Abstract
Introduction:
Cobalamin C (cblC) deficiency is a rare hereditary disorder affecting intracellular cobalamin metabolism, primarily caused by mutations in MMACHC. This condition is characterized by combined methylmalonic acidemia and hyperhomocysteinemia, displaying a wide range of clinical manifestations involving multiple organs. Owing to its uncommon occurrence and diverse clinical phenotypes, diagnosing cblC deficiency is challenging and often leads to delayed or missed diagnoses.
Case description:
In this report, we present a case of late-onset cblC deficiency with brown desquamating dermatitis on the buttocks. Magnetic resonance imaging (MRI) of the brain revealed bilateral cerebellar abnormalities. The suspicion of an inherited metabolic disorder was raised by abnormal serum amino acid and acylcarnitine levels, along with increased urine methylmalonic acid and serum homocysteine levels. Whole-exome sequencing helped identify a homozygous variant (c.482G>A) in MMACHC, confirming the diagnosis of cblC deficiency. However, despite receiving treatment with hydroxocobalamin and betaine, the patient did not experience clinical improvement, which may be attributed to the delayed diagnosis as indicated by the declining homocysteine and methylmalonic acid levels.
Conclusion:
Collectively, we emphasize the significance of recognizing the skin lesions and observing serial MRI changes in patients with cblC deficiency. Our case underscores the importance of early diagnosis and timely therapeutic intervention for this severe yet frequently manageable condition.
Introduction
Combined with methylmalonic acidemia and homocystinuria, cobalamin C (cblC) is the most common inborn error in cobalamin metabolism. Newborn genetic screening studies for metabolic errors revealed that the estimated incidence of CblC deficiency is 1/3,920 in Shandong Province, China (1), and 1/100,000 in New York, United States (2). In 2006, Lerner-Ellis et al. (3) cloned the disease-causing MMACHC gene. To date, over 50 mutations have been identified in patients with MMACHC. Mutations in MMACHC disrupt the intracellular metabolism of cobalamin, causing the accumulation of methylmalonic acid and homocysteine and a lack of methionine. However, the underlying pathophysiological mechanisms are not well understood.
cblC deficiency is associated with systemic involvement and complex clinical manifestations, including progressive encephalopathy, subacute combined degeneration of the spinal cord, maculopathy, anemia, thromboembolic complications, pulmonary arterial hypertension, and kidney injury (4, 5). Skin manifestations, although rare, are often overlooked (6). Brain magnetic resonance imaging (MRI) findings commonly include hydrocephalus, progressive white matter abnormalities, and atypical lesions in the basal ganglia and cerebellum (4, 7). Late-onset cblC deficiency, which occurs in approximately 10% of affected individuals aged above 12 months, generally presents with milder symptoms, more evident clinical presentations and improvement, and a more favorable prognosis than that observed in the early-onset form. However, some patients do not respond well to treatment, particularly those with severe complications or delayed or insufficient initiation of therapy. In this case report, we describe a 21-year-old man who presented with psychiatric symptoms, skin lesions, and bilateral cerebellar abnormalities on brain MRI. The diagnosis of cblC deficiency was confirmed using whole-exome sequencing.
