 Paula Garcia-Esparcia1,2
Paula Garcia-Esparcia1,2 Irene López-González1,2
Irene López-González1,2 Oriol Grau-Rivera3
Oriol Grau-Rivera3 María Francisca García-Garrido1Anusha Konetti1
María Francisca García-Garrido1Anusha Konetti1 Franc Llorens4
Franc Llorens4 Saima Zafar4Margarita Carmona1,2
Saima Zafar4Margarita Carmona1,2 José Antonio del Rio2,5
José Antonio del Rio2,5 Inga Zerr4Ellen Gelpi3
Inga Zerr4Ellen Gelpi3 Isidro Ferrer1,2,6*
Isidro Ferrer1,2,6*
- 1Institute of Neuropathology, Service of Pathologic Anatomy, IDIBELL-Hospital Universitari de Bellvitge, Hospitalet de Llobregat, Barcelona, Spain
- 2CIBERNED, Network Centre for Biomedical Research of Neurodegenerative Diseases, Institute Carlos III, Madrid, Spain
- 3Neurological Tissue Bank of the Biobanc-Hospital Clínic-Institut d’Investigacions Biomèdiques August Pi I Sunyer (IDIBAPS), Barcelona, Spain
- 4Department of Neurology, Clinical Dementia Center, University Medical School, Georg-August University, German Center for Neurodegenerative Diseases (DZNE), Göttingen, Germany
- 5Molecular and Cellular Neurobiotechnology, Department of Cell Biology, Institute of Bioengineering of Catalonia (IBEC), Parc Científic de Barcelona, University of Barcelona, Barcelona, Spain
- 6Department of Pathology and Experimental Therapeutics, L’Hospitalet de Llobregat, University of Barcelona, Barcelona, Spain
Objectives: The goal of this study was to assess mitochondrial function, energy, and purine metabolism, protein synthesis machinery from the nucleolus to the ribosome, inflammation, and expression of newly identified ectopic olfactory receptors (ORs) and taste receptors (TASRs) in the frontal cortex of typical cases of dementia with Lewy bodies (DLB) and cases with rapid clinical course (rpDLB: 2 years or less) compared with middle-aged non-affected individuals, in order to learn about the biochemical abnormalities underlying Lewy body pathology.
Methods: Real-time quantitative PCR, mitochondrial enzymatic assays, and analysis of β-amyloid, tau, and synuclein species were used.
Results: The main alterations in DLB and rpDLB, which are more marked in the rapidly progressive forms, include (i) deregulated expression of several mRNAs and proteins of mitochondrial subunits, and reduced activity of complexes I, II, III, and IV of the mitochondrial respiratory chain; (ii) reduced expression of selected molecules involved in energy metabolism and increased expression of enzymes involved in purine metabolism; (iii) abnormal expression of nucleolar proteins, rRNA18S, genes encoding ribosomal proteins, and initiation factors of the transcription at the ribosome; (iv) discrete inflammation; and (v) marked deregulation of brain ORs and TASRs, respectively. Severe mitochondrial dysfunction involving activity of four complexes, minimal inflammatory responses, and dramatic altered expression of ORs and TASRs discriminate DLB from Alzheimer’s disease. Altered solubility and aggregation of α-synuclein, increased β-amyloid bound to membranes, and absence of soluble tau oligomers are common in DLB and rpDLB. Low levels of soluble β-amyloid are found in DLB. However, increased soluble β-amyloid 1–40 and β-amyloid 1–42, and increased TNFα mRNA and protein expression, distinguish rpDLB.
Conclusion: Molecular alterations in frontal cortex in DLB involve key biochemical pathways such as mitochondria and energy metabolism, protein synthesis, purine metabolism, among others and are accompanied by discrete innate inflammatory response.
Introduction
Dementia with Lewy bodies (DLB) is the second most common neurodegenerative dementia in the elderly, clinically manifested by fluctuating cognition with pronounced variation in attention and alertness, recurrent visual hallucinations which are typically well formed and detailed, and spontaneous motor features of parkinsonism; repeated falls, syncope, transient loss of consciousness, systematized delusions, hallucinations in other modalities, and neuroleptic sensitivity are not uncommon (1–4). These symptoms are preceded by rapid eye movement sleep behavior disorder, psychiatric symptoms, loss of smell, and dysautonomia, together with occipital hypo-metabolism, hallucinations, and cognitive impairment (5, 6).
Dementia with Lewy bodies is pathologically characterized by Lewy bodies and Lewy neurites in the brainstem, limbic system, and cortical areas (2, 7, 8). The main pathological change is the production and accumulation, in Lewy bodies and neurites, of abnormal α-synuclein, which is phosphorylated, nitrated, and truncated, has abnormal solubility, prompts the production of oligomeric species, aggregates into fibrils and is ubiquitinated (9–19). For these reasons, DLB is classified among α-synucleinopathies with Lewy bodies or Lewy body diseases (LBDs), together with Parkinson’s disease (PD) (11). Other changes in DLB are neuron loss, microvacuolation, and Alzheimer’s disease (AD) pathology distinguished by β-amyloid deposition in the form of diffuse and senile plaques, as well as early changes of neurofibrillary tangle (NFT) pathology (8).
Whether the amount of α-synuclein pathology (i.e., Lewy bodies and neurites) in cerebral cortex is predictable of dementia in LBDs is a matter of controversy (20–23). Cholinergic and dopaminergic denervation of the neocortex probably accounts at least in part for cognitive deficits in LBDs (24–29). Concomitant pathologies have also been suggested to explain variations in the degree of cognitive impairment in DLB (30–32).
However, deficits in neurotransmission are not limited to dopaminergic and cholinergic systems. Synapses are primarily damaged in the neocortex in DLB (33–38). Synaptic damage is probably related to toxic α-synuclein oligomers and pore formation (39, 40). Moreover, synaptic alterations are accompanied by abnormalities in neurotransmitter signaling (41, 42) in a way similar to that reported in other α-synucleinopathies (43–45).
Additional molecular alterations converge in the pathogenesis of DLB, including impaired autophagy and ubiquitin-proteasome system of protein (46–50), as well as altered responses to protein misfolding (51). Preliminary studies have shown impaired mitochondrial activity and oxidative damage involving proteins, lipids, and DNA in the neocortex in DLB (52, 53); α-synuclein is one of the targets of oxidative damage in the frontal cortex in DLB (54).
The average survival time for typical DLB from the beginning of symptoms is about 5–8 years (55). However, some cases have a rapid course and are considered clinically to be in the group of rapidly progressive dementias (56, 57). DLB with rapid progression has been named rapid DLB (rpDLB) (55, 58). The mean duration of rpDLB is about 9 months; delirium, visual hallucinations, delusions, and fluctuating cognitive impairment, followed by parkinsonism and myoclonus, are the predominant symptoms (58).
Our hypothesis is that alterations of several metabolic pathways converge in the pathogenesis of DLB and that impaired mitochondria and energy metabolism, purine metabolism, protein synthesis, and inflammation may be important factors in the pathogenesis of DLB. In the same line, although neuropathological studies have not shown differences between DLB and rpDLB (58), biochemical alterations probably discriminate between DLB and rpDLB.
The present DLB-centered study was undertaken to analyze (i) levels of selected mRNAs and proteins of subunits of the five mitochondrial complexes and genes linked to energy metabolism; (ii) expression of genes encoding enzymes involved in purine metabolism; (iii) mRNA and protein expression of selected nucleolar proteins, rRNAs, and genes encoding ribosomal proteins, and protein expression of initiation and elongation factors of protein transcription at the ribosome; (iv) cytokines and mediators of the inflammatory response; and (v) gene expression of particular brain receptors involved in olfaction and taste known to be altered in other neurodegenerative diseases with abnormal protein aggregates. All these pathways were assessed in the frontal cortex of cases with typical course (DLB) and in cases with rapid course (rpDLB) compared with middle-aged (MA) individuals to identify molecular factors linked to Lewy body pathology.
Materials and Methods
Human Cases
Brain tissue was obtained from the Institute of Neuropathology HUB-ICO-IDIBELL Biobank and the Hospital Clinic-IDIBAPS Biobank following the guidelines of Spanish legislation on this matter (Real Decreto de Biobancos 1716/2011) and approval of the local ethics committees. Processing of brain tissue has been detailed elsewhere (59, 60). The postmortem interval between death and tissue processing was between 3 and 15.30 h. One hemisphere was immediately cut in coronal sections, 1 cm thick, and selected areas of the encephalon were rapidly dissected, frozen on metal plates over dry ice, placed in individual air-tight plastic bags and stored at −80°C until use for biochemical studies. The other hemisphere was fixed by immersion in 4% buffered formalin for 3 weeks for morphological studies. Neuropathological diagnosis in all cases was based on the routine study of 20 selected de-waxed paraffin sections of representative regions of the cerebral cortex, diencephalon, thalamus, brain stem, and cerebellum, which were stained with hematoxylin and eosin, and Klüver-Barrera, or processed for immunohistochemistry for microglia (antibodies Iba1 and CD68), glial fibrillary acidic protein, β-amyloid with antibodies Aβ clone 6 F/3D (diluted 1:50, Dako, Carpinteria, CA, USA), Aβ40 (diluted 1:100, Merck Millipore, Billerica, MA, USA), and Aβ42 (diluted 1:50, Merck Millipore), phospho-tau (clone AT8), α-synuclein, TDP-43, ubiquitin, and p62 using EnVision + System peroxidase (Dako), and diaminobenzidine and H2O2. DLB cases (n = 13) were neuropathologically categorized following current staging classifications for LBD (22, 61, 62), stages of of NFT pathology (21, 63), and phases of AD-related β-amyloid plaques (64), and a final ABC score was assigned according to current consensus guidelines (65). Neuropathological data were scored by two independent observers. Based on clinical criteria DLB cases were categorized as typical DLB (DLB, n = 9: eight men, one woman, age 76.4 ± 5.7 years) or rapid DLB (rpDLB, n = 4, two men and two women, age 73.7 ± 2.2) on the basis of the natural clinical course. rpDLB here was defined as 2 years or less of disease duration from the first symptom to death (58). Cases with associated pathologies such as vascular diseases excepting mild atherosclerosis and arteriolosclerosis, TDP-43 proteinopathy, infections of the nervous system, brain neoplasms, systemic and central immune diseases, metabolic syndrome, and hypoxia were excluded from the present study. MA cases (n = 12: seven men and five women, age 59.9 ± 15.59) had not suffered from neurologic, psychiatric, or metabolic diseases (including metabolic syndrome) and did not have abnormalities in the neuropathological examination excepting stages I–II of NFT pathology and phases 1–2 of β-amyloid plaques.
Quantification of β-amyloid burden was carried out with a Nikon Eclipse E800 microscope (4× objective; Nikon Imaging Inc., Tokyo, Japan). The cortical total Aβ burden was calculated as the percentage of the area of Aβ deposition in plaques with respect to the total area in 9–10 pictures taken at random from frontal cortex in every case. β-Amyloid quantification was assessed using the Adobe Photoshop CS5 software (Adobe Systems Inc., San Jose, CA, USA).
A summary of cases is shown in Table 1.
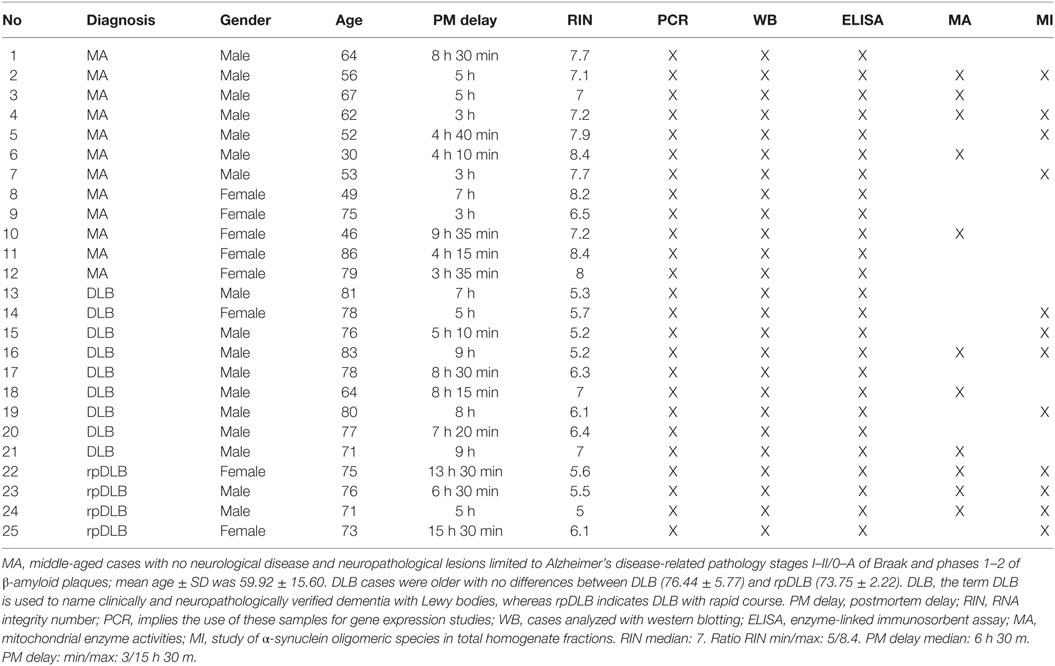
Table 1. Summary of cases used in the present study.
RNA Purification
Purification of RNA from the right frontal cortex area 8 was carried out using RNeasy Lipid Tissue Mini Kit (Qiagen, Hilden, Germany) following the protocol provided by the manufacturer combined with DNase digestion to avoid extraction and later amplification of genomic DNA. The concentration of each sample was obtained from A260 measurements with NanoDrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA, USA). RNA integrity was tested using the Agilent 2100 BioAnalyzer (Agilent, Santa Clara, CA, USA) (66). Values of RNA integrity number (RIN) are shown in Table 1. Special care was taken to assess pre-mortem and postmortem factors, which may interfere with RNA processing (67).
Bivariate analyses were carried out to detect association of our variables with potential confounding factors (age, postmortem delay, and RIN) using Spearman or Pearson correlations for quantitative variables. Stastical analysis was performed with GraphPad Prism version 5.00 and SPSS 19. Postmortem delay had no effect on RIN values in the present series.
