 Gao-Hui Cao
Gao-Hui Cao Mei-Fang Zhao1†
Mei-Fang Zhao1† Yi Dong
Yi Dong Liang-Liang Fan
Liang-Liang Fan Yi-Hui Liu
Yi-Hui Liu Lu-Lu Tang
Lu-Lu Tang- 1Department of Cell Biology, School of Life Sciences, Central South University, Changsha, China
- 2Department of Neurology, Affiliated Hospital of Yangzhou University, Yangzhou, China
- 3Department of Cardiovascular Surgery, National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha, China
This report presents a case of Charcot–Marie–Tooth dominant intermediate D (CMTDID), a rare subtype of Charcot–Marie–Tooth disease, in a 52 years-old male patient. The patient exhibited mobility impairment, foot abnormalities (pes cavus), and calf muscle atrophy. Whole exome sequencing and Sanger sequencing suggested that a novel variant (NM_000530.8, c.145C>A/p.His49Asn) of MPZ may be the genetic lesion in the patient. The bioinformatic program predicted that the new variant (p.His49Asn), located at an evolutionarily conserved site of MPZ, was neutral. Our study expands the variant spectrum of MPZ and the number of identified CMTDID patients, contributing to a better understanding of the relationship between MPZ and CMTDID.
1 Introduction
Charcot–Marie–Tooth disease (CMTD) encompasses a genetically heterogeneous group of disorders called hereditary sensory and motor neuropathies that damage the peripheral nerves (1, 2). The typical symptoms of CMTD include muscle atrophy in the feet, pes cavus, and decreased sensitivity to touch, heat, and cold in the feet and lower legs (3). Other symptoms, including hearing loss, scoliosis, hip dysplasia, restless legs syndrome, and tremor, can also be present in CMTD patients (3). As the most common inherited disorder involving the peripheral nerves, the prevalence of CMTD is about 1 in 2,500 worldwide (4). Currently, variants in four genes (Peripheral Myelin Protein 22, Gap Junction Beta 1, Myelin Protein Zero, and Mitofusin 2), are responsible for over 90% of CMT patients (5).
The Myelin Protein Zero (MPZ) gene is located on chromosome 1q23.3, and it consists of 6 exons, spanning approximately 6,369 kilobases. This gene is specifically expressed in Schwann cells of the peripheral nervous system and encodes a type I transmembrane glycoprotein that is a major structural component of the peripheral myelin sheath (6, 7). Acting as an adhesion molecule, the MPZ protein functions like molecular glue, playing a role in tightly packing the myelin around nerve cells, which wrap around and insulate peripheral nerves (7). Currently, approximately 5% of CMTD patients result from variants in MPZ (8–10). Additionally, some studies have also reported that MPZ variants can lead to other polyneuropathies, such as Dejerine–Sottas syndrome and congenital hypomyelinating nesuropathy (11, 12).
Here, we studied a Chinese family presenting with distal atrophy and weakness. Whole exome sequencing revealed a novel variant (NM_000530.8, c.145C>A/p.His49Asn) in the MPZ gene within the proband. Sanger sequencing additionally confirmed the presence of this novel variant in other affected family members, suggesting co-segregation. Furthermore, bioinformatics software predicted that this newly identified MPZ variant is deleterious.
2 Case report
2.1 Clinical description
The family, including seven people were investigated in this study (Figure 1A). The proband (II-3), a 52 years-old male from Jiangsu province in eastern China. The proband came to our clinic 1 year ago (November 2022). According to his own account, he began to realize that exercise was more difficult seven years ago (at the age of 45). The condition slowly worsened until it was difficult to walk, so he came to our hospital for consultation.