Case description
A 21-year-old man with no significant family history presented with a two-year history of weakness, numbness, and painful sensations in his lower limbs. Over the course of 1 month, he also experienced psychiatric disturbances. Figure 1 provides a timeline of the events. The patient reported additional symptoms, including urinary and bowel incontinence, as well as intellectual and motor developmental delays. He exhibited a motionless posture with a distressed facial expression, occasionally crying out loudly. Seborrheic dermatitis was observed on his face and trunk, and the buttocks had prominent brown hyperpigmentation with desquamating dermatitis (Figure 2A). Tendon reflexes were normal, with no signs of pyramidal neurological deficits. However, the patient’s impaired consciousness hindered further examination. Laboratory tests revealed hypochromic microcytic anemia (hemoglobin level: 91 g/L), elevated transaminase levels (glutamic-pyruvic transaminase: 168.7 U/L; aspartate aminotransferase: 84.1 U/L), and significantly increased serum homocysteine levels (258.4 μmol/L, normal range, 5–15.0 μmol/L). Urine organic acid analysis and serum amino acid and acylcarnitine analysis showed substantially elevated urine methylmalonic acid levels (24.8 μmol/L, normal range, <4.0 μmol/L) and serum propionyl carnitine/acetylcarnitine ratio (0.33, normal range, 0.02–0.2) (Figures 2B,C). No signs of infection, inflammation, or toxicity were present, and serum and cerebrospinal fluid (CSF) anti-cerebellitis antibodies were negative. Spinal cord MRI scan results were normal. Electromyogram (EMG) findings indicated widespread motor and sensory demyelination with accompanying axonal injuries in the limbs, particularly affecting the lower limbs. The first MRI, obtained 2 months after symptom onset, revealed diffuse cerebral atrophy (Figures 3A–C). Subsequent MRI performed at this visit (2 years after symptom onset) revealed bilateral symmetrical lesions in the cerebellum, characterized by hyperintensity on T2-weighted images, restricted diffusion on diffusion-weighted imaging (DWI), and no enhancement (Figures 3D–F). Based on the abnormal serum amino acid and acylcarnitine levels, elevated urine methylmalonic acid level, and increased serum homocysteine level, inherited metabolic diseases were strongly suspected. The diagnosis of late-onset cblC deficiency was further supported by whole-exome genetic sequencing, revealing