 Ying Zhao1†
Ying Zhao1† YaRu Lu1†
YaRu Lu1† FengYu Wang1
FengYu Wang1 YaDan Wang2
YaDan Wang2 YaQiong Li1
YaQiong Li1 RuiHua Sun1
RuiHua Sun1 JunKui Shang1
JunKui Shang1 Chao Jiang1
Chao Jiang1 Jiewen Zhang1*
Jiewen Zhang1*- 1Department of Neurology, Zhengzhou University People's Hospital, Henan Provincial People's Hospital, Zhengzhou, Henan, China
- 2Department of Neurology, Henan University People's Hospital, Henan Provincial People's Hospital, Zhengzhou, Henan, China
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is a monogenic cerebral small-vessel disease caused by mutations in NOTCH3 and is the most common hereditary cerebral small-vessel disease in adults. The clinical manifestations of CADASIL include migraines, recurrent ischemic stroke, progressive cognitive deterioration, and psychiatric symptoms. The most prevalent and earliest imaging alterations in CADASIL are white matter hyperintensities in the periventricular white matter, temporal pole, external capsule, frontoparietal white matter, and other areas on magnetic resonance imaging. Despite the substantial variations in the clinical phenotypes and disease severity in patients with CADASIL, the specific mechanisms underlying these differences remain unclear. Exploring these underlying mechanisms is crucial for enhancing our understanding of CADASIL and offering insights into its early diagnosis and treatment. This review explores the advances in research on the molecular mechanisms contributing to the variability in clinical phenotypes and disease severity among CADASIL patients with different mutations.
1 Introduction
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is the most common inherited form of single-gene cerebral small vessel disease (CSVD) in adults. It is caused by mutations in the NOTCH3 gene located on chromosome 19, which results in the number of cysteines within one of the 34 epidermal growth factor-like repeats (EGFr) in the extracellular structural domain of the NOTCH3 protein (1, 2). Clinical manifestations are characterized by migraine with aura, recurrent subcortical ischemic stroke, transient ischemic attacks (TIAs), psychiatric symptoms, and progressive cognitive impairments (3). Additional symptoms include Parkinsonian syndrome, atypical paraplegia, and seizures (4). On imaging, the hallmark and earliest feature of CADASIL is the presence of white matter hyperintensities (WMH) predominantly in the periventricular white matter, temporal poles, external capsule, and frontal and parietal regions on magnetic resonance imaging (MRI) (5). Lacunar infarcts, cerebral microbleeds (CMBs), and enlarged perivascular spaces are also characteristic imaging findings in CADASIL. Abnormal WMH on MRI may occur earlier than clinical symptoms. The diagnostic gold standard comprises the hallmark pathological feature—granular osmiophilic material (GOM) deposited on arterial vascular smooth muscle cells (VSMCs)—combined with NOTCH3 gene mutations (6). Although the characteristic clinical and imaging manifestations have been delineated, accurate identification and early diagnosis of the disease remain challenging owing to the high variability of the phenotype and the incompleteness of clinical manifestations, resulting in a high rate of missed diagnosis and misdiagnosis. Moreover, the actual prevalence of NOTCH3 mutations in the general population is higher than expected, further contributing to increased social burden (7).
Mutations in the NOTCH3 gene have been identified as the major causative factor for CADASIL. However, phenotypic variations among patients suggest a complex interplay of genetic and non-genetic factors, such as the environment, lifestyle, and genetic modifiers. The potential interaction of these factors with the NOTCH3 mutation on disease onset, severity, and progression has been previously documented. This review provides an overview of the potential factors that influence the severity of the clinical phenotype of CADASIL, from molecular mechanisms to genetic modifiers, offering a comprehensive overview of the phenotypic variability of CADASIL.
2 Molecular pathogenesis of CADASIL
The pathogenic mechanism of CADASIL is primarily attributed to missense mutations in exons 2–24 of NOTCH3, which lead to an abnormal number of cysteine residues, resulting in either an increase or a decrease to the odd number (8). NOTCH3, a member of the NOTCH family located on chromosome 19, consists of 33 exons that encode the highly conserved NOTCH3 transmembrane receptor. The NOTCH3 protein comprises three key domains: an intracellular domain (NOTCH3ICD) essential for downstream signaling, an extracellular domain (NOTCH3ECD) involved in ligand binding, and a single transmembrane region (9). The NOTCH3 signaling pathway plays a crucial role in regulating cell differentiation, proliferation, and apoptosis and is a critical determinant in the development of various organs (9, 10).
The NOTCH3ECD contains 34 EGFr and 3 Notch/Lin12 repeat fragments, where EGFr is essential for ligand-receptor binding (11). Each EGFr consists of ~40 amino acids, including six cysteine residues that pair to form three disulfide bonds, which are vital for maintaining and stabilizing its secondary structure (12). NOTCH3 is predominantly expressed in VSMCs and pericytes of small arteries (9). The classical NOTCH3 signaling pathway is activated by members of the Delta/Jagged transmembrane ligand family (8). After endoplasmic reticulum synthesis, NOTCH3 is processed through three sequential regulated proteolytic cleavage steps following ligand binding. This cascade culminates in the nuclear translocation of active NOTCH3ICD, which interacts with CSL transcription factors and mastermind (MAM) co-activators through its RBP-Jκ-associated domain to regulate downstream target gene expression (13, 14) (Figure 1).
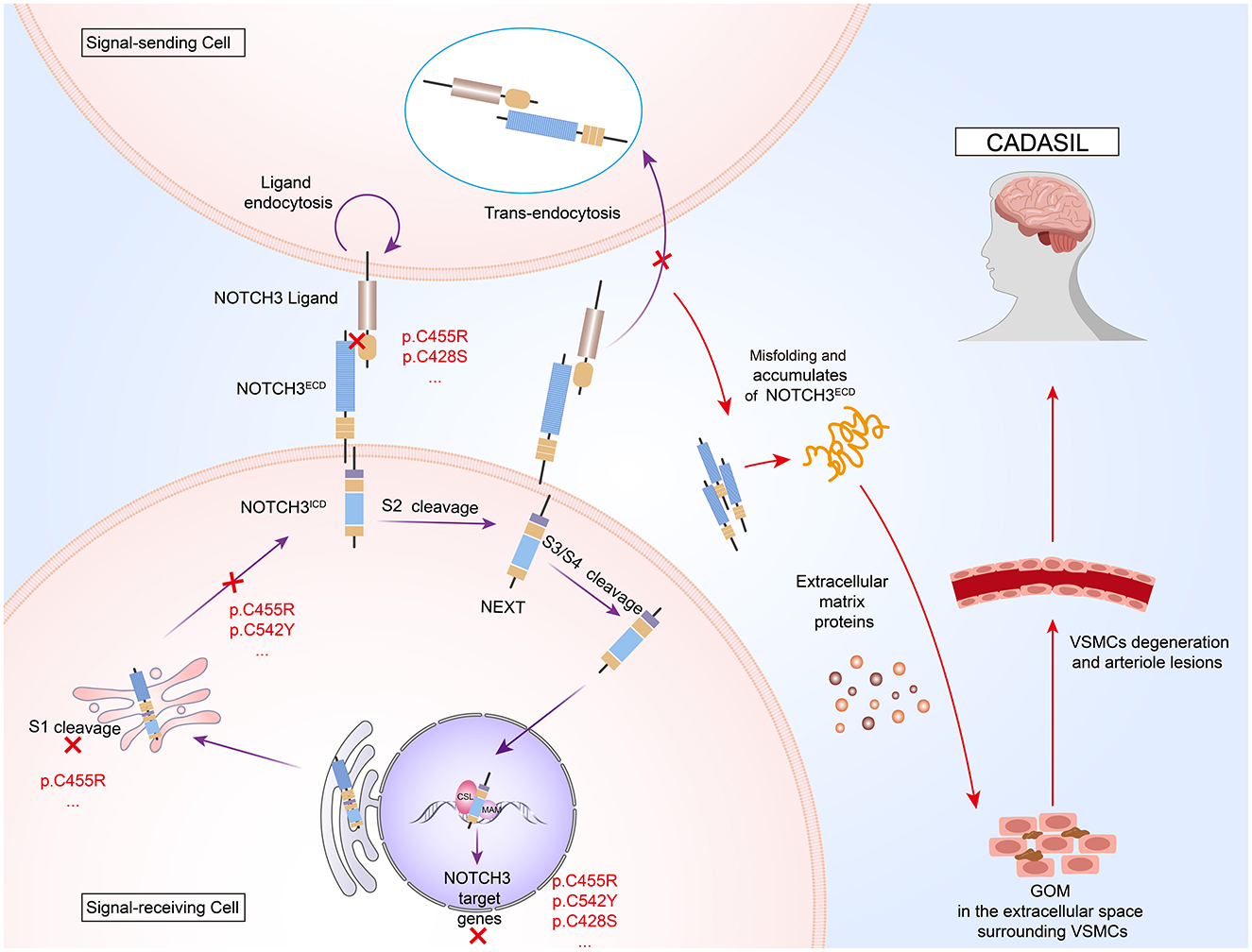
Figure 1. Pathogenic mechanisms of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). NOTCH3 is a transmembrane protein comprising an extracellular domain (NOTCH3ECD), a transmembrane region, and an intracellular domain (NOTCH3ICD). After S1 processing in the Golgi apparatus, NOTCH3 protein binds to ligands on the surface of signal-sending cells to activate S2 cleavage. Ligands bind NOTCH3ECD and are endocytosed into signal-sending cells. Notch extracellular truncation (NEXT) generated by S2 cleavage is further processed at the S3 and S4 sites. The released NOTCH3ICD is translocated to the nucleus to bind to core binding factor 1/suppressor of hairless/longevity assurance gene 1 (CSL) and mastermind (MAM), where the three-protein complex recruits co-activators (Co-A) to activate target gene transcription. NOTCH3 mutations result in the formation of granular osmiophilic material (GOM), extracellular aggregates that are hallmark of CADASIL, by inducing NOTCH3ECD misfolding and aberrant tertiary structure conformation, resulting in aberrant dimerization of the NOTCH3ECD and abnormal interactions with other proteins. Additionally, these mutations may interfere with the reduced signaling activity of ligand binding by affecting the processing of the NOTCH protein at the S1 cleavage site by Furin-like convertase in the Golgi apparatus.
