Abstract
Background:
Progressive multifocal leukoencephalopathy (PML) is a rare and often lethal brain disorder caused by the common, typically benign polyomavirus 2, also known as JC virus (JCV). In a small percentage of immunosuppressed individuals, JCV is reactivated and infects the brain, causing devastating neurological defects. A wide range of immunosuppressed groups can develop PML, such as patients with: HIV/AIDS, hematological malignancies (e.g., leukemias, lymphomas, and multiple myeloma), autoimmune disorders (e.g., psoriasis, rheumatoid arthritis, and systemic lupus erythematosus), and organ transplants. In some patients, iatrogenic (i.e., drug-induced) PML occurs as a serious adverse event from exposure to immunosuppressant therapies used to treat their disease (e.g., hematological malignancies and multiple sclerosis). While JCV infection and immunosuppression are necessary, they are not sufficient to cause PML.
Methods:
We hypothesized that patients may also have a genetic susceptibility from the presence of rare deleterious genetic variants in immune-relevant genes (e.g., those that cause inborn errors of immunity). In our prior genetic study of 184 PML cases, we discovered 19 candidate PML risk variants. In the current study of another 152 cases, we validated 4 of 19 variants in both population controls (gnomAD 3.1) and matched controls (JCV+ multiple sclerosis patients on a PML-linked drug ≥ 2 years).
Results:
The four variants, found in immune system genes with strong biological links, are: C8B, 1-57409459-C-A, rs139498867; LY9 (alias SLAMF3), 1-160769595-AG-A, rs763811636; FCN2, 9-137779251-G-A, rs76267164; STXBP2, 19-7712287-G-C, rs35490401. Carriers of any one of these variants are shown to be at high risk of PML when drug-exposed PML cases are compared to drug-exposed matched controls: P value = 3.50E-06, OR = 8.7 [3.7–20.6]. Measures of clinical validity and utility compare favorably to other genetic risk tests, such as BRCA1 and BRCA2 screening for breast cancer risk and HLA-B*15:02 pharmacogenetic screening for pharmacovigilance of carbamazepine to prevent Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis.
Conclusion:
For the first time, a PML genetic risk test can be implemented for screening patients taking or considering treatment with a PML-linked drug in order to decrease the incidence of PML and enable safer use of highly effective therapies used to treat their underlying disease.
Introduction
Progressive multifocal leukoencephalopathy (PML) is a rare brain disease caused by the reactivation of JC virus (JCV) in immunosuppressed individuals. As an aggressive demyelinating disorder, PML can be fatal and is often severe and debilitating; almost 70% of survivors experience ongoing neurological disability and there is no approved treatment once PML develops (1). While PML is quite rare, infection with JCV is common, with most patients being asymptomatic. Based on serological testing, JCV has an estimated worldwide prevalence of 40–70% (2). More recent studies in Asian populations showed even higher rates of seropositivity, ranging from 70 to 80% (3–5). We note that JCV is formally named human polyomavirus 2 (HPyV-2 or HuPyV2) (6, 7) but, for simplicity, will be referred to as JCV in the present study.
Immunosuppression in JCV-seropositive (JCV+) individuals that develop PML can be due to a wide range of underlying diseases and/or drugs but is broadly related to three underlying disease states (1, 8): Human Immunodeficiency Virus (HIV)-infected, hematological malignancies (i.e., lymphoproliferative diseases such as leukemias, lymphomas, and multiple myeloma); and autoimmune disorders, such as rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE). HIV-infected acquired immunodeficiency syndrome (AIDS) patients represent the largest proportion of PML cases (~50%). Rates in this population substantially dropped after the 1996 introduction of highly active antiretroviral therapy (HAART), although at least 10% of HIV patients who are considered “immunological non-responders” to antiviral therapies (9) could continue to have elevated PML risk similar to the pre-1996 era. Conversely, iatrogenic PML (i.e., resulting from drug exposure) is on the rise with the growing number of immunosuppressant therapies used to treat various immune disorders (1, 10). Historically, iatrogenic PML risk was sufficiently high for psoriasis patients taking efalizumab (brand name Raptiva) that the therapy was withdrawn from the market worldwide in 2009 based upon a PML incidence rate of 0.158% or ~16 in 10,000 (11). Today, multiple sclerosis (MS) patients on disease-modifying therapies are the largest proportion of iatrogenic PML cases (1).
There are over three dozen drugs that include a PML warning in their prescribing information (USA) and/or mention PML in their product characteristics (European Medicines Agency). Examples include alemtuzumab, brentuximab vedotin, dimethyl fumarate, efalizumab, fingolimod, ibrutinib, and natalizumab; as well as anti-CD20 antibodies (also known as B cell depletion therapies) such as obinutuzumab, ocrelizumab, ofatumumab, and rituximab (12, 13). Of recent note, the PML warning in ocrelizumab's prescribing information was substantially expanded in August 2022.
Given the large number of drugs linked to the development of PML (1, 10, 14, 15), it is critical to identify additional risk factors that can be taken into consideration when patients and their clinicians are selecting a therapy for treatment of the underlying disorder. Since JCV infection is a requirement for developing PML (although most JCV-infected individuals will not develop PML), testing patients with a JCV antibody test (including assessing their index level) can be useful for informing PML risk (16, 17). For example, testing every 6 months (18) is recommended by the European Medicines Agency for patients on natalizumab who are JCV-negative or have a low index value. However, development of other PML risk biomarkers continues to be an area of high unmet need (19), especially since the specificity of the JCV antibody test is low (40–70% of the population are seropositive for JCV) (2, 20), the test's false negative rate is reported to be 3% (manufacturer's prescribing information for natalizumab, Dec-2021), and index levels may be unreliable for anti-CD20 therapies because of their mechanism of action (i.e., reduced antibody levels may result in lower anti-JCV antibody levels) (21, 22). Another suggested biomarker is serum neurofilament light chain (NfL) levels (23, 24), but it is only useful in verifying PML onset and resolution of the disease (in natalizumab-treated MS patients) as opposed to predicting who may get PML in the future (i.e., before a patient decides to take a PML-linked therapy). This is an important distinction given the seriousness of the condition and its limited treatment options once it develops.
Host genetic predisposition to PML (i.e., an individual has one or more genetic variants in their genome that increases their risk of developing PML) was proposed to be a significant risk factor (25); see also Mills and Mao-Draayer (26). This hypothesis is supported by a growing number of PML case reports (25, 27–42) in which the patients were found to have mutation(s) in known immunodeficiency disorder genes (43, 44). We previously explored the possibility of genetic predisposition to PML in the largest genetic study to date, whole-exome sequencing (WES) of 184 PML cases (45). That work identified 19 rare genetic variants in known immune-modulating genes that were significantly more common in PML patients compared to populations in the Genome Aggregation Database (gnomAD) database (46).
This study reports the frequency of these variants in additional PML cases (152 new, 336 total). By far, this is the largest ever assembled set of DNA samples from PML cases, providing a unique resource for studying germline genetic links to the disease. Importantly, this work, for the first time, compares 110 drug-exposed PML cases to 718 drug-exposed controls who took PML-linked drugs for ≥2 years. In this case-control analysis, four variants show a particularly strong association with PML; two of these variants only appear in cases and are never observed in the drug-exposed controls.
Due to the severity of PML as a serious adverse event, eight currently marketed drugs have PML in a Boxed Warning in their prescribing information (the FDA's strongest drug label warning) and numerous other drugs have a warning about PML in their product labeling in the USA and similar warnings in the EU, while one drug was withdrawn from the market due to its PML risk. Our reported measures of clinical validity and utility for the identified four variants show that utilization of a simple and inexpensive genotyping test in patients considering treatment with PML-linked immunosuppressant therapies has the potential to reduce the incidence of PML and save lives.
Methods
IRB approvals
Written informed consent was obtained from all patients (PML cases and MS controls) participating in this study under IRB approved protocols from the following institutions: Accelerated Cure Project, Comitato Etico Provinciale of Brescia (PI Imberti), Beth Israel Deaconess Medical Center (PI Koralnik), Icahn School of Medicine at Mount Sinai (BioMe Biobank), NINDS/NIH (PIs Major and Cortese), Paris-Sud/INSERM (PI Taoufik), University of California San Francisco (PI Oksenberg), University of Münster (PIs Schwab and Wiendl), Université Toulouse (PIs Brassat, Martin-Blondel, and Liblau), and Vanderbilt University (BioVU Biobank).