Retrotranscription Reaction
Retrotranscription reaction of RNA samples was carried out with the High-Capacity cDNA Archive kit (Applied Biosystems, Foster City, CA, USA) following the guidelines provided by the supplier, and using Gene Amp® 9700 PCR System thermocycler (Applied Biosystems). A parallel reaction for one RNA sample was processed in the absence of reverse transcriptase to rule out DNA contamination.
Real-time PCR
Real-time quantitative PCR (RT-qPCR) assays were conducted in duplicate on 1,000 ηg of cDNA samples obtained from the retrotranscription reaction, diluted 1:20 in 384-well optical plates (Kisker Biotech, Steinfurt, GE) utilizing the ABI Prism 7900 HT Sequence Detection System (Applied Biosystems). Parallel amplification reactions were carried out using 20× TaqMan Gene Expression Assays and 2× TaqMan Universal PCR Master Mix (Applied Biosystems) (66). Genes analyzed with the corresponding abbreviations and TaqMan probes used in the study are shown in Table 2.
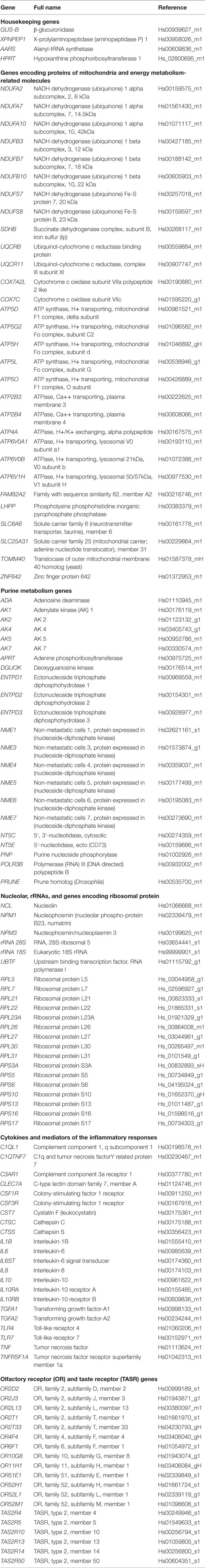
Table 2. Abbreviations, full names, and TaqMan probes used to assess gene expression in the frontal cortex of MA and DLB cases in the present study.
The selection of probes was based on criteria covering a larger project geared to analyzing similar expression of the same molecules related to inflammation, mitochondria, energy metabolism, purine metabolism, protein synthesis and selected receptors in several diseases including AD, PD, Creutzfeldt-Jakob’s disease, certain tauopathies, in addition to DLB, in all cases using the same probes and methods in an attempt to learn about commonalities and differences among these diseases in a particular brain region. Our idea is that all these pathways are altered in most neurodegenerative diseases with abnormal protein aggregates, but alterations are region-, stage-, and disease-specific.
Parallel assays for each sample were carried out using β-glucuronidase (GUS-β), X-prolyl aminopeptidase (aminopeptidase P) 1 (XPNPEP1), AARS (alanyl-transfer RNA synthase), and HPRT (hypoxanthine-guanine phosphoribosyltransferase) probes for normalization. The selection of these housekeeping genes was based on previous data showing low vulnerability in the brain of several human neurodegenerative diseases (68, 69). The reactions were performed using the following parameters: 50°C for 2 min, 95°C for 10 min, 40 cycles at 95°C for 15 s, and 60°C for 1 min. TaqMan PCR data were captured using the Sequence Detection Software (SDS version 2.2, Applied Biosystems). Subsequently, threshold cycle (CT) data for each sample were analyzed with the double-delta CT (ΔΔCT) method (66). First, delta CT (ΔCT) values were calculated as the normalized CT values for each target gene in relation to the mean values of GUS-β, XPNPEP1, AARS, and HPRT. Second, ΔΔCT values were obtained with the ΔCT of each sample minus the mean ΔCT of the population of control samples (calibrator samples). The fold change was determined using the equation 2−ΔΔCT.
Gel Electrophoresis and Western Blotting from Total Homogenate
Tissue was processed as reported elsewhere (66). A total of 0.1 g of tissue of samples from frontal cortex area 8 were lysed with a glass homogenizer in Mila lysis buffer (0.5M Tris at pH 7.4 containing 0.5 methylenediaminetetraacetic acid at pH 8.0, 5M NaCl, 0.5% Na deoxycholic acid, 0.5% Non-idet P-40, 1 mM phenylmethylsulfonyl fluoride, bi-distilled water, and protease and phosphatase inhibitor cocktails (Roche Molecular Systems, Pleasanton, CA, USA)), and then centrifuged for 15 min at 13,000 rpm at 4°C (Ultracentrifuge Beckman with 70Ti rotor, CA, USA). Protein concentration was measured with SmartspectTMplus spectrophotometer (Bio-Rad, CA, USA) using the Bradford method (Merck, Darmstadt, Germany). Samples containing 15 μg of protein and the standard Precision Plus ProteinTM Dual Color (Bio-Rad) were loaded onto 10–15% acrylamide gels. Proteins were separated with sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electrophoretically transferred to nitrocellulose membranes using the Trans-Blot®TurboTM transfer system (Bio-Rad) at 200 mA/membrane for 40 min. Non-specific bindings were blocked by incubation with 5% milk in Tris-buffered saline (TBS) containing 0.1% Tween for 1 h at room temperature. After washing, the membranes were incubated at 4°C overnight with one of the primary antibodies (Table S1 in Supplementary Material) in TBS containing 3% albumin and 0.1% Tween. These membranes were incubated for 1 h with the appropriate HRP-conjugated secondary antibody (1:2,000, Dako, Glostrup, Denmark), and the immune complexes were revealed with a chemiluminescence reagent (ECL, Amersham, GE Healthcare, Buckinghamshire, UK). Monoclonal anti-β-actin antibody diluted 1:30,000 (β-Actin, A5316; Sigma-Aldrich, St. Louis, MO, USA) was blotted to control protein loading.
Mitochondrial Enzymatic Activities
The activities of mitochondrial complexes I, II, III, IV, and V were analyzed using commercial kits following the manufacturers’ instructions (mitochondrial complex I, II, IV, and V: Novagen, Merck Biosciences, Darmstadt, Germany; mitochondrial complex III: MyBiosource, CA, USA). Isolation of mitochondria was carried out as reported in detail elsewhere (70). Briefly, mitochondria were extracted from frozen human brain tissue (100 mg) under ice-cold conditions. Tissues were minced in ice-cold isolation buffer (IB), and then homogenized and centrifuged at 1,000 × g for 10 min. The supernatant (S1) was conserved. The pellet was washed with two volumes of IB and centrifuged again under the same conditions. This last supernatant (S2) was combined with S1. Centrifugation at 10,000 × g for 10 min at 4°C resulted in the mitochondria-enriched pellet. The pellet was finally re-suspended in one volume of IB and stored at −80°C. Protein concentration was measured by Smartspect™ plus spectrophotometer (Bio-Rad, CA, USA) using the Bradford method (Merck, Darmstadt, Germany). Twenty-five micrograms of mitochondria was loaded into each well. Activity of citrate synthase was evaluated following validated protocols (71) with slight modifications. The activity of citrate synthase was determined as the rate of reduction of DTNB [5′, 5′-dithiobis (2-nitrobenzoic acid)] to thionitrobenzoic acid at 412 nm. For this purpose, 25 μg of mitochondria was added to a 1ml mixture containing 500 μl of Tris (200mM, pH 8.0) with Triton X-100 [0.2% (vol/vol)], 100 μl of DTNB, and 30 μl of 10mM Acetyl CoA, and then the final volume was adjusted to 950 l with distilled water. The reaction was started by the addition of 50 l of 10 mM oxalacetic acid. The increase in absorbance at 412 nm was read for 3 min at room temperature with a DU 800UV/Visible spectrophotometer (Beckman Coulter, CA, USA) in 1 ml polystyrene or methacrylate cuvettes (72).
Concentration of β-Amyloid 1–40 (Aβ40) and β-Amyloid 1–42 (Aβ42)
Frozen brain samples were homogenized in TBS buffer composed of 140 mM NaCl, 3 mM KCl, 25 mM Tris–HCl pH 7.4, and 5 mM ethylene-diamine-tetra-acetic acid (EDTA) with a cocktail of protease inhibitors (Roche Molecular Systems, Pleasanton, CA, USA), and ultra-centrifuged at 100,000 × g for 1 h at 4°C. The supernatant was the soluble fraction used for amyloid quantification, and the protein of this fraction was measured with BCA. The detection and measurement of β-amyloid 1–40 (Aβ40) and β-amyloid 1–42 (Aβ42) were carried out by enzyme-linked immune-absorbent assay with the corresponding detection kits (Invitrogen, Camarillo, CA, USA), following the instructions of the supplier. TBS-soluble Aβ40 and Aβ42 levels were normalized to the total amount of protein from each individual sample (73).
Quantification of Membrane-Associated β-Amyloid
Frozen samples were homogenized in TBS with a cocktail of protease and phosphatase inhibitors (Roche Molecular Systems). Homogenates were centrifuged at 14,000 × g for 30 min at 4°C. The pellet was re-suspended in 2% SDS and centrifuged at 14,000 × g for 30 min at 4°C. The supernatant was membrane-associated Aβ and the protein of this fraction was measured with BCA method (Thermo Scientific, USA). Proteins were separated in SDS-polyacrylamide gel electrophoresis. Thirty-five micrograms of protein was loaded onto a precast NuPAGE 4–12% Bis-Tris gel system (Invitrogen, MA, USA) with MES buffer (Invitrogen, MA, USA). The proteins were transferred to nitrocellulose membranes, 200 mA/membrane, for 90 min. Then membranes were boiled with PBS 1× for 15 min, and non-specific bindings were blocked by incubation in 5% non-fat dry milk in TBS containing 0.2% Tween for 1 h at room temperature. After washing, the membranes were incubated at 4°C overnight with primary antibodies to human amyloid-beta protein, clone: 4G8 (1:500, Signet, MA, USA) and 6E10 (1:2,000, Covance, NJ, USA) in TBS containing 5% albumin and 0.2% Tween. After washing, the membranes were incubated at 4°C overnight with the primary antibody to human amyloid-beta protein, clone: 4G8 (1:500, Signet, MA, USA) in TBS containing 5% albumin and 0.2% Tween. Membranes were then incubated for 1 h with the appropriate HRP-conjugated secondary antibody (1:2,000, Dako, DK), and the immune complexes were visualized with a chemiluminescence reagent (ECL, Amersham, GB). β-Actin was used as a control of protein loading (73). Using this protocol, membrane-enriched fractions may also contain small amounts of TBS-soluble β-amyloid.
Oligomeric Tau Species
Frozen samples were homogenized in lysis buffer: 100 mM Tris (pH 7.0), 100 mM NaCl, 10 mM EDTA, 0.5% NP-40, and 0.5% sodium deoxycholate plus protease and phosphatase inhibitors (Roche Molecular Systems). After centrifugation at 14,000 × g for 20 min at 4°C (Ultracentrifuge Beckman with 70Ti rotor), supernatants were quantified for protein concentration (BCA), mixed with SDS-PAGE sample buffer, boiled, and separated to 8% SDS-PAGE gels. The proteins were transferred to nitrocellulose membranes, 200 mA/membrane, for 90 min. The membranes were blocked with 5% non-fat milk in TBS containing 0.2% Tween for 1 h at room temperature. After washing, the membranes were incubated at 4°C overnight with the primary antibody anti-tau-5 (1:1,000, Thermo-Fisher, MA, USA) in TBS containing 5% albumin and 0.2% Tween. Membranes were then incubated for 1 h with the HRP-conjugated secondary anti-mouse antibody (1:2,000, Dako, DK), and the immune complexes were visualized with ECL. β-Actin was used as a control of protein loading (74). No attempt was made to analyze sarkosyl-insoluble fractions as oligomeric tau species are visualized with the utilized protocol and the identification of tau bands was not an objective of the present study.
α-Synuclein Oligomeric Species in Total Homogenate Fractions
Frozen samples were homogenized in a glass homogenizer, in 750 μl of ice-cold PBS+ (sodium phosphate buffer pH 7.0, plus protease inhibitors), sonicated, and centrifuged at 2,700 × g at 4°C for 10 min. The pellet was discarded and the resulting supernatant was ultra-centrifuged at 43,000 × g at 4°C for 1 h. The supernatant (S2) was kept as the PBS-soluble fraction. The resulting pellet was re-suspended in a solution of PBS, pH 7.0, containing 0.5% sodium deoxycholate, 1% Triton and 0.1% SDS, and this was ultra-centrifuged at 43,000 × g at 4°C for 1 h. The resulting supernatant (S3) was kept as the deoxycholate-soluble fraction. The corresponding pellet was re-suspended in a solution of 2% SDS in PBS and maintained at room temperature for 30 min. Afterward, the samples were centrifuged at 133,000 × g at 25°C for 1 h and the resulting supernatant (S4) was the SDS-soluble fraction. Equal amounts of each fraction were mixed with reducing sample buffer and processed in parallel for 10% SDS-PAGE electrophoresis and western blotting. Membranes were incubated with anti-α-synuclein oligomer-specific antibody (Agrisera, Vännäs, Sweden) at a dilution of 1:1,000. The protein bands were visualized with the ECL method (66).
Statistical Analysis
The normality of distribution of the mean fold-change values obtained by RT-qPCR for every region and stage between controls and DLB cases was analyzed with the Kolmogorov–Smirnov test. The non-parametric Kruskal–Wallis test was performed to compare groups when the samples did not follow a normal distribution, whereas the one-way ANOVA with post hoc Tukey’s range test for multiple comparisons was used for normal variables. Statistical analysis was performed with GraphPad Prism version 5.01 (La Jolla, CA, USA) and Statgraphics Statistical Analysis and Data Visualization Software version 5.1 (Warrenton, VA, USA). Differences between groups were considered statistically significant at p-values: *p < 0.05, **p < 0.01, and ***p < 0.001.
Because of the large number of parameters and regions analyzed, multiple comparison tests and false discovery rate might be employed (75, 76). However, due to the limited number of cases, the possibility to obtain significant results minimizes (77). Therefore, present quantitative results must be considered exploratory.
