 Heng Zhang
Heng Zhang Longfei Jia1
Longfei Jia1 Jianping Jia
Jianping Jia- 1Innovation Center for Neurological Disorders and Department of Neurology, Xuanwu Hospital, Capital Medical University, Beijing, China
- 2Beijing Key Laboratory of Geriatric Cognitive Disorders, Beijing, China
- 3Clinical Center for Neurodegenerative Disease and Memory Impairment, Capital Medical University, Beijing, China
- 4Center of Alzheimer's Disease, Beijing Institute for Brain Disorders, Beijing, China
Background: Alzheimer's disease (AD) is characterized by amyloid beta (Aβ) accumulation in the brain, which triggers the activation of microglia; in turn, microglia release neuroinflammatory factors capable of damaging neurons. Thus, a therapeutic approach targeting this sustained microglia-induced inflammatory response deserves investigation. Here, we examined whether oxiracetam (ORC), a nootropic of the racetam family, can indirectly prevent Aβ-induced neurotoxicity by attenuating microglial activation.
Methods: Aβ42 oligomers were used to stimulate BV2 microglial cells, and the morphological changes and phagocytic capacity of BV2 cells were evaluated using fluorescence microscopy. We used quantitative reverse transcription polymerase chain reaction to assess the inhibitory effects of ORC on Aβ-induced mRNA levels of interleukin-1β (IL-1β), IL-6, and tumor necrosis factor-α (TNF-α); enzyme-linked immunosorbent assay was used to examine the productions of these cytokines. We also assessed the mRNA level of inducible nitric oxide synthase and the production of nitric oxide (NO). The conditioned medium from BV2 cells was used to culture hippocampal HT22 cells to assess indirect toxicity using the MTT assay.
Results: ORC prevented the Aβ-induced activation of BV2 cells, as reflected by reduced morphological changes and phagocytic ability. In addition, ORC downregulated the expression of Aβ-induced cytokines (IL-1β, IL-6, and TNF-α) and the production of NO in BV2 cells. Furthermore, ORC protected HT22 cells from indirect damage evoked by Aβ-treated BV2 cell-conditioned medium, but not from direct Aβ-induced toxicity.
Conclusions: ORC suppressed the activation of BV2 cells, decreased the production of Aβ-induced inflammatory molecules and NO in BV2 cells, and protected HT22 cells against indirect toxicity mediated by Aβ-treated BV2 cell-conditioned medium. Thus, ORC may exert a protective role in AD through attenuating the damage caused by inflammation and oxidative stress.
Introduction
Alzheimer's disease (AD) is a chronic, progressive neurological disorder associated with a decline of cognitive function (1). Histopathologically, the brain of the patients with AD has two hallmarks: the extracellular accumulation of amyloid-β (Aβ) to form senile plaques and the intracellular hyperphosphorylation of tau to form neurofibrillary tangles (2). Aβ peptides, particularly Aβ oligomers, play a primary role in the pathogenesis of AD (3). Aβ oligomers are considered the most toxic form of Aβ peptides; they can trigger neuroinflammation (4), cause neuronal death (5), and impair synaptic plasticity (6).
Multiple studies suggest that there is a sustained inflammatory response, oxidative stress, and activated microglial clustering around Aβ accumulations in the brain of patients with AD (7, 8). Evidence has emerged to suggest that this sustained inflammatory response is another core feature of AD and that microglia are important mediators of Aβ-induced neuroinflammation and oxidative stress. When stimulated with Aβ, microglia are activated and release pro-inflammatory and neurotoxic factors such as interleukin-1β (IL-1β), IL-6 tumor necrosis factor-α (TNF-α), and nitric oxide (NO). In turn, these factors promote neuronal degeneration, ultimately inducing reactive microgliosis (9–11). Other evidence supports the idea that activated microglia can directly damage neurons (e.g., microglia can mediate the loss of synapses by engulfing synaptic components via the complement system) (12); they can also exacerbate the phosphorylation, aggregation, and spread of misfolded tau (13, 14). Thus, preventing Aβ-induced microglial activation, neuroinflammation, and oxidative stress may be a promising therapeutic strategy to improve the symptoms of AD pathology.
Oxiracetam (ORC) is a nootropic of the racetam family; it has been examined for its potential use in the treatment of cognitive impairment (15), cerebrovascular diseases (16), and multi-infarct dementia (17), because it can readily pass through the blood–brain barrier (BBB) and act selectively on the cortex and hippocampus (18). Recent reports have suggested that ORC improves memory in a rat model of vascular dementia and promotes recovery of cognitive function in a rat model of cerebral hypoperfusion (19). Furthermore, ORC remarkably reverses cognitive decline in older human subjects (20). Another study has suggested that ORC reduces the release of inflammatory cytokines in a rat model of stroke (21). However, whether ORC improves cognitive decline by preventing Aβ-induced inflammation and oxidative stress in AD models remains unknown; moreover, the mechanisms underlying its effects should be explored in more detail.
Here, we aimed to investigate whether ORC can prevent Aβ-induced microglial activation, inflammation, oxidative stress, and protect against Aβ neurotoxicity.
Materials and Methods
Materials
Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were purchased from Gibco (Grand Island, NY, USA). ORC was supplied by Shijiazhuang Pharmaceutical Group Ouyi Pharma Co., Ltd. (Shijiazhuang, China). Enzyme-linked immunosorbent assay (ELISA) kits were supplied by Cusabio Biotech (Wuhan, China). The NO assay kit was obtained from Jiancheng Bioengineering Institute (Nanjing, China). Dimethyl sulfoxide (DMSO), MTT, and latex beads were supplied by Sigma-Aldrich (Saint Louis, Missouri, USA). Aβ42 oligomer powder was obtained from ChinaPeptides Co., Ltd. (Suzhou, China). The RNA extraction kit was obtained from Sangon Biotech (Shanghai, China). TB Green® Premix Ex Taq™ II and PrimeScript™ RT reagent kits were obtained from Takara (Beijing, China). ActinRed was purchased from KeyGEN BioTECH (Nanjing, China).
Preparation of Aβ42 Oligomer Solution
Briefly, 1 mg of Aβ42 oligomer powder was dissolved in DMSO to obtain 1 mM of stock solution, which was further diluted with DMEM to a final concentration of 5 μM. The soluble fraction was stored at −80°C.
Cell Culture and Treatments
Murine microglial cells (BV2) were supplied by the National Infrastructure of Cell Line Resource (Beijing, China). The hippocampal neuronal cell line HT22 was obtained from LMAI Bio (Shanghai, China). Cells were maintained in DMEM containing 10% FBS at 37°C and 5% CO2.
