 Bryan J. Neth1
Bryan J. Neth1 Brent A. Bauer
Brent A. Bauer Rodolfo Savica
Rodolfo Savica- 1Departments of Neurology, Mayo Clinic, Rochester, MN, United States
- 2Internal Medicine, Mayo Clinic, Rochester, MN, United States
Introduction
Parkinson disease (PD) is the second most common neurodegenerative disease, affecting ~2% of the population over age 70. Disease prevalence increases with age and, given the aging population, may triple in the next few years (1). The neurodegenerative mechanism leading to PD is still not completely elucidated. Alpha-synuclein may drive the neurodegenerative process of PD. When aggregated in neurons as intracellular Lewy bodies, it constitutes the pathologic hallmark of PD (2). On the other hand, mitochondrial dysfunction, oxidative stress, and selective neuronal loss each contribute to PD pathology (3).
Unfortunately, there remains no disease-modifying treatment in PD despite multiple trials of promising preclinical targets. Supplements and dietary interventions have been periodically considered as possible therapeutic approaches to impact disease progression and severity in related neurodegenerative disorders (3). One such intervention is intermittent fasting (IF). This viewpoint seeks to describe the putative pathophysiologic relationships among mitochondria, alpha-synuclein and PD risk genes and to provide a background for the rationale or the use of IF and similar mitochondrial-targeting therapies in PD. Finally, we propose an outline for determining the efficacy of an IF intervention in PD.
Pathologic Hallmarks of PD
Alpha-Synuclein
Alpha-synuclein is a 140-amino acid protein existing in several forms and is believed to contribute to PD pathogenesis. In PD, Lewy bodies typically concentrate in dopaminergic neurons of the substantia nigra, with alpha-synuclein-containing neurites more evenly distributed throughout the brain (2, 4). The oligomeric form of alpha-synuclein has been associated with various forms of toxicity. Key observations suggest alpha-synuclein is toxic to mitochondria and may promote PD pathogenesis (2). Likely mechanisms of pathogenicity include increased reactive oxygen species (ROS) generation and oxidative stress (5), increased neuroinflammation (2), modified membrane permeability (6), disrupted intracellular calcium homeostasis (7), altered autophagy and ubiquitin-proteasome function (8), endoplasmic reticulum-related stress (9), cytochrome c release (10), disrupted mitochondrial dynamics and altered bioenergetics, which lead to reduced synaptic transmission and cell death (11).
Mitochondrial and Bioenergetic Dysfunction
Overview
Energy production in the form of adenosine triphosphate (ATP) is crucial for the brain, which seems to use ~20–25% of glucose and oxygen of the entire body (12). Importantly, oxidative phosphorylation is the most ATP-producing mechanism of the brain and is dependent on mitochondrial respiration.
Hereditary Forms of PD
Although the clinical symptoms of early and late-onset PD appear similar, it is important to differentiate likely differences in underlying pathophysiology (4). Several gene mutations that confer increased risk for hereditary PD are related to mitochondrial function, further supporting a role of mitochondrial dysfunction in PD.
Implicated genes include: alpha-synuclein (SNCA or PARK1), Parkin (PARK2), PINK-1 (PARK6), DJ-1 (PARK7), LRRK2 (PARKS8), and HTRA2 (PARK13) (2). Mutations in these genes either confer impaired mitochondrial function and/or increase of oxidative stress and inflammation resulting from this impairment. Notably, given the multifaceted role of mitochondria in cellular metabolism and cell death, several downstream sequelae may result from disrupted normal function. Mutations in alpha-synuclein, PINK-1, DJ-1, or LRRK2 may inhibit complex I of the ETC (2, 13). PINK-1 and Parkin mutations may also impair mitophagy and disrupt normal apoptotic pathways by interference with cytochrome c release and Bax translocation (14, 15). LRRK2 mutation affects mitochondrial fission; patients with this mutation have higher rates of mutation in mtDNA (16). HtrA2 alters the mitochondrial membrane potential and affects mitochondrial morphology (swelling) (17, 18). Additional gene mutations in UCHL-1, NURR1, and VPS35 correlate with PD and may also impact mitochondrial function. Bose and Beal (18) and Jin and Youle (19) offer a more complete review of the mitochondrial implications of PD-related genes. See Figure 1 for a visual representation of the impact of alpha-synuclein and PD-risk genes on mitochondrial function.