a homozygous variant (c.482 G > A) in MMACHC (Figure 2D). Further genetic analysis proved that the patient in our case was maternal uniparental disomy (mUPD). Following treatment with adequate betaine (9 g/day, administered intragastrically), cobamamide (1.5 mg/day, administered via intramuscular injection), mecobalamin (1 mg/day, administered via intramuscular injection), and B vitamins, the patient’s serum homocysteine levels substantially decreased (60.9 μmol/L), as did urine methylmalonic acid levels (12.5 μmol/L). At discharge, the patient did not experience significant clinical improvement, as well as observed changes on brain MRI (Figures 3G–I). Within 1.5 years of follow-up, he relieved slightly but relapsed after treatment discontinuation.
Figure 1

A timeline of events.
Figure 2
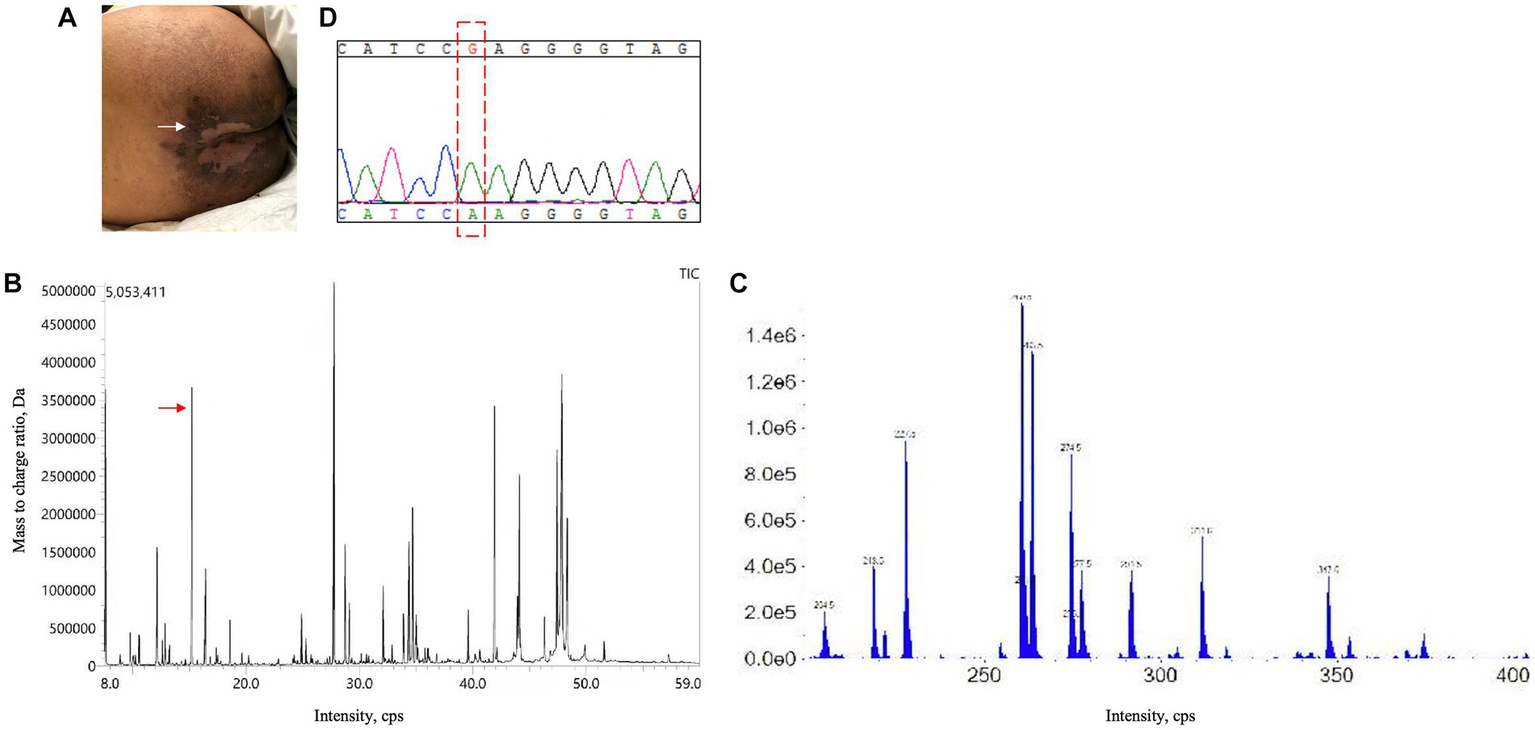
Skin lesion, urine organic acid analysis, serum amino acid and acylcarnitine analysis, and genetic testing results. (A) Superficial, erosive desquamating dermatitis on the buttocks of the present patient. (B) Urine organic acid analysis revealed an evident increase in methylmalonic acid levels (24.8 μmol/L; normal range: <4.0 μmol/L). (C) Serum amino acid and acylcarnitine analysis exhibited an increased propionyl carnitine/acetylcarnitine ratio (0.33; normal range: 0.02–0.2). (D) Next-generation sequencing identified a homozygous variant (c.482 G > A) in MMACHC, which was further validated by sanger sequencing.
Figure 3
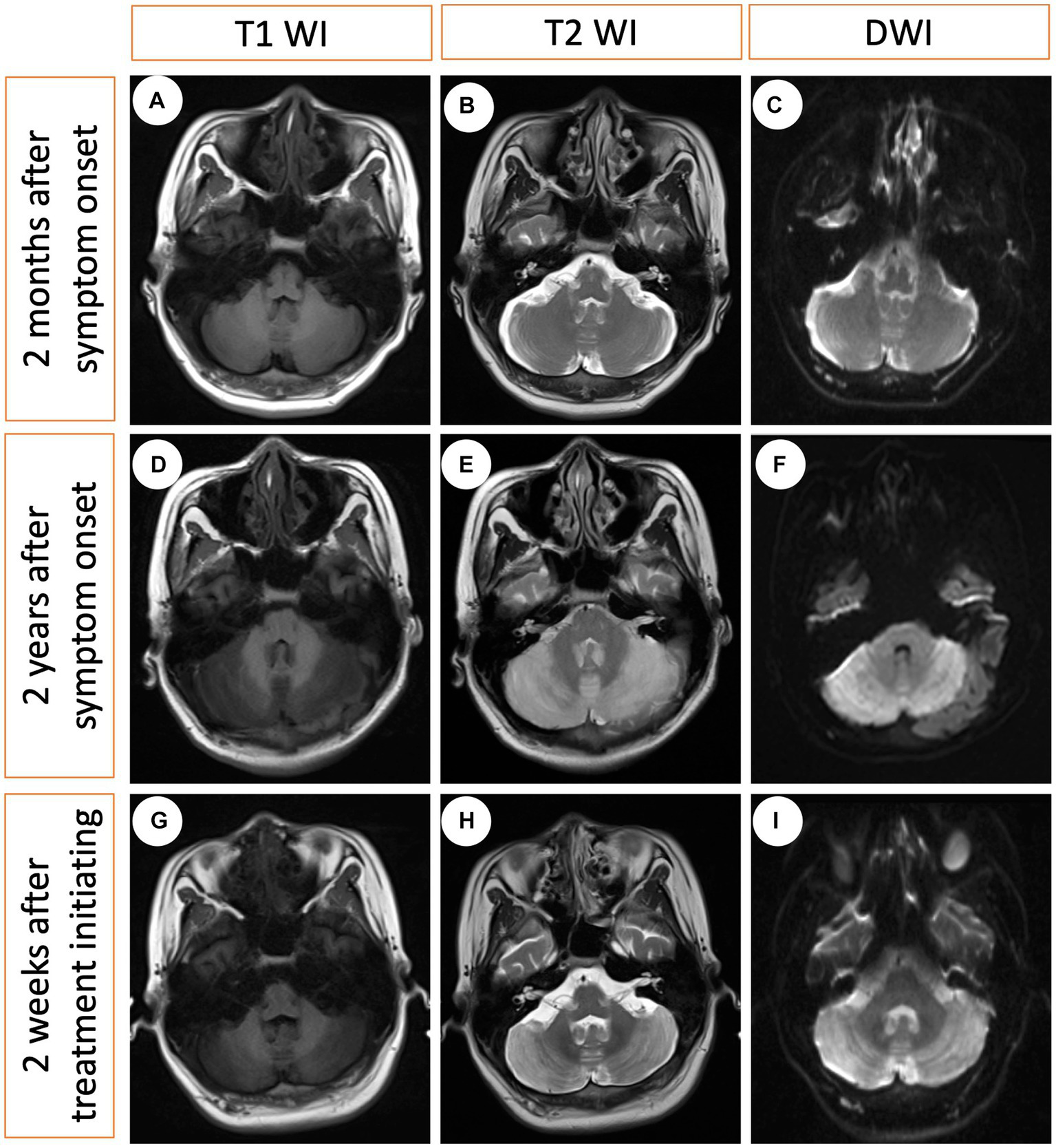
Serial MRI changes at three different time points. (A–C) The first MRI obtained 2 months after symptom onset (no treatment), indicating modest cerebral atrophy. (D–F) The second MRI obtained 2 years after symptom onset (no treatment), revealing bilateral hyperintensity in the cerebellum on the T2-weighted image and diffusion-weighted imaging (DWI), with hypointensity on the T1-weighted image. (G–I) The third MRI obtained 2 weeks after treatment initiation indicating minimal changes.
Discussion
cblC deficiency is the most prevalent disorder associated with cobalamin metabolism (5) and is a rare autosomal recessive disease induced by homozygous recessive or compound heterozygous mutations in MMACHC (3, 8). These mutations disrupt cobalamin metabolism, leading to the accumulation of toxic levels of metabolites, homocysteine and methylmalonic acid, which cause oxidative injury. While the central and peripheral nervous system are