All experimental procedures involving BV2 cells were conducted after overnight seeding and subsequent serum starvation for 0.5 h. For the phagocytosis assay and for assessing changes in microglial morphology, cells were cultured in 24-well plates (2.5 × 104 cells/well). For analysis of pro-inflammatory cytokine and inducible nitric oxide synthase (iNOS) mRNA levels, cells were cultured in six-well plates (2 × 105 cells/well). Cells were pretreated with ORC for 2 h after serum starvation and then co-cultured with Aβ for 10 h. For the analysis of pro-inflammatory cytokine and NO production, cells were pretreated with ORC for 2 h after serum starvation and then co-cultured with Aβ for 22 h. For the experiments mentioned above, four groups were classified as follows: (1) control BV2 cells; (2) Aβ-stimulated BV2 cells; (3) Aβ-stimulated BV2 cells treated with ORC; and (4) control BV2 cells treated with ORC.
For examination of indirect toxicity, conditioned media were obtained from three groups: (1) control BV2 cells; (2) Aβ-stimulated BV2 cells; and (3) Aβ-stimulated BV2 cells treated with ORC. BV2 cells in six-well plates were pretreated with ORC for 2 h and then co-cultured with Aβ for 10 h. After intervention, the medium was replaced with fresh medium without Aβ or ORC; 12 h later, the supernatant was collected as the conditioned medium.
For the MTT assay, 3 × 103 HT22 cells were cultured in 96-well plates and then treated with conditioned medium for 24 h. For the direct toxicity experiments, HT22 cells were treated with different concentrations (5, 10, 20, and 100 μM) of ORC for 2 h after serum starvation for 2 h and finally co-cultured with Aβ for another 22 h.
MTT Assay
Cell viability was examined using the MTT assay. After relevant incubations, cells were exposed to MTT for 4 h, after which DMSO (150 μl per well) was added, and optical density at 570 nm was recorded. The data are represented as a percentage of the viability in the control groups.
Microglial Phagocytosis Assay and Morphological Characterization
The procedures for the microglial phagocytosis assay were referenced from the study by Lian et al. (22). Latex beads were pre-incubated in FBS for 1 h at 37°C at a ratio of 1:5 before dilution to a final concentration of 0.01% (v/v) and 0.05% (v/v) in DMEM. After BV2 cells were treated with Aβ with/without ORC, the BV2 cell culture medium was replaced by bead-containing DMEM, and cells were incubated at 37°C for 1 h. Thereafter, cells were washed and fixed in 4% paraformaldehyde for 15 min. The cytoskeleton was then probed using ActinRed (1:50) to obtain composite images that would allow the counting of phagocytic cells. The number of phagocytic cells/the total cell number was calculated to show the scale of activation of BV2 cells. Phagocytic efficiency, which was calculated based on a weighted average of engulfed beads per cell, was also determined to evaluate phagocytic ability as previously described (23): phagocytic efficiency (%) = (1 × X1 + 2 × X2 + 3 × X3 + 4 × X4 + 5 × X5 + 6 × X6)/(total number of cells) × 100%, where Xn represents the number of cells containing n beads. We chose distinct single BV2 cells to analyze the relevant morphological characteristics. The parameters quantified were as follows: morphology, area, perimeter, and Feret's diameter using Image J (Version 1.53a).
Quantitative Reverse Transcription Polymerase Chain Reaction
Quantitative reverse transcription polymerase chain reaction (qRT-PCR) was used to evaluate levels of IL-1β, IL-6, IL-10, TNF-α, and iNOS mRNA. Total BV2 cell RNA was extracted and then reverse-transcribed into cDNA using the PrimeScript™ RT Reagent Kit. Quantitative PCR was performed using TB Green® Premix Ex Taq™ II, as follows: pre-incubation at 95°C for 30 s followed by 40 cycles of denaturation at 95°C for 5 s and annealing at 60°C for 30 s. The oligonucleotide primer sequences are shown in Table 1.

Table 1. Primers used for quantitative reverse transcription polymerase chain reaction.
ELISA Analysis
ELISA kits were used to evaluate the effects of ORC on the levels of IL-1β, IL-6, and TNF-α. In brief, after the corresponding treatments, the levels of pro-inflammatory factors in the cell supernatant were measured following the manufacturer's instructions.
Nitric Oxide Assay
The concentrations of NO in culture supernatants were examined by measuring nitrate and nitrite, the major products of NO, following the manufacturer's instructions of the NO assay kit. The optical density was assessed at 550 nm.
Statistical Analysis
For all group comparisons, one-way analysis of variance followed by Tukey's post-hoc test was performed. GraphPad Prism V8.0 (GraphPad Software Inc., California, USA) was used to analyze the data and images. All results are represented as the mean ± standard deviation. p < 0.05 was considered to be statistically significant.
Results
Oxiracetam Is Not Toxic to BV2 or HT22 Cells, Even at the Maximum Concentrations Tested
The cytotoxicity of various concentrations of ORC on BV2 (0.1–100 μM) and HT22 (1–100 μM) cells was assessed using the MTT assay. Compared with controls, none of the concentrations of ORC exerted significant cytotoxic effects on BV2 and HT22 cells (p > 0.05 for all comparisons; Figures 1A,B). Therefore, the maximum concentration (100 μM of ORC) was used in further experiments.
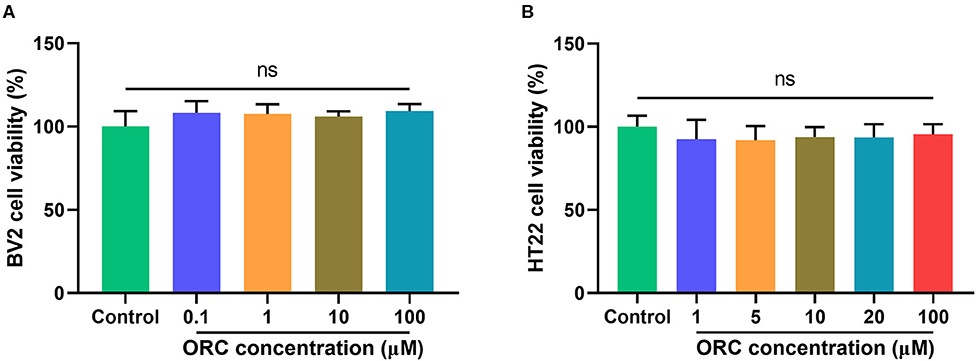
Figure 1. Cytotoxicity of oxiracetam (ORC) to BV2 microglial cells (A) and HT22 hippocampal cells (B). The results are presented as the percentage of cell viability vs. controls, with control viability regarded as 100% (n = 6 for all groups). ns, not significant (p > 0.05).
