 Danielle Golub1*†
Danielle Golub1*† Faith Williams2†
Faith Williams2† Taylor Wong1Nishanth Iyengar1Hannah Jolley3Sakinah Sabadiah4David Rhee3Gabrielle Gold-von Simson3,5
Taylor Wong1Nishanth Iyengar1Hannah Jolley3Sakinah Sabadiah4David Rhee3Gabrielle Gold-von Simson3,5- 1New York University School of Medicine, New York, NY, United States
- 2School of Medicine, Washington University School of Medicine, Saint Louis, MO, United States
- 3Department of Pediatrics, New York University School of Medicine, New York, NY, United States
- 4Department of Neurology, New York University School of Medicine, New York, NY, United States
- 5Health and Hospitals, Clinical Translational Science Institute, New York University, New York, NY, United States
Longitudinally extensive spinal cord lesions (LECL) restricted to gray matter are poorly understood as are their neurodevelopmental repercussions in children. We herein report the critical case of a 13-year-old male presenting with progressive quadriparesis found to have cervical LECL restricted to the anterior horns. Challenged with a rare diagnostic dilemma, the clinical team systematically worked through potential vascular, genetic, infectious, rheumatologic, and paraneoplastic diagnoses before assigning a working diagnosis of acute inflammatory myelopathy. Nuanced consideration of and workup for both potential ischemic causes (arterial dissection, fibrocartilaginous embolism, vascular malformation) and specific inflammatory conditions including Transverse Myelitis, Neuromyelitis Optica Spectrum Disorders (NMOSD), Multiple Sclerosis (MS), Acute Disseminated Encephalomyelitis (ADEM), and Acute Flaccid Myelitis (AFM) is explained in the context of a comprehensive systematic review of the literature on previous reports of gray matter-restricted longitudinally extensive cord lesions in children. Treatment strategy was ultimately based on additional literature review of treatment-refractory acute inflammatory neurological syndromes in children. A combination of high-dose steroids and plasmapheresis was employed with significant improvement in functional outcome, suggesting a potential benefit of combination immune-modulatory treatment in these patients. This case furthermore highlights quality clinical reasoning with respect to the elusive nature of diagnosis, nuances in neuroimaging, and multifocal treatment strategies in pediatric LECL.
Introduction
Longitudinally extensive spinal cord lesions (LECL) are a phenomenon of rapidly progressive and wide-spread spinal cord inflammation with potentially devastating clinical consequences. While LECL is best described in the literature as a key diagnostic criterion in Neuromyelitis Optica Spectrum Disorders (NMOSD), it is also an infrequent, but critical, sequela of a broad range of inflammatory central nervous system syndromes (1, 2). Inflammatory LECL is most commonly described as an autoimmune-mediated swelling of myelin sheaths directed by anti-myelin basic protein (MBP) antibodies, anti-aquaporin 4 (AQP-4) antibodies, and anti-myelin oligodendrocyte glycoprotein (MOG) antibodies, among others (3). Gray matter involvement in LECL, although rare in early manifestations of disease, is an acute finding indicative of either more progressive and refractory presentations of common autoimmune syndromes (multiple sclerosis, acute disseminated encephalomyelitis, NMOSD, sarcoidosis), or under-recognized entities such as pediatric peri-infectious acute flaccid myelitis (AFM) and paraneoplastic syndromes (4–9).
LECLs demonstrate abnormal hyperintense signal on T2-weighted magnetic resonance imaging (MRI) in the spinal cord that spans at least three vertebral segments (1). Differentiating between lesions to make a diagnosis can be complicated by the ambiguity of MRI findings among inflammatory, infectious, and vascular etiologies. In particular, gray matter-restricted LECL may closely resemble anterior spinal artery (ASA) infarction, simulating the “owl's eye” or “snake eye” appearance on T2-weighted axial MRI (10). A prospective study comparing imaging in early neuromyelitis optica spectrum disorders (NMOSD) and ASA infarction found that the “owl's eye” sign tended to complicate the diagnosis (6). Furthermore, a variable and indeterminate appearance of centrally located, T2-hyperintense LECL has been described in both MOG and AQP-4-related NMOSD, infarction, viral myelitis, sarcoidosis, and spondylitic compressive myelopathies, and most recently in paraneoplastic disorders such as GFAP autoimmunity (11–14). In pediatric patients especially, LECL limited to the gray matter generates a non-specific differential diagnosis and imparts a risk of delayed diagnosis and possible long-term morbidity (3, 15, 16). The following case of a pediatric patient with an acute and debilitating presentation of LECL sharply restricted to the gray matter illustrates the importance of a multidisciplinary, clinical reasoning team approach to diagnosis and treatment in an elusive and rare pediatric lesion.
Case Report
History
A 13-year-old male presented to the Pediatric Emergency Room at a tertiary care academic medical center due to progressive quadriparesis that reportedly began after a prolonged episode of coughing 3 days earlier. His neurological symptoms manifested as bilateral hand numbness with persistent back and neck pain, but rapidly progressed to quadriparesis and widespread tactile and proprioceptive sensory loss over 2 days. The day prior to his presentation, he was unable to walk without support. He was subsequently admitted to the Pediatric Intensive Care Unit (PICU) due to concern for potential rapid respiratory compromise.
Other than a cough, the patient reported no recent acute illness and denied travel, trauma, exposures, and vaccinations (all childhood vaccinations were up to date). Additionally, there was no history of developmental or cognitive impairment (was performing well in eighth grade), no drug or alcohol use, and no history of smoking or e-cigarette use. He had a history of mild persistent asthma, and his only medication was his albuterol inhaler that he had been using about twice daily for a week prior to admission. There were no reported allergies.
Presentation
At the time of admission, he was hemodynamically stable with normal vital signs. He was afebrile and in no acute distress. Heart and lung exams were normal. Neurological exam revealed diffuse hypotonia, diminished deep tendon reflexes in all extremities, persistent quadriparesis with most marked weakness in the radial, median, and ulnar nerve distributions (C5-T1 levels), and decreased sensation at the C4-L2 levels. Additionally, the patient had diffuse, severe hyperesthesia in response to sharp stimuli. There was no evidence of acutely altered mental status, visual or other cranial nerve deficit, nystagmus, or overt ataxia. Babinski and Hoffman's signs were negative and there was no ankle clonus. There was no spinal or paraspinal tenderness to palpation.
