 Ana Victoria Marco Hernández1,2*
Ana Victoria Marco Hernández1,2* Miguel Tomás Vila1Alfonso Caro Llopis2,3Sandra Monfort3
Miguel Tomás Vila1Alfonso Caro Llopis2,3Sandra Monfort3 Francisco Martinez2,3
Francisco Martinez2,3- 1Neuropediatrics Section, Hospital Universitari i Politècnic La Fe, Valencia, Spain
- 2Genetics Unit, Hospital Universitari i Politècnic La Fe, Valencia, Spain
- 3Genomics Unit, La Fe Health Research Institute, Valencia, Spain
Dominant pathogenic variations in the SCN1A gene are associated with several neuro developmental disorders with or without epilepsy, including Dravet syndrome (DS). Conversely, there are few published cases with homozygous or compound heterozygous variations in the SCN1A gene. Here, we describe two siblings from a consanguineous pedigree with epilepsy phenotype compatible with genetic epilepsy with febrile seizures plus (GEFS+) associated with the homozygous likely pathogenic variant (NM_001165963.1): c.4513A > C (p.Lys1505Gln). Clinical and genetic data were compared to those of other 10 previously published patients with epilepsy and variants in compound heterozygosity or homozygosity in the SCN1A gene. Most patients (11/12) had missense variants. Patients in whom the variants were located at the cytoplasmic or the extracellular domains frequently presented a less severe phenotype than those in whom they are located at the pore-forming domains. Five of the patients (41.7%) meet clinical criteria for Dravet syndrome (DS), one of them associated acute encephalopathy. Other five patients (41.7%) had a phenotype of epilepsy with febrile seizures plus familial origin, while the two remaining (17%) presented focal epileptic seizures. SCN1A-related epilepsies present in most cases an autosomal dominant inheritance; however, there is growing evidence that some genetic variants only manifest clinical symptoms when they are present in both alleles, following an autosomal recessive inheritance.
Introduction
SCN1A gene encodes the alpha 1 subunit of the sodium channel, Nav1.1, an important protein that makes up the voltage-dependent sodium channels. Pathogenic variants cause a reduction in sodium currents in gamma-aminobutyric acid (GABA)-ergic inhibitory interneurons, leading to hyperexcitability of neuronal network and the appearance of seizures (1–3). SCN1A is associated with several epilepsy syndromes and a range of other diseases (4, 5). The most common is Dravet syndrome (DS), but it is also associated with generalized epilepsy with febrile seizures plus (GEFS+), familial hemiplegic migraine (FHM), autism spectrum disorder (ASD), Sudden Unexpected Death Epilepsy (SUDEP), and epilepsy of infancy with migrating focal seizures (EIMFS) and very rarely involved in infantile spasms (4, 6–9). Genetic epilepsy with febrile seizure plus (GEFS+) is a familial epilepsy syndrome in which affected individuals within a family typically have a variety of epilepsy phenotypes, varying from simple febrile seizures and febrile seizures plus with a good outcome to severe epileptic encephalopathies (10).
About 1,871 SCN1A pathogenic variants have been identified so far (11), almost all of them under an autosomal-dominant fashion.
An attempt has been made to explain the phenotypic variability associated with SCN1A based on the type of mutation or functional alteration; in general, missense mutations are negatively correlated with a severe phenotype, and mutations with severe phenotypes are more frequently located in the pore region (12). Pathogenic variants that lead to a complete loss of function of the channel are virtually always associated with severe phenotypes, whereas milder impairments in channel function usually cause milder phenotypes (5, 12). Mutations that produce gain of function have also been described, as missense p.Thr226Met variant, which associates a severe clinical phenotype of early-onset epileptic encephalopathy, with profound developmental delay and hyperkinetic movement disorder (13, 14). However, in clinical practice, it remains difficult to fully predict the effects of novel variants on channel function. Recently, Jaber et al. have published original research reporting three patients with arthrogryposis multiplex congenita (AMC) in relation to other heterozygous missense variants of SCN1A (15).
To date, only 10 patients with SCN1A-related epilepsy due to autosomal recessive inheritance have been identified. In 2015, Brunklaus et al. published for the first time two novel homozygous missense mutations of the SCN1A gene in four children with infantile epilepsies from two consanguineous pedigrees. In both families, the heterozygous-carrier parents remained unaffected while their homozygous children developed SCN1A-related epilepsies, including DS and GEFS + (14). Since then, six more patients have been published (16–20). Here, we report two siblings, sons of a family with high consanguinity, with GEFS+ associated with the homozygous c.4513A > C variant in the SCN1A gene.