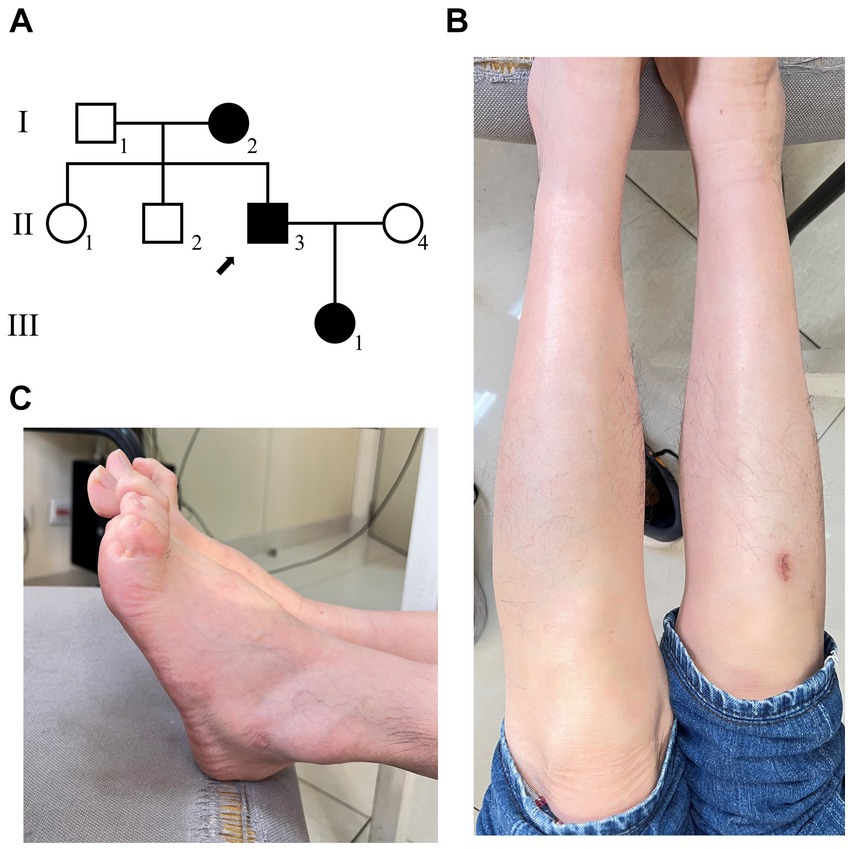
Figure 1. The clinical diagnosis of the proband. (A) Family diagram of patients with disease, and the arrow shows the proband. (B) The morphology of the patient’s feet has significantly high arches. (C) The patient has calf muscle atrophy.
Clinical examination reveals: the proband’s lower limbs exhibit an inverted bottle shape, noticeable atrophy of the calf muscles, and spinal curvature. The proband has no dislocation, but the arches are markedly elevated, displaying claw-like deformities in the toes. The patient experiences difficulties in movement, demonstrating an abnormal striding gait and poor limb balance. The patient has weaknesses in the lower limb muscles, reduced strength in the arms, but no limb tremors. Both Achilles tendon reflex and knee-jerk reflex are diminished, and there is a decrease in pinprick sensation (Figures 1B,C). Electromyography (EMG) indicated multiple symmetrical peripheral nerve lesions, particularly myelin damage. Peripheral motor nerve conduction velocity (MNCV) was moderately impaired (Table 1), alongside reduced sensory nerve conduction velocities (SNCV) in the upper limb (Table 2). In addition, autoimmune peripheral neuropathy and paraneoplastic nerve syndrome were excluded by examining ganglioside antibody spectrum (GM1, GD1b, GQ1b, GM2, GM3, GD1a, GT1b, Sulfatide, GM4, GD2, GD3, GT1a) and paraneoplastic nerve syndrome spectrum [Hu, Yo, Ri, CV2, PNMA2 (Ma-2/Ta), amphiphysin, recoverin, SOX1, titin, Zic4, GAD65, Tr (DNER)] in serum and cerebrospinal fluid. A family history investigation indicated that his mother (I-2) also suffered from mobility impairment and calf muscle atrophy. Additionally, his daughter (III-1) occasionally experiences muscle weakness in her limbs.
