Abstract
Copper, an essential trace element for the human body, plays a key role in energy metabolism, mitochondrial respiration, redox reactions, and neural signal transmission. The recently proposed concept of “cuproptosis” has further revealed the unique status of copper in cellular regulation: when copper abnormally accumulates within cells, it can directly bind to the lipoylated proteins of the mitochondrial TCA cycle, triggering protein aggregation and metabolic disorders, ultimately leading to cell death. This form of cell death plays an important role in various neurodegenerative diseases of the central nervous system, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD), and stroke. This review summarizes recent research on the mechanisms of cuproptosis, providing new perspectives and a theoretical basis for understanding the pathogenesis of these neurodegenerative diseases.
Introduction
Copper, an essential trace element, is primarily absorbed through dietary intake and enters intestinal epithelial cells (1). From there, it is distributed and transported throughout the body by various transport proteins (e.g., Ctr1 and Ctr2) and copper chaperone proteins (e.g., ATOX1, CCS, and COX17) (2). The liver, as the primary storage site for copper, regulates its distribution to tissues or excretion via bile (3). Copper is essential in physiological processes such as mitochondrial respiration, cellular energy metabolism, electron transport, and neurotransmitter synthesis (4). Copper levels are closely linked to immune function and typically remain stable within a certain range under normal physiological conditions, primarily in the brain, liver, and bones (5, 6). However, both excess and deficiency of copper can lead to pathological changes, adversely affecting health (7).
Cuproptosis is a newly identified form of cell death first reported and named by Tsvetkov et al. (8). The study found that copper ions bind directly to the lipoylated proteins of the mitochondrial TCA cycle, causing protein aggregation and metabolic disruptions that ultimately result in cell death (8). Among these key proteins, lipoylated proteins (particularly the PDH complex) and Fe–S cluster proteins serve as copper-binding sites and play critical roles in the cell (9). When copper ion levels rise, the functions of these proteins are compromised, accompanied by protein toxicity stress (such as HSP70 induction) and the collapse of mitochondrial metabolism, ultimately leading to cell death (7). FDX1, identified as a central regulatory gene of cuproptosis, promotes copper-dependent cell death by regulating the protein lipoylation process (10). This copper ion overload-induced programmatic cell death mechanism differs from all known forms of programmed cell death (such as apoptosis, ferroptosis, pyroptosis, and necroptosis) (9). The key events involve abnormal copper accumulation in cells and mitochondria, leading to impaired mitochondrial function, enhanced oxidative stress, and metabolic dysregulation (11).
Neurodegenerative diseases (NDD) are caused by degeneration or demyelination of neurons in the brain or spinal cord and can result from various etiologies, such as abnormal protein aggregation, misfolded protein propagation, genetic mutations, and epigenetic modifications (12). Examples of NDDs include Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), spinal muscular atrophy (SMA), multiple sclerosis (MS), traumatic brain injury (TBI), and stroke. Studies have shown that between 1996 and 2016, the number of people with Parkinson’s disease (PD) doubled (13). Currently, approximately 47 million people worldwide live with dementia, with projections suggesting this number could triple to around 131 million by 2050 (14). The precise pathogenesis of these diseases remains incompletely understood (14).
This article reviews the role of cuproptosis in central nervous system immune responses and its potential mechanisms in neurodegenerative diseases. By investigating these mechanisms, this review provides new insights into disease pathogenesis and suggests potential directions for prevention and treatment through the lens of cuproptosis.
Copper absorption
Copper in the human body is primarily obtained from organ meats and shellfish, with adults recommended to consume between 0.8 to 2.4 mg of copper daily to maintain balance (15). Copper is primarily absorbed through the duodenum, where it enters intestinal epithelial cells via copper transporter 1 (Ctr1) and divalent metal transporter DMT1 (DMT1) (2). Once inside these cells, copper is either distributed to copper chaperone proteins or stored as metallothionein (MT) and glutathione (GSH) (16). An animal study demonstrated that in mice lacking the copper transporter Ctr1, although copper accumulated significantly in intestinal epithelial cells, it could not be converted into a bioavailable form and thus failed to perform its normal physiological functions (17). These Ctr1-deficient mice exhibited marked growth retardation and reduced survival rates early after birth, likely due to insufficient copper absorption and subsequent impairment of copper-dependent enzymes and proteins (18). Partial correction of growth and survival defects was achieved by copper supplementation, underscoring the vital role of copper in early development (17).
Research indicates that when copper levels are insufficient, Ctr1 expression increases in key tissues (e.g., intestine, kidney, and brain), thereby promoting copper uptake and recycling to meet the body’s copper needs (19). Conversely, when copper levels are excessive, Ctr1 expression decreases, limiting excessive copper absorption and preventing copper toxicity (18). Such regulation allows cells and the organism to maintain copper homeostasis under varying copper supply conditions (20). Another study reached a similar conclusion, demonstrating that copper homeostasis is maintained by regulating the expression of Sp1 (Specificity protein1) and hCtr1 (21). In copper excess, hCtr1 is upregulated to enhance copper uptake while Sp1 expression is inhibited; when copper is deficient, Sp1 is upregulated to increase hCtr1 expression and thus boost copper intake (22). By means of a feedback mechanism, Sp1 and hCtr1 regulate each other, maintaining a balanced intracellular copper level (21).
Copper storage and transportation
Copper absorbed by intestinal epithelial cells via the transporter Ctr1 is guided by the copper chaperone Atox1 to ATPase copper transporter 7A (ATP7A) (23). ATP7A, located on the basolateral side of the epithelial cells, actively transports copper into the bloodstream (16). In the bloodstream, copper ions bind to proteins rather than circulating as free ions (23). Human serum albumin (HSA) and ceruloplasmin (CP) maintain the dynamic equilibrium of copper in the serum via distinct binding properties: CP stabilizes and stores most of the copper, while HSA is the primary carrier for exchangeable copper, aiding in copper transport and distribution (24). Specifically, about 75% of copper ions are bound to ceruloplasmin in a non-exchangeable form, whereas approximately 25% are bound to HSA in an exchangeable form (24).
Subsequently, the majority of copper ions travel via the portal vein to the liver, the central organ for copper metabolism by regulating absorption and biliary excretion (25). Metallothionein (MT), a molecule rich in thiol groups and with high affinity for copper ions, helps maintain copper homeostasis by storing and regulating copper ions (26). When cells require copper, MT releases these ions (26). This mechanism allows MT to prevent oxidative stress caused by copper overload while preserving physiological copper balance (27). ATP7B supports copper homeostasis, especially in the kidney, by facilitating copper efflux and modulating intracellular copper levels, redistributing copper from the liver to the bloodstream as needed (28).
Copper elimination
ATP7A and ATP7B are P1B-type ATPases responsible for the intracellular distribution, excretion, and storage of copper (29). When copper levels rise, these transporters relocate from the trans-Golgi network (TGN) to the cell membrane or lysosomes, promoting copper excretion or storage; when copper levels fall, they return to the TGN to restore the supply of copper for enzyme activity (25). Under normal physiological conditions, copper is primarily transported into bile by ATP7B and excreted with bile, with only about 2% excreted through urine (30).
An animal experiment demonstrated that deficiency of MURR1 led to copper accumulation in the liver and reduced copper excretion into bile, ultimately causing cirrhosis (31). This result suggests that MURR1 may participate in extracellular copper excretion by regulating ATP7B-mediated copper discharge (32). In Wilson’s disease (WD), mutations in the ATP7B gene impair its function, hindering copper excretion (33). Certain mutations cause ATP7B to accumulate in the endoplasmic reticulum, impairing its proper localization to the Golgi apparatus or cell membrane, preventing copper from being excreted in bile and leading to its buildup in the liver, brain, and other tissues (29). This accumulation leads to copper toxicity symptoms characteristic of Wilson’s disease (34) (Figure 1).
Figure 1

Schematic of normal copper metabolism. Copper from food is first reduced from Cu2+ to Cu+ by STEAP, then absorbed by enterocytes through the transporter Ctr1. In certain cases, DMT1 also participates in copper transport and absorption. Subsequently, copper is transported to the portal vein via the ATP7A transporter, where it binds to plasma proteins such as albumin and ceruloplasmin for further transport to the liver. After processing or transporting copper, the liver releases it into the bloodstream, which then enters the brain circulation, while excess copper is excreted through bile. Created with BioGDP.com (236). ATP7A, Cu-transporting ATPase 1; ATP7B, ATPase Cu-transporting beta; Ctr1, Cu transporter 1; Cu, copper; DMT1, divalent metal transporter 1; STEAP, six segment transmembrane epithelial antigen of prostate; Atox1, antioxidant 1; TGN, trans-Golgi network.
Systemic copper homeostasis
Copper is an essential trace nutrient in the human body, forming an integral part of various key metabolic enzymes like copper-zinc superoxide dismutase (SOD1), cytochrome c oxidase (CCO), and lysyl oxidase (LOX) (35). Copper is widely distributed in the body, primarily in the skeleton, liver, and kidneys (19). The skeleton contains the highest copper concentration, about 46 mg, while the kidneys contain relatively less, around 3 mg (36). Because copper exists in two oxidation states (Cu+ and Cu2+) and plays a critical cofactor role in the body’s redox systems (37). Excessively high or low copper levels can result in cytotoxicity and pathological changes (30). Maintaining copper levels within an appropriate range is essential (37).
Intracellular copper homeostasis
Copper uptake, distribution, and excretion within cells is a dynamic equilibrium regulated by metabolic demands, differentiation status, and environmental factors (38). Copper chaperone proteins like Atox1 and CCS transfer copper from Ctr1 to ATP7A/ATP7B or to copper-dependent enzymes, ensuring the efficient allocation of copper (39). An animal study showed that copper, via Atox1 as a transcription factor within the cell nucleus, binds to the Cyclin D1 promoter and promotes Cyclin D1 expression, thereby driving cell-cycle progression and cell proliferation (40). These findings indicate that Atox1’s nuclear translocation and structural domain integrity are essential for these processes (41).
CCS forms a heterodimer with superoxide dismutase 1 (SOD1), facilitating copper ion transfer from CCS to SOD1 and activating its superoxide dismutase function (42). SOD1 is a key antioxidant enzyme widely present in eukaryotic cells (43). It catalyzes the conversion of superoxide radicals (O2−) into hydrogen peroxide (H2O2) and oxygen (O2), a reaction critical for maintaining redox balance in cells (44). By doing so, it prevents oxidative stress and cellular damage caused by the accumulation of superoxide radicals (44). A recent study has shown that in SOD1-knockout mice, the absence of CuZn superoxide dismutase (CuZnSOD) led to excessive superoxide accumulation, causing oxidative stress in mitochondria and the cytoplasm (45). This oxidative stress impaired multiple antioxidant enzymes (e.g., MnSOD and GPx) and led to widespread oxidative damage in the liver, including protein oxidation, lipid peroxidation, and DNA damage (46). Significant pathological changes, including the formation of hepatocellular carcinoma (HCC), occurred in hepatocytes (47).
The protein encoded by Cox17 is crucial for copper ion transport, delivering copper ions from the cytoplasm to mitochondria, aiding in cytochrome c oxidase (CCO) assembly and activity (48). The mitochondrial inner-membrane protein PiC2 (SLC25A3) can bind copper ions and acts as a mitochondrial copper transporter, delivering copper into the organelle and promoting the metallation of cytochrome c oxidase (CCO) in eukaryotic cells (49). Although there is not yet sufficient evidence to definitively classify COA6 as a copper chaperone, studies indicate that COA6 can indirectly facilitate copper binding by promoting the reduction of disulfide bonds in the copper-coordinating cysteine residues of COX2 and SCO1 (49). Another study reported that Cox17 specifically transfers copper ions to Sco1 and Cox11 during the assembly of cytochrome c oxidase (CCO), functioning as a key copper donor (50). This mechanism ensures proper metallation of CCO and the maintenance of its normal biological function (51). An animal experiment revealed that loss of COX17p function leads not only to the failure of the mitochondrial respiratory chain but also to severe developmental defects in early embryogenesis (52). COX17p deficiency does not cause immediate lethality in early embryonic development, but as development progresses, impaired copper ion transport into mitochondria disrupts cellular energy supply, ultimately proving fatal (53).
Copper-regulated kinases (e.g., ULK1, ULK2, and MEK1) and the cAMP-degrading enzyme PDE3B mediate copper-dependent signaling that influences autophagosome formation, cell proliferation, and metabolism (54). Cytoplasmic glutathione affects both the rate of copper uptake and the oxidation state of Atox1, thereby modulating copper distribution (25). SLC31A1 (Ctr1) is the principal high-affinity copper transporter responsible for most cellular copper uptake (19). When intracellular copper levels rise, Ctr1 is internalized from the plasma membrane into endosomes through clathrin- and dynein-dependent endocytosis, thus reducing copper uptake; when copper levels fall, Ctr1 is redirected back to the plasma membrane via the retromer complex, reinstating its copper-uptake function (25) (Figure 2).