2.1 GOM-associated toxicity
Cysteine-altered NOTCH3 mutations disrupt the formation of disulfide bonds, leading to misfolding of EGFr by interfering with glycosylation of its distal structural domains (15). These structural changes alter the spatial conformation of EGFr and promote abnormal segmentation and dimerization of NOTCH3. This abnormality leads to the aggregation of NOTCH3ECD in the interstitial space of the VSMCs (15, 16). Under normal conditions, NOTCH3ECD enters ligand-expressing cells via endocytosis and is degraded through the transendocytic pathway. However, mutant NOTCH3ECD resists effective degradation, resulting in impaired clearance (17, 18). Immunoelectron microscopy studies employing domain-specific NOTCH3 antibodies have identified NOTCH3ECD as the predominant component of GOM in CADASIL patient skin biopsies (19). The NOTCH3ECD cascade hypothesis posits that the elevation or accumulation of NOTCH3ECD levels in vasculature serves as the central event initiating the cascade. Cellular models have demonstrated that NOTCH3ECD recruits extracellular matrix proteins, including tissue inhibitors of metalloproteinase 3 (TIMP3) and vitronectin (VTN), via protein-protein interactions to form stable, extracellular complexes, which exacerbate multifactorial cytotoxic effects (20). The reduction of TIMP3 or VTN has been shown to improve the CADASIL phenotype in a transgenic mouse model (21). Furthermore, studies using a transgenic mouse model of CADASIL have suggested that the pathogenesis of the disease is likely associated with the acquisition of a toxic function of NOTCH3 (12). Structural alterations from mutations impair physiological receptor-ligand interactions while promoting pathological protein aggregation. This ultimately leads to degeneration of VSMCs and fibrosis in small arteries, contributing to disease progression (15, 22). Notably, NOTCH3ECD aggregation precedes neurological symptom onset in both mouse models and human specimens, representing an early pathogenic event (23, 24). Additionally, NOTCH3 expression levels correlated with the age of onset in mice and the severity of perivascular NOTCH3ECD accumulation in mouse models (25). These findings support the role of GOM-associated toxicity in the pathogenesis of CADASIL.
2.2 Abnormalities in the NOTCH3 signaling pathway
The most widely accepted hypothesis is that the mutant NOTCH3 protein exerts a toxic effect on VSMC membranes, contributing to the pathogenesis of CADASIL. However, the alternative hypothesis suggests that the pathogenic impact of mutations may be linked to a defect in signaling within the mutant receptor (26, 27). Some case reports have demonstrated that NOTCH3 loss-of-function mutations can induce a CADASIL-like phenotype even in the absence of GOM deposition, indicating that dysregulated signaling alone may be sufficient to cause disease (28). A previous study analyzed the signaling induced by the ligand Delta1/Jagged1 via RBP-Jκ to investigate the effects of three pathogenic NOTCH3 mutations on receptor bioactivity. Notably, R133C and C183R mutations [outside the ligand-binding domain (LBD)] showed normal Jagged1-induced signaling in A7r5 VSMCs, while the C455R mutation (within the LBD) significantly reduced signaling activity. Ligand binding assays further revealed that the C455R mutation disrupts LBD structure, impairing Delta1/Jagged1 binding (29). Studies involving C455R NOTCH3 gene knockout mice demonstrated that NOTCH3 signaling is both necessary and sufficient to support the coverage of arterial wall cells, and restoring NOTCH3 signaling in these wall cells effectively prevented CSVD phenotypes in mice (30). Another study investigated five naturally occurring mutations in CADASIL, of which C212S and R90C were located in the previously identified mutation hotspot region EGFr25, C428S was located in the LBD of EGFr10–11, and the remaining two mutations, C542Y and R1006C, were located in EGFr13 and 26, respectively. The study found that two mutations (C428S and C542Y) impaired receptor activity through distinct molecular mechanisms involving the RBP-Jκ transcription factor, whereas the activities of the other three mutations were unaffected (8). These findings reveal that mutations in the EGFr10–11 domain—the LBD region for delta/serrate/LAG-2 (DSL) receptors of NOTCH3—and those disrupting membrane localization significantly impair NOTCH3-mediated downstream signaling transduction, thereby compromising NOTCH3-dependent cellular functionality. However, a recent study employing a murine model demonstrated that the R169C mutation increased NOTCH3 activity. This enhancement potentially involves the disruption of endosomal trafficking induced by aberrant deposition of NOTCH3ECD, subsequently driving vascular structural lesions via processes including the regulation of VSMCs cytoskeleton remodeling (31). Notably, inhibition of NOTCH signaling effectively alleviates luminal stenosis, whereas conditional activation of NOTCH3 directly induces analogous structural alterations independent of hemodynamic changes. These findings suggested that excessive activation of NOTCH3 signaling constitutes the pivotal pathogenic mechanism. Based on these studies, it is hypothesized that NOTCH3 signaling functions as a signaling threshold (or Goldilocks) pathway in the context of CSVD (32). Both loss of function and excessive activation of NOTCH3 signaling are hypothesized to facilitate mural cell degeneration through downstream effects on the expression of cell survival-related genes and proteins, consequently disrupting vascular homeostasis.
2.3 NOTCH3 non-enzymatic cleavage and fragments
In various chronic neurodegenerative diseases, such as Alzheimer's disease, frontotemporal lobar degeneration, and amyotrophic lateral sclerosis, pathological protein fragments resulting from proteolytic cleavage, including amyloid-beta (Aβ), tau, and TAR DNA-binding protein of 43 kDa (TDP-43), are implicated in the onset and progression of these conditions (33, 34). A previous study has elucidated a novel molecular mechanism underlying vascular disease in CADASIL, diverging from the traditional NOTCH signaling pathway, which typically involves a protein hydrolysis cascade to release its transcriptionally active intracellular structural domains (35). In CADASIL patients with NOTCH3 mutations, a specific spontaneous cleavage between EGFr1 and EGFr2 has been identified, occurring independently of the proteolytic enzymes (36). This investigation revealed site-specific fragmentation of the NOTCH3 protein through pathological analysis of the cerebral arterial vasculature in CADASIL patients and localized the fragmentation site to the NOTCH3 N-terminus at the peptide bond connecting Asp80 and Pro81. This non-enzymatic cleavage occurred within the extracellular matrix at the ultrastructural level, effectively separating EGFr1 from the remainder of the protein. The process depends on acidic pH and a reducing environment, and the enrichment of cleavage products N-terminal NOTCH3 fragments (NTF) in GOM within the media of cerebral arteries of CADASIL patients was confirmed using specific antibodies. Electron microscopy analysis revealed that the cleavage products colocalized with the basement membrane, collagen fibers, and GOM, while mutations of multiple cysteine residues of NOTCH3 significantly accelerated the cleavage by disrupting the stability of disulfide bonds. Subsequently, another study identified a comparable non-enzymatic cleavage of the NOTCH3 protein at Asp121, located between EGFr2 and EGFr3. This cleavage generates a peptide known as NTF2, which is markedly enriched in leptomeningeal arteries and can be inhibited by phosphate, EDTA, and other anions (37). This suggests that ion concentration in the vascular microenvironment may regulate lytic activity. Recently, a third Asp-Pro site (Asp964), located between EGFr24 and EGFr25, was identified, and its cleavage product NTF3 was found to affect disulfide integrity by altering the REDOX state of the NOTCH3ECD (38). Moreover, abnormal interactions with proteins such as voltage-gated potassium channel subunits may drive vascular matrix remodeling.
All three studies highlighted that the cleavage of Asp-Pro sites shared the following common characteristics: (1) dependence on a reducing environment and specific pH conditions (Asp80/Asp121 favor acidity, whereas Asp964 cleavage remains active at physiological pH); (2) regulation of NOTCH3 by its own concentration, forming a “positive feedback loop”; (3) accumulation of cleavage products (NTF, NTF2, and NTF3) in the extracellular matrix and colocalization with matrix proteins such as collagen, potentially promoting pathological deposition by altering the local protein interaction network. These data collectively support the potential pathogenic mechanism of multi-site non-enzymatic cleavage of the NOTCH3 protein in the progression of CADASIL. The non-enzymatic cleavage of NOTCH3 at sites such as Asp80, Asp121, and Asp964 may drive the progression of CADASIL through a “multi-stage amplification” mechanism: initial cleavage products (such as NTF2) promote subsequent cleavage by disrupting redox homeostasis, forming a positive feedback loop.
However, it should be emphasized that the current evidence for these non-enzymatic cleavage phenomena is mainly based on the analysis of patient vascular tissues and in vitro recombinant protein experiments. The role of these phenomena in the pathophysiological process still needs to be further confirmed through models such as gene-edited animals. Future research should clarify the functional heterogeneity of different cleavage fragments and their interaction networks with matrix proteins and establish a direct dose–response relationship between the levels of cleavage products and clinical severity (such as the volume of WMH) to promote their application as prognostic markers and further explore targeted therapeutic sites.
3 Factors associated with variations in CADASIL genotypes and clinical phenotypes
3.1 Genetic determinants
3.1.1 Location of NOTCH3 mutations in EGFr
Many cohort studies have consistently demonstrated regional mutation-dependent variability in clinical manifestations and prognosis among CADASIL patients. A retrospective cohort study of 664 CADASIL patients in the Netherlands and Europe and a general population database found that NOTCH3 mutations located in EGFr1–6 (HR-EGFr) were associated with early stroke onset, poor survival, and severe WMH (39). This study proposes that the NOTCH3 mutation location is a central factor in the phenotypic heterogeneity of CADASIL. This concept was further confirmed by another large cohort study, which found that patients with mutations in EGFr1–6 had a higher risk of stroke, dementia, and daily life dependence than those with mutations in EGFr7–34. Importantly, this association is independent of age, sex, and vascular risk factors (40). The C117F and C174Y variants within EGFr1–6 exemplify this pattern, demonstrating accelerated disease progression and mortality in longitudinal studies (41). Notably, EGFr7–34 mutation carriers under 50 years of age frequently show minimal CSVD burden, often presenting with normal brain MRI findings (42). Mutations in EGFr7–34 are associated with milder clinical symptoms, potentially leading to misdiagnosis or non-diagnosis. While conventional estimates suggest prevalence rates of 2–5 per 100,000, emerging population-level data indicate actual prevalence is expected to be 100 times higher than previously predicted (39, 43–45). To systematically compare the clinical and imaging distinctions between EGFr1–6 and EGFr7–34 mutations, we have synthesized key findings in Table 1.