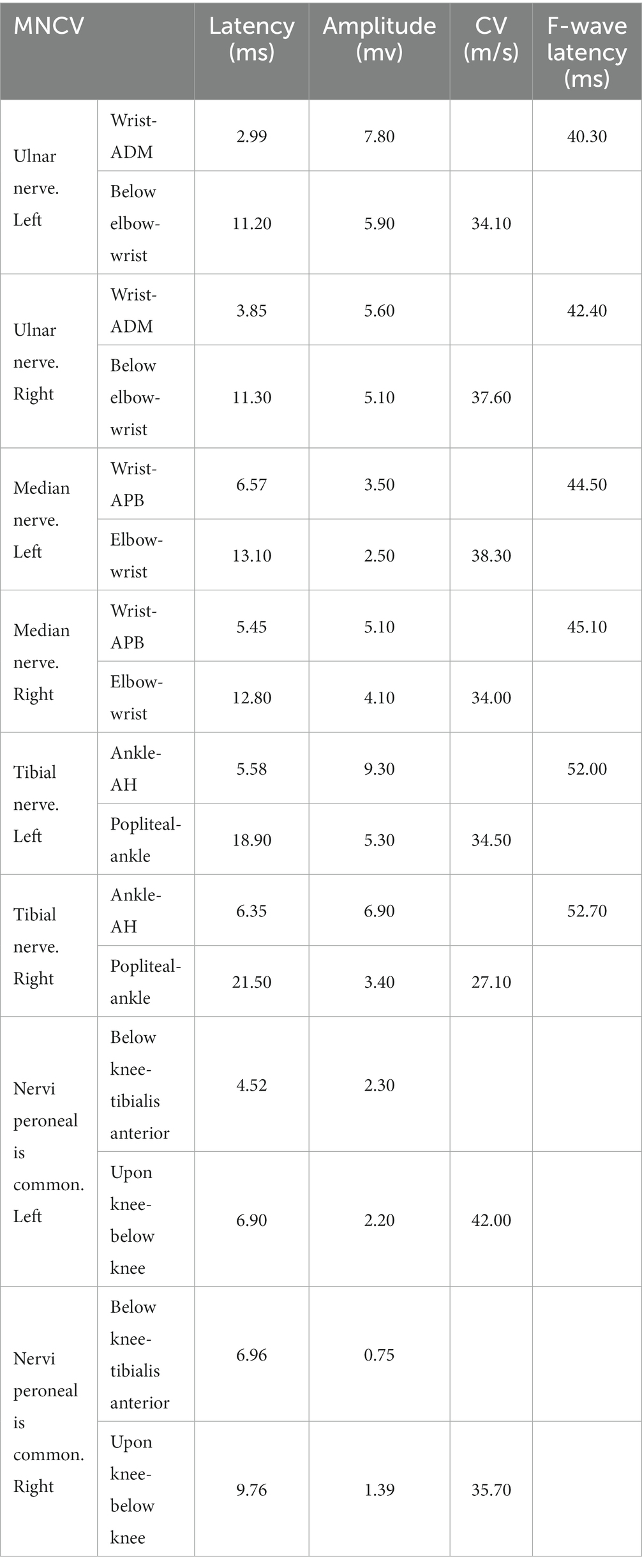
Table 1. Motor nerve conduction velocities of EMG result.
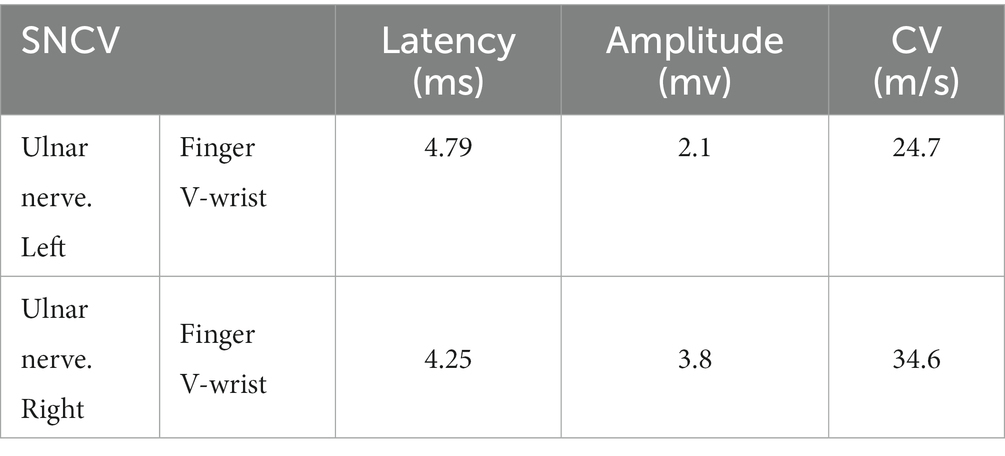
Table 2. Sensory nerve conduction velocities of EMG result.