Figure 2
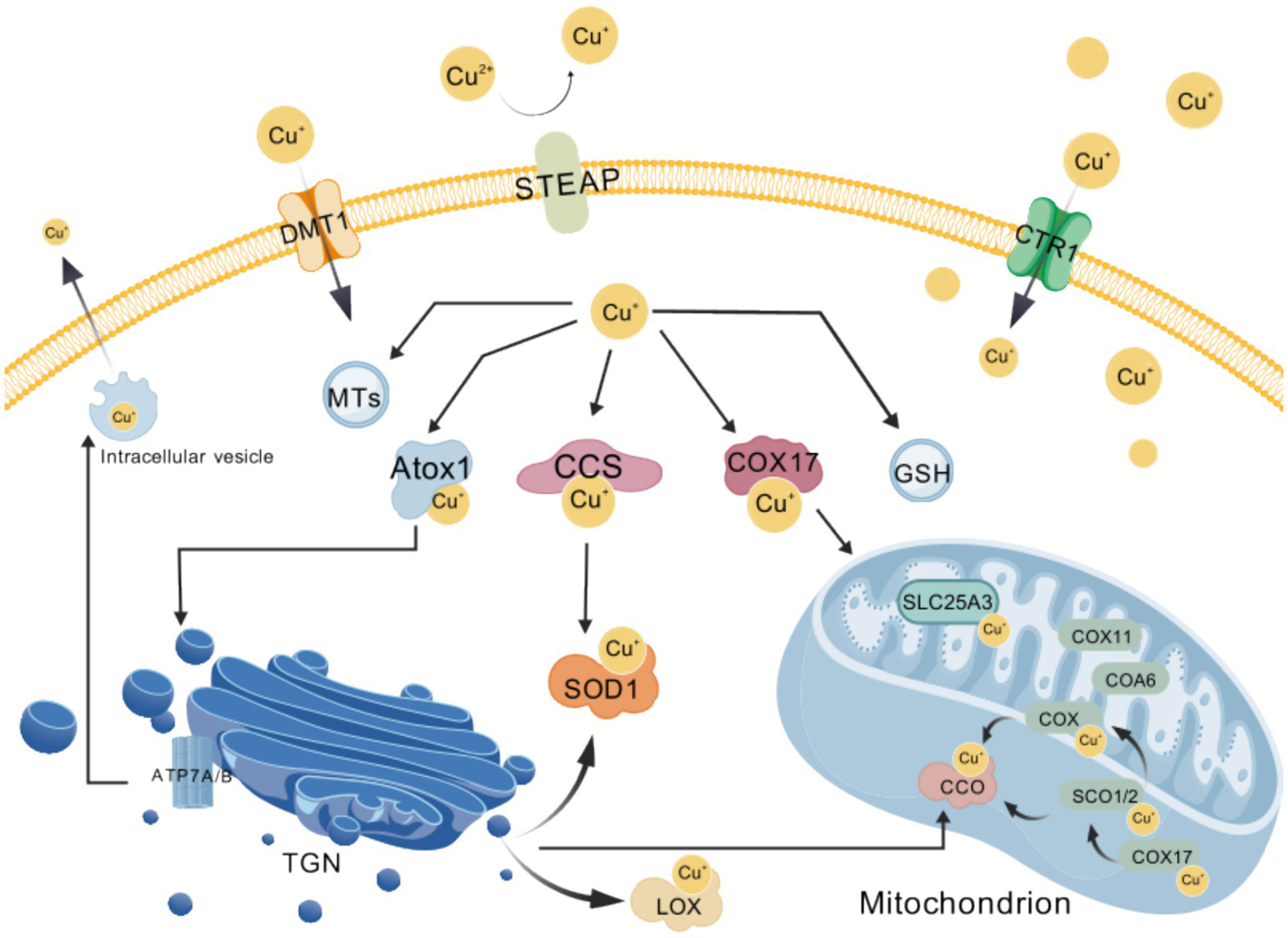
Overview of cellular copper homeostasis. Exogenous cupric ions (Cu2+) are first reduced to cuprous ions (Cu+) by the STEAP family of metalloreductases and subsequently imported into the cytosol via the membrane transporters CTR1 and DMT1; the resulting Cu+ is then precisely delivered to distinct targets by the copper chaperones ATOX1, CCS and COX17, while excess free copper is transiently sequestered by metallothioneins (MTs) and glutathione (GSH) to buffer cellular levels; when copper must be expelled, the P-type ATPases ATP7A and ATP7B pump Cu+ either out of the cell or into secretory vesicles; within the mitochondrial intermembrane space, SCO2 and COA6 cooperatively adjust the redox state of SCO1 so that it can bind Cu+ and hand the metal to cytochrome-c oxidase (CCO), whereas the inner-membrane carrier SLC25A3 transports Cu+ into the matrix to support this metallation process; these complementary mechanisms of uptake, storage, trafficking and efflux collectively ensure that copper-dependent enzymes—including CCO, lysyl oxidase (LOX) and superoxide dismutase 1 (SOD1)—acquire their essential metal cofactor, thereby sustaining cellular energy metabolism and antioxidant homeostasis. Created with BioGDP.com (236). CCS, copper chaperone for superoxide dismutase; COX17, cytochrome-c oxidase copper chaperone 17; MTs, metallothioneins; GSH, glutathione; SCO2, synthesis of cytochrome-c oxidase 2; COA6, cytochrome-c oxidase assembly factor 6; SCO1, synthesis of cytochrome-c oxidase 1; CCO, cytochrome-c oxidase, complex IV; SLC25A3, solute carrier family 25 member 3, mitochondrial phosphate carrier; LOX, lysyl oxidase; SOD1, superoxide dismutase 1.
Copper homeostasis and cuproptosis in neurodegenerative diseases of the central nervous system
Alzheimer’s disease
Alzheimer’s disease (AD) is a neurodegenerative disorder of the central nervous system, mainly characterized by progressively worsening cognitive impairment and behavioral abnormalities (55). It is the most common form of dementia (56). Studies predict that by 2050, AD will affect 115 million people worldwide (57).
While the exact cause of Alzheimer’s disease remains unclear, multiple hypotheses have been proposed to explain its pathogenesis (55). An ATN (amyloid-β, tau protein, and neurodegeneration) framework based on biomarkers, emphasize the presence of β-amyloid (Aβ) and phosphorylated tau protein to confirm AD (58). According to the “amyloid cascade” hypothesis, the initial event in AD is the misfolding and aggregation of Aβ, which triggers a series of pathological changes, including abnormal tau phosphorylation, neuroinflammation, vascular abnormalities, and neurodegeneration (59). Accumulation of Aβ within neuronal mitochondria increases ROS levels and disrupts the mitochondrial membrane potential, resulting in energy metabolic disorders that lead to apoptosis or necroptosis, severely compromising neuronal function and survival (60).
Other studies suggest that oxidative damage—through excessive reactive oxygen species (ROS)—leads to metabolic disturbances in neurons and is closely associated with Aβ accumulation, creating a vicious cycle (61). Additionally, Aβ promotes the abnormal phosphorylation of tau protein, causing microtubule disassembly and neuronal dysfunction (62, 63). Hyperphosphorylated tau is strongly associated with neurofibrillary tangle (NFT) formation, with tau phosphorylation directly affecting its binding to microtubules (64, 65). Hyperphosphorylated tau loses its microtubule-stabilizing function and forms NFTs, further exacerbating neurodegenerative changes (66, 67). Moreover, Aβ can activate signaling pathways such as p38MAPK to induce abnormal tau phosphorylation; once Aβ deposition reaches a certain level, it accelerates tau pathology and promotes the formation of amyloid plaques, ultimately causing a gradual decline in cognitive function (68, 69). In addition, theories such as the neuroinflammation hypothesis and the cholinergic hypothesis have been proposed to further explain the pathogenesis of AD (70).
Dysregulated copper metabolism is closely linked to AD progression (71). Studies show that in the brains of AD patients, copper distribution is abnormal; both copper excess and copper deficiency have been observed in various regions of the brain (72). Excess copper primarily exacerbates cognitive impairment by increasing the expression of amyloid precursor protein (APP) and β-site APP cleaving enzyme 1 (BACE1), promoting the accumulation of amyloid β (Aβ) (73). In particular, copper exposure significantly increased the mRNA and protein expression of APP and BACE1 in the hippocampus, leading to the accumulation of Aβ42, which further inhibited hippocampal long-term potentiation (LTP), a mechanism crucial for learning and memory (74). Copper exposure also disrupted synaptic plasticity by generating oxidative stress, leading to memory loss (74). In the cortex, although copper exposure did not significantly increase the expression of ADAM10, copper negatively affected the Aβ clearance mechanism (75). Copper may influence the expression of Aβ degrading enzymes such as neprilysin (NEP) and insulin-degrading enzyme (IDE), leading to insufficient Aβ clearance, which in turn affects decision-making, planning, and executive functions (76). Studies also suggest that reduced copper levels in the hippocampus may impair synaptic plasticity and neuronal growth, leading to memory decline (77). A randomized controlled trial found that copper concentration in the frontal cortex was significantly lower in Alzheimer’s disease (AD) patients compared to healthy controls, with this change mainly occurring in soluble components, leading to impaired cognitive control, such as difficulties in decision-making and attention deficits (78). This indicates a localized reduction in copper in the brains of AD patients, affecting only soluble components, while other regions such as peripheral membranes, vesicular material, and membrane components showed no significant differences (79). In the frontal cortex, copper overload induces oxidative stress and cell damage by increasing free copper, which impacts cognitive abilities and memory function (80). Although the antioxidant HT can restore copper levels and improve behavioral performance, the increase in calcium ion levels may limit these improvements, suggesting that copper metabolic imbalance affects cognitive function through a series of biochemical reactions (80). Excess copper in the medial temporal cortical region exacerbates oxidative stress, promoting the aggregation and deposition of β-amyloid, thereby worsening cognitive decline (55). An animal study showed that copper concentrations in the brainstem of AD model mice were significantly increased, particularly in transgenic AD mice, where copper levels were significantly higher in both young and old mice compared to healthy controls (Mann–Whitney U, p = 0.016), indicating that the brainstem may be a key area for early pathological changes in AD (81). Copper concentrations in the cerebellum also increased with age, but in AD mice, while copper levels did not significantly change (Mann–Whitney U, p = 0.063), significant changes in copper isotope ratios were observed (Mann–Whitney U, p = 0.016), with a notable enrichment in copper isotope ratios in AD transgenic mice (Δ65Cu = 0.33‰) (81). This suggests that copper metabolism imbalance may alter the redox status of the cerebellum, thereby affecting its function (81). In the TgCRND8 mouse model, β-amyloid plaques predominantly accumulate in various regions of the cerebral cortex, where copper concentrations were significantly higher, with copper binding to Aβ peptides to promote Aβ aggregation and neurotoxicity (82). However, in areas surrounding the plaques, copper concentrations were lower, which may be related to the upregulation of ATP7A copper transport protein in activated microglia, particularly in the regions around the plaques (78). Activated microglia, through the secretion of inflammatory cytokines like interferon-γ (IFN-γ), participate in inflammatory responses, affecting copper uptake and transport. After IFN-γ stimulation, ATP7A proteins are redistributed to intracellular vesicles, suggesting copper accumulation inside the cell. Microglia may “sequester” copper to reduce the negative interaction between copper and Aβ, thereby exerting a protective effect (82). In AD patients, the concentration of free copper (i.e., copper not bound to ceruloplasmin) is elevated, and free copper can cross the blood-brain barrier (BBB) to enter the cerebrospinal fluid (CSF) and cerebral cortex, where it interacts with Aβ to promote its aggregation (83). This copper metabolic dysfunction is closely associated with clinical symptoms, neuronal damage, and changes in biomarkers in the CSF (83). The increase in free copper is also closely linked to neuroinflammation, as it promotes the activation of microglia and astrocytes, triggering an inflammatory response in the brain, which further exacerbates neuronal damage and cognitive decline (83). Studies suggest that copper deficiency in the basal forebrain and thalamus may impair cholinergic system function, negatively affecting behavior and cognition (84). Furthermore, abnormalities in copper levels in the anterior cingulate cortex, a key brain area involved in emotional regulation and cognitive conflict, may also be closely related to cognitive symptoms and emotional changes in AD (85). Although the cerebellum is less affected in AD pathology, abnormal copper levels in this region may be associated with impaired motor coordination and cerebellar function in some AD patients (86). Other studies have found that abnormal copper distribution in the cerebellum, by affecting ion transport activity, membrane potential changes, and intracellular calcium levels, may directly influence the electrophysiological activity of neurons, leading to cognitive and behavioral changes (87). This phenomenon of copper imbalance is part of the AD pathology, referred to as the “CuAD hypothesis,” which suggests that both copper excess and deficiency lead to neuronal damage and functional impairment (88). Moreover, abnormal copper accumulation in the basal ganglia may disrupt the normal function of neurotransmitter systems, particularly those related to dopamine (89). Dopaminergic neurons in the basal ganglia play an important role in motor control, reward mechanisms, and emotional regulation (90). Research has shown that copper overload may alter dopamine synthesis, release, and clearance processes through interaction with dopamine receptors or by affecting dopamine transporter function, leading to dopamine metabolism abnormalities and impacting basal ganglia function (91). Insufficient or excessive activation of dopamine can directly lead to motor dysfunctions such as tremors and bradykinesia, which are common motor abnormalities observed in AD patients (92). Copper overload in the basal ganglia not only affects motor functions but may also further interfere with emotional and motivational regulation (93). Another important function of the basal ganglia is regulating emotions and decision-making (94). Copper overload may influence this function in two ways: first, the direct toxicity of copper to neurons may damage basal ganglia circuits, particularly those related to emotion and motivation; second, copper may alter the balance of neurotransmitter systems, leading to emotional regulation disorders, further contributing to depression, anxiety, and other emotional issues, which are very common in AD patients (95). AD patients often exhibit emotional changes, which are closely related to basal ganglia dysfunction (94).