Materials and Methods
Participants
The study was carried out in patients affected by homozygous SCN1A pathogenic or likely pathogenic variants, according to the American College of Medical Genetics and Genomics criteria (21), from Hospital La Fe. One family was recruited for inclusion. Informed consent was obtained from the parents of both participants. A systematic search was carried out in the scientific literature for other patients diagnosed with SCN1A-related epilepsy with recessive inheritance. The search terms in Pubmed were “SCN1A,” “homozygous,” and “recessive inheritance.”
A brief clinical description of this cohort can be seen in Table 1.
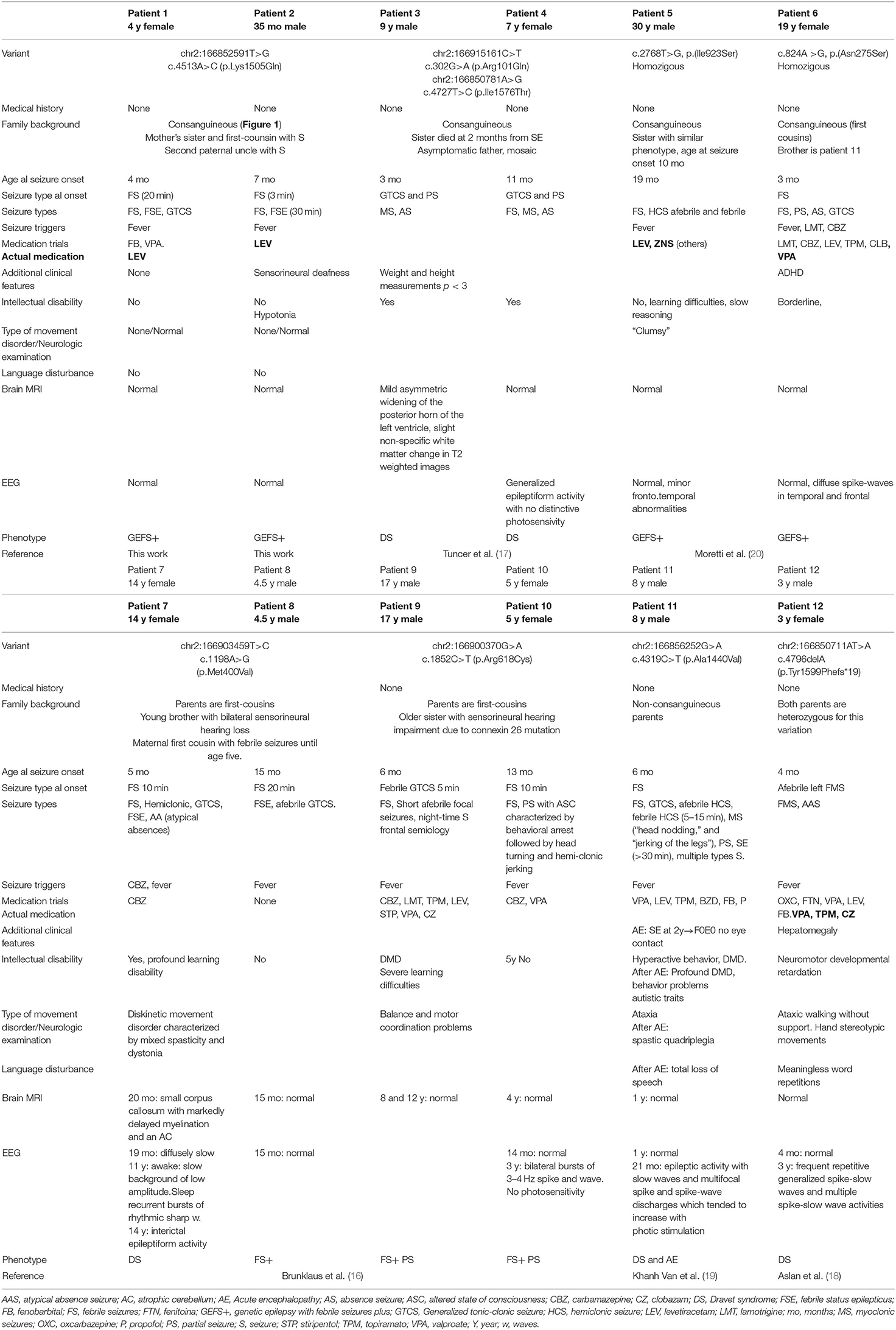
Table 1. Clinical information on 12 patients with pathogenic or likely pathogenic SCN1A variants with autosomal recessive inheritance NM_001165963.1 (see Figure 2).