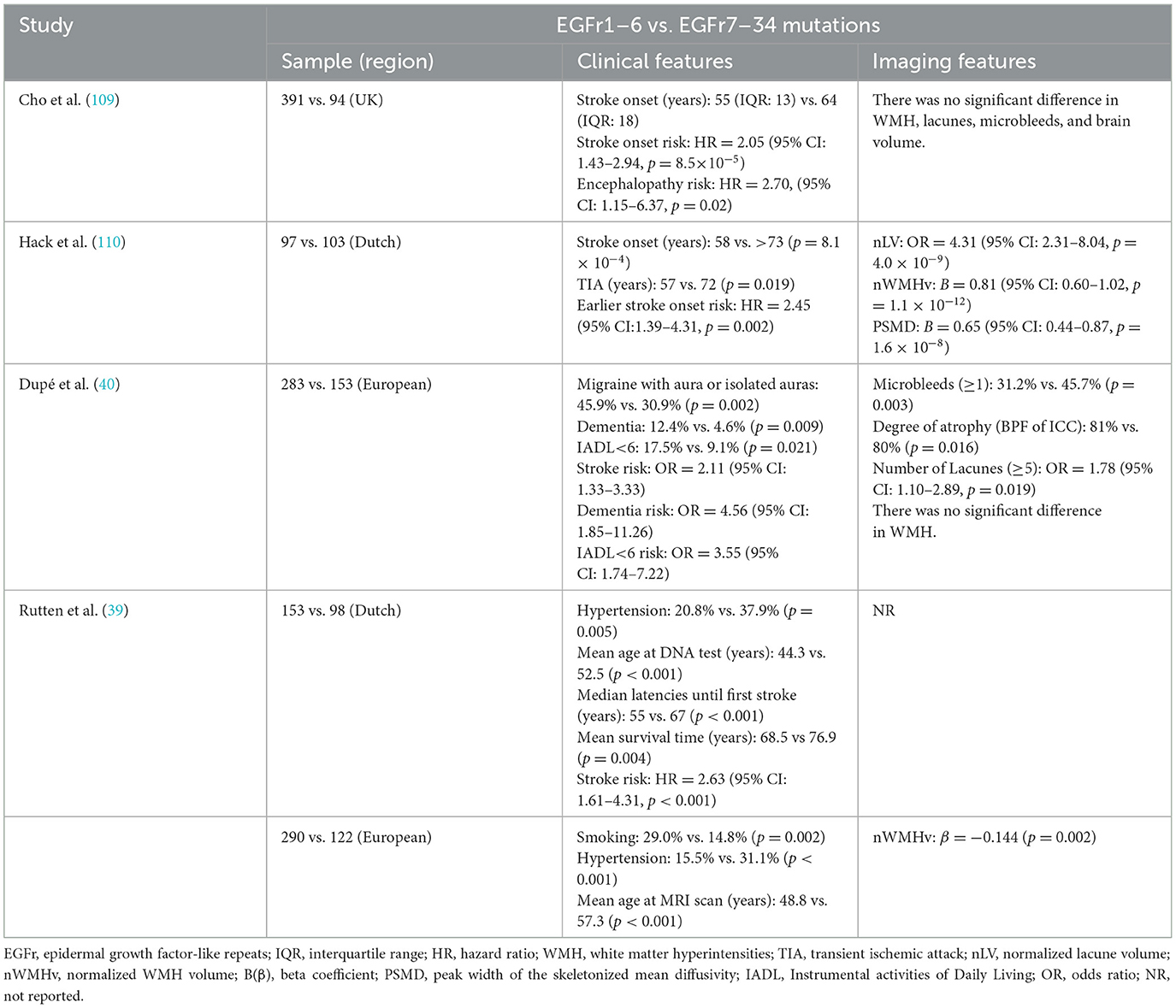
Table 1. Comparison of clinical and imaging features of mutations in EGFr1–6 and 7–34 in CADASIL.
Studies analyzing the NOTCH3 variant frequency odds ratio across EGFr domains in large CADASIL cohorts and population databases have stratified patients into three prognostic categories: high-risk (HR-EGFr: 1–6, 8, 11, 26), medium-risk (MR-EGFr: 9–10, 12–15, 17, 25, 27, 32), and low-risk (LR-EGFr: 16, 18–20, 23–24, 28–31, 33) (46). Results indicated that HR-EGFr variants show strong associations with severe stroke risk, elevated normalized WMH volume, and excessive NOTCH3 protein aggregation in VSMCs. Notably, although EGFr11 mutations impair ligand-dependent signaling activity, this reduction shows no direct correlation with clinical severity, further supporting the protein aggregation hypothesis rather than a signaling pathway dysfunction. Additionally, a prospective longitudinal study demonstrated accelerated disease progression in patients with HR-EGFr NOTCH3 mutations compared to patients with MR-EGFr (47). These findings collectively underscore NOTCH3 aggregation as a central driver of NOTCH3-small vessel disease (NOTCH3-SVD) pathogenesis, suggesting that the extent of aggregation critically influences phenotypic severity. Patients with an EGFr7–34 variant have significantly lower accumulation of NOTCH3ECD in the skin and brain vessels than patients with an EGFr1–6 variant. Furthermore, the levels of NOTCH3 accumulation in the EGFr7–34 group showed a positive correlation with lacune count and WMH volume (48). This study, for the first time, reveals the potential mechanism by which NOTCH3 variant location affects disease severity by regulating protein aggregation load. However, the limited availability of postmortem brain tissue samples in this cohort highlights the need for validation in large-scale studies. This spatial pattern suggests that compared with mutations in the C-terminal region, the unpaired cysteines in EGFr1–6 mutations at the N-terminal region of the NOTCH3ECD are more likely to interact with other proteins (46). These N-terminal domains demonstrate a particular affinity for extracellular matrix components, including HTRA1, VTN, TIMP3, and latent transforming growth factor beta-binding protein 1 (LTBP1). These interactions can substantially enhance the multimerization of CADASIL-related proteins, leading to a heavier vascular aggregate load and severe disease manifestations. Additionally, a study investigating NOTCH3 fragmentation in a cohort of individuals with NOTCH3 mutations identified a strong correlation between the mutation location and the generation of the neo-epitope (36). This suggested that pathogenic mutations near these cleavage sites may enhance the cleavage propensity of the NOTCH3 protein, resulting in a more severe disease phenotype.
A retrospective study conducted in France indicated that compared with common mutations in the EGFr2–5, mutations in the EGFr10–11 were associated with milder cognitive deficits and a tendency toward a reduction in lacunar infarct volume. However, genotype-phenotype correlation analysis revealed that patients with EGFr10–11 mutations exhibited a significantly higher prevalence of cerebral WMH in brain MRI (24). In vivo studies have shown that stroke in patients with a mutation located in EGFr11 (C455R) occurs at an abnormally early age compared to other CADASIL populations (49). Therefore, downregulation of NOTCH3 signaling may play a role in modifying the clinical phenotype of CADASIL. These findings suggest that signaling activity is differentially influenced by various NOTCH3 mutations in CADASIL. Although CADASIL mutations do not primarily disrupt the major functions of the classical NOTCH signaling pathway, alterations in NOTCH signaling can influence the disease phenotype to some extent. One possible explanation for this phenomenon is that NOTCH receptor activity is tightly regulated and highly dependent on the cellular context, with certain mutations only impacting receptor activity under specific conditions (50). Additionally, the pathogenicity of the mutation may arise from impaired receptor activity independent of the typical RBP-Jκ signaling pathway (8), warranting further investigation to determine whether impairment of the signaling pathway in NOTCH3 mutants contributes to disease severity and to elucidate the specific mechanisms linking NOTCH3 signaling to the disease phenotype.
3.1.2 Atypical CADASIL variant types
Most mutations (>95%) associated with CADASIL are heterozygous missense mutations in NOTCH3, along with splice-site mutations, insertion mutations, frameshift mutations, nonsense mutations, and small in-frame deletions (51, 52). Patients with atypical NOTCH3 mutations often present with clinical symptoms at a later stage of life and typically exhibit incomplete penetrance. Notably, these patients often lack characteristic WMH on MRI and show only non-specific vascular injury signs—without GOM deposits—on skin biopsy (51, 53). CADASIL is characterized by NOTCH3 mutations involving changes in cysteine residues; however, ~5% of NOTCH3 mutations are cysteine-sparing NOTCH3 mutations (54).
Recent studies have gradually revealed that atypical NOTCH3 mutations (such as cysteine-sparing NOTCH3 missense mutations) cause phenotypic heterogeneity in CADASIL through unique molecular mechanisms. A systematic review of CADASIL cases carrying suspected cysteine-sparing NOTCH3 missense mutations found that four mutations, namely p.R61W, p.R75P, p.D80G, and p.R213K, met the preset pathogenicity criteria. Clinical phenotype analysis showed that patients carrying such mutations presented typical CADASIL symptoms (such as stroke, migraine, and dementia). Still, the proportion of WMH in the temporal pole on imaging was significantly lower than that in patients with traditional cysteine mutations (55). A Korean cohort study found that the cysteine-sparing mutation group had less involvement of the anterior temporal lobe white matter (56), and a Chinese study further confirmed that this type of mutation had a later onset age and milder temporal lobe lesions (57). This imaging finding suggests that this type of NOTCH3 mutation may cause differences in vascular pathological damage in specific brain regions (such as the anterior temporal lobe) by affecting the tendency of protein aggregation. Another cross-population systematic review revealed region-specific genotype-phenotype associations, with significantly increased frequencies of cognitive impairment and CMBs in Asian patients with cysteine-sparing NOTCH3 missense mutations, which is a significant difference from classic mutation CADASIL patients (58). Additionally, the clinical and radiological phenotype characteristics of NOTCH3 cysteine-sparing mutation patients vary in different regions, reflecting the interaction between the mutation site and the genetic background of the population. In vitro studies using p.R75P, p.D80G, and delta88-91 showed significantly enhanced aggregation similar to cysteine mutations, further leading to VSMC degeneration. Still, the degree of GOM deposition was milder than that of classic mutations, which may explain the milder anterior temporal lobe injury (59). Moreover, functional studies have shown that such mutations may cause vascular lesions by introducing proline to disrupt β-sheet folding or promote abnormal aggregation of the NOTCH3ECD rather than relying on the traditional mechanism of cysteine residue deletion (55). In addition to NOTCH3ECD aggregation, mechanisms such as receptor misfolding, spatial conformational changes induced by these mutations, and impaired NOTCH3 signaling may contribute to the pathogenesis of cysteine-sparing NOTCH3 missense mutations. Further studies are required to elucidate the specific mechanisms involved.