Hospital Course
On hospital day (HD) 1, still unable to support himself while standing, the patient developed urinary retention requiring multiple straight catheterizations, representing a symptomatic nadir 2–3 days after initial symptom onset. He otherwise remained hemodynamically and neurologically stable during his stay in the PICU. He showed steady recovery after the following workup and treatment on HD 3–10 and was discharged to the Pediatric Acute Rehabilitation Unit on HD 22 in good condition.
Initial Diagnostic Studies
Given the acute nature of the patient's neurological symptoms, on HD 1 brain and spine imaging were performed. Brain CT and MRI were unremarkable—there were no lesions, structural abnormalities, or edematous changes of the parenchyma or cranial nerves, and there was no contrast enhancement or leptomeningeal disease. Total spine MRI, however, showed a longitudinally extensive, non-enhancing, T2-hyperintense central cord lesion strictly involving the gray matter from C2-T2 with mild edema, intervertebral disc intensity changes in the cervical levels (Figures 1A–C) and robust diffusion restriction (Figure 1D). These findings were initially thought to be suggestive of spinal cord infarct, although no abnormal flow void, cord compression, mass lesion, discrete disc herniation or desiccation, or other spinal or foraminal stenosis was noted. Follow-up MRA of the thoracic aorta was inconclusive, and concern for spinal ischemia necessitated high-resolution intravascular imaging. On HD 3, interventional angiogram showed brisk, robust filling and collateralization of the vascular supply of the spinal cord, and canal after left vertebral artery injection (Figure 1E). The anterior spinal artery was patent and of normal caliber. There was no evidence for aortic dissection or other vascular abnormality. Furthermore, serial fibrinogen, coagulation, and liver function tests showed no evidence of coagulopathy.
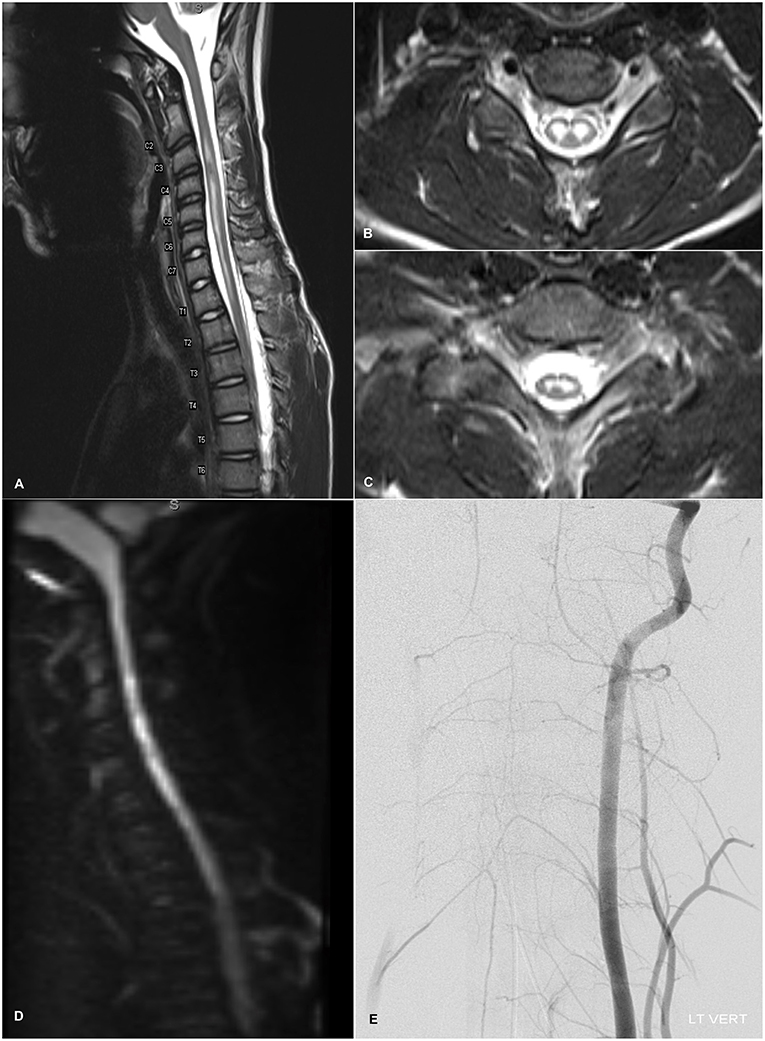
Figure 1. (A) T2-weighted sagittal admission MRI image of the cervical spine showing longitudinally extensive, non-enhancing, mildly expansile hyperintensity within the central gray matter of the spinal cord spanning levels C2-T2 with normal appearance of the vertebral flow voids. Mild intervertebral disc intensity changes are observable at C2-C6. (B,C) Axial T2-weighted MRI images at levels C6 and C8 respectively demonstrating localization of T2-hyperintensity to the anterior horns with an “owl's eye” appearance. (D) Diffusion-weighted imaging demonstrating diffusion restriction within the cervical cord lining up with the T2-hyperintense lesion. (E) Diagnostic angiogram showing the cervical view of the left vertebral artery catheterization. Brisk and robust collateralization to the vascular supply of the spinal cord, including the anterior spinal artery, and spinal canal is seen. There is no evidence of infarction or dissection of the spinal vasculature.
Further Work Up and Treatment
Out of secondary concern for an autoimmune or infectious etiology, the patient was started on Intravenous Immunoglobulin (IVIG) (400 mg/kg daily), high-dose steroids (methylprednisolone 1 g daily), vancomycin, ceftriaxone, and acyclovir for empiric meningoencephalitis coverage, and aspirin (81 mg daily) for secondary prevention of possible cord ischemia. Initial laboratory workup, however, was not indicative of an infectious or an acute inflammatory etiology with a normal white blood cell count (7.1*103/μL), negative blood cultures and respiratory viral panel, and normal sedimentation rate and C-reactive protein levels (1 mm/h and <0.1mg/L, respectively). Urine studies, including toxicology, were likewise unremarkable. Cerebrospinal fluid (CSF) obtained by lumbar puncture also proved unrevealing (WBC 0, RBC 9, glucose 65, protein 27.3). CSF infectious studies for enterovirus, HSV 1/2, syphilis, Lyme disease, ehrlichiosis, Cryptococcus, Epstein-Barr virus, and cytomegalovirus were negative, and serum titers for additional pathogens including mycoplasma and West Nile virus did not support acute infection. Additional workup for common autoimmune myelopathies including neuromyelitis optica spectrum disorders (NMOSD) and multiple sclerosis (MS) was non-diagnostic (serum was negative for aquaporin-4 antibodies (AQP-4) (titer <1:10), myelin oligodendrocyte glycoprotein antibodies (MOG) (titer <1:10), and CSF was negative for oligoclonal bands). Serum tests for myositis (aldolase, creatine kinase) were negative.
