juan Capistrano, CA, USA) flush-mounted at the exit of the shock tube and oriented perpendicular to the direction of propagation recorded side-on pressure. Analog outputs from the transducers were conditioned using instrumentation amplifiers (gain of 50) and low-pass filters (corner frequency of 40 kHz, Alligator Technologies, Costa Mesa, CA, USA). Signals were digitized with an X-series data acquisition card at 125 kHz using LabVIEW™ 2010 (National Instruments, Austin, TX, USA). Peak overpressure, duration, and impulse were calculated with custom MATLAB code (MathWorks, Natick, MA, USA). Room temperature, ambient pressure, tank pressure, and regulator settings were recorded for each blast.
in vitro Receiver
Cultures were placed in a fluid-filled blast receiver designed for use with the shock tube (Panzer et al., 2012). The in vitro blast injury receiver was composed of a polyethylene 57 L reservoir with a polycarbonate test column. The test column extended into the reservoir through a diverging nozzle. The geometry of the receiver resulted in the mitigation of the majority of the internally reflected pressure waves to reduce subsequent mechanical loading of the tissue. The end of the shock tube was placed flush with the top of the receiver and centered on its vertical axis. Turnbuckles were used to align the test column with the axis of the shock tube (Figure 1).
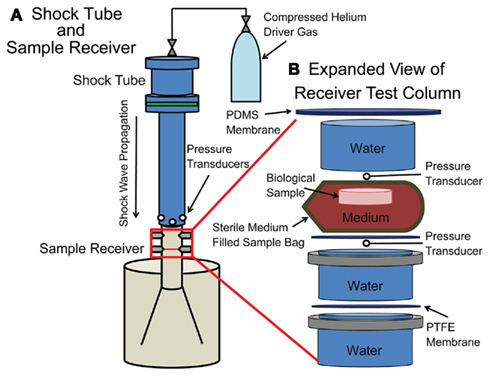
Figure 1. Schematic of the shock tube with receiver. (A) A compressed helium source was connected to an adjustable driver section of the shock tube, which was aligned vertically over the sample receiver. In-air transducers were located at the exit of the shock tube. (B) The sample within a bag rested on top of a PTFE membrane within the fluid-filled sample receiver. Submersible transducers were located directly above the sample bag and below the PTFE membrane.
The test column was separated from the reservoir by a 250-μm thick polytetrafluoroethylene (PTFE) membrane to restrict bulk fluid motion. A PTFE membrane was secured in the middle of the test column for a reproducible sample location in the receiver. A silicone membrane (Specialty Manufacturing Inc., Saginaw, MI, USA) was secured to the open end of the test column with a hose clamp to prevent spray. Membrane materials used in the receiver were carefully chosen for impedance matching with water to prevent unintended reflections. Inclusion of air bubbles was meticulously prevented.
The receiver was filled with water maintained at 37°C with a heating element (Innomax, Denver, CO, USA) affixed to the receiver for all in vitro blast injuries and blast characterizations. The water temperature in the test column was verified and recorded prior to and following each blast injury.
To characterize the loading of the culture sample, two submersible pressure transducers (Millar Instruments, Houston, TX, USA) were inserted into the test column adjacent to the sample. The face of each transducer was oriented into the direction of wave propagation. Data from these transducers were recorded as described above.
OHSC Culture
All animal procedures were approved by the Columbia University Institutional Animal Care and Use Committee (IACUC). According to previously published culture methods, P8-10 Sprague-Dawley rat pups were decapitated, and their brains were removed (Morrison et al., 2002, 2006; Cater et al., 2006, 2007; Elkin and Morrison, 2007). Hippocampi were excised and sectioned into 400 μm thick slices using a McIlwain tissue chopper (Harvard Apparatus, Holliston, MA, USA). Slices were separated aseptically in ice-cold Gey’s salt solution supplemented with 25 mM D-glucose (Sigma, St. Louis, MO, USA) using blunt, plastic spatulas (Fisher, Pittsburgh, PA, USA). Slices were plated onto porous Millipore Millicell cell culture membranes (Millipore, Billerica, MA, USA). Slices were initially fed with Neurobasal medium supplemented with 1 mM L-glutamine, 1× B27 supplement, 10 mM HEPES, and 25 mM D-glucose (Invitrogen, Carlsbad, CA, USA). Culture medium was changed to conditioned full-serum medium (50% Minimum Essential Medium, 25% Hank’s Balanced Salt Solution, 25% heat inactivated horse serum, 2 μM L-glutamine, 25 mM D-glucose, 10 mM HEPES, Sigma) 3 days following plating. OHSC were subsequently fed with conditioned full-serum medium every 2–3 days. OHSC were cultured at 37°C and 5% CO2.
After 10–14 days in culture, the baseline health of OHSC immediately prior to injury was assessed by quantifying pre-injury cell death with the fluorescent stain propidium iodide (PI, Invitrogen). OHSC with PI fluorescence greater than 10% in any region (DG, CA3, CA1) were not included in the study (see OHSC Cell Death Quantitative Analysis).
BBB Model
Using an endothelial monoculture model of the BBB (bEnd.3, mouse brain microvascular cell line, ATCC, Manassas, VA, USA) a total of 38,000 bEnd.3 cells were seeded on 1.12 cm2, poly-L-lysine coated Transwell inserts in a 12-well plate (Corning Costar, Corning, NY, USA) and were cultured for 7 days according to published methods (Simon et al., 2010, 2011). Cells were grown in serum-containing medium [DMEM supplemented with 10% newborn calf serum (Invitrogen) and 4 mM glutamine (Sigma)] and fed every 2–3 days. bEnd.3 cells were cultured at 37°C and 5% CO2.
Following 7 days in culture, to assess the baseline health of the monolayer immediately prior to injury, images of both bright-field and PI fluorescence were recorded, and baseline trans-endothelial electrical resistance (TEER) was measured. Cell monolayers with TEER less than 12 Ω cm2 were not included in the study (see BBB Cell Death Quantitative Analysis; see Trans-endothelial Electrical Resistance).