3.2 Phenotypic modifiers
Genetic analysis revealed a strong association between the CADASIL loci and various families, demonstrating the genetic homogeneity of CADASIL (60). However, notable variations in the disease phenotypes have been observed among different families. Among Caucasians, migraines are reported as the first symptom in approximately one-third of patients (61), often accompanied by imaging findings revealing abnormalities in the temporal lobe (62). Conversely, in the mainland Chinese population, migraine with aura accompanied by abnormal white matter in the temporal pole is rare (63), whereas lacunar infarction in the brainstem is more common than in their Caucasian counterparts (63, 64). A systematic review indicated that the age at the onset of CADASIL in China ranges between 20 and 73 years. The main clinical manifestations include stroke/TIA and cognitive decline; other rare symptoms include migraine, primary cerebral hemorrhage, vertigo, sensory aphasia, alopecia, tinnitus, and deafness (65). The variability in clinical phenotypes across region-specific populations cannot be fully attributed to the genotypic differences across various regions and the founder effect. This suggests that environmental factors substantially influence disease phenotypes, highlighting the need for further research to elucidate the relationship between geographical location and phenotype.
3.2.1 Environmental factors
In a study on monozygotic twins with CADASIL, individuals with the same genetic background exhibited distinct clinical phenotypes. This finding underscores the potential influence of environmental factors and lifestyle on the clinical progression of CADASIL (66). CADASIL is primarily associated with heterozygous mutations in the NOTCH3 gene; however, homozygous mutations have also been identified. Compared with age-matched CADASIL patients with the R133C mutation, homozygous patients experienced an earlier first stroke, exhibited severe findings on most neuropsychological tests and MRI, and increased accumulation of GOM. However, one heterozygous patient in this study showed more rapid progression and severity than that of the homozygous patient (67). These findings highlight the critical role of environmental and genetic factors in influencing the CADASIL disease phenotype (68–70).
Studies have shown that smoking is independently associated with an earlier age of onset and may increase the risk of stroke (71). The mechanism may involve oxidative stress components in tobacco that aggravate vascular endothelial dysfunction. Hypertension increases the risk of cerebral infarction, intracerebral hemorrhage, lacunar infarcts, and CMBs in CADASIL patients through hemodynamic changes (72). The severity and frequency of WMH increase dramatically with age and are higher in symptomatic individuals (73). Diabetes mellitus was confirmed to be associated with early stroke onset in CADASIL patients (74). In addition, individuals with affected sisters with diabetes exhibited a more severe phenotype than those with the same mutation (70). Studies of glucose metabolism in CADASIL revealed downregulation of glucose transporters (GLUTs), specifically GLUT4 and GLUT2 in VSMCs, leading to impaired glucose uptake, a mechanism that may further contribute to blood flow restriction (75). In a large-scale, community-based controlled study conducted in the United Kingdom, homocysteine was identified as an independent risk factor in Caucasian patients with CSVD, particularly ischemic leukoaraiosis (76). Additionally, high homocysteine is associated with early migraine attacks (71). Homocysteine may cause disease by impairing vascular endothelium and atherothrombosis and interfering with the synthesis and metabolic hierarchy of neurotransmitters (77). Furthermore, the extent of atherosclerosis was related to the clinical severity of CADASIL (78). In addition, the mean age of stroke onset and the median age of death are earlier in men than in women (41, 79, 80). The prevalence and phenotype of pre-monocular symptoms differed between sexes. These findings suggest that hormonal status may play a role in modulating susceptibility in CADASIL patients. These findings collectively highlight that CADASIL manifestations arise not only from genetic determinism but also from dynamic interactions among NOTCH3 mutations, vascular risk exposures, and metabolic dysregulation. As summarized in Table 2, prospective studies across diverse populations consistently report high prevalence rates of modifiable vascular risk factors in CADASIL cohorts, further validating their role as critical accelerators of disease severity.
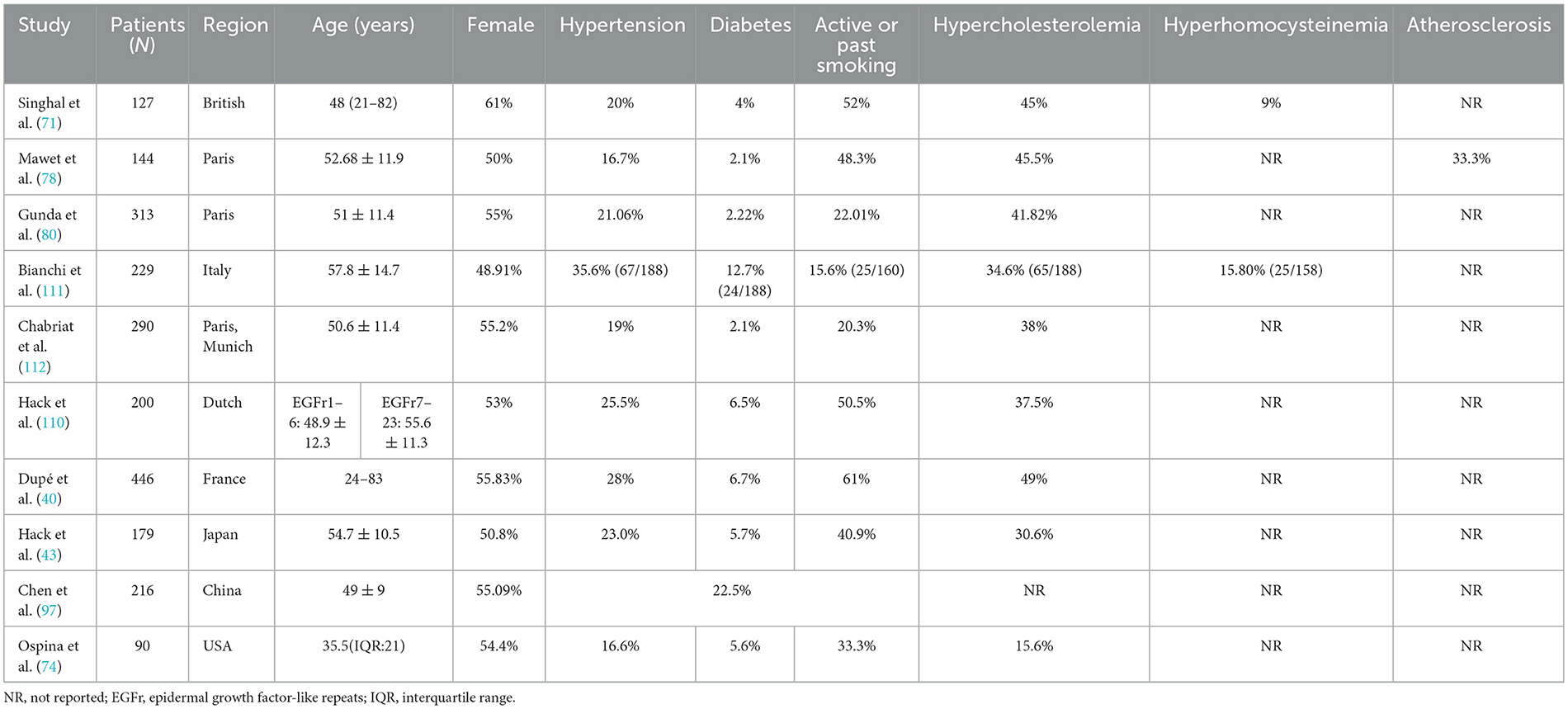
Table 2. Prevalence of vascular risk factors in patients with CADASIL as reported in prospective studies.
3.2.2 Epigenetic regulation
In addition to NOTCH3 mutations and environmental factors, epigenetic modifiers play a significant role in the clinical symptoms, imaging findings, and pathological changes observed in CADASIL patients (68). These genetic modifiers participate in autoregulation and neuronal responses to ischemia, as well as in repair processes and other functions at various levels of vascular endothelial and smooth muscle cells, thereby exerting a regulatory influence on CADASIL (81). Epigenetic modifications, including DNA methylation and RNA or histone modification, regulate gene expression through mechanisms such as methylation, phosphorylation, and ubiquitination. Consequently, these modifications are crucial for the growth, differentiation, and proliferation of various cell types (82). Epigenetic modifications play a critical role in the initiation and progression of neurodegenerative disorders such as Alzheimer's disease and Parkinson's disease (83–86). In a naturally occurring exon 9 skipping CADASIL family, individuals with exon 9 skipping exhibited a milder phenotype of small-vessel disease than that observed in most CADASIL patients, with significantly reduced NOTCH3ECD protein aggregation in the skin. Inducing exon 9 skipping in a cellular model revealed that cysteine-corrected NOTCH3 exon skipping reduced NOTCH3 aggregation and resulted in an attenuated phenotype (87). Arginine methylation is a common post-translational modification that plays a crucial role in various biological processes, including signal transduction, metabolism, and development within the human body. Elevated levels of asymmetric dimethylarginine following the methylation of arginine residues are significantly associated with an increased risk of adverse vascular events in CADASIL patients (88). Additionally, D-loop methylation is markedly reduced in the mitochondrial DNA (mtDNA) of CADASIL patients, further contributing to increase in mtDNA and mitochondrial dysfunction (89).
