Abstract
Background:
Myocardial injury is a common complication of aneurysmal subarachnoid hemorrhage (aSAH) and is associated with poor outcomes. While RNA plays a critical role in pathophysiological processes, its expression patterns and functions in myocardial injury after aSAH (aSAH-MI) remain poorly understood.
Objective:
To construct the RNA expression profile of aSAH-MI patients, explore their biological functions, and establish a gene expression regulatory network for aSAH-MI. These findings provide a theoretical basis for understanding the RNA-level mechanisms underlying aSAH-MI.
Methods:
This study included 12 patients, comprising 6 aSAH-MI patients and 6 aSAH-nonMI patients (aSAH patients without myocardial injury). RNA sequencing was performed on three patients from each group to construct an RNA expression matrix. Differentially expressed genes (lncRNAs, miRNAs, mRNAs) were identified using the limma package in R. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses were performed. miRNA, lncRNA, and mRNA interactions were predicted using miRanda and RNAhybrid. An lncRNA-miRNA-mRNA interaction network was constructed with Cytoscape, and qRT-PCR validated selected genes in an additional six patients.
Results:
In aSAH-MI patients, 617 lncRNAs, 20 miRNAs, and 510 mRNAs were significantly differentially expressed, with 258, 13, and 244 being upregulated, and 359, 7, and 266 being downregulated, respectively (P < 0.05). Bioinformatic analysis revealed that the differentially expressed mRNAs were involved in biological processes such as ion transport, immune regulation, and myocardial contraction, and were associated with pathways related to vasodilation, nerve conduction, and cardiac function regulation. ceRNA analysis identified hsa-miR-4707-3p and hsa-miR-25-5p as potential network hubs. The lncRNAs with the highest connectivity were CELSR1-204, SLCO2B1-212, AEN-204, PPFIA4-205, and MIAT-219, while the mRNAs with the highest connectivity were CHI3L1, ADORA2A, PAX8, VWA3B, and KCNE1. These findings suggest these differentially expressed genes may serve as key regulators in mediating aSAH-MI. Validation through qRT-PCR in an additional cohort of six subjects confirmed the differential expression of selected genes.
Conclusions:
This study successfully constructed the RNA expression profiles in the blood of patients with aSAH-MI through transcriptome sequencing, identifying significant differentially expressed miRNAs, mRNAs, and lncRNAs. Bioinformatic analysis suggests these genes may play critical roles in the pathogenesis of aSAH-MI.
1 Introduction
Subarachnoid hemorrhage (SAH) is a life-threatening neurological emergency, with aneurysmal subarachnoid hemorrhage (aSAH) accounting for approximately 85% of spontaneous cases due to the rupture of intracranial aneurysms (1–3). Numerous studies have identified advanced age, elevated clinical severity scores, wide-necked aneurysms, and cardiopulmonary complications as independent predictors of poor outcomes in aSAH patients (4). Among these prognostic factors, cardiac complications are particularly critical yet often underrecognized (5). This is largely because such complications frequently present with subtle or non-specific symptoms, which may be overshadowed by more severe neurological impairments. Despite this, the incidence of cardiac complications—including electrocardiographic abnormalities, myocardial injury, and cardiac dysfunction—is notably high in aSAH patients and is closely associated with adverse outcomes.
Myocardial injury is defined by elevated cardiac troponin (cTn) levels exceeding the 99th percentile upper reference limit specific to the assay used (6). Myocardial injury following aSAH (aSAH-MI) represents a common manifestation of neurocardiac syndrome, with 20%−40% of aSAH patients exhibiting elevated cTn levels (7). This elevation is associated with increased clinical severity, higher rates of neurological complications, greater mortality, and poorer overall outcomes. Despite advances in secondary prevention, the incidence of post-stroke cardiac events remains significant, highlighting the urgent need to elucidate the mechanisms underlying aSAH-MI.
Advances in genomics have underscored the importance of the relationship between genes and disease. The majority of the human transcriptome consists of non-coding RNA (ncRNA), which, despite not encoding proteins, plays critical roles in various biological processes (8). Non-coding RNAs are classified based on length into short (< 200 nucleotides) and long (>200 nucleotides) categories, including microRNAs (miRNAs) and long non-coding RNAs (lncRNAs) (9, 10). These ncRNAs are essential for maintaining cellular homeostasis and have been implicated in a wide range of diseases. They are also thought to act as mediators of inter-organ and inter-cellular communication, although their roles in neurocardiac syndromes like aSAH-MI remain poorly understood.
Competing endogenous RNA (ceRNA) refers to RNA molecules that participate in complex transcriptional regulation networks by binding to microRNA response elements, thereby modulating gene expression (11). The analysis of ceRNA networks provides a novel framework for understanding RNA interactions and their regulatory functions in gene expression. Transcriptome sequencing (RNA-Seq) offers a comprehensive approach to analyze differentially expressed mRNAs and ncRNAs, facilitating the construction of ceRNA networks that can illuminate gene regulatory mechanisms in disease states. However, studies employing RNA-Seq to investigate aSAH-MI are currently limited.
This study aims to construct the RNA expression profile of aSAH-MI patients, focusing on lncRNAs, miRNAs, and mRNAs. By utilizing RNA sequencing, we will develop an RNA expression matrix specific to myocardial injury following aSAH. The study will identify significantly differentially expressed genes and conduct extensive bioinformatics analyses, including pathway, functional, and ceRNA network analyses, to establish a comprehensive gene expression regulatory network for aSAH-MI.
2 Materials and methods
2.1 Patients and specimens
This study involved the collection of peripheral blood samples from 12 patients diagnosed with aneurysmal subarachnoid hemorrhage (aSAH), of which 6 patients developed secondary myocardial injury (aSAH-MI group), while the remaining 6 patients did not exhibit myocardial injury (aSAH-nonMI group). The diagnosis of myocardial injury secondary to aSAH was based on the criteria established by the American Heart Association/American Stroke Association (AHA/ASA) (12, 13). All participants were inpatients at the First Affiliated Hospital of Guangxi Medical University from October 2022 to October 2023. Informed consent was obtained from all participants or their legal representatives in compliance with the ethical standards outlined in the Declaration of Helsinki. Exclusion criteria included a history of heart disease, a history of cancer, severe renal or hepatic dysfunction, or other significant comorbid conditions. Four milliliters of fresh peripheral blood samples were collected using EDTA tubes, immediately mixed with five volumes of TRIzol reagent, and thoroughly homogenized. The samples were then transported on dry ice to a −80°C freezer for storage. The entire process was completed within 30 min to preserve RNA integrity.
2.2 Total RNA isolation
Total RNA was isolated from whole blood samples using the TRIzol® reagent (Takara, Japan) following the manufacturer's protocol. The RNA concentration and purity were determined using a NanoDrop ND-2000 spectrophotometer (Thermo Scientific, USA), and the RNA integrity was evaluated through 1% agarose gel electrophoresis as well as with the Agilent 5400 Bioanalyzer system (Agilent Technologies, USA). All procedures, including centrifugation and plasma aliquoting, were conducted under temperature-controlled conditions (4°C) to maintain sample stability. The separated plasma was aliquoted into cryotubes and immediately frozen at −80°C to preserve integrity. Until further processing, all samples were stored in a biobank facility at −80°C, with temperature monitored continuously to ensure optimal conditions.
2.3 Library preparation for RNA sequencing
Total RNA with an RNA integrity number (RIN) ≥7.0, extracted from blood samples, was used for transcriptome library construction. The RNA sequencing libraries were prepared and sequenced by Beijing Novogene Bioinformatics Technology (http://www.novogene.com). Five micrograms of RNA from each peripheral blood sample served as the starting material for library preparation. RNA sequencing was conducted using next-generation high-throughput sequencing technology, with two distinct types of libraries constructed from the same sample to capture a broad spectrum of RNA species. These libraries included a ribosomal RNA-depleted strand-specific library for long RNA fragments, enabling the sequencing of lncRNA, circRNA, and mRNA, and a small RNA library for short RNA fragments, facilitating the sequencing of miRNA and other small RNAs.
2.4 Clustering and sequencing
Clustering of the indexed samples was performed using the TruSeq PE Cluster Kit v3-cBot-HS (Illumina) on the cBot Cluster Generation System, following the manufacturer's protocol. Post-cluster generation, libraries were sequenced on the Illumina HiSeq™ 4,000 platform (Illumina, San Diego, CA, USA) with 150 bp single-end reads. The raw sequencing data, obtained in FASTQ format, were processed using an in-house Perl script. To ensure the integrity of downstream analyses, the raw reads were subjected to a stringent filtering process. This included the removal of low-quality reads, adapter sequences, short reads, and sequences with a high rate of ambiguous bases (N). The filtered high-quality clean data were evaluated using quality metrics such as Q20, Q30, and GC content. Only data meeting these quality standards were retained for subsequent analyses.
2.5 Reference genome
The reference genome and gene model annotation files were downloaded from a recognized genomic database. The reference genome was indexed using Bowtie2 v2.2.8, and paired-end clean reads were aligned to the reference genome utilizing Bowtie2, ensuring high alignment accuracy (14).