Densitometry of western blot bands was assessed with the TotalLab program (TotalLab Quant, Newcastle, UK) and then subsequently analyzed with GraphPad Prism, Statgraphics Statistical Analysis and Data Visualization Software version 5.1 (VA, USA) by one-way ANOVA with post hoc Tukey’s range test for multiple comparisons. Differences were considered statistically significant at p-values: *p < 0.05, **p < 0.01, and ***p < 0.001.
The enzymatic activities for each mitochondrial complex were expressed as a rate of nanomoles per minute per milligram of mitochondrial protein per protein concentration normalized with the mitochondrial complex activity rate of citrate synthase activity. Data were presented as mean ± SEM for all the experiments. All the data were analyzed with Student’s t-test using GraphPad Prism version 5.01 (La Jolla, CA, USA) and Statgraphics Statistical Analysis and Data Visualization Software version 5.1 (Warrenton, VA, USA). In all experimental procedures the significance level was set at *p < 0.05, **p < 0.01, and ***p < 0.001.
Results
General Neuropathological Findings
Brain weight and neuropathological characteristics in the frontal cortex including neuron loss, astrocytic gliosis, microgliosis, spongiosis, diffuse plaques, senile plaques, β-amyloid angiopathy, and α-synuclein aggregates (Lewy bodies and Lewy neurites) were assessed in DLB and rpDLB cases. Thal phase of β-amyloid deposition, Braak stages of NFT pathology, CERAD global, ABC classification, and LBD stage were also considered to frame AD- and LBD-related pathology in every case. Results are summarized in Table 3. A remarkable observation was the discrete microglial response, as revealed with Iba1 and CD68 antibodies, in the frontal cortex in DLB and rpDLB.
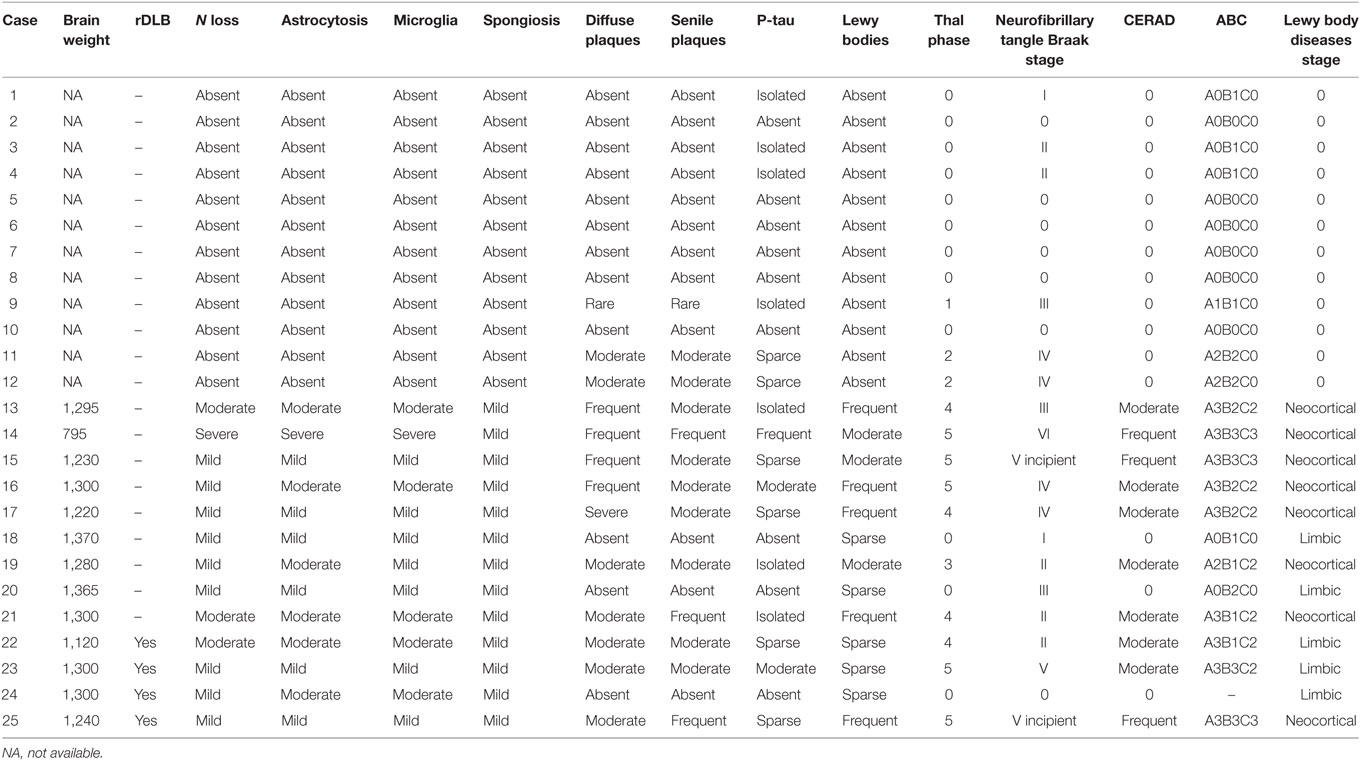
Table 3. Summary of the main neuropathological findings of MA (cases 1–12), DLB (cases 13–21), and rpDLB cases (cases 22–25) in the present series.
No differences in the amount of fibrillar β-amyloid, considering diffuse and senile plaques, were observed between the two groups; the total plaque burden varied from absent to frequent in both groups. The average percentage of Aβ40 of the total β-amyloid in plaques was 7.24% in DLB and 6.03% in rpDLB, and the percentage of Aβ42 was 68.49% in DLB and 67.70% in rpDLB. NFT pathology varied from stage I to stage VI in DLB and from stage 0 to stage V in rpDLB. NFTs in frontal cortex occurred in two DLB and two rpDLB cases.
Astrocytic gliosis was variable from one case to another and varied from mild to moderate in DLB and rpDLB excepting one DLB in which astrocytic gliosis was considered severe.
A remarkable observation was the discrete microglial response, as revealed with Iba1 and CD68 antibodies, in the frontal cortex in DLB and rpDLB.
Neuropathological findings are summarized in Table 3.
mRNA Expression Levels of Selected Mitochondrial Subunits and Energy Metabolism-Related Molecules in MA, DLB, and rpDLB
The expression levels of five of twenty-seven genes analyzed was deregulated in DLB when compared with MA cases including increase expression of ATP5G2 and ATP5H involved in mitochondrial complex (p < 0.05). Deregulation of energy metabolism genes included decrease in ATP6VOB and increase in ATP4A and SLC6A6 (p < 0.05). Regarding rpDLB, the expression levels of ATP6VOB and TOMM40, involved in energy metabolism, was decreased in rpDLB (p < 0.01 and p < 0.05, respectively) (Table 4). Since the trend of altered gene expression was in the same direction in DLB and rpDLB, no significant differences were observed when comparing DLB and rpDLB (Table 4).
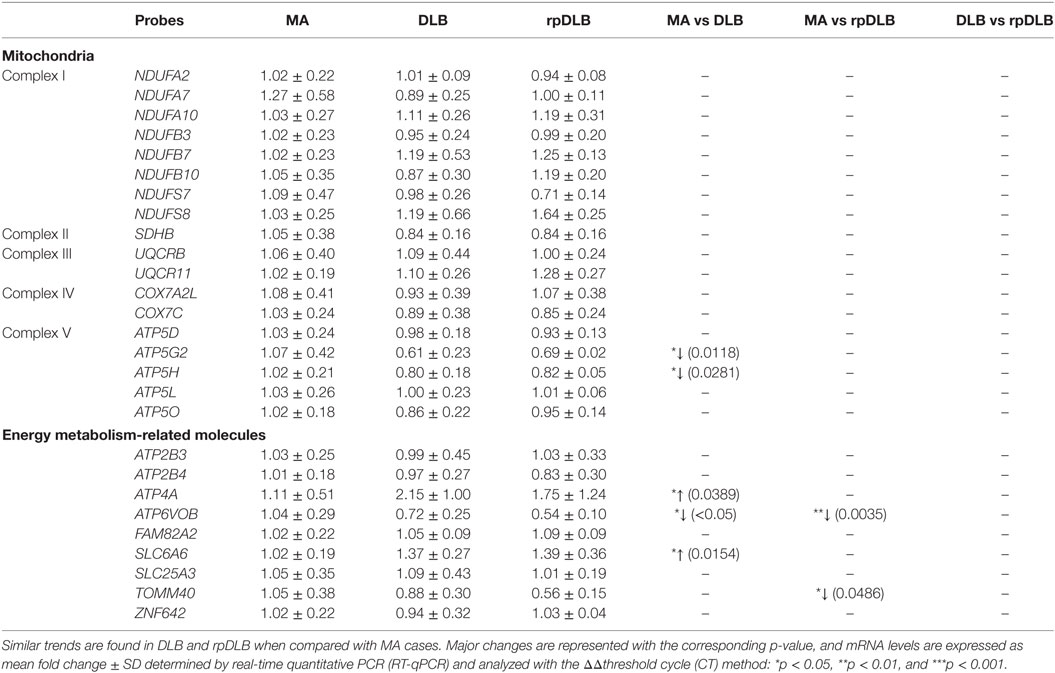
Table 4. mRNA expression of selected subunits of the mitochondrial respiratory chain and genes encoding proteins linked to energy metabolism in MA (n = 12), DLB (n = 9), and rpDLB (n = 4).
Protein Expression of Mitochondrial Subunits of the Respiratory Chain in MA, DLB, and rpDLB
Western blots showed marked alterations in DLB when compared with MA cases. The expression levels of complex I subunits NDUFA7, NDUFA10, NDUFB10, and NDUFB8 were significantly reduced (p ranging from <0.05 to <0.001) when compared with MA cases and using β-actin for protein normalization, expression of which was maintained in DLB and rpDLB when compared with MA cases. NDUFS8 protein expression was not modified in DLB when compared with MA. Protein levels of SDHB (complex II), UQCRC2 (complex III), MTCO1 (complex IV), and ATP5A and ATP50 (complex V) were also significantly decreased in DLB when compared with MA (p < 0.05) using β-actin for protein loading normalization (Figure 1). In contrast, only NDUFA7 was significantly reduced in rpDLB when compared with MA (p < 0.01). However, the expression of several subunits showed a trend toward decrease in rpDLB; therefore, significant differences between DLB and rpDLB were restricted to NDUFB8, SDHB, ATP5A, and ATP50 (p < 0.01) (Figure 1).
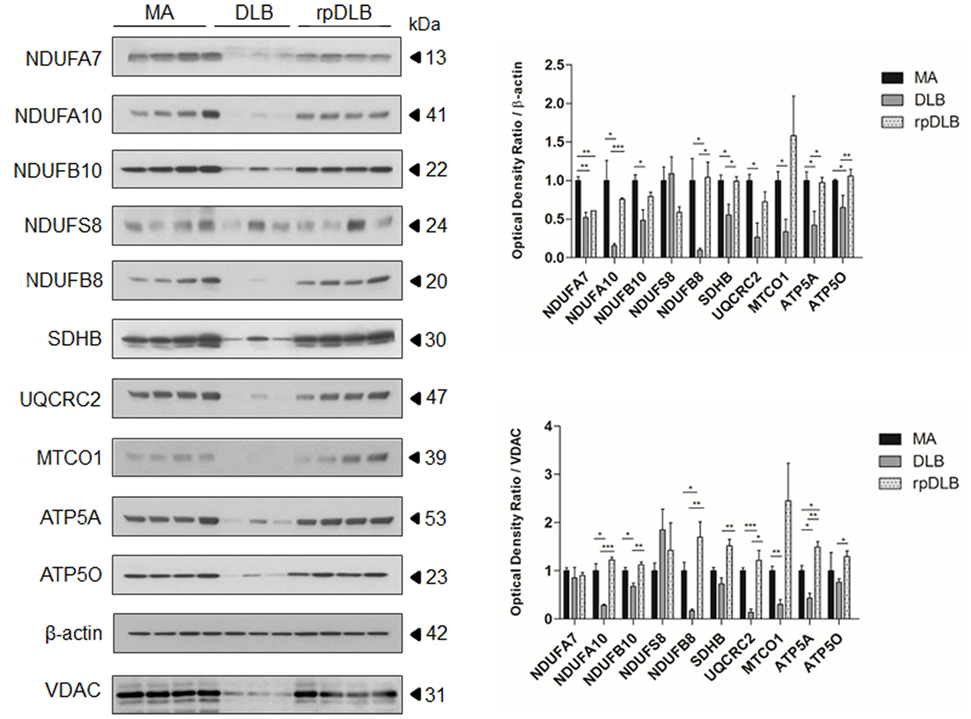
Figure 1. Protein expression in middle-aged (MA) (n = 12), dementia with Lewy bodies (DLB) (n = 9) and rapid DLB (rpDLB) (n = 4) of subunits of mitochondrial complexes I (NDUFFA7, NDUFA10, NDUFB10, NDUFS8, NDUFB8), II (SDHB), III (UQCRC2), IV (MTCO1), and V (ATP5A, ATP50) normalized with both β-actin- and voltage-dependent anion channel (VDAC). Three representative cases are shown in western blots. Diagrams show quantitative values of all assessed cases. Significant decrease in the expression levels of the majority of these subunits is seen in the frontal cortex in DLB and less markedly in rpDLB when compared with MA cases when normalized with β-actin and in most cases with VDAC. Decreased expression of VDAC in DLB probably reflects decrease in the number of mitochondria: *p < 0.05, **p < 0.01, and ***p < 0.001.
Voltage dependent anion channel (VDAC) expression was reduced in DLB and less markedly in rpDLB when compared with MA thus suggesting a reduced number of mitochondria or reduced mitochondria size. Nevertheless, NDUFA10, NDUFB10, NDUFB8, UQCRC2, MTCO1, and AT5A protein expression was also reduced in DLB cases when using VDAC for normalization (p ranging from <0.05 to <0.001). NDUFA7, NDUFS8, and ATP50 levels were similar in DLB and MA cases using VDAC for normalization. Curiously, expression levels of all the assessed subunits were preserved in rpDLB when normalized with VDAC. As a result, levels of NDUFA10, NDUFB10, NDUFB8, SDHB, UQCRC2, ATP5A, and ATP50 were significantly higher in rpDLB when compared with DLB (p values ranging from <0.05 to <0.001) (Figure 1).