3.2.3 Polygenic interactions
Emerging evidence underscores the role of polygenic factors in modulating CADASIL severity beyond NOTCH3 mutations. The coexistence of the NOTCH3 pathogenic variant (p.G420C) and the SQSTM1 pathogenic variant was identified in two siblings with CADASIL. The presence of the SQSTM1 gene as a genetic modifier may play a role in modulating NOTCH3 signaling, ultimately resulting in pronounced deterioration of the clinical manifestations associated with CADASIL in these siblings (90). Furthermore, the apolipoprotein E (APOE) ε2 allele is associated with a higher WMH volume, and individuals carrying the APOE ε2 allele may have more severe cognitive impairment (91, 92). A genome-wide association study reported that the polygenic score of CADASIL was associated with WMH volume, and multiple common genetic variants with small effect sizes affected WMH burden (93). Additionally, specific CADASIL alleles may increase the likelihood of corpus callosum lesions or the rate of disease progression (41, 94). It is noteworthy that CADASIL patient populations exhibit region- and ethnicity-specific mutation hotspots. For instance, in most European patients, such as British and German patients, NOTCH3 mutations are predominantly located in exons 2–6, particularly exon 4 (95, 96). In contrast, mutations in exons 11 and 4 are more prevalent in Asian regions such as China and South Korea (63, 72, 97). Clinically, Asian patients are more prone to TIA/ischemic stroke and cognitive impairment, while Caucasians have a higher incidence of migraine and mental disorders (98). Similar to typical CADASIL, the clinical and radiological phenotypic features of NOTCH3 cysteine retention mutation patients vary in different regions. Asian patients with atypical mutations often present a “radiological phenotype preceding clinical symptoms” pattern, with more significant imaging brain phenotypes, especially lacunes and CMBs, while Caucasians mainly exhibit typical clinical phenotypes (58). This highlights the interplay between founder effects and localized environmental pressures in shaping regional genetic landscapes. These findings collectively emphasize that CADASIL heterogeneity arises from a complex interplay of major NOTCH3 mutations, modifier genes, and population-specific genetic drift. Future studies should integrate multi-dimensional analytical approaches, including gene-gene interactions, epigenetic regulation, and environmental influences, to elucidate the precise mechanisms underlying genotype-phenotype associations.
4 Modifications in clinical diagnostic criteria for CADASIL
Due to the absence of reliable biomarkers, CADASIL diagnosis primarily relies on clinician-developed criteria, necessitating a thorough understanding of its clinical heterogeneity. Given CADASIL's phenotypic variability, the diagnostic criteria initially proposed by Davous have been iteratively refined to capture its diverse manifestations (3). The screening of patients for CADASIL typically involves a combination of characteristic clinical manifestations, imaging findings, and family history. A CADASIL scale scoring system was proposed for the genetic testing population, including eight categories: migraine (with or without aura), stroke/TIA, early-onset symptoms (≤ 50 years), psychiatric disorders, cognitive decline/dementia, leukoencephalopathy (temporal pole or external capsule involvement), subcortical infarction, and family history. This scale is mainly used to identify patients with a high probability of being affected by genetic testing, with a sensitivity of 96.7% and a specificity of 74.2% (99). Over time, the original CADASIL scale has been adapted by researchers to better address the specific needs of different populations and clinical settings. The CADASIL Scale-J, adapted for Japanese cohorts, comprises eight items: hypertension, diabetes mellitus, age at onset (≤ 50 years), pseudobulbar palsy, stroke/TIA, family history, subcortical infarction, and temporal pole lesions. The sensitivity and specificity of the CADASIL Scale-J were 78.9% and 85.7%, respectively (79). These modifications aim to enhance the scale's applicability and diagnostic accuracy within specific demographic groups. Currently, typical neuroimaging hallmarks of CADASIL include T2-WMHs in the outer temporal capsule and anterior pole, multiple lacunar infarcts, and extensive demyelination (73, 100). Nevertheless, some atypical imaging findings of CADASIL, such as corpus callosum lesions (101) and CMBs (102), have been detected in an increasing number of patients, and their influence on disease severity and prognosis should not be overlooked (103, 104). Additionally, a study conducted in Slovakia identified 23 pathogenic variants in 35 unrelated families. Among them, the mutation causing the genetic defect was found in 10.2% of patients with clinically suspected CADASIL who were eventually diagnosed with CADASIL (105). Therefore, NOTCH3 gene screening criteria should not be restricted to patients with high risk for CADASIL, especially those without typical imaging findings but presenting with early-onset stroke, a family history of stroke or dementia, and no hypertension. Such patients may benefit from a more comprehensive analysis of their clinical phenotype. In cases where diagnosis remains challenging, a skin biopsy should be performed to assist in the diagnosis.
Furthermore, the assessment of disease severity frequently depends on standardized clinical metrics (e.g., neuroimaging scores) and the assessor's personal judgment. To better analyze and compare the relevant data, a relatively consistent criterion is necessary to measure the severity in all patients. Some researchers have proposed a CADASIL severity grading system that encompasses five levels: grade 0 (absence of symptoms), grade 1 (migraine alone), grade 2 (stroke, TIA, or mild cognitive impairment), grade 3 (requiring walking assistance or dementia), and grade 4 (bedridden or advanced stage) for patients with known pathogenic NOTCH3 mutations, characteristic ischemic lesions on brain MRI, or characteristic intravascular deposits on skin biopsy (106). Some studies have validated the scoring system regionally, with relatively promising outcomes (107). More recently, a NOTCH3-SVD staging system was developed for CADASIL, the classic cysteine-altered NOTCH3 variant, to better assess disease severity and monitor disease progression. The NOTCH3-SVD staging system further refines this into nine substages (stages 0 to 4B), offering a standardized framework for monitoring progression (108). However, the reliability and applicability of the scoring system require further validation in more diverse patient populations.
5 Conclusion
CADASIL is the most common genetic cause of dementia in adults, characterized by significant phenotypic variability among patients. Over recent decades, studies utilizing animal models, cellular systems, and patient cohorts have provided valuable insights into the pathogenic mechanisms underlying CADASIL. This review primarily focused on elucidating the relationship between CADASIL and its clinical phenotypes and identified at least five factors associated with the severity of clinical manifestations in CADASIL: mutation location, variant types, environmental factors, epigenetic regulation, and polygenic interactions. Additionally, the review emphasizes the importance of refining clinical diagnostic criteria for CADASIL to improve the assessment of disease severity. Despite these advances, several potential mechanisms remain unexplored. Future studies should prioritize combining gene editing methods with multi-omics analysis to dissect the downstream molecular mechanisms caused by NOTCH3 mutations. In addition, related studies of biomarkers are also of great significance for predicting patient phenotype and prognosis. Continuous research on the genotype-phenotype correlations in CADASIL is crucial for unraveling its heterogeneity, identifying therapeutic targets, enabling early diagnosis, and mitigating disease progression. These efforts are expected to support the development of precision medicine approaches tailored to CADASIL.
Data availability statement
This review does not involve original data collection. However, the data used to support the findings of this review are publicly available from the following sources: Gene Expression Omnibus (GEO): all relevant gene expression data discussed in this review can be accessed from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). PubMed: relevant articles and publications used in this review are available through PubMed (https://pubmed.ncbi.nlm.nih.gov/). Protein Data Bank (PDB): structural data referenced in this review can be accessed through the PDB (https://www.rcsb.org/). Data sharing and accessibility were considered in the preparation of this review, and all cited data are publicly available for further research and analysis.
Author contributions
YZ: Conceptualization, Writing – original draft. YLu: Conceptualization, Writing – original draft. FW: Conceptualization, Writing – review & editing. YW: Data curation, Writing – review & editing. YLi: Visualization, Writing – review & editing. RS: Visualization, Writing – review & editing. JS: Funding acquisition, Writing – review & editing. CJ: Supervision, Writing – review & editing. JZ: Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Key Research and Development Program of Henan Province, China (241111313500) and Henan Provincial Major Project of Medical Science and Technology Key Research Program under Provincial-Ministerial Co-Construction (SBGJ202401002).
Acknowledgments
We express our gratitude to all participants for their involvement in the review, as well as to all other contributors for their efforts and support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
APOE, Apolipoprotein E; CADASIL, Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; CMBs, Cerebral microbleeds; CSVD, Cerebral small vessel diseases; ECD, Extracellular domain; EGFr, Epidermal growth factor-like repeats; GLUTs, Glucose transporters; GOM, Granular osmiophilic material; ICD, Intracellular domain; LBD, Ligand-binding domain; MAM, Mastermind; MRI, Magnetic resonance imaging; mtDNA, mitochondrial DNA; NTF, N-terminal NOTCH3 fragments; RBP-Jκ, Recombination signal-binding protein for immunoglobulin kappa J region; TIA, Transient ischemic attack; TIMP3, Tissue inhibitor of metalloproteinase 3; VSMC, Vascular smooth muscle cells; VTN, Vitronectin; WMH: White matter hyperintensities.
