 Asma Malik
Asma Malik Ghazala Hayat
Ghazala Hayat Junaid S. Kalia
Junaid S. Kalia Miguel A. Guzman
Miguel A. Guzman- 1Neurology, Saint Louis University, Saint Louis, MO, USA
- 2Department of Neurology and Neurotherapeutics, The University of Texas Southwestern, Dallas, TX, USA
- 3Department of Pathology, Saint Louis University, Saint Louis, MO, USA
Idiopathic inflammatory myopathies (IIM) are a group of chronic, autoimmune conditions affecting primarily the proximal muscles. The most common types are dermatomyositis (DM), polymyositis (PM), necrotizing autoimmune myopathy (NAM), and sporadic inclusion body myositis (sIBM). Patients typically present with sub-acute to chronic onset of proximal weakness manifested by difficulty with rising from a chair, climbing stairs, lifting objects, and combing hair. They are uniquely identified by their clinical presentation consisting of muscular and extramuscular manifestations. Laboratory investigations, including increased serum creatine kinase (CK) and myositis specific antibodies (MSA) may help in differentiating clinical phenotype and to confirm the diagnosis. However, muscle biopsy remains the gold standard for diagnosis. These disorders are potentially treatable with proper diagnosis and initiation of therapy. Goals of treatment are to eliminate inflammation, restore muscle performance, reduce morbidity, and improve quality of life. This review aims to provide a basic diagnostic approach to patients with suspected IIM, summarize current therapeutic strategies, and provide an insight into future prospective therapies.
Introduction
The Idiopathic inflammatory myopathies (IIM) are a heterogeneous group of rare systemic diseases that leads to muscle weakness, muscle enzyme elevations, inflammation on muscle biopsy, and extra muscular manifestations (1, 2).
The IIM are classified on the basis of patterns of presentation, age of onset, immunohistopathologic features, and response to treatment (3–6).The major types of IIM include: DM, PM, NAM, and sIBM. Within the last decade, NAM was made a separate subtype and was previously off-classed with PM. There is an increased risk of malignancy in specific subtypes of DM, PM, and NAM (7, 8).
DM, PM, and NAM are usually responsive to immunotherapies. sIBM is typically refractory to these agents. Since IIM are potentially treatable, proper diagnosis and early initiation of therapy are necessary (1, 2, 9–11). Consensus does not exist among experts regarding therapy and management (12).
Classifications
Over the course of time, several different criteria have emerged to classify the IIM. Bohan and Peter criteria are the earliest and still widely used (3) (Table 1).
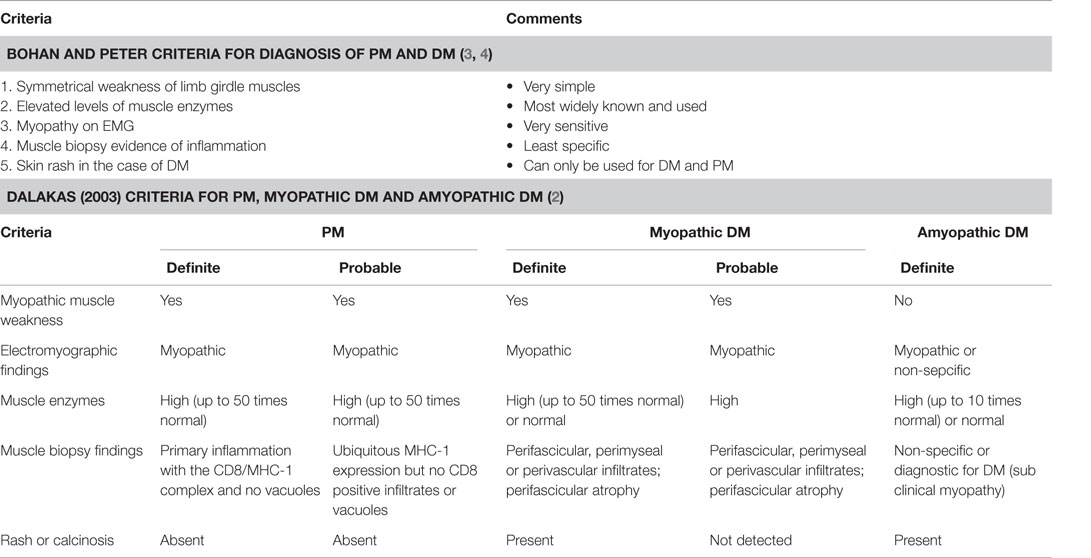
Table 1. Diagnostic criteria for IIM.
Dalakas proposed initial criteria in 1991 (1), and revised it in 2003 (2). It is very specific and sensitive and has been found to have best inter-rater reliability (13, 14). Experts have reclassified IIM based purely on their histopathological features (15).
Neither of the above mentioned criteria took into account sIBM. The Griggs criteria were proposed in 1995 and were implemented as a diagnostic guide for sIBM. These relied more on the histopathological features of the disease. Recent studies underscore the importance of the clinical features as the major determining factor in diagnosing sIBM (16). Subsequently, the European neuromuscular center (ENMC) at its 188th international workshop introduced a comprehensive set of measures that addressed both the clinical and pathophysiological manifestations of the disease (17) (Table 2). Since these criteria are fairly recent, long-term studies are required to assess their utility. A recent study proposed the ENMC criteria as being effective in diagnosing with a sensitivity of 84% (18).
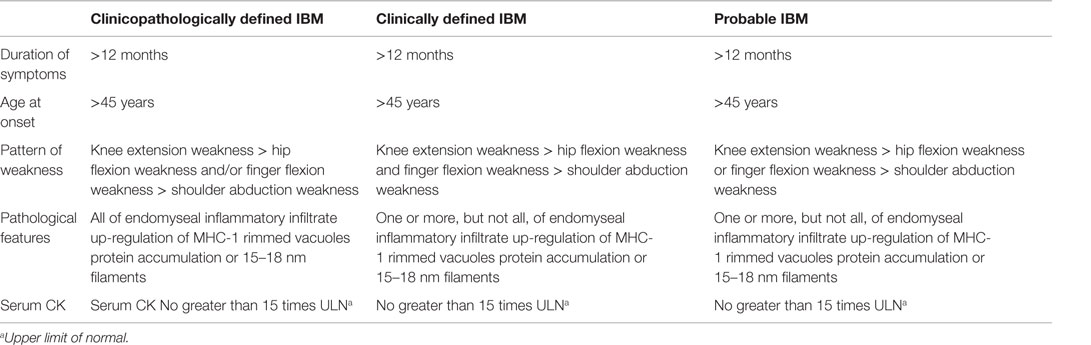
Table 2. Revised ENMC criteria (2011) for diagnosis of sIBM (17).
Idiopathic inflammatory myopathies are commonly a part of overlap myopathy (OM) and/or antisynthetase syndrome. OM are myositis associated with another defined collagen vascular disease, and a presence of “overlap autoantibodies” including MSA or myositis-associated autoantibodies (MAA). Antisynthetase syndrome is a constellation of a usually acute disease with antisynthetase antibodies, fever, interstitial lung disease (ILD) (~80%), mechanic’s hands (~70%), Raynaud’s phenomenon (60%), and polyarthritis (60%) sometimes with erosions. These patients may have clinical/pathological features of DM, PM, or NAM, but sometimes clinically evident myositis can be missing (14). Cancer-associated myositis is also considered in some literature as a separate entity that can be clinically or pathologically classified as DM, PM, or NAM. This accounts for almost half of all non-inclusion body IIM after age 65 years, but <10% in younger populations (2). A recent meta analysis involving patients with either PM or DM showed a strong association between PM/DM and malignancy (19).
Epidemiology
As IIM are rare, few epidemiologic studies have been published. There is a growing need for accurate and reliable epidemiological studies. Between 1947 and the 1990s, the reported annual incidence of IIM from studies using older diagnostic criteria ranged 0.4–1.0 cases per 100,000 (2, 20–24). Recent study in U.S. showed that the incidence and prevalence of DM is 1.4 and 5.8 cases per 100,000 persons, respectively; with female preponderance and a higher prevalence among older age group (25). PM age- and gender-adjusted incidence was 3.8 and the prevalence is 9.7 per 100,000 people. In the opinion of the authors and other experts, PM is over-diagnosed as not all studies were based on diagnostic muscle biopsies (25).
In a retrospective study, NAM represented 19% of the IIM, while DM and non-specific myositis accounted for 36 and 39%, respectively. This study excluded sIBM (26). A Mayo Clinic study showed PM as the most common clinical phenotype (27). The incidence of DM and PM increased with advancing age and reached a peak at age 50–59 years (28). However, sIBM is still considered to be the most frequent acquired myopathy after 50 years of age. The prevalence of sIBM in Australia is 9.3 per million people in the general population and 51.3 in people over 50 years, with a male preponderance (29, 30).
Clinical Features
Dermatomyositis
Dermatomyositis (DM) typically presents as an acute or insidiously progressive proximal weakness that is accompanied or preceded by a characteristic skin rash (31–33). Patients complain of difficulty getting up from a chair, climbing stairs, lifting things, and combing hair. It is usually painless, but pain can be a significant feature with acute disease and subcutaneous calcifications. Some patients develop dyspnea related to ILD or ventilatory muscle weakness, dysphagia due to esophageal or pharyngeal involvement, congestive heart failure or arrhythmia from myocarditis, and gastrointestinal bleeding due to vasculopathy of the gut.
The typical skin rashes include: erythematous, photosensitive rash on the neck, back, and shoulders (shawl sign) (Figure 1); Malar and facial erythema along with purplish discoloration of eyelids (heliotrope rash) that is often associated with periorbital edema (Figure 2); and erythematous lichenoid papular scaly rash over the knuckles (Gottron’s papules) (Figure 3). Less commonly, rash may affect the anterior chest (V-sign) and the volar aspect of hands (inverse Gottron’s papules). Other skin manifestations include dilated capillary loops at the nail beds with periungual telangiectasias (Figure 4) and thickened, cracked skin on the dorsal and ventral surfaces of the hands (mechanic’s hands) in which case it is more often than not associated with the “antisynthetase syndrome.”
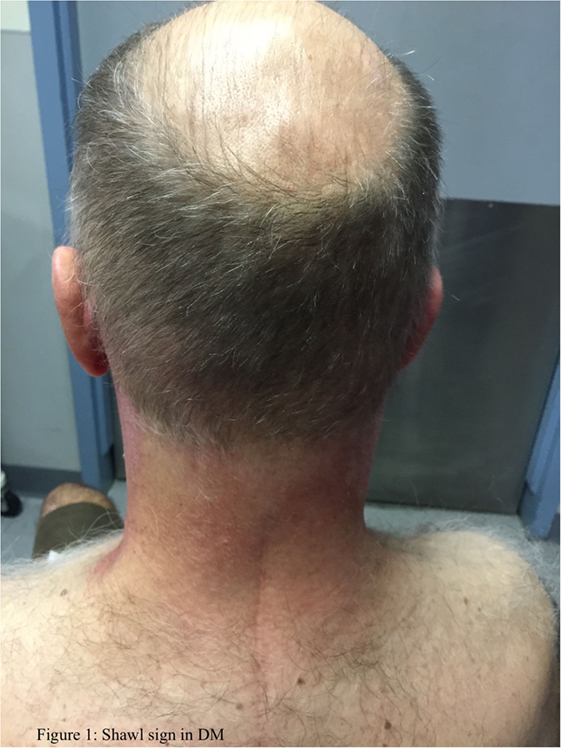
Figure 1. Shawl sign in DM.
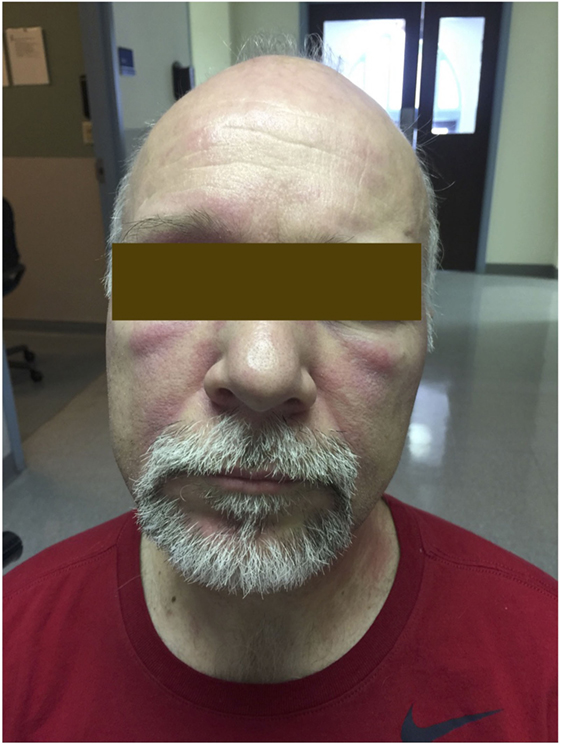
Figure 2. Heliotrope rash of dermatomyositis.
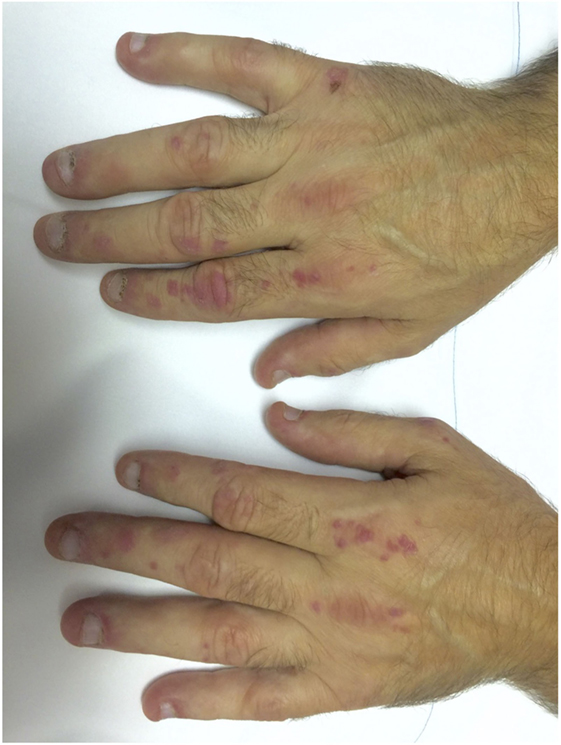
Figure 3. Gottron’s papules in a case of dermatomyositis.