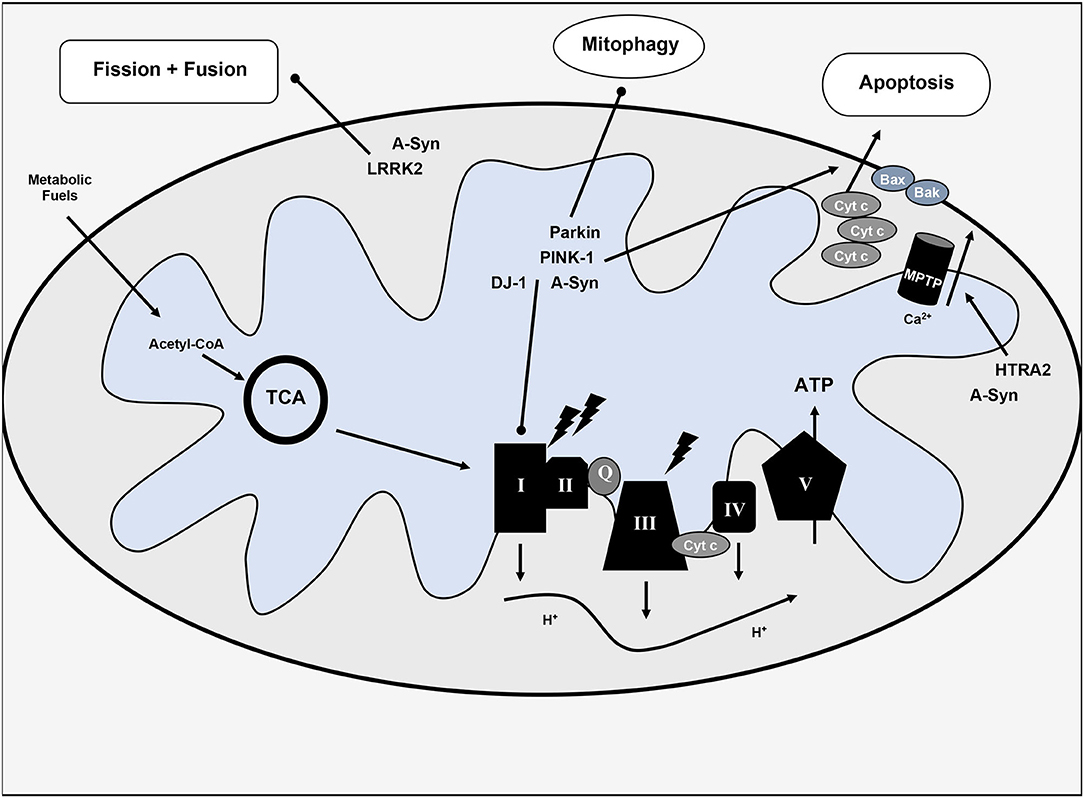
Figure 1. Overview of mitochondrial dysfunction in Parkinson's disease. Various hereditary forms of Parkinson's disease with implicated genes are included with their pathophysiologic mechanisms. TCA, tricarboxylic acid cycle or citric acid cycle; Cyt c, cytochrome c; Q, coenzyme Q; ATP, adenosine triphosphate; I-V, complexes I-V of the electron transport chain; MPTP, mitochondrial permeability transition pore; H+, protons; Ca2+, calcium, lighting bolt signifies oxidative stress.
Metabolic Risk Factors for PD
Given the prevalence of vascular-metabolic disease and the rising incidence of neurodegenerative disorders, many epidemiologic studies have tried to define the link between systemic metabolic risk factors such as obesity and PD (20). Midlife triceps skinfold thickness was associated with a 3-fold increased risk to develop PD independent of other lifestyle and adiposity measures (21). Greater body mass index (BMI) has been related to increased PD risk, which was influenced by sex. On the other hand, other studies have shown no link between adiposity or BMI and PD risk (22). The link between type 2 diabetes mellitus and PD also remains controversial. Yet, it seems that insulin resistance or impaired insulin signaling may increase PD risk, linked to mitochondrial dysfunction (18, 23).