As a result, patients are diagnosed with Charcot–Marie–Tooth disease. CMT cannot be effectively treated, so none of the patients were hospitalized. Patients are advised to increase their intake of vitamins B1 and B12. A follow-up was conducted 1 year later. There was no significant change in the patient’s condition.
2.2 Genetic analysis
Initially, multiplex ligation-dependent probe amplification was employed to exclude copy number variants in two candidate genes, Kinesin Family Member 1B and Peripheral Myelin Protein 22, which are commonly associated with copy number variations in CMTD patients (Supplementary Figure S1). Subsequently, the proband underwent whole sequencing to detect potential gene variants. A total of 9.16 GB of data, encompassing 70,012 SNVs/Indels, were identified in the proband. Following the aforementioned data filtering process, 12 variants were retained (Supplementary Table S1). Among these 12 variants, only the novel variant (NM_000530.8, c.145C>A/p.His49Asn) in MPZ was deemed to be the underlying genetic anomaly for the family. Sanger sequencing further confirmed the co-segregation of this variant with the affected family members (Figure 2A) and its absence in our 200 control cohorts. This novel variant, resulting in the substitution of histidine with asparagine, was located at a neutral tolerant site and Immunoglobulin-like domain of protein zero (IgV_P0) (Figure 2B). We predicted and compared the protein structure after the p.His49Asn variant (Figure 2C), based on the latest reports of IgV_P0 domain (PDBid: 8iia (13)). Surface potential analysis additionally revealed that variant altered the surface charge of the MPZ protein (Figure 2C). According to ACMG guideline, the variant belongs to Likely pathogenic (PM1 + PM2 + PP1 + PP3) (14).
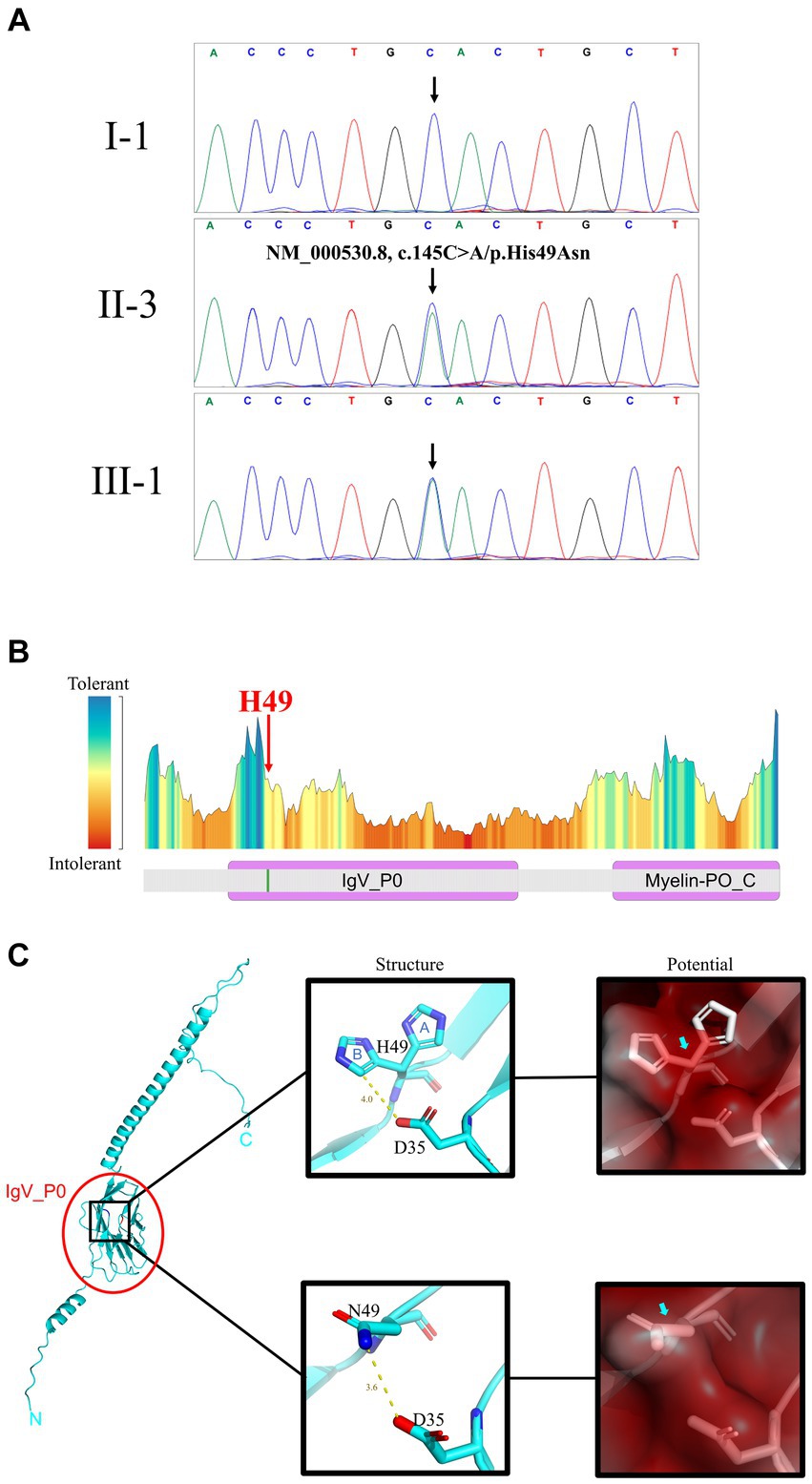