PML cases
In addition to the 184 cases previously studied using WES (45), new cases were assembled for genetic validation via genotyping. A total of 156 new DNA samples were collected from the following collaborating institutions: Accelerated Cure Project (n = 1), Comitato Etico Provinciale of Brescia (n = 11), NINDS/NIH (n = 32), Paris-Sud/INSERM (n = 9), Université Toulouse (n = 57), University of Münster (n = 44), and University of California San Francisco (n = 2). Potential cases were assessed using the consensus PML diagnostic criteria (47) and only “Definite” or “Probable” PML cases were retained. Wherever possible, drug exposures for immunosuppressant drugs (approved or used off-label) were noted. Primary underlying diseases were recorded and were then categorized as blood cancer (BC), HIV, MS, or Other. The BC subgroup includes: acute myeloid leukemia, anaplastic plasmacytoma, B-cell lymphoma, chronic lymphocytic leukemia, follicular lymphoma, Hodgkin lymphoma, leukemia, lymphoma, marginal zone lymphoma, myelodysplastic syndrome, multiple myeloma, and non-Hodgkin lymphoma. The Other subgroup includes: alcoholic cirrhosis, anti-synthetase syndrome, aplastic anemia, B-cell deficiency, Behcet's disease, cancer (non-hematological: colon and liver), common variable immunodeficiency, dermatomyositis, dermatopolymyositis, granulomatosis, idiopathic CD4 lymphocytopenia, immune thrombocytopenia, lymphopenia, microscopic polyangiitis, ocular pemphigoid, polycythemia vera, primary CD8 lymphopenia, psoriasis, RA, sarcoidosis (kidney, pulmonary, and unspecified), severe combined immunodeficiency, silicosis, thymoma with immunodeficiency, transplants (bone marrow, kidney, and liver), vasculitis, or unknown.
Control subjects
For comparison to drug-exposed PML cases, a set of drug-exposed controls with MS (called “matched controls”) were assembled from two laboratories: Université Toulouse and University of California San Francisco (UCSF). Inclusion criteria for controls were as follows: 1) JCV seropositivity, 2) exposure to an immunosuppressant/PML-linked drug for at least 2 years as PML risk increases after 2 years in MS patients (17), and 3) absence of a PML diagnosis. The JCV antibody status was already determined to be positive for all Université Toulouse controls; for the UCSF controls, JCV antibody status experiments were performed by Lytic Solutions (Madison, WI, USA). Detection of anti-JCV IgGs in serum samples was performed according to manufacturer instructions using the ELISA-VIDITEST anti-JCV IgG diagnostic kit (Catalog # ODZ-450) from Vidia spol. s.r.o. (Vestec, Czech Republic; distributed by Boca Scientific Inc., Dedham, MA, USA). All serum samples (diluted 1:100) were run in duplicate and 96-well plates included control human serum samples of known JCV infection status. After color development (using kit-supplied stop solution), absorbance values at 450 nm (with a reference reading at 650 nm) were measured using a Molecular Devices Spectra Max Plus plate reader (San Jose, CA, USA). Background-subtracted values were averaged for each sample. The qualitative interpretation procedure for data analysis was performed according to the manufacturer instructions using the internal plate calibrator value and the plate lot correction factor. Samples with absorbances lower than 90% of the cut-off value were considered negative and samples with absorbances higher than 110% of the cut-off value were considered positive. Samples with values between these two cut-offs were considered indeterminable. Only JCV+ samples were retained for further analyses and all had MS as their primary disease.
To assess PML risk across all primary disease subgroups (BC, HIV, MS, and Other) in the context of population-level data, we used the most recent version (3.1) of gnomAD (46). This release consists of Whole Genome Sequencing (WGS) data for ~76,000 genomes corresponding to a variety of ethnicities; results are also reported by ethnic subgroups for European (EUR, ~34,000 non-Finnish European genomes), African (AFR, ~21,000 genomes), and EUR plus AFR (~55,000 genomes). In addition to the functional prediction methods PolyPhen and SIFT, gnomAD 3.1 also reports the results for other prediction measures of deleteriousness, such as CADD scores.
Genetic analyses
Ancestry and duplicate sample analyses were assessed for all PML cases and matched controls using previously described methods (45) with the exception that WGS (0.1x read depth) of newly acquired PML cases and matched controls was performed by Psomagen (Rockville, MD, USA). Ancestry analysis was performed by Gencove (New York, NY, USA) using 0.1x read depth WGS data based on implementation of a supervised version of the STRUCTURE model (48), which is trained on a panel of 7,345 individuals grouped in 49 populations. Primary ethnicities were assigned as AFR or EUR based on the majority percentage of ancestry.
For duplicate sample analyses, the low coverage WGS VCF files from Psomagen were filtered using bcftools (v1.10) to include only biallelic single nucleotide variants (SNVs) with exactly two alleles and PASS quality. The filtered variants were then annotated and evaluated for relatedness using plink (v1.9) and KING software (v2.2.6). Duplicate samples were excluded from further analysis.
Previously published WES data on the 19 PML risk variants (45) was reanalyzed in the context of new PML cases and JCV+ matched controls. We note that one of the previously published 185 PML cases was a bone marrow transplant patient whose DNA sample was acquired post-transplant; therefore we excluded this patient from the present analyses. For new PML cases and controls, the 19 variants were genotyped by a service provider (LGC Genomics, UK) with custom designed assays that use kompetitive allele specific PCR (KASP) chemistry. Sex was confirmed via genotyping.
To verify that previously published PML risk variants (45) were associated with PML and not with MS, we assessed 12 of 19 variants in a large genome-wide association study (GWAS) conducted by the International Multiple Sclerosis Genetics Consortium. This MS study used an exome chip (Illumina HumanExome Beadchip) containing 137,007 genome-wide common (12%) and rare (88%) variants to identify MS-associated loci in 32,367 MS cases vs. 36,012 healthy controls. The seven variants that were not assessed were either not found on the Illumina exome array or were not reported in the study (49).
Statistical and pharmacogenetic test analyses
Association statistics, Odds Ratio (OR) values and P values (two-tailed Fisher's Exact Test), were calculated as previously described (45). To avoid infinite ORs for variants that were not present in matched controls, 0.5 was added to all cells of the contingency table (50). The 19 previously identified variants for PML risk were evaluated in drug-exposed PML cases compared to drug-exposed controls and gnomAD population controls. Several PML cases had mixed ancestry (i.e., one or more other ethnicities present at >5%). Therefore, statistical analyses using gnomAD population controls included all ethnicities (i.e., all ~76,000 WGS data sets).
Following individual variant association testing, combinations of the highest-risk variants (as identified in the case-control analyses) were explored for use in a panel test. Pharmacogenetic test parameters (clinical validity and clinical utility) for this panel test were calculated using the method of Tonk et al. (51). Clinical validity measures are sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV). Clinical utility measures are population attributable fraction (PAF), number needed to treat (NNT), and number needed to genotype (NNG). As the incidence of drug-induced PML varies by drug type/exposure, for the adverse event frequency we used the best-established long term rate reported for JCV+ MS patients on natalizumab, which is 3% (17).
Results
Assembly of PML cases and matched controls to further validate candidate PML risk genetic variants
A total of 340 potential PML cases were assembled for the present study. Following ancestry and duplicate sample analyses, four newly acquired samples were found to be identical to previous samples and were therefore removed. The final PML cohort includes a total of 336 PML cases: 184 from our previous study (45) and 152 new, unique cases. Using consensus PML diagnostic criteria (47), 287 (85%) cases were Definite PML and 49 (15%) cases were Probable PML. Eleven PML cases (3.3%) had neither predominantly AFR nor EUR ancestry and were assigned EUR. Additionally, 60% (33/55) of AFR and 22% (62/281) of EUR cases had one or more other ethnicities present at >5%. Sex, primary ethnicity, primary disease, and drug exposures for these cases are summarized in Table 1 and a workflow of the recruitment and study design is shown in Figure 1. Of the assembled PML cohort, 110 of 336 (32.7%) PML cases were drug-exposed.