Blast Loading
OHSC injury
Prior to placing cultures in the receiver, individual culture wells were sealed inside sterile bags to prevent contamination, maintain medium pH, minimize bulk flow immediately around the sample, and minimize waste of culture medium. Small sterile bags made of 57 μm thick, low density polyethylene (Whirl Pak, Fort Atkinson, WI, USA) were filled with pre-warmed, serum-free culture medium that had been equilibrated with 5% CO2/95% O2 for 10 min. Care was taken to prevent entrapment of air bubbles. The culture and the bag were submerged in the test column of the receiver and oriented perpendicular to pressure wave propagation.
Injured cultures were subjected to a single blast exposure. Following blast exposure, the bag with the culture was immediately removed from the receiver, and the culture was returned to fresh, serum-free medium and incubated. Sham-exposed samples were sealed into bags with equilibrated, warmed, serum-free medium, and submerged in the receiver for an equivalent period, but the shock tube was not fired. Resultant cell death was measured 4 days following blast injury given the delayed cell death response of OHSC exposed to stretch-injury (Morrison et al., 1998, 2002, 2003, 2006; Cater et al., 2006, 2007).
BBB injury
Prior to placing cultures in the receiver, individual Transwells were sealed inside sterile bags to prevent contamination, maintain medium pH, minimize bulk flow immediately around the sample, and minimize waste of culture medium (Whirl Pak). Sample bags were filled with pre-warmed, serum-containing culture medium that had been equilibrated with 5% CO2/95% O2 for 10 min. Care was taken to prevent entrapment of air bubbles. The culture and the bag were submerged in the test column of the receiver and oriented perpendicular to the direction of wave propagation.
Injured cultures were subjected to a single blast exposure. Following blast exposure, the bag with the culture was immediately removed from the receiver, and the culture was returned to the incubator in fresh medium. Sham-exposed samples were sealed into bags and submerged in the receiver for an equivalent period, but the shock tube was not fired.
Excitotoxic Injury
After OHSC had been imaged for blast-induced cell death, total cell death resultant from an excitotoxic injury was induced. OHSC medium was switched to a 10-mM glutamate solution in serum-free medium. Cultures were incubated for 3 h and then returned to fresh serum-free medium. Cell death resulting from excitotoxic injury was determined 24 h later with PI staining and imaging (see OHSC Cell Death Quantitative Analysis).
Cell Death Analysis
OHSC cell death quantitative analysis
PI fluorescence was used to quantify cell death prior to and 4 days following injury and 1 day following excitotoxic injury. OHSC were incubated in 2.5 μM PI in serum-free medium for 1 h before imaging. Images were acquired using an Olympus IX81 microscope with 568/24 excitation and 610/40 emission filters. Following imaging, cultures were returned to fresh, serum-free medium. Cell death was determined for specific OHSC regions (DG, CA1, CA3), as previously described, using MetaMorph [Molecular Devices, Downingtown, PA, USA (Morrison et al., 2002, 2003, 2006; Cater et al., 2006; Elkin and Morrison, 2007)]. In brief, the same threshold for fluorescence was used to analyze all images at each time point. Tissue damage at a given time point was quantified as the percentage area of a specific region exhibiting fluorescence above the threshold. Changes in percent cell death were normalized to maximum cell death resulting from excitotoxic injury.
BBB cell death quantitative analysis
PI fluorescence was used to quantify cell death prior to, 2 and 8 h after injury. Cell monolayers were incubated in 2.5 μM PI for 1 h in serum-free medium before imaging. Images were acquired using an Olympus IX81 microscope with 568/24 excitation and 610/40 emission filters. Images were examined using MetaMorph (Molecular Devices, Downingtown, PA, USA). In brief, the same threshold for fluorescence was used to analyze all images at each time point. Tissue damage at a given time point was quantified as the number of dead cells per area of a specific region exhibiting fluorescence above the threshold. Following imaging, PI medium was aspirated completely, and the cultures were returned to fresh, supplemented culture medium.
Trans-Endothelial Electrical Resistance
Changes in TEER were quantified using an Endohm-12 chamber electrode connected to an EVOMX Epithelial Voltohmmeter (World Precision Instruments), taking into account the TEER of cell-free Transwell filters. TEER values were normalized to the membrane surface area. TEER measurements were taken immediately prior to and after injury. Sham-exposed samples were processed identically to blast-exposed cultures.
Statistical Analysis
A univariate general linear model was used to analyze the complete data set for each region of the OHSC with cell death as the dependent variable and experimental group (sham, injured) as the fixed factor (SPSS v. 19, IBM, Armonk, NY, USA, significance *p < 0.05). Independent samples t-tests were used to analyze the results of injured and sham-exposed BBB cultures, with TEER as the dependent variable (SPSS v. 19, significance *p < 0.05).
Results
Characterization of the shock tube was performed without the receiver in place (Figure 2). Pressure time-histories recorded at the end of the shock tube were typical of a Friedlander wave (Figure 2B) and demonstrated good inter-test consistency (Figure 2A). Duration of the positive pressure phase was correlated with peak overpressure, and the relationship was well-defined by a second-order polynomial fit (Figure 2C). The impulse was also correlated with peak overpressure and was well-defined by a second-order polynomial as well (Figure 2D).