Figure 2. Gene and protein analysis. (A) Sanger sequencing results for I-1 (unaffected +/+), II-3 (affected +/−), and III-1 (affected +/−) patients (see Figure 1A for pedigree). (B) Predicted residue tolerance in the MPZ protein obtained using MetaDome. H49 is predicted to be of neutral tolerance to mutations. MPZ contains two domains: immunoglobulin-like domain of protein zero (IgV_P0) and Myelin-PO glycoprotein cytoplasmic C-term (Myelin-PO_C) by Conserved Domain Search prediction. (C) H49 is located on the IgV_P0 domain shown on the full length MPZ model (left panel). The atomic conformation (middle panels) and the surface electrostatic potential energy (right panels) change with the H49N variant. In the middle panels, blue “A” and “B” indicate 2 conformations of H49 in the crystal structure. Yellow line and text indicate the distance between atoms on side chains at positions 35 and 49 (angstrom). The blue and red atoms represent negative and positive charges, respectively. The variation of H49N causes the side chain charge from negative to become neutral. In the right panels, red means positive, blue means negative, and white means neutral. The surface potential prediction is slight changed from positive to neutral.
3 Discussion and conclusion
Charcot–Marie–Tooth disease (CMTD) comprises several subtypes, including CMT dominant intermediate D (CMTDID), a rare form defined by motor nerve conduction velocity (MNCV) falling within the intermediate range of 25–45 m/s (1, 15). This subtype was initially reported in 1999 within a 4-generation Macedonian family. The family exhibited a symmetrical pattern of distal muscle atrophy, weakness, and sensory impairment, more pronounced in the lower limbs and to a lesser extent in the upper limbs, besides, the youngest patients only 34 years old (16). In our study, the proband showed myelin damage in both motor and sensory nerves, with MNCV ranging from 27–42 m/s. Whole exome sequencing and Sanger sequencing further confirmed that the MPZ variant (NM_000530.8, c.145C>A/p.His49Asn) was the genetic anomaly responsible for the family’s condition. Our research may broaden the variant spectrum of MPZ and aid in genetic counseling and early diagnosis for CMT disease patients.