Table 1
| Drug-exposed | |||
|---|---|---|---|
| Total PML cases | PML cases | Matched controlsa | |
| Subjects | 336 | 110 | 718 |
| Primary diseaseb | |||
| HIV | 156 | 0 | n/a |
| MS | 94 | 94 | 718 |
| Other | 45 | 8 | n/a |
| BC | 41 | 8 | n/a |
| Drug exposurec | |||
| Natalizumab | 86 | 604 | |
| Rituximab | 13 | 25 | |
| Unknownd | 4 | 0 | |
| Dimethyl fumarate | 3 | 43 | |
| Alemtuzumab | 1 | 0 | |
| Fingolimod | 1 | 55 | |
| Glatiramer acetate | 1 | 0 | |
| Mycophenolate mofetil | 1 | 0 | |
| Ocrelizumab | 0 | 12 | |
| TOTAL, non-redundant | 110 | 718 | |
| Sex | |||
| Male | 184 | 31 | 194 |
| Female | 152 | 79 | 524 |
| Primary ethnicitye | |||
| EUR | 281 | 109 | 645 |
| AFR | 55 | 1 | 73 |
Summary of PML cases and drug-exposed controls: primary disease group, MS drug exposure, and demographics.
Drug-exposed matched controls are JCV+ MS patients on an MS drug ≥ 2 years who did not develop PML.
Primary disease: BC, blood cancer; HIV, human immunodeficiency virus infected; MS, multiple sclerosis; Other, various. See Methods for list of diseases under the BC and Other subgroups.
All drugs have PML listed in the prescribing information (Boxed Warning and/or Warnings and Precautions) with the exception of glatiramer acetate. Drug exposure times were unavailable for PML cases but are ≥ 2 years for controls (a subset were exposed to two or more drugs for ≥ 2 years). Of the 110 drug-exposed PML cases, four had multiple reported drug exposures: 1 glatiramer acetate (also exposed to interferon beta-1a, but no exposure to natalizumab) and 3 rituximab (also exposed to bendamustine, cyclophosphamide-fludarabine, or cyclosporine-methotrexate-mycophenolate mofetil-steroids-tacrolimus).
Four PML cases had unknown drug exposures but were assumed to be drug-exposed since all were MS patients, a patient group that is not known to develop PML in the absence of treatment with a disease-modifying therapy.
A primary ethnicity was assigned as AFR or EUR (see Methods) for statisical analyses. An Other ethnicity was annotated if > 5% of one or more other ethnicities was found: EUR, 62/281 PML cases and 128/645 drug-exposed controls; AFR, 33/55 PML cases and 67/73 drug-exposed controls.
Figure 1

Case and control recruitment and study design. (A) Prior study for genetic discovery and validation using Whole Exome Sequencing (WES), 669 candidate immune response genes, and gnomAD 2.1 (WES + WGS) population controls (45). (B) New recruitment of PML cases and matched controls (JCV+ MS patients exposed to a PML-linked drug ≥2 years). All PML cases and matched controls were genotyped for the prior study's 19 candidate PML risk variants. Matched controls without JCV serostatus were assayed (see Methods). Excluded cases: four were found to be duplicates of the prior study (see Methods). Excluded controls: 152 JCV seronegative (JCV-) patients; one QC failure for genotyping assays due to low quality DNA. (C) Drug-exposed analysis is the pooled subgroup (n = 110) of total PML cases (n = 336) compared to matched controls. Drug-exposed study results are reported in Tables 2, 5 and genes for the top 4 variants are listed.
A total of 879 potential controls were assembled from Université Toulouse and UCSF. Of these, 152 samples (all from UCSF) were removed for lack of JCV seropositivity and 9 Toulouse samples were removed for either relatedness or incomplete drug-exposure data. This yielded a final drug-exposed control cohort of 718 individuals, all of whom had MS as a primary disease (hereafter referred to as drug-exposed controls). According to ancestry analysis, 24 (3.3%) controls had neither predominantly AFR nor EUR ancestry and were assigned as EUR (Table 1 and Figure 1).
Association of top PML risk variants in drug-exposed PML cases vs. matched controls
The presence of the 19 previously identified PML risk variants was assessed in drug-exposed PML cases (n = 110) vs. drug-exposed controls (n = 718) and vs. gnomAD population controls (n = ~76,000). As summarized in Table 2, four variants showed strong association with PML risk in this analysis. Variants in genes C8B, FCN2, and STXBP2 were found to be significant (P value <0.05) compared to both drug-exposed controls and gnomAD population controls. The LY9 variant was only significant when compared to gnomAD controls, likely a consequence of its very low frequency (11 out of 76,504 subjects). Of note, the STXBP2 and LY9 variants were absent in drug-exposed controls and had large effect sizes (OR = 33.1 and 19.6, respectively).
Table 2
| Drug-exposed controlsb | gnomAD controlsc | |||||
|---|---|---|---|---|---|---|
| (n = 718) | ( n = 76,071) | |||||
| Gene symbol | dbSNP ID | Variant (GRCh37, hg19) | OR (95% CI) | P value | OR (95% CI) | P value |
| STXBP2 | rs35490401 | 19-7712287-G-C | 33.1 (1.6 - 694.4) | 0.0175 | 6.8 (1.7 - 27.6) | 0.0373 |
| LY9 | rs763811636 | 1-160769595-AG-A | 19.6 (0.8 - 484.3) | 0.1330 | 63.4 (8.1 - 495.5) | 0.0172 |
| C8B | rs139498867 | 1-57409459-C-A | 6.7 (1.7 - 27.3) | 0.0135 | 4.3 (1.6 - 11.8) | 0.0159 |
| FCN2 | rs76267164 | 9-137779251-G-A | 5.7 (1.7 - 18.8) | 0.0090 | 7.0 (2.9 - 17.3) | 0.0001 |
Association statisticsa for PML risk variants: drug-exposed PML cases (n = 110) vs. drug-exposed controls and gnomAD 3.1 population controls.
The subset of total PML cases that were drug-exposed (110 of 336) were compared to drug-exposed controls and population controls (gnomAD). P values were calculated using Fisher's Exact Test; OR, odds ratio; CI, confidence interval. Variants are ordered by descending OR and bold-highlighted P values denote significance <0.05.
Drug-exposed (matched) controls are JCV+ MS patients on a MS drug ≥ 2 years who did not develop PML.
All ethnicities in gnomAD 3.1 population controls were used (see Methods). Sample size (subject number) varies slightly by variant, n = 76,071 is an average of the 4 variants: STXBP2, n = 76,099; LY9, n = 76,054; C8B, n = 76,081; FCN2, n = 76,050.
As summarized in Table 1, natalizumab-exposed PML cases (n = 86) represent the largest subgroup of PML cases with a PML-linked drug exposure history in our study. Similarly, natalizumab-exposed controls (n = 604) were also the largest subgroup for matched controls. Therefore, we also assessed the association of the PML risk variants in the natalizumab subgroup (see Supplementary Table 6) and found comparable results to the full set of of drug-exposed cases and controls (Table 2 and Supplementary Table 5). The association statistics for three of the four top variants were slightly improved for the natalizumab subgroup (lower P values and higher ORs) but were less significant for the FCN2 variant.
PML risk variants are rare and predicted to be pathogenic
All four variants are rare in the general population (Table 3), with gnomAD 3.1 allele frequencies <0.5% and thus providing supporting evidence of their pathogenicity (52). Three variants are missense and are predicted to be probably or possibly damaging by PolyPhen and deleterious by SIFT. The LY9 variant is a frameshift predicted to cause loss of function of the protein (pLOF). A third prediction method, the CADD score (as reported in gnomAD 3.1), is also reported in Table 3. The CADD score range was 22.8–26.0, indicating that all four variants are predicted to be detrimental (CADD score >20 is the highest category of deleteriousness in gnomAD 3.1 annotation).
Table 3
| gnomAD 3.1 in silico predictions | |||||||
|---|---|---|---|---|---|---|---|
| Gene symbol | dbSNP ID |
Variant
(GRCh37, hg19) |
gnomAD
3.1 AFb |
Consequencec | Polyphen | SIFT | CADDe |
| STXBP2 | rs35490401 | 19-7712287-G-C | 0.001367 | missense | probably damaging | deleterious | 26.0 |
| LY9 | rs763811636 | 1-160769595-AG-A | 0.000072 | frameshift (pLOF) | n/ad | n/ad | 22.8 |
| C8B | rs139498867 | 1-57409459-C-A | 0.004331 | missense | possibly damaging | deleterious | 23.1 |
| FCN2 | rs76267164 | 9-137779251-G-A | 0.003393 | missense | probably damaging | deleterious | 24.0 |
PML risk variant functional impact predictions.a
These PML risk variants are a subset of the 19 previously reported variants (45). Gray-shading denotes severity of functional predictions: no shading = low impact (none for these 4 variants), light gray, moderate impact, dark gray, high impact.
AF, allele frequency of the variant in gnomAD 3.1 for all ethnicities (Total).