These specific manifestations of copper abnormalities in different pathological stages and brain regions of AD suggest that copper’s role in AD is not merely confined to simple concentration changes within brain regions but rather influences neuronal function and the stability of neural circuits through complex biochemical mechanisms, thereby playing a critical role in the cognitive impairment and behavioral changes observed in AD (96).
Cu2+ coordinates with the histidine (His) and tyrosine (Tyr) residues of Aβ, affecting its aggregation pathway and forming two main aggregate types: amyloid-like fibrils and Cu2+-induced aggregates (97, 98). Cu2+-induced aggregates generate dityrosine, triggering oxidative stress and increasing neurotoxicity (99). When the Cu2+:Aβ molar ratio is below 1:1, Cu2+ primarily promotes the formation of amyloid-like fibrils; at higher molar ratios, it favors Cu2+-induced aggregates, which hinder fibrillization (100). Cu2+ binding also promotes the conversion of tau from its native structure into a conformation that is more prone to aggregation, providing a basis for pathological tau aggregations such as neurofibrillary tangles (NFTs) (30). Furthermore, Cu2+ induces nitrotyrosination of lipoprotein receptor-related protein 1 (LRP1), marking it for proteasomal degradation and thereby reducing LRP1 levels, which in turn lowers Aβ clearance efficiency (101).
A meta-analysis revealed significant alterations in trace elements in AD patients, notably copper (serum), iron (plasma), zinc (hair), and selenium (plasma), with standardized mean differences (SMD) of 0.37, −0.68, −0.35, and −0.61, respectively (102). Another meta-analysis showed that copper content in the brain tissue of AD patients decreases, while copper and non-ceruloplasmin bound copper (non-Cp Cu) levels in serum/plasma increase (103). Moreover, the AG haplotype of the ATP7B gene is associated with increased susceptibility to AD, indicating that AD patients may fail to maintain proper copper metabolism and that certain individuals carrying the ATP7B AG haplotype may be more prone to copper imbalance (103). This suggests that copper dyshomeostasis could represent a disease subtype in AD.
Research indicates that iron, copper, and zinc each bind to β-amyloid (Aβ) to modulate its aggregation and toxicity, inducing oxidative stress and programmed cell death (including ferroptosis and cuproptosis) and further aggravating neuronal damage (70, 104). At the same time, iron and copper contribute to neurofibrillary tangle (NFT) formation by activating tau-protein kinases (e.g., GSK3β) and facilitating excessive tau phosphorylation (70). One study revealed that copper ions bind to the histidine residues of Aβ, forming a non-β-folded Aβ/Cu complex that inhibits fibrillization and blocks the formation of typical amyloid fibrils (70). This process may, in early stages of AD, influence the aggregation patterns of Aβ and the development of senile plaques, thereby driving disease pathology (105).
The role of copper in Alzheimer’s disease (AD) extends beyond the accumulation of Aβ and tau; copper also plays a significant role in disease progression by regulating immune system functions (58). Copper’s role in AD is first manifested in its regulation of immune molecules (59). Studies show that molecules like Aβ, tau, and ApoE maintain neural homeostasis under normal conditions, but in AD, their functions change, and they shift to exert antimicrobial effects (106). For example, Aβ, as a natural antimicrobial peptide (AMP), forms fibrous structures through aggregation, interacting with pathogens to defend against infections (106). In this process, copper binds to Aβ, promoting its aggregation and enhancing Aβ’s antimicrobial activity (106). However, prolonged copper accumulation can lead to excessive aggregation of Aβ, which in turn triggers neurotoxicity, causing immune system dysfunction and exacerbating neurodegenerative changes (106).
Additionally, copper significantly affects the activity of immune cells (107). In the brains of AD patients, abnormal distribution of copper leads to overactivation of microglial cells and astrocytes (107). The abnormal response of these immune cells to pathogen invasion aggravates neuroinflammation (107). The loss of function of copper-dependent enzymes, such as cytochrome c oxidase and ceruloplasmin, in AD further disrupts iron homeostasis and redox reactions, thereby intensifying neuroinflammatory responses (107).
Copper’s role in immune regulation is also reflected in its control over immune cell function (59). In AD, copper binds to Aβ to form a more stable redox complex, which not only enhances Aβ’s antimicrobial activity but also leads to copper depletion, further affecting the normal function of the nervous system (101). Copper deficiency reduces the activity of copper enzymes such as ceruloplasmin, which are closely related to neuroimmune functions and iron homeostasis, thereby exacerbating neurodegenerative changes (106).
Copper interacts with key proteins in the immune system to regulate immune responses (103). Studies have shown that copper promotes the release of interferon-γ (IFN-γ), increases copper uptake in microglial cells, and enhances ATP7A expression, which helps limit Aβ aggregation and improves its clearance (103). At the same time, copper regulates the M1/M2 shift in microglial cells, exerting its dual effect (107). In some cases, copper suppresses the generation of nitric oxide (NO), promoting the transformation of microglial cells from the pro-inflammatory M1 type to the neuroprotective M2 type, thus alleviating neuroinflammation and promoting neuroprotection (107). This indicates that the immune regulatory role of copper depends on its physiological concentration and distribution, with appropriate copper levels helping to suppress excessive immune responses and exert protective effects (107).
In summary, copper’s immune regulatory role in AD is dual in nature, potentially promoting harmful inflammatory responses but also, under appropriate conditions, exerting anti-inflammatory and neuroprotective effects (107). The dysregulation of copper homeostasis leads to immune dysfunction by affecting the function of immune molecules, the activation state of immune cells, and the deficiency of copper enzymes, which in turn drives the progression of neurodegenerative changes (59). Both copper overload and deficiency can affect the activity of microglial cells, astrocytes, and other immune cells, intensifying neuroinflammation and ultimately influencing the course of AD (106). Therefore, regulating copper homeostasis is a critical component of the pathological mechanism of AD, offering new directions and potential targets for AD treatment (59).
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease marked by the degeneration of motor neurons (108). Common symptoms include skeletal muscle weakness, atrophy, fasciculations, and bulbar palsy (109). Progressive paralysis eventually leads to respiratory failure, with death typically occurring within about 3 years of onset (110). The exact etiology of ALS remains unclear, potentially involving genetic factors, immune system dysfunction, lifestyle choices, and metabolic imbalances (111). Despite extensive studies on its pathogenesis, challenges in ALS diagnosis and treatment persist, with therapeutic outcomes still suboptimal (108). In recent years, an increasing number of studies have underscored the importance of targeted therapy for maintaining copper homeostasis (112, 113).
Research has shown that misfolded mutant SOD1 can bind to proteins on the mitochondrial outer membrane (OMM), such as Bcl-2 and VDAC1, thereby causing mitochondrial dysfunction, increased oxidative stress, and activation of apoptotic pathways (114, 115). These changes promote cell apoptosis and ultimately lead to the death of motor neurons, contributing to the onset of ALS (116). Meanwhile, copper—regulating SOD1 activity—has gained recognition for its role in the aggregation and abnormal function of SOD1 (115). Copper metabolism imbalances may exacerbate SOD1 misfolding, forming insoluble aggregates and accelerating ALS pathology (117).
An animal study demonstrated that tetrathiomolybdate (TTM) provides protective effects in ALS by modulating superoxide dismutase 1 (SOD1) and copper metabolism, restoring abnormally elevated copper levels in the spinal cord (118). TTM significantly reduced insoluble SOD1 aggregates and suppressed SOD1 enzyme activity, thereby slowing the pathological progression of ALS (119). A randomized controlled trial showed significantly lower copper levels in the cerebrospinal fluid (CSF) of ALS patients compared to healthy controls, particularly in spinal-onset ALS (109). Research reports that certain SOD1 mutations (e.g., A4V) reduce enzymatic activity and copper-binding affinity (120). Under zinc-deficient conditions, mutant SOD1 can generate highly active hydroxyl radicals and peroxynitrite via abnormal copper chemistry, further exacerbating neuronal oxidative damage and protein nitration (120). This same study indicated that dysregulated copper metabolism promotes SOD1 aggregation and disulfide bond instability, further aggravating ALS pathology (121).
Copper chaperone for SOD1 (CCS) regulates copper uptake and structural maturation of SOD1 (122). One study found a positive correlation between CCS expression and survival in ALS patients, suggesting that higher CCS expression may prolong survival and confer protective effects in ALS pathogenesis (123). A randomized controlled study revealed higher expression of copper metabolism-related genes (CuRGs) in ALS patients, highlighting their pivotal role in ALS onset (111). Moreover, by analyzing differing CuRG expression profiles, the study classified ALS patients into two distinct pathological subtypes with marked differences in immune characteristics and biological processes, highlighting the complex interplay between abnormal copper metabolism and immune responses in ALS pathogenesis (111).
Some research indicates that copper metabolism dysfunction in ALS may increase oxidative stress, foster lipid peroxidation and ferroptosis, and thus injure neurons (122). Copper modulates the cell’s antioxidant defenses via interactions with enzymes such as thioredoxin reductase (TRXND) and glutathione peroxidase (GPX) (111). Another study found that copper is associated with membrane lipids such as sphingomyelins and fatty acid acyl glycosides, both essential for neuronal function and integrity (124, 125). Copper imbalance not only heightens oxidative stress but may also interfere with lipid metabolism and signaling, further advancing the neurodegenerative processes in ALS (111).
In one study, tetrathiomolybdate (TTM), used as a copper chelator, lowered spinal-cord copper levels and SOD1 activity in mice, delaying ALS onset and progression and extending survival (126). Another study showed that triazines, also acting as copper chelators, significantly improved survival in an ALS mouse model, suggesting that copper chelation can slow disease progression (127). Ceruloplasmin, a multi-copper oxidase involved in iron metabolism regulation and systemic copper transport, was found to increase in ALS model mice during disease progression, yet its enzymatic activity substantially declined (128). This indicates that ALS-related copper dysregulation not only affects metal loading in a single protein but also systematically reduces the functionality of copper-dependent enzymes, further disrupting metabolic balance in the central nervous system (129).
In the pathogenesis of amyotrophic lateral sclerosis (ALS), copper metabolism imbalance not only affects the function of SOD1 but also plays a key role in regulating immune responses in the central nervous system (CNS), thereby promoting the development of neurodegeneration and the pathological progression of the disease (119).
First, in ALS, the mutant SOD1 interacts with mitochondrial membrane proteins such as Bcl-2 and VDAC1, leading to conformational changes in these proteins that expose their death domains, triggering apoptosis (116). The abnormal activity of VDAC1 also affects the mitochondrial membrane potential and permeability, leading to an increase in intracellular Ca2+ concentrations, which further exacerbates the activation of immune cells and the inflammatory response (116).
Secondly, copper imbalance also directly participates in the regulation of immune cell function (119). Studies have shown that copper ions are important regulatory factors for the function of immune cells such as microglia and astrocytes (119). In ALS, excessive copper accumulation promotes the activation of these immune cells, particularly by enhancing their oxidative stress response and the secretion of pro-inflammatory cytokines (130). Copper interacts with antioxidant enzymes such as SOD1 within these immune cells, enhancing the oxidative stress response in neutrophils (123). Cytokines such as interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α) secreted by these immune cells not only promote the further expansion of the immune response but also increase copper uptake and accumulation, forming a vicious cycle that exacerbates neuronal damage (111).
Copper metabolism imbalance may also worsen immune responses through mechanisms such as lipid peroxidation (130). In immune responses, oxidative stress is an important pathogenic mechanism (129). The excessive accumulation of copper promotes the generation of reactive oxygen species (ROS) and lipid peroxides, which can directly damage neurons, further activate the immune system, and lead to neurodegeneration (129). Therefore, copper not only alters the function of SOD1, leading to oxidative stress and cell death, but also activates inflammatory responses in immune cells, promoting the progression of ALS (130).