Consequences of Mitochondrial Dysfunction
Many downstream effects of mitochondrial dysfunction have been associated to the development of PD (Figure 2), as reviewed in Rocha et al. (2); these include decreased respiratory capacity and bioenergetic function, increased production of pro-oxidant species and oxidative stress, disrupted mitochondrial dynamics, altered mitochondrial membrane potential, and calcium homeostasis – provoking further inflammatory response and cell death.
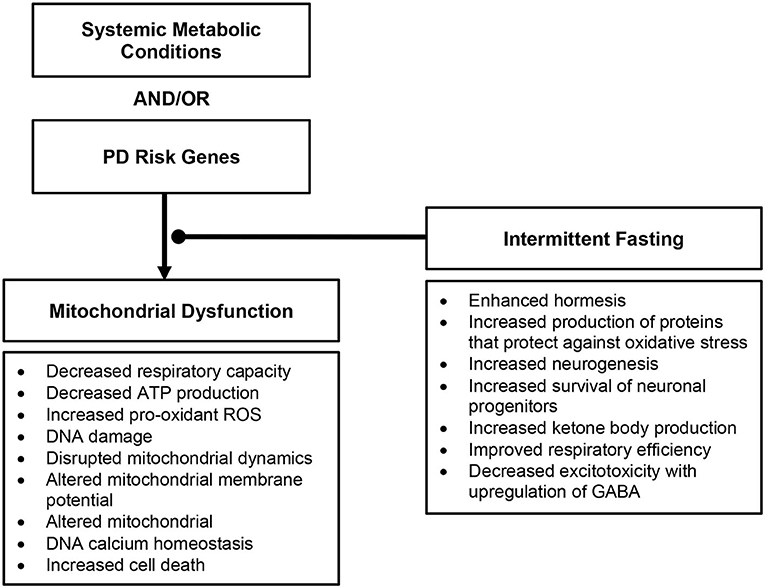
Figure 2. Overview of downstream effects of mitochondrial dysfunction in Parkinson's disease and the role intermittent fasting may play.
The ultimate sequelae of mitochondrial dysfunction and disrupted mitochondrial dynamics is cellular death, occurring in vulnerable cell populations precipitating the clinical phenotype of parkinsonism (24).The pathophysiologic hallmarks of mitochondrial dysfunction found in hereditary and sporadic PD offer important therapeutic potential to impact disease course.
Overview of Therapies Targeting Mitochondrial Dysfunction
Background
There is increasing focus on interventions targeting mitochondrial dysfunction in neurodegenerative disease (25). Many mitochondrial-targeted therapies are based on the concept of hormesis, defined as any process by which there is a biphasic response to increasing quantities and frequency of a given exposure (26). Hormesis is beneficial in conditions where smaller exposure leads to a beneficial or favorable response, and larger exposure leads to negative or toxic consequences (2). Mattson et al. review intermittent metabolic switching from glucose to ketones and back to glucose, through periods of short fasts and/or exercise (negative energy balance) and periods of eating and rest (positive energy balance) (27).
Evidence in PD
Models
Current evidence regarding mitochondrial therapies in PD is from experimental models of disease. Anson et al. showed that C57BL/6 mice (susceptible to diet-induced obesity, diabetes mellitus, and atherosclerosis), maintained on an IF regimen consisting of fasting every other day, had improved insulin sensitivity (as evidenced by reduced serum glucose and insulin levels), increased BHB levels, and improved neuronal resistance to excitotoxicity, relative to mice on an ad libitum diet. These findings were independent of any caloric deficit or even weight loss hypothesized to contribute to the beneficial effect of fasting (28). Additionally, benefits of fasting are not limited to early therapeutic (pre-symptomatic) interventions. Elfawy and Das show improved motor and cognitive performance in rats that started an IF intervention (every other day feeding schedule) after 70% of their expected lifespan, later than many studies focusing on earlier intervention (29).
Both IF and CR have proven protective against neuronal excitotoxicity in rats and mice (27), potentially mediated through direct effects on mitochondrial function with inhibited mitochondrial permeability transition pore and retained mitochondrial calcium (27). A 6-month study of CR (70% baseline food intake) in a primate PD model led to better locomotor activity with higher striatal dopamine levels relative to ad libitum-fed controls (30).