Missense variants are amino acid substitutions; for the LY9 variant, pLOF denotes protein loss-of-function.
Polyphen and SIFT are prediction methods for missense variants and are not applicable (n/a) to other types of variants (e.g., the high impact frameshift for the LY9 variant).
CADD scores >20 are the highest category of deleteriousness in gnomAD 3.1 annotation.
No association of PML risk variants with MS
Since iatrogenic PML cases are on the rise and MS patients are one of the intended patient groups for a PML risk genetic test, we checked if any of our top four PML risk variants were associated with MS. Previously reported MS genome-wide association study (GWAS) data from a large international study (49) were used for this analysis and included 32,367 MS cases vs. 36,012 healthy controls. Table 4 shows the association results for the top four PML risk variants. Three of the four top variants in genes C8B, FCN2, and STXBP2 show no association with MS. All three had an OR of 1.0 and uncorrected genome-wide P values of 0.07–0.71. The fourth top variant (in the LY9 gene) is very rare in the general population (gnomAD 3.1 allele frequency = 0.000072) and has not been reported in the literature to be associated with disease (including MS).
Table 4
| Association with MSa | Association with PMLb | |||||
|---|---|---|---|---|---|---|
| Gene symbol | dbSNP ID | Variant (GRCh37, hg19) | OR | P value | OR | P value |
| STXBP2 | rs35490401 | 19-7712287-G-C | 1.0 | 0.6853 | 33.1 | 0.0175 |
| LY9 | rs763811636 | 1-160769595-AG-A | n/ac | n/ac | 19.6 | 0.1330 |
| C8B | rs139498867 | 1-57409459-C-A | 1.0 | 0.0670 | 6.7 | 0.0135 |
| FCN2 | rs76267164 | 9-137779251-G-A | 1.0 | 0.7088 | 5.7 | 0.0090 |
PML risk variant association with MS vs. drug-exposed PML cases.
MS association results (32,367 MS cases vs. 36,012 healthy controls) were previously reported (49), see Methods for details.
Drug-exposed results (110 PML cases vs. 718 matched controls) are from Table 2 (as a comparator to the MS association results); P values were calculated using Fisher's Exact Test; OR, odds ratio.
The LY9 variant was not evaluated (n/a, not applicable) in the MS association study, likely because it is very rare in the general population (gnomAD 3.1 allele frequency = 0.000072) and therefore not included on the exome chip (Illumina Exome BeadChip).
Utilization of a genetic risk test to reduce the incidence of PML with immunosuppressant therapies
Based on the results of the association analysis in the drug-exposed PML cases, a panel of four rare variants in genes (C8B, FCN2, STXBP2, and LY9) with strong immune-linked biology was identified as being potentially useful to identify patients at high risk of PML (see Supplementary Table 9 for analysis of the four individual variants vs. the 4-variant panel test in three different groups of PML cases: All, any Drug-exposed, and Natalizumab-exposed). Clinical validity and population impact measures (i.e., clinical utility) are shown in Table 5. No subject in either cases or controls presented with more than one of the four variants in the panel. Presence of any one of these four variants was 10.9% in the drug-exposed PML cases vs. only 1.4% in the drug-exposed controls. Association statistics for the 4-variant panel were strong, with a P value of 3.50E-06 and high effect size (OR = 8.67). The population attributable fraction (PAF), or percentage of drug-induced PML cases that could be avoided with preventative genetic testing, is 9.4%.
Table 5
| Association statisticsa | |
| Frequency in PML cases (12/110)b | 10.9% |
| Frequency in matched controls (10/718) | 1.4% |
| P value | 3.50E-06 |
| OR (95% CI) | 8.7 (3.7–20.6) |
| Clinical validityc | |
| Sensitivity | 10.9% |
| Specificity | 98.6% |
| PPV | 19.5% |
| NPV | 97.3% |
| Clinical utilityc | |
| PAF | 9.4% |
| NNT | 6 |
| NNG | 355 |
Clinical validity and utility of a 4-variant PML genetic risk test in drug-exposed cases vs. matched controls.
Frequencies and statistics for drug-exposed PML cases and drug-exposed controls testing positive with the 4-variant PML genetic risk test: P values were calculated using Fisher's Exact Test; OR, odds ratio; CI, confidence interval.
Details for the 12 genotype-positive PML cases are as follows: C8B variant 1-57409459-C-A (4 total), 4 natalizumab-treated MS patients; FCN2 variant 9-137779251-G-A (5 total), 2 natalizumab-treated MS patients, 1 dimethyl fumarate-treated MS patient (natalizumab-naïve), 1 rituximab-treated B cell lymphoma patient, and 1 rituximab-treated Behcet's disease patient that also had immune thrombocytopenia; LY9 variant 1-160769595-AG-A (1 total), 1 natalizumab-treated MS patient; STXBP2 variant 19-7712287-G-C (2 total), 2 natalizumab-treated MS patients.
Clinical validity and utility (also known as population impact) measures were calculated as described in Tonk et al. (51): PPV, positive predictive value; NPV, negative predictive value; PAF, population attributable fraction; NNT, number needed to treat; NNG, number needed to genotype. Values were calculated using a 3% adverse event frequency (PML incidence rate): JCV+ patients taking natalizumab and receiving at least 72 infusions (17).
In the total cohort of PML cases (n = 336), three of the four variants were found in both EUR and AFR cases (Table 6). All four variants were distributed across multiple primary disease subgroups, further supporting their association with PML rather than any one of the underlying disease groups (BC, HIV, MS, Other). In the drug-exposed PML cases (n = 110), three of the variants were found only in the MS subgroup (Table 6, footnote a), likely due to the high proportion of MS cases (Table 1, 86/110). However, the FCN2 variant was found in three primary disease subgroups (BC, MS, Other) and in PML cases exposed to one of three different drugs (1 dimethyl fumarate case, 2 natalizumab cases, and 2 rituximab cases; see Supplementary Table 8). Taken together, these results suggest that the 4-variant PML risk genetic test could be used for advising on PML risk in general and for preventing iatrogenic PML cases.
Table 6
| Gene symbol | dbSNP ID | Variant (GRCh37, hg19) | Primary ethnicityb | Primary diseasec |
|---|---|---|---|---|
| STXBP2 | rs35490401 | 19-7712287-G-C | 4 EUR | 1 HIV, 2 MS, 1 Other |
| LY9 | rs763811636 | 1-160769595-AG-A | 1 EUR, 2 AFR | 1 HIV, 1 MS, 1 Other |
| C8B | rs139498867 | 1-57409459-C-A | 7 EUR, 2 AFR | 1 BC, 4 HIV, 4 MS |
| FCN2 | rs76267164 | 9-137779251-G-A | 9 EUR, 1 AFR | 2 BC, 4 HIV, 3 MS, 1 Other |
Distribution of genotype-positive PML casesa across ethnicities and primary diseases.
Results are shown for all PML cases (n = 336). Results for each of the 4 variants in the drug-exposed PML cases (n = 110): STXBP2, 2 MS; LY9, 1 MS; C8B, 4 MS; FCN2, 1 BC, 3 MS, 1 Other.
Number of genotype-positive PML cases assigned to European (EUR) or African (AFR) ancestry (see Methods).
Number of genotype-positive PML cases assigned to 1 of 4 primary disease subgroups (see Methods): BC, blood cancer; HIV, Human immunodeficiency virus infected; MS, multiple sclerosis; Other, various other diseases/conditions (see Methods).
Discussion
Four actionable risk variants identified from case-control analysis
With the addition of 152 PML cases to our previously studied 184 PML cases (45), we have now assembled the largest collection of PML DNA samples (n = 336) for studying germline genetics to identify variants associated with PML risk. One crucial improvement to our previous work is the assembly of drug-exposed matched controls (n = 718), defined as JCV+ MS patients who did not develop PML after being exposed to an immunosuppressant therapy with PML risk for ≥2 years. This cohort enabled us to conduct a targeted, case-control analysis on the previously identified set of 19 PML genetic risk variants. From this analysis we demonstrate the clinical validity and utility of four immune-linked, high effect size, rare variants for use in an iatrogenic PML risk genetic test in the following genes: C8B, FCN2, STXBP2, and LY9 (Tables 2–5).