This study was approved by the local ethics committee of the Hospital Universitari i Politècnic La Fe (Valencia, Spain).
Genetic Studies
Peripheral blood samples from individuals and their parents were collected by standard methods for the genetic studies. Genomic DNA was isolated using the Qiacube extractor (QIAGEN, Hilden, Germany). DNA purity and concentration were measured using the NanoDrop 8000 spectrophotometer (Thermo Scientific) and a Qubit 2.0 fluorometer (Invitrogen), respectively.
Massive parallel sequencing for exome sequencing was performed by using the SureSelect Clinical Research Exome (Agilent Technologies). The libraries were sequenced on an Illumina NextSeq 500 following the manufacturer's protocol to get a mean reading depth of 100 X. Sequence read alignments, variant calling, and annotation were performed in the Alissa Interpret platform (Agilent Technologies). All disease-causing genes related with epilepsy and/or neurodevelopmental disorders described in different databases were analyzed. To evaluate the clinical impact and to assess the pathogenicity of variants in exome sequencing, we applied the previously described criteria (22). All clinically relevant genetic variants detected were confirmed by Sanger sequencing from purified PCR products. Primers for amplification and Sanger sequencing were designed with exon-primer (primers and PCR conditions are available on request).
Case Studies
Patient 1
Patient 1 is a 4-year-old female born to healthy consanguineous parents (see Figure 1). During pregnancy, no antenatal problems were detected, neither any problem during delivery. The patient lived in her country of origin (Romania) for up to 12 months. The family reported that, at 4 months of age, she presented the first febrile status epilepticus (SE) (20 min). At 7 months, she presented a 10-min febrile seizure (FS), for which phenobarbital was prescribed. She presented two more FS up to 11 months. At 11 months, she presented first generalized tonic-clonic seizure (GTCS) lasting 5 min without fever. So, treatment with valproic acid was established. At 12 months, she was assessed for the first time in our center. She presented age-appropriate psychomotor development. Brain MRI and electroencephalogram study were normal. In the metabolic study, lactic acid, ammonium, and organic acids were normal. In the acylcarnitine profile, a slight decrease in free carnitine 11.95 μmol/L (12.92–36.88) was detected, probably secondary to treatment with valproate, so it was decided to suspend phenobarbital and valproic acid to start treatment with levetiracetam. Since then, she has presented four more FS in the context of infectious illness, and her psychomotor development and neurological examination are normal. The clinical phenotype of the patient is GEFS+.
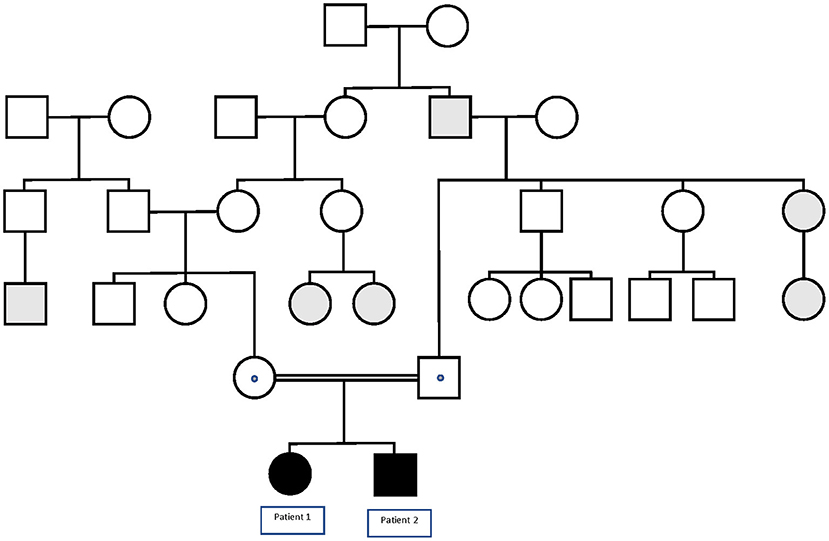
Figure 1. Pedigree of the family in which the p.Lys1505Gln variant in the SCN1A gene segregates with GEFS+. Black symbols denote homozygous patients, and dotted symbols denote obligate or confirmed heterozygous healthy carriers. Gray symbols denote patients with FS or FS+.