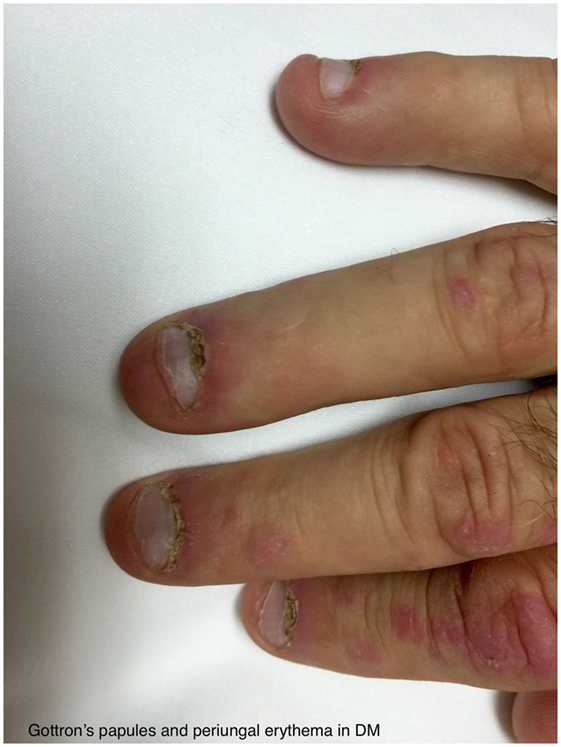
Figure 4. Gottron’s papules and periungal erythema in DM.
Dermatomyositis may present by itself or be a part of other syndromes, e.g., antisynthetase syndrome and overlap syndromes.
Antisynthetase syndrome is the constellation of Raynaud’s phenomenon, arthritis, and ILD. It presents with mechanic’s hands (as mentioned above). It is characterized by the presence of antibodies to aminoacyl transfer ribonucleic acid (RNA) synthetases (34).
Overlap syndrome is an entity that satisfies criteria of at least two connective tissue diseases most notably systemic sclerosis, PM/DM, Sjogrens syndrome, and SLE. Some retrospective studies have showed presence and prevalence of IIM in combination with other autoimmune diseases (35, 36).
Amyopathic DM presents with cutaneous manifestations without the muscle involvement (37), while adermatopathic DM presents with isolated myositis and has pathological features of DM on muscle biopsy. Juvenile dermatomyositis (JDM) affects children younger than 18 years of age; commonly presents after a febrile episode and skin rash. Multisystem involvement is common in JDM and is associated with calcinosis cutis and vasculopathy affecting the gastrointestinal tract (38, 39). The presence of calcinosis cutis suggests active disease in JDM and may be associated with delay to diagnosis and treatment (40). Classically, calcinosis is found at the subcutaneous level, but it may be seen intramuscularly.
Polymyositis
Polymyositis (PM) is a rare entity and an exclusionary diagnosis. It presents with muscular and extra muscular organ involvement similar to DM, without a rash (6, 41, 42).
It usually manifests in adults, more commonly in women, over the age of 20 years (2, 3, 32). Unlike DM, PM is usually not seen in childhood. It presents typically with progressive neck flexor and symmetric proximal limb muscle weakness, which develops over weeks to months. Myalgias and tenderness are common complaints. Dysphagia occurs in one-third of patients. The most common extra muscular involvement is ILD and myocarditis.
Necrotizing Autoimmune Myopathy
Necrotizing autoimmune myopathy (NAM) presents in adults with a sub acute, progressive proximal muscle weakness without a rash. Weakness generally develops more rapidly than PM, and is markedly severe (26). There may be associated myalgias and dysphagia. CK is usually higher than seen with other IIM. NAM is thought to be immune mediated with a trigger such as drugs (43–46). NAM has several variants including paraneoplastic-necrotizing myopathy, which is a severe and rapidly progressive disease that affects adults over the age of 40. Necrotizing myopathy with pipestem capillaries affects a similar age group and is associated with sub-acute weakness, brain infarction due to vasculitis, or connective tissue disease. Signal recognition particle (SRP) autoantibodies affect younger NAM patients, women more than men. It results in fulminant weakness and congestive heart failure. Statin-induced autoimmune NAM (SANAM) affects individuals between 46 and 89 years of age. The onset may be delayed up to 10 years following statin initiation and may occur several months after statin discontinuation (24, 45).
Sporadic Inclusion Body Myositis
Sporadic inclusion body myositis (sIBM) presents in-patients over the age of 40 years, with a male to female ratio of 3:1 (47, 48). It presents in an insidious fashion with progression over several years. Unlike other IIM, it is unique in that it involves both the proximal and distal musculature in a symmetric or an asymmetric fashion. The weakness starts in flexor forearm muscles in two-thirds of patients along with significant atrophy, particularly the deep finger flexors (Figure 5). The quadriceps and anterior tibial muscles are also affected early in IBM leading to tripping and falling. Dysphagia is very common in sIBM and may be the presenting feature (1, 49). In contrast to PM and DM, mild facial weakness is common (10). The Griggs criteria (50) address several clinical, laboratory, and histopathological features: duration of illness longer than 6 months; age at onset older than 30 years; weakness of proximal and distal muscles of the upper and lower extremities and either finger flexor weakness, wrist flexor greater than wrist extensor weakness, or quadriceps weakness; serum CK level less than 12 times normal; and muscle biopsy with evidence of invasion of non-necrotic fibers by mononuclear cells, vacuolated muscle fibers, and intracellular amyloid deposits or 15- to 18-nm tubulofilaments on muscle biopsy.

Figure 5. Wasting of the small muscles of the hands and forearm in sIBM.
Diagnostic Evaluations
Laboratory Studies
Serum Creatine Kinase
Serum Creatine Kinase (CK) level is the most sensitive measure but does not correlate with the severity of the symptoms; it might improve with treatment. In DM, 70–80% will have up to 50-fold levels, while 20% of DM patients will have normal CK levels (51). These patients might rarely have isolated elevated aldolase levels. In PM the elevated levels range 5 to 50-fold above normal. CK levels in NAM can be extremely high and reaching 100-fold. In sIBM, CK levels can be normal or only mildly elevated, less than 10 times the upper limits of normal.
Other Muscle Enzymes
Other muscle enzymes include lactate dehydrogenase (LDH), aspartate aminotransferase (AST), alanine aminotransferase (ALT), and aldolase levels. They are less sensitive, and may not be elevated. Aldolase can be selectively high in myositis with perimysial pathology (15).
Erythrocyte Sedimentation Rate
Erythrocyte sedimentation rate (ESR) is not a reliable indicator as it is usually normal or only mildly elevated. The same holds true for C reactive protein (CRP).
Connective Tissue Disease Autoantibodies
The presence of these antibodies suggests that the myopathy may be secondary to a connective tissue disease (overlap syndrome). However, it does not necessarily establish a connective tissue disease diagnosis. Antinuclear antibodies (ANA) are detected in 24–60% of DM, 16–40% of PM, and in as many as 20% of patients with IBM (31, 47, 51, 52). Anti-Ro(SSA) and Anti-La(SSB), anti-Smith, anti-RNP, anti-Scl70, and anti-centromere antibodies should be also checked.
Myositis Specific Antibodies
Myositis specific antibodies have a controversial pathogenic role. They may occasionally define the clinical phenotype, and offer a prognosis for a subset of patients; most are predictors of poor treatment response (53–56).
The MSA include: (1) Cytoplasmic antibodies directed against Mi-2 and Mas antigens. (2) Antibodies targeting translational proteins such as tRNA synthetases, anti (SRP), transcriptional intermediary factor-1 gamma (TIF-1; anti-155/140 Ab), and the melanoma differentiation-associated gene-5 (MDA5; anti-CADM140 Ab).
Jo-1 autoantibody is the most common tRNA synthetase antibody (up to 20% of IIM). The other antisynthetases (PL-7, PL-12, EJ, KS, OJ, Ha, and Zo) occur in <5% of IIM (57, 58). They all lead to a similar phenotype with ILD, arthritis, Raynaud’s phenomenon, and mechanics hands (59).
Antibody to nuclear matrix protein NXP2 (or MJ antibody) is one of the most common MSA in JDM, but occur in <2% of adult DM cases with up to 50% having an associated malignancy.
The anti-155/140 autoantibodies target TIF-1 and are strongly associated with malignancy in adults (89% specificity) (60, 61). However, in JDM patients, they are associated with calcinosis rather than cancer (62).
The anti-MDA5 antibodies mostly described in Asians is associated with amyopathic DM and aggressive ILD (63).
Antibodies to Mi-2, a 240-kDa nuclear helicase, are found in 15–30% of DM patients and associated with a favorable prognosis (64, 65) and suggestions of environmental trigger in adult DM (66).
The anti-SRP antibodies are associated with NAM or maybe non-specifically positive (67). Patients present with acute and severe proximal weakness, dilated cardiomyopathy, ILD, and are often steroid resistant (68).
Anti 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMG-CoAR) antibodies (200/100 autoantibodies) have been described in patients with NAM and statin use (69).
Screening for Malignancy
Malignancies are associated with DM (25%), PM (10%), and have been noted with NAM. Most are diagnosed within 1 year, but can take up to 5 years (7, 70). Breast and ovarian cancers are common in women, whereas lung and prostate cancers predominate in men. Other malignancies include lymphoma, colon, pancreatic, and bladder cancer (71).
Data on cancer screening are limited, and there are no guidelines. The European federation of Neurological societies recommended that DM patients have computed tomography (CT) of the chest/abdomen, pelvic ultrasound and mammography in women, ultrasound of testes in men, and colonoscopy in men and women over 50. If primary screening is negative, repeat screening is recommended after 3–6 months; thereafter screening is recommended every 6 months for 4 years (72). Periodic tumor markers (prostate specific antigen PSA, CA-125, CA19-9, CEA, and AFP) can also be checked.
Electrophysiology
Nerve conduction study (NCV) should be done to rule out a neuropathic process. Low compound muscle action potentials (CMAP) amplitudes may not necessarily indicate a neuropathic process, as it may reflect muscle atrophy and fibrosis.
Electromyography (EMG) must be done on one side of the body, so that the muscle biopsy is done on the contralateral side. Almost always, it shows “irritable myopathic process” (73) characterized by:
(1) Increased insertional and spontaneous activity with fibrillation potentials, positive sharp waves. Muscle fibrosis in advanced cases may lead to reduced insertional activity. Worsening strength with no abnormal spontaneous activity suggests steroid induced myopathy. Pseudomyotonic discharges can be seen in NAM.
(2) Polyphasic motor unit action potentials (MUAPs) of short duration and low amplitude, with early recruitment patterns. In sIBM, there are long-duration, large-amplitude, and polyphasic MUAPs secondary to the chronicity of the disease as opposed to a neurogenic process (74).
Skeletal Muscle Imaging
Magnetic resonance imaging (MRI) with T1 weighted, T2 weighted, fat suppression, and short tau inversion recovery (STIR) sequences have become a diagnostic modality routinely used to confirm the diagnosis, identify a muscle site for biopsy, and monitor treatment response (75–77). It is commonly used to diagnose children with JDM as it can spare the invasive testing; EMG or muscle biopsy (78). It also may help to identify subclinical involvement of muscles, which may point to another disease such as a muscular dystrophy.
In early stages, T2-weighted images and STIR sequences show patchy or diffuse increased signal in proximal muscles suggesting edema. In DM, the connective tissue septa and muscle fascia may be involved. These signal changes correlate with the degree of muscle inflammation and disease activity. After few months, T1-weighted images may show muscle atrophy, fatty transformation, and chronic muscle damage. This usually selectively involves the hamstrings with relative sparing of the adductor and obturator muscles (79).
Histopathology and Pathogenesis
Muscle biopsy is the gold standard to make the diagnosis. It also helps to differentiate inflammatory myopathy from some muscular dystrophies that can mimic it clinically. To maximize the yield a moderately weak muscle should be chosen for biopsy. A mildly weak muscle biopsy may not be as sensitive, and a severely weak muscle may show fibrosis. MRI can help identify the affected muscles. Vastus medialis is commonly chosen; care should be taken to avoid needle EMG at the site to be biopsied.
Dermatomyositis and Juvenile Dermatomyositis
Dermatomyositis and JDM are humoral-mediated vasculopathies of the small vessels in muscle tissue. This may be caused by overexpression of type 1 interferons α/β (INF-1) by plasmacytoid dendritic cells (DCs) (80) as well as increased major histocompatibility-I (MHC-I) and immunoglobulin gene transcript (81). The DCs produce type 1 interferon in response to viral nucleic acid that binds to their toll-like receptors (TLR-7 and TLR-9) (82). The activated TLR leads to generation of cytokines and chemokines including TNF-α, IL-4, IL-6, IL-15, and IL-17. Cytokines lead to cell migration and mononuclear cell infiltration in muscle fibers. The cell infiltration consists of B cells and CD4+T cells in the perimysial and perivascular area, and plasmacytoid DCs in perifascicular areas (4–6, 83–85) (Figure 6). On immunohistochemical stain, aggregates of B lymphocytes positive for CD20 are found (Figure 7).