MPZ protein, an integral membrane glycoprotein, primarily connects adjacent lamellae to stabilize myelin assembly (17). It serves as the principal structural component of peripheral myelin and is exclusively expressed in Schwann cells (18). The MPZ protein is composed of three domains: a singular Immunoglobulin V-Type-like extracellular domain, a lone transmembrane domain, and a single cytosolic domain (19). Previous studies have indicated that the majority of pathogenic MPZ variants can trigger the unfolded protein response and endoplasmic reticulum retention (7). In our investigation, the novel variant (NM_000530.8, c.145C>A/p.His49Asn) in MPZ was situated in the extracellular IgV_P0 domain (Figure 2B). Crystallographic analysis of the extracellular domain of MPZ revealed its capacity to form interactions, resulting in homotetramer structures which are supported by recent solution-based studies using SEC, SAXS, and NMR (13, 20). Further, one recent study indicated that the extracellular domains of the MPZ protein form an 8-mer responsible with a potential involvement in membrane adhesion (13). The novel variant’s alteration of the MPZ protein’s charge may potentially influence the stabilization of membrane layers in compact myelin and adhesion between layers, further leading to demyelination (Figure 2C). Also, MPZ plays a crucial role in the development of myelin structure. Variants in MPZ could potentially impact the normal formation of myelin, consequently disturbing the interactions between Schwann cells and axons, ultimately resulting in abnormal axon (7). Interestingly, the earliest identified CMTDID patients carried the D35Y variant (16). The shortest distance between D35 and H49 (B conformation) is 4 angstroms, and the shortest distance between D35 and N49 is only 3.6 angstroms (Figure 2C). Therefore, there may be a relationship between CMTDID disease and residue contact of these two positions. In addition, Veneri et al. (21) found that the increase of glycosylation sites in MPZ can impair its function and lead to loosen myelin. Mutations in H49N produce an NCS sequence that belongs to the glycosylation motif (N-X-S/T), resulting in excessively glycosylation of MPZ. CMTDID reported in this study belongs to the intermediate type (16), showing both mild demyelinating lesions and mild axonal abnormalities.
Currently, a total of 180 variants in the MPZ gene have been reported in patients displaying various phenotypes. Through summarizing these reported MPZ variants, we observed that the majority of cases (78.2%) carrying MPZ variants exhibited Charcot–Marie–Tooth (CMT) phenotypes. Additionally, 7.4% of carriers presented with Dejerine–Sottas syndrome, and 0.8% displayed Roussy–Levy syndrome. Within 78.2% of carriers manifesting CMT diseases, a mere 0.4% of patients showed the CMT dominant intermediate D (CMTDID) subtype (HGMD database: https://www.hgmd.cf.ac.uk/ac/index.php). This scarcity underscores the rarity of CMTDID subtypes identified among MPZ variant carriers. In this context, we identified a novel MPZ variant (NM_000530.8, c.145C>A/p.His49Asn) in a CMTDID patient, thereby reporting a unique case arising from a novel MPZ variant. This contributes to the expanding pool of recognized CMTDID patients and furthers our understanding of this subtype.
In MPZ+/− mice, neuropathy develops in adulthood, displaying minimal nerve conduction slowing and mild demyelination, akin to patients with MPZ variants (22, 23). Recently, Shackleford et al. created a new MPZ variant (p.T124M) knock-in mouse model, revealing impaired motor performance, reduced compound motor action potential amplitudes, and axonal damage, albeit with normal nerve conduction velocities (24). The distinctions between MPZ+/− mice and MPZ (p.T124M) knock-in mice underscore the intricate role of MPZ in CMT disease development, implying that this study’s primary constraint lies in its absence of functional research.
In summation, our study employed whole exome sequencing and Sanger sequencing to identify a novel MPZ variant (NM_000530.8, c.145C>A/p.His49Asn) in a Chinese family afflicted by CMT disease. Subsequent analysis validated this variant as the cause of a rare CMT subtype known as CMTDID. Our work enhances the diversity of MPZ variant profiles and the roster of recognized CMTDID patients, contributing to a deeper comprehension of the relationship between MPZ and CMTDID.
Data availability statement
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by the Ethics Committee of the Affiliated Hospital of Yangzhou University, Yangzhou, China. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
GH-C: Data curation, Formal analysis, Investigation, Software, Validation, Visualization, Writing – original draft. M-FZ: Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft. YDo: Writing – review & editing. L-LF: Methodology, Writing – review & editing. Y-HL: Writing – review & editing. YDe: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Validation, Writing – review & editing. L-LT: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported by the National Natural Science Foundation of China (82201394); the Open Research Fund of State Key Laboratory of Hybrid Rice (Wuhan University) (KF202202); Research Project of the Hunan Provincial Health Commission (202103012102); Central South University Graduate Students’ Independent Exploration and Innovation Project (2023ZZTS0571).