Pharmacologic therapies targeting cellular metabolic and energetic activities also have been explored in PD. Oral anti-diabetic agents have been tested in animal models of PD. Metformin has been shown to have beneficial effects on PD-related pathology. Administration of metformin (3 weeks) led to improved motor activity, reduced oxidative stress and decreased neurodegeneration in a MPTP/probenecid mouse model of PD (31), potentially mediated through BDNF. Administration of metformin (5 weeks) led to decreased cell loss of dopaminergic neurons in the substantia nigra, increased dopamine levels, improved motor performance in a MPTP/probenecid mouse model of PD (32). Pioglitazone has been shown to be neuroprotective in MPTP mouse models of PD through peroxisome proliferator-activated receptor (PPAR) activation and inhibition of inducible nitric oxide synthase (iNOS) (33). Glucagon-like peptide 1 (GLP-1) has been targeted through GLP-1 agonist, Exenatide, showing neuroprotective effects (34). In a clinical study on PD patients, Exenatide provided benefit in off-medication scores on part 3 of the MDS-UPDRS at 60 weeks relative to controls with moderate PD (35). Unfortunately, despite that pharmacologic agents and supplements may provide therapeutic benefit, these are limited in targeting a solitary pathophysiologic mechanism, whereas dietary intervention that may have multiple targets.
Clinical
Few clinical studies have assessed the impact of dietary interventions or supplementation on mitochondrial dysfunction in PD. A 28-day ketogenic diet (4:1, fat:carbohydrates) trial in a limited number of PD patients (n = 7) showed varying adherence levels with two participants leaving the trials within the first week. However, patients completing the trial (mean decrease 43.4%) had an improved Unified Parkinson's Disease Rating Scale (UPDRS) score, with noted improvement in motor symptoms, mood, and energy level (36). Amigo et al. showed the feasibility of a dietary intervention aimed at ketosis in PD patients; 38/44 participants (86%) completed the 8-week study period: 18/22 randomized to the ketogenic diet and 20/22 randomized to the control low-fat/high-carbohydrate diet (37). Both groups showed improvement from baseline MDS-UPDRS Parts 1–4 scores; however, the KD group had the most significant improvement with greater changes in non-motor, daily-living experiences scores (UPDRS part I 4.58 vs. −0.99, p < 0.001) relative to the control group. These findings supported that KD also improved non-motor PD symptoms (37). Notwithstanding limited sample sizes and lacking confirmatory results, these trials highlight the feasibility of dietary intervention in PD.
Proposed Intervention
Why Use Intermittent Fasting in PD?
Without any available disease-modifying therapeutic options, one potential approach to slow progression of PD is to target key pathophysiologic changes in PD. Mitochondrial function is a possible target, especially in the PD cases when the mitochondria dysfunction seems to be the main and/or crucial mechanism. The benefits of IF likely originate from controlled amounts of small stress and recovery, or hormesis (38). In PD models, IF has resulted in improving insulin sensitivity (39), decreased excitotoxicity (40), reduced neurodegeneration (40), and protection against autonomic dysfunction (27, 41), and motor and cognitive decline (30). IF counteracts other pathologic features of PD by enhancing neurogenesis (37) and improving survival of neuronal progenitors (42). Moreover, the resulting ketosis may promote decreased excitotoxicity with the upregulation of GABA (43).
Importantly, IF may provide benefit for the non-motor symptoms of PD in addition to the motor symptoms. Griffioen and colleagues showed that intermittent fasting (energy restriction relative to a high energy diet) led to a decreased burden of alpha-synuclein in the brainstem that contributes to autonomic dysfunction (elevated resting heart rate, impaired cardiovascular stress response, reduced parasympathetic activity) commonly seen in PD (43). As autonomic dysfunction contributes to worse functional status, it remains an important therapeutic target in addition to motor symptoms (44, 45).
An IF dietary intervention is a more encompassing approach to mitochondrial dysfunction and its downstream consequences than nutritional supplementation and, thus, may be more beneficial to target similar pathology. IF likely influences several physiologic pathways, unlike supplementation, which only affects a narrower target (36). See Figure 3 for proposed dietary intervention.