Recovery and Follow Up
After two doses of high-dose steroids (total 5 day course) and IVIG on HD 3–5, the patient's lower extremity strength began to improve, urinary retention resolved, and deep tendon reflexes returned to baseline. Given the negative infectious workup, antibiotics and acyclovir were discontinued on HD 3. However, the patient demonstrated a persistent weakness of the bilateral upper extremities that prompted a 5 day plasmapheresis course (HD 5–10) as salvage therapy for medically-refractory myelitis. Over the next few days, the patient achieved significant gains in strength, sensation, and balance.
The patient was transferred to a pediatric acute rehabilitation unit on HD 22. His exam remained notable for diminished hand, wrist, biceps, and triceps strength bilaterally, but he was able to walk with minimal assistance and his muscle strength normalized. Sensation was intact in the extremities but remained slightly diminished in the T4-T10 dermatomes. Paresthesias were no longer present. At his 3 month follow-up with the pediatric neurologist, his only residual finding was bilateral hand weakness, although greatly improved. He was back at school on a full-day, regular schedule. Diagnosis remained “unspecified inflammatory spinal cord syndrome.”
Discussion
LECL is a rare phenomenon and presents a critical diagnostic dilemma in pediatric patients. Possible etiologies for LECL include traumatic, ischemic, vascular, inflammatory, and infectious causes or even a genetic polymorphism, each requiring rapid diagnosis and unique treatment for successful recovery (17). LECLs are well-described as a characteristic features of NMOSD. However, in contrast to this patient, the lesions of NMOSD are typically centrally located, diffuse or patchy, extend to the medulla, demonstrate contrast enhancement, and are only rarely seen restricted solely to the anterior horns (18, 19). Furthermore, most cases of NMOSD and transverse myelitis are associated with a central, expansive, and contrast-enhancing lesion (20, 21). Our patient's presentation was particularly enigmatic in that his LECL was isolated to the anterior horns yielding the “owl's eye” appearance on T2-weighted imaging with restricted diffusion but without gadolinium enhancement. Initial unfamiliarity with the potential diagnoses in a non-traumatic pediatric acute anterior spinal cord myelopathy resulted in delayed diagnosis and the use of numerous empiric treatments. A systematic review of PubMed/MEDLINE and EMBASE for reports sharing MRI findings and pathology consistent with gray matter-restricted LECL in pediatric patients is shown in Table 1. The search strategy used in PubMed is defined in Appendix A and was adapted for EMBASE.
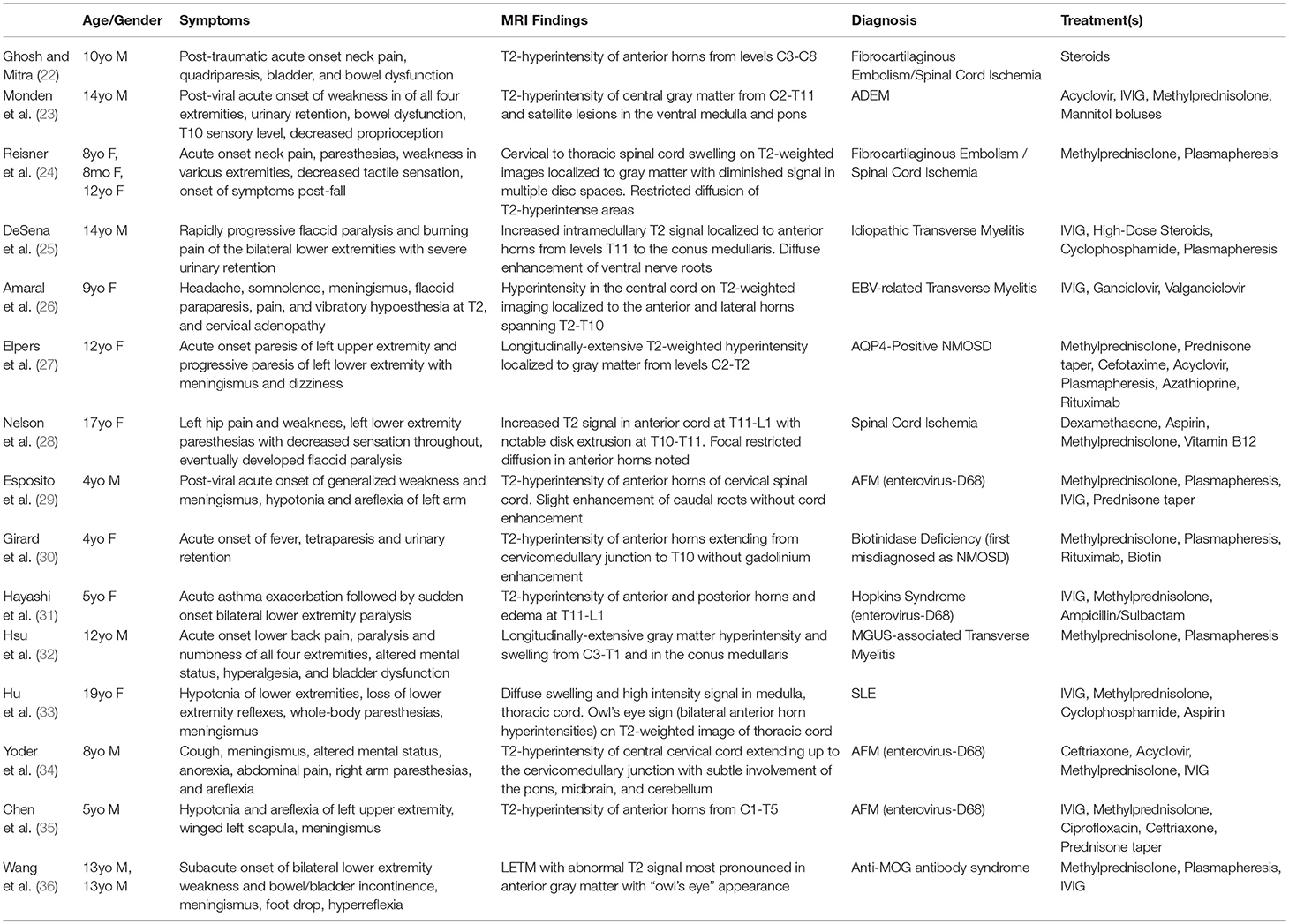
Table 1. Previous Reports of Isolated Longitudinally Extensive Gray Matter Cord Lesions in Pediatrics.