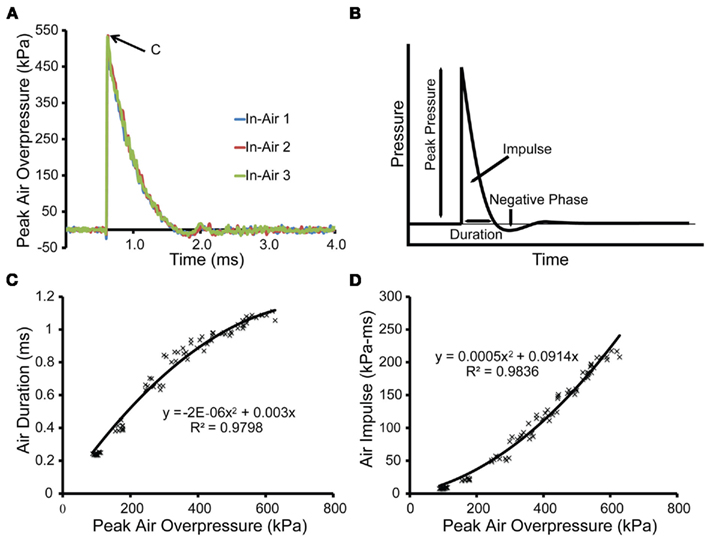
Figure 2. Characterization of the open shock tube. (A) Three in-air pressure transducers located equidistant around the exit to the shock tube recorded pressure transients in-air for blast of a 1.5-mm thick PET burst membrane. The peak overpressure [denoted by point (C)] for this blast was 534 kPa with a duration of 1.040 ms and an impulse of 184 kPa-ms. (B) The output of the shock tube was similar in shape to the Friedlander wave, which models the primary blast produced from an explosion in the free-field. (C) For the open shock tube, the durations were plotted as a function of the peak overpressures for each blast and fit to a second-order polynomial (n = 78). (D) For the open tube, the impulses were plotted as a function of peak overpressures for each blast and fit to a second-order polynomial (n = 78).
Placement of the receiver below the shock tube significantly altered the pressure recorded by the transducers at the end of the shock tube (Figure 3A). Similar to the principles of transmission for acoustic waves, the pressure history revealed two waves produced from the interaction of the incident shock wave with the top surface of the fluid-filled receiver. The incident pressure of the shock wave exiting the shock tube (Figure 3A, point “I”) remained discernable in the pressure trace as the first peak. However, the subsequent, larger peak (Figure 3A, point “R”) was due to the shock wave reflecting off the liquid surface and re-entering the shock tube. The presence of the receiver effectively altered the pressure history, affecting the peak pressure, duration, and impulse. These parameters were highly sensitive to the placement and interaction of the receiver with the shock wave, so we have chosen to characterize the applied blast loading by the pressure history in the open tube configuration and by the pressure experienced directly by the biological sample (Chavko et al., 2011). The close temporal relationship and the significantly higher magnitude of the pressure of the reflected wave can make it difficult to identify the peak pressure of the incident shock wave. Misappropriation of the reflection as the incident shock could confound interpretation of the loading conditions and lead to the erroneous conclusion that the presence of an object amplified the incident shock wave.
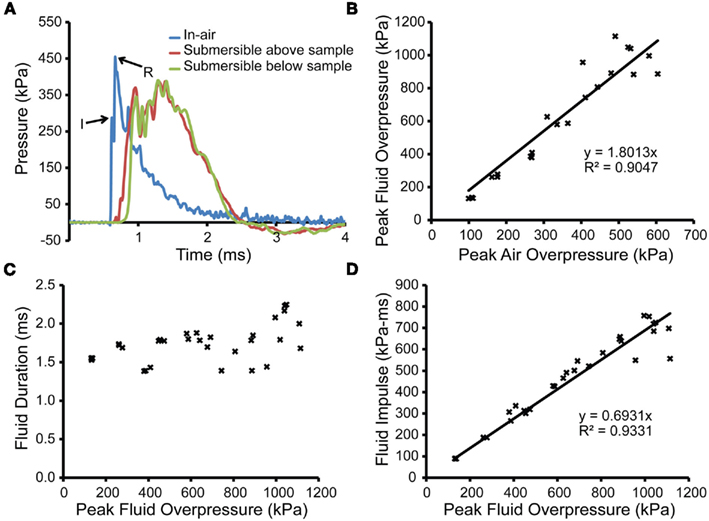
Figure 3. Characterization of the shock tube and fluid pressures with receiver in place. (A) An in-air pressure transducer (blue trace) located at the exit of the shock tube recorded pressure transients in-air for a shock produced from a 508-μm thick burst membrane with the receiver in place. The peak pressure in-air of the incident shock wave upon exiting the shock tube is denoted by “I,” and the peak pressure of the reflection is denoted as “R.” Two submersible transducers, located above or below the sample holder (including culture well, bag, and PTFE membrane; red and green traces, respectively) demonstrated the absence of attenuation through the in vitro set-up. (B) The peak overpressures from the submersible transducer below the sample holder were correlated to peak pressures measured by the in-air transducers. (C) The durations within the fluid-filled receiver were plotted against peak overpressures measured within the fluid-filled receiver, with no correlation to pressure found. (D) The impulses within the fluid-filled receiver were correlated to peak overpressures measured within the fluid-filled receiver and approximated using a linear fit.
Transducers located on the upstream and downstream side of the sample holder (culture well, bag, and PTFE membrane) recorded the pressure transients within the fluid-filled receiver (Figure 3A). The peak overpressures measured by the upstream and downstream transducers were 405 ± 17.0 and 405 ± 15.0 kPa with durations of 1.8 ± 0.036 and 1.8 ± 0.004 ms, respectively (n = 3). These results indicated that the propagation of the pressure wave was not affected by the presence of the culture well, bag, or PTFE membrane. The relationship between the peak incident pressure (in-air) and the peak pressure in the sample receiver was linear (R2 = 0.90474) over the pressure range tested (Figure 3B). As peak pressure increased, the duration values remained consistent, increasing slightly over the upper range of pressures but did not correlate with the incident pressure (Figure 3C). The durations ranged from 1.384 to 2.248 ms. Impulse correlated linearly (R2 = 0.9331) to peak overpressure measured in the fluid-filled receiver (Figure 3D). Impulse values ranged from 265.6 to 757.6 kPa-ms.