In conclusion, there is a complex interaction between copper metabolism imbalance and the immune response in ALS (111). Copper regulates the function and folding of SOD1, maintaining normal immune cell function, while excessive copper accumulation or deficiency changes immune cell activation and pro-inflammatory responses, leading to increased neuroinflammation, thereby promoting the occurrence of neurodegeneration (116). Copper homeostasis imbalance in ALS affects not only the redox state and immune cell function but also potentially modulates immune responses, influencing the progression of neurodegenerative diseases (123). Restoring copper metabolic balance, especially in the immune response, may provide a new direction for the treatment of ALS and other neurodegenerative diseases (111).
Huntington’s disease
Huntington’s disease (HD) is a progressive neurodegenerative condition caused by the expansion of the polyglutamine (polyQ) region in the huntingtin (Htt) protein (131). Symptoms include movement, cognitive, and psychiatric disturbances, typically manifesting in midlife and progressively worsening over the subsequent two decades (132). Current treatments mainly focus on reducing HTT gene expression, nucleic acid therapies, gene therapy, DNA repair, and cell replacement strategies (133).
Research has shown that altering the expression of copper transporters (e.g., Ctr1B and DmATP7) or regulating dietary copper intake can significantly influence HD pathology (131). Both copper excess and copper deficiency affect the aggregation and fibrillization of huntingtin protein (Htt exon1-polyQ), thereby influencing disease progression (131). Studies further indicate that removing potential copper-binding sites on Htt can eliminate copper’s pathogenic effect in HD, suggesting that Huntington’s disease is not only a polyglutamine disease but also involves copper-regulated pathogenic mechanisms (131).
One experiment demonstrated that copper ions markedly enhance the toxic aggregation of mutant huntingtin protein (Htt) associated with HD (134). In a fruit fly model, dietary copper promoted Htt oligomer formation, increased β-amyloid structures, and strengthened the interaction between Ref(2)P and Htt aggregates, contributing to neurotoxicity (134). Copper intensified synaptic degeneration, shown by elevated caspase-3-mediated cell death, reduced BRP expression, and thinner neuronal structures (134). Moreover, increased dietary copper led to more Htt aggregates, especially smaller ones (<5 μm2) (135). These results highlight the significant contribution of copper in HD pathogenesis by exacerbating Htt aggregation and neurotoxicity (136). Further research revealed that the copper chelator D-penicillamine (DPA) effectively alleviated this copper-induced Htt aggregation and β-amyloid accumulation, thereby reducing synaptic damage and cytotoxicity (137, 138). These findings provide new insights into copper’s potential role in HD and support copper chelation as a possible therapeutic avenue (134).
Some studies suggest that copper’s interaction with metals like zinc (Zn) may inhibit DMT1, causing imbalances in metals and worsening HD pathology (134, 136). Meanwhile, research indicates that natural antioxidants like rutin can mitigate copper-induced toxicity, reduce protein aggregation, and prevent neurodegenerative injury through chelating effects, suggesting a potential therapeutic application in HD (136).
Studies show that in early-stage HD, especially in the pre-symptomatic phase, copper levels in CSF are elevated while zinc (Zn) levels decrease (139). This rise in copper may reflect changes in blood-brain barrier integrity or metal transport mechanisms (139). Copper directly interacts with mutant huntingtin protein (mHTT), promoting its aggregation and hastening neuronal death (134). Excess free copper ions can increase reactive oxygen species (ROS) levels through the Fenton reaction, enhancing oxidative stress and cellular damage (140). Moreover, copper affects the regulation of neurotransmitters by promoting the synthesis and release of GABA, disturbing inhibitory neurotransmission and worsening HD-related motor and cognitive symptoms (141). Excess copper may also displace zinc in enzymatic sites, altering enzyme activity and interfering with vital biochemical processes such as mitochondrial function and antioxidant defense (139).
Conversely, copper deficiency could reduce its chelation of iron, leading to increased free iron participating in ROS generation and thereby exacerbating oxidative stress (142). Additionally, copper is a key component of cytochrome c oxidase, so inadequate copper might impair mitochondrial function and ATP production, further diminishing energy output (143). Because copper is also an essential cofactor of superoxide dismutase 1 (SOD1), insufficient copper may lower antioxidant activity and render the brain more vulnerable to oxidative damage (144).
An animal study showed that copper supplementation in the drinking water of mice with quinolinic acid (QUIN)-induced HD had a neuroprotective effect (139). After 28 days of copper supplementation, there was no impact on the mice’s food intake, body weight, or water consumption, but striatal copper levels significantly increased (145). Notably, following QUIN-induced injury, copper supplementation elevated striatal copper content compared to the group receiving only QUIN injury, implying a potential protective role for copper in neural damage (146). Copper supplementation prevented QUIN-induced reductions in GABA levels and reduced circling behavior, suggesting copper protects by inhibiting NMDA receptor activity and enhancing antioxidant enzymes (e.g., Cu–Zn SOD) (146). Copper supplementation significantly decreased oxidative stress indicators [e.g., ROS and lipid peroxidation (LP)], supporting the idea that copper mitigates neuronal damage by activating endogenous antioxidant systems (147, 148). Copper showed protective effects in the QUIN-induced HD model, but further research is needed to fully understand its role in HD, particularly whether increased copper levels compensate for free radical damage (146).
The role of copper in Huntington’s disease (HD) goes beyond directly binding to mutant huntingtin (mHTT) and promoting its aggregation; it also involves exacerbating the pathological process through the regulation of immune responses and oxidative stress mechanisms (131). When copper metabolism is disrupted, excessive accumulation of copper generates reactive oxygen species (ROS) via the Fenton reaction (133). These free radicals damage lipids, proteins, and DNA within cells, leading to cell death (133). Additionally, oxidative stress activates immune cells, particularly microglia, which promote neuroinflammatory responses (133). The activation of these immune cells not only promotes further accumulation of copper in the nervous system but also exacerbates local neuronal damage (134).
Specifically, copper accumulation leads to abnormal activation of immune response cells (136). Copper directly affects the function of immune cells, altering their activation, proliferation, and migration behavior (136). When copper levels are excessive, immune cells such as microglia and macrophages become overactivated and release large amounts of pro-inflammatory cytokines, such as interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), and interleukins (IL-1, IL-4, etc.) (139). These cytokines not only enhance local inflammation but also further promote copper uptake and accumulation through feedback mechanisms, thereby creating a vicious cycle that exacerbates neuronal damage (134).
Furthermore, copper regulates the expression of metallothioneins (such as MtnA and MtnB), which affect copper homeostasis within cells (131). Metallothioneins are intracellular copper regulators involved in copper absorption, storage, and excretion (131). When copper levels are excessive, the expression of these metallothioneins changes, which may lead to an ineffective cellular response to oxidative stress, further promoting neuronal damage (131). The excessive accumulation of copper not only exacerbates immune responses through these direct oxidative damage pathways but may also influence immune cell function by altering the metal ion balance in immune cells, thus advancing neurodegenerative processes (131).
Copper metabolism disruption may also impact immune responses through interactions with other metals, such as zinc and iron (136). Copper and metals like zinc share the same transport proteins and regulate similar signaling pathways (136). Therefore, excessive copper may compete with zinc for transport channels, altering zinc’s function in immune cells (136). Zinc is an essential metal for various antioxidant enzymes, and its deficiency reduces immune cells’ ability to defend against oxidative stress, thereby worsening immune responses and neuronal damage (136). The interaction between copper metabolism disruption and the imbalance of metals like zinc and iron complicates immune responses, further accelerating the neurodegenerative progression of HD (136).
In summary, copper’s role in HD is not limited to directly promoting mHTT aggregation and toxic expression but also exacerbates neurodegeneration by intensifying oxidative stress, altering immune cell function, and modulating immune responses (131). The excessive accumulation of copper plays a key role in HD’s pathological process through its dual impact on oxidative stress and immune responses (136, 144). Therefore, regulating copper metabolism and immune responses could provide new intervention strategies for HD treatment, especially in terms of copper homeostasis restoration and immune modulation (139).
Parkinson’s disease
Parkinson’s disease (PD) is a common neurodegenerative disorder and currently the fastest-growing neurological condition worldwide (149). Its etiology is complex. In 3–5% of cases, single-gene mutations are responsible, while others correlate with genetic risk variants, family history, constipation, nonsmoking status, and exposure to environmental toxins (e.g., pesticides and trichloroethylene) (149). Core manifestations include bradykinesia, resting tremor, and muscular rigidity, but non-motor symptoms (such as constipation, hyposmia, depression, cognitive decline, and sleep disturbances) also significantly impact quality of life (150). PD typically progresses slowly, with non-motor symptoms often appearing years before motor symptoms, leading to delayed diagnosis (151). Diagnosis primarily relies on clinical presentation and is further supported by a positive response to levodopa treatment; additional tests help rule out atypical presentations (152). Current trends in PD diagnosis focus on early detection and individualized approaches (150). Integrative methods—combining highly sensitive technologies, specific molecular markers, and artificial intelligence—aim to improve early diagnostic accuracy and enhance patient treatment and quality of life (153).
The hallmark feature of PD is the gradual degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc), leading to reduced dopamine levels in the striatum (particularly in the putamen) and motor symptoms (154). Loss of nigral neurons and the dopaminergic denervation of the basal ganglia underlie the onset of these motor deficits. Decreased expression of the dopamine transporter (DAT), aromatic L-amino acid decarboxylase (AADC), and vesicular monoamine transporter-2 (VMAT-2) in the striatum are important indicators of disease progression (152).
Lewy bodies—intraneuronal inclusions composed primarily of α-synuclein (α-Syn)—are another core pathological hallmark of PD; these α-Syn aggregates (oligomers and Lewy bodies) can be detected in neurons, glial cells, and peripheral tissues (152, 155). The aberrant aggregation of α-Syn leads to neuronal dysfunction and disrupts key processes, including synaptic transmission, lipid metabolism, vesicular transport, and dopamine metabolism, exacerbating neuronal damage (156). Misfolded α-Syn can propagate between neurons, inducing the aggregation of endogenous α-Syn in healthy cells and spreading pathology (157). This mechanism is considered a major driver of PD pathogenesis and interacts with mitochondrial dysfunction, lysosomal pathway abnormalities, and neuroinflammation to facilitate disease progression (158).
To date, six monogenic forms of PD have been identified, arising from mutations in SNCA, LRRK2, Parkin, PINK1, DJ-1, and ATP13A2 (159). Additional genes—such as GBA, MAPT, and APOE—also raise PD risk via polymorphisms (157). The Bax protein promotes dopaminergic neuron apoptosis in PD through multiple mechanisms, including interactions with antiapoptotic Bcl-2 family proteins and translocation to the mitochondrial membrane to inhibit other antiapoptotic factors; this mechanism may function independently of cytochrome c release (160). Bax can exert either proapoptotic or antiapoptotic effects in different cell types, acting as a critical cofactor in the apoptotic cascade (161).
A randomized controlled trial demonstrated that copper levels in dopaminergic neurons of the substantia nigra are marginally lower in PD patients than in controls (162). Another mouse study showed that copper levels are abnormally elevated during the prodromal stage of PD and that ATH434 can regulate copper homeostasis and improve olfactory function (163). As the disease progresses, metal dyshomeostasis in PD may shift from copper-dominated abnormalities to iron-centered pathology (163). Copper-chelating agents like PBT2 and TDMQ20 bind and transport excessive copper in the brain, reducing copper toxicity while preserving essential levels for normal neuronal physiology (164). These chelators can partially reverse or slow pathological damage associated with Aβ aggregation and PD (165). Proteins such as FDX1, DLAT, and LIAS play pivotal roles in the tricarboxylic acid (TCA) cycle; these proteins can undergo lipoylation and, under conditions of copper excess, abnormally aggregate, triggering cell death (166).
Research suggests that ceruloplasmin (Cp) is centrally involved not only in copper and iron metabolism in the liver and bloodstream but also in regulating metal homeostasis and neuronal function in the brain (167). Cp deficiency and mutations in copper-transporting genes such as ATP7B can cause abnormal metal distribution in the brain and induce (or exacerbate) neurodegenerative processes, including PD (168). Genes such as ATP7A, SLC31A1, and DBT, closely linked to copper homeostasis, neuronal function, and immunometabolism, display significantly altered expression in the substantia nigra of PD patients, correlating with disease staging (169). Other studies indicate that ATP7B maintains cellular copper homeostasis by transporting copper to the Golgi apparatus for integration into copper-dependent enzymes, NFE2L2 (NRF2) activates various antioxidant enzymes to mitigate oxidative stress and inflammation caused by copper excess, and MTF1 detects abnormal metal concentrations and induces detoxification and stress-response gene expression (170). These three factors together form a core network for copper regulation; dysfunctional expression or activity can exacerbate PD and other neurodegenerative diseases (170, 171).