2.6 Differential expression analysis
Differential expression analysis was performed using the DESeq R package (version 4.2.1), which implements statistical routines for assessing differential expression in digital gene expression (DGE) data based on a negative binomial distribution. To control for false discovery, p-values were adjusted using the Benjamini & Hochberg method. Genes with an adjusted p-value (q-value) < 0.05 were considered differentially expressed (DE). All analyses were conducted using the default parameters of DESeq to ensure consistency and reproducibility.
2.7 Bioinformatic analysis of differentially expressed mRNA
Gene Ontology (GO) enrichment analysis was performed on the differentially expressed (DE) mRNA using the GOseq R package to predict gene functions. These functions were categorized into three primary subgroups: Biological Process (BP), Molecular Function (MF), and Cellular Component (CC). The results of the GO enrichment analysis were visualized using DE mRNA GO Functional Enrichment bubble plot to represent the most enriched categories. Additionally, pathways associated with the DE mRNA were identified through the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis.
Target genes of significantly differentially expressed miRNAs were predicted using two bioinformatics tools: miRanda and RNAhybrid. The intersection of significantly differentially expressed miRNAs and mRNAs was determined based on the principle of miRNA-mediated mRNA inhibition. Consequently, significantly downregulated miRNAs paired with significantly upregulated mRNAs, and significantly upregulated miRNAs paired with significantly downregulated mRNAs, were identified as potential regulatory pairs.
To explore the regulation of target gene mRNA expression by lncRNA through co-localization or co-expression, we performed an intersection analysis between the target genes of differentially expressed lncRNAs and differentially expressed mRNAs. When the target genes of differentially expressed lncRNAs were also identified as significantly differentially expressed mRNAs across samples, it suggested a higher likelihood of direct or indirect regulatory effects of the lncRNA on the DE mRNA. The union of target genes influenced by both co-localization and co-expression with DE mRNAs was selected for further analysis. The prediction of miRNAs targeting lncRNAs was also conducted using the miRanda software. Finally, Cytoscape software was employed to construct a regulatory interaction network diagram illustrating lncRNA-miRNA-mRNA interactions.
2.8 qPCR validation of differential RNA
To validate the differentially expressed (DE) RNAs identified by RNA-seq, quantitative real-time PCR (qRT-PCR) was performed using six total RNA samples. A total of nine DE RNAs were selected for validation. For the analysis of lncRNAs and mRNAs, reaction mixtures were prepared following the instructions provided with the PrimeScript™ RT reagent Kit, which includes gDNA Eraser. Complementary DNA (cDNA) templates were synthesized from 2 μg of total RNA. qRT-PCR amplification was conducted using TB Green™ Premix Ex Taq™ II (Takara, Japan) on an ABI 7500 real-time PCR system (Applied Biosystems, USA). The optimal reaction conditions were set as follows: initial denaturation at 95°C for 30 seconds, followed by 40 cycles of denaturation at 95°C for 5 seconds, and annealing/extension at 60°C for 30 seconds. The specificity of the amplification was confirmed by a single peak in the melting curve analysis, indicating accurate and specific amplification. The verification analysis of miRNAs were performed according to the instructions of the miRNA reverse transcription kit (Takara, Japan). All primers used for the RNA analysis were designed and synthesized by Genesis Biotechnology (Nanning, China). Actin was employed as an internal control, and the relative gene expression levels were calculated using the 2–ΔΔCt method.
2.9 Statistical analysis
All statistical analyses were conducted using SPSS software (version 26.0). Graphical representations were created using GraphPad Prism software (version 9.5) and Cytoscape (version 3.10.0). Image processing were performed with Adobe Photoshop (version 25.0.0) and sangerbox online tool (http://sangerbox.com/). For comparisons between two independent groups, Student's t-test was employed. Data are presented as mean ± standard error (SE). Each experiment was conducted in triplicate to ensure reproducibility. A p-value of < 0.05 was considered statistically significant.
3 Results
3.1 Clinical information of the study population
In the experimental group (aSAH-MI), the initial cardiac troponin I (cTnI) levels of all six samples exceeded the 99th percentile upper reference limit (0.033 ng/ml), with values of 1.045, 0.638, 0.278, 0.211, 0.203, and 0.058 ng/ml, respectively. Subsequent re-examinations showed a decrease in cTnI levels to 0.170, 0.365, 0.236, 0.037, 0.069, and 0.002 ng/ml, respectively, corresponding to reductions of 0.875, 0.273, 0.042, 0.174, 0.134, and 0.056 ng/ml. The experimental group (aSAH-MI) meets the diagnostic criteria for myocardial injury. In contrast, all six samples in the control group (aSAH-nonMI) had cTnI levels below the 99th percentile upper reference limit (0.033 ng/ml), with values of 0.015, 0.010, 0.007, 0.003, 0.002, and 0.000 ng/ml, respectively, indicating that the control group (aSAH-nonMI) does not meet the diagnostic criteria for myocardial injury. There were no statistically significant differences in baseline characteristics between the two groups, including age, sex, hypertension, and diabetes (all P > 0.05; see Table 1), indicating comparability of the groups. Table 1 presents key clinical information for both the experimental and control groups.
Table 1
| Variables | MI | Control | p-value |
|---|---|---|---|
| Age (years) | 60(47, 62.75) | 41.75(41.75, 61.25) | 0.334 |
| Male gender (n (%)) | 2(33.33%) | 2(33.33%) | 1 |
| History of heart disease (n (%)) | 0(0) | 0(0) | 1 |
| Hypertension (n (%)) | 4(66.7%) | 3(50%) | 0.575 |
| Diabetes (n (%)) | 1(16.7%) | 0(0) | 0.317 |
| Neoplasm (n (%)) | 0(0) | 0(0) | 1 |
| Acute respiratory failure (n (%)) | 0(0) | 0(0) | 1 |
| Uncontrolled metabolic disorders (n (%)) | 0(0) | 0(0) | 1 |
| Immune system diseases (n (%)) | 0(0) | 0(0) | 1 |
| Severe infections (n (%)) | 0(0) | 0(0) | 1 |
| Current smoking (n (%)) | 0(0) | 1(16.7%) | 0.317 |
| Alcohol intake (n (%)) | 0(0) | 1(16.7%) | 0.317 |
| Liver dysfunction (AST or ALT≥120U/L) (n (%)) | 0(0) | 0(0) | 1 |
| Renal failure (creatine≥120μmol/L) (n (%)) | 0(0) | 0(0) | 1 |
| LVEF < 50% (n (%)) | 0(0) | 0(0) | 1 |
| Arrhythmology (tachycardia or bradycardia) (n (%)) | 2(33.33%) | 2(33.33%) | 1 |
| GCS (points) | 14(12.25, 15) | 15(11.25, 15) | 0.432 |
| Hunt-Hess grades≥III ratio (n (%)) | 3(50%) | 2(33.33%) | 0.575 |
| WFNS grades≥III ratio (n (%)) | 3(50%) | 2(33.33%) | 0.575 |
| Aneurysm location | |||
| ICA (n (%)) | 3(50%) | 2(33.33%) | 0.575 |
| MCA (n (%)) | 2(33.33%) | 2(33.33%) | 1 |
| ACA (n (%)) | 1(16.7%) | 0(0) | 0.317 |
| AComA (n (%)) | 0(0) | 2(33.33%) | 0.138 |
Clinical information of the study population.
AST/ALT, Aspartate aminotransferase/Alanine aminotransferase; LVEF, Left ventricular ejection fraction; GCS, Glasgow coma scale; WFNS, World Federation of Neurosurgical Societies; ICA, Internal Carotid Artery, MCA, middle cerebral artery; ACA, arteriae cerebral anterior; AComA, anterior communicating artery.
3.2 Expression profiles of lncRNA, miRNA, and mRNA in aSAH-MI patients
Significant DE RNAs between aSAH-MI patients and controls were identified using a p-value threshold of < 0.05. A total of 617 lncRNAs, 20 miRNAs, and 510 mRNAs were found to be significantly differentially expressed. Among these, 258 lncRNAs, 13 miRNAs, and 244 mRNAs were significantly upregulated, while 359 lncRNAs, 7 miRNAs, and 266 mRNAs were significantly downregulated in aSAH-MI patients compared to controls. Volcano plots visualized the significant DE RNAs in aSAH-MI, with red dots representing significantly upregulated RNAs and green dots representing downregulated RNAs (Figures 1A, 2A, 3A). Hierarchical clustering analysis revealed that the RNA expression patterns of aSAH-MI patients were distinguishable from those of the control group (Figures 1B, 2B, 3B).