Acknowledgments
The authors are grateful to the patients who agreed to participate in the study for their assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2024.1319962/full#supplementary-material
References
1. Berciano, J, García, A, Gallardo, E, Peeters, K, Pelayo-Negro, AL, Álvarez-Paradelo, S, et al. Intermediate Charcot–Marie–Tooth disease: an electrophysiological reappraisal and systematic review. J Neurol. (2017) 264:1655–77. doi: 10.1007/s00415-017-8474-3
2. Volodarsky, M, Kerkhof, J, Stuart, A, Levy, M, Brady, LI, Tarnopolsky, M, et al. Comprehensive genetic sequence and copy number analysis for Charcot–Marie–Tooth disease in a Canadian cohort of 2517 patients. J Med Genet. (2021) 58:284–8. doi: 10.1136/jmedgenet-2019-106641
3. Pareyson, D, and Marchesi, C. Diagnosis, natural history, and management of Charcot–Marie–Tooth disease. Lancet Neurol. (2009) 8:654–67. doi: 10.1016/S1474-4422(09)70110-3
4. Barreto, LCLS, Oliveira, FS, Nunes, PS, de França Costa, IMP, Garcez, CA, Goes, GM, et al. Epidemiologic study of Charcot–Marie–Tooth disease: a systematic review. Neuroepidemiology. (2016) 46:157–65. doi: 10.1159/000443706
5. Herrmann, DN. Experimental therapeutics in hereditary neuropathies: the past, the present, and the future. Neurotherapeutics. (2008) 5:507–15. doi: 10.1016/j.nurt.2008.07.001
6. Moss, KR, Bopp, TS, Johnson, AE, and Höke, A. New evidence for secondary axonal degeneration in demyelinating neuropathies. Neurosci Lett. (2021) 744:135595. doi: 10.1016/j.neulet.2020.135595
7. Shy, ME, Jáni, A, Krajewski, K, Grandis, M, Lewis, RA, Li, J, et al. Phenotypic clustering in MPZ mutations. Brain. (2004) 127:371–84. doi: 10.1093/brain/awh048
8. Hayasaka, K, Himoro, M, Sato, W, Takada, G, Uyemura, K, Shimizu, N, et al. Charcot–Marie–Tooth neuropathy type 1B is associated with mutations of the myelin P0 gene. Nat Genet. (1993) 5:31–4. doi: 10.1038/ng0993-31
9. Auer-Grumbach, M, Strasser-Fuchs, S, Robl, T, Windpassinger, C, and Wagner, K. Late onset Charcot–Marie–Tooth 2 syndrome caused by two novel mutations in the MPZ gene. Neurology. (2003) 61:1435–7. doi: 10.1212/01.WNL.0000094197.46109.75
10. Seeman, P, Mazanec, R, Huehne, K, Suslíková, P, Keller, O, and Rautenstrauss, B. Hearing loss as the first feature of late-onset axonal CMT disease due to a novel P0 mutation. Neurology. (2004) 63:733–5. doi: 10.1212/01.WNL.0000134605.61307.DE
11. Hayasaka, K, Himoro, M, Sawaishi, Y, Nanao, K, Takahashi, T, Takada, G, et al. De novo mutation of the myelin P0 gene in Dejerine–Sottas disease (hereditary motor and sensory neuropathy type III). Nat Genet. (1993) 5:266–8. doi: 10.1038/ng1193-266
12. Auer-Grumbach, M, Strasser-Fuchs, S, Wagner, K, Körner, E, and Fazekas, F. Roussy–Lévy syndrome is a phenotypic variant of Charcot–Marie–Tooth syndrome IA associated with a duplication on chromosome 17p11.2. J Neurol Sci. (1998) 154:72–5. doi: 10.1016/S0022-510X(97)00218-9
13. Sakakura, M, Tanabe, M, Mori, M, Takahashi, H, and Mio, K. Structural bases for the Charcot–Marie–Tooth disease induced by single amino acid substitutions of myelin protein zero. Structure. (2023) 31:1452–1462.e4. doi: 10.1016/j.str.2023.08.016