α-Synuclein (α-Syn) binds Cu2+ with high affinity via its N-terminal amine, the side chain of Asp2, and the imidazole group of His50, inducing conformational changes that modulate copper redox activity, reduce oxidative stress, and promote membrane recycling and SNARE-complex membrane fusion (172). In PD, Cu2+ binding may weaken the interaction of α-Syn with membranes, increasing its soluble fraction, accelerating fibrillization, and promoting Lewy body formation, thereby driving disease progression (172). Copper ions have a high affinity for α-Syn, enhancing α-Syn fibril formation and directly leading to cytotoxicity or inducing α-Syn aggregation (173). Studies show that copper ions can promote the formation of α-Syn short fibrils (<0.2 μm) via an atypical pathway; these short fibrils regulated by copper display heightened intracellular transmissibility and toxicity (174). Other research has found that negatively charged membrane proteins, such as heparan sulfate proteoglycans (HSPGs), mediate copper/iron-induced fibril internalization via electrostatic interactions (175). Concurrently, changes in α-Syn secondary structure and fibril morphology induced by copper/iron ions enhance its aggregation and cellular toxicity (175). Clinically used chelators TETA and DF effectively hinder the damaging effects of copper/iron ions on α-Syn fibril propagation and prolong the lifespan of PD model nematodes (175). Furthermore, copper/iron ions have been found to affect the aggregation or toxicity of other disease-associated amyloid proteins (including β-amyloid, tau, and prion proteins), suggesting new avenues for investigating other protein misfolding disorders (175).
Copper ions (Cu2+) alter α-Syn conformation and aggregation by directly binding to critical residues (His50 and Asp121) and promoting reactive oxygen species (ROS) generation (176, 177). Studies indicate that upon Cu2+ binding, the hydrophobic non-amyloid-β component (NAC) domain becomes more solvent-accessible, facilitating initial hydrophobic interactions that form toxic oligomers (178, 179). Moreover, Cu2+ binding can disrupt long-range intramolecular interactions (e.g., His50-Val71 and Asp121-Lys96), weakening the barrier effect among NAC domains and accelerating α-Syn fibrillization (180). Copper binding results in a more compact α-Syn peptide chain, reinforcing intramolecular hydrogen bonds and residue interactions (181). Such changes likely promote protein aggregation in PD by influencing the tightness of the NAC (non-amyloid-β component) region and the affinity of the peptide chain for membranes (182).
Studies demonstrate that copper triggers abnormal aggregation by directly binding lipoylated proteins, leading to protein toxicity stress and cell death (183). During this process, lipoylated TCA-cycle enzymes are key targets that, under copper’s influence, aggregate and destabilize iron–sulfur (Fe–S) cluster proteins, ultimately compromising mitochondrial metabolic function (184, 185). Glutathione (GSH), a natural intracellular copper chaperone, is critical in this context (186). When GSH is depleted, cells become markedly more sensitive to copper, presenting decreased lipoylation, increased DLAT aggregation, and further destabilization of Fe–S cluster proteins (8). These changes induce severe protein toxicity stress and cell death (8). An animal study using a novel fluorescent probe (R13) showed significantly reduced GSH levels in the brains of a PD mouse model, whereas oxidized glutathione (GSSG) levels increased, indicating that PD is associated with oxidative stress and that lower GSH levels reflect weakened radical-scavenging capacity (187). Other research has demonstrated that polymorphisms in GST (glutathione S-transferase) are closely related to PD risk; specifically, the deletion (null) genotypes of GSTM1 and GSTT1 significantly increase PD risk, and a combined deletion further heightens this association (188). In animal experiments, copper ions binding to GSH caused intracellular GSH depletion, impairing antioxidant capacity, exacerbating oxidative stress and protein nitration, and leading to neuronal damage (8). Meanwhile, GSH depletion disrupts the ubiquitin-proteasome system (UPS) and autophagic pathways, resulting in aggregation of proteins such as α-Syn—an important mechanism of neuronal injury in PD (189).
DLAT is a key enzyme in protein lipoylation and plays a vital role in the TCA cycle (8). Under copper overload, DLAT expression significantly increases, promoting abnormal protein lipoylation and causing aggregated lipoylated proteins as well as the loss of iron–sulfur (Fe–S) proteins (190). These changes induce protein toxicity stress, further exacerbating cell apoptosis and tissue damage (148). Studies show that 4-amino-TEMPO (4-AT) provides antioxidant effects and may activate Nrf2, enhancing cellular antioxidant capacity and restoring GSH levels to improve neuronal viability (191). Another study found that hypoxanthine boosts cysteine uptake via EAAC1 (a cysteine transporter) in HEK293 and SH-SY5Y cells, increasing intracellular GSH synthesis and exhibiting neuroprotective effects against oxidative stress (e.g., H₂O₂ treatment) (192).
The relationship between copper metabolism abnormalities and central nervous system (CNS) immune responses has gained increasing attention in the pathological mechanisms of Parkinson’s disease (PD) (151). One of the pathological features of PD is the loss of dopaminergic neurons and the accumulation of Lewy bodies, with neuroinflammation being considered a key factor in this process (149). Abnormal copper accumulation plays an important role in PD pathology, and through its regulation of the immune system, particularly its impact on immune cells, it enhances neuroinflammatory responses and further exacerbates neurodegenerative damage (154).
Firstly, abnormalities in copper metabolism lead to the overactivation of immune cells, especially microglia and peripheral immune cells such as macrophages, monocytes, and neutrophils (168). Studies have shown that copper accumulation can stimulate microglial activation, as these cells are the primary immune cells in the CNS and play a critical role in neuroinflammation (168). Overactivation of microglia releases inflammatory cytokines, further promoting neuronal damage and exacerbating neurodegenerative lesions (170). The infiltration and activation of peripheral immune cells are also regulated by copper, which increases their ability to enter the CNS, thus intensifying the local inflammatory response (170). Copper not only directly affects the functions of these immune cells but also regulates their migration and activation, further driving the progression of PD (182).
Using the 6-hydroxydopamine (6-OHDA) mouse model, combined with TSPO PET and TREM1 PET imaging techniques, researchers further explored the role of copper in PD. TSPO is a marker for microglia and peripheral immune cells (193). The study found that copper accumulation is closely related to the activation of immune cells in the 6-OHDA mouse model (193). TREM1 PET imaging showed that the infiltration of peripheral immune cells, particularly neutrophils, enhanced this process, suggesting that the role of copper in immune activation should not be underestimated (193). The high expression of TREM1 as a pro-inflammatory immune cell marker, correlated with copper metabolism abnormalities, further reveals that copper may enhance neuroinflammatory responses by influencing immune cell dynamics, thus promoting the neurodegenerative process of PD (193).
Additionally, copper imbalance also affects the metal homeostasis within neurons, influencing the aggregation of α-synuclein (152). Studies have shown that copper can bind to α-synuclein and promote its aggregation, forming Lewy bodies, which is a hallmark pathological feature of PD (156). Copper accumulation not only promotes α-synuclein aggregation but also induces oxidative stress, further enhancing neuroimmune responses (157). The aggregation of α-synuclein promotes neuroinflammation, and this immune activation is closely related to copper regulation (170).
In conclusion, copper metabolism imbalance not only directly affects the functions of immune cells but also influences the aggregation of α-synuclein by altering metal homeostasis within neurons, thereby exacerbating neuroinflammation and neurodegenerative lesions (172). Copper accumulation drives neuronal damage and PD progression through immune cell activation and increased oxidative stress (175). This finding reveals the complex interplay between copper and immune responses, providing potential therapeutic targets for future PD treatment strategies, particularly in copper homeostasis regulation and immune response control (182).
Stroke
Stroke is caused by acute focal injury to the central nervous system (CNS), leading to neurological deficits from vascular lesions (194). Strokes are generally classified as ischemic or hemorrhagic; the vast majority are ischemic, attributable to reduced blood flow (usually due to arterial occlusion) (195). During stroke, blood flow is interrupted or a cerebral artery ruptures, discontinuing energy supply to brain tissue (194). This interruption results in metabolic imbalance within neurons and heightened oxidative stress responses (194). In this process, copper contributes to neuronal damage and death via multiple mechanisms, closely tied to reactive oxygen species (ROS) production, mitochondrial dysfunction, and regulation of apoptotic signaling pathways (196).
Copper serves as an essential cofactor for numerous redox enzymes—including superoxide dismutase (SOD)—that are critical for redox balance within neurons (197). When copper levels are dysregulated (deficient or excessive), ROS levels surge, collapsing mitochondrial membrane potential and accumulating ROS (198). Excessive ROS generation triggers lipid peroxidation, protein damage, and DNA breaks, initiating endogenous apoptotic pathways (2). These effects are particularly pronounced in ischemic stroke: oxygen and nutrient supply plummet, forcing neurons to rely on anaerobic glycolysis for ATP production, an inefficient pathway insufficient to meet metabolic demands (199). This shortfall further worsens intracellular calcium overload and redox imbalance (200).
Studies show that SOD1 overexpression substantially reduces ROS levels by enhancing the antioxidant capacity of neural stem cells (NSCs) against ischemia-reperfusion injury (201). Consequently, SOD1 overexpression lessens oxidative stress-mediated cell death and improves NSC survival and reparative capacity for ischemic brain damage both in vitro and in vivo (201). In addition to promoting vascular endothelial growth factor (VEGF) secretion and angiogenesis in ischemic regions, SOD1 overexpression supports the release of neuroprotective factors, reinforcing endogenous repair mechanisms that shrink infarct volume and improve brain function (202).
Research suggests that combined deficiency of copper-transporting proteins (e.g., ATP7A) and metallothioneins (MTs) abolishes cellular tolerance to copper toxicity (203, 204). Even at extremely low copper concentrations, these cells experience severe copper overload, resulting in rapid ROS accumulation, collapse of the mitochondrial membrane potential, and disruption of the glutathione (GSH) redox system, ultimately activating various oxidative stress-mediated cell death pathways (e.g., apoptosis, necrosis, or cuproptosis) (205). Another study indicates that high mobility group protein B1 (HMGB1), acting as an inflammatory mediator induced by cuproptosis, is rapidly released extracellularly post-cerebral ischemia (206). HMGB1 then activates danger-associated molecular pattern (DAMP) signaling, triggering microglial activation and proinflammatory cytokine release, thereby intensifying neuroinflammation and causing further disruption of the blood-brain barrier (205).
Metallothioneins (MTs) and the glutathione (GSH) system both modulate copper toxicity through their antioxidant properties following cerebral ischemia (207). MTs bind free copper via thiol groups, preventing copper ions from damaging other intracellular molecules, whereas GSH maintains redox homeostasis by binding copper ions (27). Under ischemic conditions, GSH is rapidly oxidized, reducing the GSH/GSSG ratio and weakening antioxidant capacity, rendering cells more vulnerable to the combined damage of copper and ROS (205). Additionally, copper overload inhibits the synthesis of iron–sulfur cluster proteins, further amplifying metabolic imbalance (208).
In ischemic stroke, autophagy can serve as a protective mechanism, clearing damaged mitochondria and proteins; yet excessive autophagic activation may hasten cell death (209). Copper modulates autophagy levels through the AMPK-mTOR axis and also directly binds to and activates autophagy-related kinases ULK1/ULK2. In cerebral ischemia, copper-driven autophagy may not only counter cellular damage but also exacerbate neuronal loss by selectively degrading antiapoptotic or antioxidant factors such as GPX4 (205).
Copper regulation of inflammatory responses is closely interwoven with stroke pathogenesis (210). Studies suggest that copper-mediated redox reactions may escalate inflammatory responses by promoting proinflammatory cytokine release (210, 211). In the setting of ischemia-reperfusion injury, copper accumulation may activate the NF-κB signaling pathway, intensifying inflammatory cascades (212). Moreover, disrupted copper metabolism can undermine extracellular matrix remodeling around blood vessels, triggering cerebrovascular dysfunction (213). For instance, copper enhances lysyl oxidase (LOX) activity, promoting collagen cross-linking and increasing vascular stiffness (214). This alteration compromises the integrity of the blood-brain barrier, creating a potential pathological foundation for vascular stroke (2).
Copper has dual roles in modulating excitotoxicity and neuronal death (215). By regulating N-methyl-D-aspartate receptor (NMDAR) activity, copper influences glutamate neurotoxicity (216). Under normal physiological conditions, copper released at the synapse suppresses NMDAR activity, thus exerting neuroprotective effects (216). However, in the presence of copper deficiency or ATP7A dysfunction, this regulatory mechanism falters, leading to NMDAR overactivation, calcium overload, and neuronal apoptosis (217, 218). In hemorrhagic stroke or following brain trauma, copper can accumulate directly in brain tissue, generating harmful hydroxyl radicals through the non-enzymatic Fenton reaction, which induces apoptosis or necrosis (219). Furthermore, copper can bind directly to key proteins (e.g., prion protein, Aβ, or tau), forming abnormal aggregates that magnify brain tissue injury in a vicious cycle (2).
Copper’s functions are not limited to the onset of brain injury but also extend to neurological recovery (220). Copper participates in the maturation of various neuropeptides, including neuropeptide Y and corticotropin-releasing hormone, both of which are vital for neuroprotection and modulating neuroinflammation after stroke (196, 199). Additionally, copper-dependent metalloproteins play regulatory roles in angiogenesis and neural regeneration, influencing the functional remodeling of brain tissue (2).