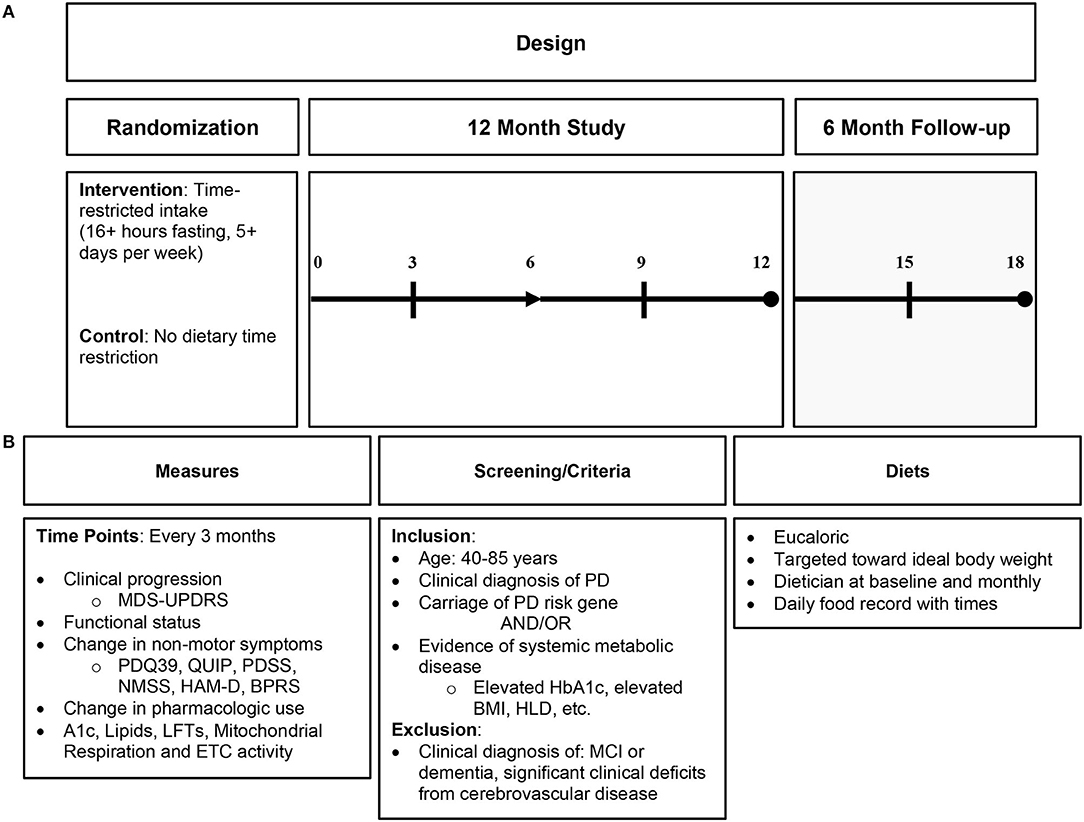
Figure 3. Proposed intermittent fasting intervention in Parkinson's disease. (A) Proposed study design; (B) proposed study details including measures of clinical endpoint with biomarkers, study criteria, and diet characteristics. PD, Parkinson's disease; HbA1c, hemoglobin A1c; HLD, hyperlipidemia; MCI, mild cognitive impairment; ETC, electron transport chain.
Is Intermittent Fasting Feasible in PD?
In theory, IF differs traditional restrictive diets (e.g., low fat or ketogenic diet). The only restriction in IF is a time-limited feeding interval. There is no limitation in foods to be consumed, although lean proteins, complex carbohydrates, and unsaturated fats are preferable. Importantly, the feeding phase of an IF diet is fundamental to the beneficial response (46). The fasting period may also vary. To improve adherence, fasting may be from 16 to 18 h daily or for a full 24-h period every other day with a normal eating pattern on opposite days. Because the diet does not restrict particular foods, there should be more consistent intake of quality foods and higher likelihood of long-term adherence, which is questionable with traditional restrictive diets. While ketosis is a sequela of IF, it is not the primary objective and thus is more amenable to dietary fluctuations.
Participants in any proposed trial should be carefully selected. An important question is whether IF may be more beneficial to those with sporadic PD or those with early-onset PD. As many genes implicated in early-onset PD relate to mitochondrial function, it seems logical that IF is useful in hereditary forms of PD. This may be a more readily targeted subset of PD patients. Nonetheless, mitochondrial dysfunction due to the presence of alpha-synuclein-related effects remains an important finding in sporadic PD, and IF should have beneficial effects in sporadic PD as well, particularly if there are metabolic risk factors or clear evidence for mitochondrial dysfunction in selection criteria for a given trial.