Oxiracetam Inhibits Aβ-Induced Morphological Changes and Increase in Phagocytosis in BV2 Cells
On stimulation with Aβ, the morphology of BV2 cells changed from a short and compact state to an extended and elongated one (Figure 2A); however, ORC treatment reduced the ratio of elongated cells (Figure 2B). We further quantified three other parameters of BV2 morphology: cell area, perimeter, and Feret's diameter. As the results reveal, when BV2 cells were stimulated with Aβ, the cell area, perimeter, and Feret's diameter increased significantly. However, ORC treatment significantly reversed these morphological changes (Figures 2C–E). Given that activated microglia proliferate and concentrate around Aβ plaques and respond to neuroinflammation via phagocytosis, we further analyzed the morphological alterations of activated BV2 cells using the phagocytosis assay. To investigate the effect of ORC on the number of phagocytic cells and the weighted average of ingested beads per cell, which reflect the scale of BV2 cell activation and the phagocytic efficiency, respectively, a microglial phagocytosis assay was performed (Figure 3A). Fluorescence analysis shows that, compared with that of the control group, the percentage of phagocytic cells in the Aβ-treated BV2 cell group was increased by approximately 15% (26.90 ± 4.15% vs. 40.09 ± 1.34%, p < 0.001; Figure 3B). Moreover, ORC treatment significantly reduced the number of phagocytic cells by approximately 20% than did Aβ treatment alone (21.16 ± 6.88% vs. 40.09 ± 1.34%, p < 0.001; Figure 3B). We also analyzed the phagocytic efficiency of Aβ-treated BV2 cells with or without ORC treatment. Compared with Aβ treatment only, ORC treatment significantly reduced the phagocytic efficiency of BV2 cells by approximately 25% (50.89 ± 3.50% vs. 24.96 ± 9.30%, p < 0.001; Figure 3C). These results demonstrated that ORC can inhibit the activation and phagocytic ability of Aβ-treated BV2 cells.
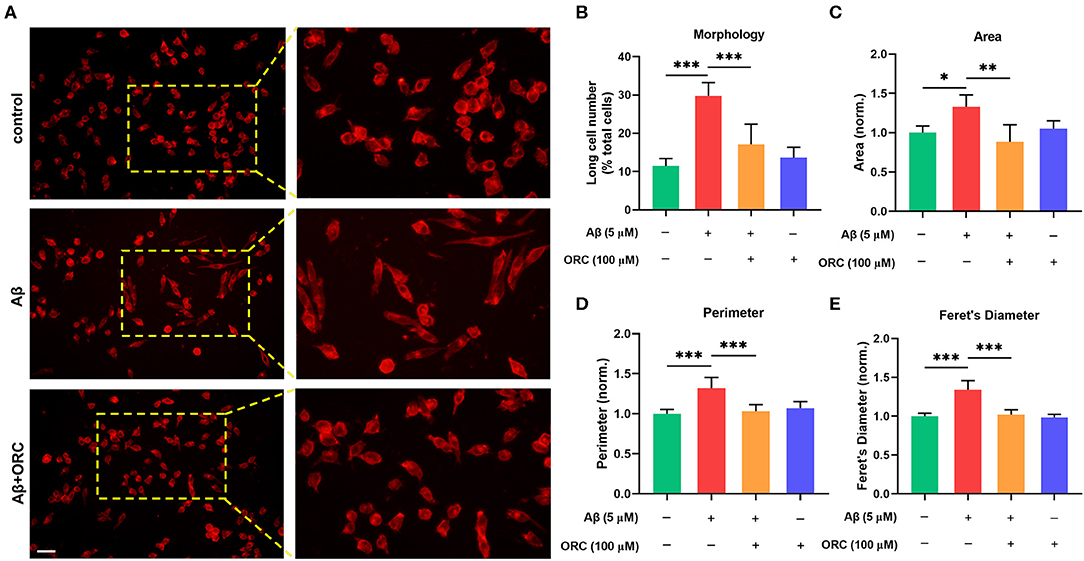
Figure 2. Oxiracetam (ORC) inhibits morphological changes induced by Aβ. The morphological changes in BV2 cells were assessed using ActinRed, and the “short” and “long” morphological phenotypes within BV2 cell populations were observed. Scale bar: 50 μm (A). Stimulation with Aβ led to an increased ratio of cells with the “long” phenotype. ORC attenuated this change (B). ORC modulated Aβ-induced BV2 cell morphological alterations in terms of cell area (C), perimeter (D), and Feret's diameter (E). *p < 0.05, **p < 0.01, and ***p < 0.001.
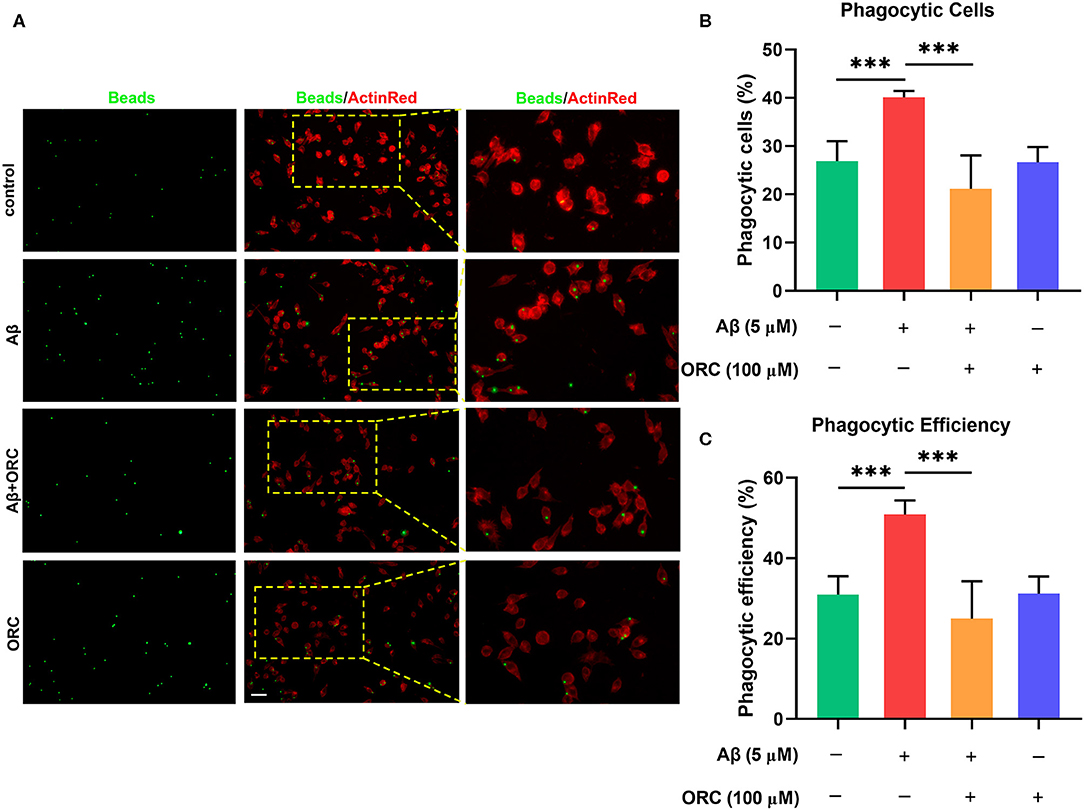
Figure 3. Inhibitory effects of oxiracetam (ORC) on Aβ-induced phagocytosis by BV2 microglial cells. (A) Representative immunofluorescence images of latex beads phagocytosed by ActinRed-marked BV2 cells. Scale bar: 50 μm. (B) Quantitative analysis of the percentage of phagocytic BV2 cells treated with Aβ and/or ORC. (C) Quantitative analysis of the phagocytic efficiency of BV2 cells based on a weighted average of ingested beads per cell. Data represent the mean ± standard deviation (n = 6 for all groups). ***p < 0.001.