Our patient's history of a coughing paroxysm, back pain, and clinician awareness of the more common association of the “owl's eye” radiographic sign and restricted diffusion with ischemia (reflected by the greatest number of cases in Table 1) prioritized aortic dissection or other vascular pathology on the differential. Dissection and vascular malformation were ruled out by angiogram, but transient ischemic injury from a clot or fibrocartilaginous embolism dislodged after the prodromal coughing paroxysm remained under consideration. One of the largest case series of patients with suspected fibrocartilaginous embolism reported normal angiography in nearly all patients for whom imaging was available (37). While no explicit evidence of disc desiccation or herniation was seen, intervertebral disc intensity changes in our patient's cervical spine on T2-weighted imaging may be suggestive of premature disc disease, a critical risk factor for fibrocartilaginous embolism (38, 39). Furthermore, the risk of fibrocartilaginous embolism is thought to be elevated in children due to increased presence of small spinal arteries that would allow for retrograde migration of emboli (38, 40). Our patient's presentation is also generally congruent with recently proposed diagnostic criteria for spinal cord infarction: while symptoms worsened in a stepwise manner over 2–3 days and not within the expected 12 h timeframe, CSF was non-inflammatory and spinal MRI showed diffusion restriction without cord compression (41). Although fibrinogen and other coagulation testing was negative, given the heightened concern for possible embolic ischemic injury, the patient remained on aspirin throughout his hospitalization.
Infectious or inflammatory etiologies were also highly considered, especially since the patient reported recent cough. Since 2014, the incidence in the United States of a virally-induced spinal cord disease known as AFM has slowly increased (8). Much like our patient, AFM presents with acute flaccid limb weakness following a viral prodrome and MRI findings of LECL on T2-weighted imaging largely restricted to the anterior horns with a predilection for the cervical spine (8, 42) Positive serum, CSF or respiratory viral panel for enteroviruses D68 and 71 are heavily associated with recent cases, but AFM historically has been linked to a host of viruses including West Nile, coxsackie, and adenovirus (8) Furthermore, there are a handful of reports of a heterogenous entity linking asthma exacerbation and AFM in children, Hopkins Syndrome, most of which describe similar imaging findings as in our patient (31, 43, 44). However, in our patient, a causative viral agent or definitive prodrome was not identified and therefore a diagnosis of AFM could not be made.
Further workup for a possible inflammatory etiology included testing for AQP-4 and MOG-associated NMOSD, multiple sclerosis (MS), and other autoimmune disorders. MOG antibody-associated myelitis is associated with gray-matter restricted T2-signal changes coupled with a lack of gadolinium enhancement and an AFM-like phenotype, as was seen in our patient (45). Serum anti-MOG and anti-AQP4 tests, however, were negative and are known to have a high degree of sensitivity in NMOSD and other anti-MOG syndromes (46, 47). It is important to note, however, that our patient received two treatments of intravenous immunoglobulin (IVIG) prior to AQP-4 and MOG antibody testing, which is known to diminish test sensitivity (48). Multiple sclerosis (MS), the most common idiopathic inflammatory neurodegenerative disease of the central nervous system, was also considered and can be associated with longitudinally extensive lesions such as the one in this case in up to 14% of children with MS (3, 49). However, our patient's single clinical event with one MRI lesion demonstrates neither dissemination in space nor time and does not meet IPMSSG criteria for pediatric multiple sclerosis (50). Furthermore, the chance that our patient experienced a clinically isolated syndrome preceding a future diagnosis of MS is <4% given negative CSF testing for oligoclonal bands (51). Systemic rheumatologic causes, such as systemic lupus erythematous (SLE), were ruled out because diagnostic clinical features were lacking and laboratory testing was negative. Additionally, lack of contrast enhancement of the LECL on MRI is inconsistent with the majority of reports of sarcoid myelitis or a covert spondylitic myelopathy (13, 14).
Acute disseminated encephalomyelitis (ADEM) is a poorly defined syndrome of multifocal CNS inflammation that represents 22–32% of acquired monophasic demyelinating syndromes in children and has a slight male predominance (52–54). It is a diagnosis of exclusion, and while typically seen as a post-viral or post-vaccinal syndrome, it can occur in up to 25% of children with no predisposing history (55). Unlike in most inflammatory myelopathies, up to 61.5% of ADEM patients have normal CSF findings, consistent with our patient's workup (56). A handful of cases of ADEM isolated to the brainstem and spinal cord, including one with MRI findings of LECL restricted to the anterior horns, have been reported (23, 25, 57). Furthermore, ADEM is known to more often involve the gray matter than either MS or NMOSD (58). While some key differentiating features of ADEM such as encephalopathy, multifocal large brain lesions of the same age, and anti-MOG seropositivity were not seen in our patient, a diagnosis of spinal-cord isolated ADEM could not be effectively ruled out (56). Furthermore, approximately only 40% of ADEM cases demonstrate anti-MOG seropositivity (and only extremely rare cases have shown AQP-4 seropositivity) (59–61).