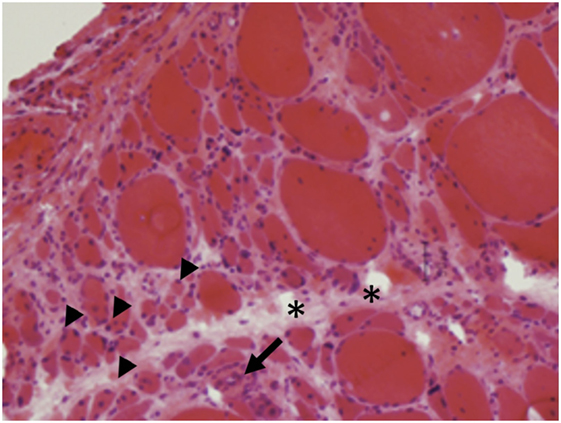
Figure 6. Perifascicular atrophy (arrowheads) with increased endomysial connective tissue (asterisks) and inflammatory infiltrates (arrows) are characteristics of dermatomyositis (A, H&E, 40×).
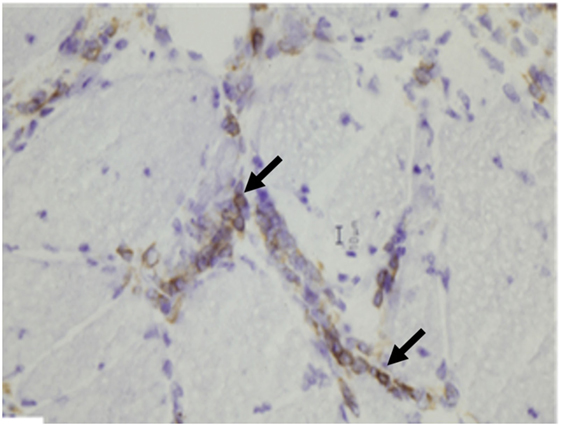
Figure 7. Aggregates of B lymphocytes (arrows) positive for CD20 immunohistochemical stain are found in dermatomyositis (B, CD20 immunostatin, 40×).
The earliest histological abnormality is the deposition of the C5b-9 membrane attack complex (MAC) around the microvasculature (86, 87). This will lead to abnormalities in both perimysial intermediate-sized vessels and endomysial capillaries within regions of perifascicular myofibers. Chronic immune vascular damage may cause ischemia, myofiber atrophy, and capillary damage in “watershed” regions (88). As a result, muscle biopsies demonstrate perifascicular atrophy, often without an inflammatory infiltrate. On electron microscopy (EM), the earliest recognized changes are tubuloreticular inclusions in the intramuscular arterioles and capillaries (89).
The typical histological findings on skin biopsy are vacuolar interface dermatitis with vacuolar changes of the epidermal basal layer, apoptosis, necrotic keratinocytes, and perivascular lymphocytic infiltrate and mucin deposition in the dermis (90, 91).
Polymyositis
Polymyositis is a cell-mediated cytotoxic immune response. CD8+ cytotoxic T cells and macrophages clonally expand and infiltrate the endomysium. They surround and invade non-necrotic muscle fiber cells expressing MHC class I, attack myocytes through the perforin pathway, causing muscle fiber necrosis and regeneration. (6, 92, 93) The microvasculature is not involved. The sarcoplasmic reticular pattern of internal MHC-1 reactivity is characteristic (94). There are abundant myeloid DCs that surround non-necrotic fibers and act as antigen presenting cells (APCs) (95). There is an increased immunoglobulin gene expression, with no deposition in the muscle blood vessels (96).
The main muscle biopsy features are fiber size variability, cellular invasion of non-necrotic muscle fibers expressing MHC-1 antigens, and scattered necrotic and regenerating fibers (Figures 8 and 9).
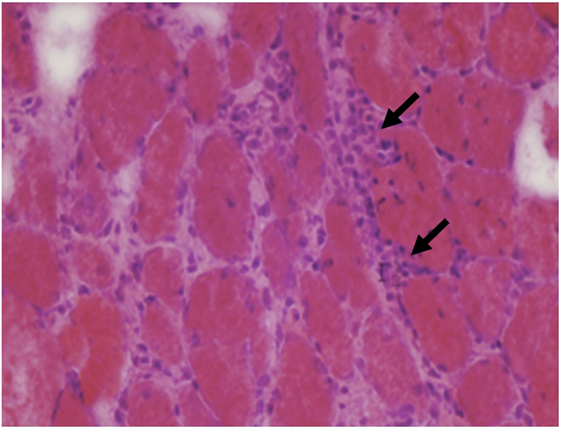
Figure 8. Muscle biopsy from a patient with polymyositis showing endomysial inflammatory cells (arrows) and variations of fiber size without a specific pattern (C, H&E 40×).
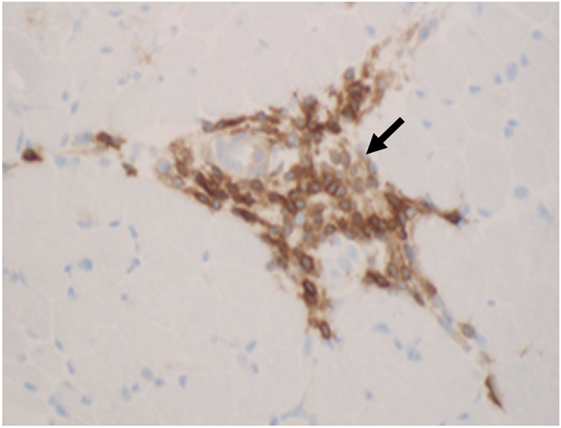
Figure 9. The inflammatory cells (arrows) in polymyositis are mostly T cells that are highlighted by CD3 immunohistochemistry (D, CD3 immunostatin, 40×).
Sporadic Inclusion Body Myositis
Sporadic inclusion body myositis has inflammatory and degenerative mechanisms. It remains controversial if the inflammatory mechanisms are cause or consequence of the degeneration.
The inflammatory process is similar to PM with an invasion of non-necrotic fibers by macrophages and cytotoxic CD8+ T cells. The degenerative process is characterized by rimmed vacuoles that can be highlighted on modified trichrome stain, and sometimes, ragged red fibers with mitochondrial excess, eosinophilic inclusions, and cytochrome oxidase negative fibers (50, 97, 98) (Figures 10 and 11). On EM, nuclear and cytoplasmic inclusions are detected. Macroautophagic processing has been attributed to the accumulation of aberrant proteins, such as congophilic intracellular β-amyloid deposits, presenilin 1, apolipoprotein E, γ-tubulin, α-synucline, and phosphorylated tau proteins (99). They accumulate as 12–16 nm filamentous masses, reported to be identical to the paired helical filaments found in the brains of Alzheimer’s disease (100).
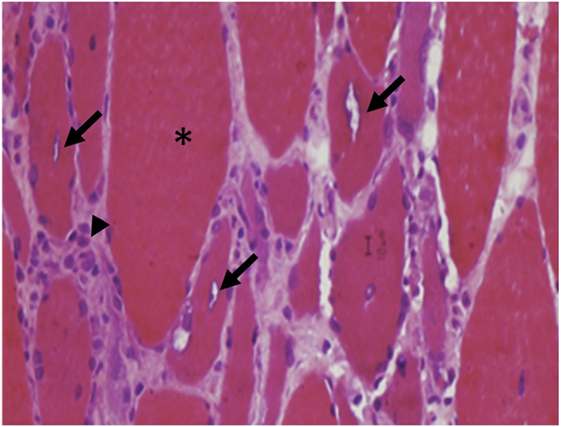
Figure 10. Atrophic and hypertrophic fibers (asterisk) with internal, rimmed vacuoles (arrows) are typical findings in inclusion body myositis.
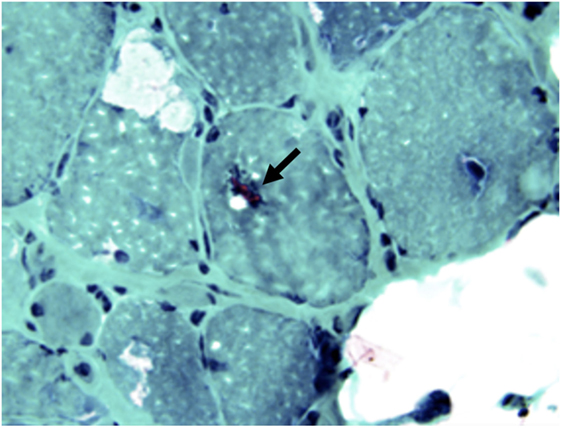
Figure 11. Rimmed vacuoles (arrow) in sIBM can be highlighted by modified trichrome stain (F, Modified trichrome stain, 40×).
Inducible nitric oxide synthase under pro-inflammatory conditions is upregulated and causes myofiber death (101).
Necrotizing Autoimmune Myopathy
Necrotizing autoimmune myopathy is thought to be macrophage-mediated immune response (43, 44). Other findings suggest an antibody-mediated mechanism in some subtypes (anti-SRP and anti-HMG-CoAR) (45, 69). The major findings on biopsy are scattered necrotic muscle fibers (46), surrounded by sparse inflammatory cells, predominately lymphocytes and occasionally some CD4+ and CD8+ T cells (102). These are detected through immunostaining (CD3, CD68). The overexpression of MHC-I in necrotic and regenerating fibers is variable (103, 104). NAM shares some features with DM of MAC deposition on micro vessels, but without perivascular inflammation, perifascicular atrophy, or tubuloreticular inclusion in endothelium. A variety of distinctive findings occur in specific subtypes of NAM. Demonstration of thickened basement membranes and enlarged pipestem capillaries of normal number is diagnostic of NAM with pipestem capillaries (43).
Treatment
The main goals of IIM therapy are to restore muscle strength, limit/eliminate the inflammation, and prevent other organs damage. It is ideal if the treatment involved a multidisciplinary approach; neurology, rheumatology, dermatology, pulmonary, physical occupational, and speech therapy.
There are only a few published prospective, double-blind, placebo-controlled trials for the treatment of IIM.
In general, NAM is more resistant to immunosuppressive therapy than DM and PM, particularly if there is an underlying malignancy or a statin trigger. The vast majority of sIBM patients are poorly responsive to immunotherapy.
Initial treatment Approach in Adults
Corticosteroids
Corticosteroids (high-dose) is the first line of treatment for adult onset DM, PM, and NAM (51). Its effect has never been formally proved in a prospective double-blind study, but the therapy is based on early reports suggesting a positive effect of corticosteroids on muscle strength (105). The initial prednisone dose is 0.5–1 mg/kg/day (60–100 mg once daily). Depending on patient response, taper usually take place after 4–6 weeks or when strength improvement reaches a plateau. Multiple tapering regimens have been used; one of the tapering regimens is 10 mg q2weeks to reach 30 mg/day, then 5 mg q2weeks to reach 20 mg/day, then 2.5 mg q2weeks until taper is completed or until reaching the lowest dose that will keep the patient in sustained remission (12). Others use taper to alternate day dosing over 2–3 months. However, some patients may not tolerate the swings, especially diabetics (74). Monthly 4-day course of 40 mg dexamethasone as an oral pulse therapy displayed a comparable efficacy as daily prednisone, but significantly less side effects. (106) Serum CK levels should be monitored, but adjustments of treatment should be based on objective clinical examination.
No response after an adequate trial of high dose prednisone should raise suspicion of alternative diagnoses like sIBM or inflammatory muscular dystrophy. Repeat muscle biopsy should be considered. Increasing weakness after an initial response may be due to a relapse or corticosteroids related myopathy. Relapse is more common to happen during taper. An EMG can be helpful to differentiate, as it does not show muscle membrane irritability with steroid myopathy.
Common side effects of high-dose corticosteroids include sleep disturbances, exacerbation of mood disorders, psychosis, glaucoma, cataract, avascular necrosis, osteoporosis and pathological fractures, hypertension, and hyperglycemia. Dual energy x-ray absorptiometry (DEXA) should be obtained at baseline and yearly while patients are receiving corticosteroids. Calcium (1 g/day) and vitamin D (400–800 IU/day) are initiated for prophylaxis against steroid-induced osteoporosis (107). Bisphosphonate can be added in postmenopausal women. Proton pump inhibitors are helpful in prevention of gastrointestinal complications. Periodic ophthalmological monitoring for glaucoma and cataract is recommended. Also, fasting blood glucose and serum potassium levels should be monitored. Potassium supplementation may be required if therapy leads to hypokalemia.
The decision regarding the timing of adding a second line agent may vary according to the severity of weakness, the initial response to prednisone, relapse, and in patients with increased risk of steroid complications (diabetics, osteoporosis). In most cases, starting an immunosuppressive drug at the time of initiating steroid treatment is necessary. The usual choices are methotrexate (MTX), azathioprine, and mycophenolate mofetil (MMF). There were no trials that compared these agents head to head, therefore, there is no superiority of choosing one of them over the others.
Methotrexate
Methotrexate is an antifolate that inhibits lymphocyte proliferation. The initial dose is 7.5 mg once weekly. Folic acid 1 mg daily or leucovorin 5 mg weekly on the subsequent day are important to limit some side effects (108). The dose can be increased by 2.5 mg weekly to reach target dose of 25 mg once weekly. With higher doses MTX can be given in three divided doses 12 h apart. Therapeutic effects of oral MTX are often noticeable after 4–8 weeks. If there is no improvement after 1 month of 25 mg weekly of oral MTX, some experts switch to weekly subcutaneous MTX and increase the dose by 5 mg every week up to 60 mg weekly (74).
Gastrointestinal side effects and alopecia are common. Painful stomatitis can happen and may respond to higher dose of folic acid. MTX is an oncogenic and teratogenic drug, and women of childbearing potential should be advised to use two forms of birth control while taking it. It is also hepatotoxic, nephrotoxic, and myelotoxic. Therefore, baseline liver function tests (LFTs) and measurement of Hepatitis B virus (HBV) and Hepatitis C virus HCV antibodies should be done before initiating the treatment. LFTs, complete blood count (CBC), and creatinine should be checked monthly for the first 3 months, then every 3 months (109). Gamma glutamyltransferase (GGT) is a specific hepatic marker that can help determine whether transaminitis is due to hepatotoxicity or muscle involvement alone. MTX can rarely induce pneumonitis, which may be indistinguishable from myositis associated ILD. This side effect makes some experts hesitant to start MTX on patients with ILD or Jo-1 antibodies. Screening for lung disease with pulmonary function tests (PFT) periodically is advised.