14. Miller, DT, Lee, K, Abul-Husn, NS, Amendola, LM, Brothers, K, Chung, WK, et al. ACMG SF v3.2 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. (2023) 25:100866. doi: 10.1016/j.gim.2023.100866
15. Young, P, De Jonghe, P, Stögbauer, F, and Butterfass-Bahloul, T. Treatment for Charcot–Marie–Tooth disease. Cochrane Database Syst Rev. (2008) 2008:CD006052. doi: 10.1002/14651858.CD006052.pub2
16. Mastaglia, FL, Nowak, KJ, Stell, R, Phillips, BA, Edmondston, JE, Dorosz, SM, et al. Novel mutation in the myelin protein zero gene in a family with intermediate hereditary motor and sensory neuropathy. J Neurol Neurosurg Psychiatry. (1999) 67:174–9. doi: 10.1136/jnnp.67.2.174
17. Howard, P, Feely, SME, Grider, T, Bacha, A, Scarlato, M, Fazio, R, et al. Loss of function MPZ mutation causes milder CMT1B neuropathy. J Peripher Nerv Syst. (2021) 26:177–83. doi: 10.1111/jns.12452
18. Lei, L, Han, D, Gong, S, Zheng, J, and Xu, J. Mpz gene suppression by shRNA increases Schwann cell apoptosis in vitro. Neurol Sci. (2010) 31:603–8. doi: 10.1007/s10072-010-0341-2
19. Lemke, G, and Axel, R. Isolation and sequence of a cDNA encoding the major structural protein of peripheral myelin. Cell. (1985) 40:501–8. doi: 10.1016/0092-8674(85)90198-9
20. Ptak, CP, Peterson, TA, Hopkins, JB, Ahern, CA, Shy, ME, and Piper, RC. Homomeric interactions of the MPZ Ig domain and their relation to Charcot–Marie–Tooth disease. Brain. (2023) 146:5110–23. doi: 10.1093/brain/awad258
21. Veneri, FA, Prada, V, Mastrangelo, R, Ferri, C, Nobbio, L, Passalacqua, M, et al. A novel mouse model of CMT1B identifies hyperglycosylation as a new pathogenetic mechanism. Hum Mol Genet. (2022) 31:4255–74. doi: 10.1093/hmg/ddac170
22. Giese, KP, Martini, R, Lemke, G, Soriano, P, and Schachner, M. Mouse P0 gene disruption leads to hypomyelination, abnormal expression of recognition molecules, and degeneration of myelin and axons. Cell. (1992) 71:565–76. doi: 10.1016/0092-8674(92)90591-Y
23. Wrabetz, L, Feltri, ML, Quattrini, A, Imperiale, D, Previtali, S, D'Antonio, M, et al. P0 glycoprotein overexpression causes congenital hypomyelination of peripheral nerves. J Cell Biol. (2000) 148:1021–34. doi: 10.1083/jcb.148.5.1021
Keywords: Myelin Protein Zero , Charcot–Marie–Tooth disease, CMT dominant intermediate D, missense variant, whole exome sequencing
Citation: Cao G-H, Zhao M-F, Dong Y, Fan L-L, Liu Y-H, Deng Y and Tang L-L (2024) Case report: A novel variant (H49N) in Myelin Protein Zero gene is responsible for a patient with Charcot–Marie–Tooth disease. Front. Neurol. 15:1319962. doi: 10.3389/fneur.2024.1319962
Edited by:
Huifang Shang, Sichuan University, ChinaReviewed by:
Masayoshi Sakakura, Yokohama City University, Tsurumi, JapanChristopher P. Ptak, The University of Iowa, United States
Copyright © 2024 Cao, Zhao, Dong, Fan, Liu, Deng and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi-Hui Liu, Y3lnbDA3MjJAZ21haWwuY29t; Yao Deng, NjE5MDY1Mzc0QHFxLmNvbQ==; Lu-Lu Tang, bGx0YW5nQGNzdS5lZHUuY24=
†These authors have contributed equally to this work