In a recent informatics-based study, Barreras et al. describe a predictive model for determining etiology of myelopathy based on the time differential from a patient's symptom onset to symptom nadir; in our patient's case, his symptom nadir fell between 2 and 3 days from symptom onset, placing him at Barreras' cutoff between acute and subacute presentations—supporting our differential diagnosis and suggesting either an ischemic (acute) or inflammatory (subacute) etiology for our patients LECL (62). Since AQP-4 and MOG-associated NMOSD, MS, and systemic autoimmune causes such as SLE or sarcoidosis were considered unlikely based on imaging and laboratory testing, clinical management of our patient was directed by existing evidence for treatment of acute disseminated encephalomyelitis (ADEM) and other acute inflammatory myelopathies more generally (while keeping in mind the possibility of a transient or spontaneously resolving ischemic etiology and maintaining the patient on aspirin throughout the admission). High-dose corticosteroids are widely considered first-line therapy for acute inflammatory central nervous system processes including ADEM and other autoimmune myelopathies (63–65). Furthermore, a large observational study comparing functional outcomes of children with ADEM strongly favored methylprednisolone over dexamethasone as the primary agent (66). In our patient, the urgency of a rapidly progressive quadriparesis with a likely immune-mediated etiology demanded adjuvant treatment. IVIG, well-described in autoantibody-mediated acute inflammatory demyelinating polyneuropathies, is frequently a first choice for adjuvant or second-line immunomodulatory therapy in inflammatory CNS disease (especially in pediatrics) because of its ease of administration through a peripheral intravenous line and its high tolerability (67, 68). However, more recent data suggests that IVIG is inferior to similar treatments in acute inflammatory myelopathy, such as plasmapheresis or cyclophosphamide (69, 70).
Additionally, plasmapheresis is acknowledged in the inflammatory myelopathy literature mostly for its use as a salvage therapy (71–73). There is increasing evidence, however, for a synergistic effect of plasmapheresis and corticosteroids when administered early in the disease course (70, 74). While the modulatory effect is temporary, plasmapheresis effectively eliminates pathogenic autoantibodies, inflammatory cytokines, and complement proteins, thereby enhancing the longer-lasting molecular and genetic effects of corticosteroids on macrophage activation and cytokine production. Targeting both innate and humoral immune responses with early administration of combination therapy proved successful in achieving a rapid functional improvement in our patient.
Multiple large cohort studies in NMOSD patients additionally support early intervention with apheresis therapies during flares and found that reduced delay to apheresis improved the chance of a complete therapeutic response (75, 76). Our experience and literature review suggest consideration of early initiation of plasmapheresis in inflammatory LECL, despite its logistical challenges. Further investigation of the merit of early combination therapy vs. corticosteroids alone as first-line treatment is warranted in this context.
Conclusion
Swift and accurate diagnosis of the etiology of a T2-hyperintense gray matter LECL in pediatrics is a complex and critical task. Systematic literature review supports our experience that while inflammatory, infectious, and vascular etiologies should all be considered and worked up, aggressive immunomodulatory treatment should be maximized early in the disease course, especially in cases that present a time-consuming diagnostic challenge. Furthermore, plasmapheresis may contribute to more comprehensive functional improvement and should be further explored as part of a primary combination treatment strategy with high-dose steroids.
This case of LECL restricted to the anterior horns in a child emphasizes the use of an evidence based, thoughtful and multidisciplinary clinical reasoning team approach to inform the diagnosis, work up, and treatment of a rare, critical and elusive diagnosis in pediatrics where time is of the essence.
Ethics Statement
Written informed consent regarding the submission and potential publication of this manuscript was obtained from the case study patient and his legal guardian. Additionally, consent for treatment was likewise obtained in the usual fashion during the course of the patient's hospitalization.
Author Contributions
DG, FW, NI, TW, HJ, and SS drafted the manuscript. DG and FW designed figure and table. DG, FW, NI, TW, HJ, SS, DR, and GG edited and revised manuscript. DG, FW, TW, DR, and GG reviewed systematic literature. DG, FW, DR, and GG research team management and oversight.
Funding
This research is supported in part by the following grants: NYU CTSA/NCATS: UL1TR001445 NIDDK: T35DK007421.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2019.00270/full#supplementary-material
References
1. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. (2015) 85:177–89. doi: 10.1212/WNL.0000000000001729
2. Kim HJ, Paul F, Lana-Peixoto MA, Tenembaum S, Asgari N, Palace J, et al. MRI characteristics of neuromyelitis optica spectrum disorder: an international update. Neurology. (2015) 84:1165–73. doi: 10.1212/WNL.0000000000001367
3. Flanagan EP. Autoimmune myelopathies. Handb Clin Neurol. (2016) 133:327–51. doi: 10.1016/B978-0-444-63432-0.00019-0