Clinically, modulating copper metabolism abnormalities shows therapeutic promise (221). Under copper deficiency, copper supplementation restores copper-dependent enzyme function and bolsters antioxidant defenses (196, 222). Conversely, when copper is elevated or abnormally localized, copper-chelating agents (e.g., penicillamine or trientine) alleviate oxidative stress and neurotoxicity by reducing copper load (2). The fine-tuned regulation of copper metabolism and distribution in the brain, via lipophilic metal-protein-attenuating compounds (MPACs) that redistribute copper, might be a potential strategy for treating stroke and related neurodegenerative diseases (71, 223). This approach provides an avenue to intervene in copper metabolism and precisely modulate copper distribution to restore brain function and reduce pathological damage (2). Thus, targeted copper regulation provides new avenues for clinical therapy (71).
An animal study showed that in a mouse model treated with tetrathiomolybdate (TM), microvessel density (MVD) decreased by 50% (224). A case-control study indicated a nonlinear, L-shaped relationship between increased copper intake and reduced stroke risk: stroke risk gradually fell with greater copper intake before leveling off beyond a certain intake threshold (225). Other research has found that elevated plasma copper concentration is significantly associated with increased ischemic stroke risk (226). The same study also discovered a strong correlation between plasma copper levels and hyperlipidemia, a known risk factor for carotid atherosclerosis and ischemic stroke (227). A meta-analysis corroborated these findings: across eight high-quality studies, serum copper levels were significantly higher in patients with ischemic stroke than in controls (228).
An animal experiment showed that mice consuming trace amounts of copper (one-tenth the EPA’s upper limit) for 14 weeks had increased infarct volume and worse neurologic outcomes in a MCAO-induced ischemic brain injury model (229). This damage was closely tied to endothelial progenitor cell (EPC) dysfunction, as copper intake impaired EPC migration, tube formation, and adhesion, while markedly reducing phosphorylated eNOS and MnSOD levels, lowering nitric oxide (NO) production, and raising TSP-1 expression—ultimately inhibiting angiogenesis (230–232). Brain tissue analyses revealed that copper-exposure-induced EPC dysfunction led to significantly lower microvessel density in ischemic regions, hindering vascular growth factor (e.g., VEGF) secretion and endothelial repair mechanisms, and aggravating ischemic brain injury (229, 233, 234). These findings illuminate the core molecular and cellular processes by which trace copper intake exacerbates cerebral ischemic damage (229).
Copper plays a key role in immune responses in the central nervous system (CNS), primarily through its involvement in redox reactions and its regulation of immune system function (196). Copper not only participates in neurotransmitter synthesis, energy metabolism, and antioxidant defense but also directly influences immune responses (198). When copper metabolism is disrupted, either through excess copper or copper deficiency, it can trigger abnormal immune responses, thereby exacerbating neuronal damage, especially in neurodegenerative diseases such as ischemic stroke (200).
Firstly, the role of copper in the immune response within the nervous system is closely related to oxidative stress (2). Copper, through its participation in redox reactions, helps maintain the redox balance within cells (2). For example, copper acts as a cofactor for antioxidant enzymes such as copper/zinc superoxide dismutase (Cu/Zn SOD), helping to clear reactive oxygen species (ROS) and thus mitigating oxidative damage (2). However, excessive copper accumulation leads to increased ROS generation, resulting in oxidative stress and cellular damage (196). This oxidative stress not only directly harms neurons but also activates immune cells in the CNS, particularly microglia, which play a critical role in neuroinflammation (227). Excess copper accumulation can stimulate microglial activation, prompting the release of more pro-inflammatory cytokines such as TNF-α and IL-1β, which further exacerbate local neuroinflammatory responses (235).
The mechanism by which copper excess activates immune cells through oxidative stress is especially important during stroke (196). Oxidative stress increases the accumulation of free radicals and activates signaling pathways such as MAPK and NF-κB, promoting the production of pro-inflammatory cytokines (196). In this process, copper exacerbates oxidative damage by enhancing ROS generation and promoting the overactivation of immune cells, further amplifying local inflammation (2). These interactions create a vicious cycle, where the continuously enhanced inflammatory response not only causes neuronal damage but also accelerates the development of neurodegenerative lesions (196).
In addition to oxidative stress, copper’s role in immune modulation within the nervous system is also crucial (2). During neuro-pathological conditions like stroke, copper homeostasis directly impacts immune cell function (205). Copper regulates the intensity of immune responses through interactions with immune-modulatory factors such as metallothioneins (MTs) (2). When copper is deficient, the activation of microglia and immune responses is suppressed, leading to a weakened immune response and making the nervous system more vulnerable to damage (196). In contrast, excess copper enhances immune cell activation and pro-inflammatory cytokine release, thus promoting neuroinflammation and accelerating pathological progression (196).
Furthermore, the neuroimmune mechanisms related to stroke also involve copper’s relationship with atherosclerosis (227). Excess copper accumulation may promote lipid deposition in blood vessel walls through the oxidation of low-density lipoprotein (LDL), leading to the development of atherosclerosis (229). Copper exacerbates vascular damage through this mechanism and may also stimulate immune responses in the vascular endothelium, increasing local inflammation and thereby raising the risk of ischemic stroke (228).
Dysregulation of copper metabolism, especially copper excess, has been confirmed as a significant factor in the occurrence and progression of neurodegenerative diseases like stroke (235). Excess copper not only causes direct oxidative damage but also alters immune cell function, changing the neuroimmune response and further promoting inflammation within the nervous system (235). Moreover, maintaining copper homeostasis is crucial for normal immune function in the CNS, protecting neurons from damage, and promoting cerebrovascular repair (228). Disrupted copper metabolism may become a potential factor in the worsening of neurodegenerative diseases and cerebral ischemic injuries, suggesting that special attention should be given to copper balance in the treatment of stroke and other neuroimmune diseases (227). Proper copper intake and homeostasis are vital for stroke prevention, neuronal damage repair, and the normal regulation of immune responses (229).
Condensing content
Copper homeostasis is crucial for normal physiological functioning, playing pivotal roles in energy metabolism, antioxidant defense, and immune regulation, while also being intimately involved in the onset and progression of neurodegenerative diseases. The recent conceptualization of cuproptosis has elevated copper from its conventional classification as an “oxidative stress factor” and “enzyme cofactor” to a central determinant of cell fate. Existing studies indicate that in the central nervous system, copper can drive protein misfolding and aggregation to exacerbate neuronal injury, but it can also participate in the activation of antioxidant enzymes and repair factors to inhibit excessive cell death and inflammation, demonstrating a “double-edged sword” effect. As research into the pathological mechanisms of Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, Huntington’s disease, and stroke intensifies, emerging causal connections between copper homeostasis and neuronal damage, protein aggregation, mitochondrial dysfunction, and immune-inflammatory processes provide new insights into critical disease pathogenesis and clinical intervention strategies (72, 119, 131, 157, 196). Nevertheless, the precise mechanisms and pathological significance of cuproptosis in neurodegenerative conditions require further investigation. First, copper dysregulation involves dynamic, multilayered regulation—encompassing transmembrane transport, chaperone-mediated protein allocation, and redox coupling—leaving many key regulatory factors and interactions incompletely understood. Second, cuproptosis may intersect with ferroptosis, apoptosis, and autophagy, potentially synergizing to amplify damage, underscoring the importance of clarifying its predominant role at various disease stages for targeted therapy and prognostic assessment. Third, current evaluations of interventions such as copper chelators, metal-protein-attenuating compounds (MPACs), and gene therapy remain constrained by dosage windows, blood-brain barrier permeability, and individual variability, necessitating large-scale clinical trials to confirm their safety and efficacy. Future research might focus on real-time, high-resolution molecular imaging and multi-omics platforms that monitor copper dynamics in the brain and its binding states with critical proteins, on systematically screening and optimizing cellular and animal models to identify drug targets and small-molecule compounds that can balance removal of excess copper or replenishment of copper deficiency without disrupting the overall metal equilibrium, and on leveraging gene editing and immune modulation to reconstruct copper homeostasis and inflammatory pathways in the neural microenvironment for personalized therapies. As our understanding of cuproptosis and the pathophysiology of the nervous system deepens, interventions centered on copper homeostasis may pave the way for next-generation treatments of neurodegenerative diseases, offering novel possibilities for slowing disease progression and improving patient quality of life.
Statements
Author contributions
LLi: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. LLv: Conceptualization, Project administration, Supervision, Writing – review & editing. ZW: Conceptualization, Project administration, Supervision, Writing – review & editing. XL: Conceptualization, Project administration, Supervision, Writing – review & editing. QW: Conceptualization, Project administration, Supervision, Writing – review & editing. HZ: Project administration, Supervision, Writing – review & editing. BJ: Project administration, Supervision, Writing – review & editing. YH: Project administration, Supervision, Writing – review & editing. XP: Supervision, Writing – review & editing. XZ: Supervision, Writing – review & editing. LR: Supervision, Writing – review & editing. ZC: Resources, Writing – review & editing, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Natural Science Foundation of China (NSFC), Youth Science Fund Project under the lump sum funding system (Grant No. 82205163); the National Natural Science Foundation of China (NSFC), Budget-Based Project (Grant No. 82174421); and the Heilongjiang Provincial Administration of Traditional Chinese Medicine, Heilongjiang Traditional Chinese Medicine Research Project (Grant No. 2HY2022-110), Heilongjiang Provincial Natural Science Foundation (Grant No. PL2024H220), and Heilongjiang Provincial Chinese Medicine Scientific Research Project (Grant No. HYZ2022-128).
Acknowledgments
The authors are deeply grateful to the Key Laboratory of Basic Research on Traditional Chinese Medicine of Heilongjiang Province for their invaluable support and contributions to this research.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Correction note
A correction has been made to this article. Details can be found at: 10.3389/fneur.2025.1664184.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
Schulte NB Reznik N Chacon KN Fass D Franz KJ . Simultaneous binding of Cu+ and Cu2+ at the two-tiered copper binding site of the intestinal mucin MUC2. Inorg Chem. (2025) 64:5568–78. doi: 10.1021/acs.inorgchem.5c00016
2.
Chen J Jiang Y Shi H Peng Y Fan X Li C . The molecular mechanisms of copper metabolism and its roles in human diseases. Pflugers Arch. (2020) 472:1415–29. doi: 10.1007/s00424-020-02412-2
3.
Guan D Zhao L Shi X Ma X Chen Z . Copper in cancer: from pathogenesis to therapy. Biomed Pharmacother. (2023) 163:114791. doi: 10.1016/j.biopha.2023.114791
4.
Gao J Wu X Huang S Zhao Z He W Song M . Novel insights into anticancer mechanisms of elesclomol: more than a prooxidant drug. Redox Biol. (2023) 67:102891. doi: 10.1016/j.redox.2023.102891
5.
Zeinali T Salmani F Naseri K . Dietary intake of cadmium, chromium, copper, nickel, and lead through the consumption of meat, liver, and kidney and assessment of human health risk in Birjand, southeast of Iran. Biol Trace Elem Res. (2019) 191:338–47. doi: 10.1007/s12011-019-1637-6
6.
Collins JF . Copper nutrition and biochemistry and human (patho)physiology. Adv Food Nutr Res. (2021) 96:311–64. doi: 10.1016/bs.afnr.2021.01.005
7.
Blades B Ayton S Hung YH Bush AI La Fontaine S . Copper and lipid metabolism: a reciprocal relationship. Biochim Biophys Acta Gen Subj. (2021) 1865:129979. doi: 10.1016/j.bbagen.2021.129979
8.
Tsvetkov P Coy S Petrova B Dreishpoon M Verma A Abdusamad M et al . Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. (2022) 375:1254–61. doi: 10.1126/science.abf0529
9.
Feng Q Huo C Wang M Huang H Zheng X Xie M . Research progress on cuproptosis in cancer. Front Pharmacol. (2024) 15:1290592. doi: 10.3389/fphar.2024.1290592
10.
Gao L Zhang A . Copper-instigated modulatory cell mortality mechanisms and progress in oncological treatment investigations. Front Immunol. (2023) 14:1236063. doi: 10.3389/fimmu.2023.1236063
11.
Xie J Yang Y Gao Y He J . Cuproptosis: mechanisms and links with cancers. Mol Cancer. (2023) 22:46. doi: 10.1186/s12943-023-01732-y
12.
Uttara B Singh AV Zamboni P Mahajan RT . Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol. (2009) 7:65–74. doi: 10.2174/157015909787602823
13.
GBD 2016 Neurology Collaborators . Global, regional, and national burden of neurological disorders, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. (2019) 18:459–80. doi: 10.1016/S1474-4422(18)30499-X
14.
Tiwari S Atluri V Kaushik A Yndart A Nair M . Alzheimer’s disease: pathogenesis, diagnostics, and therapeutics. Int J Nanomedicine. (2019) 14:5541–54. doi: 10.2147/IJN.S200490
15.
Bost M Houdart S Oberli M Kalonji E Huneau JF Margaritis I . Dietary copper and human health: current evidence and unresolved issues. J Trace Elem Med Biol. (2016) 35:107–15. doi: 10.1016/j.jtemb.2016.02.006
16.