Figure 1
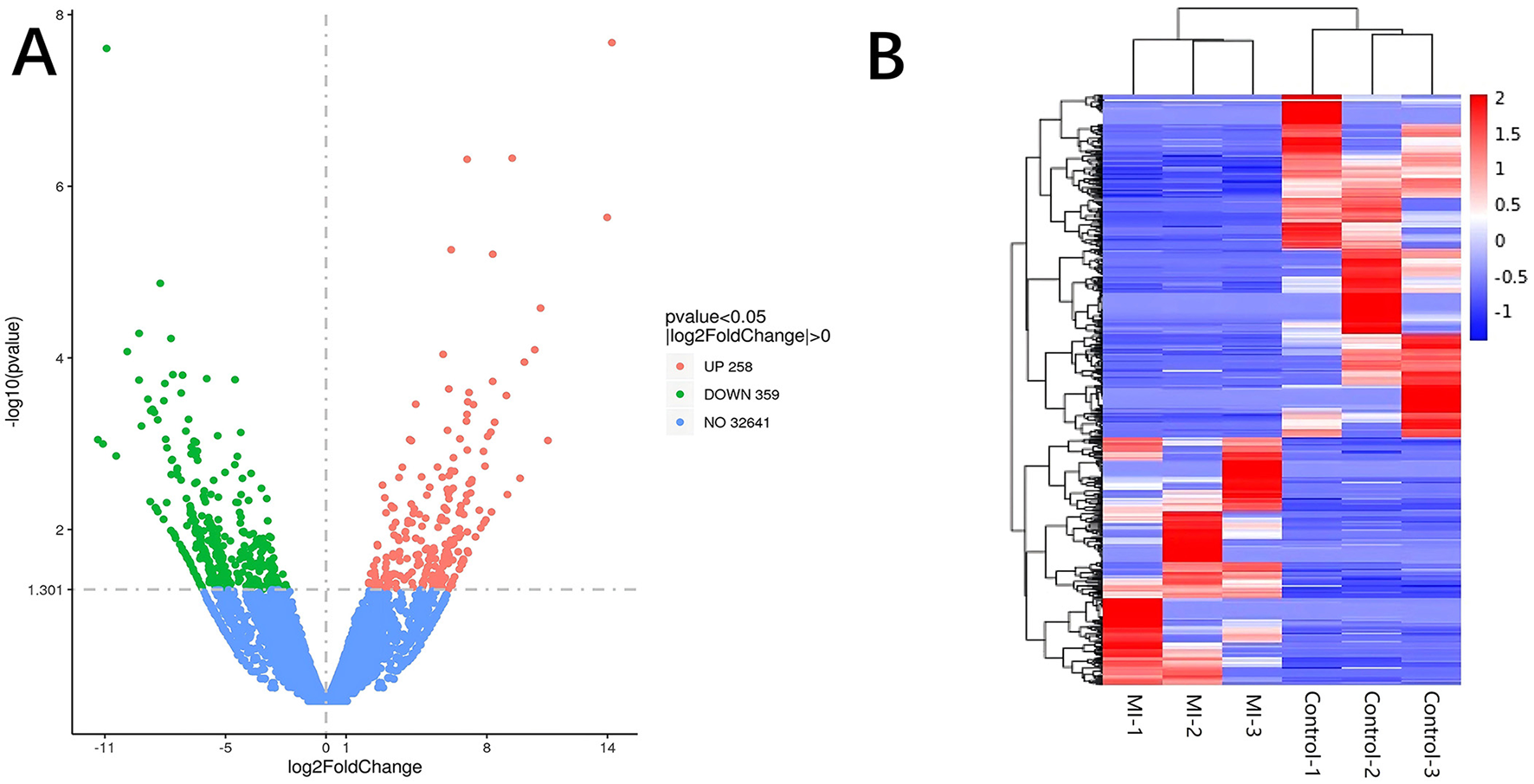
DE lncRNA volcano plot (A) and clustering heatmap (B).
Figure 2
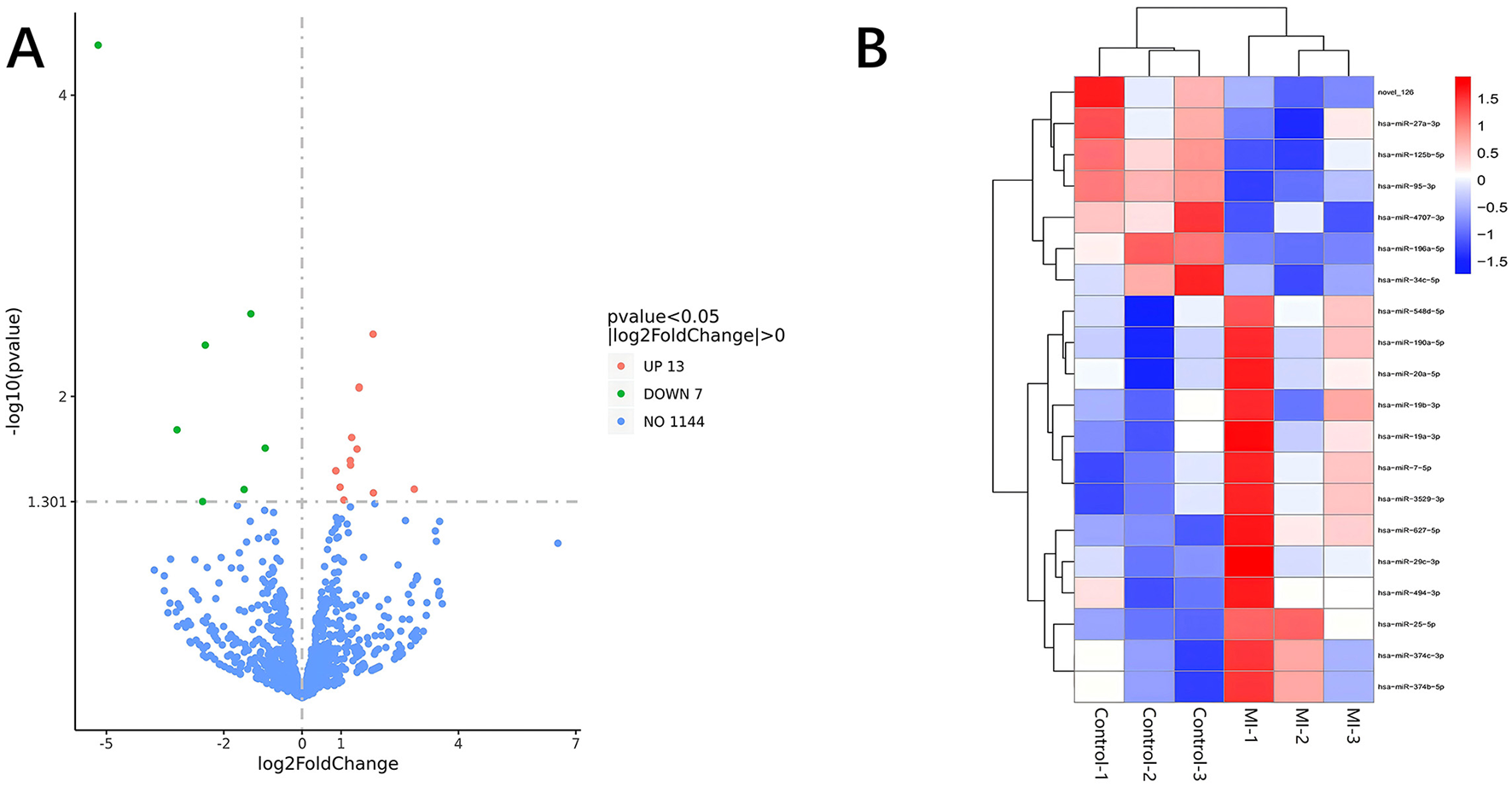
DE miRNA volcano plot (A) and clustering heatmap (B).