Oxiracetam Downregulates the Expression of Inflammatory Cytokines
It has been proved that Aβ oligomers stimulate the secretion of inflammatory molecules from microglial cells. Thus, to further investigate whether ORC has any inhibitory effects on the Aβ-induced increase in pro-inflammatory cytokine levels, we assessed the mRNA levels of IL-1β, IL-6, and TNF-α using qRT-PCR. The results showed that expression of IL-1β, IL-6, and TNF-α was significantly upregulated following treatment with Aβ compared with control. However, treatment with ORC downregulated the mRNA level of IL-1β (fold over control, Aβ = 1.25 ± 0.18; Aβ + ORC = 1.02 ± 0.08, p < 0.05; Figure 4A), IL-6 (fold over control, Aβ = 1.50 ± 0.20; Aβ + ORC = 1.14 ± 0.26, p < 0.05; Figure 4B), and TNF-α (fold over control, Aβ = 2.19 ± 0.10; Aβ + ORC = 1.68 ± 0.16, p < 0.001; Figure 4C). We also analyzed the mRNA level of IL-10, considered as a major anti-inflammatory cytokine, under Aβ stimulation, in the presence or absence of ORC. The results showed that Aβ did not significantly affect the mRNA level of IL-10 and that ORC did not affect the levels of this anti-inflammatory cytokine (fold over control, Aβ = 1.18 ± 0.2, p > 0.70 vs. control; Aβ + ORC = 0.98 ± 0.24, p > 0.60 vs. Aβ; Supplementary Figure 1).
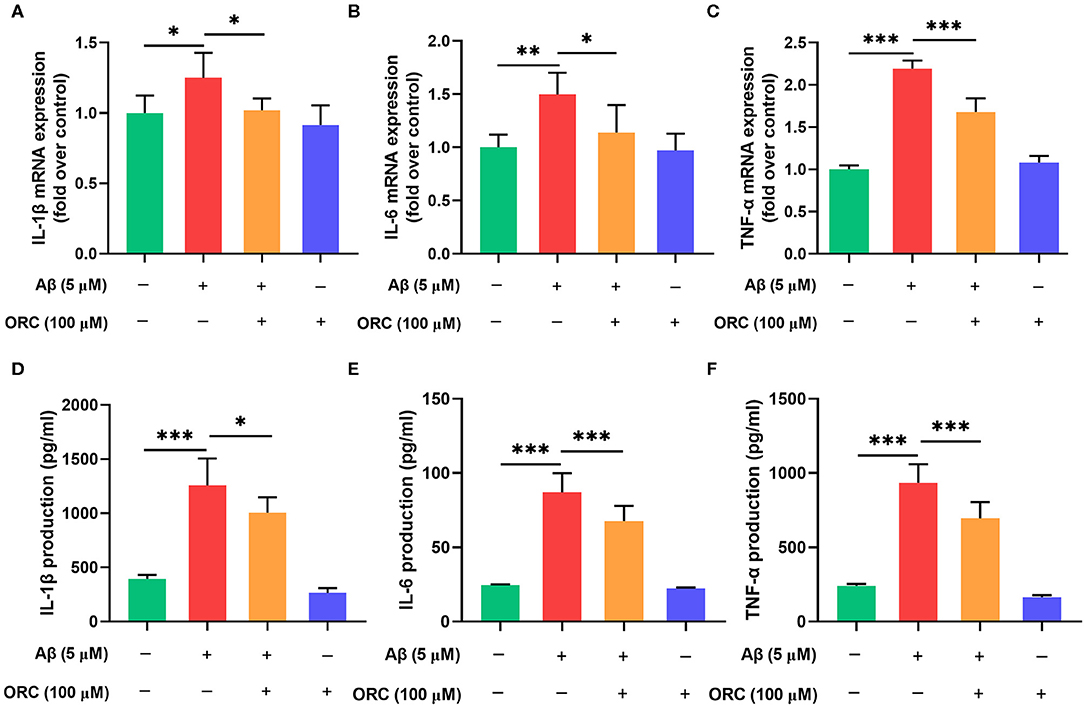
Figure 4. Inhibitory effects of oxiracetam (ORC) on pro-inflammatory cytokine mRNA and protein levels. BV2 microglial cells were treated with Aβ in the presence or absence of ORC. The mRNA levels of IL-1β (A), IL-6 (B), and TNF-α (C) were determined using qRT-PCR (n = 6 for all groups). The levels of IL-1β (D), IL-6 (E), and TNF-α (F) proteins were examined using ELISA (n = 7 for IL-1β and TNF-α, n = 9 for IL-6). All values are presented as the mean ± standard deviation. *p < 0.05, **p < 0.01, and ***p < 0.001.
In addition, we used ELISA to examine the expression of these proteins in the supernatant of BV2 cells stimulated with Aβ. Consistent with the results of the mRNA level analyses, the production of IL-1β (1,256.80 ± 248.48 vs. 395.28 ± 35.95 pg/ml, p < 0.001), IL-6 (87.11 ± 12.84 vs. 24.62 ± 0.50 pg/ml, p < 0.001), and TNF-α (933.80 ± 125.90 vs. 240.80 ± 14.01 pg/ml, p < 0.001) was increased upon Aβ induction than in the control group (Figures 4D–F). Pretreatment with ORC attenuated the production of IL-1β (p < 0.05), IL-6 (p < 0.001), and TNF-α (p < 0.001) by ~ 20, 23, and 25%, respectively, than did Aβ treatment only. These results show that ORC suppresses Aβ-triggered secretion of pro-inflammatory cytokines.