References
1. Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. (1996) 383:707–10. doi: 10.1038/383707a0
2. Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, et al. Notch3 mutations in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), a mendelian condition causing stroke and vascular dementia. Ann NY Acad Sci. (1997) 826:213–7. doi: 10.1111/j.1749-6632.1997.tb48472.x
3. Davous P. CADASIL: a review with proposed diagnostic criteria. Eur. J. Neurol. (1998) 5:219–33. doi: 10.1046/j.1468-1331.1998.530219.x
4. Carvalho MDSDN, Santos JADO, Carvalho MDR, Barbosa ND, Melo LCD, Costa FHMD, et al. Clinical manifestations, laboratorial findings and mechanisms of CADASIL: an integrative review. Res. Soc. Dev. (2022) 11:e21311528005. doi: 10.33448/rsd-v11i5.28005
5. Schoemaker D, Quiroz YT, Torrico-Teave H, Arboleda-Velasquez JF. Clinical and research applications of magnetic resonance imaging in the study of CADASIL. Neurosci Lett. (2019) 698:173–9. doi: 10.1016/j.neulet.2019.01.014
6. Ampuero I, Alegre-Abarrategui J, Rodal I, España A, Ros R, Sendón JLL, et al. On the diagnosis of CADASIL. J Alzheimer's Dis. (2009) 17:787–94. doi: 10.3233/JAD-2009-1112
7. Cho BPH, Nannoni S, Harshfield EL, Tozer D, Gräf S, Bell S, et al. NOTCH3 variants are more common than expected in the general population and associated with stroke and vascular dementia: an analysis of 200 000 participants. J Neurol Neurosurg Psychiat. (2021) 92:694–701. doi: 10.1136/jnnp-2020-325838
8. Joutel A, Monet M, Domenga V, Riant F, Tournier-Lasserve E. Pathogenic mutations associated with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy differently affect Jagged1 binding and Notch3 activity via the RBP/JK signaling Pathway. Am J Hum Genet. (2004) 74:338–47. doi: 10.1086/381506
9. Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. (1999) 284:770–6. doi: 10.1126/science.284.5415.770
10. Ables JL, Breunig JJ, Eisch AJ, Rakic P. Not(ch) just development: notch signalling in the adult brain. Nat Rev Neurosci. (2011) 12:269–83. doi: 10.1038/nrn3024
11. Rebay I, Fleming RJ, Fehon RG, Cherbas L, Cherbas P, Artavanis-Tsakonas S. Specific EGF repeats of notch mediate interactions with delta and serrate: implications for notch as a multifunctional receptor. Cell. (1991) 67:687–99. doi: 10.1016/0092-8674(91)90064-6
12. Manini A, Pantoni L. CADASIL from bench to bedside: disease models and novel therapeutic approaches. Mol Neurobiol. (2021) 58:2558–73. doi: 10.1007/s12035-021-02282-4
13. Schweisguth F. Regulation of notch signaling activity. Curr Biol. (2004) 14:R129–138. doi: 10.1016/S0960-9822(04)00038-7
14. Campos AH, Wang W, Pollman MJ, Gibbons GH. Determinants of Notch-3 receptor expression and signaling in vascular smooth muscle cells: implications in cell-cycle regulation. Circ Res. (2002) 91:999–1006. doi: 10.1161/01.RES.0000044944.99984.25
15. Joutel A, Vahedi K, Corpechot C, Troesch A, Chabriat H, Vayssière C, et al. Strong clustering and stereotyped nature of Notch3 mutations in CADASIL patients. Lancet. (1997) 350:1511–5. doi: 10.1016/S0140-6736(97)08083-5
16. Duering M, Karpinska A, Rosner S, Hopfner F, Zechmeister M, Peters N, et al. Co-aggregate formation of CADASIL-mutant NOTCH3: A single-particle analysis. Hum Mol Genet. (2011) 20:3256–65. doi: 10.1093/hmg/ddr237
17. Takahashi K, Adachi K, Yoshizaki K, Kunimoto S, Kalaria RN, Watanabe A. Mutations in NOTCH3 cause the formation and retention of aggregates in the endoplasmic reticulum, leading to impaired cell proliferation. Hum Mol Genet. (2010) 19:79–89. doi: 10.1093/hmg/ddp468
18. Kopan R, Ilagan MXG. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. (2009) 137:216–33. doi: 10.1016/j.cell.2009.03.045
19. Ishiko A, Shimizu A, Nagata E, Takahashi K, Tabira T, Suzuki N. Notch3 ectodomain is a major component of granular osmiophilic material (GOM) in CADASIL. Acta Neuropathol. (2006) 112:333–9. doi: 10.1007/s00401-006-0116-2
20. Monet-Leprêtre M, Haddad I, Baron-Menguy C, Fouillot-Panchal M, Riani M, Domenga-Denier V, et al. Abnormal recruitment of extracellular matrix proteins by excess Notch3 ECD: a new pathomechanism in CADASIL. Brain. (2013) 136:1830–45. doi: 10.1093/brain/awt092
21. Capone C, Cognat E, Ghezali L, Baron-Menguy C, Aubin D, Mesnard L, et al. Reducing Timp3 or vitronectin ameliorates disease manifestations in CADASIL mice. Ann Neurol. (2016) 79:387–403. doi: 10.1002/ana.24573
22. Miao Q, Paloneva T, Tuominen S, Pöyhönen M, Tuisku S, Viitanen M, et al. Fibrosis and stenosis of the long penetrating cerebral arteries: the cause of the white matter pathology in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Brain Pathol. (2004) 14:358–64. doi: 10.1111/j.1750-3639.2004.tb00078.x
23. Mayer M, Straube A, Bruening R, Uttner I, Pongratz D, Gasser T, et al. Muscle and skin biopsies are a sensitive diagnostic tool in the diagnosis of CADASIL. J Neurol. (1999) 246:526–32. doi: 10.1007/s004150050398
24. Monet-Leprêtre M, Bardot B, Lemaire B, Domenga V, Godin O, Dichgans M, et al. Distinct phenotypic and functional features of CADASIL mutations in the Notch3 ligand binding domain. Brain. (2009) 132:1601–12. doi: 10.1093/brain/awp049
25. Rutten JW, Klever RR, Hegeman IM, Poole DS, Dauwerse HG, Broos LAM, et al. The NOTCH3 score: a pre-clinical CADASIL biomarker in a novel human genomic NOTCH3 transgenic mouse model with early progressive vascular NOTCH3 accumulation. Acta Neuropathol Commun. (2015) 3:89. doi: 10.1186/s40478-015-0268-1
26. Karlström H, Beatus P, Dannaeus K, Chapman G, Lendahl U, Lundkvist J, et al. CADASIL-mutated Notch 3 receptor exhibits impaired intracellular trafficking and maturation but normal ligand-induced signaling. Proc Natl Acad Sci USA. (2002) 99:17119–24. doi: 10.1073/pnas.252624099
27. Arboleda-Velasquez JF, Manent J, Lee JH, Tikka S, Ospina C, Vanderburg CR, et al. Hypomorphic notch 3 alleles link notch signaling to ischemic cerebral small-vessel disease. Proc Natl Acad Sci. (2011) 108:E128–35. doi: 10.1073/pnas.1101964108
28. Moccia M, Mosca L, Erro R, Cervasio M, Allocca R, Vitale C, et al. Hypomorphic NOTCH3 mutation in an italian family with CADASIL features. Neurobiol Aging. (2015) 36:547.e5-11. doi: 10.1016/j.neurobiolaging.2014.08.021
29. Peters N, Opherk C, Zacherle S, Capell A, Gempel P, Dichgans M. CADASIL-associated Notch3 mutations have differential effects both on ligand binding and ligand-induced Notch3 receptor signaling through RBP-jk. Exp Cell Res. (2004) 299:454–64. doi: 10.1016/j.yexcr.2004.06.004
30. Machuca-Parra AI, Bigger-Allen AA, Sanchez AV, Boutabla A, Cardona-Vélez J, Amarnani D, et al. Therapeutic antibody targeting of Notch3 signaling prevents mural cell loss in CADASIL. J Exp Med. (2017) 214:2271–82. doi: 10.1084/jem.20161715
31. Baron-Menguy C, Domenga-Denier V, Ghezali L, Faraci FM, Joutel A. Increased Notch3 activity mediates pathological changes in structure of cerebral arteries. Hypertension. (2017) 69:60–70. doi: 10.1161/HYPERTENSIONAHA.116.08015
32. Schoemaker D, Arboleda-Velasquez JF. Notch3 signaling and aggregation as targets for the treatment of CADASIL and other NOTCH3-associated small-vessel diseases. Am J Pathol. (2021) 191:1856–70. doi: 10.1016/j.ajpath.2021.03.015
33. Andrew RJ, Kellett KAB, Thinakaran G, Hooper NM. A Greek tragedy: the growing complexity of Alzheimer amyloid precursor protein proteolysis. J Biol Chem. (2016) 291:19235–44. doi: 10.1074/jbc.R116.746032
34. Berning BA, Walker AK. The Pathobiology of TDP-43 C-Terminal fragments in ALS and FTLD. Front Neurosci. (2019) 13:335. doi: 10.3389/fnins.2019.00335
35. Kovall RA, Gebelein B, Sprinzak D, Kopan R. The canonical notch signaling pathway: structural and biochemical insights into shape, sugar, and force. Dev Cell. (2017) 41:228–41. doi: 10.1016/j.devcel.2017.04.001
36. Young KZ, Lee SJ, Zhang X, Cartee NMP, Torres M, Keep SG, et al. NOTCH3 is non-enzymatically fragmented in inherited cerebral small-vessel disease. J Biol Chem. (2020) 295:1960–72. doi: 10.1074/jbc.RA119.007724
37. Zhang X, Lee SJ, Wang MM. Hydrolysis of a second Asp-Pro site at the N-terminus of NOTCH3 in inherited vascular dementia. Sci Rep. (2021) 11:17246. doi: 10.1038/s41598-021-96679-9
38. Lee SJ, Zhang X, Xu G, Borjigin J, Wang MM. A midposition NOTCH3 truncation in inherited cerebral small vessel disease may affect the protein interactome. J Biol Chem. (2023) 299:102772. doi: 10.1016/j.jbc.2022.102772
39. Rutten JW, Van Eijsden BJ, Duering M, Jouvent E, Opherk C, Pantoni L, et al. The effect of NOTCH3 pathogenic variant position on CADASIL disease severity: NOTCH3 EGFr 1–6 pathogenic variant are associated with a more severe phenotype and lower survival compared with EGFr 7–34 pathogenic variant. Genet Med. (2019) 21:676–82. doi: 10.1038/s41436-018-0088-3