Azathioprine
Azathioprine is an antimetabolite that blocks T-lymphocyte proliferation. The initial dose is 25–50 mg daily; with increment of 25 mg qweek to goal does of 2–3 mg/kg of ideal body weight (100–250 mg daily). The dose can be given daily or can be divided into twice daily regimen. Azathioprine has a delayed onset of response that begins in 4–8 months and peaks at 1–2 years.
Common side effects of azathioprine include nausea and loose stools. A flulike reversible systemic reaction affects 12% in the first 2 weeks of therapy (110). Thiopurine methyltransferase (TPMT) should be checked before initiating treatment. This enzyme deficiency may lead to myelosuppression if standard dosage of azathioprine was given. In heterozygous TPMT mutation, azathioprine dose should be reduced, while in homozygous TPMT mutation, azathioprine should be avoided (111, 112). Azathioprine is teratogenic and oncogenic and can also cause pancreatitis. Hepatotoxicity may develop within few months. Myelosuppression can develop within weeks, but may also take 2 years. Hence, as with MTX, LFTs CBCs and creatinine should be monitored monthly for the first 3 months, then every 3 months. Dose should be decreased if white blood count (WBC) falls below 4000/mm3 and held if WBC declines to 2500/mm3 or LFTs increases more than twice baseline. This toxicity may take few months to reverse. Some patients may tolerate a rechallenge when laboratory values return to baseline (110).
Mycophenolate Mofetil
Mycophenolate mofetil (MMF) is an inosine monophosphate dehydrogenase inhibitor that blocks purine synthesis and inhibits T- and B-cell proliferation. Patients with anti-synthetase syndrome may respond favorably to MMF. It is initiated at 500 mg twice daily and increased by 500 mg weekly to goal of 1000 mg twice daily, or sometimes 1500 mg twice daily. The dose should be decreased in patients with renal insufficiency (1000 mg total dose).
Mycophenolate mofetil is a well-tolerated drug, with some side effects at higher doses, mainly nausea and loose stools. Severe infections may happen. Patients on MMF should be monitored for leukopenia and transaminitis. Women of childbearing potential should use two forms of birth control. Pregnancy should not be planned before withdrawal from immunosuppressant for several months (113).
Treatment Approach for Severe or Refractory Disease
Sometimes patients with IIM can be refractory to standard treatment or can have severe disease including severe cutaneous symptoms, severe muscle weakness, dysphagia, or significant weight loss. More aggressive treatment should be chosen for these groups.
Intravenous Methylprednisone
Intravenous methylprednisone (IVMP) can be used. Dose is usually 500–1000 mg daily for 3 days, followed by high-dose oral prednisone with taper.
Intravenous Immunoglobulin
Intravenous immunoglobulin (IVIG) has multiple indications. It can be used for refractory cases or as an add-on during relapses. IVIG is a good alternative to immunosuppressant agents in patients suffering from side effects or in childbearing women. There is little evidence that IVIG is effective as a monotherapy. It has a complex immunomodulatory mechanism of action including: (1) inhibition of complement activation, (2) reduction of autoantibody production and binding, (3) enhancement of antigen recognition by sensitized T cells, (4) blockade of Fc receptors, (5) downregulation of phagocytosis, and (6) inhibition of cell transmigration into the muscle (114). Dose is 2 g/kg given over 2 or 5 days once a month up to 6 months.
Serum IgA should be checked before starting treatment because IgA deficiency may lead to severe anaphylaxis caused by complexes formed between infused IgA and anti-IgA antibodies (115). In cases of IgA-deficiency, IVIG with reduced IgA levels can be given. Renal function should be checked due to the risk of IVIG induced renal failure. Flu-like symptoms including headaches, myalgias, fever, chills, nausea, and vomiting are common (up to 50%) and can be premeditated with acetaminophen and diphenhydramine. Aseptic meningitis, myocardial infarction, and stroke can also complicate IVIG administration.
Rituximab
Rituximab is a human monoclonal antibody to CD20 that act through depletion of B cells in circulation. It is effective in treating refractory IIM including SRP positive patients. It is given in two doses course, 1000 mg dose 2 weeks apart. This course can be repeated in 6–9 months. Baseline immunoglobulins, HBV and HCV antibodies, and tuberculosis screen should be done prior to starting the treatment. Recent trials have shown that rituximab is an effective choice for treatment resistant IIM, especially that in association with antisynthetase syndrome (116, 117). A recent retrospective review of charts of patients with refractory PM and DM treated with rituximab showed an objective improvement in regards to the CPK values and lung function tests. It also showed that patients with antisynthetase syndrome frequently required retreatment, and that, infections were a major limiting factor in treatment (118). Adverse effects can range from fever, chills, to bronchospasm, neutropenia, and thrombocytopenia. Severe infections can happen. Progressive multifocal leukoencephalopathy has been reported (119).
Cyclophosphamide
Cyclophosphamide (CYC) is a nitrogen mustard-alkylating agent that blocks both T- and B-cell proliferation. It is used when all other treatments fail or with severe multi-organ manifestations. This is due to the serious adverse effects including cytopenia, hemorrhagic cystitis due to metabolizing to acrolein, premature ovarian failure, and severe infections. Other major side effects are GI upset, alopecia, teratogenicity, and oncogenicity. It can be given as weekly intravenous infusion of 0.6–1 g/m2 after adequate oral and intravenous hydration, antiemetics, and mesna. The weekly infusions can be given up to 6 months, and sometimes 12 months. Mesna should be given with and after 4 and 8 h of CYC infusion to reduce the incidence of hemorrhagic cystitis. CYC can be also given orally as 1–2 mg/kg daily dose. Patients should maintain good hydration and frequent urination. Urinalysis and CBC are monitored every other week. A nadir of WBC that occurs 2 weeks after the IV infusion should not fall <3000/mm3.
CyclosporineA and Tacrolimus
CyclosporineA (CSA) and tacrolimus are calcineurin inhibitors that suppress helper T lymphocytes and block the production and secretion of interleukin-2. Pulmonary improvement was noted in patients with anti-Jo-1 and anti-SRP antibodies who were treated with tacrolimus. Both drugs should be started at a low dose and titrated slowly to 6 mg/kg/day for CSA and to 0.2 mg/kg/day for tacrolimus. The use of CSA and tacrolimus is limited because of serious adverse effects including hypertension, electrolytes imbalance, and renal insufficiency (120). Other side effects include GI upset, gum hyperplasia, hirsutism, oncogenicity, tremor, and risk of infection. Serum CSA and tacrolimus trough levels are monitored routinely to avoid renal toxicity.
Tumor Necrosis Factor Alpha Blockers
Tumor necrosis factor alpha (TNF-a) blockers like infliximab and etanercept were tried and showed steroid sparing effect (121).
Emerging Therapies
Anakinra
Anakinra is a recombinant human IL-1 receptor antagonist used commonly for the treatment of Rheumatoid arthritis and is an emerging drug in the treatment of IIM (122, 123). A 12-month follow-up study showed the beneficial effects of Anakinra. Fifteen patients with refractory myositis were treated with Anakinra for 12 months and treatment response was gaged by the parameters set by the International Myositis Assessment and Clinical Studies (IMACS) group and the functional Index (FI) (123). A recent study was performed on 15 patients treated with Anakinra for 12 months at the end of which a beneficial clinical response was noted in seven of these subjects. Repeat muscle biopsies were also done to explore possible predictive biomarkers (124). There is still room for large-scale randomized trials to show the efficacy of anakinra in IIM.
Alemtuzumab
Alemtuzumab is a humanized monoclonal anti-CD52 antibody that causes an immediate and severe depletion in the peripheral blood lymphocytes. It has shown to be a promising drug for the treatment of sIBM. As sIBM is largely resistant to treatment with steroids and immunosuppressive agents and has a rapidly deteriorating course. In a “proof-of-principle” study on thirteen sIBM patients, Dalakas et al. showed that alemtuzumab infusions slowed down disease progression up to 6 months and improved muscle strength (125). Recently, a long-term follow-up case study on a treatment-resistant case treated with alemtuzumab showed marked improvement in muscle strength 12 weeks into a single treatment cycle with alemtuzumab and this lasted for ~3 years (126).
Belimumab
Belimumab is a human monoclonal antibody directed against B lymphocyte stimulator (BLys), which is a TNF-related cytokine implicated in B cell maturation and development. It was approved for treatment of Systemic lupus erythematosus (SLE) in March 2011 (127).
Its role is currently being studied in the management of PM DM.1
Sifalimumab
Sifalimumab is an anti-IFN-monoclonal antibody Neutralization of the type I IFN gene signature by sifalimumab resulted in coordinated suppression of T cell-related proteins such as soluble IL-2RA, TNF receptor 2 (TNFR2), and IL-18 (128).
Treatment Approach to JDM
The strategy is similar to the one used for adults, except that initial prednisone dose is 2 mg/kg; maximum of 60 mg daily. MTX is the main steroid sparing agent and is added at onset 15 mg/m2 subcutaneously weekly. Other immunosuppressive agents that can be used are azathioprine, cyclosporine, and tacrolimus. MMF is not used routinely in children. IVIG is efficacious and safe for refractory cases. Rituximab 575–750 mg/m2 is recently gaining acceptance. IV CYC at 0.5–1.0 g/m2 can be used as a last resort.
Management of Skin Disease
To prevent skin flairs, patients with DM and JDM should avoid UV rays; use sunscreen in addition to appropriate coverage. Topical steroids and tacrolimus have been used (129).
Hydroxycholoroquine
Hydroxycholoroquine is an antimalarial drug that is used for cutaneous manifestations in DM and JDM and is given 200 mg twice daily. Baseline electrocardiogram should be obtained to screen and monitor for QT prolongation. Also a baseline ophthalmology testing is important to rule out macular disease, and subsequent annual screening after 5 years of treatment should be done to monitor for retinal toxicity (130).
Management of Calcinosis
Therapy of calcinosis shows lack of meaningful response. Diltiazem may produce a partial response (131). There was an improvement of calcinosis with abatacept and sodium thiosulfate – a vasodilator that chelates calcium in a case report. (132) Surgical excision is an option.
Management of Dysphagia
Dysphagia can occur in all subtypes of myositis. A high percentage of patients with IBM are affected (133), which leads to the risk for malnutrition and aspiration pneumonia. Treatment with IVIG improves swallowing in IBM (134–136) as well as prednisone-resistant DM or PM (137).
Cricopharyngeomyotomy is used when the underlying mechanisms of dysphagia is failed relaxation of the upper esophageal sphincter. This intervention may not be beneficial with normal relaxation and other mechanisms of dysphagia, i.e., delayed swallow initiation and decreased hyolaryngeal excursion (138). Other interventions include percutaneous endoscopic gastrostomy (PEG), pharyngoesophageal dilatation, and injection of botulinum toxin (139–141).
Treatment of Associated ILD
Most patients require adjuvant immune modulating drugs with the first-line corticosteroids (57). MMF is the favored drug. Also, cyclosporine and tacrolimus are effective as second-line agents (142). Cyclophosphamide is considered a third-line agent (143). Rituximab has recently emerged as a promising effective agent in treatment of antisynthetase syndrome, which is notoriously associated with severe ILD (116). Factors predictive of poor ILD prognosis include older age, lower values of PFTs, CT scan with a pattern of interstitial pneumonia, and treatment-refractory.
Physical Therapy
Physical and occupation therapy are essential and along with orthotic devices if needed, help patients improve mobility, retain motor function, prevent contractures that can arise especially in JDM and may help prevent steroids side effects like weight gain, osteoporosis, and type 2 fiber atrophy. Strengthening programs twice weekly can be started as early as 2–3 weeks from the acute phase (144). With severe cases, passive range of motion exercises can be done for 3 months, until strength improve; at which point strengthening exercises are initiated. There is growing evidence for safety and beneficial effects of physiotherapy and home exercise programs in myositis (145).
A recent study demonstrated the effect of a 12-week aerobic exercise program in 10 children with chronic JDM. At the end of this longitudinal study, the subjects showed an improvement in muscle strength and function, aerobic conditioning, and a better quality of life (146).
Dastmalchi et al. showed that muscle biopsies in patients with PM/DM contained a low population of oxygen dependent type 1 fibers in comparison to healthy individuals. After 5 days a week to 12-week home exercise program, repeat muscle biopsies showed a higher frequency of oxygen-dependent type 1 fiber. Through this study, a molecular basis for the beneficial effects of exercise training was established (147).
Treatment of sIBM
Treatment of sIBM is challenging, as the disease typically is resistant to standard immunotherapy. Prednisone is usually not effective (47) and may lead to more rapid progression (30). However, some patients may experience at least a temporary improvement. Some treat newly diagnosed patients with immunosuppression (12). Rationale is that early suppression of the inflammatory cascade may prevent downstream effects leading to muscle degeneration (148).
Studies with MTX (149), anti-T-lymphocyte globulin (150), etanercept (151), oxandrolone (152), or β-interferon (153, 154) failed to identify clinical efficiency. Although IVIG can help with dysphagia, it was not found effective for muscle strength (155).
The most encouraging study used alemtuzumab, a monoclonal antibody that deplete peripheral lymphocytes. It seemed to delay the disease progression for up to 6 months, and in some patients, the muscle strength was improved (125).
The concept that modulating the action of the Heat shock response proteins (HSR) in cells can dampen the detrimental aspects of both degeneration and inflammation has recently been tested through a phase two placebo-controlled trial. For this purpose, arimoclomol, an agent that amplifies heat shock protein expression was used. The results have been encouraging and provide a platform for further large scale trials (156).