Lonnerdal B . Intestinal regulation of copper homeostasis: a developmental perspective. Am J Clin Nutr. (2008) 88:846S–50S. doi: 10.1093/ajcn/88.3.846S
17.
Nose Y Kim BE Thiele DJ . Ctr1 drives intestinal copper absorption and is essential for growth, iron metabolism, and neonatal cardiac function. Cell Metab. (2006) 4:235–44. doi: 10.1016/j.cmet.2006.08.009
18.
Nose Y Wood LK Kim BE Prohaska JR Fry RS Spears JW et al . Ctr1 is an apical copper transporter in mammalian intestinal epithelial cells in vivo that is controlled at the level of protein stability. J Biol Chem. (2010) 285:32385–92. doi: 10.1074/jbc.M110.143826
19.
Haberkiewicz O Lipinski P Starzynski RR Jonczy A Kurowska P Ogorek M et al . Decreased expression of the Slc31a1 gene and cytoplasmic relocalization of membrane CTR1 protein in renal epithelial cells: a potent protective mechanism against copper nephrotoxicity in a mouse model of Menkes disease. Int J Mol Sci. (2022) 23:11441. doi: 10.3390/ijms231911441
20.
Kuo YM Gybina AA Pyatskowit JW Gitschier J Prohaska JR . Copper transport protein (Ctr1) levels in mice are tissue specific and dependent on copper status. J Nutr. (2006) 136:21–6. doi: 10.1093/jn/136.1.21
21.
Liang ZD Tsai WB Lee MY Savaraj N Kuo MT . Specificity protein 1 (sp1) oscillation is involved in copper homeostasis maintenance by regulating human high-affinity copper transporter 1 expression. Mol Pharmacol. (2012) 81:455–64. doi: 10.1124/mol.111.076422
22.
Kuo MT Huang YF Chou CY Chen HHW . Targeting the copper transport system to improve treatment efficacies of platinum-containing drugs in cancer chemotherapy. Pharmaceuticals. (2021) 14:549. doi: 10.3390/ph14060549
23.
Perkal O Qasem Z Turgeman M Schwartz R Gevorkyan-Airapetov L Pavlin M et al . Cu(I) controls conformational states in human Atox1 metallochaperone: an EPR and multiscale simulation study. J Phys Chem B. (2020) 124:4399–411. doi: 10.1021/acs.jpcb.0c01744
24.
Kirsipuu T Zadoroznaja A Smirnova J Friedemann M Plitz T Tougu V et al . Copper(II)-binding equilibria in human blood. Sci Rep. (2020) 10:5686. doi: 10.1038/s41598-020-62560-4
25.
Lutsenko S . Dynamic and cell-specific transport networks for intracellular copper ions. J Cell Sci. (2021) 134:jcs240523. doi: 10.1242/jcs.240523
26.
Nezhad RM Shahpiri A Mirlohi A . Heterologous expression and metal-binding characterization of a type 1 metallothionein isoform (OsMTI-1b) from rice (Oryza sativa). Protein J. (2013) 32:131–7. doi: 10.1007/s10930-013-9469-2
27.
Santoro A Calvo JS Peris-Diaz MD Krezel A Meloni G Faller P . The glutathione/metallothionein system challenges the design of efficient O2-activating copper complexes. Angew Chem Int Ed Engl. (2020) 59:7830–5. doi: 10.1002/anie.201916316
28.
La Fontaine S Ackland ML Mercer JF . Mammalian copper-transporting P-type ATPases, ATP7A and ATP7B: emerging roles. Int J Biochem Cell Biol. (2010) 42:206–9. doi: 10.1016/j.biocel.2009.11.007
29.
Bitter RM Oh S Deng Z Rahman S Hite RK Yuan P . Structure of the Wilson disease copper transporter ATP7B. Sci Adv. (2022) 8:eabl5508. doi: 10.1126/sciadv.abl5508
30.
Yang Y Wu J Wang L Ji G Dang Y . Copper homeostasis and cuproptosis in health and disease. MedComm. (2020) 5:e724. doi: 10.1002/mco2.724
31.
Singla A Chen Q Suzuki K Song J Fedoseienko A Wijers M et al . Regulation of murine copper homeostasis by members of the COMMD protein family. Dis Model Mech. (2021) 14:dmm045963. doi: 10.1242/dmm.045963
32.
Wijmenga C Klomp LW . Molecular regulation of copper excretion in the liver. Proc Nutr Soc. (2004) 63:31–9. doi: 10.1079/PNS2003316
33.
Gul B Firasat S Tehreem R Shan T Afshan K . Analysis of Wilson disease mutations in copper binding domain of ATP7B gene. PLoS One. (2022) 17:e0269833. doi: 10.1371/journal.pone.0269833
34.
Huster D Kuhne A Bhattacharjee A Raines L Jantsch V Noe J et al . Diverse functional properties of Wilson disease ATP7B variants. Gastroenterology. (2012) 142:947–956.e5. doi: 10.1053/j.gastro.2011.12.048
35.
Liu T Liu Y Zhang F Gao Y . Copper homeostasis dysregulation promoting cell damage and the association with liver diseases. Chin Med J. (2023) 136:1653–62. doi: 10.1097/CM9.0000000000002697
36.
Yang L Yang P Lip GYH Ren J . Copper homeostasis and cuproptosis in cardiovascular disease therapeutics. Trends Pharmacol Sci. (2023) 44:573–85. doi: 10.1016/j.tips.2023.07.004
37.
Zelinka SL Kirker GT Sterbinsky GE Bourne KJ . Oxidation states of copper in preservative treated wood as studied by X-ray absorption near edge spectroscopy (XANES). PLoS One. (2022) 17:e0263073. doi: 10.1371/journal.pone.0263073
38.
Kirk FT Munk DE Swenson ES Quicquaro AM Vendelbo MH Larsen A et al . Effects of tetrathiomolybdate on copper metabolism in healthy volunteers and in patients with Wilson disease. J Hepatol. (2024) 80:586–95. doi: 10.1016/j.jhep.2023.11.023
39.
Yang D Xiao P Qiu B Yu HF Teng CB . Copper chaperone antioxidant 1: multiple roles and a potential therapeutic target. J Mol Med. (2023) 101:527–42. doi: 10.1007/s00109-023-02311-w
40.
Jana A Das A Krett NL Guzman G Thomas A Mancinelli G et al . Nuclear translocation of Atox1 potentiates activin A-induced cell migration and colony formation in colon cancer. PLoS One. (2020) 15:e0227916. doi: 10.1371/journal.pone.0227916
41.
Itoh S Kim HW Nakagawa O Ozumi K Lessner SM Aoki H et al . Novel role of antioxidant-1 (Atox1) as a copper-dependent transcription factor involved in cell proliferation. J Biol Chem. (2008) 283:9157–67. doi: 10.1074/jbc.M709463200
42.
Rosenzweig AC . Copper delivery by metallochaperone proteins. Acc Chem Res. (2001) 34:119–28. doi: 10.1021/ar000012p
43.
de Souza ERP Braz M Castro RN Pereira MD Riger CJ . Influence of microbial fermentation on the antioxidant activity of phenolic substances in Saccharomyces cerevisiae. J Appl Microbiol. (2023) 134:lxad148. doi: 10.1093/jambio/lxad148
44.
Dong X Zhang Z Zhao J Lei J Chen Y Li X et al . The rational design of specific SOD1 inhibitors via copper coordination and their application in ROS signaling research. Chem Sci. (2016) 7:6251–62. doi: 10.1039/C6SC01272H
45.
Hwang J Jin J Jeon S Moon SH Park MY Yum DY et al . SOD1 suppresses pro-inflammatory immune responses by protecting against oxidative stress in colitis. Redox Biol. (2020) 37:101760. doi: 10.1016/j.redox.2020.101760
46.
Cao Z Chen C Wang C Li T Chang L Si L et al . Enterocytozoon hepatopenaei (EHP) infection alters the metabolic processes and induces oxidative stress in Penaeus vannamei. Animals. (2023) 13:3661. doi: 10.3390/ani13233661
47.
Elchuri S Oberley TD Qi W Eisenstein RS Jackson Roberts L Van Remmen H et al . CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene. (2005) 24:367–80. doi: 10.1038/sj.onc.1208207
48.
Ding Y Chen Y Wu Z Yang N Rana K Meng X et al . SsCox17, a copper chaperone, is required for pathogenic process and oxidative stress tolerance of Sclerotinia sclerotiorum. Plant Sci. (2022) 322:111345. doi: 10.1016/j.plantsci.2022.111345
49.
Chen Y Li C Li M Han B . Roles of copper transport systems members in breast cancer. Cancer Med. (2024) 13:e70498. doi: 10.1002/cam4.70498
50.
Chojnacka M Gornicka A Oeljeklaus S Warscheid B Chacinska A . Cox17 protein is an auxiliary factor involved in the control of the mitochondrial contact site and cristae organizing system. J Biol Chem. (2015) 290:15304–12. doi: 10.1074/jbc.M115.645069
51.
Horng YC Cobine PA Maxfield AB Carr HS Winge DR . Specific copper transfer from the Cox17 metallochaperone to both Sco1 and Cox11 in the assembly of yeast cytochrome C oxidase. J Biol Chem. (2004) 279:35334–40. doi: 10.1074/jbc.M404747200
52.
Typas D . MOF acetylates COX17 to preserve functional mitochondria: a KAT with two lives. Nat Struct Mol Biol. (2023) 30:1620. doi: 10.1038/s41594-023-01158-6
53.
Takahashi Y Kako K Kashiwabara S Takehara A Inada Y Arai H et al . Mammalian copper chaperone Cox17p has an essential role in activation of cytochrome C oxidase and embryonic development. Mol Cell Biol. (2002) 22:7614–21. doi: 10.1128/MCB.22.21.7614-7621.2002
54.
Ge EJ Bush AI Casini A Cobine PA Cross JR DeNicola GM et al . Connecting copper and cancer: from transition metal signalling to metalloplasia. Nat Rev Cancer. (2022) 22:102–13. doi: 10.1038/s41568-021-00417-2
55.
Agarwal P Ayton S Agrawal S Dhana K Bennett DA Barnes LL et al . Brain copper may protect from cognitive decline and Alzheimer’s disease pathology: a community-based study. Mol Psychiatry. (2022) 27:4307–13. doi: 10.1038/s41380-022-01802-5
56.
Graff-Radford J Yong KXX Apostolova LG Bouwman FH Carrillo M Dickerson BC et al . New insights into atypical Alzheimer’s disease in the era of biomarkers. Lancet Neurol. (2021) 20:222–34. doi: 10.1016/S1474-4422(20)30440-3
57.
Arvanitakis Z Shah RC Bennett DA . Diagnosis and management of dementia: review. JAMA. (2019) 322:1589–99. doi: 10.1001/jama.2019.4782
58.
Scheltens P De Strooper B Kivipelto M Holstege H Chetelat G Teunissen CE et al . Alzheimer’s disease. Lancet. (2021) 397:1577–90. doi: 10.1016/S0140-6736(20)32205-4
59.
Jucker M Walker LC . Alzheimer’s disease: from immunotherapy to immunoprevention. Cell. (2023) 186:4260–70. doi: 10.1016/j.cell.2023.08.021
60.
Moujalled D Strasser A Liddell JR . Molecular mechanisms of cell death in neurological diseases. Cell Death Differ. (2021) 28:2029–44. doi: 10.1038/s41418-021-00814-y
61.
Kim SY Chae CW Lee HJ Jung YH Choi GE Kim JS et al . Sodium butyrate inhibits high cholesterol-induced neuronal amyloidogenesis by modulating NRF2 stabilization-mediated ROS levels: involvement of NOX2 and SOD1. Cell Death Dis. (2020) 11:469. doi: 10.1038/s41419-020-2663-1
62.
Massaro M Baudo G Lee H Liu H Blanco E . Nuclear respiratory factor-1 (NRF1) induction drives mitochondrial biogenesis and attenuates amyloid beta-induced mitochondrial dysfunction and neurotoxicity. Neurotherapeutics. (2025) 22:e00513. doi: 10.1016/j.neurot.2024.e00513
63.
Zhang H Li X Wang X Xu J Elefant F Wang J . Cellular response to beta-amyloid neurotoxicity in Alzheimer’s disease and implications in new therapeutics. Animal Model Exp Med. (2023) 6:3–9. doi: 10.1002/ame2.12313
64.
Horie K Li Y Barthelemy NR Gordon B Hassenstab J Benzinger TLS et al . change in cerebrospinal fluid tau microtubule binding region detects symptom onset, cognitive decline, tangles, and atrophy in dominantly inherited Alzheimer’s disease. Ann Neurol. (2023) 93:1158–72. doi: 10.1002/ana.26620
65.
Abasi LS Elathram N Movva M Deep A Corbett KD Debelouchina GT . Phosphorylation regulates tau's phase separation behavior and interactions with chromatin. Commun Biol. (2024) 7:251. doi: 10.1038/s42003-024-05920-4
66.