Oxiracetam Inhibits Aβ-Induced Overproduction of Nitric Oxide
Previous studies have demonstrated that Aβ can increase the levels of iNOS, which promotes the production of NO, in microglial cells. Oxidative stress is another damaging pathway that can lead to neuronal apoptosis via the overproduction of NO. Therefore, we next examined whether ORC has any effect on the levels of iNOS and NO. The results showed that level of iNOS mRNA was significantly upregulated upon stimulation with Aβ compared with that with control. However, treatment with ORC downregulated the iNOS mRNA level (fold over control, Aβ = 1.48 ± 0.06; Aβ + ORC = 1.01 ± 0.05, p < 0.05; Figure 5A). Furthermore, we showed that Aβ-stimulated BV2 cells significantly overproduced NO. However, ORC treatment reduced NO production significantly vs. Aβ alone (30.14 ± 7.2 vs. 53.09 ± 6.3 μM/L, p < 0.001; Figure 5B). Thus, ORC can inhibit Aβ-induced overproduction of NO in BV2 cells.
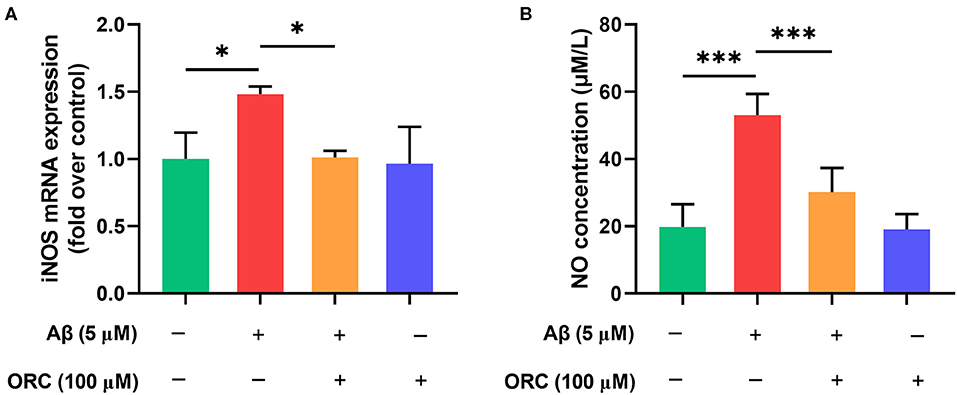
Figure 5. Inhibitory effects of oxiracetam (ORC) on the oxidative stress induced by Aβ in BV2 cells. BV2 microglial cells were treated with Aβ in the presence or absence of ORC. (A) The inducible nitric oxide synthase (iNOS) mRNA level was determined using qRT-PCR (n = 3 for all groups). (B) The level of nitric oxide (NO) in supernatants was examined using the NO assay (n = 5 for all groups). *p < 0.05 and ***p < 0.001.
Oxiracetam Protects HT22 Cells Against Aβ-Induced Neurotoxicity Indirectly
To determine whether the decreased production of pro-inflammatory cytokines and NO in Aβ-induced BV2 cells in the presence of ORC exerted a neuroprotective role, we next tested whether ORC could prevent the indirect toxicity of Aβ-stimulated BV2 cells on HT22 cells (Figure 6A). The viability of HT22 cells in the conditioned medium from Aβ-stimulated BV2 cells significantly decreased by almost 16% (vs. control, p < 0.01; Figure 6B). However, the viability of HT22 cells in conditioned medium from Aβ-induced BV2 cells co-cultured with ORC was increased (Aβ conditioned medium, 84.49 ± 5.44%; Aβ + ORC conditioned medium, 96.02 ± 9.86%, p < 0.05; Figure 6B). Thus, ORC protected HT22 cells against indirect Aβ-triggered toxicity. Next, we investigated whether ORC protected HT22 cells against direct Aβ toxicity. The results of the MTT assay revealed that treatment of HT22 cells with Aβ significantly reduced their viability (p < 0.001; Figure 6C). However, we did not find any direct protective effects ORC on Aβ-treated HT22 cell viability (p > 0.05; Figure 6C).
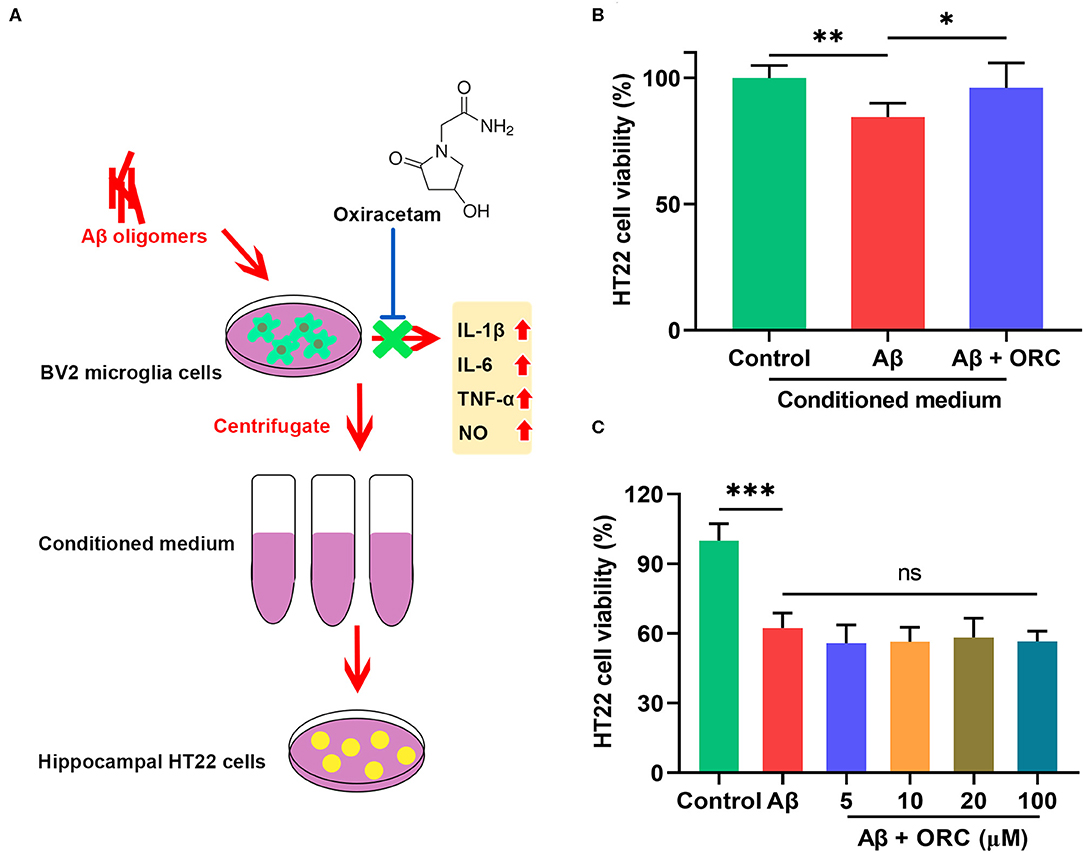
Figure 6. Neuroprotective effects of oxiracetam (ORC) against indirect toxicity of microglia-conditioned medium but not direct toxicity of Aβ. (A) Schematic of the experimental procedure to test the indirect toxicity of Aβ-induced BV2 cells in the presence or absence of ORC. (B) The results of the MTT assay revealed that ORC rescued the reduction of HT22 cell viability induced by conditioned medium from Aβ-induced BV2 cells. (C) The Aβ-induced decrease in HT22 cell viability was not affected by ORC (n = 8 for all groups). There was no significant difference between Aβ and Aβ + ORC groups. ns, not significant; *p < 0.05, **p < 0.01, and ***p < 0.001.