Exposure of OHSC to 530 ± 17.7 kPa peak incident pressure, 1.026 ± 0.017 ms duration, and 190 ± 10.7 kPa-ms impulse in-air increased cell death 4 days following blast (Figure 4). Injured tissue cultures experienced significantly more cell death 4 days following blast as compared to sham-exposed samples in all regions of the hippocampus (Figure 4A). Injured tissue appeared darker in bright-field images (Figure 4B), indicative of ultrastructural changes inducible by cell death such as mitochondrial swelling (Muller and Somjen, 1999). Additionally, resultant cell death in injured tissue was isolated to the principal cell layers (pyramidal and granule cells) as has been seen 4 days following in vitro stretch-injury of OHSC (Morrison et al., 2002, 2003; Cater et al., 2006, 2007). OHSC exposed to the sham injury maintained healthy morphology and experienced minimal cell death over the same time-course (Figure 4C).
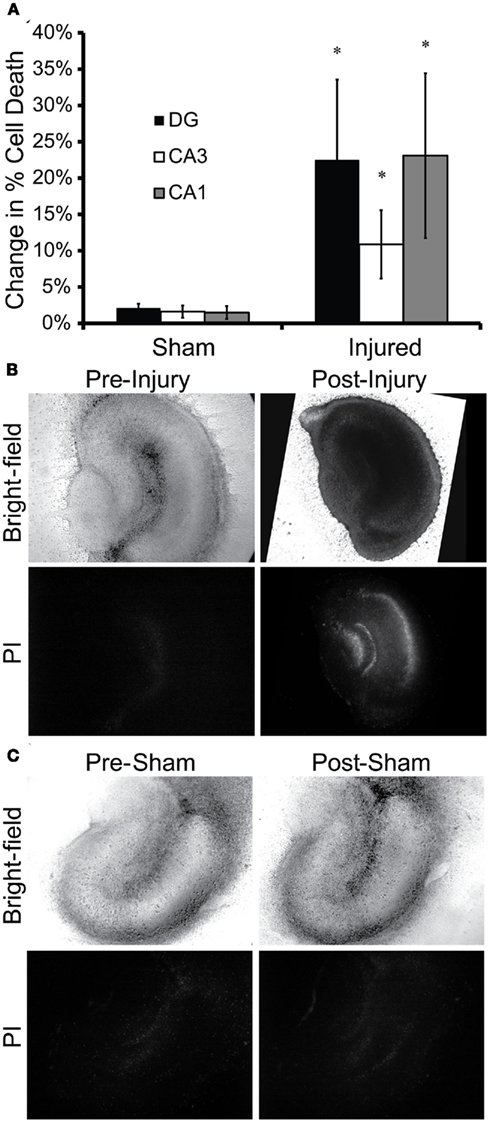
Figure 4. In vitro blast injury of OHSC. OHSC were exposed to 530 ± 17.7 kPa peak incident overpressure, 1.026 ms duration ± 0.017 ms, and 190 ± 10.7 kPa-ms impulse in-air. (A) Injured OHSC experienced increased cell death 4 days following blast. Cell death increased significantly in all regions of injured OHSC as compared to sham-exposed OHSC (*p < 0.05; SEM; Sham n = 21; Injured n = 13). (B) Bright-field and fluorescent images of injured tissue revealed significant PI staining 4 days following injury. (C) Sham-exposed cultures maintained normal morphology with minimal cell death over the 4-day period following injury.
Exposure of bEnd.3 cultures to 581 ± 10.0 kPa peak incident overpressure, 1.067 ± 0.006 ms duration, and 222 ± 6.9 kPa-ms impulse in-air significantly decreased TEER to 75 ± 7% of pre-exposure levels immediately following blast (Figure 5A). Bright-field images revealed healthy cell morphology consistent across all time points for both injured and sham-exposed cultures. Minimal cell death was observed in blast-exposed cultures (Figure 5C) as compared to sham-exposed cultures (Figure 5D) at 2 and 8 h following the injury time point, which served as a qualitative indicator of endothelial monolayer health. The number of dead cells per millimeter squared was not significantly higher in injured cultures as compared to sham-exposed cultures at the pre-injury (0.7 ± 0.6 injured vs. 0.2 ± 0.1 sham), 2 h post-injury (15.9 ± 7.6 injured vs. 11.4 ± 8.8 sham), and 8 h post-injury (3.3 ± 1.2 injured vs. 2.7 ± 1.4 sham) time points tested (Figure 5B). The increase of cell death in all cultures at the 2-h time point was likely due to medium changes and physical manipulations required to transfer cultures into the receiver.
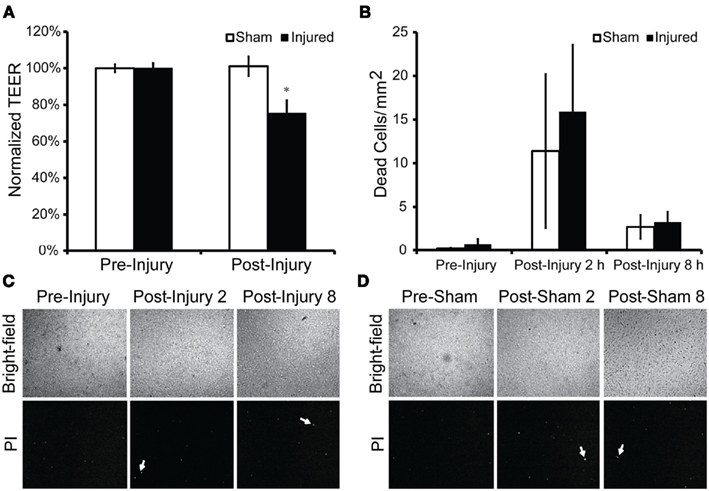
Figure 5. Trans-endothelial electrical resistance (TEER) and bEnd.3 monolayer viability following blast injury. Cultures were exposed to 581 ± 10.0 kPa peak overpressure with a 1.067 ± 0.006-ms duration and 222 ± 6.9 kPa-ms impulse in-air. (A) An acute decrease in TEER was observed for the injured cultures immediately following injury to 75 ± 7% of pre-exposure levels (*p < 0.05; SEM; Sham n = 6; Injured n = 6). Bright-field and PI fluorescence images were taken immediately prior to, 2 and 8 h following injury. (B) The number of dead cells in injured cultures as compared to controls was not significantly higher at the pre-injury, 2 h post-injury, and 8 h post-injury time points tested. (C) Blast exposure resulted in no change in morphology and only minimal cell death (white arrows) at 2 and 8 h post-exposure. (D) Sham exposure resulted in no change in morphology or PI staining, indicating a lack of cell death.