Individually, the four variants show strong associations in the drug-exposed cases vs. matched controls and gnomAD population controls (Table 2). Notably, the LY9 and STXBP2 variants were absent in the 718 drug-exposed controls. There was no association with MS for three of the variants (Table 4) and the fourth variant was not evaluated in the study, presumably due to its rarity (49). When combined as a single PML risk test, the top four variants show robust statistical associations, with a P value = 3.5E-06 and OR = 8.7 (Table 5). They were present in 10.9% of PML cases vs. only 1.4% of drug-exposed matched controls. As such, testing for these four variants could prevent a substantial number of patients from developing PML without deterring most patients from their treatment plan. Finally, each of the four variants appears to be individually predictive of PML risk, as no PML case or matched control had more than one of these variants. This is consistent with the hypothesis that rare, deleterious variants in immune-regulating genes confer risk of PML.
Pharmacovigilance with a PML risk test is supported by clinical validity and utility measures
Clinical validity (sensitivity, specificity, PPV, NPV) refers to a test's ability to accurately predict a disorder while clinical utility (PAF, NNT, NNG), also referred to as population impact, measures its impact on the disorder (in this situation, PML cases prevented). See Tonk et al. (51) for further background information on pharmacogenetic test measures. The clinical impact of screening patients considering PML-linked drugs is shown in Table 5. The pharmacogenetic test measures shown are based on the results of this study and the rate of PML (3%) observed in JCV+ long duration natalizumab patients (17). One PML case would be prevented for every 355 patients genotyped (NNG). For every six patients (NNT) who carry one of these variants, one case of PML can be avoided. Additionally, the PAF of 9.4% suggests that nearly 10% of drug-exposed PML cases could be prevented. Taken together, preventative genotyping of patients considering treatment with a PML-linked drug would eliminate a significant portion of iatrogenic PML cases without deterring otherwise tolerant users (98.6% of the patient population who are not carriers of any of the top four variants) from starting or continuing treatment.
Comparison to other clinically important genetic tests
Comparisons to other clinically important genetic tests suggest that pre-treatment screening with our PML risk test would be appropriate and could reduce the occurrence of PML for any therapy with known or suspected PML risk. As shown in Figure 2, the results of a large study (95,961 patients) published in 2017 (53) reports lower OR values for the association of breast cancer with all known pathogenic variants in either BRCA gene (OR = 5.9 for BRCA1, OR = 3.3 for BRCA2) than the proposed 4-variant PML risk test (OR = 8.7). Moreover, this PML risk panel test was positive for 10.9% of PML cases in our study (Table 5), which is higher than the presence of BRCA1 and BRCA2 variants in breast cancer patients (2.8 and 2.7%, respectively) (53).
Figure 2

Predictive risk comparison to BRCA screening tests, odds ratio (OR) vs. diagnostic (Dx) yield. Results for a 4-variant PML risk test are shown in comparison to the BRCA1/BRCA2 breast cancer risk prediction test. The proposed 4-variant PML risk test data point (●) is based on the total drug-exposed PML cases (Table 5). The BRCA1 (Δ) and BRCA2 (□) risk test data points are based on results for over 95,000 women reported in Kurian et al. (53). More Utility is defined as higher OR and higher Dx yield and Less Utility is defined as lower OR and lower Dx yield.
Another relevant comparison is carbamazepine, an anticonvulsant drug. The FDA added a Boxed Warning to its prescribing information requiring pre-treatment genetic testing in certain populations for HLA-B*15:02 due to Stevens-Johnson syndrome/Toxic Epidermal Necrolysis (SJS/TEN) risks. In Figure 3, a comparison of pharmacogenetic testing measures (PPV vs. NNT) as indicators of clinical utility shows the proposed PML risk panel test has the potential to provide greater utility than the currently recommended test for carbamazepine's HLA-B*15:02 association with SJS/TEN (55). Also note that mortalities associated with SJS and TEN are estimated at 1–5% and 25–35%, respectively (56). Whereas PML-associated mortality is higher, reported as 23–65% (1, 15).
Figure 3
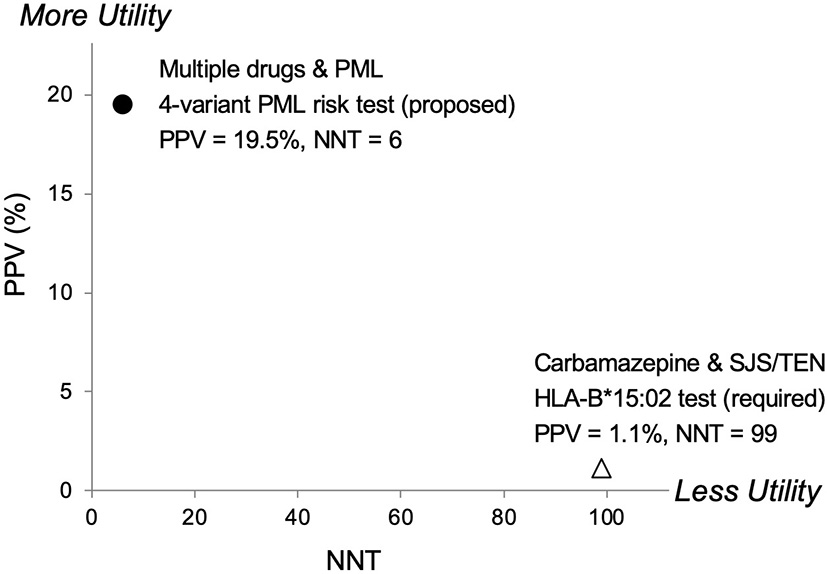
Predictive risk comparison to HLA-B*1502, positive predictive value (PPV) vs. number needed to treat (NNT). Results for a 4-variant PML risk test are shown in comparison to the HLA-B*15:02 test that is required in Asian populations before administering carbamazepine (CBZ). CBZ is a cause of the serious adverse event Stevens-Johnson Syndrome (SJS) and Toxic Epidermal Necrolysis (TEN). The proposed 4-variant PML risk test data point (●) is based on the total drug-exposed PML cases (Table 5) and a PML incidence rate of 3% (17). The HLA-B*15:02 SJS/TEN risk test data point (Δ) is based on results reported in Shi et al. (54). More Utility is defined as higher PPV and lower NNT and Less Utility is defined as lower PPV and higher NNT.
The top four variants are predicted to be pathogenic and have strong biological connections
In addition to being supported by strong statistical, clinical validity, and clinical utility measures, the four variants proposed for inclusion in the PML risk panel are predicted to be deleterious (Table 3) and their rarity further supports that they are pathogenic. All of the genes in which the four variants are located are linked to the immune system's viral defense mechanisms. Two genes (C8B and FCN2) are part of the complement system (lectin and terminal pathways) (57–60). The other two genes (LY9 and STXBP2) cause or are linked to hemophagocytic lymphohistiocytosis (HLH) disorders, including macrophage activation syndrome (MAS) (61–65). Two genes (C8B and STXBP2) with PML risk variants are among the 437 genes designated by the International Union of Immunological Societies (IUIS) to cause inborn errors of immunity (43, 44), thereby supporting our original hypothesis (25) of host genetics as an additonal risk factor for development of PML.
Study limitations
A few areas of limitation are noted. Of the 336 PML cases, 49 had insufficient information to confirm them as Definite PML (47). Since PML is rare, assembling patient cohorts for research studies is very challenging and several of our cases were documented before consensus diagnostic criteria were implemented. Therefore, we decided to include both Definite and Probable PML cases in our study. Of note, Probable cases were almost entirely collected from PML centers of excellence, increasing the likelihood that they are in fact genuine PML cases.
The ethnic diversity for our PML cases is somewhat limited (Table 1 and Methods). While 11/336 (3%) PML cases assigned as EUR ancestry to simplify association analyses (see Methods) formally belonged to another majority ancestry, our study is lacking in predominantly East Asian, South Asian, and Latino/Admixed American ancestries. For example, studying germline genetics in East Asians may help to explain why PML incidence rates are about 8-fold higher in Japan for fingolimod-treated MS patients compared to the US and worldwide rates (66, 67). Presently, a higher rate of JCV seropositivity (2–5) and HLA-DRB1 alleles have been suggested as potential factors for this higher rate of PML development in fingolimod-treated Japanese MS patients (68).
We note that 95/336 (28%) PML cases had mixed ancestry (i.e., at least one other ancestry present at > 5%) and all four of our top variants are globally rare for all gnomAD ethnicities (allele frequency range 0.000072 to 0.004331). Furthermore, variant associations were significant whether analyzed by primary ethnicity (EUR or AFR) or using all ethnicities (All pooled analysis). While it is important to continue to study PML cases in underrepresented ancestries, we believe the global rarity of these variants—combined with the strength of the associations observed here—obviates the need to assess the variants in specific populations and enables the use of our PML risk test in the general population.