Figure 3
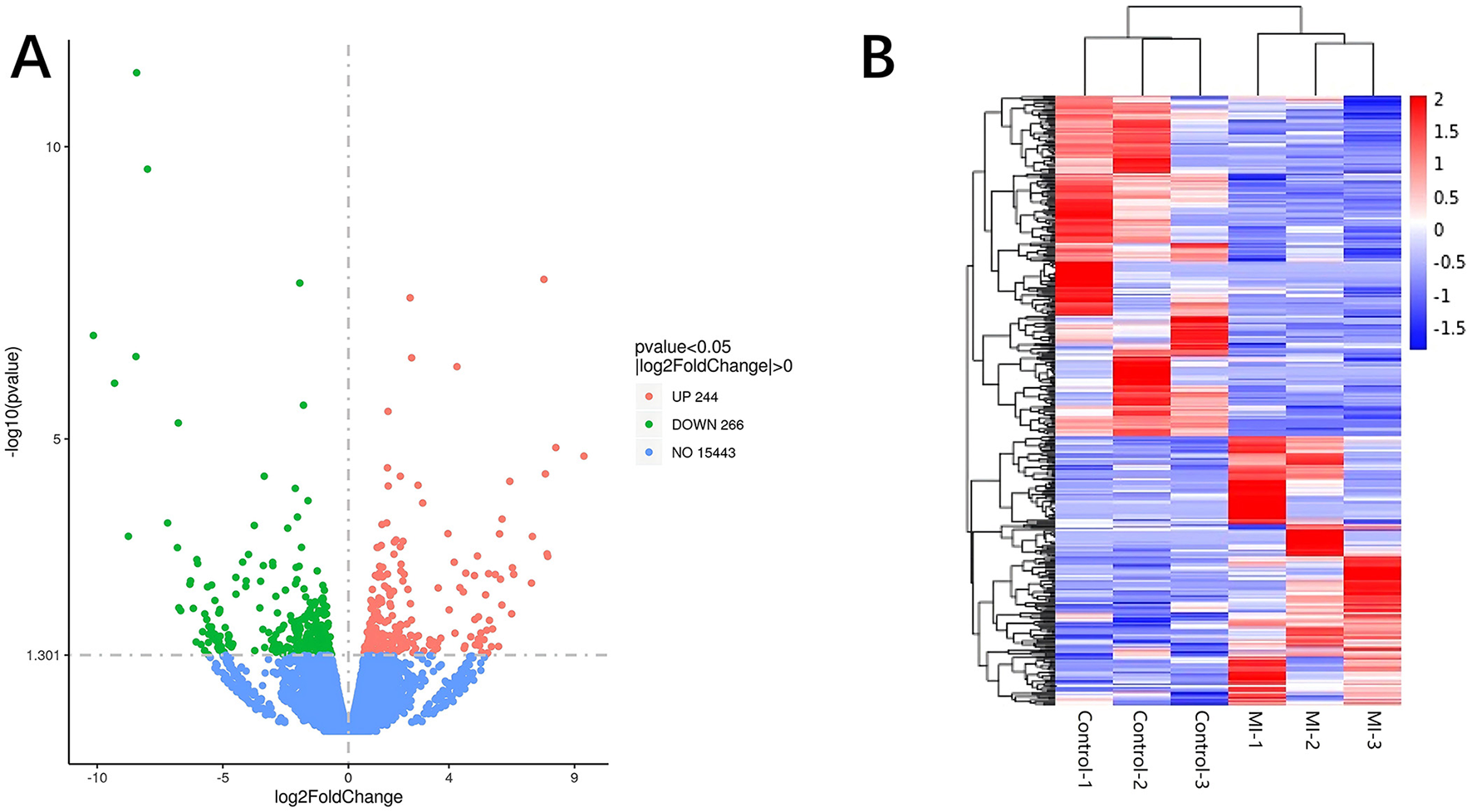
DE mRNA volcano plot (A) and clustering heatmap (B).
3.3 GO and KEGG analyses of mRNA
Differentially expressed (DE) mRNAs identified in aSAH-MI patients were subjected to Gene Ontology (GO) annotation and enrichment analysis, as well as Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, to elucidate their potential biological roles. GO analysis categorized the DE mRNAs into three main classes: Biological Process (BP), Cellular Component (CC), and Molecular Function (MF). The analysis revealed that these mRNAs were significantly associated with a range of biological processes, including ion transport, immune regulation, and myocardial cell contraction. Regarding cellular components, the dysregulated mRNAs were predominantly localized to cytosolic parts, intracellular organelles, protein complexes, and muscle fibers. The molecular functions of these mRNAs were primarily involved in oxygen carrier activity, oxygen binding, oxidoreductase activity, ion channel activity, protein binding, cytokine binding, and enzyme activity (Figure 4). These findings suggest significant alterations in cellular and molecular functions, as well as peripheral blood metabolism, in aSAH-MI patients compared to controls. Such changes are closely linked to the pathological mechanisms underlying brain injury.
Figure 4
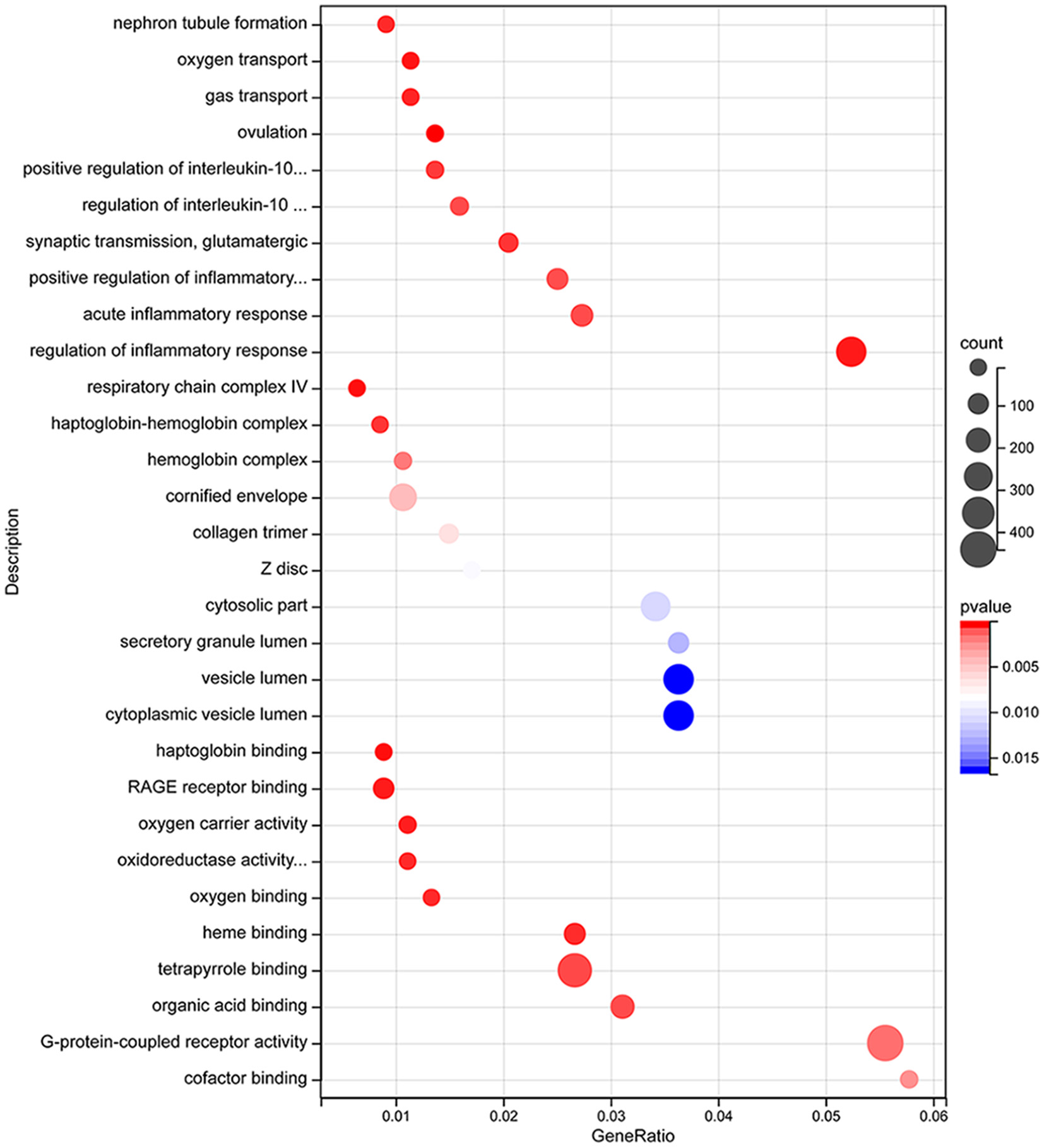
DE mRNA GO functional enrichment bubble plot.
Subsequent KEGG pathway enrichment analysis further highlighted the involvement of DE mRNAs in various signaling pathways, particularly those related to vascular dilation and constriction, neurotransmission, and the regulation of cardiac function (Figure 5). These pathways, which include those regulating endothelial function and neurotransmitter release, are critical in maintaining cardiovascular homeostasis and could play significant roles in the pathophysiology of myocardial injury following aSAH.