40. Dupé C, Guey S, Biard L, Dieng S, Lebenberg J, Grosset L, et al. Phenotypic variability in 446 CADASIL patients: impact of NOTCH3 gene mutation location in addition to the effects of age, sex and vascular risk factors. J Cereb Blood Flow Metab. (2023) 43:153–66. doi: 10.1177/0271678X221126280
41. Opherk C, Peters N, Herzog J, Luedtke R, Dichgans M. Long-term prognosis and causes of death in CADASIL: a retrospective study in 411 patients. Brain. (2004) 127:2533–9. doi: 10.1093/brain/awh282
42. Hack RJ, Gravesteijn G, Cerfontaine MN, Hegeman IM, Mulder AA, Lesnik Oberstein SAJ, et al. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy family members with a pathogenic NOTCH3 variant can have a normal brain magnetic resonance imaging and skin biopsy beyond age 50 years. Stroke. (2022) 53:1964–74. doi: 10.1161/STROKEAHA.121.036307
43. Mukai M, Mizuta I, Watanabe-Hosomi A, Koizumi T, Matsuura J, Hamano A, et al. Genotype-phenotype correlations and effect of mutation location in japanese CADASIL patients. J Hum Genet. (2020) 65:637–46. doi: 10.1038/s10038-020-0751-9
44. Liu JY, Yao M, Dai Y, Han F, Zhai FF, Zhang DD, et al. Rare NOTCH3 variants in a Chinese population-based cohort and its relationship with cerebral small vessel disease. Stroke. (2021) 52:3918–25. doi: 10.1161/STROKEAHA.120.032265
45. Rutten JW, Dauwerse HG, Gravesteijn G, van Belzen MJ, van der Grond J, Polke JM, et al. Archetypal NOTCH3 mutations frequent in public exome: implications for CADASIL. Annal Clin Trans Neurol. (2016) 3:844–53. doi: 10.1002/acn3.344
46. Hack RJ, Gravesteijn G, Cerfontaine MN, Santcroos MA, Gatti L, Kopczak A, et al. Three-tiered EGFr domain risk stratification for individualized NOTCH3-small vessel disease prediction. Brain. (2023) 146:2913–27. doi: 10.1093/brain/awac486
47. Cerfontaine MN, Hack RJ, Gesierich B, Duering M, Witjes-Ané MNW, Rodríguez-Girondo M, et al. Association of NOTCH3 variant risk category with 2-year clinical and radiologic small vessel disease progression in patients with CADASIL. Neurology. (2024) 102:e209310. doi: 10.1212/WNL.0000000000209310
48. Gravesteijn G, Hack RJ, Mulder AA, Cerfontaine MN, Doorn R, Hegeman IM, et al. NOTCH3 variant position is associated with NOTCH3 aggregation load in CADASIL vasculature. Neuropathol. Appl. Neurobiol. (2022) 48:e12751. doi: 10.1111/nan.12751
49. Arboleda–Velasquez JF, Lopera F, Lopez E, Frosch MP, Sepulveda–Falla D, Gutierrez JE, et al. C455R notch3 mutation in a Colombian CADASIL kindred with early onset of stroke. Neurology. (2002) 59: 277–9. doi: 10.1212/WNL.59.2.277
50. Li Y, Lei L, Irvine KD, Baker NE. Notch activity in neural cells triggered by a mutant allele with altered glycosylation. Development. (2003) 130:2829–40. doi: 10.1242/dev.00498
51. Dotti MT, De Stefano N, Bianchi S, Malandrini A, Battisti C, Cardaioli E, et al. A novel NOTCH3 frameshift deletion and mitochondrial abnormalities in a patient with CADASIL. Arch Neurol. (2004) 61:942–5. doi: 10.1001/archneur.61.6.942
52. Joutel A. Pathogenesis of CADASIL: transgenic and knock-out mice to probe function and dysfunction of the mutated gene, Notch3, in the cerebrovasculature. Bioessays. (2011) 33:73–80. doi: 10.1002/bies.201000093
53. Rutten JW, Boon EMJ, Liem MK, Dauwerse JG, Pont MJ, Vollebregt E, et al. Hypomorphic NOTCH3 alleles do not cause CADASIL in humans. Hum Mutat. (2013) 34:1486–9. doi: 10.1002/humu.22432
54. Huang L, Li W, Li Y, Song C, Wang P, Wang H, et al. A novel cysteine-sparing G73A mutation of NOTCH3 in a Chinese CADASIL family. Neurogenetics. (2020) 21:39–49. doi: 10.1007/s10048-019-00592-3
55. Muiño E, Gallego-Fabrega C, Cullell N, Carrera C, Torres N, Krupinski J, et al. Systematic review of cysteine-sparing NOTCH3 missense mutations in patients with clinical suspicion of CADASIL. Int J Mol Sci. (2017) 18:1964. doi: 10.3390/ijms18091964
56. Kim H, Lim YM, Lee EJ, Oh YJ, Kim KK. Clinical and imaging features of patients with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy and cysteine-sparing NOTCH3 mutations. PLoS ONE. (2020) 15:e0234797. doi: 10.1371/journal.pone.0234797
57. Hu Y, Sun Q, Zhou Y, Yi F, Tang H, Yao L, et al. NOTCH3 variants and genotype-phenotype features in Chinese CADASIL patients. Front Genet. (2021) 12:705284. doi: 10.3389/fgene.2021.705284
58. Cao Y, Zhang DD, Han F, Jiang N, Yao M, Zhu YC. Phenotypes associated with NOTCH3 cysteine-sparing mutations in patients with clinical suspicion of CADASIL: a systematic review. Int J Mol Sci. (2024) 25:8796. doi: 10.3390/ijms25168796
59. Wollenweber FA, Hanecker P, Bayer-Karpinska A, Malik R, Bäzner H, Moreton F, et al. Cysteine-sparing CADASIL mutations in NOTCH3 show proaggregatory properties in vitro. Stroke. (2015) 46:786–92. doi: 10.1161/STROKEAHA.114.007472
60. Chabriat H, Vahedi K, Iba-Zizen MT, Joutel A, Nibbio A, Nagy TG, et al. Clinical spectrum of CADASIL: a study of 7 families. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Lancet. (1995) 346:934–9. doi: 10.1016/S0140-6736(95)91557-5
61. Kalimo H, Ruchoux MM, Viitanen M, Kalaria RN. CADASIL a common form of hereditary arteriopathy causing brain infarcts and dementia. Brain Pathol. (2006) 12:371–84. doi: 10.1111/j.1750-3639.2002.tb00451.x
62. Singhal S, Rich P, Markus HS. The spatial distribution of MR imaging abnormalities in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy and their relationship to age and clinical features. AJNR Am J Neuroradiol. (2005) 26:2481–7.
63. Wang Z, Yuan Y, Zhang W, Lv H, Hong D, Chen B, et al. NOTCH3 mutations and clinical features in 33 mainland Chinese families with CADASIL. J Neurol Neurosurg Psychiat. (2011) 82:534–9. doi: 10.1136/jnnp.2010.209247
64. Chabriat H, Mrissa R, Levy C, Vahedi K, Taillia H, Iba-Zizen MT, et al. Brain stem MRI signal abnormalities in CADASIL. Stroke. (1999) 30:457–9. doi: 10.1161/01.STR.30.2.457
65. Liu X, Zuo Y, Sun W, Zhang W, Lv H, Huang Y, et al. The genetic spectrum and the evaluation of CADASIL screening scale in Chinese patients with NOTCH3 mutations. J Neurol Sci. (2015) 354:63–9. doi: 10.1016/j.jns.2015.04.047
66. Mykkänen K, Junna M, Amberla K, Bronge L, Kääriäinen H, Pöyhönen M, et al. Different clinical phenotypes in monozygotic CADASIL twins with a novel NOTCH3 mutation. Stroke. (2009) 40:2215–8. doi: 10.1161/STROKEAHA.108.528661
67. Tuominen S, Juvonen V, Amberla K, Jolma T, Rinne JO, Tuisku S, et al. Phenotype of a homozygous CADASIL patient in comparison to 9 age-matched heterozygous patients with the same R133C Notch3 mutation. Stroke. (2001) 32:1767–74. doi: 10.1161/01.STR.32.8.1767
68. Abou Al-Shaar H, Qadi N, Al-Hamed MH, Meyer BF, Bohlega S. Phenotypic comparison of individuals with homozygous or heterozygous mutation of NOTCH3 in a large CADASIL family. J Neurol Sci. (2016) 367:239–43. doi: 10.1016/j.jns.2016.05.061
69. Liem MK, Lesnik Oberstein SAJ, Vollebregt MJ, Middelkoop HAM, van der Grond J, Helderman-van den Enden ATJM. Homozygosity for a NOTCH3 mutation in a 65-year-old CADASIL patient with mild symptoms: a family report. J Neurol. (2008) 255:1978–80. doi: 10.1007/s00415-009-0036-x
70. Soong BW, Liao YC, Tu PH, Tsai PC, Lee IH, Chung CP. et al. A homozygous NOTCH3 mutation pR544C and a heterozygous TREX1 variant pC99MfsX3 in a family with hereditary small vessel disease of the brain. J Chin Med Assoc. (2013) 76:319–24. doi: 10.1016/j.jcma.2013.03.002
71. Singhal S, Bevan S, Barrick T, Rich P, Markus HS. The influence of genetic and cardiovascular risk factors on the CADASIL phenotype. Brain. (2004) 127:2031–8. doi: 10.1093/brain/awh223
72. Choi JC, Song SK, Lee JS, Kang SY, Kang JH. Diversity of stroke presentation in CADASIL: study from patients harboring the predominant NOTCH3 mutation R544C. J Stroke Cerebrovasc Dis. (2013) 22:126–31. doi: 10.1016/j.jstrokecerebrovasdis.2011.07.002
73. Chabriat H, Levy C, Taillia H, Iba-Zizen MT, Vahedi K, Joutel A, et al. Patterns of MRI lesions in CADASIL. Neurology. (1998) 51:452–7. doi: 10.1212/WNL.51.2.452
74. Ospina C, Arboleda-Velasquez JF, Aguirre-Acevedo DC, Zuluaga-Castaño Y, Velilla L, Garcia GP, et al. Genetic and nongenetic factors associated with CADASIL: a retrospective cohort study. J Neurol Sci. (2020) 419:117178. doi: 10.1016/j.jns.2020.117178
75. Panahi M, Rodriguez PR, Fereshtehnejad SM, Arafa D, Bogdanovic N, Winblad B, et al. Insulin-independent and dependent glucose transporters in brain mural cells in CADASIL. Front Genet. (2020) 11:1022. doi: 10.3389/fgene.2020.01022
76. Hassan A, Hunt BJ, O'Sullivan M, Bell R, D'Souza R, Jeffery S, et al. Homocysteine is a risk factor for cerebral small vessel disease, acting via endothelial dysfunction. Brain. (2004) 127:212–9. doi: 10.1093/brain/awh023