commonly affected, cblC deficiency can also involve more organs and tissues, including the kidneys (9), lung (10), micrangium (11), retina (12), and skin (6). Early-onset cblC deficiency, with an age of onset <12 months, is more prevalent and typically manifests with feeding difficulties, somnolence, developmental delays, seizures, muscular hypotonia, hydrocephalus, and microcephaly (1, 4, 8, 13). Late-onset cblC deficiency is characterized by neuropsychiatric symptoms, cognitive dysfunction, myelopathy, rental function abnormality, and pulmonary arterial hypertension (14, 15). The present case exhibited psychiatric symptoms, cognitive decline, peripheral neuropathy, dermatitis, anemia, and hepatic dysfunction were found.
Diffuse cerebral atrophy and bilateral hyperintensity in the deep white matter and cerebellum are common brain MRI findings in patients with late-onset cblC deficiency (15, 16). The patient presented with symmetrical bilateral cerebellar lesions. The differential diagnoses of cerebellar lesions are broad and include vascular causes (occlusion of lateral posterior inferior cerebellar artery), infectious agents (such as bacteria, viruses, and mycoplasma), autoimmune factors (such as anti-glutamate decarboxylase 65 antibody-associated cerebellitis, paraneoplastic), hereditary conditions (hereditary spinocerebellar ataxia, fragile X–associated tremor ataxia syndrome), as well as metabolic and toxic factors (such as alcohol, cytotoxicity drugs, organic solvent) (17–20). The clinical history and laboratory analyzes narrowed the diagnostic possibilities, and genetic testing confirmed the final diagnosis.
Skin lesions owing to nutritional deficiency are rare manifestations of extra-nervous system involvement in cblC deficiency. They can present as cheilitis, diffuse erythema with erosions and desquamation, and superficial erosive desquamating dermatitis, which may be attributed to enzymatic deficiency or nutritional restriction (6, 21, 22). Researchers have suggested that erosive desquamating dermatitis with histopathological characteristics resembling acrodermatitis enteropathica may be an initial systemic sign of cblC deficiency. The presence of brown desquamation dermatitis should prompt clinicians to consider this diagnosis when nutritional deficiency is suspected. Renal examination was normal, but screening for optic neuropathy and retina was not performed for the patient’s poor psychiatric status.