Patient 2
Patient 2 is a 35-month-old male, the brother of Patient 1 (see Figure 1). The child was born from uneventful eutocic delivery after a 37-week regular gestation, with a birth weight 3,580 g; Apgar score, 8/9/10. In the screening with otoacoustic emissions carried out at birth, hearing loss was detected, which was confirmed by performing auditory-evoked potentials of the trunk (response, 70 dB in both ears with dysmorphic curves). At 7 months of age, he presented the first FS (3 min). Furthermore, at that age, significant axial hypotonia was detected, without maintaining head support and little visual contact. For this reason, it was decided to perform an MRI study and electromyogram, which were normal. Metabolic study was extended to ammonium, amino acids in serum and cerebrospinal fluid, acylcarnitines, and organic acids, all in the normal range. At 10 months of age, hearing aids were placed, and early stimulation was started with very good evolution. In the following months, the patient presented a total of four more typical FS. At 20 months of life, he presented a febrile SE of 30 min in the context of high fever, so it was decided to start treatment with levetiracetam. Subsequently, he has only presented a more typical FS.
Currently, psychomotor development is at the lower limit of normality, and the neurological examination is normal. The clinical phenotype of the Patient 2 is compatible with GEFS+.
Genetics
Initially, Patient 2 was genetically studied due to hearing loss; only the heterozygous variant c.269T > C (p.Leu90Pro) in the GJB2 gene was considered of relevance. The inheritance pattern of hearing loss due to this gene is autosomal recessive and requires the presence of a second mutation in this gene to confirm the genetic diagnosis of hearing loss. It is possible that there might be a second undetected mutation or that there is another genetic or non-genetic cause of the hearing loss. On the other hand, Patient 2 presents the homozygous variant c.4513A > C in the SCN1A gene as the only clinically relevant result. This variant causes the substitution of the highly conserved amino acid lysine by glutamine in position 1,505 of the protein (p.Lys1505Gln), predicted as likely pathogenic by most of the in silico predictors (see Table 2). This variant has not been previously described. The variant is located between the repeat Domains III and IV at the cytoplasmic domain (see Figure 2). According to the ACMG criteria, this variant was classified as likely pathogenic (PP3, PM1, PM2, and PP2).
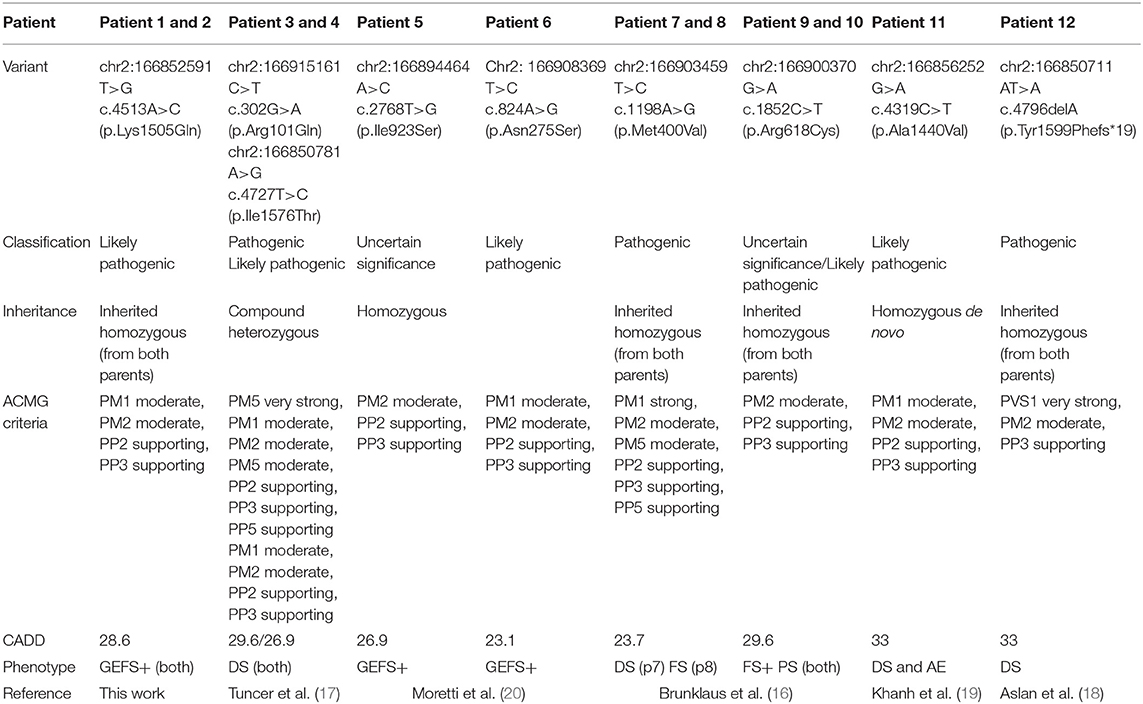
Table 2. Genetic information on 12 patients with pathogenic or likely pathogenic SCN1A variants with autosomal recessive inheritance NM_001165963.1.