Conclusion
PD is an incurable neurodegenerative disorder with hallmark pathologic features including alpha-synuclein accumulation, mitochondrial dysfunction, and oxidative stress. Targeting mitochondrial activity and oxidative stress may promote beneficial effects on PD. IF may positively impact the pathological mitochondrial changes seen in PD. It has minimal side effects and is less restrictive than other commonly used dietary interventions. Without disease-modifying treatment, we must continue to explore novel therapeutic approaches that target clinically relevant pathology. Lastly, IF may be an adjunctive treatment to counter pathophysiologic disease mechanisms and enhance therapeutic response.
Author Contributions
BN: research conception and execution and writing first draft. BB: research organization and review and critique. EB: research execution and review and critique. RS: research conception, organization, and execution and review and critique. All authors contributed to the article and approved the submitted version.
Conflict of Interest
BB receives support from the Department of Energy, Thorne Research, iPEx5 GmbH, and Reulay, Inc. RS receives support from the National Institute on Aging, the National Institute of Neurological Disorders and Stroke, the Parkinson's Disease Foundation, and Acadia Pharmaceuticals.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Savica R, Grossardt BR, Rocca WA, Bower JH. Parkinson disease with and without dementia: a prevalence study and future projections. Mov Disord. (2018) 33:537–43. doi: 10.1002/mds.27277
2. Rocha EM, De Miranda B, Sanders LH. Alpha-synuclein: pathology, mitochondrial dysfunction and neuroinflammation in Parkinson's disease. Neurobiol Dis. (2018) 109:249–57. doi: 10.1016/j.nbd.2017.04.004
3. Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, et al. Parkinson disease. Nat Rev Dis Primers. (2017) 3:1–21. doi: 10.1038/nrdp.2017.13
4. Roberts HL, Brown DR. Seeking a mechanism for the toxicity of oligomeric α-synuclein. Biomolecules. (2015) 5:282–305. doi: 10.3390/biom5020282
5. Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, et al. In vivo demonstration that α-synuclein oligomers are toxic. Proc Natl Acad Sci USA. (2011) 108:4194–9. doi: 10.1073/pnas.1100976108
6. Junn E, Mouradian MM. Human α-synuclein over-expression increases intracellular reactive oxygen species levels and susceptibility to dopamine. Neurosci Lett. (2002) 320:146–50. doi: 10.1016/S0304-3940(02)00016-2
7. Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W, et al. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia. (2013) 61:349–60. doi: 10.1002/glia.22437
8. Gallegos S, Pacheco C, Peters C, Opazo CM, Aguayo LG. Features of alpha-synuclein that could explain the progression and irreversibility of Parkinson's disease. Front Neurosci. (2015) 9:59. doi: 10.3389/fnins.2015.00059
9. Luth ES, Stavrovskaya IG, Bartels T, Kristal BS, Selkoe DJ. Soluble, prefibrillar α-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J Biol Chem. (2014) 289:21490–507. doi: 10.1074/jbc.M113.545749
10. Choubey V, Safiulina D, Vaarmann A, Cagalinec M, Wareski P, Kuum M, et al. Mutant A53T α-synuclein induces neuronal death by increasing mitochondrial autophagy. J Biol Chem. (2011) 286:10814–24. doi: 10.1074/jbc.M110.132514
11. Parihar M, Parihar A, Fujita M, Hashimoto M, Ghafourifar P. Mitochondrial association of alpha-synuclein causes oxidative stress. Cell Mol Life Sci. (2008) 65:1272–84. doi: 10.1007/s00018-008-7589-1
12. Xie W, Chung KK. Alpha-synuclein impairs normal dynamics of mitochondria in cell and animal models of Parkinson's disease. J Neurochem. (2012) 122:404–14. doi: 10.1111/j.1471-4159.2012.07769.x