Through a pilot trial, it has recently been suggested that TGFb signaling through activin receptors may be responsible for the pathological changes seen in sIBM (157). In this trial, an actRII inhibitory antibody, bimagrumab was shown to result in improvement in thigh muscle volume in patients with sIBM at 8 weeks.
An ongoing phase II trial is being conducted by Novartis to prove bimagrumab’s efficacy in treating patients with sIBM2.
Different studies conducted for IIM therapies are outlined in Table 3. The efficacy/side effects among different IIM are briefly mentioned.
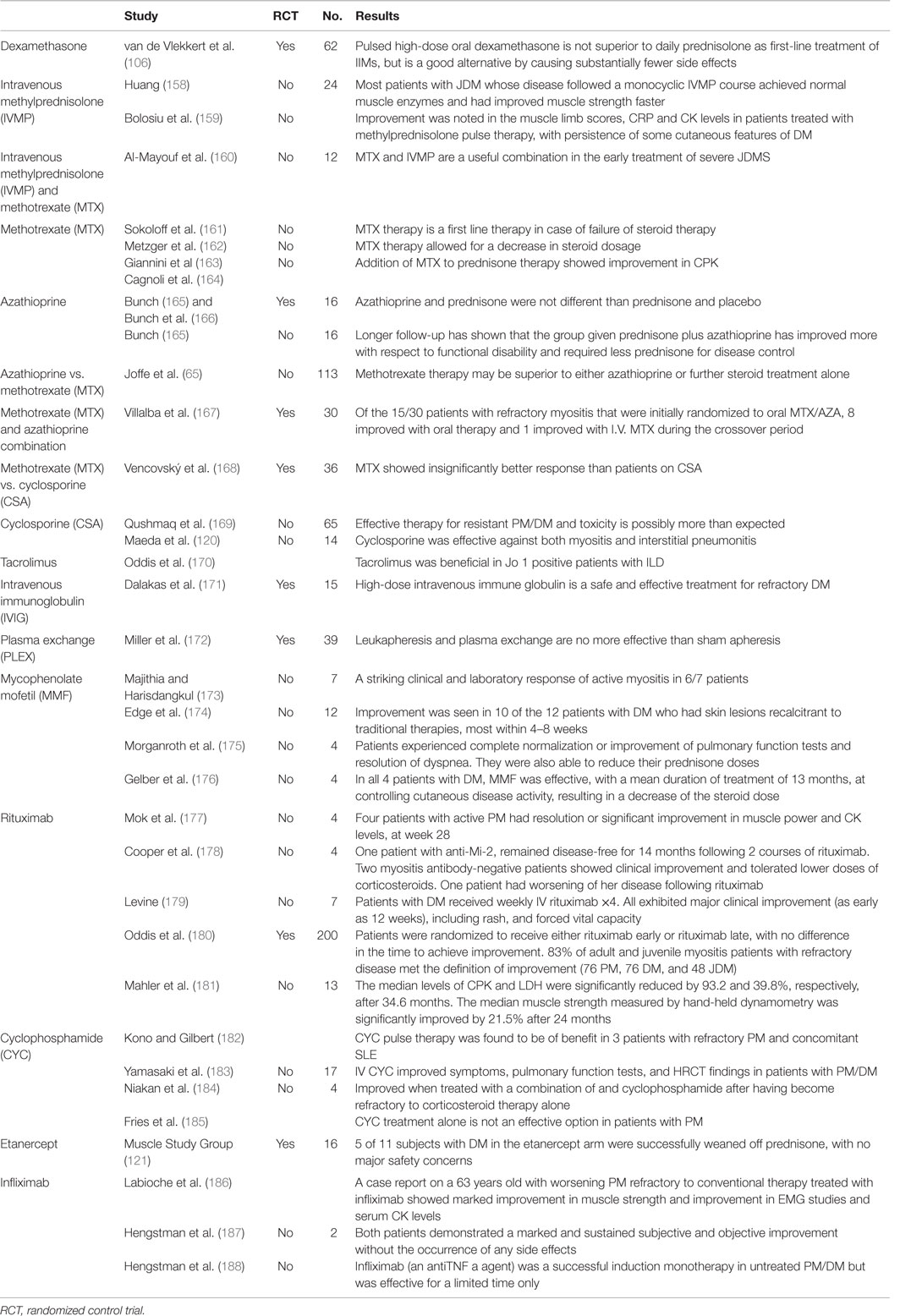
Table 3. Various treatments, modalities and corresponding studies.
Table 4 summarizes clinical presentation, laboratory investigations, therapies, and prognosis.
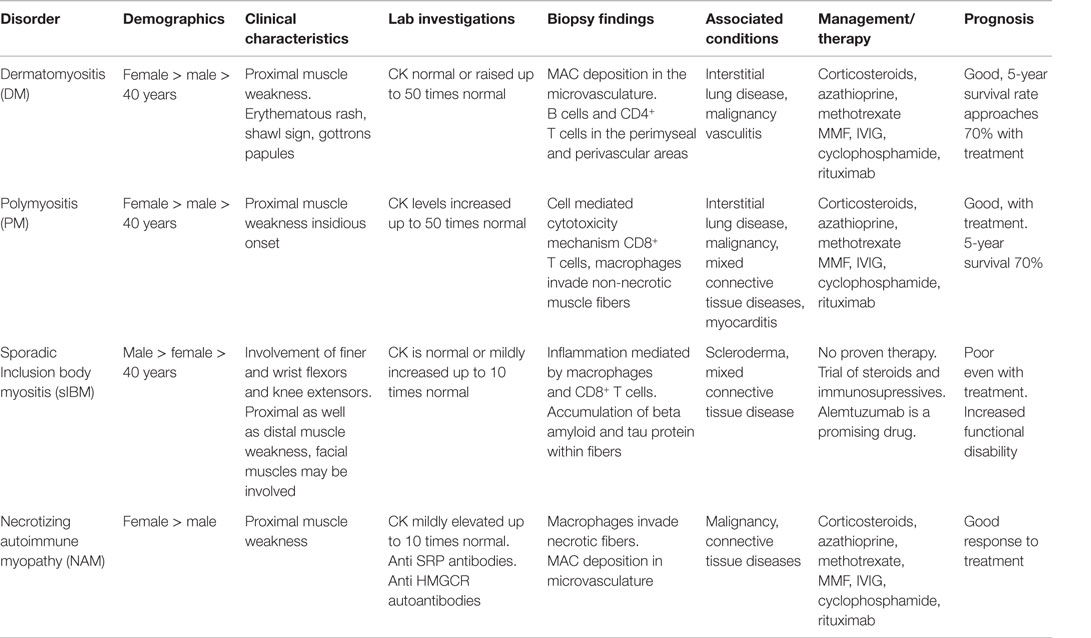
Table 4. Idiopathic inflammatory myopathies (IIM), Summary.
Conclusion
Idiopathic inflammatory myopathies are a group of varied presentations and pathogenesis. Different classifications over time have enhanced the diagnostic yield. Enhanced pathological processing and imaging of the muscles have improved the diagnosis. Newer agents are being investigated for the therapy of IIM and may lead to better results. Early diagnosis and therapy remains the gold standard for optimal prognosis. sIBM has been grouped with IIM and may eventually be established a neurodegenerative disorder due to pathological features and poor response to immune modulating therapies. A standardized classification will help with the relatively consistent diagnosis among different neuromuscular specialist and be meaningful for randomized studies.
Author Contributions
AM contributed to the body and design of the write up and made required changes. JK wrote the abstract, helped with initial edits, and constructed the tables. Prof. GH did the critical review, editing, and approved the final manuscript. Dr. MG provided the figures and the figure legends.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer RNB and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
Footnotes
References
1. Dalakas MC. Polymyositis, dermatomyositis, and inclusion-body myositis. N Engl J Med (1991) 325(21):1487–98. doi:10.1056/NEJM199111213252107
2. Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet (2003) 362(9388):971–82. doi:10.1016/S0140-6736(03)14368-1
3. Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med (1975) 292(7):344–7. doi:10.1056/NEJM197502132920706
4. Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med (1975) 292(8):403–7. doi:10.1056/NEJM197502202920807
5. Emslie-Smith AM, Arahata K, Engel AG. Major histocompatibility complex class I antigen expression, immunolocalization of interferon subtypes, and T cell-mediated cytotoxicity in myopathies. Hum Pathol (1989) 20(3):224–31. doi:10.1016/0046-8177(89)90128-7
6. Dalakas MC, Sivakumar K. The immunopathologic and inflammatory differences between dermatomyositis, polymyositis and sporadic inclusion body myositis. Curr Opin Neurol (1996) 9(3):235–9. doi:10.1097/00019052-199606000-00015
7. Sigurgeirsson B, Lindelof B, Edhag O, Allander E. Risk of cancer in patients with dermatomyositis or polymyositis. A population-based study. N Engl J Med (1992) 326(6):363–7. doi:10.1056/NEJM199202063260602
8. Callen JP. Relationship of cancer to inflammatory muscle diseases. Dermatomyositis, polymyositis, and inclusion body myositis. Rheum Dis Clin North Am (1994) 20(0889–857X):943–53.
9. Mastaglia FL, Phillips BA. Idiopathic inflammatory myopathies: epidemiology, classification, and diagnostic criteria. Rheum Dis Clin North Am (2002) 28(4):723–41. doi:10.1016/S0889-857X(02)00021-2
10. Dalakas MC. Inflammatory muscle diseases: a critical review on pathogenesis and therapies. Curr Opin Pharmacol (2010) 10(3):346–52. doi:10.1016/j.coph.2010.03.001
11. Schmidt J, Dalakas MC. Pathomechanisms of inflammatory myopathies: recent advances and implications for diagnosis and therapies. Expert Opin Med Diagn (2010) 4(3):241–50. doi:10.1517/17530051003713499
12. Ernste FC, Reed AM. Idiopathic inflammatory myopathies: current trends in pathogenesis, clinical features, and up-to-date treatment recommendations. Mayo Clin Proc (2013) 88:83–105. doi:10.1016/j.mayocp.2012.10.017
13. Hoogendijk JE, Amato AA, Lecky BR, Choy EH, Lundberg IE, Rose MR, et al. 119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10-12 October 2003, Naarden, The Netherlands. Neuromuscul Disord (2004) 14(5):337–45. doi:10.1016/j.nmd.2004.02.006
14. Lazarou IN, Guerne P-A. Classification, diagnosis, and management of idiopathic inflammatory myopathies. J Rheumatol (2013) 40(5):550–64. doi:10.3899/jrheum.120682
15. Pestronk A. Acquired immune and inflammatory myopathies: pathologic classification. Curr Opin Rheumatol (2011) 23(6):595–604. doi:10.1097/BOR.0b013e32834bab42
16. Brady S, Squier W, Hilton-Jones D. Clinical assessment determines the diagnosis of inclusion body myositis independently of pathological features. J Neurol Neurosurg Psychiatry (2013) 84:1240–6. doi:10.1136/jnnp-2013-305690
17. Rose MR, ENMC IBM Working Group. 188th ENMC International Workshop: Inclusion Body Myositis, 2–4 December 2011, Naarden, The Netherlands. Neuromuscul Disord (2013) 23(12):1044–55.
18. Lloyd TE, Mammen AL, Amato AA, Weiss MD, Needham M, Greenberg SA. Evaluation and construction of diagnostic criteria for inclusion body myositis. Neurology (2014) 83(5):426–33. doi:10.1212/WNL.0000000000000642
19. Yang Z, Lin F, Qin B, Yan L, Renqian Z. Polymyositis/dermatomyositis and malignancy risk: a metaanalysis study. J Rheumatol (2015) 42(2):282–91. doi:10.3899/jrheum.140566
21. Briani C, Doria A, Sarzi-Puttini P, Dalakas MC. Update on idiopathic inflammatory myopathies. Autoimmunity (2006) 39(3):161–70. doi:10.1080/08916930600622132
22. Wilson FC, Ytterberg SR, St. Sauver JL, Reed AM. Epidemiology of sporadic inclusion body myositis and polymyositis in Olmsted County, Minnesota. J Rheumatol (2008) 35(3):445–7.