Discussion
Tissue-level injury criteria for the brain from blast loading have yet to be published. In vitro models of the brain have proven to be highly predicative of the brain’s response to injury in vivo and additionally allow for precise control and characterization of injury biomechanics (Morrison et al., 2011). The choice of the blast injury model and its characterization are critical for reproducing operationally relevant loading histories. With a realistic injury model, understanding the energy transfer to the tissue and the resultant biological response can begin. The in vitro blast injury methodology described here benefits from twofold utility to this end: (1) an easily modifiable receiver to accommodate various in vitro biological models and (2) a high degree of characterization for correlating primary blast exposure to biological outcomes. Development of an in vitro injury risk-function coupled with a strong understanding of damage mechanisms will supplement in vivo studies to facilitate an understanding of the intrinsic and extrinsic signals essential in the overall, neurological outcome following blast injury.
Organotypic hippocampal slice culture were chosen for this study because they have proven to be biofidelic in recapitulating the progressive neurodegenerative cascades and delayed cell death observed in animal models of neurodegeneration (Morrison et al., 1998, 2002, 2011; Sundstrom et al., 2005; Cater et al., 2006, 2007). After blast exposure, cell death in OHSC was largely isolated to the principal cell layers in all regions of the hippocampus (i.e., pyramidal and granule cells), similar to cell death patterns observed in in vitro models of inertial injuries (i.e., stretch or shear). Although the biomechanics of blast (low strain, high strain rate) and inertial injuries (large strain, relatively low strain rates) are fundamentally different, the similar pattern observed suggests that similar cell types are vulnerable to both loading conditions (Morrison et al., 1998, 2002, 2003, 2006; Cater et al., 2006, 2007). Future studies will determine whether the pathobiology initiated by both injuries differs. Studies in the rat have observed dose-dependent induction of axonal damage, activation of apoptotic transcription factors, and cell death in the hippocampus after exposure to 154 and 240 kPa incident overpressure with associated 1.7 and 2 ms durations, respectively (Kaur et al., 1995; Saljo et al., 2002a,b). These changes occurred between 2 h and 21 days following injury, suggesting that blast initiates an extended pathobiology that results in progressive neurodegenerative changes (Saljo et al., 2003). One limitation of these findings reported by Kaur et al. (1995) was that the overpressure history was exceedingly complex, being formed from an explosive charge detonated within a closed bunker. Saljo et al. (2002a,b) utilized the same blast parameters under more controlled conditions, using an explosive-driven blast tube; however, a limitation of their studies was that the thorax was not protected during exposure such that the response could have been due to pulmonary effects of blast. A primary conclusion from our study was that principal cells of the hippocampus were vulnerable to primary blast injury without a complex overpressure history. One limitation of our study is that we did not investigate the time-course of this cell death response; therefore we were unable to report whether cell death was acute or delayed. Future studies are necessary to explore the induced pathobiology in more detail, specifically to determine OHSC tolerance and mechanisms of cell death. However, our results do establish the feasibility of inducing hippocampal cell death in response to a pressure transient that mimics operationally relevant primary blast loading.
Previous studies have reported an acute increase in BBB permeability as observed by IgG immunoreactivity in rat brain at 3 and 24 h following exposure to a shock wave of 120 kPa (duration not reported) or 240 kPa peak overpressure with 4 ms duration (Readnower et al., 2010; Garman et al., 2011). This acute disruption of the barrier was consistent with our findings that TEER of endothelial monolayers – an indicator of BBB integrity – decreased immediately after exposure to overpressure. In vivo, BBB damage was reported to be more severe in the cerebral cortex and underlying striatum contralateral to the direction of the impinging shock wave (Garman et al., 2011). The authors speculated that diffraction of the shock wave around the skull produced a localized region of imploding shock amplification on the contralateral side, suggesting that their reported pressures may have been underestimates of the in situ pressures required to induce BBB damage. One limitation of our data presented for disruption of endothelial monolayer integrity is the lack of a TEER time-course recorded following exposure to blast overpressure. Previous studies have reported restoration of the compromised BBB 72 h following blast, evidenced by the return of IgG immunoreactivity to control levels (Readnower et al., 2010; Garman et al., 2011). In addition, we found that cell death of blast-exposed cultures was not significantly higher than that of sham-exposed cultures at each time point tested, further ruling out the possibility of cell death as the underlying cause for acute changes in TEER following injury. The slightly elevated number of dead cells in both sham-exposed and injured groups at 2 h post-injury may have been due to sample handling involved with sham and blast injury. Less propidium iodide staining at the 8-h time point following injury is thought to be the result of detachment of dead cells during the staining process. Together, these data suggest that BBB disruption could be a transient phenomenon not caused solely by cell death, with a time-course and mechanisms of repair that warrant further investigation.