CblC deficiency is often curable. Most patients with late-onset cblC deficiency respond well to hydroxocobalamin and betaine treatment. The goals of treatment are to improve clinical presentation, normalize serum methionine levels, reduce urine methylmalonic acid levels, and lower homocysteine levels by promoting homocysteine-to-methionine conversion, facilitating re-methylation, and accelerating acylcarnitine clearance (5, 23). The patient in our case only received betaine, cobamamide, and mecobalamin, not hydroxycobalamin, because hydroxycobalamin injection was not available in many provinces of China. Delayed diagnosis and therapeutic intervention, low compliance to maintenance treatment, and no hydroxycobalamin was given contributed to the poor outcomes in this case. Constant and aggressive treatment seems to be required.
In conclusion, we reported a rare case of late-onset cblC deficiency with a delayed diagnosis, presenting with brown desquamation dermatitis and bilateral cerebellar MRI abnormalities. Owing to its rarity and heterogeneous clinical presentation, the diagnosis of cblC deficiency poses challenges and is often delayed or missed. Our case highlights the importance of early diagnosis and timely therapeutic intervention for this severe but often treatable disease. Otherwise, irreversible damage to multiple organs, particularly neurological complications, is unavoidable, ultimately resulting in poor outcomes.
Funding
This work was supported by grant from the National Natural Science Foundation of China (No. 82101342 to LQ), the Provincial Natural Science Foundation (No. 2022JJ30833 to LQ), and Scientific Research Launch Project for new employees of the Second Xiangya Hospital of Central South University.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Data availability statement
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the Second Xiangya Hospital, Central South University. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
QC: Writing – original draft, Writing – review & editing. JT: Writing – original draft, Writing – review & editing. HZ: Writing – original draft, Writing – review & editing. LQ: Writing – original draft, Writing – review & editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1.
Han B Cao Z Tian L Zou H Yang L Zhu W et al . Clinical presentation, gene analysis and outcomes in young patients with early-treated combined methylmalonic Acidemia and Homocysteinemia (Cblc type) in Shandong Province. China Brain Dev. (2016) 38:491–7. doi: 10.1016/j.braindev.2015.10.016
2.
Weisfeld-Adams JD Morrissey MA Kirmse BM Salveson BR Wasserstein MP McGuire PJ et al . Newborn screening and early biochemical follow-up in combined methylmalonic aciduria and homocystinuria, Cblc type, and utility of methionine as a secondary screening analyte. Mol Genet Metab. (2010) 99:116–23. doi: 10.1016/j.ymgme.2009.09.008
3.
Lerner-Ellis JP Tirone JC Pawelek PD Doré C Atkinson JL Watkins D et al . Identification of the gene responsible for methylmalonic aciduria and homocystinuria. Nat Genet. (2006) 38:93–100. doi: 10.1038/ng1683
4.
Carrillo-Carrasco N Venditti CP . Combined methylmalonic Acidemia and homocystinuria, Cblc type. Ii. Complications, pathophysiology, and outcomes. J Inherit Metab Dis. (2012) 35:103–14. doi: 10.1007/s10545-011-9365-x
5.
Carrillo-Carrasco N Chandler RJ Venditti CP . Combined methylmalonic Acidemia and homocystinuria, Cblc type. I. Clinical presentations, diagnosis and management. J Inherit Metab Dis. (2012) 35:91–102. doi: 10.1007/s10545-011-9364-y
6.
Gilson RC Wallis L Yeh J Gilson RT . Dementia, Diarrhea, desquamating shellac-like dermatitis revealing late-onset Cobalamin C deficiency. JAAD Case Rep. (2018) 4:91–4. doi: 10.1016/j.jdcr.2017.09.016
7.
Longo D Fariello G Dionisi-Vici C Cannatà V Boenzi S Genovese E et al . Mri and 1h-Mrs findings in early-onset Cobalamin C/D defect. Neuropediatrics. (2005) 36:366–72. doi: 10.1055/s-2005-873057
8.
Wang F Han L Yang Y Gu X Ye J Qiu W et al . Clinical, biochemical, and molecular analysis of combined methylmalonic Acidemia and Hyperhomocysteinemia (Cblc type) in China. J Inherit Metab Dis. (2010) 33:435–42. doi: 10.1007/s10545-010-9217-0
9.
Lemoine M Grangé S Guerrot D . Kidney disease in Cobalamin C deficiency. Nephrol Ther. (2019) 15:201–14. doi: 10.1016/j.nephro.2019.03.011
10.