13. Di Maio R, Barrett PJ, Hoffman EK, Barrett CW, Zharikov A, Borah A, et al. α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson's disease. Sci Transl Med. (2016) 8:342ra378. doi: 10.1126/scitranslmed.aaf3634
14. Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab. (2001) 21:1133–45. doi: 10.1097/00004647-200110000-00001
15. Bélanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. (2011) 14:724–38. doi: 10.1016/j.cmet.2011.08.016
16. Ahlskog JE. Parkin and PINK1 parkinsonism may represent nigral mitochondrial cytopathies distinct from Lewy body Parkinson's disease. Parkinson Relat Disord. (2009) 15:721–7. doi: 10.1016/j.parkreldis.2009.09.010
17. Esteves A, Arduino D, Silva D, Oliveira C, Cardoso S. Mitochondrial dysfunction: the road to alpha-synuclein oligomerization in PD. Parkinson Dis. (2011). doi: 10.4061/2011/693761
18. Bose A, Beal MF. Mitochondrial dysfunction in Parkinson's disease. J Neurochem. (2016) 139:216–31. doi: 10.1111/jnc.13731
19. Jin SM, Youle RJ. PINK1-and Parkin-mediated mitophagy at a glance. J Cell Sci. (2012) 125:795–9. doi: 10.1242/jcs.093849
20. Abbott R, Ross G, White L, Nelson J, Masaki K, Tanner C, et al. Midlife adiposity and the future risk of Parkinson's disease. Neurology. (2002) 59:1051–7. doi: 10.1212/WNL.59.7.1051
21. Wang X, Yan MH, Fujioka H, Liu J, Wilson-Delfosse A, Chen SG, et al. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum Mol Genet. (2012) 21:1931–44. doi: 10.1093/hmg/dds003
22. Strauss KM, Martins LM, Plun-Favreau H, Marx FP, Kautzmann S, Berg D, et al. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson's disease. Hum Mol Genet. (2005) 14:2099–111. doi: 10.1093/hmg/ddi215
23. Henchcliffe C, Beal MF. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat Clin Pract Neurol. (2008) 4:600–9. doi: 10.1038/ncpneuro0924
24. Savica R, Grossardt BR, Ahlskog JE, Rocca WA. Metabolic markers or conditions preceding Parkinson's disease: a case-control study. Mov Disord. (2012) 27:974–9. doi: 10.1002/mds.25016
25. Hong C-T, Chen K-Y, Wang W, Chiu J-Y, Wu D, Chao T-Y, et al. Insulin resistance promotes parkinson's disease through aberrant expression of α-synuclein, mitochondrial dysfunction, and deregulation of the polo-like kinase 2 signaling. Cells. (2020) 9:740. doi: 10.3390/cells9030740
26. Potashkin J, Huang X, Becker C, Chen H, Foltynie T, Marras C. Understanding the links between cardiovascular disease and Parkinson's disease. Mov Disord. (2020) 35:55–74. doi: 10.1002/mds.27836
27. Mattson MP, Moehl K, Ghena N, Schmaedick M, Cheng A. Intermittent metabolic switching, neuroplasticity and brain health. Nat Rev Neurosci. (2018) 19:63. doi: 10.1038/nrn.2017.156
28. Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. (2006) 443:787–95. doi: 10.1038/nature05292
29. Elfawy HA, Das B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: etiologies and therapeutic strategies. Life Sci. (2019) 218:165–84. doi: 10.1016/j.lfs.2018.12.029
30. Anson RM, Guo Z, De Cabo R, Iyun T, Rios M, Hagepanos A, et al. Intermittent fasting dissociates beneficial effects of dietary restriction on glucose metabolism and neuronal resistance to injury from calorie intake. Proc Natl Acad Sci USA. (2003) 100:6216–20. doi: 10.1073/pnas.1035720100
31. Patil SP, Jain PD, Ghumatkar PJ, Tambe R, Sathaye S. Neuroprotective effect of metformin in MPTP-induced Parkinson's disease in mice. Neuroscience. (2014) 277:747–54. doi: 10.1016/j.neuroscience.2014.07.046
32. Lu M, Su C, Qiao C, Bian Y, Ding J, Hu G. Metformin prevents dopaminergic neuron death in MPTP/P-induced mouse model of Parkinson's disease via autophagy and mitochondrial ROS clearance. Int J Neuropsychopharmacol. (2016) 19:1–11. doi: 10.1093/ijnp/pyw047