Previously published in vitro models of bTBI have reported the incident overpressure of the blast wave but not the loading conditions at the tissue-level (Leung et al., 2008; Arun et al., 2011; Connell et al., 2011). Without a tissue-level biomechanical context, it is difficult to make quantitative comparisons between biological outcomes from different studies given the potential for vastly different loading conditions. In a high-throughput model of blast, rodent- and human-derived neuroblastoma cell lines were injured by exposure to a single overpressure of 145 kPa; however, the study reported the paradoxical finding of reduced injury after multiple exposures (Arun et al., 2011). For this study, cell cultures in 96-well plates were exposed to blast inside the shock tube. The injury biomechanics at the sample-level were not reported. In a different in vitro blast injury model, the excised spinal cord was subjected to strains as high as 60% with a jet of gas produced by a blast tube, which did not accurately reproduce the low strain, high strain rate biomechanics of blast (Connell et al., 2011). In each of these previous studies, overpressures were applied directly to the culture preparations, so the loading conditions were not representative of internal physiological loading conditions. Our studies benefited from a novel fluid-filled receiver, which propagated a fast-rising pressure wave through the tissue to reproduce in situ intracranial biomechanics (Panzer et al., 2012). In addition, the receiver allowed for measurement of the injury parameters that directly interact with the tissue.
Combining tissue culture models with blast modeling held additional challenges related to the maintenance of important culture conditions during injury. Many of these challenges were surmounted by encasing the cultures within sterile, media-filled bags. Critically, we showed that the bag, sample holder, and culture wells did not alter the passage of the pressure wave. Isolating the cultures from the fluid in the receiver prevented infections, which was absolutely required for analysis of cell death over 4 days. To maintain physiologic pH during injury, culture medium was equilibrated with 5% CO2/95% O2, which was critical since pH changes can induce excitotoxicity of pyramidal neurons (Pringle et al., 2000) and interneurons (Wang and Xu, 2011; Zhao et al., 2011). The fluid in the receiver was maintained at 37°C to prevent hypothermia, which is highly neuroprotective in multiple models of neurodegeneration (Dietrich et al., 1994; Tymianski et al., 1998; Lawrence et al., 2005). The sample bags also reduced bulk fluid flow in the immediate vicinity of the cultures, thereby reducing unwanted tissue deformation after passage of the pressure wave.
Development and implementation of a simplified model of blast injury for the definition of blast tolerance criteria is only a first step toward an understanding of the acute and long-term pathobiology of bTBI. However, there were some limitations associated with this study. Blast exposure was simulated as a Friedlander wave without the complexity caused by reflections that occur operationally. Currently our device is not configured to reproduce more complex blasts but could be modified to do so (Panzer et al., 2012). Preparation of OHSC was a lengthy and technically challenging culture process with a culture period of 2 weeks prior to injury to ensure tissue health and maturation (Morrison et al., 2003). bEnd.3 cells were chosen to model the BBB; however, bEnd.3 cells are a cell line, and therefore may respond differently to blast as compared to primary endothelial cells. Although the shock tube requires minimal set-up, the preparation of the in vitro biological samples to prevent infection and control physiologic parameters during exposure decreased throughput.
In vitro models of the brain parenchyma (OHSC) and BBB were exposed to simulated blast loading with our unique system consisting of a shock tube and a specialized receiver, resulting in cell death in OHSC and disruption of tight junction integrity in our BBB cultures. Our in vitro blast model benefited from the ability to measure the loading conditions at the level of the cell or tissue sample. Future studies will utilize our well-characterized blast injury model and methods along with biomarkers previously shown to be valuable for bTBI to correlate injury biomechanics to biological responses.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors gratefully acknowledge funding support from the Army Research Office under the Multidisciplinary University Research Initiative (W911MF-10-1-0526; University of Pennsylvania as prime institution).
References
Arun, P., Spadaro, J., John, J., Gharavi, R. B., Bentley, T. B., and Nambiar, M. P. (2011). Studies on blast traumatic brain injury using in-vitro model with shock tube. Neuroreport 22, 379–384.
Bass, C., Rafaels, K. A., and Salzar, R. S. (2008). Pulmonary injury risk assessment for short-duration blasts. J. Trauma 65, 604–615.
Cassen, B., Curtis, L., and Kistler, K. (1950). Initial studies of the effect of laboratory produced air blast on animals. J. Aviat. Med. 21, 38–47.
Cater, H. L., Gitterman, D., Davis, S. M., Benham, C. D., Morrison, B. III, and Sundstrom, L. E. (2007). Stretch-induced injury in organotypic hippocampal slice cultures reproduces in vivo post-traumatic neurodegeneration: role of glutamate receptors and voltage-dependent calcium channels. J. Neurochem. 101, 434–447.
Cater, H. L., Sundstrom, L. E., and Morrison, B. III (2006). Temporal development of hippocampal cell death is dependent on tissue strain but not strain rate. J. Biomech. 39, 2810–2818.
Celander, H., Clemedson, C. J., Ericsson, U. A., and Hultman, H. I. (1955). The use of a compressed air operated shock tube for physiological blast research. Acta Physiol. Scand. 33, 6–13.
Cernak, I., Wang, Z., Jiang, J., Bian, X., and Savic, J. (2001a). Cognitive deficits following blast injury- induced neurotrauma: possible involvement of nitric oxide. Brain Inj. 15, 593–612.
Cernak, I., Wang, Z., Jiang, J., Bian, X., and Savic, J. (2001b). Ultrastructural and functional characteristics of blast injury-induced neurotrauma. J. Trauma 50, 695–706.
Chavko, M., Koller, W. A., Prusaczyk, W. K., and Mccarron, R. M. (2007). Measurement of blast wave by a miniature fiber optic pressure transducer in the rat brain. J. Neurosci. Methods 159, 277–281.
Chavko, M., Watanabe, T., Adeeb, S., Lankasky, J., and Ahlers, S. T. (2011). Relationship between orientation to a blast and pressure wave propagation inside the rat brain. J. Neurosci. Methods 195, 61–66.
Chen, Y. C., Smith, D. H., and Meaney, D. F. (2009). In-vitro approached for studying blast-induced traumatic brain injury. J. Neurotrauma 26, 861–876.
Clemedson, C. J., and Pettersson, H. (1956). Propagation of a high explosive air shock wave through different parts of an animal body. Am. J. Physiol. 184, 119–126.
Connell, S., Gao, J., Chen, J., and Shi, R. (2011). Novel model to investigate blast injury in the central nervous system. J. Neurotrauma 28, 1229–1236.