23. Prieto S, Grau JM. The geoepidemiology of autoimmune muscle disease. Autoimmun Rev (2010) 9(5):A330–4. doi:10.1016/j.autrev.2009.11.006
24. Dimachkie MM, Barohn RJ. Idiopathic inflammatory myopathies. Semin Neurol (2012) 32(3):227–36. doi:10.1055/s-0032-1329201
25. Furst DE, Amato AA, Iorga SR, Gajria K, Fernandes AW. Epidemiology of adult idiopathic inflammatory myopathies in a U.S. Managed care plan. Muscle Nerve (2012) 45(5):676–83. doi:10.1002/mus.23302
26. van der Meulen MFG, Bronner IM, Hoogendijk JE, Burger H, van Venrooij WJ, Voskuyl AE, et al. Polymyositis: an overdiagnosed entity. Neurology (2003) 61(3):316–21. doi:10.1212/WNL.61.3.316
27. Chahin N, Engel AG. Correlation of muscle biopsy, clinical course, and outcome in PM and sporadic IBM. Neurology (2008) 70(6):418–24. doi:10.1212/01.wnl.0000277527.69388.fe
28. Kuo CF, See LC, Yu KH, Chou IJ, Chang HC, Chiou MJ, et al. Incidence, cancer risk and mortality of dermatomyositis and polymyositis in Taiwan: a nationwide population study. Br J Dermatol (2011) 165(6):1273–9. doi:10.1111/j.1365-2133.2011.10595.x
29. Needham M, Corbett A, Day T, Christiansen F, Fabian V, Mastaglia FL. Prevalence of sporadic inclusion body myositis and factors contributing to delayed diagnosis. J Clin Neurosci (2008) 15(12):1350–3. doi:10.1016/j.jocn.2008.01.011
30. Benveniste O, Guiguet M, Freebody J, Dubourg O, Squier W, Maisonobe T, et al. Long-term observational study of sporadic inclusion body myositis. Brain (2011) 134(11):3176–84. doi:10.1093/brain/awr213
31. Hochberg MC, Feldman D, Stevens MB. Adult onset polymyositis/dermatomyositis: an analysis of clinical and laboratory features and survival in 76 patients with a review of the literature. Semin Arthritis Rheum (1986) 5(3):168–78. doi:10.1016/0049-0172(86)90014-4
32. Amato AA, Barohn RJ. Idiopathic inflammatory myopathies. Neurol Clin (1997) 15(3):615–48. doi:10.1016/S0733-8619(05)70337-6
33. Sontheimer RD. Cutaneous features of classic dermatomyositis and amyopathic dermatomyositis. Curr Opin Rheumatol (1999) 11(6):475–82. doi:10.1097/00002281-199911000-00005
34. Chatterjee S, Prayson R, Farver C. Antisynthetase syndrome: not just an inflammatory myopathy. Cleve Clin J Med (2013) 80(10):655–66. doi:10.3949/ccjm.80a.12171
35. Balbir-Gurman A, Braun-Moscovici Y. Scleroderma overlap syndrome. Isr Med Assoc J (2011) 13(1):14–20.
36. Vancsa A, Gergely L, Ponyi A, Lakos G, Németh J, Szodoray P, et al. Myositis-specific and myositis-associated antibodies in overlap myositis in comparison to primary dermatopolymyositis: relevance for clinical classification: retrospective study of 169 patients. Jt Bone Spine (2010) 77(2):125–30. doi:10.1016/j.jbspin.2009.08.008
37. Euwer RL, Sontheimer RD. Amyopathic dermatomyositis: a review. J Invest Dermatol (1993) 100(1):124S–7S. doi:10.1111/1523-1747.ep12356896
38. Rider LG, Miller FW. Classification and treatment of the juvenile idiopathic inflammatory myopathies. Rheum Dis Clin North Am (1997) 23:619–55. doi:10.1016/S0889-857X(05)70350-1
39. Lorenzoni PJ, Scola RH, Kay CSK, Prevedello PG, Espindola G, Werneck LC. Idiopathic inflammatory myopathies in childhood: a brief review of 27 cases. Pediatr Neurol (2011) 45(1):17–22. doi:10.1016/j.pediatrneurol.2011.01.018
40. Pachman LM, Hayford JR, Chung A, Daugherty CA, Pallansch MA, Fink CW, et al. Juvenile dermatomyositis at diagnosis: clinical characteristics of 79 children. J Rheumatol (1998) 25(6):1198–204.
41. Marie I, Hachulla E, Chérin P, Dominique S, Hatron P-Y, Hellot M-F, et al. Interstitial lung disease in polymyositis and dermatomyositis. Arthritis Rheum (2002) 47(6):614–22. doi:10.1002/art.10794
42. Yazici Y, Kagen LJ. Cardiac involvement in myositis. Curr Opin Rheumatol (2002) 14(6):663–5. doi:10.1097/00002281-200211000-00006
43. Emslie-Smith AM, Engel AG. Necrotizing myopathy with pipestem capillaries, microvascular deposition of the complement membrane attack complex (MAC), and minimal cellular infiltration. Neurology (1991) 41(6):936–9. doi:10.1212/WNL.41.6.936
44. Bronner IM, Hoogendijk JE, Wintzen AR, Van Der Meulen MFG, Linssen WHJP, Wokke JHJ, et al. Necrotising myopathy, an unusual presentation of a steroid-responsive myopathy. J Neurol (2003) 250(4):480–5. doi:10.1007/s00415-003-1027-y
45. Grable-Esposito P, Katzberg HD, Greenberg SA, Srinivasan J, Katz J, Amato AA. Immune-mediated necrotizing myopathy associated with statins. Muscle Nerve (2010) 41(2):185–90. doi:10.1002/mus.21486
46. Liang C, Needham M. Necrotizing autoimmune myopathy. Curr Opin Rheumatol (2011) 23(6):612–9. doi:10.1097/BOR.0b013e32834b324b
47. Lotz BP, Engel AG, Nishino H, Stevens JC, Litchy WJ. Inclusion body myositis. Observations in 40 patients. Brain (1989) 112(Pt 3):727–47. doi:10.1093/brain/112.3.727
48. Amato AA, Gronseth GS, Jackson CE, Wolfe GI, Katz JS, Bryan WW, et al. Inclusion body myositis: clinical and pathological boundaries. Ann Neurol (1996) 40(4):581–6. doi:10.1002/ana.410400407
49. Sekul EA, Dalakas MC. Inclusion body myositis: old and new concepts. J Neurol Neurosurg Psychiatry (1993) 80:256–63.
50. Griggs RC, Askanas V, DiMauro S, Engel A, Karpati G, Mendell JR, et al. Inclusion body myositis and myopathies. Ann Neurol (1995) 38:705–13. doi:10.1002/ana.410380504
51. Bohan A, Peter JB, Bowman RL, Pearson CM. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore) (1977) 56(4):255–86. doi:10.1097/00005792-197707000-00001
52. Tymms KE, Beller EM, Webb J, Schrieber L, Buchanan WW. Correlation between tests of muscle involvement and clinical muscle weakness in polymyositis and dermatomyositis. Clin Rheumatol (1990) 9(4):523–9. doi:10.1007/BF02030515
53. Love LA, Leff RL, Fraser DD, Targoff IN, Dalakas M, Plotz PH, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (1991) 70(6):360–74. doi:10.1097/00005792-199111000-00002
54. Hengstman GJD, van Engelen BGM, van Venrooij WJ. Myositis specific autoantibodies: changing insights in pathophysiology and clinical associations. Curr Opin Rheumatol (2004) 16(6):692–9.
55. Miller T, Al-Lozi M, Lopate G, Pestronk A. Myopathy with antibodies to the signal recognition particle: clinical and pathological features. J Neurol Neurosurg Psychiatry (2002) 73(4):420–8. doi:10.1136/jnnp.73.4.420
56. Ichimura Y, Matsushita T, Hamaguchi Y, Kaji K, Hasegawa M, Tanino Y, et al. Anti-NXP2 autoantibodies in adult patients with idiopathic inflammatory myopathies: possible association with malignancy. Ann Rheum Dis (2012) 71(5):710–3. doi:10.1136/annrheumdis-2011-200697
57. Mimori T, Nakashima R, Hosono Y. Interstitial lung disease in myositis: clinical subsets, biomarkers, and treatment. Curr Rheumatol Rep (2012) 14(3):264–74. doi:10.1007/s11926-012-0246-6
58. Gunawardena H, Betteridge ZE, McHugh NJ. Myositis-specific autoantibodies: their clinical and pathogenic significance in disease expression. Rheumatology (Oxford) (2009) 48(6):607–12. doi:10.1093/rheumatology/kep078
59. Targoff IN, Miller FW, Medsger TA, Oddis CV. Classification criteria for the idiopathic inflammatory myopathies. Curr Opin Rheumatol (1997) 9(6):527–35. doi:10.1097/00002281-199711000-00008
60. Casciola-Rosen L, Mammen AL. Myositis autoantibodies. Curr Opin Rheumatol (2012) 24(6):602–8. doi:10.1097/BOR.0b013e328358bd85
61. Chinoy H, Fertig N, Oddis CV, Ollier WE, Cooper RG. The diagnostic utility of myositis autoantibody testing for predicting the risk of cancer-associated myositis. Ann Rheum Dis (2007) 66(10):1345–9. doi:10.1136/ard.2006.068502
62. Gunawardena H, Wedderburn LR, Chinoy H, Betteridge ZE, North J, Ollier WE, et al. Autoantibodies to a 140-kd protein in juvenile dermatomyositis are associated with calcinosis. Arthritis Rheum (2009) 60(6):1807–14. doi:10.1002/art.24547
63. Hoshino K, Muro Y, Sugiura K, Tomita Y, Nakashima R, Mimori T. Anti-MDA5 and anti-TIF1-γ antibodies have clinical significance for patients with dermatomyositis. Rheumatology (2010) 49(9):1726–33. doi:10.1093/rheumatology/keq153
64. Joffe MM, Love LA, Leff RL, Fraser DD, Targoff IN, Hicks JE, et al. Drug therapy of the idiopathic inflammatory myopathies: predictors of response to prednisone, azathioprine, and methotrexate and a comparison of their efficacy. Am J Med (1993) 94(4):379–87. doi:10.1016/0002-9343(93)90148-I
65. Hamaguchi Y, Kuwana M, Hoshino K, Hasegawa M, Kaji K, Matsushita T, et al. Clinical correlations with dermatomyositis-specific autoantibodies in adult Japanese patients with dermatomyositis: a multicenter cross-sectional study. Arch Dermatol (2011) 147:391–8. doi:10.1001/archdermatol.2011.52
66. Love LA, Weinberg CR, McConnaughey DR, Oddis CV, Medsger TA, Reveille JD, et al. Ultraviolet radiation intensity predicts the relative distribution of dermatomyositis and anti-Mi-2 autoantibodies in women. Arthritis Rheum (2009) 60(8):2499–504. doi:10.1002/art.24702
67. Hanisch F, Müller T, Stoltenburg G, Zierz S. Unusual manifestations in two cases of necrotizing myopathy associated with SRP-antibodies. Clin Neurol Neurosurg (2012) 114(7):1104–6. doi:10.1016/j.clineuro.2011.12.055
68. Kao AH, Lacomis D, Lucas M, Fertig N, Oddis CV. Anti-signal recognition particle autoantibody in patients with and patients without idiopathic inflammatory myopathy. Arthritis Rheum (2004) 50(1):209–15. doi:10.1002/art.11484
69. Christopher-Stine L, Casciola-Rosen LA, Hong G, Chung T, Corse AM, Mammen AL. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune mediated necrotizing myopathy. Arthritis Rheum (2010) 62(9):2757–66. doi:10.1002/art.27572
70. Buchbinder R, Forbes A, Hall S, Dennett X, Giles G. Incidence of malignant disease in biopsy-proven inflammatory myopathy: a population-based cohort study. Ann Intern Med (2001) 134(12):1087–95. doi:10.7326/0003-4819-134-12-200106190-00008
71. Hill CL, Zhang Y, Sigurgeirsson B, Pukkala E, Mellemkjaer L, Airio A, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet (2001) 357(9250):96–100. doi:10.1016/S0140-6736(00)04588-8
72. Titulaer MJ, Soffietti R, Dalmau J, Gilhus NE, Giometto B, Graus F, et al. Screening for tumours in paraneoplastic syndromes: report of an EFNS task force. Eur J Neurol (2011) 18(1):19–27. doi:10.1111/j.1468-1331.2010.03220.x
73. Blijham PJ, Hengstman GJD, Hama-Amin AD, Van Engelen BGM, Zwarts MJ. Needle electromyographic findings in 98 patients with myositis. Eur Neurol (2006) 55(4):183–8. doi:10.1159/000093866
74. Amato AA, Barohn RJ. Evaluation and treatment of inflammatory myopathies. J Neurol Neurosurg Psychiatry (2009) 80(10):1060–8. doi:10.1136/jnnp.2008.169375
75. Adams EM, Chow CK, Premkumar A, Plotz PH. The idiopathic inflammatory myopathies: spectrum of MR imaging findings. Radiographics (1995) 15(3):563–74. doi:10.1148/radiographics.15.3.7624563
76. Schulze M, Kötter I, Ernemann U, Fenchel M, Tzaribatchev N, Claussen CD, et al. MRI findings in inflammatory muscle diseases and their noninflammatory mimics. Am J Roentgenol (2009) 192(6):1708–16. doi:10.2214/AJR.08.1764
77. Maillard SM, Jones R, Owens C, Pilkington C, Woo P, Wedderburn LR, et al. Quantitative assessment of MRI T2 relaxation time of thigh muscles in juvenile dermatomyositis. Rheumatology (2004) 43(5):603–8. doi:10.1093/rheumatology/keh130
78. Ruperto N, Ravelli A, Pistorio A, Ferriani V, Calvo I, Ganser G, et al. The provisional Paediatric Rheumatology International Trials Organisation/American College of Rheumatology/European League Against Rheumatism Disease activity core set for the evaluation of response to therapy in juvenile dermatomyositis: a prospective validation study. Arthritis Rheum (2008) 59(1):4–13. doi:10.1002/art.23248
79. Degardin A, Morillon D, Lacour A, Cotten A, Vermersch P, Stojkovic T. Morphologic imaging in muscular dystrophies and inflammatory myopathies. Skeletal Radiol (2010) 39(12):1219–27. doi:10.1007/s00256-010-0930-4
80. Greenberg SA, Bradshaw EM, Pinkus JL, Pinkus GS, Burleson T, Due B, et al. Plasma cells in muscle in inclusion body myositis and polymyositis. Neurology (2005) 65(11):1782–7. doi:10.1212/01.wnl.0000187124.92826.20
81. Zhou X, Dimachkie MM, Xiong M, Tan FK, Arnett FC. cDNA microarrays reveal distinct gene expression clusters in idiopathic inflammatory myopathies. Med Sci Monit (2004) 10(7):BR191–7.