Activity of Mitochondrial Complexes I, II, III, IV, and V in Frontal Cortex Area 8 in DLB and rpDLB Cases
Significant decrease in the activity of complexes I, II, III, and IV was detected in frontal cortex area 8 in DLB and rpDLB when compared with MA cases and normalized with the ratio of citrate synthase activity (p < 0.05) (Figure 2). No differences were observed when comparing DLB and rpDLB cases. The activity of complex V showed a trend toward decrease in DLB and more clearly in rpDLB but without statistical significance (Figure 2).
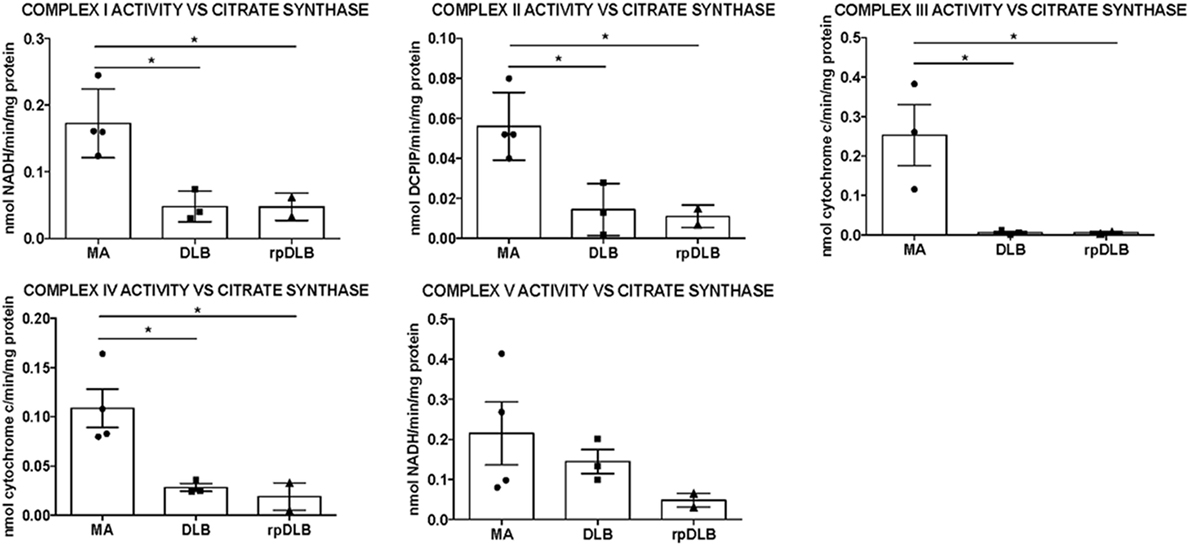
Figure 2. Mitochondrial enzymatic activities in complex I, II, III, IV, and V in middle-aged (MA), dementia with Lewy bodies (DLB), and rapid DLB (rpDLB). All the mitochondrial activities are corrected with the appropriate values of citrate synthase for each sample. Significant decreased activity of complex I, II, III, and IV is observed in DLB and rpDLB when compared with MA. Complex V activity showed a trend toward reduction in DLB and rpDLB: *p < 0.05, **p < 0.01, ***p < 0.001.
mRNA Expression of Genes Involved in Purine Metabolism in MA, DLB, and rpDLB
The expression of all the assessed genes involved in purine metabolism was altered in the same direction in DLB and rpDLB when compared with MA cases. Changes were greater in rpDLB than in DLB. ADA, AK1, ENTPD1, NME6, and PNP (p < 0.05); NME1, NME3, NME4 (all of them with p < 0.01) and PRUNE (p < 0.001) were upregulated in rpDLB when compared with MA cases (Table 5). Finally, three genes, ENTPD2, NME3, and PRUNE were significantly upregulated in rpDLB when compared with DLB (p values varied from <0.05 to <0.001) (Table 5).
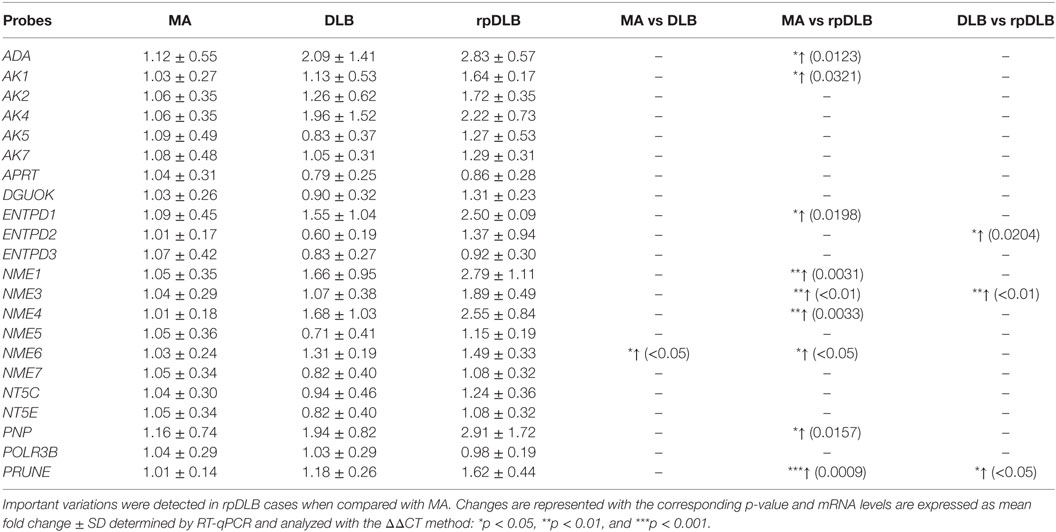
Table 5. mRNA expression of genes encoding proteins linked to purine metabolism in MA (n = 12), DLB (n = 9), and rpDLB (n = 4).
mRNA Expression Levels of Genes Encoding Nucleolar Proteins and Ribosomal Proteins, and rRNAs18S and 28S in MA, DLB, and rpDLB
Nucleoplasmin 1 (NPM1) mRNA was significantly decreased in DLB when compared with MA (p < 0.05) (Table 6).
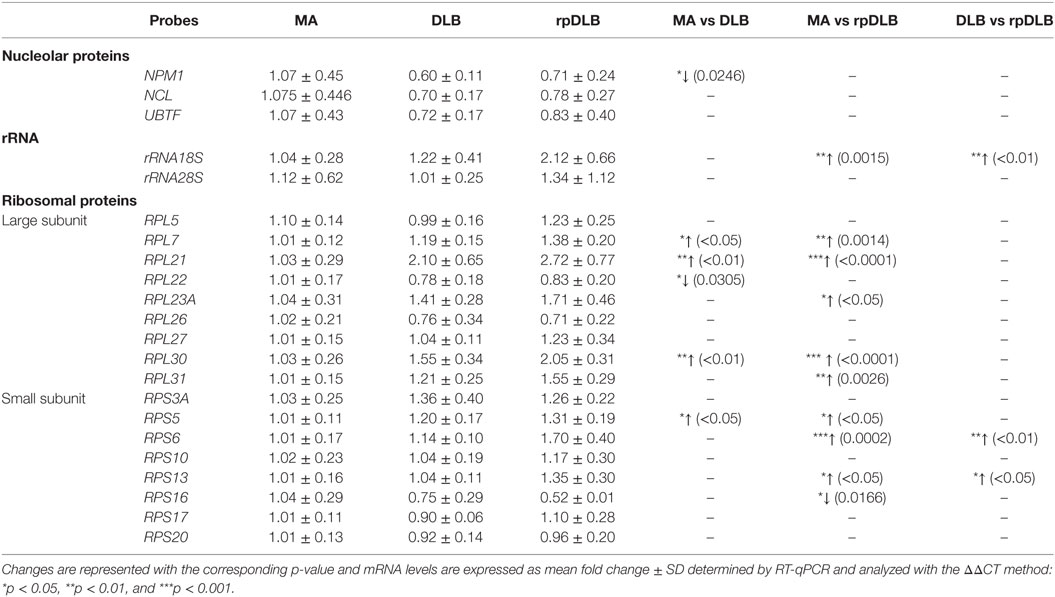
Table 6. mRNA expression levels of genes encoding nucleolar proteins, ribosomal proteins, and 18S and 28S rRNAs in MA (n = 12), DLB (n = 9), and rpDLB (n = 4).
rRNA 18S expression was increased in rpDLB (p < 0.01) when compared with MA and also when compared with DLB (p < 0.01) (Table 6).
Genes encoding ribosomal proteins of the large (L) and the small (S) subunits were upregulated in DLB when compared with MA control cases including RPL7, RPL21, RPL30, and RPS5 (p values varied from <0.05 to <0.001), but RPL22 was downregulated in DLB when compared with MA (p < 0.05) (Table 6).
RPL7, RPL21, RPL23A, RPL30, RPL31, RPS5, RPS6, and RPS13 were upregulated in rpDLB in comparison to MA (p values varied from <0.05 to <0.001); RPS16 was downregulated (p < 0.05) (Table 6). Due to similar trends of gene expression in DLB and rpDLB, only RPS6 and RPS13 were significantly upregulated (p values ranged from <0.05 to <0.001) in rpDLB when compared with DLB (Table 6).
Expression of Proteins Involved in Ribosomal Transcription in MA, DLB, and rpDLB
Only eIF5 was significantly reduced in DLB when compared with MA cases (p < 0.05). However, the expression levels of initiation factors eIF2α and eIF5 were significantly decreased in rpDLB when compared with MA cases (p < 0.01 and p < 0.05, respectively). eIF2α, p-eIF2α, and eIF3η were significantly decreased (p < 0.05) in rpDLB when compared with DLB (Figure 3).
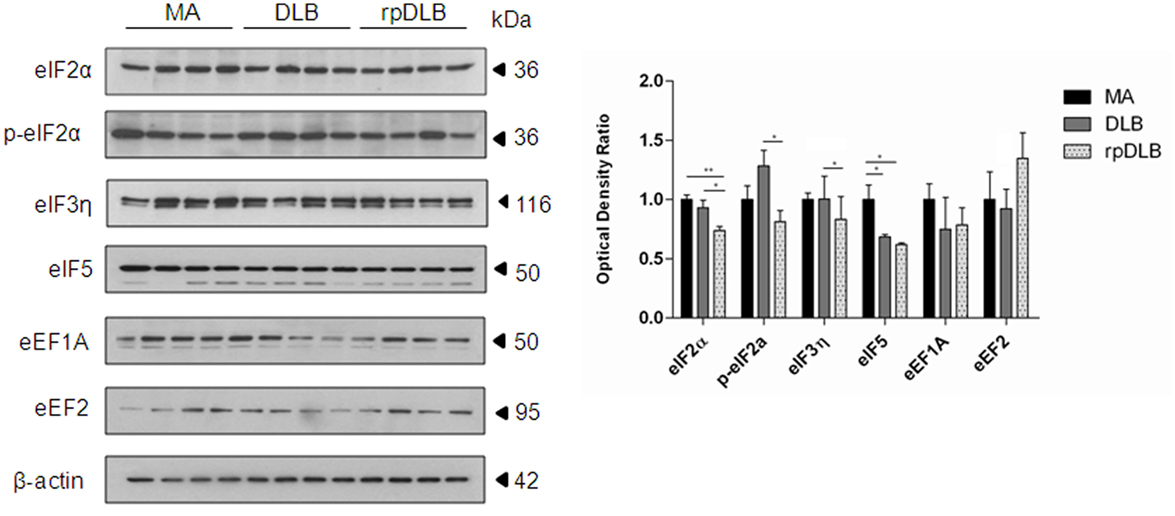
Figure 3. Protein expression, as revealed by western blotting, of initiation and elongation factors of protein transcription at the ribosome in MA (n = 12), dementia with Lewy bodies (DLB) (n = 9) and rapid DLB (rpDLB) (n = 4) using β-actin for normalization. Reduced expression of initiation factors is more marked in rpDLB than in DLB, whereas the expression of elongation factors eEF1A and eEF2 is not modified in DLB and rpDLB: *p < 0.05, **p < 0.01, and ***p < 0.001.
In contrast, no alterations in the expression levels of elongation factors eEF1A and eEF2 were observed in DLB and rpDLB when compared with MA cases (Figure 3).
mRNA Expression Levels of Cytokines and Mediators of the Innate Inflammatory Response in MA, DLB, and rpDLB
No differences in the expression of twenty-three genes encoding cytokines and mediators of the inflammatory response were detected in DLB when compared with MA. However, significant increase in the levels of TNFα (p < 0.001) and CST7 (p < 0.05) was found in rpDLB cases when compared with MA (Table 7). TNFα and C1QL1 were significantly upregulated in rpDLB when compared with DLB (p < 0.01 and p < 0.05, respectively) (Table 7).
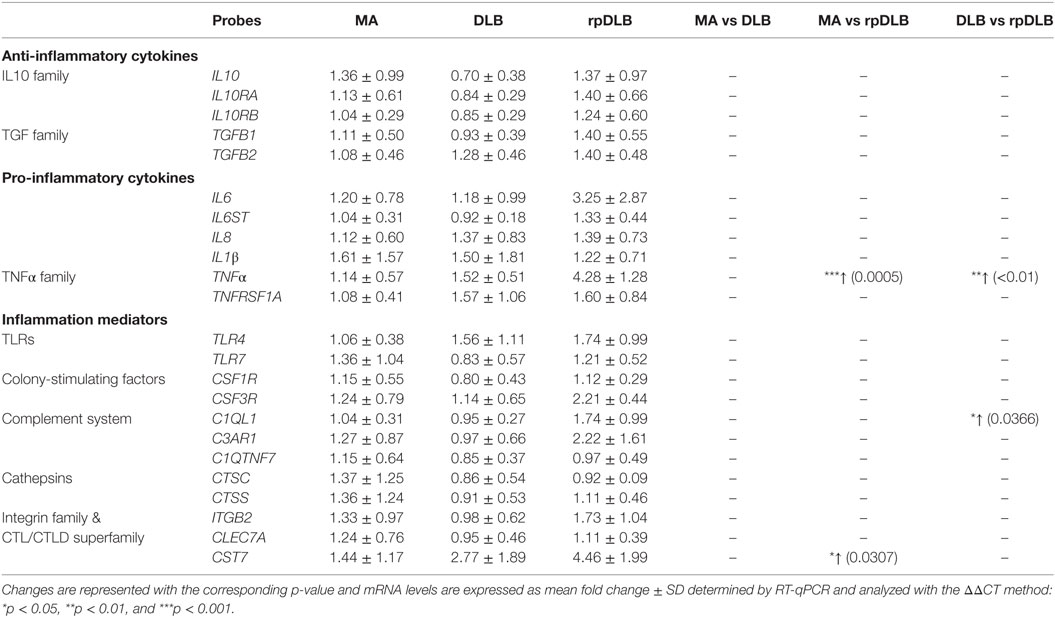
Table 7. mRNA expression levels of genes encoding cytokines and mediators of the innate inflammatory response in MA (n = 12), DLB (n = 9), and rpDLB (n = 4).