Discussion
Accumulating clinical evidence indicates that ORC is beneficial for patients with cognitive impairment resulting from primary degenerative or multi-infarct dementia (20, 24). However, a previous study has shown that ORC does not significantly reduce cognitive impairment due to AD (25). Therefore, it is important to confirm whether ORC could play a protective role in AD. In our present study, we found that ORC inhibited Aβ-induced activation of microglia and attenuated the release of pro-inflammatory markers and NO. In addition, ORC protected hippocampal (HT22) against indirect toxicity from BV2 microglial cells. These findings are in line with the previously reported anti-inflammatory effects of ORC.
As the most important factor correlated with AD, activated microglia have been suggested to be related to amyloid plaque types and to contribute to neuroinflammation (26). Alterations in the phagocytic activity of microglia reflect the dynamic changes in microglial activation. During early AD pathogenesis, extracellular Aβ oligomers trigger a series of cascade reactions that lead to neuronal apoptosis and loss of neurons. Some studies have demonstrated that microglia can protect the brain by phagocytic clearance of damaged cells, debris, and Aβ aggregates, in part because this phagocytosis reduces inflammation (27). However, the deleterious role of phagocytosis in AD has also been reported. Recent studies investigated whether neuronal death mediated by Aβ-activated microglia resulted from the phagocytosis of viable neurons (28, 29). Activated microglia release TNF-α or other oxidants that cause the exposure of “eat-me” signals on the surface of live neurons, which evoked their phagocytosis by microglia (28, 30). In a mouse model of early phase AD, soluble Aβ oligomers induced the engulfment of synapses to contribute to cognitive decline in a CR3 pathway-dependent manner (12). Furthermore, microglia actively contribute to the amplification of tau aggregates via phagocytosis during AD pathogenesis (14). In the present study, we observed Aβ-induced BV2 phagocytosis of latex beads, which conceivably mimics the early microglial phagocytosis process; this process was significantly suppressed by ORC. To the best of our knowledge, no studies have explored the effects of ORC on the activation and phagocytosis of microglia in models of AD. Our results illustrate that ORC modulates Aβ-enhanced microglial phagocytosis. This modulation might be beneficial to the survival of neurons and synapses in AD.
As a response to Aβ stimulation, cultured microglial cells increase the expression of various cytokines, including IL-1β, IL-6, and TNF-α, as well as nitrogen species, some of which promote the sustained production of Aβ and subsequent continuous microglial activation in a vicious cycle that can cause deteriorative neuronal damage (31, 32). IL-1β, considered as a key modulator of the inflammatory response that can promote the release of other cytokines, including IL-6 (33), has been shown to exacerbate the typical pathologies of Aβ and tau accumulation (34, 35). In clinical practice, IL-6 has also been shown to be increased in the cerebrospinal fluid (36). A cohort study reported that individuals with elevated IL-6 level are at a greater risk of cognitive impairment (37). Anti-TNF-α drugs can reduce Aβ deposition and inflammation and rescue the behavioral performance in mouse models of AD, which suggests that TNF-α has an adverse effect on the progress of AD (38). Moreover, in response to Aβ, the overproduction of NO by activated microglia has also been verified in the progression of AD. NO can trigger inflammation and induce neuronal death via oxidative stress (39); therefore, reducing the levels of pro-inflammatory cytokines and NO released by activated microglia would be a promising strategy for the treatment of AD. In our study, ORC inhibited the elevation of IL-1β, IL-6, TNF-α, and NO levels in Aβ-stimulated BV2 cells; however, the levels were not restored to the baseline levels of control cultures. These results suggest that ORC may interfere with only a few inflammatory signaling pathways to block Aβ-induced increase in microglia-expressed cytokine levels. Other molecular mechanisms underlying the effects of ORC in AD models should be explored. For example, studies indicate that Aβ-induced prolonged activation of the toll-like receptor (TLR) pathway may be responsible for not only the aberrant phagocytosis process of microglia (28, 29, 40) but also the threatening oxidative stress and inflammatory responses (41). In addition, several studies have demonstrated the activation of PI3Kδ inhibited pro-inflammatory cytokine secretion through inhibition of TLR-mediated inflammation (42–45). A study has also demonstrated that S-ORC can reduce neuronal apoptosis by activating the PI3K signaling pathway (46). Collectively, we hypothesized that ORC may suppress Aβ-induced phagocytosis, microglial activation, and inflammation via modulating the TLR and/or PI3K signal pathway. We will verify this hypothesis in a further study.