33. Dehmer T, Heneka MT, Sastre M, Dichgans J, Schulz JB. Protection by pioglitazone in the MPTP model of Parkinson's disease correlates with I kappa B alpha induction and block of NF kappa B and iNOS activation. J Neurochem. (2004) 88:494–501. doi: 10.1046/j.1471-4159.2003.02210.x
34. Aviles-Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Ell P, Soderlund T, et al. Exenatide and the treatment of patients with Parkinson's disease. J Clin Invest. (2013) 123:2730–6. doi: 10.1172/JCI68295
35. Athauda D, Maclagan K, Skene SS, Bajwa-Joseph M, Letchford D, Chowdhury K, et al. Exenatide once weekly versus placebo in Parkinson's disease: a randomised, double-blind, placebo-controlled trial. Lancet. (2017) 390:1664–75. doi: 10.1016/S0140-6736(17)31585-4
36. Singh R, Lakhanpal D, Kumar S, Sharma S, Kataria H, Kaur M, et al. Late-onset intermittent fasting dietary restriction as a potential intervention to retard age-associated brain function impairments in male rats. Age. (2012) 34:917–33. doi: 10.1007/s11357-011-9289-2
37. Amigo I, Menezes-Filho SL, Luévano-Martínez LA, Chausse B, Kowaltowski AJ. Caloric restriction increases brain mitochondrial calcium retention capacity and protects against excitotoxicity. Aging Cell. (2017) 16:73–81. doi: 10.1111/acel.12527
38. Maswood N, Young J, Tilmont E, Zhang Z, Gash DM, Gerhardt GA, et al. Caloric restriction increases neurotrophic factor levels and attenuates neurochemical and behavioral deficits in a primate model of Parkinson's disease. Proc Natl Acad Sci USA. (2004) 101:18171–6. doi: 10.1073/pnas.0405831102
39. Vanitallie TB, Nonas C, Di Rocco A, Boyar K, Hyams K, Heymsfield S. Treatment of Parkinson disease with diet-induced hyperketonemia: a feasibility study. Neurology. (2005) 64:728–30. doi: 10.1212/01.WNL.0000152046.11390.45
40. Phillips MC, Murtagh DK, Gilbertson LJ, Asztely FJ, Lynch CD. Low-fat versus ketogenic diet in Parkinson's disease: a pilot randomized controlled trial. Mov Disord. (2018) 33:1306–14. doi: 10.1002/mds.27390
41. Mattson MP, Calabrese EJ editors. Hormesis: what it is and why it matters. In: Hormesis. Totowa, NJ: Springer (2010). p. 1–13.
42. Bayliss JA, Andrews ZB. Ghrelin is the metabolic link connecting calorie restriction to neuroprotection. Neural Regen Res. (2016) 11:1228. doi: 10.4103/1673-5374.189171
43. Griffioen KJ, Rothman SM, Ladenheim B, Wan R, Vranis N, Hutchison E, et al. Dietary energy intake modifies brainstem autonomic dysfunction caused by mutant α-synuclein. Neurobiol Aging. (2013) 34:928–35. doi: 10.1016/j.neurobiolaging.2012.07.008
44. Cersosimo MG, Benarroch EE. Autonomic involvement in Parkinson's disease: pathology, pathophysiology, clinical features and possible peripheral biomarkers. J Neurol Sci. (2012) 313:57–63. doi: 10.1016/j.jns.2011.09.030
45. Palma JA, Kaufmann H. Treatment of autonomic dysfunction in Parkinson disease and other synucleinopathies. Mov Disord. (2018) 33:372–90. doi: 10.1002/mds.27344
Keywords: intermittent fasting, Parkinson's disease, mitochondrial dysfunction, oxidative stress, neuronal loss, alpha synuclein
Citation: Neth BJ, Bauer BA, Benarroch EE and Savica R (2021) The Role of Intermittent Fasting in Parkinson's Disease. Front. Neurol. 12:682184. doi: 10.3389/fneur.2021.682184
Received: 17 March 2021; Accepted: 27 April 2021;
Published: 01 June 2021.
Edited by:
Piotr Janik, Medical University of Warsaw, PolandReviewed by:
Mark Mattson, Johns Hopkins University, United StatesCopyright © 2021 Neth, Bauer, Benarroch and Savica. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rodolfo Savica, c2F2aWNhLnJvZG9sZm9AbWF5by5lZHU=