Microglia and TNFα in MA, DLB, and rpDLB
Western blotting revealed a significant increase in GFAP protein levels in DLB and rpDLB when compared with MA cases (p < 0.01), whereas the levels of Iba-1 were maintained when compared with MA cases (Figure 4). TNFα protein levels were also significantly increased in rpDLB when compared with MA (p < 0.05) (Figure 4).
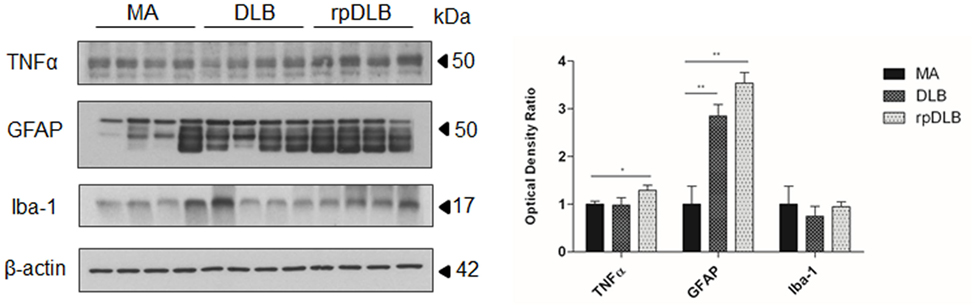
Figure 4. TNFα, GFAP, and Iba-1 protein levels in middle-aged (MA), dementia with Lewy bodies (DLB), and rapid DLB (rpDLB) as revealed by western blotting using β-actin for normalization. Significant increased GFAP expression occurs in DLB and rpDLB when compared with MA (p < 0.01). A significant increase also occurs in TNFα levels in rpDLB when compared with MA (p < 0.05): *p < 0.05, **p < 0.01, and ***p < 0.001.
mRNA Expression Levels of Olfactory Receptors (ORs) and Taste Receptors (TASRs) in MA, DLB, and rpDLB
Increased expression of OR2D2, OR4F4, OR11H1, and OR52H1 was observed in DLB when compared with MA cases (p < 0.05). Upregulation of OR2D2 and OR2T33 was significantly higher in rpDLB compared with MA (p < 0.05). Significant differences between DLB and rpDLB were restricted to OR2T33 and OR4F4 (p < 0.001 and p < 0.05, respectively) (Table 8).
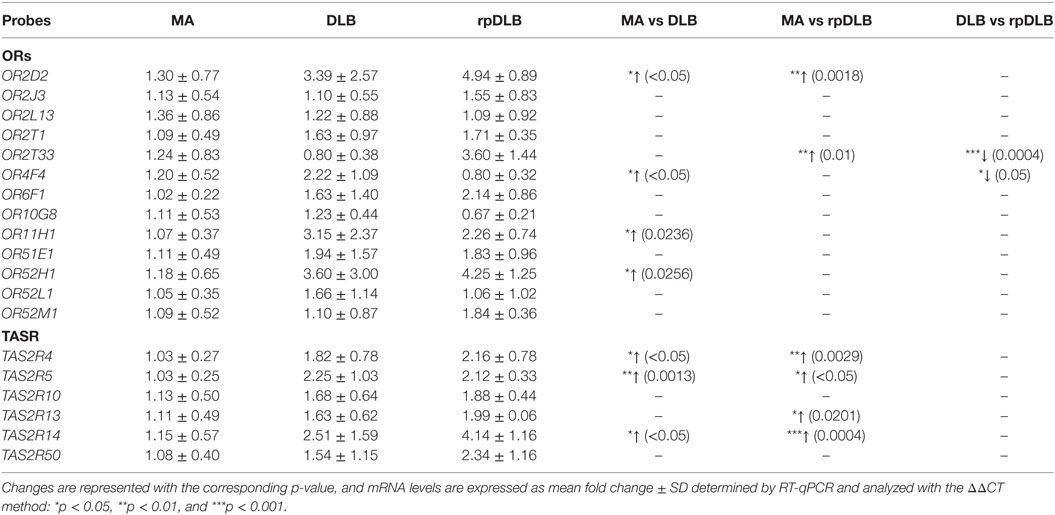
Table 8. mRNA expression levels of genes encoding olfactory receptors (ORs) and taste receptors (TASRs) in MA (n = 12), DLB (n = 9), and rpDLB (n = 4).
TAS2R4, TAS2R5, and TAS2R14 were upregulated in DLB when compared with MA (p values varied from <0.05 to <0.01). TAS2R4, TAS2R5, TAS2R13, and TAS2R14 expression was significantly increased in rpDLB in comparison to MA cases (p values from <0.05 to <0.001) (Table 8).
Soluble Aβ40 and Aβ42, Membrane-Associated β-Amyloid, Tau Oligomeric Species, and α-Synuclein Oligomeric Species in Total Homogenate Fractions
In spite of there being no apparent differences in β-amyloid plaque burden in DLB and rpDLB, significant increases in soluble Aβ40 and Aβ42 levels were seen in rpDLB when compared with DLB (p < 0.05 and p < 0.01, respectively) (Figure 5A). Curiously, levels in DLB were similar to those in controls. However, no differences between DLB and rpDLB were seen regarding β-amyloid associated with membranes (Figure 5B), which in AD has a close correlation with fibrillar β-amyloid in amyloid plaques (78).
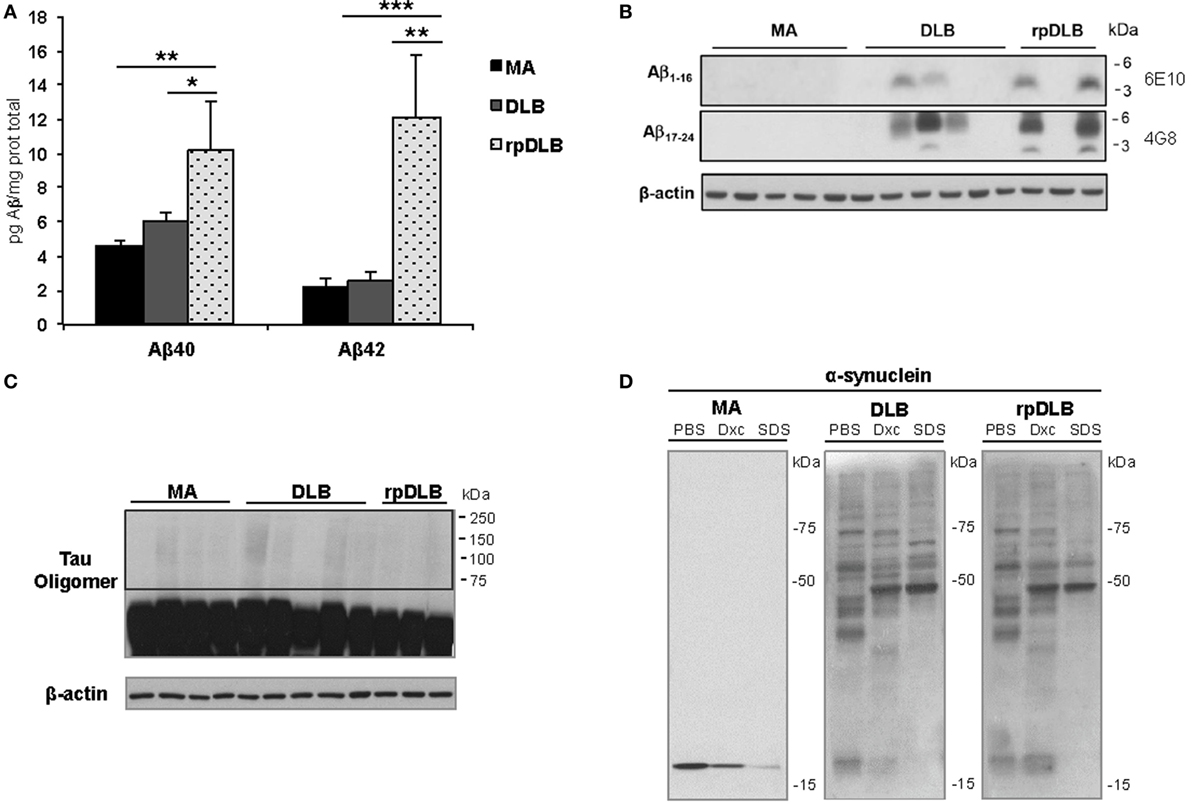
Figure 5. Soluble Aβ40 and Aβ42, membrane-associated β-amyloid, tau oligomers, and α-synuclein oligomeric species in total homogenate fractions in middle-aged (MA) (n = 10), dementia with Lewy bodies (DLB) (n = 10) and rapid DLB (rpDLB) (n = 4). (A) Soluble Aβ40 and Aβ42 levels are similar in MA and DLB cases but soluble Aβ40 and Aβ42 are significantly increased in rpDLB when compared with MA and DLB; (B) membrane-associated β-amyloid is detected in DLB and rpDLB but not in MA cases as revealed with human amyloid-beta protein antibodies 4G8 and 6E10; (C) no tau oligomers are detected in DLB and rpDLB even after membrane over-exposition; (D) α-synuclein oligomers are present equally in DLB and rpDLB, which is in contrast with the lack of α-synuclein oligomers in MA. The figures are representative of four MA, four DLB, and four rpDLB; statistical values represent the totality of samples: *p < 0.05, **p < 0.01, and ***p < 0.001.
Regarding tau pathology, western blots of total homogenates disclosed no tau oligomers in frontal cortex in DLB and rpDLB (Figure 5C) thus being in accordance with the small quantity of NFT in the frontal cortex in the present series.
Finally, α-synuclein oligomers were observed in DLB and rpDLB in contrast with the lack of α-synuclein oligomers in MA cases. Importantly, the band pattern and the density of the bands of oligomers were similar in DLB and rpDLB (Figure 5D).
Discussion
The present study was undertaken to identify alterations of several metabolic pathways, which may participate in age-related DLB pathology including mitochondrial function and energy metabolism, purine metabolism, protein synthesis machinery, inflammation and certain recently discovered new ectopic ORs and TASRs expressed in brain.
The focus of the study was to learn about biochemical alterations beyond the well-known modifications of target proteins in DLB such as α-synuclein and β-amyloid. Present biochemical studies have shown similar percentages of Aβ40 and Aβ42 in plaques in DLB and rpDLB abnormal solubility and aggregation of α-synuclein and increased β-amyloid bound to membranes in the frontal cortex in DLB and rpDLB. In contrast, no differences in tau oligomers were found between MA and DLB cases. Whether levels of soluble Aβ40 and Aβ42 are within control values in DLB in spite of the presence of plaques (73, 74) needs further study.
Mitochondria and Energy Metabolism
Mitochondrial alterations in the frontal cortex are prominent in DLB. ATP5G2 and ATP5H expression is decreased in DLB when compared with MA individuals; protein expression of NDUFA7, NDUFA10, NDUFB8, SDHB, UQCRC2, MTCO1, ATP5A, and ATP50 is reduced. No differences in expression are found in rpDLB in contrast to DLB. NDUFA7 protein levels are significantly reduced in rpDLB and significant differences between DLB and rpDLB are restricted to NDUFB8, SDHB, ATP5A, and ATP50. It is worth stressing that these alterations are not the mere consequence of mitochondrial loss in DLB. Expression levels of VDAC are decreased in DLB but the expression levels of the mentioned subunits are reduced even considering VDAC for normalization of protein levels. It can be argued that the rapid course of the disease results in a reduced harmful impact on mitochondria in rpDLB when compared with DLB. Despite differences in gene and protein expression, mitochondrial enzymatic activity of complexes I, II, III, and IV is significantly decreased in frontal cortex area 8 in DLB and rpDLB. Therefore, the present observations point to altered mitochondrial function in frontal cortex as a major factor in the pathogenesis of DLB and rpDLB.
Regarding energy metabolism, ATP4A and SLC6A6 are upregulated in DLB, while ATP6V0B is downregulated in DLB and rpDLB. TOMM40 is downregulated in rpDLB ATP4A encodes a membrane-bound P-type ATPase, which permits ion transport through cell membranes (79). SLC6A6 encodes a taurine transporter (SLc6a6/TauT) involved in the uptake of gamma-aminobutyric acid (GABA) (80). Whether these changes have implications in GABA metabolism in DLB is not known although GABA levels in the CSF in DLB are not altered when compared with normal individuals (81). ATP6V0B encodes ATPase H+ transporting Vo subunit b, which is involved in protein sorting, zymogen activation, receptor-mediated endocytosis, and synaptic vesicle proton gradient generation (82). TOMM40 encodes a translocase of outer mitochondrial membrane which is required for protein transfer into mitochondria (83). Together, these results suggest energy metabolism impairment in frontal cortex in DLB and rpDLB.
Purine Metabolism
Several genes encoding enzymes linked to purine metabolism are upregulated in DLB and rpDLB although significant values are only obtained when comparing rpDLB with MA cases. These include ADA, AK1, ENTPD1, NME1, NME3, NME4, NME6, PNP, and PRUNE. However, only three genes, ENTPD2, NME3, and PRUNE, are significantly upregulated in rpDLB when compared with DLB.