4. Kearney H, Miszkiel KA, Yiannakas MC, Altmann DR, Ciccarelli O, Miller DH. Grey matter involvement by focal cervical spinal cord lesions is associated with progressive multiple sclerosis. Mult Scler. (2016) 22:910–20. doi: 10.1177/1352458515604905
5. Kearney H, Schneider T, Yiannakas MC, Altmann DR, Wheeler-Kingshott CA, Ciccarelli O, et al. Spinal cord grey matter abnormalities are associated with secondary progression and physical disability in multiple sclerosis. J Neurol Neurosurg Psychiatry. (2015) 86:608–14. doi: 10.1136/jnnp-2014-308241
6. Kister I, Johnson E, Raz E, Babb J, Loh J, Shepherd TM. Specific MRI findings help distinguish acute transverse myelitis of neuromyelitis optica from spinal cord infarction. Mult Scler Relat Disord. (2016) 9:62–7. doi: 10.1016/j.msard.2016.04.005
7. Abu Libdeh A, Goodkin HP, Ramirez-Montealegre D, Brenton JN. Acute disseminated encephalomyelitis: a gray distinction. Pediatr Neurol. (2017) 68:64–7. doi: 10.1016/j.pediatrneurol.2016.12.006
8. Hopkins SE. Acute flaccid myelitis: etiologic challenges, diagnostic and management considerations. Curr Treat Options Neurol. (2017) 19:48. doi: 10.1007/s11940-017-0480-3
9. Hazenfield JM, Gaskill-Shipley MF. Neoplastic and paraneoplastic involvement of the spinal cord. Semin Ultrasound CT MR. (2016) 37:482-497. doi: 10.1053/j.sult.2016.05.009
10. Yadav N, Pendharkar H, Kulkarni GB. Spinal cord infarction: clinical and radiological features. J Stroke Cerebrovasc Dis. (2018) 27:2810–21. doi: 10.1016/j.jstrokecerebrovasdis.2018.06.008
11. Ciccarelli O, Cohen JA, Reingold SC, Weinshenker BG. Spinal cord involvement in multiple sclerosis and neuromyelitis optica spectrum disorders. Lancet Neurol. (2019) 18:185–97. doi: 10.1016/S1474-4422(18)30460-5
12. Fang B, McKeon A, Hinson SR, Kryzer TJ, Pittock SJ, Aksamit AJ, et al. Autoimmune glial fibrillary acidic protein astrocytopathy: a novel meningoencephalomyelitis. JAMA Neurol. (2016) 73:1297–307. doi: 10.1001/jamaneurol.2016.2549
13. Flanagan EP, Krecke KN, Marsh RW, Giannini C, Keegan BM, Weinshenker BG. Specific pattern of gadolinium enhancement in spondylotic myelopathy. Ann Neurol. (2014) 76:54–65. doi: 10.1002/ana.24184
14. Flanagan EP, Kaufmann TJ, Krecke KN, Aksamit AJ, Pittock SJ, Keegan BM, et al. Discriminating long myelitis of neuromyelitis optica from sarcoidosis. Ann Neurol. (2016) 79:437–47. doi: 10.1002/ana.24582
15. Banwell B, Tenembaum S, Lennon VA, Ursell E, Kennedy J, Bar-Or A, et al. Neuromyelitis optica-IgG in childhood inflammatory demyelinating CNS disorders. Neurology. (2008) 70:344–52. doi: 10.1212/01.wnl.0000284600.80782.d5
16. Spencer TS, Campellone JV, Maldonado I, Huang N, Usmani Q, Reginato AJ. Clinical and magnetic resonance imaging manifestations of neurosarcoidosis. Semin Arthritis Rheum. (2005) 34:649–61. doi: 10.1016/j.semarthrit.2004.07.011
17. Kitley JL, Leite MI, George JS, Palace JA. The differential diagnosis of longitudinally extensive transverse myelitis. Mult Scler. (2012) 18:271–85. doi: 10.1177/1352458511406165
18. Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology. (2006) 66:1485–9. doi: 10.1212/01.wnl.0000216139.44259.74
19. Iorio R, Damato V, Mirabella M, Evoli A, Marti A, Plantone D, et al. Distinctive clinical and neuroimaging characteristics of longitudinally extensive transverse myelitis associated with aquaporin-4 autoantibodies. J Neurol. (2013) 260:2396–402. doi: 10.1007/s00415-013-6997-9
20. Andronikou S, Albuquerque-Jonathan G, Wilmshurst J, Hewlett R. MRI findings in acute idiopathic transverse myelopathy in children. Pediatr Radiol. (2003) 33:624–9. doi: 10.1007/s00247-003-1004-8
21. Wolf VL, Lupo PJ, Lotze TE. Pediatric acute transverse myelitis overview and differential diagnosis. J Child Neurol. (2012) 27:1426–36. doi: 10.1177/0883073812452916
22. Ghosh PS, Mitra S. Owl's eye in spinal magnetic resonance imaging. Arch Neurol. (2012) 69:407–8. doi: 10.1001/archneurol.2011.1132
23. Monden Y, Yamagata T, Kuroiwa Y, Takahashi T, Mori M, Fukuda T, et al. A case of ADEM with atypical MRI findings of a centrally-located long spinal cord lesion. Brain Dev. (2012) 34:380–3. doi: 10.1016/j.braindev.2011.06.010
24. Reisner A, Gary MF, Chern JJ, Grattan-Smith JD. Spinal cord infarction following minor trauma in children: fibrocartilaginous embolism as a putative cause. J Neurosurg Pediatr. (2013) 11:445–50. doi: 10.3171/2013.1.PEDS12382
25. DeSena A, Graves D, Morriss MC, Greenberg BM. Transverse myelitis plus syndrome and acute disseminated encephalomyelitis plus syndrome: a case series of 5 children. JAMA Neurol. (2014) 71:624–9. doi: 10.1001/jamaneurol.2013.5323
26. Amaral DM, Parreira T, Sampaio M. Longitudinally extensive transverse myelitis and meningitis due to a rare infectious cause. BMJ Case Rep. (2015) 2015:bcr2015211761. doi: 10.1136/bcr-2015-211761
27. Elpers C, Gross CC, Fiedler B, Meuth SG, Kurlemann G. A case report on juvenile neuromyelitis optica: early onset, long remission period, and atypical treatment response. Neuropediatrics. (2015) 46:292–5. doi: 10.1055/s-0035-1554101
28. Nelson JA, Ho CY, Golomb MR. Spinal cord stroke presenting with acute monoplegia in a 17-year-old tennis player. Pediatr Neurol. (2016) 56:76–9. doi: 10.1016/j.pediatrneurol.2015.11.007
29. Esposito S, Chidini G, Cinnante C, Napolitano L, Giannini A, Terranova L, et al. Acute flaccid myelitis associated with enterovirus-D68 infection in an otherwise healthy child. Virol J. (2017) 14:4. doi: 10.1186/s12985-016-0678-0