77. Herrmann W, Obeid R. Homocysteine: a biomarker in neurodegenerative diseases. Clin Chem Lab Med. (2011) 49:435–41. doi: 10.1515/CCLM.2011.084
78. Mawet J, Vahedi K, Aout M, Vicaut E, Duering M, Touboul PJ, et al. Carotid atherosclerotic markers in CADASIL. Cerebrovasc Dis. (2011) 31:246–52. doi: 10.1159/000321932
79. Takashi Koizumi, Mizuta I, Watanabe-Hosomi A, Mukai M, Hamano A, Matsuura J, et al. The CADASIL Scale-J, a modified scale to prioritize access to genetic testing for Japanese CADASIL-suspected patients. J Stroke Cerebrovasc Dis. (2019) 28: 1431–9. doi: 10.1016/j.jstrokecerebrovasdis.2019.03.026
80. Gunda B, Hervé D, Godin O, Bruno M, Reyes S, Alili N, et al. Effects of gender on the phenotype of CADASIL. Stroke. (2012) 43:137–41. doi: 10.1161/STROKEAHA.111.631028
81. Opherk C, Peters N, Holtmannspötter M, Gschwendtner A, Müller-Myhsok B, Dichgans M. Heritability of MRI lesion volume in CADASIL: evidence for genetic modifiers. Stroke. (2006) 37:2684–9. doi: 10.1161/01.STR.0000245084.35575.66
82. Cavalli G, Heard E. Advances in epigenetics link genetics to the environment and disease. Nature. (2019) 571:489–99. doi: 10.1038/s41586-019-1411-0
83. Di Francesco A, Arosio B, Falconi A, Micioni Di Bonaventura MV, Karimi M, Mari D, et al. Global changes in DNA methylation in Alzheimer's disease peripheral blood mononuclear cells. Brain Behav Immun. (2015) 45:139–44. doi: 10.1016/j.bbi.2014.11.002
84. Batista PJ. The RNA modification N6-methyladenosine and its implications in human disease. Genomics Proteomics Bioinformatics. (2017) 15:154–63. doi: 10.1016/j.gpb.2017.03.002
85. Marshall LL, Killinger BA, Ensink E, Li P, Li K, Cui W, et al. Epigenomic analysis of Parkinson's disease neurons identifies Tet2 loss as neuroprotective. Nat Neurosci. (2020) 23:1203–14. doi: 10.1038/s41593-020-0690-y
86. Nativio R, Lan Y, Donahue G, Sidoli S, Berson A, Srinivasan AR, et al. An integrated multi-omics approach identifies epigenetic alterations associated with Alzheimer's disease. Nat Genet. (2020) 52:1024–35. doi: 10.1038/s41588-020-00733-7
87. Gravesteijn G, Dauwerse JG, Overzier M, Brouwer G, Hegeman I, Mulder AA, et al. Naturally occurring NOTCH3 exon skipping attenuates NOTCH3 protein aggregation and disease severity in CADASIL patients. Hum Mol Genet. (2020) 29:1853–63. doi: 10.1093/hmg/ddz285
88. Rufa A, Blardi P, De Lalla A, Cevenini G, De Stefano N, Zicari E, et al. Plasma levels of asymmetric dimethylarginine in cerebral autosomal dominant arteriopathy with subcortical infarct and leukoencephalopathy. Cerebrovasc Dis. (2008) 26:636–40. doi: 10.1159/000166840
89. Zhang J, Shang J, Wang F, Huo X, Sun R, Ren Z, et al. Decreased mitochondrial D-loop region methylation mediates an increase in mitochondrial DNA copy number in CADASIL. Clin Epigenet. (2022) 14:2. doi: 10.1186/s13148-021-01225-z
90. Almeida MR, Silva AR, Elias I, Fernandes C, Machado R, Galego O, et al. SQSTM1 gene as a potential genetic modifier of CADASIL phenotype. J Neurol. (2021) 268:1453–60. doi: 10.1007/s00415-020-10308-5
91. Gesierich B, Opherk C, Rosand J, Gonik M, Malik R, Jouvent E, et al. APOE ε2 is associated with white matter hyperintensity volume in CADASIL. J Cereb Blood Flow Metab. (2016) 36:199–203. doi: 10.1038/jcbfm.2015.85
92. Cheng YW, Liao YC, Chen CH, Chung CP, Fann CSJ, Chang CC, et al. Contribution of the APOE genotype to cognitive impairment in individuals with NOTCH3 cysteine-altering variants. J Am Heart Assoc. (2023) 12:e032689. doi: 10.1161/JAHA.123.032689
93. Opherk C, Gonik M, Duering M, Malik R, Jouvent E, Hervé D, et al. Genome-wide genotyping demonstrates a polygenic risk score associated with white matter hyperintensity volume in CADASIL. Stroke. (2014) 45:968–72. doi: 10.1161/STROKEAHA.113.004461
94. Murakami T, Iwatsuki K, Hayashi T, Sato K, Matsubara E, Nagano I, et al. Two Japanese CADASIL families with a R141C mutation in the Notch3 gene. Intern Med. (2001) 40:1144–8. doi: 10.2169/internalmedicine.40.1144
95. Peters N, Opherk C, Bergmann T, Castro M, Herzog JR, Dichgans M. Spectrum of mutations in biopsy-proven CADASIL - Implications for diagnostic strategies. Arch Neurol. (2005) 62:1091–4. doi: 10.1001/archneur.62.7.1091
96. Markus HS, Martin RJ, Simpson MA, Dong YB, Ali N, Crosby AH, et al. Diagnostic strategies in CADASIL. Neurology. (2002) 59:1134–8. doi: 10.1212/WNL.59.8.1134
97. Chen S, Ni W, Yin XZ, Liu HQ, Lu C, Zheng QJ, et al. Clinical features and mutation spectrum in Chinese patients with CADASIL: a multicenter retrospective study. CNS Neurosci Ther. (2017) 23:707–16. doi: 10.1111/cns.12719
98. Ni W, Zhang Y, Zhang L, Xie JJ Li HF, Wu ZY. Genetic spectrum of NOTCH3 and clinical phenotype of CADASIL patients in different populations. CNS Neurosci Ther. (2022) 28:1779–89. doi: 10.1111/cns.13917
99. Pescini F, Nannucci S, Bertaccini B, Salvadori E, Bianchi S, Ragno M, et al. The cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) scale: a screening tool to select patients for NOTCH3 gene analysis. Stroke. (2012) 43:2871–6. doi: 10.1161/STROKEAHA.112.665927
100. Bersano A, Bedini G, Markus HS, Vitali P, Colli-Tibaldi E, Taroni F, et al. The role of clinical and neuroimaging features in the diagnosis of CADASIL. J Neurol. (2018) 265:2934–43. doi: 10.1007/s00415-018-9072-8
101. Coulthard A, Blank SC, Bushby K, Kalaria RN, Burn DJ. Distribution of cranial MRI abnormalities in patients with symptomatic and subclinical CADASIL. Br J Radiol. (2000) 73:256–65. doi: 10.1259/bjr.73.867.10817040
102. Oberstein S, van den Boom R, van Buchem MA, van Houwelingen HC, Bakker E, Vollebregt E, et al. Cerebral microbleeds in CADASIL. Neurology. (2001) 57:1066–70. doi: 10.1212/WNL.57.6.1066
103. Chen CH, Cheng YW, Chen YF, Tang SC, Jeng JS. Plasma neurofilament light chain and glial fibrillary acidic protein predict stroke in CADASIL. J Neuroinflammation. (2020) 17:124. doi: 10.1186/s12974-020-01813-5
104. Chung CP, Chen JW, Chang FC Li WC, Lee YC, Chen LF, et al. Cerebral microbleed burdens in specific brain regions are associated with disease severity of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. J Am Heart Assoc. (2020) 9:e016233. doi: 10.1161/JAHA.120.016233
105. Juhosová M, Chandoga J, Cisárik F, Dallemule S, Durina P, Jarásková D, et al. Influence of different spectra of NOTCH3 variants on the clinical phenotype of CADASIL - experience from slovakia. Neurogenetics. (2023) 24:1–16. doi: 10.1007/s10048-022-00704-6
106. Meschia JF. Severity grading of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Neurol Neurochir Pol. (2022) 56:193–4. doi: 10.5603/PJNNS.a2022.0019
107. Anisetti B, Greco E, Stojadinovic E, Goldstein ED, Sakusic A, Badi MK, et al. Novel grading system for CADASIL severity: a multicenter cross-sectional study. Cereb Circ Cogn Behav. (2023) 5:100170. doi: 10.1016/j.cccb.2023.100170
108. Gravesteijn G, Rutten JW, Cerfontaine MN, Hack RJ, Liao YC, Jolly AA, et al. Disease severity staging system for NOTCH3-associated small vessel disease, including CADASIL. JAMA Neurol. (2024) 82:49–60. doi: 10.1001/jamaneurol.2024.4487
109. Cho BPH, Jolly AA, Nannoni S, Tozer D, Bell S, Markus HS. Association of NOTCH3 variant position with stroke onset and other clinical features among patients with CADASIL. Neurology. (2022) 99:e430–9. doi: 10.1212/WNL.0000000000200744
110. Hack RJ, Cerfontaine MN, Gravesteijn G, Tap S, Hafkemeijer A, van der Grond J, et al. Effect of NOTCH3 EGFr group, sex, and cardiovascular risk factors on CADASIL clinical and neuroimaging outcomes. Stroke. (2022) 53:3133–44. doi: 10.1161/STROKEAHA.122.039325
111. Bianchi S, Zicari E, Carluccio A, Di Donato I, Pescini F, Nannucci S, et al. CADASIL in central italy: a retrospective clinical and genetic study in 229 patients. J Neurol. (2015) 262:134–41. doi: 10.1007/s00415-014-7533-2
Keywords: CADASIL, NOTCH3, granular osmiophilic material, epidermal growth factor-like repeats, phenotype
Citation: Zhao Y, Lu Y, Wang F, Wang Y, Li Y, Sun R, Shang J, Jiang C and Zhang J (2025) Mechanistic advances in factors influencing phenotypic variability in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy: a review. Front. Neurol. 16:1573052. doi: 10.3389/fneur.2025.1573052
Received: 10 February 2025; Accepted: 29 April 2025;
Published: 21 May 2025.
Edited by:
Félix Javier Jiménez-Jiménez, Hospital Universitario del Sureste, SpainReviewed by:
Yong Chen, Peking University Third Hospital, ChinaChih-Hao Chen, National Taiwan University Hospital, Taiwan
Copyright © 2025 Zhao, Lu, Wang, Wang, Li, Sun, Shang, Jiang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiewen Zhang, emhhbmdqaWV3ZW45OTAwQDEyNi5jb20=
†These authors have contributed equally to this work and share first authorship