82. Swiecki M, Colonna M. Accumulation of plasmacytoid DC: roles in disease pathogenesis and targets for immunotherapy. Eur J Immuno (2010) 40(8):2094–8. doi:10.1002/eji.201040602
83. Arahata K, Engel AG. Monoclonal antibody analysis of mononuclear cells in myopathies. I: quantitation of subsets according to diagnosis and sites of accumulation and demonstration and counts of muscle fibers invaded by T cells. Ann Neurol (1984) 16(2):193–208. doi:10.1002/ana.410160206
84. Lopez De Padilla CM, Vallejo AN, McNallan KT, Vehe R, Smith SA, Dietz AB, et al. Plasmacytoid dendritic cells in inflamed muscle of patients with juvenile dermatomyositis. Arthritis Rheum (2007) 56(5):1658–68. doi:10.1002/art.22558
85. Liu Y-J. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol (2005) 23:275–306. doi:10.1146/annurev.immunol.23.021704.115633
86. Kissel JT, Mendell JR, Rammohan KW. Microvascular deposition of complement membrane attack complex in dermatomyositis. N Engl J Med (1986) 314(6):329–34. doi:10.1056/NEJM198602063140601
87. Emslie-Smith AM, Engel AG. Microvascular changes in early and advanced dermatomyositis: a quantitative study. Ann Neurol (1990) 27(4):343–56. doi:10.1002/ana.410270402
88. Pestronk A, Schmidt RE, Choksi R. Vascular pathology in dermatomyositis and anatomic relations to myopathology. Muscle Nerve (2010) 42(1):53–61. doi:10.1002/mus.21651
89. De Visser M, Emslie-Smith AM, Engel AG. Early ultrastructural alterations in adult dermatomyositis. Capillary abnormalities precede other structural changes in muscle. J Neurol Sci (1989) 94(1–3):181–92. doi:10.1016/0022-510X(89)90228-1
90. Santmyire-Rosenberger B, Dugan EM. Skin involvement in dermatomyositis. Curr Opin Rheumatol (2003) 15(6):714–22. doi:10.1097/00002281-200311000-00006
91. Callen JP, Gruber GG, Hodge SJ, Owen LG. Cutaneous manifestations of gastrointestinal disorders. J Ky Med Assoc (1978) 76(12):603–7.
92. Engel AG, Arahata K. Mononuclear cells in myopathies: quantitation of functionally distinct subsets, recognition of antigen-specific cell-mediated cytotoxicity in some diseases, and implications for the pathogenesis of the different inflammatory myopathies. Hum Pathol (1986) 17(7):704–21. doi:10.1016/S0046-8177(86)80180-0
93. Engel AG, Arahata K, Emslie-Smith A. Immune effector mechanisms in inflammatory myopathies. Res Publ Assoc Res Nerv Ment Dis (1990) 68:141–57.
94. Salaroli R, Baldin E, Papa V, Rinaldi R, Tarantino L, De Giorgi LB, et al. Validity of internal expression of the major histocompatibility complex class I in the diagnosis of inflammatory myopathies. J Clin Pathol (2012) 65(1):14–9. doi:10.1136/jclinpath-2011-200138
95. Greenberg SA, Pinkus GS, Amato AA, Pinkus JL. Myeloid dendritic cells in inclusion-body myositis and polymyositis. Muscle Nerve (2007) 35(1):17–23. doi:10.1002/mus.20649
96. Greenberg SA, Pinkus JL, Pinkus GS, Burleson T, Sanoudou D, Tawil R, et al. Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol (2005) 57(5):664–78. doi:10.1002/ana.20464
97. Dalakas MC. Sporadic inclusion body myositis – diagnosis, pathogenesis and therapeutic strategies. Nat Clin Pract Neurol (2006) 2(8):437–47. doi:10.1038/ncpneuro0261
98. Karpati G, O’Ferrall EK. Sporadic inclusion body myositis: pathogenic considerations. Ann Neurol (2009) 65(1):7–11. doi:10.1002/ana.21622
99. Askanas V, Engel WK, Nogalska A. Pathogenic considerations in sporadic inclusion-body myositis, a degenerative muscle disease associated with aging and abnormalities of myoproteostasis. J Neuropathol Exp Neurol (2012) 71(8):680–93. doi:10.1097/NEN.0b013e31826183c8
100. Askanas V, Engel WK, Nogalska A. Inclusion body myositis: a degenerative muscle disease associated with intra-muscle fiber multi-protein aggregates, proteasome inhibition, endoplasmic reticulum stress and decreased lysosomal degradation: mini-symposium: protein aggregate myopathies. Brain Pathol (2009) 19(3):493–506. doi:10.1111/j.1750-3639.2009.00290.x
101. Schmidt J, Barthel K, Zschuntzsch J, Muth IE, Swindle EJ, Hombach A, et al. Nitric oxide stress in sporadic inclusion body myositis muscle fibres: inhibition of inducible nitric oxide synthase prevents interleukin-1beta-induced accumulation of? beta-amyloid and cell death. Brain (2012) 135(4):1102–14. doi:10.1093/brain/aws046
102. Preuße C, Goebel HH, Held J, Wengert O, Scheibe F, Irlbacher K, et al. Immune-mediated necrotizing myopathy is characterized by a specific Th1-M1 polarized immune profile. Am J Pathol (2012) 181(6):2161–71. doi:10.1016/j.ajpath.2012.08.033
103. Ellis E, Tan JA, Lester S, Tucker G, Blumbergs P, Roberts-Thomson P, et al. Necrotizing myopathy: clinicoserologic associations. Muscle Nerve (2012) 45(2):189–94. doi:10.1002/mus.22279
104. Needham M, Fabian V, Knezevic W, Panegyres P, Zilko P, Mastaglia FL. Progressive myopathy with up-regulation of MHC-I associated with statin therapy. Neuromuscul Disord (2007) 17(2):194–200. doi:10.1016/j.nmd.2006.10.007
105. Micks RH, Mullaney J. Dermatomyositis successfully treated by prednisone. Ir J Med Sci (1958) 33(391):333–4. doi:10.1007/BF02950398
106. van de Vlekkert J, Hoogendijk JE, de Haan RJ, Algra A, van der Tweel I, van der Pol WL, et al. Oral dexamethasone pulse therapy versus daily prednisone in subacute inflammatory myositis, a randomised clinical trial. Neuromuscul Disord (2010) 20(6):382–9. doi:10.1016/j.nmd.2010.03.011
107. Pereira RMR, de Carvalho JF, Paula AP, Zerbini C, Domiciano DS, Gonçalves H, et al. Guidelines for the prevention and treatment of glucocorticoid-induced osteoporosis. Rev Bras Reumatol (2012) 52(4):580–93. doi:10.1590/S0482-50042012000400009
108. Levitt M, Mosher MB, DeConti RC, Farber LR, Skeel RT, Marsh JC, et al. Improved therapeutic index of methotrexate with “leucovorin rescue”. Cancer Res (1973) 33(7):1729–34.
109. Saag KG, Teng GG, Patkar NM, Anuntiyo J, Finney C, Curtis JR, et al. American College of Rheumatology 2008 recommendations for the use of nonbiologic and biologic disease-modifying antirheumatic drugs in rheumatoid arthritis. Arthritis Rheum (2008) 59(6):762.784. doi:10.1002/art.23721
110. Kissel JT, Levy RJ, Mendell JR, Griggs RC. Azathioprine toxicity in neuromuscular disease. Neurology (1986) 36(1):35–9. doi:10.1212/WNL.36.1.35
111. Evans WE, Hon YY, Bomgaars L, Coutre S, Holdsworth M, Janco R, et al. Preponderance of thiopurine S-methyltransferase deficiency and heterozygosity among patients intolerant to mercaptopurine or azathioprine. J Clin Oncol (2001) 19(8):2293–301.
112. Booth RA, Ansari MT, Loit E, Tricco AC, Weeks L, Doucette S, et al. Assessment of thiopurine S-methyltransferase activity in patients prescribed thiopurines: a systematic review. Ann Intern Med (2011) 154(12):814–24. doi:10.7326/0003-4819-154-12-201106210-00009
113. Ostensen M, Forger F. Management of RA medications in pregnant patients. Nat Rev Rheumatol (2009) 5(7):382–90. doi:10.1038/nrrheum.2009.103
114. Dalakas MC. Intravenous immunoglobulin in autoimmune neuromuscular diseases. JAMA (2004) 291(19):2367–75. doi:10.1001/jama.291.19.2367
115. Dalakas MC. Mechanism of action of intravenous immunoglobulin and therapeutic considerations in the treatment of autoimmune neurologic diseases. Neurology (1998) 51(6 Suppl 5):S2–8. doi:10.1212/WNL.51.6_Suppl_5.S2
116. Nalotto L, Iaccarino L, Zen M, Gatto M, Borella E, Domenighetti M, et al. Rituximab in refractory idiopathic inflammatory myopathies and antisynthetase syndrome: personal experience and review of the literature. Immunol Res (2013) 56(2–3):362–70. doi:10.1007/s12026-013-8408-9
117. Limaye V, Hissaria P, Liew CL, Koszyka B. Efficacy of rituximab in refractory antisynthetase syndrome. Intern Med J (2012) 42(3):4–7. doi:10.1111/j.1445-5994.2011.02702.x
118. Aringer M. Rituximab therapy in patients with refractory dermatomyositis or polymyositis: differential effects in a real-life population. Rheumatology (Oxford) (2014) 53(9):1630–8. doi:10.1093/rheumatology/keu024
119. Vulliemoz S, Lurati-Ruiz F, Borruat F-X, Delavelle J, Koralnik IJ, Kuntzer T, et al. Favourable outcome of progressive multifocal leucoencephalopathy in two patients with dermatomyositis. J Neurol Neurosurg Psychiatry (2006) 77(9):1079–82. doi:10.1136/jnnp.2006.092353
120. Maeda K, Kimura R, Komuta K, Igarashi T. Cyclosporine treatment for polymyositis/dermatomyositis: is it possible to rescue the deteriorating cases with interstitial pneumonitis? Scand J Rheumatol (1997) 26(1):24–9. doi:10.3109/03009749709065660
121. Amato A. A randomized, pilot trial of etanercept in dermatomyositis. Ann Neurol (2011) 70(3):427–36. doi:10.1002/ana.22477
122. Zong M, Malmström V, Lundberg IE. Anakinra effects on T cells in patients with refractory idiopathic inflammatory myopathies. Ann Rheum Dis (2011) 70:A80–1. doi:10.1136/ard.2010.149013.31
123. Dorph C, Dastmalchi M, Alexanderson H, Ottosson C, Lindroos E, Nennesmo I, et al. Anakinra in patients with refractory idiopathic inflammatory myopathies. Arthritis Rheum (2009) 60:589. doi:10.1002/art.25669
124. Zong M, Dorph C, Dastmalchi M, Alexanderson H, Pieper J, Amoudruz P, et al. Anakinra treatment in patients with refractory inflammatory myopathies and possible predictive response biomarkers: a mechanistic study with 12 months follow-up. Ann Rheum Dis (2014) 73(5):913–20. doi:10.1136/annrheumdis-2012-202857
125. Dalakas MC, Rakocevic G, Schmidt J, Salajegheh M, McElroy B, Harris-Love MO, et al. Effect of Alemtuzumab (CAMPATH 1-H) in patients with inclusion-body myositis. Brain (2009) 132(6):1536–44. doi:10.1093/brain/awp104
126. Ruck T, Bittner S, Kuhlmann T, Wiendl H, Meuth SG. Long-term efficacy of alemtuzumab in polymyositis. Meuth Rheumatol (Oxford) (2015) 54(3):560–2. doi:10.1093/rheumatology/keu484
127. Sanz I, Yasothan U, Kirkpatrick P. Belimumab. Nat Rev Drug Discov (2011) 10(5):335–6. doi:10.1038/nrd3436
128. Higgs BW, Zhu W, Morehouse C, White WI, Brohawn P, Guo X, et al. A phase 1b clinical trial evaluating sifalimumab, an anti-IFN-monoclonal antibody, shows target neutralisation of a type I IFN signature in blood of dermatomyositis and polymyositis patients. Ann Rheum Dis (2013) 73(1):256–62. doi:10.1136/annrheumdis-2012-202794
129. Yoshimasu T, Ohtani T, Sakamoto T, Oshima A, Furukawa F. Topical FK506 (tacrolimus) therapy for facial erythematous lesions of cutaneous lupus erythematosus and dermatomyositis. Eur J Dermatol (2002) 12(1):50–2.