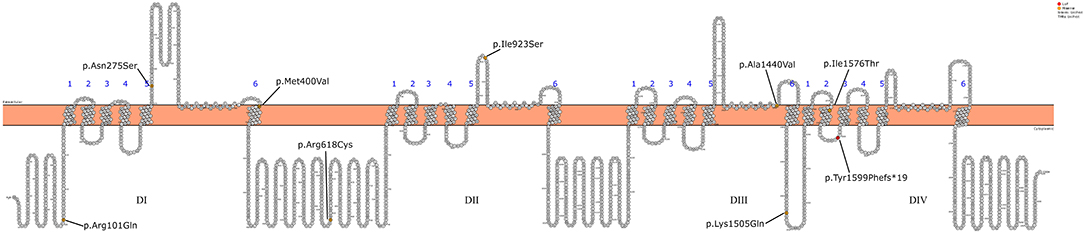
Figure 2. Schematic representation of the SCN1A gene mutations detected in the 12 patients.
Familial segregational study by Sanger sequencing confirmed that Patient 1 shares the variant c.4513A > C in SCN1A gene in homozygous state, while the healthy parents of the two patients were heterozygous carriers.
Discussion
So far, thousands of patients with heterozygous variants in the SCN1A gene have been published. The spectrum of the clinical phenotype associated with variations in the SCN1A gene is wide. Numerous studies have attempted to correlate the clinical phenotype according to the genotype (5, 12, 23), and there is evidence that, depending on the type and location of the mutation in the gene, the severity of the clinical picture varies. However, predicting the clinical consequences of novel genetic variants found in routine clinical practice is not an easy task. On the other hand, new phenotypes associated with SCN1A are continually being published (13, 15, 24–27).
The majority of published patients with pathogenic variants in SCN1A have autosomal dominant inheritance, more frequently by de novo mutation, although there are also many inherited cases (28). However, in recent years, several individuals who presented epilepsy in relation to homozygous or compound heterozygous variants in SCN1A, that is, with autosomal recessive inheritance, have been published. We have collected all the published cases and added our patients in order to broaden the phenotypic and genotypic spectrum associated with SCN1A.
In 2015, Brunklaus et al. published for the first time two novel homozygous missense mutations of the SCN1A gene in four children. Then, Tuncer et al. published two brothers with compound heterozygosity in SCN1A with symptoms compatible with DS (17). Subsequently, Aslan et al. and Van et al. have published two more cases with homozygous variants in patients with DS (18, 19), and Moretti et al. published two novel homozygous missense variations of the SCN1A gene in two individuals from two different families from consanguineous pedigrees. Both of them encompassed with GEFS+ (20) (see Tables 1, 2).
Ten of the total 12 patients were children of families with consanguinity. Patient 11, published by Van et al. presented the variant p.Ala1440Val in homozygosity, but the authors verified that it was a de novo mutation, since, despite having verified the paternity of the parents, none of them were carriers (19).
Regarding the clinical phenotype, five of the patients (41.7%) are clinically encompassed by Dravet syndrome (DS), one of them associated acute encephalopathy (AE). Five (41.7%) had a phenotype of GEFS+ of familial origin. Two (16.7%) of the patients presented FS+ and focal epileptic seizures (10). In Table 1, we have collected the description of all the patients.
Patient 2 presented sensorineural deafness, of unknown etiology, despite having carried out an extensive etiological study. Furthermore, Patients 7 and 8 had a younger brother with sensorineural hearing loss without a specified cause, and Patients 9 and 10 had an older sister with sensorineural hearing impairment due to connexin 26 mutation. Therefore, of the eight families in the series, three of them had individuals with sensorineural deafness. There are no publications linking SCN1A variants with sensorineural deafness, despite the large number of published patients with variants in this gene. For this reason, we think that this finding is probably related to consanguinity of the family rather than to SCN1A gene. The concurrence in a family of two autosomal recessive inherited diseases is not so uncommon when it comes to consanguineous families (29). Sensorineural deafness of genetic cause is mostly (about 80%) due to genetic variants of autosomal recessive inheritance (30, 31).
In the same way, we think that, possibly, the hepatomegaly that Patient 12 presented is unrelated to the variants in SCN1A gene.