Defense and Veterans Brain Injury Center. (2012). TBI Numbers by Severity – All Armed Forces. Available at: http://www.dvbic.org/TBINumbers.aspx
Dietrich, W. D., Alonso, O., Busto, R., Globus, M. Y. T., and Ginsberg, M. D. (1994). Post-traumatic brain hypothermia reduces histopathological damage following concussive brain injury in the rat. Acta Neuropathol. 87, 250–258.
Dodd, K. T., Mundie, T. G., Lagutchik, M. S., and Morrison, J. R. (1997). Cardiopulmonary effects of high-impulse noise exposure. J. Trauma 43, 656–666.
Elkin, B. S., and Morrison, B. III. (2007). Region-specific tolerance criteria for the living brain. Stapp Car Crash J. 51, 127–138.
Garman, R., Jenkins, L. W., Bauman, R., Swauger, P., Parks, S., Switzer, R., Dixon, C. E., Clark, R. S. B., Bayir, H., Kagan, V., Jackson, E. K., and Kochanek, P. M. (2009). Blast overpressure injury in rats with body protection produces acute and sub-acute axonal, dendritic and synaptic neuropathology. J. Neurotrauma 26, A-53.
Garman, R., Jenkins, L. W., Switzer, R. C., Bauman, R. A., Tong, L. C., Swauger, P., Parks, S., Ritzel, D. V., Dixon, C. E., Clark, R. S. B., Bayir, H., Kagan, V., Jackson, E. K., and Kochanek, P. M. (2011). Blast exposure in rats with body shielding is characterized primarily by diffuse axonal injury. J. Neurotrauma 28, 947–959.
Gorbunov, N. V., Mcfaul, S. J., Januskiewicz, A., and Atkins, J. L. (2005). Pro-inflammatory alterations and status of blood plasma iron in a model of blast-induced lung trauma. Int. J. Immunopathol. Pharmacol. 18, 547–556.
Kaur, C., Singh, J., Lim, M. K., Ng, B. L., and Ling, E. A. (1997). Macrophages/microglia as ‘sensors’ of injury in the pineal glad of rats following a non-penetrative blast. Neurosci. Res. 27, 317–322.
Kaur, C., Singh, J., Lim, M. K., Ng, B. L., Yap, E. P. H., and Ling, E. A. (1995). The response of neurons and microglia to blast injury in the rat brain. Neuropathol. Appl. Neurobiol. 21, 369–377.
Lawrence, E. J., Dentcheva, E., Curtis, K. M., Roberts, V. L., Siman, R., and Neumar, R. W. (2005). Neuroprotection with delayed initiation of prolonged hypothermia after in vitro transient global brain ischemia. Resuscitation 64, 383–388.
Leung, L., Vandevord, P. J., Dal Cengio, A. L., Bir, C., Yang, K. H., and King, A. I. (2008). Blast related neurotrauma: a review of cellular injury. Mol. Cell Biomech. 5, 155–168.
Livingston, I. S., and O’hanlon, M. E. (2011). Afghanistan Index Tracking Variables of Reconstruction & Security in Post-9/11 Afghanistan. Brookings, Washington, D.C.
Long, J. B., Bentley, T. L., Wessner, K. A., Cerone, C., Sweeney, S., and Bauman, R. A. (2009). Blast overpressure in rats: recreating a battlefield injury in the laboratory. J. Neurotrauma 26, 827–840.
Moochhala, S. M., Md, S., Lu, J., Teng, C. H., and Greengrass, C. (2004). Neuroprotective role of aminoguanidine in behavioral changes after blast injury. J. Trauma 56, 393–403.
Morrison, B. III, Cater, H. L., Benham, C. D., and Sundstrom, L. E. (2006). An in vitro model of traumatic brain injury utilising two-dimensional stretch of organotypic hippocampal slice cultures. J. Neurosci. Methods 150, 192–201.
Morrison, B. III, Cater, H. L., Wang, C. C., Thomas, F. C., Hung, C. T., Ateshian, G. A., and Sundstrom, L. E. (2003). A tissue level tolerance criterion for living brain developed with an in vitro model of mechanical loading. Stapp Car Crash J. 47, 93–105.
Morrison, B. III, Elkin, B. S., Dolle, J. P., and Yarmush, M. L. (2011). In vitro models of traumatic brain injury. Annu. Rev. Biomed. Eng. 13, 91–126.
Morrison, B. III, Pringle, A. K., Mcmanus, T., Ellard, J., Bradley, M., Signorelli, F., Ianotti, F., and Sundstrom, L. E. (2002). L-arginyl-3,4-spermidine is neuroprotective in several in vitro models of neurodegeneration and in vivo ischaemia without suppressing synaptic transmission. Br. J. Pharmacol. 137, 1255–1268.
Morrison, B. III, Saatman, K. E., Meaney, D. F., and Mcintosh, T. K. (1998). In vitro central nervous system models of mechanically induced trauma: a review. J. Neurotrauma 15, 911–928.
Muller, M., and Somjen, G. G. (1999). Intrinsic optical signals in rat hippocampal slices during hypoxia-induced spreading depression-like depolarization. J. Neurophysiol. 82, 1818–1831.
O’Hanlon, M. E., and Livingston, I. S. (2011). Iraq Index Tracking Variables of Reconstruction & security in Post-Saddam Iraq. Brookings, Washington, D.C.
Owens, B. D., Kragh, J. F., Wenke, J. C., Macaitis, J., Wade, C. E., and Holcomb, J. B. (2008). Combat wounds in operation Iraqi freedom and operation enduring freedom. J. Trauma 64, 259–299.