Liu J Tang X Zhou C Xu H Yang H He R et al . Cobalamin C deficiency presenting with diffuse alveolar Hemorrhage and pulmonary microangiopathy. Pediatr Pulmonol. (2020) 55:1481–6. Epub 2020/04/16. doi: 10.1002/ppul.24781
11.
George JN Cobalamin C . Deficiency-associated thrombotic microangiopathy: uncommon or unrecognised?Lancet. (2015) 386:1012. doi: 10.1016/s0140-6736(15)00077-x
12.
Garcia-Gonzalez JM Neiweem AE Grassi MA . Cobalamin C deficiency-associated pigmentary retinopathy. JAMA Ophthalmol. (2015) 133:e152161. doi: 10.1001/jamaophthalmol.2015.2161
13.
Rosenblatt DS Aspler AL Shevell MI Pletcher BA Fenton WA Seashore MR . Clinical heterogeneity and prognosis in combined methylmalonic aciduria and homocystinuria (Cblc). J Inherit Metab Dis. (1997) 20:528–38. doi: 10.1023/a:1005353530303
14.
Thauvin-Robinet C Roze E Couvreur G Horellou MH Sedel F Grabli D et al . The adolescent and adult form of Cobalamin C disease: clinical and molecular Spectrum. J Neurol Neurosurg Psychiatry. (2008) 79:725–8. doi: 10.1136/jnnp.2007.133025
15.
Huemer M Scholl-Bürgi S Hadaya K Kern I Beer R Seppi K et al . Three new cases of late-onset Cblc defect and review of the literature illustrating when to consider inborn errors of metabolism beyond infancy. Orphanet J Rare Dis. (2014) 9:161. doi: 10.1186/s13023-014-0161-1
16.
Wang SJ Zhao YY Yan CZ . Reversible encephalopathy caused by an inborn error of Cobalamin metabolism. Lancet. (2019) 393:e29. doi: 10.1016/s0140-6736(19)30043-1
17.
Baizabal-Carvallo JF Jankovic J . Autoimmune and paraneoplastic movement disorders: an update. J Neurol Sci. (2018) 385:175–84. doi: 10.1016/j.jns.2017.12.035
18.
de Silva RN Vallortigara J Greenfield J Hunt B Giunti P Hadjivassiliou M . Diagnosis and Management of Progressive Ataxia in adults. Pract Neurol. (2019) 19:196–207. doi: 10.1136/practneurol-2018-002096
19.
Mitoma H Manto M Hampe CS . Immune-mediated cerebellar ataxias: practical guidelines and therapeutic challenges. Curr Neuropharmacol. (2019) 17:33–58. doi: 10.2174/1570159x16666180917105033
20.
Klockgether T . Sporadic ataxia with adult onset: classification and diagnostic criteria. Lancet Neurol. (2010) 9:94–104. doi: 10.1016/s1474-4422(09)70305-9
21.
Howard R Frieden IJ Crawford D McCalmont T Levy ML Rosenblatt DS et al . Methylmalonic Acidemia, Cobalamin C type, presenting with cutaneous manifestations. Arch Dermatol. (1997) 133:1563–6. doi: 10.1001/archderm.1997.03890480083012
22.
Karamifar H Shakibazad N Saki F Saki N Kardeh S . Skin manifestation of methylmalonic Acidemia: case report and review of the literature. G Ital Dermatol Venereol. (2015) 150:741–4. PMID:
23.
Gurkas E Kartal A Aydin K Kucukçongar A Dilber C Ceylaner S . Reversible clinical and magnetic resonance imaging findings in late-onset Cobalamin C defect. Genet Couns. (2015) 26:425–30. Epub 2016/02/09. PMID:
Summary
Keywords
desquamating dermatitis, cerebellum, genes, cobalamin C deficiency, skin lesions
Citation
Chen Q, Tang J, Zhang H and Qin L (2023) Case report: Desquamating dermatitis, bilateral cerebellar lesions in a late-onset methylmalonic acidemia patient. Front. Neurol. 14:1255128. doi: 10.3389/fneur.2023.1255128
Received
08 July 2023
Accepted
28 August 2023
Published
22 September 2023
Volume
14 - 2023
Edited by
Huifang Shang, Sichuan University, China
Reviewed by
Chao Yuan, Southern Medical University, China; Arushi Gahlot Saini, Post Graduate Institute of Medical Education and Research (PGIMER), India
Updates
Copyright
© 2023 Chen, Tang, Zhang and Qin.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lixia Qin, qinlixia1027@csu.edu.cn
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.