Missense variants in SCN1A gene have variable effects, depending on the functional regions in which they occur and the nature of amino acid substitutions (11).
Missense mutations leading to disease are more likely to occur in the voltage sensor (S4) and the pore region (S5-S6 segments) of the four homologous domains, as in the case of Patients 3, 4, 7, and 11 of this series (32). As an exception, Patient 8 (the brother of Patient 7) presents the variant in the pore and a mild phenotype. In some pairs of siblings, a marked heterogeneity of the phenotype is observed. It has been proposed that variations in other epilepsy genes can modify the clinical phenotype associated with variants in SCN1A (33). This phenomenon has also been described in other genetic epilepsies associated with other ion channels, such as SCN2A (34). This variability associated with the same variation in SCN1A has been described both in individuals from the same family and at the interfamily level. However, further unknown environmental or epigenetic factors have been also proposed to play a significant role in the development and evolution of the epilepsy phenotype (35, 36).
All the variants described in the series are missenses, except for the frames hit variant in Patient 12, who presents a severe DS phenotype even though it is located at the intracytoplasmic level. So, due to the characteristics of the variant, the effect on the protein can be variable. Pathogenic variants that lead to a complete loss of function of the channel are virtually always associated with severe phenotypes.
It is difficult to establish a correlation between the genotype phenotype with such a small series; however, the data suggest that the variants located at the intracytoplasmic or extracellular level (Patients 1, 2, 5, 6, 9, 10, and 12) present a less severe epileptic phenotype. Conversely, those variants found at the pore region (Patients 3, 4, 7, 8, and 11) more frequently present a more severe phenotype, with a phenotype of Dravet syndrome in most patients.
It is worth noting that heterozygous carrier parents or other relatives are asymptomatic. Therefore, the variants described in this work are probably hypomorphic, both due to their location within the gene and because of their intrinsic properties, and they are only capable of altering the function of the Nav1.1 protein when they are in homozygous state. In any case, it is also remarkable that the phenotypic spectrum of the whole series of patients presenting the autosomal recessive phenotype related to SCN1A is quite similar to the phenotypic spectrum of most patients with autosomal dominant inheritance, as well as a similar genotype-phenotype correlation.
Conclusions
The 12 patients published so far with an autosomal recessive epileptic phenotype associated with SCN1A show that not all patients with symptoms associated with this gene have autosomal dominant inheritance. On the other hand, it is worth noting that the resulting phenotypes do not differ from those usually caused by dominant heterozygous variants. Furthermore, the analysis of the variants found in each patient, correlating it with the phenotype he or she presents, allows us to know a little better the correlation of genotype-phenotype associated with SCN1A. It is possible that hypomorphic heterozygous variants only manifest when affecting the two alleles of this gene.
Data Availability Statement
The datasets presented in this article are not readily available due to ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Comité de ética del Instituto de Investigación Sanitaria La Fe. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
AM: drafting of the article, clinical examination of the patients, and bibliographic search. AC and SM: substantial contributions to analysis and interpretation of genetic data. MT: revising the article for important intellectual content and contribution to clinical data analysis. FM: analysis and interpretation of genetic data and revising of the article and final approval. All authors contributed to the article and approved the submitted version.
Funding
This work had received financial support from the CM19/00181 Grant from the Carlos III Institute (AM) and the Mutua Madrileña Foundation (holded by SM). AC was supported by a research grant by Fundación Mutua Madrileña. Support for open access publication was provided by Fundación Síndrome de Dravet (Dravet Syndrome Foundation Spain) (grant number FSD-OASP-XI-III).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We warmly thank all the patients and their families for their involvement in this study.