For iatrogenic PML cases, drug-specific association results may be informative but in our present work, this was only possible for the subgroup of PML cases and matched controls that were exposed to natalizumab (86 cases vs. 604 controls); see Supplementary Tables 6, 9. The next largest drug-specific subgroup for the drugs listed in Table 1 was rituximab (13 cases vs. 25 controls). However, given the consistent results (Supplementary Table 9) for our 4-variant PML genetic risk test among natalizumab-exposed and all drug-exposed cases and controls (110 cases vs. 718 controls), we would not expect dramatic differences across drug-specific groups. Furthermore, one of our PML risk variants (in the FCN2 gene) was found in PML cases (see Table 5, footnote b) with three different underlying diseases (MS, B cell lymphoma, and Behcet's disease with immune thrombocytopenia), but also representing exposure to three different PML-linked drugs (dimethyl fumarate, natalizumab, and rituximab).
Finally, using matched controls for other underlying disorders besides MS (e.g., leukemia/lymphoma, other autoimmune conditions, or HIV-infected patients who did not develop PML) may provide additional support to the use of our proposed PML risk test in other clinical settings. Beyond the practical limitations of performing PML risk case-control studies for other underlying disorders and drug exposures, these results suggest it is likely unnecessary. All four top variants were found in PML cases representing at least three of four primary disease subgroups, with the FCN2 variant found in all four subgroups (Table 6). This is consistent with the understanding that PML is the same clinical entity regardless of the patient's underlying disorder (1) and supports the use of our test in all patients considering the use of PML-linked therapies.
Conclusion
Identification of patients at risk of PML is an important area of unmet need given the growing number of PML-linked immunosuppressive therapies. Building on our previous work (45), this study represents what we believe to be the first case-control analysis of germline genetic variants that confer risk of PML. The association of PML risk with damaging variants in the immune-linked genes C8B, FCN2, LY9, and STXBP2 is confirmed, with two variants being completely absent in the drug-exposed controls. High OR values and statistical significance support the use of this information when assessing patient risk of PML. The underlying genetic immunodeficiency conditions linked to these variants predispose carriers to uncontrolled JCV virus reactivation (i.e. PML), a serious infection. Simple, low-cost genetic screening in patients considering drugs with known or suspected PML risk will prevent future cases. Due to the seriousness of a PML diagnosis—particularly because it often leads to life-threatening outcomes (69) and the lack of treatment options once it develops—it would seem unethical not to test individuals considering immunosuppressive therapies with PML risk for our top four variants, and advising those with a positive result to consider an alternative therapy or treatment strategy.
Statements
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: National Center for Biotechnology Information (NCBI) ClinVar, https://www.ncbi.nlm.nih.gov/clinvar/, SCV002572501.1, SCV002572502.1, SCV002572503.1, and SCV002572504.1.
Ethics statement
The studies involving human participants were reviewed and approved by IRB protocols from the following institutions: Accelerated Cure Project, Comitato Etico Provinciale of Brescia (LI), Beth Israel Deaconess Medical Center (IK), Icahn School of Medicine at Mount Sinai (BioMe Biobank), NINDS/NIH (EM and IC), Paris-Sud/INSERM (YT), University of California San Francisco (JO), University of Münster (NS and HW), Université Toulouse (DB, GM-B, and RL), and Vanderbilt University (BioVU Biobank). The patients provided their written informed consent to participate in this study.
Author contributions
EH, ES, and PE: conception and design of the study. EH, SJ, and DR: laboratory experiments. EH, ES, SJ, CB, TR, and PE: data analysis and interpretation. YT, HH-C, RL, DB, GM-B, HW, NS, IC, MM, LI, RC, JO, JG, BS, IK, BH, and EM: provision of study materials and clinical information for patients. EH, ES, CC, and PE: wrote the manuscript. All authors revised/approved the manuscript.
Funding
A portion of this work was supported by the National Institutes of Health (R01 NS074995 and NS047029 to IK) and the National Institute of Neurological Disorders and Stroke Division of Intramural Research (EM and IC). The UCSF DNA biorepository is supported by grant RG-1611-26299 (JO) from the National Multiple Sclerosis Society.
Acknowledgments
We are grateful to the PML patients that participated in this study, and the Accelerated Cure Project for providing a PML DNA sample from their MS repository. We also thank Dr. Bruce Bebo, Dr. Mark Allegretta, and Dr. Walter Kostich of the National Multiple Sclerosis Society for helpful discussions.
Conflict of interest
Authors CB and CC are employed by Emerald Lake Safety LLC. Authors EH, ES, PE, and SJ are employed by Population Bio, Inc. Author TR is employed by Richmond Bioinformatics Consulting and author DR is employed by Lytic Solutions, LLC. Authors HW, IK, NS, and RL received funding from PML Screening, LLC to partially offset the costs for collection, and clinical characterization of patient samples used in the research. Authors EH, ES, PE, and YT are inventors of genetic screening methods for PML risk and have issued and pending patents related to this work. Applicants/Assignees on issued patents are: PML Screening, LLC, Newport Beach, CA (US), a joint venture between Population Bio, Inc. and Emerald Lake Safety LLC; Université Paris-Saclay, Gif sur Yvette (FR); The Assistance Publique-Hôpitaux de Paris (APHP), Paris (FR); and The Institut National de la Santé et de la Recherche Médicale (INSERM), Paris (FR). Author IC is a shareholder in Keires AG, Nouscom AG, and PDC*line Pharma. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The authors declare that this study received funding from PML Screening, LLC. The funders had the following involvement with the study: conception and design of the study, laboratory experiments, data analysis and interpretation, and wrote the manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2022.1016377/full#supplementary-material
References
1.
Cortese I Reich DS Nath A . Progressive multifocal leukoencephalopathy and the spectrum of JC virus-related disease. Nat Rev Neurol. (2021) 17:37–51. 10.1038/s41582-020-00427-y
2.
Paz SPC Branco L Pereira MAC Spessotto C Fragoso YD . Systematic review of the published data on the worldwide prevalence of John Cunningham virus in patients with multiple sclerosis and neuromyelitis optica. Epidemiol Health. (2018) 40:e2018001. 10.4178/epih.e2018001
3.
Aoyama S Mori M Uzawa A Uchida T Masuda H Ohtani R et al . Serum anti-JCV antibody indexes in Japanese patients with multiple sclerosis: elevations along with fingolimod treatment duration. J Neurol. (2018) 265:1145–50. 10.1007/s00415-018-8813-z
4.
Kim SH Kim Y Jung JY Park NY Jang H Hyun JW et al . High seroprevalence and index of anti-john-cunningham virus antibodies in korean patients with multiple sclerosis. J Clin Neurol. (2019) 15:454–60. 10.3988/jcn.2019.15.4.454
5.
Lau A Qiu W Kermode A Au C Ng A Wong A et al . High prevalence and indexes of anti-John Cunningham virus antibodies in a cohort of Chinese patients with multiple sclerosis. Mult Scler J Exp Transl Clin. (2018) 4:2055217318788699. 10.1177/2055217318788699
6.
Ehlers B Anoh AE Ben Salem N Broll S Couacy-Hymann E Fischer D et al . Novel polyomaviruses in mammals from multiple orders and reassessment of polyomavirus evolution and taxonomy. Viruses. (2019) 11:930. 10.3390/v11100930
7.
Feltkamp MC Daugherty MD . Moens U, Ramqvist T, et al. A taxonomy update for the family Polyomaviridae. Arch Virol. (2016) 161:1739–50. 10.1007/s00705-016-2794-y
8.
Mohn N Grote-Levi L Hopfner F Eiz-Vesper B Maecker-Kolhoff B Warnke C et al . Innovative therapeutic concepts of progressive multifocal leukoencephalopathy. J Neurol. (2022) 269:2403–13. 10.1007/s00415-021-10952-5
9.
Yang X Su B Zhang X Liu Y Wu H Zhang T . Incomplete immune reconstitution in HIV/AIDS patients on antiretroviral therapy: Challenges of immunological non-responders. J Leukoc Biol. (2020) 107:597–612. 10.1002/JLB.4MR1019-189R
10.
Raisch DW Rafi JA Chen C Bennett CL . Detection of cases of progressive multifocal leukoencephalopathy associated with new biologicals and targeted cancer therapies from the FDA's adverse event reporting system. Expert Opin Drug Saf. (2016) 15:1003–11. 10.1080/14740338.2016.1198775
11.