Memantine and four cholinesterase inhibitors are the only approved symptomatic treatment drugs available for the treatment of AD. In particular, drugs meant to target Aβ pathology have faced setbacks again and again. This should serve as a reminder that the indirect toxicity caused by the Aβ cascade, involving phenomena such as inflammation and oxidative stress, deserved more attention. In the present study, ORC demonstrated the protective capacity to suppress the secondary damage derived from Aβ-induced activation of microglia, which indicates that ORC may also have a positive effect on indirect toxicity in complex in vivo systems. In addition, many previous studies have also reported that ORC improves abnormal mitochondrial oxidative phosphorylation and subsequent ATP metabolism (19, 47). ORC has also been shown to alleviate middle cerebral artery occlusion/reperfusion-induced BBB dysfunction, another pathology observed in patients with AD. So ORC may exert neuro-protection via multi-target strategies, such as improving energy metabolism, protecting the integrity of the BBB, and reducing inflammatory response, in AD. The mechanisms by which ORC confers neuroprotection should be further explored in vivo models of AD.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author Contributions
HZ performed experiments, analyzed data, and wrote the manuscript. LJ designed the study and reviewed the manuscript. JJ supervised the research, revised the manuscript, and obtained funding. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Key Project of the National Natural Science Foundation of China (81530036); the National Key Scientific Instrument and Equipment Development Project (31627803); Mission Program of Beijing Municipal Administration of Hospitals (SML20150801); Beijing Scholars Program; Beijing Brain Initiative from Beijing Municipal Science & Technology Commission (Z161100000216137); Project for Outstanding Doctor with Combined Ability of Western and Chinese Medicine; and Beijing Municipal Commission of Health and Family Planning (PXM2019_026283_000003).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2020.00623/full#supplementary-material
References
1. Selkoe D, Mandelkow E, Holtzman D. Deciphering alzheimer disease. Cold Spring Harb Perspect Med. (2012) 2:a011460. doi: 10.1101/cshperspect.a011460
2. Blennow K, de Leon MJ, Zetterberg H. Alzheimer's disease. Lancet. (2006) 368:387–403. doi: 10.1016/S0140-6736(06)69113-7
3. Gandy S, Simon AJ, Steele JW, Lublin AL, Lah JJ, Walker LC, et al. Days to criterion as an indicator of toxicity associated with human alzheimer amyloid-beta oligomers. Ann Neurol. (2010) 68:220–30. doi: 10.1002/ana.22052
4. Montoliu-Gaya L, Mulder SD, Herrebout MAC, Baayen JC, Villegas S, Veerhuis R. Abeta-oligomer uptake and the resulting inflammatory response in adult human astrocytes are precluded by an anti-Abeta single chain variable fragment in combination with an apoE mimetic peptide. Mol Cell Neurosci. (2018) 89:49–59. doi: 10.1016/j.mcn.2018.03.015
5. Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, et al. Diffusible, nonfibrillar ligands derived from abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. (1998) 95:6448–53. doi: 10.1073/pnas.95.11.6448
6. Wu HY, Hudry E, Hashimoto T, Kuchibhotla K, Rozkalne A, Fan Z, et al. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J Neurosci. (2010) 30:2636–49. doi: 10.1523/JNEUROSCI.4456-09.2010
7. Lue LF, Rydel R, Brigham EF, Yang LB, Hampel H, Murphy GM, et al. Inflammatory repertoire of alzheimer's disease and nondemented elderly microglia in vitro. Glia. (2001) 35:72–9. doi: 10.1002/glia.1072
8. Gella A, Durany N. Oxidative stress in alzheimer disease. Cell Adh Migr. (2009) 3:88–93. doi: 10.4161/cam.3.1.7402
9. Sochocka M, Diniz BS, Leszek J. Inflammatory response in the CNS: friend or foe? Mol Neurobiol. (2017) 54:8071–89. doi: 10.1007/s12035-016-0297-1
10. Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. (2010) 6:193–201. doi: 10.1038/nrneurol.2010.17
11. Chen Z, Zhong C. Oxidative stress in alzheimer's disease. Neurosci Bull. (2014) 30:271–81. doi: 10.1007/s12264-013-1423-y
12. Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in alzheimer mouse models. Science. (2016) 352:712–6. doi: 10.1126/science.aad8373
13. Li Y, Liu L, Barger SW, Griffin WS. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J Neurosci. (2003) 23:1605–11. doi: 10.1523/JNEUROSCI.23-05-01605.2003
14. Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. (2015) 18:1584–93. doi: 10.1038/nn.4132
15. Bottini G, Vallar G, Cappa S, Monza GC, Scarpini E, Baron P, et al. Oxiracetam in dementia: a double-blind, placebo-controlled study. Acta Neurol Scand. (1992) 86:237–41. doi: 10.1111/j.1600-0404.1992.tb05077.x
16. Kometani M, Okada M, Takemori E, Hasegawa Y, Nakao N, Inukai T. Effect of oxiracetam on cerebrovascular impairment in rats. Arzneimittelforschung. (1991) 41:684–9.
17. Baumel B, Eisner L, Karukin M, MacNamara R, Katz RJ, Deveaugh-Geiss J. Oxiracetam in the treatment of multi-infarct dementia. Prog Neuropsychopharmacol Biol Psychiatry. (1989) 13:673–82. doi: 10.1016/0278-5846(89)90054-7
18. Gouliaev AH, Senning A. Piracetam and other structurally related nootropics. Brain Res Brain Res Rev. (1994) 19:180–222. doi: 10.1016/0165-0173(94)90011-6
19. Li W, Liu H, Jiang H, Wang C, Guo Y, Sun Y, et al. (S)-Oxiracetam is the active ingredient in oxiracetam that alleviates the cognitive impairment induced by chronic cerebral hypoperfusion in rats. Sci Rep. (2017) 7:10052. doi: 10.1038/s41598-017-10283-4
20. Rozzini R, Zanetti O, Bianchetti A. Treatment of cognitive impairment secondary to degenerative dementia. Effectiveness of oxiracetam therapy. Acta Neurol. (1993) 15:44–52.