Figure 5
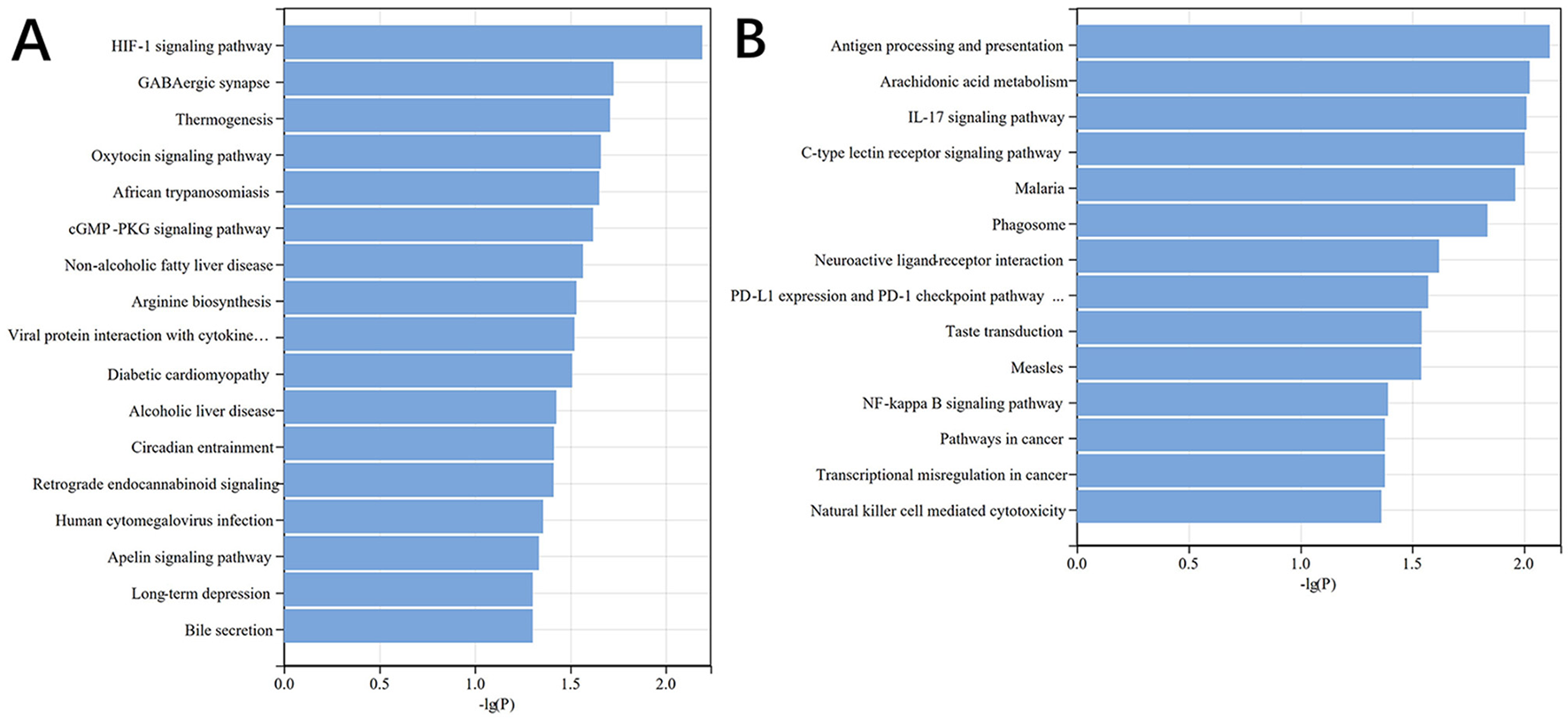
DE mRNA KEGG histogram. (A) KEGG pathways associated with upregulated genes, (B) KEGG pathways associated with downregulated genes.
3.4 Construction and analysis of the lncRNA-miRNA-mRNA network
Increasing evidence supports the role of long non-coding RNAs (lncRNAs) as miRNA sponges, regulators of splicing and transcription, and modulators of host gene expression through the sequestration of specific miRNAs. In this study, we utilized the miRanda and RNAhybrid databases to predict miRNA target genes. The intersection of predictions from both databases, combined with the significantly differentially expressed mRNAs identified through RNA sequencing, was selected as the final set of target mRNAs. By analyzing sequence complementarity between competing endogenous RNAs (ceRNAs), the negative correlation in their expression levels, and the regulatory relationships among lncRNAs, miRNAs, and mRNAs, we constructed an lncRNA-miRNA-mRNA network. This network encompassed 287 lncRNAs, 9 miRNAs, and 63 mRNAs, forming 2861 ceRNA interactions (Figure 6). Within the network, the lncRNAs exhibiting the highest degree of connectivity were CELSR1-204, SLCO2B1-212, AEN-204, PPFIA4-205, and MIAT-219. The miRNA with the greatest connectivity was hsa-miR-4707-3p, while the mRNAs with the highest connectivity included CHI3L1, ADORA2A, PAX8, VWA3B, and KCNE1. Further functional analysis of the mRNAs within this network revealed their involvement in key biological processes, including glial cell development and activation, cardiac conduction, and the regulation of NF-κB signaling (Figure 7). Additionally, these genes were linked to critical signaling pathways such as the Wnt signaling pathway (Figure 8).
Figure 6
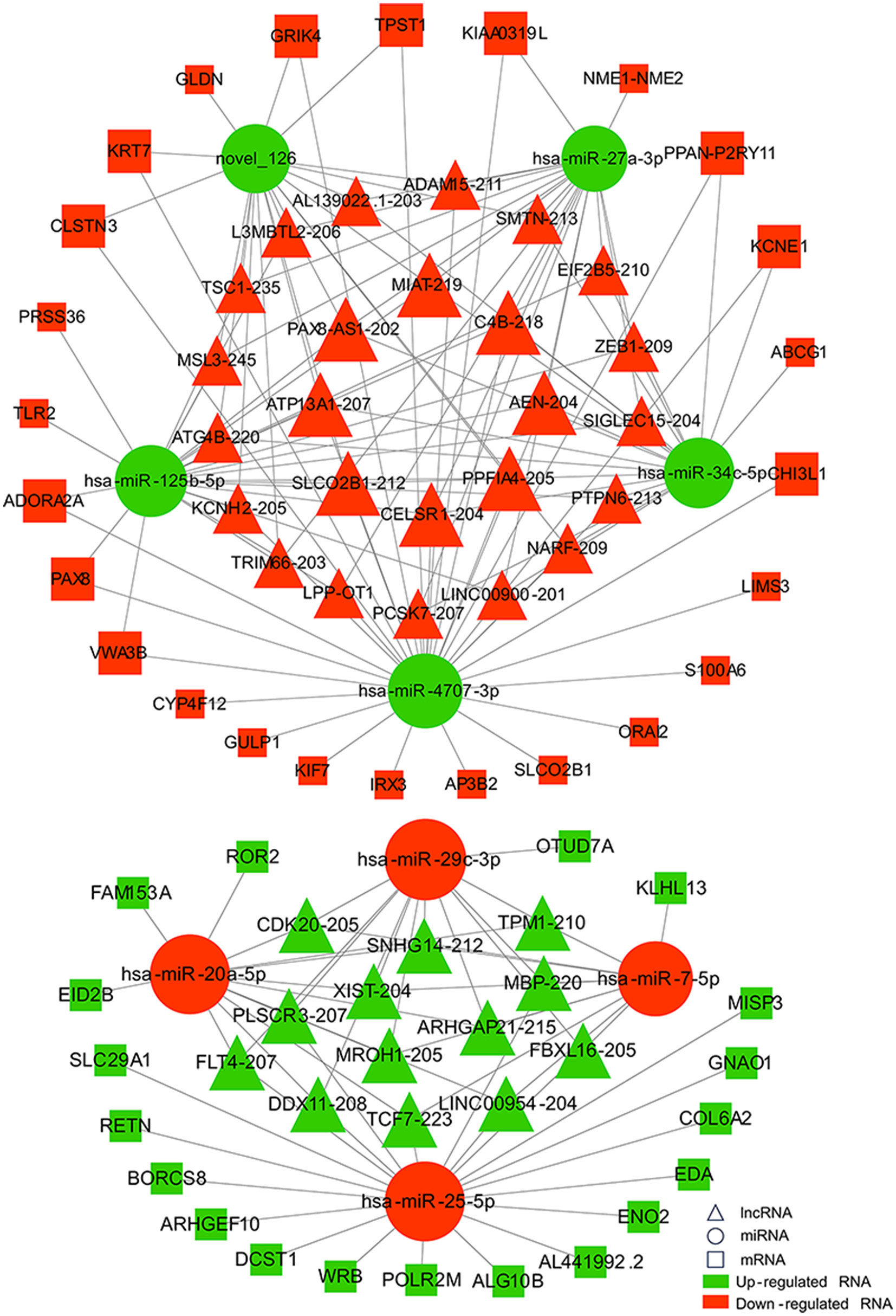
Network diagram of lncRNA-miRNA-mRNA interactions. Triangles represent lncRNAs, circles represent miRNAs, and squares represent mRNAs. Red indicates up-regulated RNAs, and green indicates down-regulated RNAs.