130. Marmor MF, Kellner U, Lai TY, Lyons JS, Mieler WF, American Academy of Ophthalmology. Revised recommendations on screening for chloroquine and hydroxychloroquine retinopathy. Ophthalmology (2011) 118(2):415–22. doi:10.1016/j.ophtha.2010.11.017
131. Balin SJ, Wetter DA, Andersen LK, Davis MD. Calcinosis cutis occurring in association with autoimmune connective tissue disease: the Mayo Clinic experience with 78 patients, 1996-2009. Arch Dermatol (2012) 148(4):455–62. doi:10.1001/archdermatol.2011.2052
132. Arabshahi B, Silverman RA, Jones OY, Rider LG. Abatacept and sodium thiosulfate for treatment of recalcitrant juvenile dermatomyositis complicated by ulceration and calcinosis. J Pediatr (2012) 160(3):520–2. doi:10.1016/j.jpeds.2011.11.057
133. Cox FM, Verschuuren JJ, Verbist BM, Niks EH, Wintzen AR, Badrising UA. Detecting dysphagia in inclusion body myositis. J Neurol (2009) 256(12):2009–13. doi:10.1007/s00415-009-5229-9
134. Dalakas MC, Sonies B, Dambrosia J, Sekul E, Cupler E, Sivakumar K. Treatment of inclusion-body myositis with IVIg: a double-blind, placebo-controlled study. Neurology (1997) 48(3):712–6. doi:10.1212/WNL.48.3.712
135. Cherin P, Pelletier S, Teixeira A, Laforet P, Simon A, Herson S, et al. Intravenous immunoglobulin for dysphagia of inclusion body myositis. Neurology (2002) 58(2):326. doi:10.1212/WNL.58.2.326
136. Walter MC, Lochmüller H, Toepfer M, Schlotter B, Reilich P, Schröder M, et al. High-dose immunoglobulin therapy in sporadic inclusion body myositis: a double-blind, placebo-controlled study. J Neurol (2000) 247(1):22–8. doi:10.1007/s004150050005
137. Marie I, Menard J-F, Hatron PY, Hachulla E, Mouthon L, Tiev K, et al. Intravenous immunoglobulins for steroid-refractory esophageal involvement related to polymyositis and dermatomyositis: a series of 73 patients. Arthritis Care Res (Hoboken) (2010) 62(12):1748–55. doi:10.1002/acr.20325
138. Langdon PC, Mulcahy K, Shepherd KL, Low VH, Mastaglia FL. Pharyngeal dysphagia in inflammatory muscle diseases resulting from impaired suprahyoid musculature. Dysphagia (2012) 27(3):408–17. doi:10.1007/s00455-011-9384-7
139. Oh TH, Brumfield KA, Hoskin TL, Stolp KA, Murray JA, Bassford JR. Dysphagia in inflammatory myopathy: clinical characteristics, treatment strategies, and outcome in 62 patients. Mayo Clin Proc (2007) 82(4):441–7. doi:10.4065/82.4.441
140. Oh TH, Brumfield KA, Hoskin TL, Kasperbauer JL, Basford JR. Dysphagia in inclusion body myositis: clinical features, management, and clinical outcome. Am J Phys Med Rehabil (2008) 87(11):883–9. doi:10.1097/PHM.0b013e31818a50e2
141. Liu LW, Tarnopolsky M, Armstrong D. Injection of botulinum toxin A to the upper esophageal sphincter for oropharyngeal dysphagia in two patients with inclusion body myositis. Can J Gastroenterol (2004) 18(6):397–9. doi:10.1155/2004/360537
142. Kotani T, Takeuchi T, Makino S, Hata K, Yoshida S, Nagai K, et al. Combination with corticosteroids and cyclosporin-A improves pulmonary function test results and chest HRCT findings in dermatomyositis patients with acute/subacute interstitial pneumonia. Clin Rheumatol (2011) 30(8):1021–8. doi:10.1007/s10067-011-1713-6
143. Marie I, Hatron PY, Dominique S, Cherin P, Mouthon L, Menard JF. Short-term and long-term outcomes of interstitial lung disease in polymyositis and dermatomyositis: a series of 107 patients. Arthritis Rheum (2011) 63(11):3439–47. doi:10.1002/art.30513
144. Varjú C, Pethö E, Kutas R, Czirják L. The effect of physical exercise following acute disease exacerbation in patients with dermato/polymyositis. Clin Rehabil (2003) 17(1):83–7. doi:10.1191/0269215503cr572oa
145. Alexanderson H, Lundberg IE. Exercise as a therapeutic modality in patients with idiopathic inflammatory myopathies. Curr Opin Rheumatol (2012) 24(2):201–7. doi:10.1097/BOR.0b013e32834f19f5
146. Omori CH, Silva CAA, Sallum AME, Rodrigues Pereira RM, Lúciade Sá Pinto A, Roschel H, et al. Exercise training in juvenile dermatomyositis. Arthritis Care Res (Hoboken) (2012) 64(8):1186–94. doi:10.1002/acr.21684
147. Dastmalchi M, Alexanderson H, Loell I, Ståhlberg M, Borg K, Lundberg IE, et al. Effect of physical training on the proportion of slow-twitch type I muscle fibers, a novel nonimmune-mediated mechanism for muscle impairment in polymyositis or dermatomyositis. Arthritis Care Res (2007) 57(7):1303–10. doi:10.1002/art.22996
148. Dalakas MC. Immunotherapy of inflammatory myopathies: practical approach and future prospects. Curr Treat Options Neurol (2011) 13(3):311–23. doi:10.1007/s11940-011-0119-8
149. Badrising UA, Maat-Schieman MLC, Ferrari MD, Zwinderman AH, Wessels JAM, Breedveld FC, et al. Comparison of weakness progression in inclusion body myositis during treatment with methotrexate or placebo. Ann Neurol (2002) 51(3):369–72. doi:10.1002/ana.10121
150. Lindberg C, Trysberg E, Tarkowski A, Oldfors A. Anti-T-lymphocyte globulin treatment in inclusion body myositis: a randomized pilot study. Neurology (2003) 61(2):260–2. doi:10.1212/01.WNL.0000071852.27182.C7
151. Barohn RJ, Herbelin L, Kissel JT, King W, McVey AL, Saperstein DS, et al. Pilot trial of etanercept in the treatment of inclusion-body myositis. Neurology (2006) 66(2 Suppl 1):S123–4. doi:10.1212/01.wnl.0000192258.32408.54
152. Rutkove SB, Parker RA, Nardin RA, Connolly CE, Felice KJ, Raynor EM. A pilot randomized trial of oxandrolone in inclusion body myositis. Neurology (2002) 58(7):1081–7. doi:10.1212/WNL.58.7.1081
153. Muscle T, Group S. Randomized pilot trial of betaINF1a (Avonex) in patients with inclusion body myositis. Neurology (2001) 57(9):1566–70. doi:10.1212/WNL.57.9.1566
154. Muscle T, Group S. Randomized pilot trial of high-dose betaINF-1a in patients with inclusion body myositis. Neurology (2004) 63(4):718–20. doi:10.1212/01.WNL.0000134675.98525.79
155. Dalakas MC, Koffman B, Fujii M, Spector S, Sivakumar K, Cupler E. A controlled study of intravenous immunoglobulin combined with prednisone in the treatment of IBM. Neurology (2001) 56(3):323–7. doi:10.1212/WNL.56.3.323
156. Machado P, Miller A, Herbelin L, He J, Noel J, Wang Y, et al. LB0002 safety and tolerability of arimoclomol in patients with sporadic inclusion body myositis: a randomised, double-blind, placebo-controlled, phase IIa proof-of-concept trial. Ann Rheum Dis (2014) 72(Suppl 3):A164–164. doi:10.1136/annrheumdis-2013-eular.527
157. Amato AA, Sivakumar K, Goyal N, David WS, Salajegheh M, Praestgaard J, et al. Treatment of sporadic inclusion body myositis with bimagrumab. Neurology (2014) 83(24):2239–46. doi:10.1212/WNL.0000000000001070
158. Huang JL. Long-term prognosis of patients with juvenile dermatomyositis initially treated with intravenous methylprednisolone pulse therapy. Clin Exp Rheumatol (1999) 17(5):621–4.
159. Bolosiu HD, Man L, Rednic S. The effect of methylprednisolone pulse therapy in polymyositis/dermatomyositis. Adv Exp Med Biol (1999) 455:349–57. doi:10.1007/978-1-4615-4857-7_54
160. Al-Mayouf S, Al-Mazyed AA, Bahabri S. Efficacy of early treatment of severe juvenile dermatomyositis with intravenous methylprednisolone and methotrexate. Clin Rheumatol (2000) 19(2):138–41. doi:10.1007/s100670050032
161. Sokoloff MC, Goldberg LS, Pearson CM. Treatment of corticosteroid-resistant polymyositis with methotrexate. Lancet (1971) 1(7688):14–6. doi:10.1016/S0140-6736(71)80005-3
162. Metzger AL, Bohan A, Goldberg LS, Bluestone R, Pearson CM. Polymyositis and dermatomyositis: combined methotrexate and corticosteroid therapy. Ann Intern Med (1971) 81(2):182–9. doi:10.7326/0003-4819-81-2-182
163. Giannini M, Callen JP. Treatment of dermatomyositis with methotrexate and prednisone. Arch Dermatol (1979) 115(10):1251–2. doi:10.1001/archderm.115.10.1251
164. Cagnoli M, Marchesoni A, Tosi S. Combined steroid, methotrexate and chlorambucil therapy for steroid-resistant dermatomyositis. Clin Exp Rheumatol (1991) 9(6):658–9.
165. Bunch TW. Prednisone and azathioprine for polymyositis: long-term followup. Arthritis Rheum (1981) 24(1):45–8. doi:10.1002/art.1780240107
166. Bunch TW, Worthington JW, Combs JJ, Ilstrup DM, Engel AG. Azathioprine with prednisone for polymyositis. A controlled, clinical trial. Ann Intern Med (1980) 92(3):365–9. doi:10.7326/0003-4819-92-3-365
167. Villalba L, Hicks JE, Adams EM, Sherman JB, Gourley MF, Leff RL, et al. Treatment of refractory myositis: a randomized crossover study of two new cytotoxic regimens. Arthritis Rheum (1998) 41(3):392–9. doi:10.1002/1529-0131(199803)41:3<392::AID-ART3>3.3.CO;2-O
168. Vencovský J, Jarosová K, Machácek S, Studýnková J, Kafková J, Bartůnková J, et al. Cyclosporine A versus methotrexate in the treatment of polymyositis and dermatomyositis. Scand J Rheumatol (2000) 29(2):95–102. doi:10.1080/030097400750001897
169. Qushmaq KA, Chalmers A, Esdaile JM. Cyclosporin A in the treatment of refractory adult polymyositis/dermatomyositis: population based experience in 6 patients and literature review. J Rheumatol (2000) 27(12):2855–9.
170. Oddis CV, Sciurba FC, Elmagd KA, Starzl TE. Tacrolimus in refractory polymyositis with interstitial lung disease. Lancet (1999) 353(9166):1762–3. doi:10.1016/S0140-6736(99)01927-3
171. Dalakas MC, Illa I, Dambrosia JM, Soueidan SA, Stein DP, Otero C, et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med (1993) 329(27):1993–2000. doi:10.1056/NEJM199312303292704
172. Miller FW, Leitman SF, Cronin ME, Hicks JE, Leff RL, Wesley R, et al. Controlled trial of plasma exchange and leukapheresis in polymyositis and dermatomyositis. N Engl J Med (1992) 326(21):1380–4. doi:10.1056/NEJM199205213262102
173. Majithia V, Harisdangkul V. Mycophenolate mofetil (CellCept): an alternative therapy for autoimmune inflammatory myopathy. Rheumatology (2005) 44(3):386–9. doi:10.1093/rheumatology/keh499
174. Edge JC, Outland JD, Dempsey JR, Callen JP. Mycophenolate mofetil as an effective corticosteroid-sparing therapy for recalcitrant dermatomyositis. Arch Dermatol (2006) 142(1):65–9. doi:10.1001/archderm.142.1.65
175. Morganroth PA, Kreider ME, Werth VP. Mycophenolate mofetil for interstitial lung disease in dermatomyositis. Arthritis Care Res (Hoboken) (2010) 62(10):1496–501. doi:10.1002/acr.20212
176. Gelber AC, Nousari HC, Wigley FM. Mycophenolate mofetil in the treatment of severe skin manifestations of dermatomyositis: a series of 4 cases. J Rheumatol (2000) 27(6):1542–5.
177. Mok CC, Ho LY, To CH. Rituximab for refractory polymyositis: an open-label prospective study. J Rheumatol (2007) 34(9):1864–8.
178. Cooper MA, Willingham DL, Brown DE, French AR, Shih FF, White AJ. Rituximab for the treatment of juvenile dermatomyositis: a report of four pediatric patients. Arthritis Rheum (2007) 56(9):3107–11. doi:10.1002/art.22856
179. Levine TD. Rituximab in the treatment of dermatomyositis: an open-label pilot study. Arthritis Rheum (2005) 52(2):601–7. doi:10.1002/art.20849
180. Oddis CV, Reed AM, Aggarwal R, Rider LG, Ascherman DP, Levesque MC, et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo-phase trial. Arthritis Rheum (2013) 65(2):314–24. doi:10.1002/art.37754
181. Mahler EAM, Blom M, Voermans NC, van Engelen BGM, van Riel PLCM, Vonk MC. Rituximab treatment in patients with refractory inflammatory myopathies. Rheumatology (Oxford) (2011) 50(12):2206–13. doi:10.1093/rheumatology/ker088
182. Kono DH, Gilbert RC. Successful IV pulse cyclophosphamide in refractory PM in 3 patients with SLE. J Rheumatol (1990) 17(7):982–3.
183. Yamasaki Y, Yamada H, Yamasaki M, Ohkubo M, Azuma K, Matsuoka S, et al. Intravenous cyclophosphamide therapy for progressive interstitial pneumonia in patients with polymyositis/dermatomyositis. Rheumatology (Oxford) (2007) 46(1):124–30. doi:10.1093/rheumatology/kel112
184. Niakan E, Pitner SE, Whitaker JN, Bertorini TE. Immunosuppressive agents in corticosteroid-refractory childhood dermatomyositis. Neurology (1980) 30(3):286–91. doi:10.1212/WNL.30.3.286
185. Fries JF, Sharp GC, McDevitt HO, Holman HR. Cyclophosphamide therapy in systemic lupus erythematosus and polymyositis. Arthritis Rheum (1973) 16(2):154162. doi:10.1002/art.1780160204
186. Labioche I, Liozon E, Weschler B, Loustaud-Ratti V, Soria P, Vidal E. Refractory polymyositis responding to infliximab: extended follow-up. Rheumatology (Oxford) (2004) 43(4):531–2. doi:10.1093/rheumatology/keh079
187. Hengstman GJD, Van Den Hoogen FHJ, Barrera P, Netea MG, Pieterse A, Van De Putte LBA, et al. Successful treatment of dermatomyositis and polymyositis with anti-tumor-necrosis-factor-alpha: preliminary observations. Eur Neurol (2003) 50(1):10–5. doi:10.1159/000070852
Keywords: idiopathic inflammatory mypathies, polymyositis, dermatomyositis, necrotizing myopathy, inclusion body myositis, antiSRP, myositis specific antibodies
Citation: Malik A, Hayat G, Kalia JS and Guzman MA (2016) Idiopathic Inflammatory Myopathies: Clinical Approach and Management. Front. Neurol. 7:64. doi: 10.3389/fneur.2016.00064
Received: 15 December 2015; Accepted: 12 April 2016;
Published: 20 May 2016
Edited by:
Aleksandar Beric, New York University School of Medicine, USAReviewed by:
Rositsa Nikolova Baltadzhieva, New York University School of Medicine, USAKiril Kiprovski, New York University Medical Center, USA
Copyright: © 2016 Malik, Hayat, Kalia and Guzman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Asma Malik, YW1hbGlrMTJAc2x1LmVkdQ==;
Ghazala Hayat, aGF5YXRtMkBzbHUuZWR1