30. Girard B, Bonnemains C, Schmitt E, Raffo E, Bilbault C. Biotinidase deficiency mimicking neuromyelitis optica beginning at the age of 4: A treatable disease. Mult Scler. (2017) 23:119–22. doi: 10.1177/1352458516646087
31. Hayashi F, Hayashi S, Matsuse D, Yamasaki R, Yonekura K, Kira JI. Hopkins syndrome following the first episode of bronchial asthma associated with enterovirus D68: a case report. BMC Neurol. (2018) 18:71. doi: 10.1186/s12883-018-1075-7
32. Hsu PC, Chen SJ. Longitudinal extensive transverse myelitis with an abnormal uFLC ratio in a pediatric patient: case report and literature review. Medicine (Baltimore). (2017) 96:e9389. doi: 10.1097/MD.0000000000009389
33. Hu B, Wu P, Zhou Y, Peng Y, Tang X, Ding W, et al. A case of neuropsychiatric lupus Erythematosus characterized by the Owl's eye sign: a case report. BMC Neurol. (2017) 17:123. doi: 10.1186/s12883-017-0902-6
34. Yoder JA, Lloyd M, Zabrocki L, Auten J. Pediatric acute flaccid paralysis: enterovirus D68-associated anterior myelitis. J Emerg Med. (2017) 53:e19–23. doi: 10.1016/j.jemermed.2017.03.020
35. Chen IJ, Hu SC, Hung KL, Lo CW. Acute flaccid myelitis associated with enterovirus D68 infection: a case report. Medicine (Baltimore). (2018) 97:e11831. doi: 10.1097/MD.0000000000011831
36. Wang C, Narayan R, Greenberg B. Anti-Myelin oligodendrocyte glycoprotein antibody associated with gray matter predominant transverse myelitis mimicking acute flaccid myelitis: a presentation of two cases. Pediatr Neurol. (2018) 86:42–5. doi: 10.1016/j.pediatrneurol.2018.06.003
37. Mateen FJ, Monrad PA, Hunderfund AN, Robertson CE, Sorenson EJ. Clinically suspected fibrocartilaginous embolism: clinical characteristics, treatments, and outcomes. Eur J Neurol. (2011) 18:218–25. doi: 10.1111/j.1468-1331.2010.03200.x
38. Manara R, Calderone M, Severino MS, Citton V, Toldo I, Laverda AM, et al. Spinal cord infarction due to fibrocartilaginous embolization: the role of diffusion weighted imaging and short-tau inversion recovery sequences. J Child Neurol. (2010) 25:1024–8. doi: 10.1177/0883073809355822
39. Han JJ, Massagli TL, Jaffe KM. Fibrocartilaginous embolism–an uncommon cause of spinal cord infarction: a case report and review of the literature. Arch Phys Med Rehabil. (2004) 85:153–7. doi: 10.1016/S0003-9993(03)00289-2
40. Tosi L, Rigoli G, Beltramello A. Fibrocartilaginous embolism of the spinal cord: a clinical and pathogenetic reconsideration. J Neurol Neurosurg Psychiatry. (1996) 60:55–60. doi: 10.1136/jnnp.60.1.55
41. Zalewski NL, Rabinstein AA, Krecke KN, Brown RD, Wijdicks EFM, Weinshenker BG, et al. Characteristics of spontaneous spinal cord infarction and proposed diagnostic criteria. JAMA Neurol. (2019) 76:56–63. doi: 10.1001/jamaneurol.2018.2734
42. Maloney JA, Mirsky DM, Messacar K, Dominguez SR, Schreiner T, Stence NV. MRI findings in children with acute flaccid paralysis and cranial nerve dysfunction occurring during the 2014 enterovirus D68 outbreak. Am J Neuroradiol. (2015) 36:245–50. doi: 10.3174/ajnr.A4188
43. Hopkins IJ, Shield LK. Letter: poliomyelitis-like illness associated with asthma in childhood. Lancet. (1974) 1:760. doi: 10.1016/S0140-6736(74)92989-4
44. Cantarín-Extremera V, González-Gutiérrez-Solana L, Ramírez-Orellana M, López-Marín L, Duat-Rodríguez A, Ruíz-Falcó-Rojas ML. Immune-mediated mechanisms in the pathogenesis of Hopkins syndrome. Pediatr Neurol. (2012) 47:373–4. doi: 10.1016/j.pediatrneurol.2012.08.006
45. Dubey D, Pittock SJ, Krecke KN, Morris PP, Sechi E, Zalewski NL, et al. Clinical, radiologic, and prognostic features of myelitis associated with myelin oligodendrocyte glycoprotein autoantibod. JAMA Neurol. (2018). doi: 10.1001/jamaneurol.2018.4053. [Epub ahead of print].
46. Jarius S, Franciotta D, Paul F, Ruprecht K, Bergamaschi R, Rommer PS, et al. Cerebrospinal fluid antibodies to aquaporin-4 in neuromyelitis optica and related disorders: frequency, origin, and diagnostic relevance. J Neuroinflammation. (2010) 7:52. doi: 10.1186/1742-2094-7-52
47. Jarius S, Paul F, Aktas O, Asgari N, Dale RC, de Seze J, et al. MOG encephalomyelitis: international recommendations on diagnosis and antibody testing. J Neuroinflammation. (2018) 15:134. doi: 10.1186/s12974-018-1144-2
48. Wingerchuk DM. Neuromyelitis optica: potential roles for intravenous immunoglobulin. J Clin Immunol. (2013) 33 (Suppl. 1):S33–7. doi: 10.1007/s10875-012-9796-72
49. Compston A, Coles A. Multiple sclerosis. Lancet. (2002) 359:1221–31. doi: 10.1016/S0140-6736(02)08220-X
50. Krupp LB, Tardieu M, Amato MP, Banwell B, Chitnis T, Dale RC, et al. International Pediatric Multiple Sclerosis Study Group criteria for pediatric multiple sclerosis and immune-mediated central nervous system demyelinating disorders: revisions to the 2007 definitions. Mult Scler. (2013) 19:1261–7. doi: 10.1177/1352458513484547
51. Miller DH, Chard DT, Ciccarelli O. Clinically isolated syndromes. Lancet Neurol. (2012) 11:157–69. doi: 10.1016/S1474-4422(11)70274-5
52. Mathey G, Michaud M, Pittion-Vouyovitch S, Debouverie M. Classification and diagnostic criteria for demyelinating diseases of the central nervous system: Where do we stand today? Rev Neurol (Paris). (2018) 174:378–90. doi: 10.1016/j.neurol.2018.01.368
53. Wingerchuk DM, Weinshenker BG. Acute disseminated encephalomyelitis, transverse myelitis, and neuromyelitis optica. Continuum (Minneap Minn). (2013) 19(4 Multiple Sclerosis):944–67. doi: 10.1212/01.CON.0000433289.38339.a2