Panzer, M. B., Bass, C. R., Rafaels, K. A., Shridharani, J., and Capehart, B. P. (2011). Primary blast survival and injury risk assessment for repeated blast exposure. J. Trauma 28, doi: 10.1097/TA.0b013e31821e8270
Panzer, M. B., Matthews, K. A., Yu, A. W., Morrison, III, B., Meaney, D. F and Bass, C. R. (2012) A multiscale approach to blast neurotrauma modeling: part I – development of novel test devices for in vivo and in vitro blast injury models. Front. Neur. 3:46. doi: 10.3389/fneur.2012.00046
Pringle, A. K., Self, J., Eshak, M., and Iannotti, F. (2000). Reducing conditions significantly attenuate the neuroprotective efficacy of competitive, but not other NMDA receptor antagonists in vitro. Eur. J. Neurosci. 12, 3833–3842.
Rafaels, K., Bass, C. R., Salazar, R. S., Panzer, M. B., Woods, W., Feldman, S., Cummings, T., and Capehart, B. (2011). Survival risk assesssment for primary blast exposure to the head. J. Neurotrauma 28, 2319–2328.
Rafaels, K. A., Bass, C. R., Panzer, M. B., and Salzar, R. S. (2010). Pulmonary injury risk assessment for long-duration blasts: a meta-analysis. J. Trauma 69, 368–374.
Readnower, R. D., Chavko, M., Adeeb, S., Conroy, M. D., Pauly, J. R., Mccarron, R. M., and Sullivan, P. G. (2010). Increase in blood-brain barrier permeability, oxidative stress, and activated microglia in a rat model of blast-induced traumatic brain injury. J. Neurosci. Res. 88, 3530–3539.
Richmond, D. R., Clare, V. R., Goldizen, V. C., Pratt, D. E., Sanchez, R. T., and White, C. S. (1961). Biological effect of overpressure. II. A shock tube utilized to produce sharp-rising overpressures of 400 milliseconds duration and its employment in biomedical experiments. Aerosp. Med. 32, 997–1008.
Risling, M., Suneson, A., Skold, M., Lai, L. J., Abo, M., Chen, Z., and Bjelke, B. (2002). Evaluation of Diffuse Brain Injury with Magnetic Resonance Imaging: Technical Report for the Swedish Defence Research Agency. Stockholm, Sweden: Defence Medicine, Experimental Traumatology, Swedish Defence Research Agency. Report No.: FOI-R–0756–SE.
Romba, J. J., Martin, P., and Dorsey, W. (1961). The Propagation of Air Shock Waves on a Biophysical Model. Aberdeen Proving Ground, MD: Human Engineering Laboratory.
Saljo, A., Arrhen, F., Bolouri, H., Mayorga, M., and Hamberger, A. (2008). Neuropathology and pressure in the pig brain resulting from low-impulse noise exposure. J. Neurotrauma 25, 1397–1406.
Saljo, A., Bao, F., Jingshan, S., Hamberger, A., Hansson, H. A., and Haglid, K. G. (2002a). Exposure to short- lasting impulse noise causes neuronal c-Jun expression and induction of apoptosis in the adult rat brain. J. Neurotrauma 19, 985–991.
Saljo, A., Bao, F., Shi, J., Hamberger, A., Hansson, H. A., and Haglid, K. G. (2002b). Expression of c-Fos and c-Myc and deposition of β-APP in neurons in the adult rat brain as a result of exposure to short-lasting impulse noise. J. Neurotrauma 19, 379–385.
Saljo, A., Huang, Y. L., and Hansson, H. A. (2003). Impulse noise transiently increased the permeability of nerve and glial cell membranes, an effect accentuated by a recent brain injury. J. Neurotrauma 20, 787–794.
Simon, M. J., Kang, W. H., Gao, S., Banta, S., and Morrison, B. III. (2010). Increased delivery of TAT across an endothelial monolayer following ischemic injury. Neurosci. Lett. 486, 1–4.
Simon, M. J., Kang, W. H., Gao, S., Banta, S., and Morrison, B. III. (2011). TAT is not capable of transcellular delivery across an intact endothelial monolayer in vitro. Ann. Biomed. Eng. 39, 394–401.
Sundstrom, L., Morrison, B. III, Bradley, M., and Pringle, A. (2005). Organotypic cultures as tools for functional screening in the CNS. Drug Discov. Today 10, 993–1000.
Tanielian, T. L., and Jaycox, L. H. (2008). Invisible Wounds of War: Psychological and Cognitive Injuries, their Consequences, and Services to Assist Recovery. Santa Monica, CA: RAND Corporation.
Tymianski, M., Sattler, R., Zabramski, J. M., and Spetzler, R. F. (1998). Characterization of neuroprotection from excitotoxicity by moderate and profound hypothermia in cultured cortical neurons unmasks a temperature-insensitive component of glutamate neurotoxicity. J. Cereb. Blood Flow Metab. 18, 848–867.
Wang, Y. Z., and Xu, T. L. (2011). Acidosis, acid-sensing ion channels, and neuronal cell death. Mol. Neurobiol. 44, doi: 10.1007/s12035-011-8204-2
Keywords: blast injury, shock tube, organotypic slice culture, hippocampus, neuron, astrocyte, blood–brain barrier, endothelial cells
Citation: Effgen GB, Hue CD, Vogel E III, Panzer MB, Meaney DF, Bass CR and Morrison B III (2012) A multiscale approach to blast neurotrauma modeling: part II: methodology for inducing blast injury to in vitro models. Front. Neur. 3:23. doi: 10.3389/fneur.2012.00023
Received: 03 November 2011;
Accepted: 07 February 2012;
Published online: 24 February 2012.
Edited by:
Ibolja Cernak, Johns Hopkins University Applied Physics Lab, USAReviewed by:
Bogdan A. Stoica, University of Maryland School of Medicine, USAMichelle LaPlaca, Georgia Institute of Technology, USA
Copyright: © 2012 Effgen, Hue, Vogel, Panzer, Meaney, Bass and Morrison. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Barclay Morrison III, Department of Biomedical Engineering, Columbia University, 351 Engineering Terrace, MC 8904, 1210 Amsterdam Avenue, New York, NY 10027, USA. e-mail:Ym0yMTE5QGNvbHVtYmlhLmVkdQ==