Seminara NM Gelfand JM . Assessing long-term drug safety: lessons (re) learned from raptiva. Semin Cutan Med Surg. (2010) 29:16–9. 10.1016/j.sder.2010.01.001
12.
Bennett CL Focosi D Socal MP Bian JC Nabhan C Hrushesky WJ et al . Progressive multifocal leukoencephalopathy in patients treated with rituximab: a 20-year review from the Southern Network on adverse reactions. Lancet Haematol. (2021) 8:e593–604. 10.1016/S2352-3026(21)00167-8
13.
Lee DSW Rojas OL Gommerman JL . B cell depletion therapies in autoimmune disease: advances and mechanistic insights. Nat Rev Drug Discov. (2021) 20:179–99. 10.1038/s41573-020-00092-2
14.
Calabrese LH Molloy E Berger J . Sorting out the risks in progressive multifocal leukoencephalopathy. Nat Rev Rheumatol. (2015) 11:119–23. 10.1038/nrrheum.2014.167
15.
Diamantopoulos PT Kalopisis K Tsatsou A Efthymiou A Giannakopoulou N Hatzidavid S et al . Progressive multifocal leukoencephalopathy in the context of newer therapies in hematology and review of new treatment strategies. Eur J Haematol. (2022) 108:359–68. 10.1111/ejh.13751
16.
Ho PR Koendgen H Campbell N Haddock B Richman S Chang I . Risk of natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: a retrospective analysis of data from four clinical studies. Lancet Neurol. (2017) 16:925–33. 10.1016/S1474-4422(17)30282-X
17.
Tugemann B Berger JR . Improving risk-stratification of natalizumab-associated PML. Ann Clin Transl Neurol. (2021) 8:696–703. 10.1002/acn3.51130
18.
Wiendl H Gold R Berger T Derfuss T Linker R Maurer M et al . Multiple Sclerosis Therapy Consensus Group (MSTCG): position statement on disease-modifying therapies for multiple sclerosis (white paper). Ther Adv Neurol Disord. (2021) 14:17562864211039648. 10.1177/17562864211039648
19.
Major EO Yousry TA Clifford DB . Pathogenesis of progressive multifocal leukoencephalopathy and risks associated with treatments for multiple sclerosis: a decade of lessons learned. Lancet Neurol. (2018) 17:467–80. 10.1016/S1474-4422(18)30040-1
20.
Schwab N Schneider-Hohendorf T Melzer N Cutter G Wiendl H . Natalizumab-associated PML: Challenges with incidence, resulting risk, and risk stratification. Neurology. (2017) 88:1197–205. 10.1212/WNL.0000000000003739
21.
Baber U Bouley A Egnor E Sloane JA . Anti-JC virus antibody index changes in rituximab-treated multiple sclerosis patients. J Neurol. (2018) 265:2342–5. 10.1007/s00415-018-8996-3
22.
Sgarlata E Chisari CG Toscano S Finocchiaro C Lo Fermo S Millefiorini E et al . Changes in john cunningham virus index in multiple sclerosis patients treated with different disease-modifying therapies. Curr Neuropharmacol. (2022) 20:1978–87. 10.2174/1570159X19666211111123202
23.
Dalla Costa G Martinelli V Moiola L Sangalli F Colombo B Finardi A et al . Serum neurofilaments increase at progressive multifocal leukoencephalopathy onset in natalizumab-treated multiple sclerosis patients. Ann Neurol. (2019) 85:606–10. 10.1002/ana.25437
24.
Fissolo N Pignolet B Rio J Vermersch P Ruet A deSeze J et al . Serum neurofilament levels and PML risk in patients with multiple sclerosis treated with natalizumab. Neurol Neuroimmunol Neuroinflamm. (2021) 8:e1003. 10.1212/NXI.0000000000001003
25.
Hatchwell E . Is there a (host) genetic predisposition to progressive multifocal leukoencephalopathy?Front Immunol. (2015) 6:216. 10.3389/fimmu.2015.00216
26.
Mills EA Mao-Draayer Y . Understanding progressive multifocal leukoencephalopathy risk in multiple sclerosis patients treated with immunomodulatory therapies: a bird's eye view. Front Immunol. (2018) 9:138. 10.3389/fimmu.2018.00138
27.
Al Shekaili L Sheikh F Al Gazlan S Al Dhekri H Al Mousa H Al Ghonaium A et al . Novel mutation in DOCK8-HIES with severe phenotype and successful transplantation. Clin Immunol. (2017) 178:39–44. 10.1016/j.clim.2016.08.002
28.
Aschermann Z Gomori E Kovacs GG Pal E Simon G Komoly S et al . X-linked hyper-IgM syndrome associated with a rapid course of multifocal leukoencephalopathy. Arch Neurol. (2007) 64:273–6. 10.1001/archneur.64.2.273
29.
Day-Williams AG Sun C Jelcic I McLaughlin H Harris T Martin R et al . Whole genome sequencing reveals a chromosome 9p deletion causing DOCK8 deficiency in an adult diagnosed with hyper IgE syndrome who developed progressive multifocal leukoencephalopathy. J Clin Immunol. (2015) 35:92–6. 10.1007/s10875-014-0114-4
30.
Dhalla F Murray S Sadler R Chaigne-Delalande B Sadaoka T Soilleux E et al . Identification of a novel mutation in MAGT1 and progressive multifocal leucoencephalopathy in a 58-year-old man with XMEN disease. J Clin Immunol. (2015) 35:112–8. 10.1007/s10875-014-0116-2
31.
Downes SM Black GC Hyman N Simmonds M Morris J Barton C . Visual loss due to progressive multifocal leukoencephalopathy in a congenital immunodeficiency disorder. Arch Ophthalmol. (2001) 119:1376–8. 10.1001/archopht.119.9.1376
32.
Engelhardt KR Gertz ME Keles S Schaffer AA Sigmund EC Glocker C et al . The extended clinical phenotype of 64 patients with dedicator of cytokinesis 8 deficiency. J Allergy Clin Immunol. (2015) 136:402–12. 10.1016/j.jaci.2014.12.1945
33.
Hadjadj J Guffroy A Delavaud C Taieb G Meyts I Fresard A et al . Progressive multifocal leukoencephalopathy in primary immunodeficiencies. J Clin Immunol. (2019) 39:55–64. 10.1007/s10875-018-0578-8
34.
Lorenzini T Fliegauf M Klammer N Frede N Proietti M Bulashevska A et al . Characterization of the clinical and immunologic phenotype and management of 157 individuals with 56 distinct heterozygous NFKB1 mutations. J Allergy Clin Immunol. (2020) 146:901–11. 10.1016/j.jaci.2019.11.051
35.
Maffucci P Filion CA Boisson B Itan Y Shang L Casanova JL et al . Genetic diagnosis using whole exome sequencing in common variable immunodeficiency. Front Immunol. (2016) 7:220. 10.3389/fimmu.2016.00220
36.
Marechal E Beel K Crols R Hernalsteen D Willekens B . Long-term survival after progressive multifocal leukoencephalopathy in a patient with primary immune deficiency and NFKB1 mutation. J Clin Immunol. (2020) 40:1138–43. 10.1007/s10875-020-00862-y
37.
Sampaio EP Hsu AP Pechacek J Bax HI Dias DL Paulson ML et al . Signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations and disseminated coccidioidomycosis and histoplasmosis. J Allergy Clin Immunol. (2013) 131:1624–34. 10.1016/j.jaci.2013.01.052
38.
Schröder C Baerlecken NT Pannicke U Dörk T Witte T Jacobs R et al . Evaluation of RAG1 mutations in an adult with combined immunodeficiency and progressive multifocal leukoencephalopathy. Clin Immunol. (2017) 179:1–7. 10.1016/j.clim.2016.12.013
39.
Seguier J Briantais A Ebbo M Meunier B Aurran T Coze S et al . Late-onset progressive multifocal leukoencephalopathy (PML) and lymphoma in a 65-year-old patient with XIAP deficiency. J Clin Immunol. (2021) 41:1975–8. 10.1007/s10875-021-01139-8
40.
Suzuki H Takahashi Y Miyajima H . Progressive multifocal leukoencephalopathy complicating X-linked hyper-IgM syndrome in an adult. Intern Med. (2006) 45:1187–8. 10.2169/internalmedicine.45.6023
41.
Teramoto T Kaneko H Funato M Sawa H Nagashima K Hirose Y et al . Progressive multifocal leukoencephalopathy in a patient with X-linked agammaglobulinemia. Scand J Infect Dis. (2003) 35:909–10. 10.1080/00365540310016673
42.