Purines and pyrimidines are the core of DNA, RNA, nucleosides, and nucleotides. Nucleotides are involved in cell signaling and energy metabolism, and purine bases are also cofactors of several enzymatic reactions (84–87). Adenylate kinases participate in the phosphorylation of AMP to ADP and dAMP to dATP (88, 89). The NME gene family encodes nucleotide diphosphate kinases, which are involved in the phosphorylation of nucleotide diphosphates to form nucleotide triphosphates. These enzymes are enriched in synapses. Increased NME mRNA expression in DLB contrasts with its decrease in AD (90, 91) and suggests a compensatory role of NME in response to reduced synapses in DLB. The product of PRUNE participates in the metabolism of guanosine pentaphosphate and tetraphosphate and is linked to NME in memory conservation (92). Deoxyguanosine kinase (encoded by DGUOK) phosphorylates purine deoxyribonucleosides in the mitochondrial matrix (93–95). Adenine phosphoribosyltransferase is related to adenine metabolism and catalyses the phosphorylation reaction, whereas ADA de-aminates adenosine.
Furthermore, ectonucleoside triphosphate diphosphohydrolases, encoded by ENTPD genes, hydrolyze the terminal phosphate group of nucleoside tri- and diphosphates to form di- and monophosphates, thus controlling extracellular ATP concentrations (96), adenosine-activated type I receptors, nucleotide-activated type 2 ligand-gated ion channels, and metabotropic P2Y receptors (97). Finally, ecto 5′-nucleotidase catalyzes the generation of adenosine from degradation of AMP in the extracellular space. In brain, ectonucleotidases are involved in several functions including modulation of synaptic transmission, ATP-mediated propagation of calcium waves in glial cells, neurogenesis, microglial function, and blood flow (98).
Present findings show marked alterations in the expression of enzymes involved in purine metabolism in the frontal cortex in DLB and rpDLB.
Protein Synthesis
Decreased expression of NPM1 is found in the frontal cortex in DLB, which may be indicative of nucleolar stress and linked to altered ribosomal biogenesis (99–103).
Alterations in the expression of genes encoding ribosomal proteins are seen in DLB and rpDLB, including up- and downregulation of RPLs, thus suggesting impaired ribosome biogenesis (104–107). This is accompanied by reduced protein expression of several initiation factors of transcription at the ribosome, which are more marked in rpDLB than in DLB. However, the expression levels of elongation factors eEF1A and eEF2 are preserved in DLB and rpDLB. Although the direct study of protein synthesis is not possible in human postmortem samples due to postmortem delay between death and tissue processing, the present findings indicate that the machinery of protein synthesis is altered in DLB (108–110). Importantly, altered expression of proteins involved in transcription at the ribosome is more severe in rpDLB than in DLB.
Inflammatory Responses
No significant differences in gene expression of several cytokines and mediators of the inflammatory response are seen in DLB. These data are in line with the observation of no major increase in Iba1 protein levels and CD68 expression in frontal cortex in DLB when compared with MA individuals. They are also in agreement with previous observations showing very limited activation of microglia in DLB (111, 112).
Subtle change refers to TNFα gene expression in rpDLB. Whereas TNFα mRNA is not altered in DLB, significant TNFα mRNA upregulation and increased TNFα protein levels occur in the frontal cortex in rpDLB. TNFα is involved in several metabolic pathways, particularly facilitating gene transcription, activating of the JNK pathway and promoting apoptosis via caspase-dependent and caspase-independent signaling (113). Therefore, TNFα upregulation in rpDLB may have functional implications. Further molecular studies are needed to elucidate activated molecules of the TNFα pathways in rpDLB.
Olfactory and Taste Brain Receptors
Olfactory and taste receptors are widely expressed in human and rodent brain including the cerebral cortex. ORs and TASRs in brain are accompanied by all downstream molecules that permit a functional signaling pathway, and they are functional, as revealed in culture neurons under appropriate stimuli (114, 115).
The expression of several genes encoding ORs is increased in DLB and rpDLB. Three out of six TASR are upregulatred in DLB and four genes in rpDLB.
The function of ORs and TASRs in brain is not known although it has been postulated that they may participate in intra- and extracellular signaling in association, or not, with other receptors. Identification of natural ligands of brain ORs and TASRs should yield insights about the function of these ectopic receptors (114).
Mitochondrial Function, Inflammation, and Deregulation of ORs and TASRs in Frontal Cortex Discriminate DLB and AD
The present study reveals disease-specific alterations when comparing the present results in DLB with available data for AD in the same region, the frontal cortex, at similar stages of disease progression.
Mitochondrial alterations and impaired activity of complex V are early events in the entorhinal cortex in AD (116). NDUFA2, NDUFB3, UQCR11, COX7C, ATPD, ATP5L, and ATP50 gene expression is reduced in the entorhinal cortex with disease progression, and this is accompanied by impaired activity of complexes I, II, and V (72). In contrast, RNA expression levels of several subunits of complexes I–V and enzymatic activities of complex I, II, IV, and V are preserved in frontal cortex in AD even at stages V–VI of Braak in individuals with mean ages similar to those of assessed DLB cases (72). Therefore, mitochondrial dysfunction in frontal cortex markedly differs in AD and DLB, with detrimental effects in most mitochondrial complexes in DLB in comparison with AD.
There is a large body of information demonstrating microglial responses and increased expression of inflammatory markers in the cerebral cortex in AD (73, 74, 117–131). In contrast, inflammatory responses are very limited in DLB. Therefore, inflammation is characteristically an important factor in AD pathogenesis, whereas it has low impact in DLB.
Regarding purine metabolism, ENTPD2, NME3, PNP, and PRUNE RNAs are deregulated in the frontal cortex in AD (91), whereas ADA, AK1, ENTPD1, NME1, NME3, NME4, NME6, PNP, and PRUNE are upregulated in rpDLB with changes less marked in DLB.
Severe alterations of the machinery involved in protein synthesis from the nucleolus to the ribosome have been observed in the entorhinal cortex in AD (132), but no similar data are available in AD frontal cortex.
Finally, OR4F4 mRNA expression levels are increased in frontal cortex area 8 at stages III–IV, and OR52L1 mRNA at Braak stages III–IV and V–VI in AD (133). Regarding TASRs, no modification in the mRNA expression levels is observed in frontal cortex area 8 at any stage of AD (133). These observations in AD are in contrast with the extensive deregulation of ORs and TASRs in the frontal cortex in DLB, thus indicating marked differences in the regulation of these brain receptors in DLB when compared with AD.
Specific Traits in rpDLB Compared with DLB
No neuropathological differences are seen between DLB and rpDLB in accordance with other studies (58). Moreover, rpDLB has similar biochemical profiles regarding mitochondrial gene and protein expression, protein synthesis, and expression of ORs and TASRs. However, gene and protein expression of subunits of mitochondrial respiratory complexes are more pronounced in DLB when compared with rpDLB, whereas alterations in purine metabolism and initiation of protein translation are more pronounced in rpDLB when compared with DLB. Due to the small number of rpDLB cases available for biochemical studies, these observations need further validation.
Two additional traits which differentiate rpDLB and DLB merit a comment. The first of these is the higher levels of soluble Aβ40 and Aβ42 in rpDLB in spite of similar plaque burden and β-amyloid associated with membranes in rpDLB when compared with DLB. In this line, modified characteristics of Aβ42 have been described in rapidly progressive AD (134), thus suggesting that β-amyloid conformers pace disease progression in AD. It can be suggested that higher levels of soluble β-amyloid oligomeric species, known to be toxic to nerve cells (135–140), can precipitate disease progression in rpDLB. The second differential aspect is the higher levels of TNFα in rpDLB when compared with DLB, which can also be toxic to nerve cells.
Conclusion
Molecular alterations in the cerebral cortex in DLB include (i) deregulated expression of several mRNAs and proteins of mitochondrial subunits and reduced activity of complexes I, II, III, and IV of the mitochondrial respiratory chain; (ii) reduced expression of selected molecules involved in energy metabolism and increased expression of enzymes involved in purine metabolism; (iii) abnormal expression of certain nucleolar proteins, rRNA18S, genes encoding ribosomal proteins and initiation factors of the transcription at the ribosome; (iv) discrete inflammation; and (v) marked deregulation of brain ORs and TASRs. Severe mitochondrial dysfunction involving activity of four complexes, minimal inflammatory responses, and dramatic altered expression of ORs and TASRs discriminate DLB from AD. Altered solubility and aggregation of α-synuclein, increased β-amyloid bound to membranes, and absence of soluble tau oligomers are common in DLB and DLB with rapid progression. Increased soluble β-amyloid 1–40 and β-amyloid 1–42, and increased TNFα mRNA and protein expression, distinguish DLB with rapid progression from typical DLB. Since high levels of soluble β-amyloid species are toxic for nerve cells and TNF-α.
Author Contributions
PG-E carried out RT-qPCR, western blots, and enzymatic activities. IL-G performed the studies of beta-amyliid and tau oligomers. OG-R participated in the selection of cases and in the neuropathological study. MG-G and AK helped PG-E. FL and SZ helped in molecular studies. MC helped in the processing of samples. JR helped in the design of molecular studies in rapid dementias. IZ helped in the design of the study of rapid dementias. EG carried out the neuropathological and supervised the clinical and pathological relations. IF directed the study, supervised the work, wrote the advanced version of the manuscript, which was circulated among the authors for comments and suggestions, and prepared the final version for submission.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank T. Yohannan for editorial help.
Funding
This study was funded by the Ministerio de Economía y Competitividad, Instituto de Salud Carlos III—Fondos FEDER, a Way to Build Europe FIS grant PI14/00757 and coordinated Intraciber 2014: Rapid Dementias.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fneur.2017.00089/full#supplementary-material.
References
1. McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology (1996) 47:1113–24. doi: 10.1212/WNL.47.5.1113
2. McKeith I, Mintzer J, Aarsland D, Burn D, Chiu H, Cohen-Mansfield J, et al. Dementia with Lewy bodies. Lancet Neurol (2004) 3:19–28. doi:10.1016/S1474-4422(03)00619-7
3. McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology (2005) 65:1863–72. doi:10.1212/01.wnl.0000187889.17253.b1
4. Mayo MC, Bordelon Y. Dementia with Lewy bodies. Semin Neurol (2014) 34:182–8. doi:10.1055/s-0034-1381741
5. Fujishiro H, Nakamura S, Sato K, Iseki E. Prodromal dementia with Lewy bodies. Geriatr Gerontol Int (2015) 2015(15):817–26. doi:10.1111/ggi.12466
6. Donaghy PC, O’Brien JT, Thomas AJ. Prodromal dementia with Lewy bodies. Psychol Med (2015) 45:259–68. doi:10.1017/S0033291714000816
7. Fujishiro H, Ferman TJ, Boeve BF, Smith GE, Graff-Radford NR, Uitti RJ, et al. Validation of the neuropathologic criteria of the third consortium for dementia with Lewy bodies for prospectively diagnosed cases. J Neuropathol Exp Neurol (2008) 67:649–56. doi:10.1097/NEN.0b013e31817d7a1d
8. Ince PG. Dementia with Lewy bodies and Parkinson’s disease with dementia. In: Dickson DW, Weller RO, editors. Neurodegeneration, the Molecular Pathology of Dementia and Movement Disorders. Oxford: Wiley-Blackwell (2011). p. 224–37.
9. Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and Dementia with Lewy Bodies. Proc Natl Acad Sci U S A (1998) 95:6469–73. doi:10.1073/pnas.95.11.6469
10. Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc Natl Acad Sci U S A (2000) 97:4897–902. doi:10.1073/pnas.97.9.4897
11. Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci (2001) 2:492–501. doi:10.1038/35081564
12. Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, et al. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol (2002) 2002(4):160–4. doi:10.1038/ncb748
13. Kaplan B, Ratner V, Haas E. Alpha-synuclein: its biological function and role in neurodegenerative diseases. J Mol Neurosci (2003) 20:83–92. doi:10.1385/JMN:20:2:83
14. Tofaris GK, Razzaq A, Ghetti B, Lilley KS, Spillantini MG. Ubiquitination of α-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. J Biol Chem (2003) 278:44405–11. doi:10.1074/jbc.M308041200
15. Hashimoto M, Kawahara K, Bar-On P, Rockenstein E, Crews L, Masliah E. The role of alpha-synuclein assembly and metabolism in the pathogenesis of Lewy body disease. J Mol Neurosci (2004) 24:343–52. doi:10.1385/JMN:24:3:343
16. Li W, West N, Colla E, Pletnikova O, Troncoso JC, Marsh L, et al. Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proc Natl Acad Sci U S A (2005) 102:2162–7. doi:10.1073/pnas.0406976102
17. Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ, et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem (2006) 281:29739–52. doi:10.1074/jbc.M600933200
18. Mukaetova-Ladinska EB, McKeith IG. Pathophysiology of synuclein aggregation in Lewy body disease. Mech Ageing Dev (2006) 127:188–202. doi:10.1016/j.mad.2005.09.014
19. Tofaris GK, Spillantini MG. Physiological and pathological properties of alpha-synuclein. Cell Mol Life Sci (2007) 64:2194–201. doi:10.1007/s00018-007-7217-5
20. Parkkinen L, Kauppinen T, Pirttilä T, Autere JM, Alafuzoff I. α-Synuclein pathology does not predict extrapyramidal symptoms or dementia. Ann Neurol (2005) 57:82–91. doi:10.1002/ana.20321
21. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol (2006) 12:389–404. doi:10.1007/s00401-006-0127-z
22. Parkkinen L, Pirttilä T, Alafuzoff I. Applicability of current staging/categorization of alpha-synuclein pathology and their clinical relevance. Acta Neuropathol (2008) 115:399–407. doi:10.1007/s00401-008-0346-6
23. Jellinger KA. Significance of brain lesions in Parkinson disease dementia and Lewy body dementia. Front Neurol Neurosci (2009) 24:114–25. doi:10.1159/000197890
24. Shimada H, Hirano S, Shinotoh H, Aotsuka A, Sato K, Tanaka N, et al. Mapping of brain acetylcholinesterase alterations in Lewy body disease by PET. Neurology (2009) 73:273–8. doi:10.1212/WNL.0b013e3181ab2b58
25. Lim SM, Katsifis A, Villemagne VL, Best R, Jones G, Saling M, et al. The 18F-FDG PET cingulate island sign and comparison to 123I-beta-CIT SPECT for diagnosis of dementia with Lewy bodies. J Nucl Med (2009) 50:1638–45. doi:10.2967/jnumed.109.065870
26. Kim HJ, Lee JE, Shin SJ, Sohn YH, Lee PH. Analysis of the substantia innominata volume in patients with Parkinson’s disease with dementia, dementia with Lewy bodies, and Alzheimer’s disease. J Mov Disord (2011) 4:68–72. doi:10.14802/jmd.11014
27. Grothe MJ, Schuster C, Bauer F, Heinsen H, Prudlo J, Teipel SJ. Atrophy of the cholinergic basal forebrain in dementia with Lewy bodies and Alzheimer’s disease dementia. J Neurol (2014) 261:1939–48. doi:10.1007/s00415-014-7439-z
28. Huang SH, Chang CC, Lui CC, Chen NC, Lee CC, Wang PW, et al. Cortical metabolic and nigrostriatal abnormalities associated with clinical stage-specific dementia with Lewy bodies. Clin Nucl Med (2015) 40:26–31. doi:10.1097/RLU.0000000000000620
29. Buddhala C, Loftin SK, Kuley BM, Cairns NJ, Campbell MC, Perlmutter JS, et al. Dopaminergic, serotonergic, and noradrenergic deficits in Parkinson disease. Ann Clin Transl Neurol (2015) 2:949–59. doi:10.1002/acn3.246
30. Jellinger KA. Morphological substrates of parkinsonism with and without dementia: a retrospective clinico-pathological study. J Neural Transm Suppl (2007) 72:91–104. doi:10.1007/978-3-211-73574-9_12
31. Jellinger KA, Attems J. Prevalence and impact of vascular and Alzheimer pathologies in Lewy body disease. Acta Neuropathol (2008) 115:427–36. doi:10.1007/s00401-008-0347-5
32. Colom-Cadena M, Gelpi E, Charif S, Belbin O, Blesa R, Martí MJ, et al. Confluence of α-synuclein, tau, and β-amyloid pathologies in dementia with Lewy bodies. J Neuropathol Exp Neurol (2013) 72:1203–12. doi:10.1097/NEN.0000000000000018
33. Hashimoto M, Masliah E. Cycles of aberrant synaptic sprouting and neurodegeneration in Alzheimer’s and dementia with Lewy bodies. Neurochem Res (2003) 28:1743–56. doi:10.1023/A:1026073324672
34. Dalfó E, Barrachina M, Rosa JL, Ambrosio S, Ferrer I. Abnormal alpha-synuclein interactions with rab3a and rabphilin in diffuse Lewy body disease. Neurobiol Dis (2004) 16:92–7. doi:10.1016/j.nbd.2004.01.001
35. Kramer ML, Schulz-Schaeffer WJ. Presynaptic alpha-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J Neurosci (2007) 27:1405–10. doi:10.1523/JNEUROSCI.4564-06.2007
36. Schulz-Schaeffer WJ. The synaptic pathology of α-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol (2010) 120:131–43. doi:10.1007/s00401-010-0711-0
37. Mukaetova-Ladinska EB, Andras A, Milne J, Abdel-All Z, Borr I, Jaros E, et al. Synaptic proteins and choline acetyltransferase loss in visual cortex in dementia with Lewy bodies. J Neuropathol Exp Neurol (2013) 72:53–60. doi:10.1097/NEN.0b013e31827c5710
38. Overk CR, Masliah E. Pathogenesis of synaptic degeneration in Alzheimer’s disease and Lewy body diseases. Biochem Pharmacol (2014) 88:508–16. doi:10.1016/j.bcp.2014.01.015
39. Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT Jr. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc Natl Acad Sci U S A (2000) 97:571–6. doi:10.1073/pnas.97.2.571
40. Lashuel HA, Petre BM, Wall J, Simon M, Nowak RJ, Walz T, et al. Alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J Mol Biol (2002) 2002(322):1089–102. doi:10.1016/S0022-2836(02)00735-0
41. Dalfó E, Albasanz JL, Martin M, Ferrer I. Abnormal metabotropic glutamate receptor expression and signaling in the cerebral cortex in diffuse Lewy body disease is associated with irregular alpha-synuclein/phospholipase C (PLCβ1) interactions. Brain Pathol (2004) 14:388–98. doi:10.1111/j.1750-3639.2004.tb00082.x
42. Albasanz JL, Dalfó E, Ferrer I, Martín M. Impaired metabotropic glutamate receptor/phospholipase C signaling pathway in the cerebral cortex in Alzheimer’s disease and dementia with Lewy bodies correlates with stage of Alzheimer’s-disease-related changes. Neurobiol Dis (2005) 20:685–93. doi:10.1016/j.nbd.2005.05.001
43. Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, et al. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle re-clustering after endocytosis. Neuron (2010) 65:66–79. doi:10.1016/j.neuron.2009.12.023
44. Scott DA, Tabarean I, Tang Y, Cartier A, Masliah E, Roy S. A pathologic cascade leading to synaptic dysfunction in alpha-synuclein-induced neurodegeneration. J Neurosci (2010) 30:8083–95. doi:10.1523/JNEUROSCI.1091-10.2010
45. Garcia-Reitböck P, Anichtchik O, Bellucci A, Iovino M, Ballini C, Fineberg E, et al. SNARE protein redistribution and synaptic failure in a transgenic mouse model of Parkinson’s disease. Brain (2010) 133:2032–44. doi:10.1093/brain/awq132
46. MacInnes N, Iravani MM, Perry E, Piggott M, Perry R, Jenner P, et al. Proteasomal abnormalities in cortical Lewy body disease and the impact of proteasomal inhibition within cortical and cholinergic systems. J Neural Transm (2008) 115:869–78. doi:10.1007/s00702-008-0027-6
47. Tanji K, Mori F, Kakita A, Takahashi H, Wakabayashi K. Alteration of autophagosomal proteins (LC3, GABARAP and GATE-16) in Lewy body disease. Neurobiol Dis (2011) 43:690–7. doi:10.1016/j.nbd.2011.05.022
48. Kragh CL, Ubhi K, Wyss-Coray T, Masliah E. Autophagy in dementias. Brain Pathol (2012) 22:99–109. doi:10.1111/j.1750-3639.2011.00545.x
49. Poehler AM, Xiang W, Spitzer P, May VE, Meixner H, Rockenstein E, et al. Autophagy modulates SNCA/α-synuclein release, thereby generating a hostile microenvironment. Autophagy (2014) 10:2171–92. doi:10.4161/auto.36436
50. Miki Y, Tanji K, Mori F, Utsumi J, Sasaki H, Kakita A, et al. Alteration of upstream autophagy-related proteins (ULK1, ULK2, Beclin1, VPS34 and AMBRA1) in Lewy body disease. Brain Pathol (2016) 26:359–70. doi:10.1111/bpa.12297
51. Baek JH, Whitfield D, Howlett D, Francis P, Bereczki E, Ballard C, et al. Unfolded protein response is activated in Lewy body dementias. Neuropathol Appl Neurobiol (2016) 42:352–60. doi:10.1111/nan.12260
52. Lyras L, Perry RH, Perry EK, Ince PG, Jenner A, Jenner P, et al. Oxidative damage to proteins, lipids, and DNA in cortical brain regions from patients with dementia with Lewy bodies. J Neurochem (1998) 71:302–12. doi:10.1046/j.1471-4159.1998.71010302.x
53. Navarro A, Boveris A, Bández MJ, Sánchez-Pino MJ, Gómez C, Muntané G, et al. Human brain cortex: mitochondrial oxidative damage and adaptive response in Parkinson disease and in dementia with Lewy bodies. Free Radic Biol Med (2009) 46:1574–80. doi:10.1016/j.freeradbiomed.2009.03.007
54. Dalfó E, Ferrer I. Early α-synuclein lipoxidation in neocortex in Lewy body diseases. Neurobiol Aging (2008) 29:408–17. doi:10.1016/j.neurobiolaging.2006.10.022
55. Williams MM, Xiong C, Morris JC, Galvin JE. Survival and mortality differences between dementia with Lewy bodies vs Alzheimer disease. Neurology (2006) 67:1935–41. doi:10.1212/01.wnl.0000247041.63081.98
56. Geschwind MD, Shu H, Haman A, Sejvar JJ, Miller BL. Rapidly progressive dementia. Ann Neurol (2008) 64:97–108. doi:10.1002/ana.21430
57. Josephs KA, Ahlskog JE, Parisi JE, Boeve BF, Crum BA, Giannini C, et al. Rapidly progressive neurodegenerative dementias. Arch Neurol (2009) 66:201–7. doi:10.1001/archneurol.2008.534
58. Gaig C, Valldeoriola F, Gelpi E, Ezquerra M, Llufriu S, Buongiorno M, et al. Rapidly progressive diffuse Lewy body disease. Mov Disord (2011) 26:1316–23. doi:10.1002/mds.23506
59. Ferrer I, Martinez A, Boluda S, Parchi P, Barrachina M. Brain banks: benefits, limitations and cautions concerning the use of post-mortem brain tissue for molecular studies. Cell Tissue Bank (2008) 2008(9):181–94. doi:10.1007/s10561-008-9077-0
60. Ravid R, Ferrer I. Brain banks as key part of biochemical and molecular studies on cerebral cortex involvement in Parkinson’s disease. FEBS J (2012) 279:1167–76. doi:10.1111/j.1742-4658.2012.08518.x
61. Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging (2003) 24:197–211. doi:10.1016/S0197-4580(02)00065-9
62. Alafuzoff I, Ince PG, Arzberger T, Al-Sarraj S, Bell J, Bodi I, et al. Staging/typing of Lewy body related alpha-synuclein pathology: a study of the BrainNet Europe Consortium. Acta Neuropathol (2009) 8117:635–52. doi:10.1007/s00401-009-0523-2
63. Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol (1991) 82:239–59. doi:10.1007/BF00308809
64. Thal DR, Rüb U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology (2002) 58:1791–800. doi:10.1212/WNL.58.12.1791
65. Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol (2012) 123:1–11. doi:10.1007/s00401-011-0910-3
66. Garcia-Esparcia P, Hernández-Ortega K, Koneti A, Gil L, Delgado-Morales R, Castaño E, et al. Altered machinery of protein synthesis is region- and stage-dependent and is associated with α-synuclein oligomers in Parkinson’s disease. Acta Neuropathol Commun (2015) 3:76. doi:10.1186/s40478-015-0257-4
67. Durrenberger PF, Fernando S, Kashefi SN, Ferrer I, Hauw JJ, Seilhean D, et al. Effects of antemortem and postmortem variables on human brain mRNA quality: a BrainNet Europe study. J Neuropathol Exp Neurol (2010) 69:70–81. doi:10.1097/NEN.0b013e3181c7e32f
68. Barrachina M, Castaño E, Ferrer I. TaqMan PCR assay in the control of RNA normalization in human post-mortem brain tissue. Neurochem Int (2006) 49:276–84. doi:10.1016/j.neuint.2006.01.018
69. Durrenberger PF, Fernando FS, Magliozzi R, Kashefi SN, Bonnert TP, Ferrer I, et al. Selection of novel reference genes for use in the human central nervous system: a BrainNet Europe Study. Acta Neuropathol (2012) 124:893–903. doi:10.1007/s00401-012-1027-z
70. Garcia-Esparcia P, Koneti A, Rodríguez-Oroz MC, Lago B, del Rio JA, Ferrer I. Mitochondrial activity in the frontal cortex area 8 and angular gyrus in Parkinson’s disease and Parkinson’s disease with dementia. Brain Pathol (2016). doi:10.1111/bpa.12474
71. Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat Protoc (2012) 7:1235–46. doi:10.1038/nprot.2012.058
72. Armand-Ugon M, Ansoleaga B, Berjaoui S, Ferrer I. Reduced mitochondrial activity is early and steady in the entorhinal cortex but it is mainly unmodified in the frontal cortex in Alzheimer’s disease. Curr Alzheimer Res (2016).