References
1. Catterall WA, Kalume F, Oakley JC. Nav1.1 channels and epilepsy. J Physiol. (2010) 588:1984–59. doi: 10.1113/jphysiol.2010.187484
2. Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. (2006) 9:1142–9. doi: 10.1038/nn1754
3. Ogiwara I, Miyamoto H, Morita N, Atapoui N, Mazaki E, Inoue I, et al. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci. (2007) 27:5903–14. doi: 10.1523/JNEUROSCI.5270-06.2007
4. Scheffer IE, Nabbout R. SCN1A-related phenotypes: epilepsy and beyond. Epilepsia. (2019) 60:S17–24. doi: 10.1111/epi.16386
5. de Lange IM, Koudijs MJ, van't Slot R, Gunning B, Sonsma ACM, van Gemert LJJM, et al. Mosaicism of de novo pathogenic SCN1A variants in epilepsy is a frequent phenomenon that correlates with variable phenotypes. Epilepsia. (2018) 59:690–703. doi: 10.1111/epi.14021
6. Mei D, Cetica V, Marini C, Gerrini R. Dravet syndrome as part of the clinical and genetic spectrum of sodium cannel epilepsies and encephalopathies. Epilepsia. (2019) 60:S2–7. doi: 10.1111/epi.16054
7. Carranza Rojo D, Hamiwka L, McMahon JM, Dibbens LM, Arsov T, Suls A, et al. De novo SCN1A mutations in migrating partial seizures of infancy. Neurology. (2011) 77:380–3. doi: 10.1212/WNL.0b013e318227046d
8. Hirose S, Scheffer IE, Marini C, De Jonghe P, Andermann E, Goldman AM, et al. SCN1A testing for epilepsy: application in clinical practice. Epilepsia. (2013) 54:946–52. doi: 10.1111/epi.12168
9. Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De Novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. (2001) 68:1327–32. doi: 10.1086/320609
10. Myers KA, Scheffer IE, Berkovic SF, on behalf of the ILAE Genetics Commission. Genetic literacy series: genetic epilepsy with febrile seizures plus. Epilepstic Disord. (2018) 20:232–8. doi: 10.1684/epd.2018.0985
11. SCN1A mutation database. Institute of Neurosciences, Guangzhou Medical University. C2021 – (2021). Available online at: http://scn1a.caae.org.cn (accessed July 8, 2021)
12. Meng H, Xu HQ, Yu L, Lin GW, He N, Su T, et al. The SCN1A mutation database: updating information and analysis of the relationships among genotype, functional alteration, and phenotype. Hum Mutat. (2015) 36:573–80. doi: 10.1002/humu.22782
13. Sadleir LG, Moutier EI, Gill D, Davis S, Joshi C, DeVile C, et al. Not all SCN1A epileptic encephalopathies are Dravet syndrome: early profound Thr226Met phenotype. Neurology. (2017) 89:1035–42. doi: 10.1212/WNL.0000000000004331
14. Beck VC, Hull JM, Isom LL. Beyond dravet syndrome: characterization of a novel, more severe SCN1A-linked epileptic encephalopathy. Epilepsy Curr. (2019) 19:266–8. doi: 10.1177/1535759719858339
15. Jaber D, Gitiaux C, Blesson S, Marguet F, Buard D, Varela Salgado M, et al. De novo mutations of SCN1A are responsible for arthrogryposis broadening the SCN1A-related phenotypes. J Med Genet. (2020) 20:1–6. doi: 10.1136/jmedgenet-2020-107166
16. Brunklaus A, Ellis R, Stewart H, Aylett S, Reavey E, Jefferson R, et al. Homozygous mutations in the SCN1A gene associated with genetic epilepsy with febrile seizures plus and Dravet syndrome in 2 families. Eur J Paediatr Neurol. (2015) 19:484–8. doi: 10.1016/j.ejpn.2015.02.001
17. Tuncer FN, Gormez Z, Calik M, Uzun GA, Sagiroglu MS, Yuceturk B, et al. A clinical variant in SCN1A inherited from a mosaic father cosegregates with a novel variant to cause Dravet syndrome in a consanguineous family. Epilepsy Res. (2015) 113:5–10. doi: 10.1016/j.eplepsyres.2015.02.020
18. Aslan M, Ozgor B, Kirik S, Gungor S. A novel SCN1A mutation: a case report. J Pediatr Neurosci. (2020) 15:120–3. doi: 10.4103/jpn.JPN_118_18
19. Khanh Van LT, Hien HTD, Kieu HTT, Hieu NLT, Vinh LS, Hoa G, et al. De novo homozygous variant of the SCN1A gene in patient with severe Dravet syndrome complicated by acute encephalopathy. Neurogenet. (2021) 22:133–6. doi: 10.1007/s10048-021-00636-7
20. Moretti R, Arnaud L, Bouteiller D, Trouillard O, Moreau P, Buratti J, et al. SCN1A-related epilepsy with recessive inheritance: two further families. Eur J Paediatric Neurol. (2021) 33:121–4. doi: 10.1016/j.ejpn.2021.05.018
21. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
22. Martinez F, Caro-Llopis A, Rosello M, Oltra S, Mayo S, Monfort S, et al. High diagnostic yield of syndromic intellectual disability by targeted next-generation sequencing. J Med Genet. (2017) 54:87–92. doi: 10.1136/jmedgenet-2016-103964
23. Brunklaus A, Schorge S, Smith AD, Ghanty I, Stewart K, Gardiner S, et al. SCN1A variants from bench to bedside-improved clinical prediction from functional characterization. Hum Mutat. (2019) 41:363–74. doi: 10.1002/humu.23943
24. Spagnoli C, Frattini D, Rizzi S, Salerno GG, Fusco C. Early infantile SCN1A epileptic encephalopathy: expanding the genotype-phenotype correlations. Seizure. (2019) 65:62–4. doi: 10.1016/j.seizure.2019.01.002
25. Berecki G, Bryson A, Terhag J, Maljevic S, Gazina EV, Hill SL, et al. SCN1A gain of function in early infantile encephalopathy. Ann Neurol. (2019) 85:514–25. doi: 10.1002/ana.25438
26. Ohashi T, Akasaka N, Kobayashi Y, Magra S, Kawashima H, Matsumoto N, et al. Infantile epileptic encephalopathy with a hyperkinetic movement disorder and hand stereotypies associated with a novel SCN1A mutation. Epileptic Disord. (2014) 16:208–12. doi: 10.1684/epd.2014.0649
27. Ohtsuka Y, Ohmori I, Ogino T, Ouchida M, Shimizu K, Oka E. Paroxysmal movement disorders in severe myoclonic epilepsy in infancy. Brain Dev. (2003) 25:401–5. doi: 10.1016/S0387-7604(03)00025-1
28. Scheffer IE, Berkovic S, Capovilla G, Connoly MB, French J, Ghilhoto L, et al. ILAE classification of the epilepsies position paper of the ILAE comission for classification and terminology. Epilepsia. (2017) 58:512–21. doi: 10.1111/epi.13709
29. Romdhane L, Mezzi N, Hamdi Y, El-Kamab G, Barakat A, Abdelhak S. Consanguinity and inbreeding in health and disease in North African populations. Annu Rev Genomics Hum Genet. (2019) 20:155–79. doi: 10.1146/annurev-genom-083118-014954
30. Yang T, Guo L, Wang L, Yu X. Diagnosis, intervention, and prevention of genetic hearing loss. Adv Exp Med Biol. (2019) 1130:73–92. doi: 10.1007/978-981-13-6123-4_5
31. Petersen MB, Willems PJ. Non-syndromic, autosomal-recessive deafness. Clin Genet. (2006) 69:371–92. doi: 10.1111/j.1399-0004.2006.00613.x
32. Zuberi SM, Brunklaus A, Birch R, Reavey E, Duncan J, Forbes GH. Genotype-phenotype associations in SCN1A-related epilepsies. Neurology. (2011) 76:594–600. doi: 10.1212/WNL.0b013e31820c309b
33. De Lange IM, Mulder F, van't Slot R, Sonsma ACM, van Kempen MJA, Nijaman IJ, et al. Modifier genes in SCN1A-related epilepsy syndromes. Mol Genet Genomic Med. (2020) 8:e1103. doi: 10.1002/mgg3.1103
34. Nieto-Barceló JJ, Gonzalez Montes N, Gonzalo Alonso I, Martínez F, Aparisi MJ, Martínez-Matilla M, et al. Variant in CACNA1G as a posible genetic modifier of neonatal epilepsy in an infant with a De Novo SCN2A mutation. J J Pediatr Genet. (2021). doi: 10.1055/s-0041-1723958
35. Suls A, Velizarova R, Yordanova I, Deprez L, Van Dyck T, Wauters J, et al. Four generations of epilepsy caused by an inherited microdelection of the epilepsy caused by an inherited microdeletion of the SCN1A gene. Neurology. (2010) 75:72–6. doi: 10.1212/WNL.0b013e3181e62088
Keywords: SCN1A, Dravet syndrome, GEFS+, homozygous, autosomal recessive inheritance
Citation: Marco Hernández AV, Tomás Vila M, Caro Llopis A, Monfort S and Martinez F (2021) Case Report: Novel Homozygous Likely Pathogenic SCN1A Variant With Autosomal Recessive Inheritance and Review of the Literature. Front. Neurol. 12:784892. doi: 10.3389/fneur.2021.784892
Received: 28 September 2021; Accepted: 20 October 2021;
Published: 30 November 2021.
Edited by:
Fernando Cendes, State University of Campinas, BrazilReviewed by:
Gaetan Lesca, Université Claude Bernard Lyon 1, FranceXiaorong Liu, Guangzhou Medical University, China
Copyright © 2021 Marco Hernández, Tomás Vila, Caro Llopis, Monfort and Martinez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ana Victoria Marco Hernández, bWFyY29fYW5haGVyQGd2YS5lcw==