Figure 7
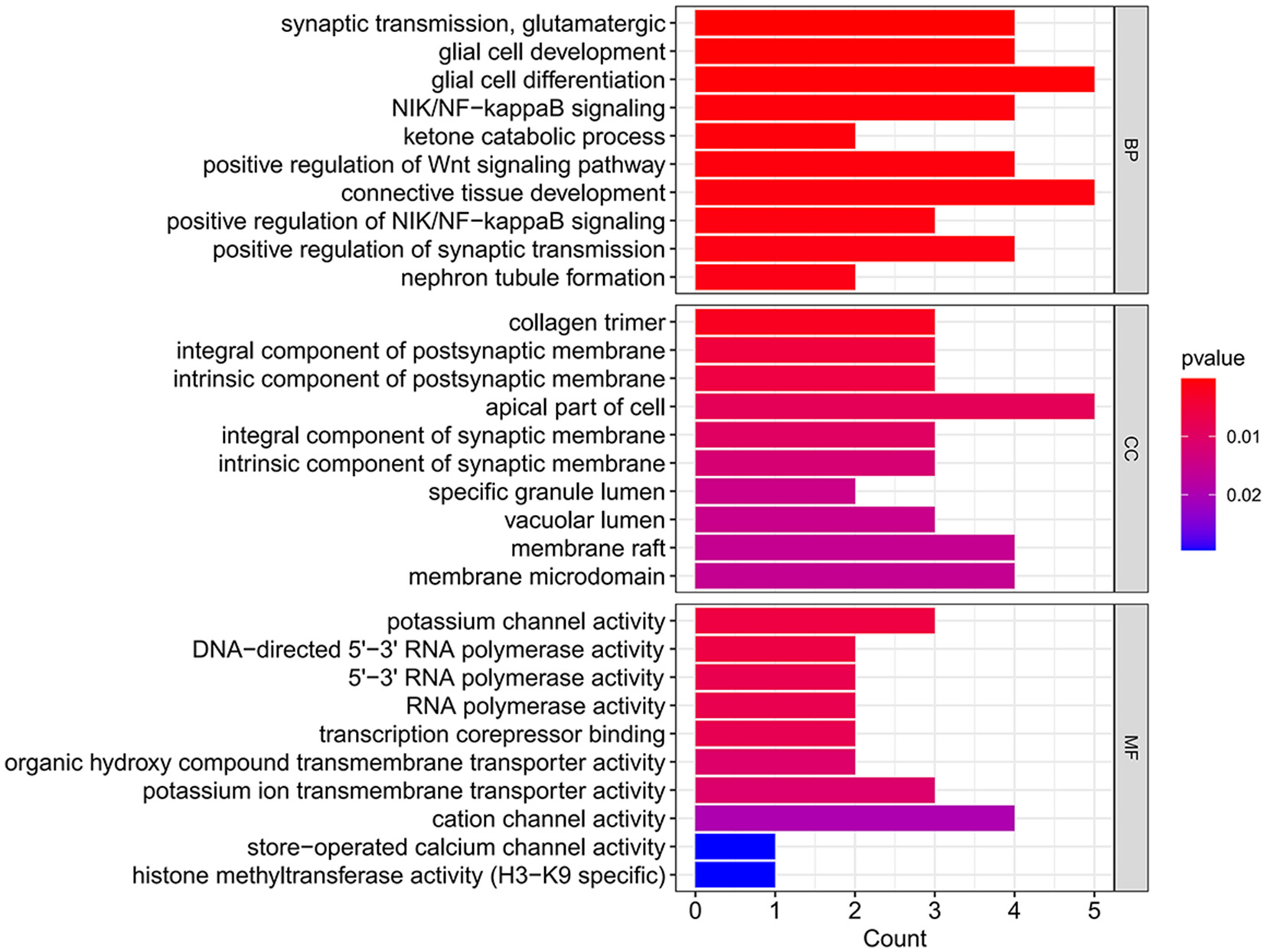
GO analysis histogram of DE mRNAs in the ceRNA network.
Figure 8
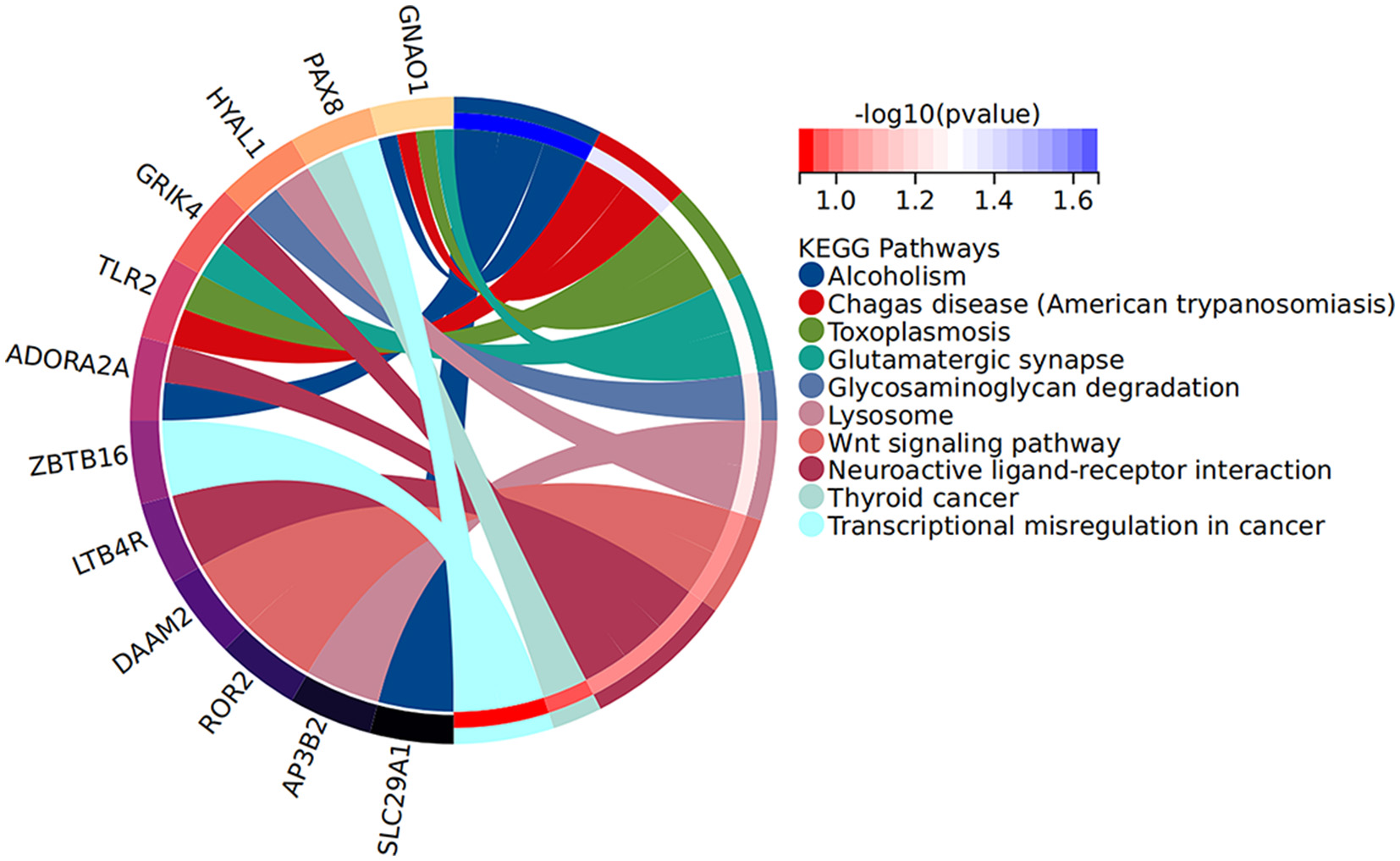
KEGG pathway histogram of DE mRNAs in the ceRNA network.
3.5 Validation of selected RNA expression levels
To confirm the accuracy of the RNA-Seq data, two upregulated and one downregulated RNA were selected from each of the three classes of differentially expressed (DE) RNAs (lncRNAs, miRNAs, and mRNAs). Quantitative real-time PCR (qRT-PCR) analysis was performed to assess the expression levels of these RNAs in three patients with aSAH-MI and three control subjects. The qRT-PCR results demonstrated expression patterns consistent with the RNA-Seq data, thereby confirming the reliability of the RNA-Seq findings. This validation reinforces the accuracy of the RNA-Seq analysis, providing robust support for the data's validity (Figure 9).