73. López-González I, Schlüter A, Aso E, Garcia-Esparcia P, Ansoleaga B, LLorens F, et al. Neuroinflammatory signals in Alzheimer disease and APP/PS1 transgenic mice: correlations with plaques, tangles, and oligomeric species. J Neuropathol Exp Neurol (2015) 74:319–44. doi:10.1097/NEN.0000000000000176
74. López-González I, Aso E, Carmona M, Armand-Ugon M, Blanco R, Naudí A, et al. Neuroinflammatory gene regulation, mitochondrial function, oxidative stress, and brain lipid modifications with disease progression in tau P301S transgenic mice as a model of frontotemporal lobar degeneration-tau. J Neuropathol Exp Neurol (2015) 74:975–99. doi:10.1097/NEN.0000000000000241
75. Nakagawa S. A farewell to Bonferroni: the problems of low statistical power and publication bias. Behav Ecol (2004) 15:1044e5. doi:10.1093/beheco/arh107
76. Glickman ME, Rao SR, Schultz MR. False discovery rate control is a recommended alternative to Bonferroni-type adjustments in health studies. J Clin Epidemiol (2014) 67:850–7. doi:10.1016/j.jclinepi.2014.03.012
77. López-González I, Tebé Cordomí C, Ferrer I. Regional gene expression of inflammation and oxidative stress responses does not predict neurodegeneration in aging. J Neuropathol Exp Neurol (2017). doi:10.1093/jnen/nlw117
78. Thal DR, Walter J, Saido TC, Fändrich M. Neuropathology and biochemistry of Aβ and its aggregates in Alzheimer’s disease. Acta Neuropathol (2015) 129:167–82. doi:10.1007/s00401-014-1375-y
79. Singh V, Mani I, Chaudhary DK. ATP4A gene regulatory network for fine-tuning of proton pump and ion channels. Syst Synth Biol (2013) 7:23–32. doi:10.1007/s11693-012-9103-1
80. Tomi M, Tajima A, Tachikawa M, Hosoya K. Function of taurine transporter (Slc6a6/TauT) as a GABA transporting protein and its relevance to GABA transport in rat retinal capillary endothelial cells. Biochim Biophys Acta (2008) 1778:2138–42. doi:10.1016/j.bbamem.2008.04.012
81. Molina JA, Gómez P, Vargas C, Ortiz S, Pérez-Rial S, Urigüen L, et al. Neurotransmitter amino acid in cerebrospinal fluid of patients with dementia with Lewy bodies. J Neural Transm (2005) 2005(112):557–63. doi:10.1007/s00702-004-0202-3
82. Lu M, Ammar D, Ives H, Albrecht F, Gluck SL. Physical interaction between aldolase and vacuolar H+-ATPase is essential for the assembly and activity of the proton pump. J Biol Chem (2007) 282:24495–503. doi:10.1074/jbc.M702598200
83. Gottschalk WK, Lutz MW, He YT, Saunders AM, Burns DK, Roses AD, et al. The broad impact of TOM40 on neurodegenerative diseases in aging. J Parkinson Dis Alzheimer Dis (2014) 1:12. doi:10.13188/2376-922X.1000003
84. Ribeiro JA, Sebastiao AM, de Mendonça A. Adenosine receptors in the nervous system: pathophysiological implications. Prog Neurobiol (2003) 68:377–92. doi:10.1016/S0301-0082(02)00155-7
85. Boison D. Adenosine as a neuromodulator in neurological diseases. Curr Opin Pharmacol (2008) 8:2–7. doi:10.1016/j.coph.2007.09.002
86. Burnstock G, Fredholm BB, Verkhratsky A. Adenosine and ATP receptors in the brain. Curr Top Med Chem (2011) 11:973–1011. doi:10.2174/156802611795347627
87. Ipata PL, Camici M, Micheli V, Tozzi MG. Metabolic network of nucleosides in the brain. Curr Top Med Chem (2011) 11:902–222. doi:10.2174/156802611795347555
88. Noma T. Dynamic of nucleotide metabolism as a supporter of life phenomena. J Med Invest (2005) 5:127–36. doi:10.2152/jmi.52.127
89. Dzeja P, Terzic A. Adenylate kinase and AMP signaling networks: metabolic monitoring, signal communication and body energy sensing. Int J Mol Sci (2009) 10:1729–72. doi:10.3390/ijms10041729
90. Kim SH, Fountoulakis M, Cairns NJ, Lubec G. Human brain nucleoside diphosphate kinase activity is decreased in Alzheimer’s disease and Down syndrome. Biochem Biophys Res Commun (2000) 296:970–5.
91. Ansoleaga B, Jové M, Schlüter A, Garcia-Esparcia P, Moreno J, Pujol A, et al. Deregulation of purine metabolism in Alzheimer’s disease. Neurobiol Aging (2015) 36:68–80. doi:10.1016/j.neurobiolaging.2014.08.004
92. Li K, Müller I, Patil S, Höger H, Pollak A, Russo-Schlaff N, et al. Strain-independent global effect of hippocampal proteins in mice trained in the Morris water maze. Amino Acids (2012) 43:1739–49. doi:10.1007/s00726-012-1258-2
93. Jüllig M, Eriksson S. Mitochondrial and submitochondrial localization of human deoxyguanosine kinase. Eur J Biochem (2000) 267:5466–72. doi:10.1046/j.1432-1327.2000.01607.x
94. Johansson K, Ramaswamy S, Ljungcrantz C, Knecht W, Piskur J, Munch-Petersen B, et al. Structural basis for substrate specificities of cellular deoxyribonucleoside kinases. Nat Struct Biol (2001) 8:616–20. doi:10.1038/89661
95. Desvignes T, Pontarotti P, Fauvel C, Bobe J. NME protein family evolutionary history, a vertebrate perspective. BMC Evol Biol (2009) 9:256. doi:10.1186/1471-2148-9-256
96. Yegutkin GG. Nucleotide- and nucleoside-converting ectoenzymes: important modulators of purinergic signalling cascade. Biochim Biophys Acta (2008) 1783:673–94. doi:10.1016/j.bbamcr.2008.01.024
97. Al-Rashida M, Iqbal J. Therapeutic potentials of ecto-nucleoside triphosphate diphosphohydrolase, ecto-nucleotide pyrophosphatase/phosphodiesterase, ecto-5’-nucleotidase, and alkaline phosphatase inhibitors. Eur J Med Chem (2013) 66:438–49. doi:10.1016/j.ejmech.2013.06.015
98. Zimmermann H. Ectonucleotidases in the nervous system. Novartis Found Symp (2006) 276:113–28. doi:10.1002/9780470032244.ch10
99. Olson MO. Sensing cellular stress: another new function for the nucleolus? Sci STKE (2004) 2004(224):e10. doi:10.1126/stke.2242004pe10
100. Boulon S, Westman BJ, Hutten S, Boisvert FM, Lamond AI. The nucleolus under stress. Mol Cell (2010) 40:216–27. doi:10.1016/j.molcel.2010.09.024
101. Baltanas FC, Casefont I, Weruaga E, Alonso JR, Berciano MT, Lafarga M. Nucleolar disruption and cajal body disassembly are nuclear hallmarks of DNA damage-induced neurodegeneration in Purkinje cells. Brain Pathol (2011) 21:374–88. doi:10.1111/j.1750-3639.2010.00461.x
102. Hetman M, Pietrzak M. Emerging roles of the neuronal nucleolus. Trends Neurosci (2012) 35:305–14. doi:10.1016/j.tins.2012.01.002
103. Parlato R, Kreiner G. Nucleolar activity in neurodegenerative diseases: a missing piece of the puzzle? J Mol Med (2013) 91:541–7. doi:10.1007/s00109-012-0981-1
104. Granneman S, Baserga SJ. Ribosome biogenesis: of knobs and RNA processing. Exp Cell Res (2004) 296:43–50. doi:10.1016/j.yexcr.2004.03.016
105. Klein DJ, Moore PB, Steitz TA. The roles of ribosomal proteins in the structure assembly, and evolution of the large ribosomal subunit. J Mol Biol (2004) 340:141–77. doi:10.1016/j.jmb.2004.03.076
106. Kressler D, Hurt E, Bassler J. Driving ribosome assembly. Biochem Biophys Acta (2010) 1803:673–83. doi:10.1016/j.bbamcr.2009.10.009
107. Korobeinikova AV, Garber MB, Gongadze GM. Ribosomal proteins: structure, function, and evolution. Biochemistry (2012) 77:562–74. doi:10.1134/S0006297912060028
108. Kapp LD, Lorsch JR. The molecular mechanisms of eukaryotic translation. Annu Rev Biochem (2004) 73:657–704. doi:10.1146/annurev.biochem.73.030403.080419
109. Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol (2010) 11:113–27. doi:10.1038/nrm2838
110. Voorhees RM, Ramakrishnan V. Structural basis of the translational elongation cycle. Annu Rev Biochem (2013) 82:203–36. doi:10.1146/annurev-biochem-113009-092313
111. Shepherd CE, Thiel E, McCann H, Harding AJ, Halliday GM. Cortical inflammation in Alzheimer disease but not dementia with Lewy bodies. Arch Neurol (2000) 57:817–22. doi:10.1001/archneur.57.6.817
112. Streit WJ, Xue QS. Microglia in dementia with Lewy bodies. Brain Behav Immun (2016) 55:191–201. doi:10.1016/j.bbi.2015.10.012
114. Garcia-Esparcia P, Schlüter A, Carmona M, Moreno J, Ansoleaga B, Torrejón-Escribano B, et al. Functional genomics reveals dysregulation of cortical olfactory receptors in Parkinson disease: novel putative chemoreceptors in the human brain. J Neuropathol Exp Neurol (2013) 72:524–39. doi:10.1097/NEN.0b013e318294fd76
115. Grison A, Zucchelli S, Urzì A, Zamparo I, Lazarevic D, Pascarella G, et al. Mesencephalic dopaminergic neurons express a repertoire of olfactory receptors and respond to odorant-like molecules. BMC Genomics (2014) 15:729. doi:10.1186/1471-2164-15-729
116. Terni B, Boada J, Portero-Otin M, Pamplona R, Ferrer I. Mitochondrial ATP-synthase in the entorhinal cortex is a target of oxidative stress at stages I/II of Alzheimer’s disease pathology. Brain Pathol (2010) 20:222–33. doi:10.1111/j.1750-3639.2009.00266.x
117. Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, et al. Glial-neuronal interactions in Alzheimer’s disease: the potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol (1998) 8:65–72. doi:10.1111/j.1750-3639.1998.tb00136.x
118. McGeer PL, McGeer EG. Inflammation, autotoxicity and Alzheimer disease. Neurobiol Aging (2001) 22:799–809. doi:10.1016/S0197-4580(01)00289-5
119. Streit WJ, Conde JR, Harrison JK. Chemokines and Alzheimer’s disease. Neurobiol Aging (2001) 22:909–13. doi:10.1016/S0197-4580(01)00290-1
120. Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging (2000) 21:383–421. doi:10.1016/S0197-4580(00)00124-X
121. Graeber MB, Streit WJ. Microglia: biology and pathology. Acta Neuropathol (2010) 2010(119):89–105. doi:10.1007/s00401-009-0622-0
122. Lee YJ, Han SB, Nam SY, Oh KW, Hong JT. Inflammation and Alzheimer’s disease. Arch Pharm Res (2010) 2010(33):1539–56. doi:10.1007/s12272-010-1006-7
123. Eikelenboom P, van Exel E, Hoozemans JJ, Veerhuis R, Rozemuller AJ, van Gool WA. Neuroinflammation – an early event in both the history and pathogenesis of Alzheimer’s disease. Neurodegener Dis (2010) 7:38–41. doi:10.1159/000283480
124. Rubio-Perez JM, Morillas-Ruiz JM. A review: inflammatory process in Alzheimer’s disease, role of cytokines. Sci World J (2012) 2012:756357. doi:10.1100/2012/756357
125. Leung E, Guo L, Bu J, Maloof M, El Khoury J, Geula C. Microglia activation mediates fibrillar amyloid-beta toxicity in the aged primate cortex. Neurobiol Aging (2011) 32:387–97. doi:10.1016/j.neurobiolaging.2009.02.025
126. Morimoto K, Horio J, Satoh H, Sue L, Beach T, Arita S, et al. Expression profiles of cytokines in the brains of Alzheimer’s disease (AD) patients, compared to the brains of non-demented patients with and without increasing AD pathology. J Alzheimer Dis (2011) 25:59–76. doi:10.3233/JAD-2011-101815
127. McGeer PL, McGeer EG. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: implications for therapy. Acta Neuropathol (2013) 126:479–97. doi:10.1007/s00401-013-1177-7
128. Prokop S, Miller KR, Heppner FL. Microglia actions in Alzheimer’s disease. Acta Neuropathol (2013) 126:461–77. doi:10.1007/s00401-013-1182-x
129. Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nature (2015) 16:358–72. doi:10.1038/nrn3880
130. Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol (2015) 14:388–405. doi:10.1016/S1474-4422(15)70016-5
131. López González I, Garcia-Esparcia P, Llorens F, Ferrer I. Genetic and transcriptomic profiles of inflammation in neurodegenerative diseases: Alzheimer, Parkinson, Creutzfeldt-Jakob and tauopathies. Int J Mol Sci (2016) 17:206. doi:10.3390/ijms17020206
132. Hernández-Ortega K, Garcia-Esparcia P, Gil L, Lucas JJ, Ferrer I. Altered machinery of protein synthesis in Alzheimer’s: from the nucleolus to the ribosome. Brain Pathol (2016) 26:593–605. doi:10.1111/bpa.12335
133. Ansoleaga B, Garcia-Esparcia P, Llorens F, Moreno J, Aso E, Ferrer I. Dysregulation of brain olfactory and taste receptors in AD, PSP and CJD, and AD-related model. Neuroscience (2013) 2013(248):369–82. doi:10.1016/j.neuroscience.2013.06.034
134. Cohen ML, Kim C, Haldiman T, ElHag M, Mehndiratta P, Pichet T, et al. Rapidly progressive Alzheimer’s disease features distinct structures of amyloid-β. Brain (2015) 138:1009–22. doi:10.1093/brain/awv006
135. Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A (1998) 1998(95):6448–53. doi:10.1073/pnas.95.11.6448
136. Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science (2002) 298:78. doi:10.1126/science.1074069
137. Klein WL, Krafft GA, Finch CE. Targeting small Abeta oligomers: the solution to an Alzheimer’s disease conundrum? Trends Neurosci (2001) 24:219–24. doi:10.1016/S0166-2236(00)01749-5
138. Gandy S, Simon AJ, Steele JW, Lublin AL, Lah JJ, Walker LC, et al. Days to criterion as an indicator of toxicity associated with human Alzheimer amyloid-beta oligomers. Ann Neurol (2010) 68:220–30. doi:10.1002/ana.22052
139. Hayden EY, Teplow DB. Amyloid β-protein oligomers and Alzheimer’s disease. Alzheimers Res Ther (2013) 5:60. doi:10.1186/alzrt226
Keywords: dementia with Lewy bodies, Alzheimer’s disease, α-synuclein, mitochondria, protein synthesis, inflammation, β-amyloid, olfactory receptors
Citation: Garcia-Esparcia P, López-González I, Grau-Rivera O, García-Garrido MF, Konetti A, Llorens F, Zafar S, Carmona M, del Rio JA, Zerr I, Gelpi E and Ferrer I (2017) Dementia with Lewy Bodies: Molecular Pathology in the Frontal Cortex in Typical and Rapidly Progressive Forms. Front. Neurol. 8:89. doi: 10.3389/fneur.2017.00089
Received: 03 October 2016; Accepted: 24 February 2017;
Published: 13 March 2017
Edited by:
Martin Ingelsson, Uppsala University, SwedenReviewed by:
Alberto Serrano-Pozo, University of Iowa, USAChristine A. F. Von Arnim, University of Ulm, Germany
Copyright: © 2017 Garcia-Esparcia, López-González, Grau-Rivera, García-Garrido, Konetti, Llorens, Zafar, Carmona, del Rio, Zerr, Gelpi and Ferrer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isidro Ferrer, ODA4MmlmYUBnbWFpbC5jb20=