21. Huang L, Shang E, Fan W, Li X, Li B, He S, et al. S-oxiracetam protect against ischemic stroke via alleviating blood brain barrier dysfunction in rats. Eur J Pharm Sci. (2017) 109:40–7. doi: 10.1016/j.ejps.2017.07.029
22. Lian H, Roy E, Zheng H. Microglial phagocytosis assay. Bio Protoc. (2016) 6:e1988. doi: 10.21769/BioProtoc.1988
23. Song S, Zhou F, Chen WR. Low-level laser therapy regulates microglial function through Src-mediated signaling pathways: implications for neurodegenerative diseases. J Neuroinflammation. (2012) 9:219. doi: 10.1186/1742-2094-9-219
24. Maina G, Fiori L, Torta R, Fagiani MB, Ravizza L, Bonavita E, et al. Oxiracetam in the treatment of primary degenerative and multi-infarct dementia: a double-blind, placebo-controlled study. Neuropsychobiology. (1989) 21:141–5. doi: 10.1159/000118567
25. Green RC, Goldstein FC, Auchus AP, Presley R, Clark WS, Van Tuyl L, et al. Treatment trial of oxiracetam in alzheimer's disease. Arch Neurol. (1992) 49:1135–6. doi: 10.1001/archneur.1992.00530350049018
26. Morgan D, Gordon MN, Tan J, Wilcock D, Rojiani AM. Dynamic complexity of the microglial activation response in transgenic models of amyloid deposition: implications for alzheimer therapeutics. J Neuropathol Exp Neurol. (2005) 64:743–53. doi: 10.1097/01.jnen.0000178444.33972.e0
27. Solito E, Sastre M. Microglia function in alzheimer's disease. Front Pharmacol. (2012) 3:14. doi: 10.3389/fphar.2012.00014
28. Neher JJ, Neniskyte U, Zhao JW, Bal-Price A, Tolkovsky AM, Brown GC. Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J Immunol. (2011) 186:4973–83. doi: 10.4049/jimmunol.1003600
29. Neniskyte U, Neher JJ, Brown GC. Neuronal death induced by nanomolar amyloid beta is mediated by primary phagocytosis of neurons by microglia. J Biol Chem. (2011) 286:39904–13. doi: 10.1074/jbc.M111.267583
30. Brown GC, Neher JJ. Microglial phagocytosis of live neurons. Nat Rev Neurosci. (2014) 15:209–16. doi: 10.1038/nrn3710
31. Lindberg C, Hjorth E, Post C, Winblad B, Schultzberg M. Cytokine production by a human microglial cell line: effects of beta-amyloid and alpha-melanocyte-stimulating hormone. Neurotox Res. (2005) 8:267–76. doi: 10.1007/BF03033980
32. Kuno R, Wang J, Kawanokuchi J, Takeuchi H, Mizuno T, Suzumura A. Autocrine activation of microglia by tumor necrosis factor-alpha. J Neuroimmunol. (2005) 162:89–96. doi: 10.1016/j.jneuroim.2005.01.015
33. Quintanilla RA, Orellana DI, Gonzalez-Billault C, Maccioni RB. Interleukin-6 induces alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp Cell Res. (2004) 295:245–57. doi: 10.1016/j.yexcr.2004.01.002
34. Griffin WS, Mrak RE. Interleukin-1 in the genesis and progression of and risk for development of neuronal degeneration in alzheimer's disease. J Leukoc Biol. (2002) 72:233–8.
35. Lee DC, Rizer J, Hunt JB, Selenica ML, Gordon MN, Morgan D. Review: experimental manipulations of microglia in mouse models of alzheimer's pathology: activation reduces amyloid but hastens tau pathology. Neuropathol Appl Neurobiol. (2013) 39:69–85. doi: 10.1111/nan.12002
36. Hull M, Berger M, Volk B, Bauer J. Occurrence of interleukin-6 in cortical plaques of alzheimer's disease patients may precede transformation of diffuse into neuritic plaques. Ann N Y Acad Sci. (1996) 777:205–12. doi: 10.1111/j.1749-6632.1996.tb34420.x
37. Wichmann MA, Cruickshanks KJ, Carlsson CM, Chappell R, Fischer ME, Klein BE, et al. Long-term systemic inflammation and cognitive impairment in a population-based cohort. J Am Geriatr Soc. (2014) 62:1683–91. doi: 10.1111/jgs.12994
38. Detrait ER, Danis B, Lamberty Y, Foerch P. Peripheral administration of an anti-TNF-alpha receptor fusion protein counteracts the amyloid induced elevation of hippocampal TNF-alpha levels and memory deficits in mice. Neurochem Int. (2014) 72:10–3. doi: 10.1016/j.neuint.2014.04.001
39. Asiimwe N, Yeo SG, Kim MS, Jung J, Jeong NY. Nitric oxide: exploring the contextual link with alzheimer's disease. Oxid Med Cell Longev. (2016) 2016:7205747. doi: 10.1155/2016/7205747
40. Neniskyte U, Brown GC. Lactadherin/MFG-E8 is essential for microglia-mediated neuronal loss and phagoptosis induced by amyloid beta. J Neurochem. (2013) 126:312–7. doi: 10.1111/jnc.12288
41. Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B, et al. TLR2 is a primary receptor for alzheimer's amyloid beta peptide to trigger neuroinflammatory activation. J Immunol. (2012) 188:1098–107. doi: 10.4049/jimmunol.1101121
42. Laird MH, Rhee SH, Perkins DJ, Medvedev AE, Piao W, Fenton MJ, et al. TLR4/MyD88/PI3K interactions regulate TLR4 signaling. J Leukoc Biol. (2009) 85:966–77. doi: 10.1189/jlb.1208763
43. Fallah MP, Chelvarajan RL, Garvy BA, Bondada S. Role of phosphoinositide 3-kinase-Akt signaling pathway in the age-related cytokine dysregulation in splenic macrophages stimulated via TLR-2 or TLR-4 receptors. Mech Ageing Dev. (2011) 132:274–86. doi: 10.1016/j.mad.2011.05.003
44. Aksoy E, Taboubi S, Torres D, Delbauve S, Hachani A, Whitehead MA, et al. The p110delta isoform of the kinase PI(3)K controls the subcellular compartmentalization of TLR4 signaling and protects from endotoxic shock. Nat Immunol. (2012) 13:1045–54. doi: 10.1038/ni.2426
45. Brown J, Wang H, Hajishengallis GN, Martin M. TLR-signaling networks: an integration of adaptor molecules, kinases, and cross-talk. J Dent Res. (2011) 90:417–27. doi: 10.1177/0022034510381264
46. Fan W, Li X, Huang L, He S, Xie Z, Fu Y, et al. S-oxiracetam ameliorates ischemic stroke induced neuronal apoptosis through up-regulating alpha7 nAChR and PI3K / Akt / GSK3beta signal pathway in rats. Neurochem Int. (2018) 115:50–60. doi: 10.1016/j.neuint.2018.01.008
Keywords: Alzheimer's disease, microglia, inflammation, oxiracetam, neuronal protection
Citation: Zhang H, Jia L and Jia J (2020) Oxiracetam Offers Neuroprotection by Reducing Amyloid β-Induced Microglial Activation and Inflammation in Alzheimer's Disease. Front. Neurol. 11:623. doi: 10.3389/fneur.2020.00623
Received: 03 April 2020; Accepted: 28 May 2020;
Published: 17 July 2020.
Edited by:
Anna R. Carta, University of Cagliari, ItalyReviewed by:
Caterina Scuderi, Sapienza University of Rome, ItalyMichael R. Nichols, University of Missouri–St. Louis, United States
Copyright © 2020 Zhang, Jia and Jia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianping Jia, ampwQGNjbXUuZWR1LmNu