Figure 9
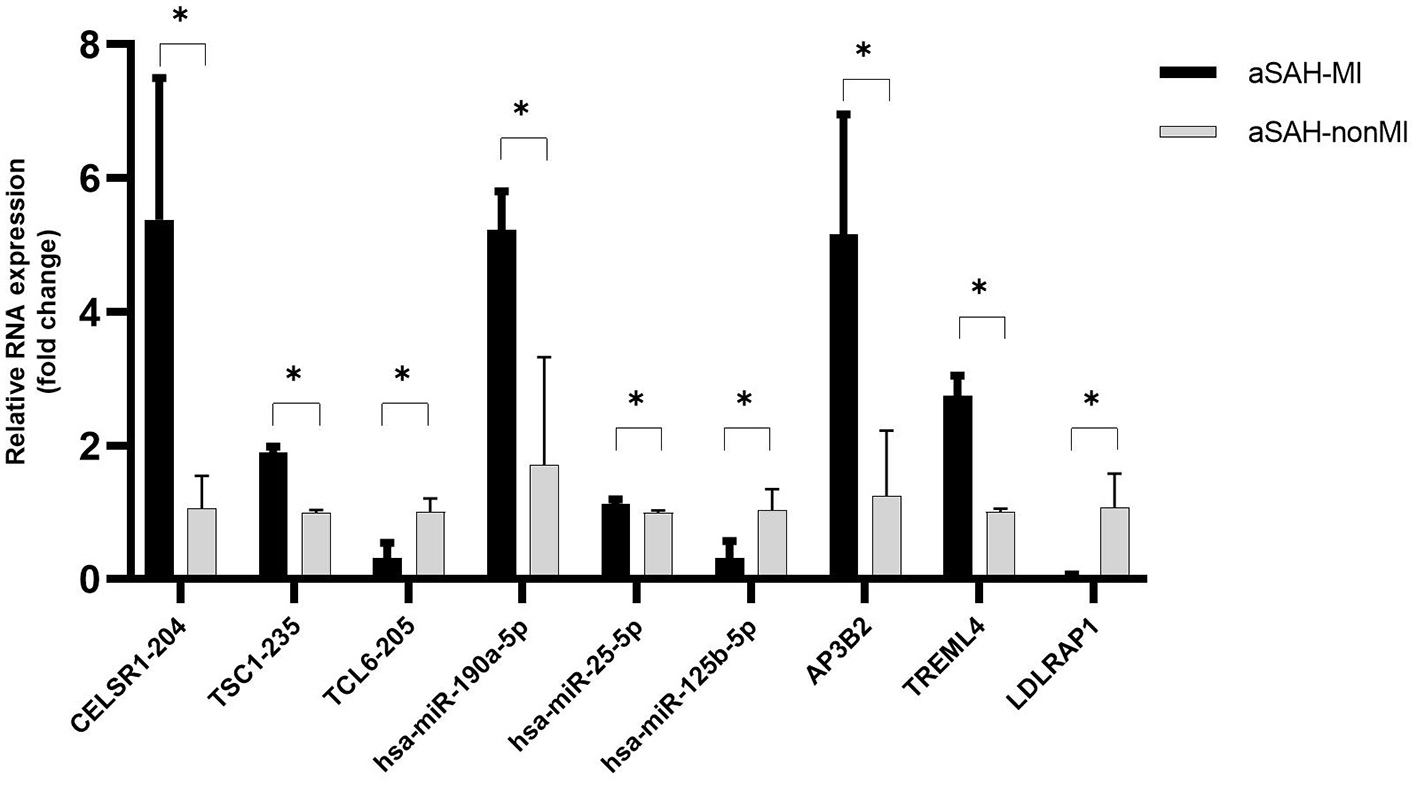
qRT-PCR validation results of DE RNAs. * indicates P < 0.05. The experiment was performed in three biological replicates, and the samples used for PCR validation were different from those used for transcriptome sequencing.
4 Discussion
In this study, we explored the expression profiles of long non-coding RNAs (lncRNAs) and messenger RNAs (mRNAs) associated with myocardial injury following aneurysmal subarachnoid hemorrhage (aSAH) using next-generation sequencing technology. A comprehensive analysis revealed 617 differentially expressed lncRNAs and 510 differentially expressed mRNAs, providing valuable insights into the molecular mechanisms underlying post-aSAH myocardial injury. To further elucidate the functional roles of these differentially expressed genes, we conducted extensive bioinformatics analyses, including Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses, as well as a competing endogenous RNA (ceRNA) network analysis.
The GO analysis identified 754 significantly enriched terms, highlighting the involvement of the differentially expressed mRNAs in key biological processes such as ion transport, immune regulation, myocardial cell contraction, and cardiac conduction. KEGG pathway analysis further revealed 31 significantly enriched pathways, implicating signal transduction processes related to vascular tone, neurotransmission, and cardiac function, including the HIF-1, Apelin, cGMP-PKG, IL-17, and NF-κB signaling pathways. Notably, the cGMP-PKG signaling pathway, which mediates interactions between cyclic guanosine monophosphate (cGMP) and protein kinase G (PKG), plays a crucial role in cardiac hypertrophy and heart failure management (15, 16). Meanwhile, NF-κB signaling pathway, as a crucial cellular signaling cascade, participates in various biological processes including immune response and apoptosis. Previous studies have identified NF-κB as a key mediator in aneurysm formation (17). Multiple genes mediate early brain injury and neuroinflammation following subarachnoid hemorrhage through the NF-κB signaling pathway (18, 19). The observed downregulation of the cGMP-PKG pathway and upregulation of the NF-κB pathway in this study underscore their potential roles in the pathogenesis of myocardial injury following aSAH. Future studies should focus on experimentally validating the involvement of these signaling pathways in this context.
LncRNAs are increasingly recognized for their ability to function as miRNA sponges, thereby modulating mRNA expression by sequestering specific miRNAs (20–22). The intricate interactions between mRNAs and ncRNAs were further explored through the construction of a ceRNA network in this study, which included 287 lncRNAs, 9 miRNAs, and 63 mRNAs, forming a total of 2,861 lncRNA-miRNA-mRNA interactions. This network suggests that differentially expressed lncRNAs may regulate gene transcription by acting as miRNA sponges, contributing to the development of myocardial injury after aSAH.
In the ceRNA network we constructed, CHI3L1 is one of the mRNAs with the highest degree, and its expression is upregulated in this study. CHI3L1 (Chitinase-3-like protein 1) is a gene encoding a secretory glycoprotein, and its encoded protein, also known as YKL-40, can be synthesized and secreted by astrocytes (23). Genome-wide association studies have shown that elevated levels of YKL-40 are associated with an increased risk of large artery stroke (24). In a prospective study involving 10,472 stroke patients, high levels of YKL-40 were also observed to be independently associated with recurrent stroke and adverse functional outcomes (25). Another study involving 4,298 patients with coronary artery disease reported that elevated serum YKL-40 levels were associated with more severe coronary artery disease, and higher YKL-40 levels were independent predictors of overall mortality and cardiovascular mortality (26). In this study, upregulation of CHI3L1 was observed, and its role in myocardial injury after aSAH is not yet clear. Subsequent investigations, such as CHI3L1 knockout/overexpression, may help elucidate the role of CHI3L1.
Further analysis of the mRNAs within the ceRNA network indicated their involvement in critical biological processes, including astrocyte development and activation, cardiac conduction, and regulation of the NF-κB signaling pathway. Additionally, these genes were implicated in key signaling pathways, such as the Wnt signaling pathway and Toll-like receptor signaling pathway. The concordance between the functions of the significantly differentially expressed mRNAs and those within the network suggests that these genes may serve as crucial regulators of myocardial injury following aSAH.
A limitation of this study is that it is confined to transcriptomic-level research. Emerging evidence suggests that metabolomics is increasingly valuable in revealing the pathophysiological processes underlying aSAH, it has been used to identify changes in glucose, lipid, and protein metabolism, which are associated with the severity of aSAH and its complications, such as cardiac dysfunction (27). For instance, alterations in taurine and ornithine levels are closely related to functional outcomes (27). Metabolomics can complement transcriptomic studies, offering a holistic view of the molecular and metabolic disturbances in aSAH. Future integrative research is expected to bridge the gap between gene expression changes and physiological responses.
In summary, this study has identified and characterized the expression profiles of lncRNAs and mRNAs associated with myocardial injury after aSAH. Our findings provide a foundation for further exploration into the biological functions and regulatory mechanisms of these differentially expressed genes, offering potential avenues for therapeutic intervention in post-aSAH cardiac complications.
5 Conclusions
This study successfully constructed the RNA expression profile in the blood of patients with myocardial injury following aSAH using transcriptome sequencing and identified significantly differentially expressed miRNAs, mRNAs, and lncRNAs. Through bioinformatic analyses, including GO, KEGG, miRNA-mRNA network analysis, and lncRNA-miRNA-mRNA network analysis, we demonstrated that these differentially expressed genes are likely to play significant roles in the pathogenesis of myocardial injury following aSAH.
Statements
Data availability statement
The data presented in the study are deposited in the Genome Sequence Archive for Human. Small RNA Library, accession: HRA011828, https://bigd.big.ac.cn/gsa-human/browse/HRA011828 and Ribosome-depleted Strand-specific RNA Library, accession: HRA011869, https://bigd.big.ac.cn/gsa-human/browse/HRA011869.
Ethics statement
The studies involving humans were approved by the Ethics Committee of the First Affiliated Hospital of Guangxi Medical University. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
ZW: Conceptualization, Data curation, Formal analysis, Methodology, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. WH: Conceptualization, Data curation, Formal analysis, Methodology, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. WY: Conceptualization, Writing – review & editing, Data curation. SX: Conceptualization, Writing – review & editing, Data curation. SW: Conceptualization, Writing – review & editing, Methodology. BH: Conceptualization, Writing – review & editing, Methodology. PL: Conceptualization, Methodology, Writing – review & editing, Software. YT: Conceptualization, Writing – review & editing, Project administration. CQ: Conceptualization, Writing – review & editing, Funding acquisition, Supervision. YL: Conceptualization, Writing – review & editing, Funding acquisition, Methodology, Validation. ZY: Conceptualization, Funding acquisition, Writing – review & editing, Methodology, Validation.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The research leading to these results received funding from the Joint Project on Regional High-Incidence Diseases Research of Guangxi Natural Science Foundation under Grant Nos. 2024GXNSFAA010221, 2024GXNSFAA010210, the National Nature and Science Foundation of China under Grant Agreement Nos. 82360456, 82360248, 82060226, 82260240, and 82301502, the Guangxi Nature and Science Foundation under Grant Agreement Nos. 2018GXNSFAA138010 and 2019GXNSFAA185029, as well as “Medical Excellent Award” funded by the Creative Research Development Grant from the First Affiliated Hospital of Guangxi Medical University under Grant Agreement Nos. 201907 and 202101. These fundings play important roles in the study design, acquisition analysis and interpretation of data, as well as in drafting the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2025.1492398/full#supplementary-material
References
1.