54. Koelman DL, Mateen FJ. Acute disseminated encephalomyelitis: current controversies in diagnosis and outcome. J Neurol. (2015) 262:2013–24. doi: 10.1007/s00415-015-7694-7
55. Scolding N. Acute disseminated encephalomyelitis and other inflammatory demyelinating variants. Handb Clin Neurol. (2014) 122:601–11. doi: 10.1016/B978-0-444-52001-2.00026-1
56. Marin SE, Callen DJ. The magnetic resonance imaging appearance of monophasic acute disseminated encephalomyelitis: an update post application of the 2007 consensus criteria. Neuroimaging Clin N Am. (2013) 23:245–66. doi: 10.1016/j.nic.2012.12.005
57. Alper G, Sreedher G, Zuccoli G. Isolated brain stem lesion in children: is it acute disseminated encephalomyelitis or not? Am J Neuroradiol. (2013) 34:217–20. doi: 10.3174/ajnr.A3176
58. Rossi A. Imaging of acute disseminated encephalomyelitis. Neuroimaging Clin N Am. (2008) 18:149–61. doi: 10.1016/j.nic.2007.12.007
59. Brilot F, Dale RC, Selter RC, Grummel V, Kalluri SR, Aslam M, et al. Antibodies to native myelin oligodendrocyte glycoprotein in children with inflammatory demyelinating central nervous system disease. Ann Neurol. (2009) 66:833–42. doi: 10.1002/ana.21916
60. Di Pauli F, Mader S, Rostasy K, Schanda K, Bajer-Kornek B, Ehling R, et al. Temporal dynamics of anti-MOG antibodies in CNS demyelinating diseases. Clin Immunol. (2011) 138:247–54. doi: 10.1016/j.clim.2010.11.013
61. Sakalauskaitė-Juodeikienė E, Armalienė G, Kizlaitienė R, Bagdonaitė L, Giedraitienė N, Mickevičienė D, et al. Detection of aquaporin-4 antibodies for patients with CNS inflammatory demyelinating diseases other than typical MS in Lithuania. Brain Behav. (2018) 8:e01129. doi: 10.1002/brb3.1129
62. Barreras P, Fitzgerald KC, Mealy MA, Jimenez JA, Becker D, Newsome SD, et al. Clinical biomarkers differentiate myelitis from vascular and other causes of myelopathy. Neurology. (2018) 90:e12–21. doi: 10.1212/WNL.0000000000004765
63. McDaneld LM, Fields JD, Bourdette DN, Bhardwaj A. Immunomodulatory therapies in neurologic critical care. Neurocrit Care. (2010) 12:132–43. doi: 10.1007/s12028-009-9274-0
64. Pohl D, Tenembaum S. Treatment of acute disseminated encephalomyelitis. Curr Treat Options Neurol. (2012) 14:264–75. doi: 10.1007/s11940-012-0170-0
65. Defresne P, Meyer L, Tardieu M, Scalais E, Nuttin C, De Bont B, et al. Efficacy of high dose steroid therapy in children with severe acute transverse myelitis. J Neurol Neurosurg Psychiatry. (2001) 71:272–4. doi: 10.1136/jnnp.71.2.272
66. Tenembaum S, Chamoles N, Fejerman N. Acute disseminated encephalomyelitis: a long-term follow-up study of 84 pediatric patients. Neurology. (2002) 59:1224–31. doi: 10.1212/WNL.59.8.1224
67. Greenberg BM. Treatment of acute transverse myelitis and its early complications. Continuum. (2011) 17:733–43. doi: 10.1212/01.CON.0000403792.36161.f5
68. Marchioni E, Marinou-Aktipi K, Uggetti C, Bottanelli M, Pichiecchio A, Soragna D, et al. Effectiveness of intravenous immunoglobulin treatment in adult patients with steroid-resistant monophasic or recurrent acute disseminated encephalomyelitis. J Neurol. (2002) 249:100–4. doi: 10.1007/PL00007836
69. Murai H, Arahata H, Osoegawa M, Ochi H, Minohara M, Taniwaki T, et al. Effect of immunotherapy in myelitis with atopic diathesis. J Neurol Sci. (2004) 227:39–47. doi: 10.1016/j.jns.2004.08.001
70. Greenberg BM, Thomas KP, Krishnan C, Kaplin AI, Calabresi PA, Kerr DA. Idiopathic transverse myelitis: corticosteroids, plasma exchange, or cyclophosphamide. Neurology. (2007) 68:1614–7. doi: 10.1212/01.wnl.0000260970.63493.c8
71. Weinshenker BG, O'Brien PC, Petterson TM, Noseworthy JH, Lucchinetti CF, Dodick DW, et al. A randomized trial of plasma exchange in acute central nervous system inflammatory demyelinating disease. Ann Neurol. (1999) 46:878–86. doi: 10.1002/1531-8249(199912)46:6<878::AID-ANA10>3.0.CO;2-Q
72. Schröder A, Linker RA, Gold R. Plasmapheresis for neuroinflammatory disorders. Clin Exp Neuroimmunol. (2010) 1:95–102. doi: 10.1111/j.1759-1961.2010.00010.x
73. Gwathmey K, Balogun RA, Burns T. Neurologic indications for therapeutic plasma exchange: 2013 update. J Clin Apher. (2014) 29:211–9. doi: 10.1002/jca.21331
74. Bigi S, Banwell B, Yeh EA. Outcomes after early administration of plasma exchange in pediatric central nervous system inflammatory demyelination. J Child Neurol. (2015) 30:874–80. doi: 10.1177/0883073814545883
75. Bonnan M, Valentino R, Debeugny S, Merle H, Fergé JL, Mehdaoui H, et al. Short delay to initiate plasma exchange is the strongest predictor of outcome in severe attacks of NMO spectrum disorders. J Neurol Neurosurg Psychiatry. (2018) 89:346–51. doi: 10.1136/jnnp-2017-316286
Keywords: myelitis, gray matter, anterior horn, ADEM, acute flaccid myelitis
Citation: Golub D, Williams F, Wong T, Iyengar N, Jolley H, Sabadiah S, Rhee D and Gold-von Simson G (2019) A Longitudinally Extensive Spinal Cord Lesion Restricted to Gray Matter in an Adolescent Male. Front. Neurol. 10:270. doi: 10.3389/fneur.2019.00270
Received: 02 January 2019; Accepted: 28 February 2019;
Published: 20 March 2019.
Edited by:
Robert Weissert, University of Regensburg, GermanyReviewed by:
Eoin Flanagan, Mayo Clinic, United StatesFriedemann Paul, Charité Medical University of Berlin, Germany
Copyright © 2019 Golub, Williams, Wong, Iyengar, Jolley, Sabadiah, Rhee and Gold-von Simson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Danielle Golub, ZGFuaWVsbGUuZ29sdWJAbnl1bGFuZ29uZS5vcmc=
†Primary Co-authors