Zerbe CS Marciano BE Katial RK Santos CB Adamo N Hsu AP et al . Progressive multifocal leukoencephalopathy in primary immune deficiencies: Stat1 gain of function and review of the literature. Clin Infect Dis. (2016) 62:986–94. 10.1093/cid/civ1220
43.
Tangye SG Al-Herz W Bousfiha A Chatila T Cunningham-Rundles C Etzioni A et al . Human inborn errors of immunity: 2019 update on the classification from the International Union of Immunological Societies expert committee. J Clin Immunol. (2020) 40:24–64. 10.1007/s10875-019-00737-x
44.
Tangye SG Al-Herz W Bousfiha A Cunningham-Rundles C Franco JL Holland SM et al . The ever-increasing array of novel inborn errors of immunity: an interim update by the IUIS committee. J Clin Immunol. (2021) 41:666–79. 10.1007/s10875-021-00980-1
45.
Eis PS Bruno CD Richmond TA Koralnik IJ Hanson BA Major EO et al . Germline genetic risk variants for progressive multifocal leukoencephalopathy. Front Neurol. (2020) 11:186. 10.3389/fneur.2020.00186
46.
Karczewski KJ Francioli LC Tiao G Cummings BB Alfoldi J Wang Q et al . The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. (2020) 581:434–43. 10.1038/s41586-020-2308-7
47.
Berger JR Aksamit AJ Clifford DB Davis L Koralnik IJ Sejvar JJ et al . PML diagnostic criteria: consensus statement from the AAN Neuroinfectious Disease Section. Neurology. (2013) 80:1430–8. 10.1212/WNL.0b013e31828c2fa1
48.
Pritchard JK Stephens M Donnelly P . Inference of population structure using multilocus genotype data. Genetics. (2000) 155:945–59. 10.1093/genetics/155.2.945
49.
International Multiple Sclerosis Genetics Consortium . Low-frequency and rare-coding variation contributes to multiple sclerosis risk. Cell. (2018) 175:1679–87 e7. 10.1016/j.cell.2018.09.049
50.
Deeks JJ Higgins JP . Statistical algorithms in review manager 5. Statistical Methods Group of The Cochrane Collaboration. (2010) 1–11.
51.
Tonk ECM Gurwitz D . Maitland-van der Zee AH, Janssens A. Assessment of pharmacogenetic tests: presenting measures of clinical validity and potential population impact in association studies. Pharmacogenom J. (2017) 17:386–92. 10.1038/tpj.2016.34
52.
Richards S Aziz N Bale S Bick D Das S Gastier-Foster J et al . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. 10.1038/gim.2015.30
53.
Kurian AW Hughes E Handorf EA Gutin A Allen B Hartman AR et al . Breast and ovarian cancer penetrance estimates derived from germline multiple-gene sequencing results in women. JCO Precis Oncol. (2017) 1:1–12. 10.1200/PO.16.00066
54.
Shi YW Min FL Zhou D Qin B Wang J Hu FY et al . HLA-A*24:02 as a common risk factor for antiepileptic drug-induced cutaneous adverse reactions. Neurology. (2017) 88:2183–91. 10.1212/WNL.0000000000004008
55.
Genin E Chen DP Hung SI Sekula P Schumacher M Chang PY et al . HLA-A*31:01 and different types of carbamazepine-induced severe cutaneous adverse reactions: an international study and meta-analysis. Pharmacogenomics J. (2014) 14:281–8. 10.1038/tpj.2013.40
56.
Harr T French LE . Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis. (2010) 5:39. 10.1186/1750-1172-5-39
57.
Agrawal P Sharma S Pal P Ojha H Mullick J Sahu A . The imitation game: a viral strategy to subvert the complement system. FEBS Lett. (2020) 594:2518–42. 10.1002/1873-3468.13856
58.
Mayilyan KR . Complement genetics, deficiencies, and disease associations. Protein Cell. (2012) 3:487–96. 10.1007/s13238-012-2924-6
59.
Murugaiah V Varghese PM Beirag N De Cordova S Sim RB Kishore U . Complement proteins as soluble pattern recognition receptors for pathogenic viruses. Viruses. (2021) 13:824. 10.3390/v13050824
60.
Pouw RB Ricklin D . Tipping the balance: intricate roles of the complement system in disease and therapy. Semin Immunopathol. (2021) 43:757–71. 10.1007/s00281-021-00892-7
61.
Angulo A Cuenca M Martinez-Vicente P Engel P . Viral CD229 (Ly9) homologs as new manipulators of host immunity. J Leukoc Biol. (2019) 105:947–54. 10.1002/JLB.2MR1018-413R
62.
Canna SW Marsh RA . Pediatric hemophagocytic lymphohistiocytosis. Blood. (2020) 135:1332–43. 10.1182/blood.2019000936
63.
Schulert GS Cron RQ . The genetics of macrophage activation syndrome. Genes Immun. (2020) 21:169–81. 10.1038/s41435-020-0098-4
64.
Sieni E Cetica V Hackmann Y Coniglio ML Da Ros M Ciambotti B et al . Familial hemophagocytic lymphohistiocytosis: when rare diseases shed light on immune system functioning. Front Immunol. (2014) 5:167. 10.3389/fimmu.2014.00167
65.
van Driel BJ Liao G Engel P Terhorst C . Responses to microbial challenges by SLAMF receptors. Front Immunol. (2016) 7:4. 10.3389/fimmu.2016.00004
66.
Nakahara J Tomaske L Kume K Takata T Kamada M Deguchi K et al . Three cases of non-carryover fingolimod-PML: Is the risk in Japan increased?Neurol Neuroimmunol Neuroinflamm. (2019) 6:e559. 10.1212/NXI.0000000000000559
67.
Oshima Y Tanimoto T Yuji K Tojo A . Drug-associated progressive multifocal leukoencephalopathy in multiple sclerosis patients. Mult Scler. (2019) 25:1141–9. 10.1177/1352458518786075
68.
Watanabe M Nakamura Y Isobe N Tanaka M Sakoda A Hayashi F et al . Two susceptible HLA-DRB1 alleles for multiple sclerosis differentially regulate anti-JC virus antibody serostatus along with fingolimod. J Neuroinflammation. (2020) 17:206. 10.1186/s12974-020-01865-7
69.
Pacanowski M Schuck RN . Evidence, in context: a regulatory perspective on pharmacogenetics. Clin Pharmacol Ther. (2022) 111:1202–4. 10.1002/cpt.2347
Summary
Keywords
immunodeficiency, JC virus, multiple sclerosis, natalizumab, pharmacovigilance, progressive multifocal leukoencephalopathy, PML, serious adverse event
Citation
Hatchwell E, Smith EB III, Jalilzadeh S, Bruno CD, Taoufik Y, Hendel-Chavez H, Liblau R, Brassat D, Martin-Blondel G, Wiendl H, Schwab N, Cortese I, Monaco MC, Imberti L, Capra R, Oksenberg JR, Gasnault J, Stankoff B, Richmond TA, Rancour DM, Koralnik IJ, Hanson BA, Major EO, Chow CR and Eis PS (2022) Progressive multifocal leukoencephalopathy genetic risk variants for pharmacovigilance of immunosuppressant therapies. Front. Neurol. 13:1016377. doi: 10.3389/fneur.2022.1016377
Received
10 August 2022
Accepted
11 November 2022
Published
14 December 2022
Volume
13 - 2022
Edited by
Luisa María Villar, Ramón y Cajal University Hospital, Spain
Reviewed by
Daniele Focosi, Pisana University Hospital, Italy; Thomas Weber, Katholisches Marienkrankenhaus GmbH, Germany
Updates
Copyright
© 2022 Hatchwell, Smith, Jalilzadeh, Bruno, Taoufik, Hendel-Chavez, Liblau, Brassat, Martin-Blondel, Wiendl, Schwab, Cortese, Monaco, Imberti, Capra, Oksenberg, Gasnault, Stankoff, Richmond, Rancour, Koralnik, Hanson, Major, Chow and Eis.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eli Hatchwell elihatchwell@populationbio.comPeggy S. Eis pegeis@populationbio.com
‡These authors have contributed equally to this work
†Present address: David Brassat, AP-HP, Hôpital Pitié-Salpêtrière, Paris, France
This article was submitted to Multiple Sclerosis and Neuroimmunology, a section of the journal Frontiers in Neurology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.