David J W Gargi BJL . Convexity subarachnoid haemorrhage. Lancet. (2023) 401:10372. 10.1016/S0140-6736(23)00007-7
2.
Stefan W Dorothee M Christoph B Vesna M Thomas K Maria W et al . Effectiveness of lumbar cerebrospinal fluid drain among patients with aneurysmal subarachnoid hemorrhage: a randomized clinical trial. JAMA Neurol. (2023) 80:833–42. 10.1001/jamaneurol.2023.3002
3.
Kong Y Wang D Jin X Liu Y Xu H . Unveiling the Significance of Trem1/2 in Hemorrhagic Stroke: Structure, Function, and Therapeutic Implications. Front Neurol. (2024) 15:1334786. 10.3389/fneur.2024.1334786
4.
Zhao B Yang H Zheng K Li Z Xiong Y Tan X et al . Preoperative and postoperative predictors of long-term outcome after endovascular treatment of poor-grade aneurysmal subarachnoid hemorrhage. J Neurosurg. (2017) 126:1764–71. 10.3171/2016.4.JNS152587
5.
Lång M Jakob SM Takala R Lyngbakken MN Turpeinen A Omland T et al . The prevalence of cardiac complications and their impact on outcomes in patients with non-traumatic subarachnoid hemorrhage. Sci Rep. (2022) 12:20109. 10.1038/s41598-022-24675-8
6.
van der Bilt IA Hasan D Vandertop WP Wilde AA Algra A Visser FC et al . Impact of cardiac complications on outcome after aneurysmal subarachnoid hemorrhage: a meta-analysis. Neurology. (2009) 72:635–42. 10.1212/01.wnl.0000342471.07290.07
7.
Lin F Chen Y He Q Zeng C Zhang C Chen X et al . Prognostic value of elevated cardiac troponin I after aneurysmal subarachnoid hemorrhage. Front Neurol. (2021) 12:677961. 10.3389/fneur.2021.677961
8.
Belete MA Tadesse S Tilahun M Gedefie A Shibabaw A Mulatie Z et al . Long noncoding rNAS and circular rnas as potential diagnostic biomarkers of inflammatory bowel diseases: a systematic review and meta-analysis. Front Immunol. (2024) 15:1362437. 10.3389/fimmu.2024.1362437
9.
Bai Y Ren H Zhu Y Chen X Yang L Xia J et al . Diagnosis and prognostic value of circdlgap4 in acute ischemic stroke and its correlation with outcomes. Front Neurol. (2022) 13:931435. 10.3389/fneur.2022.931435
10.
Long S Wu B Yang L Wang L Wang B Yan Y et al . Novel tumor necrosis factor-related long non-coding rNAS signature for risk stratification and prognosis in glioblastoma. Front Neurol. (2023) 14:1054686. 10.3389/fneur.2023.1054686
11.
Cai Z Li S Yu T Deng J Li X Jin J . Non-coding RNA regulatory network in ischemic stroke. Front Neurol. (2022) 13:820858. 10.3389/fneur.2022.820858
12.
Hoh BL Ko NU Amin-Hanjani S Chou S-Y Cruz-Flores S Dangayach NS et al . 2023 Guideline for the management of patients with aneurysmal subarachnoid hemorrhage: a guideline from the American Heart Association/American Stroke Association. Stroke. (2023) 54:e314–e70. 10.1161/STR.0000000000000436
13.
Treggiari MM Rabinstein AA Busl KM Caylor MM Citerio G Deem S et al . Guidelines for the neurocritical care management of aneurysmal subarachnoid hemorrhage. Neurocrit Care. (2023) 39:1–28. 10.1007/s12028-023-01713-5
14.
Langmead B Salzberg SL . Fast Gapped-Read Alignment with Bowtie 2. Nat Methods. (2012) 9:357–9. 10.1038/nmeth.1923
15.
Armstrong GA Rodgers CI Money TG Robertson RM . Suppression of spreading depression-like events in locusts by inhibition of the No/Cgmp/Pkg pathway. J Neurosci. (2009) 29:8225–35. 10.1523/JNEUROSCI.1652-09.2009
16.
Chen L Zhou X Deng Y Yang Y Chen X Chen Q et al . Zhenwu decoction ameliorates cardiac hypertrophy through activating Sgc (Soluble Guanylate Cyclase) - Cgmp (Cyclic Guanosine Monophosphate) - Pkg (Protein Kinase G) pathway. J Ethnopharmacol. (2023) 300:115705. 10.1016/j.jep.2022.115705
17.
Aoki T Kataoka H Shimamura M Nakagami H Wakayama K Moriwaki T et al . Nf-Kappab is a key mediator of cerebral aneurysm formation. Circulation. (2007) 116:2830–40. 10.1161/CIRCULATIONAHA.107.728303
18.
Liu C Yao K Tian Q Guo Y Wang G He P et al . Cxcr4-Btk axis mediate pyroptosis and lipid peroxidation in early brain injury after subarachnoid hemorrhage via Nlrp3 inflammasome and Nf-κb pathway. Redox Biol. (2023) 68:102960. 10.1016/j.redox.2023.102960
19.
Xie Z Huang L Enkhjargal B Reis C Wan W Tang J et al . Recombinant netrin-1 binding Unc5b receptor attenuates neuroinflammation and brain injury via Pparγ/Nfκb signaling pathway after subarachnoid hemorrhage in rats. Brain Behav Immun. (2018) 69:190–202. 10.1016/j.bbi.2017.11.012
20.
Sabet Sarvestani F Afshari A Azarpira N . The role of non-protein-coding rnas in ischemic acute kidney injury. Front Immunol. (2024) 15:1230742. 10.3389/fimmu.2024.1230742
21.
Jiang H Zhang Y Yue J Shi Y Xiao B Xiao W et al . Non-Coding Rnas: the neuroinflammatory regulators in neurodegenerative diseases. Front Neurol. (2022) 13:929290. 10.3389/fneur.2022.929290
22.
Wang F Zhang H Qiu G Li Z Wang Y . The Linc00452/Mir-204/Chst4 axis regulating thymic tregs might be involved in the progression of thymoma-associated myasthenia gravis. Front Neurol. (2022) 13:828970. 10.3389/fneur.2022.828970
23.
Li F Liu A Zhao M Luo L . Astrocytic chitinase-3-like protein 1 in neurological diseases: potential roles and future perspectives. J Neurochem. (2023) 165:772–90. 10.1111/jnc.15824
24.
Xu Q Sun L Wang Y Wang R Jia Y Guo D et al . Causal effects of Ykl-40 on ischemic stroke and its subtypes: a 2-sample mendelian randomization study. J Am Heart Assoc. (2023) 12:e029000. 10.1161/JAHA.122.029000
25.
Li J Lin J Pan Y Wang M Meng X Li H et al . Interleukin-6 and Ykl-40 predicted recurrent stroke after ischemic stroke or tia: analysis of 6 inflammation biomarkers in a prospective cohort study. J Neuroinflammation. (2022) 19:131. 10.1186/s12974-022-02467-1
26.
Kastrup J Johansen JS Winkel P Hansen JF Hildebrandt P Jensen GB et al . High serum Ykl-40 concentration is associated with cardiovascular and all-cause mortality in patients with stable coronary artery disease. Eur Heart J. (2009) 30:1066–72. 10.1093/eurheartj/ehp049
27.
Lasica N Raicevic V Stojanovic NM Djilvesi D Horvat I Jelaca B et al . Metabolomics as a potential tool for monitoring patients with aneurysmal subarachnoid hemorrhage. Front Neurol. (2023) 13:1101524. 10.3389/fneur.2022.1101524
Summary
Keywords
aSAH-MI, lncRNA, miRNA, mRNA, ceRNA
Citation
Wu Z, He W, Ye W, Xu S, Wei S, Huang B, Li P, Tang Y, Qin C, Liu Y and Ye Z (2025) Transcriptomic profiling and bioinformatic insights into myocardial injury following aneurysmal subarachnoid hemorrhage. Front. Neurol. 16:1492398. doi: 10.3389/fneur.2025.1492398
Received
06 September 2024
Accepted
12 May 2025
Published
24 June 2025
Volume
16 - 2025
Edited by
Nishant K. Mishra, Yale University, United States
Reviewed by
Nebojsa Lasica, University of Novi Sad, Serbia
Chuanchuan Wang, Second Military Medical University, China
Tijana Nastasovic, University of Belgrade, Serbia
Updates
Copyright
© 2025 Wu, He, Ye, Xu, Wei, Huang, Li, Tang, Qin, Liu and Ye.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ziming Ye yeziming1620@163.comYing Liu liuying10052024@163.com
†These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.