Stefanoska K Gajwani M Tan ARP Ahel HI Asih PR Volkerling A et al . Alzheimer’s disease: ablating single master site abolishes tau hyperphosphorylation. Sci Adv. (2022) 8:eabl8809. doi: 10.1126/sciadv.abl8809
67.
Wesseling H Mair W Kumar M Schlaffner CN Tang S Beerepoot P et al . Tau PTM profiles identify patient heterogeneity and stages of Alzheimer’s disease. Cell. (2020) 183:1699–1713.e13. doi: 10.1016/j.cell.2020.10.029
68.
Ma C Hong F Yang S . Amyloidosis in Alzheimer’s disease: pathogeny, etiology, and related therapeutic directions. Molecules. (2022) 27:1210. doi: 10.3390/molecules27041210
69.
Karran E De Strooper B . The amyloid hypothesis in Alzheimer disease: new insights from new therapeutics. Nat Rev Drug Discov. (2022) 21:306–18. doi: 10.1038/s41573-022-00391-w
70.
Chen LL Fan YG Zhao LX Zhang Q Wang ZY . The metal ion hypothesis of Alzheimer’s disease and the anti-neuroinflammatory effect of metal chelators. Bioorg Chem. (2023) 131:106301. doi: 10.1016/j.bioorg.2022.106301
71.
Zhong G Wang X Li J Xie Z Wu Q Chen J et al . Insights into the role of copper in neurodegenerative diseases and the therapeutic potential of natural compounds. Curr Neuropharmacol. (2024) 22:1650–71. doi: 10.2174/1570159X22666231103085859
72.
Wang Q Sun J Chen T Song S Hou Y Feng L et al . Ferroptosis, pyroptosis, and cuproptosis in Alzheimer’s disease. ACS Chem Neurosci. (2023) 14:3564–87. doi: 10.1021/acschemneuro.3c00343
73.
Lin R Chen X Li W Han Y Liu P Pi R . Exposure to metal ions regulates mRNA levels of APP and BACE1 in PC12 cells: blockage by curcumin. Neurosci Lett. (2008) 440:344–7. doi: 10.1016/j.neulet.2008.05.070
74.
Ishii A Pathoulas JA MoustafaFathy Omar O Ge Y Yao AY Pantalena T et al . Contribution of amyloid deposition from oligodendrocytes in a mouse model of Alzheimer’s disease. Mol Neurodegener. (2024) 19:83. doi: 10.1186/s13024-024-00759-z
75.
Kim N Lee HJ . Redox-active metal ions and amyloid-degrading enzymes in Alzheimer’s disease. Int J Mol Sci. (2021) 22:7697. doi: 10.3390/ijms22147697
76.
Mao X Ye J Zhou S Pi R Dou J Zang L et al . The effects of chronic copper exposure on the amyloid protein metabolisim associated genes’ expression in chronic cerebral hypoperfused rats. Neurosci Lett. (2012) 518:14–8. doi: 10.1016/j.neulet.2012.04.030
77.
Scholefield M Church SJ Xu J Cooper GJS . Metallomic analysis of brain tissues distinguishes between cases of dementia with Lewy bodies, Alzheimer’s disease, and Parkinson’s disease dementia. Front Neurosci. (2024) 18:1412356. doi: 10.3389/fnins.2024.1412356
78.
Mot AI Wedd AG Sinclair L Brown DR Collins SJ Brazier MW . Metal attenuating therapies in neurodegenerative disease. Expert Rev Neurother. (2011) 11:1717–45. doi: 10.1586/ern.11.170
79.
Rembach A Hare DJ Lind M Fowler CJ Cherny RA McLean C et al . Decreased copper in Alzheimer’s disease brain is predominantly in the soluble extractable fraction. Int J Alzheimers Dis. (2013) 2013:623241. doi: 10.1155/2013/623241
80.
Tabanez M Santos IR Ikebara JM Camargo MLM Dos Santos BA Freire BM et al . The impact of hydroxytyrosol on the metallomic-profile in an animal model of Alzheimer’s disease. Int J Mol Sci. (2023) 24:4950. doi: 10.3390/ijms241914950
81.
Hobin K Costas-Rodriguez M Van Wonterghem E Vandenbroucke RE Vanhaecke F . Alzheimer's disease and age-related changes in the Cu isotopic composition of blood plasma and brain tissues of the APP(NL-G-F) murine model revealed by multi-collector ICP-mass spectrometry. Biology. (2023) 12:857. doi: 10.3390/biology12060857
82.
Zheng Z White C Lee J Peterson TS Bush AI Sun GY et al . Altered microglial copper homeostasis in a mouse model of Alzheimer’s disease. J Neurochem. (2010) 114:1630–8. doi: 10.1111/j.1471-4159.2010.06888.x
83.
Squitti R . Copper dysfunction in Alzheimer’s disease: from meta-analysis of biochemical studies to new insight into genetics. J Trace Elem Med Biol. (2012) 26:93–6. doi: 10.1016/j.jtemb.2012.04.012
84.
Sun F Zhao J Zhang H Shi Q Liu Y Robert A et al . Proteomics evidence of the role of TDMQ20 in the cholinergic system and synaptic transmission in a mouse model of Alzheimer’s disease. ACS Chem Neurosci. (2022) 13:3093–107. doi: 10.1021/acschemneuro.2c00455
85.
Melek IM Kus B Kaptan Z Petekkaya E . Correlation of metal ions with specific brain region volumes in neurodegenerative diseases. Turk J Med Sci. (2023) 53:1465–75. doi: 10.55730/1300-0144.5714
86.
Verheijen MCT Krauskopf J Caiment F Nazaruk M Wen QF van Herwijnen MHM et al . iPSC-derived cortical neurons to study sporadic Alzheimer disease: a transcriptome comparison with post-mortem brain samples. Toxicol Lett. (2022) 356:89–99. doi: 10.1016/j.toxlet.2021.12.009
87.
D'Angiolini S Basile MS Mazzon E Gugliandolo A . In silico analysis reveals the modulation of ion transmembrane transporters in the cerebellum of Alzheimer’s disease patients. Int J Mol Sci. (2023) 24:13924. doi: 10.3390/ijms241813924
88.
Kirss S Reinapu A Kabin E Smirnova J Tougu V Palumaa P . Alpha-lipoic acid: a potential regulator of copper metabolism in Alzheimer’s disease. Front Mol Biosci. (2024) 11:1451536. doi: 10.3389/fmolb.2024.1451536
89.
Raj K Kaur P Gupta GD Singh S . Metals associated neurodegeneration in Parkinson’s disease: insight to physiological, pathological mechanisms and management. Neurosci Lett. (2021) 753:135873. doi: 10.1016/j.neulet.2021.135873
90.
Turk AZ Lotfi Marchoubeh M Fritsch I Maguire GA SheikhBahaei S . Dopamine, vocalization, and astrocytes. Brain Lang. (2021) 219:104970. doi: 10.1016/j.bandl.2021.104970
91.
Belloli S Morari M Murtaj V Valtorta S Moresco RM Gilardi MC . Translation imaging in Parkinson's disease: focus on neuroinflammation. Front Aging Neurosci. (2020) 12:152. doi: 10.3389/fnagi.2020.00152
92.
Watamura N Kakiya N Fujioka R Kamano N Takahashi M Nilsson P et al . The dopaminergic system promotes neprilysin-mediated degradation of amyloid-beta in the brain. Sci Signal. (2024) 17:eadk1822. doi: 10.1126/scisignal.adk1822
93.
van Dyck CH Arnsten AFT Padala PR Brawman-Mintzer O Lerner AJ Porsteinsson AP et al . Neurobiologic rationale for treatment of apathy in Alzheimer’s disease with methylphenidate. Am J Geriatr Psychiatry. (2021) 29:51–62. doi: 10.1016/j.jagp.2020.04.026
94.
Dodich A Crespi C Santi GC Luzzi S Ranaldi V Iannaccone S et al . Diagnostic accuracy of affective social tasks in the clinical classification between the behavioral variant of frontotemporal dementia and other neurodegenerative disease. J Alzheimers Dis. (2021) 80:1401–11. doi: 10.3233/JAD-201210
95.
Mondal S Vashi Y Ghosh P Roy D Barthakur M Kumar S et al . Amyloid targeting “artificial chaperone” impairs oligomer mediated neuronal damage and mitochondrial dysfunction associated with Alzheimer’s disease. ACS Chem Neurosci. (2020) 11:3277–87. doi: 10.1021/acschemneuro.0c00387
96.
Scholefield M Church SJ Xu J Kassab S Gardiner NJ Roncaroli F et al . Evidence that levels of nine essential metals in post-mortem human-Alzheimer’s-brain and ex vivo rat-brain tissues are unaffected by differences in post-mortem delay, age, disease staging, and brain bank location. Metallomics. (2020) 12:952–62. doi: 10.1039/d0mt00048e
97.
Wen L Shen L . Effect of surface-chelated Cu2+ on amyloid-beta peptide fibrillation. Langmuir. (2022) 38:174–81. doi: 10.1021/acs.langmuir.1c02322
98.
Friedemann M Tougu V Palumaa P . Copper(II) partially protects three histidine residues and the N-terminus of amyloid-beta peptide from diethyl pyrocarbonate (DEPC) modification. FEBS Open Bio. (2020) 10:1072–81. doi: 10.1002/2211-5463.12857
99.
Gu M Bode DC Viles JH . Copper redox cycling inhibits Aβ fibre formation and promotes fibre fragmentation, while generating a dityrosine Aβ dimer. Sci Rep. (2018) 8:16190. doi: 10.1038/s41598-018-33935-5
100.
Smith DP Ciccotosto GD Tew DJ Fodero-Tavoletti MT Johanssen T Masters CL et al . Concentration dependent Cu2+ induced aggregation and dityrosine formation of the Alzheimer’s disease amyloid-beta peptide. Biochemistry. (2007) 46:2881–91. doi: 10.1021/bi0620961
101.
Kitazawa M Hsu HW Medeiros R . Copper exposure perturbs brain inflammatory responses and impairs clearance of amyloid-beta. Toxicol Sci. (2016) 152:194–204. doi: 10.1093/toxsci/kfw081
102.
Li K Li A Mei Y Zhao J Zhou Q Li Y et al . Trace elements and Alzheimer dementia in population-based studies: a bibliometric and meta-analysis. Environ Pollut. (2023) 318:120782. doi: 10.1016/j.envpol.2022.120782
103.
Squitti R Ventriglia M Simonelli I Bonvicini C Costa A Perini G et al . Copper imbalance in Alzheimer’s disease: meta-analysis of serum, plasma, and brain specimens, and replication study evaluating ATP7B gene variants. Biomol Ther. (2021) 11:960. doi: 10.3390/biom11070960
104.
Liu F Zhang Z Zhang L Meng RN Gao J Jin M et al . Effect of metal ions on Alzheimer’s disease. Brain Behav. (2022) 12:e2527. doi: 10.1002/brb3.2527
105.
Singh SK Balendra V Obaid AA Esposto J Tikhonova MA Gautam NK et al . Copper-mediated beta-amyloid toxicity and its chelation therapy in Alzheimer’s disease. Metallomics. (2022) 14:mfac018. doi: 10.1093/mtomcs/mfac018
106.
Min JH Sarlus H Harris RA . MAD-microbial (origin of) Alzheimer’s disease hypothesis: from infection and the antimicrobial response to disruption of key copper-based systems. Front Neurosci. (2024) 18:1467333. doi: 10.3389/fnins.2024.1467333
107.
Choo XY Alukaidey L White AR Grubman A . Neuroinflammation and copper in Alzheimer’s disease. Int J Alzheimers Dis. (2013) 2013:145345. doi: 10.1155/2013/145345
108.
Feldman EL Goutman SA Petri S Mazzini L Savelieff MG Shaw PJ et al . Amyotrophic lateral sclerosis. Lancet. (2022) 400:1363–80. doi: 10.1016/S0140-6736(22)01272-7
109.
Chen QY Wu P Wen T Qin X Zhang R Jia R et al . Association of cerebral spinal fluid copper imbalance in amyotrophic lateral sclerosis. Front Aging Neurosci. (2022) 14:970711. doi: 10.3389/fnagi.2022.970711
110.
Al-Chalabi A Hardiman O . The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol. (2013) 9:617–28. doi: 10.1038/nrneurol.2013.203
111.
Jia F Zhang B Yu W Chen Z Xu W Zhao W et al . Exploring the cuproptosis-related molecular clusters in the peripheral blood of patients with amyotrophic lateral sclerosis. Comput Biol Med. (2024) 168:107776. doi: 10.1016/j.compbiomed.2023.107776
112.
Guan M Cheng K Xie XT Li Y Ma MW Zhang B et al . Regulating copper homeostasis of tumor cells to promote cuproptosis for enhancing breast cancer immunotherapy. Nat Commun. (2024) 15:10060. doi: 10.1038/s41467-024-54469-7
113.
Shang N Zhu L Li Y Song C Liu X . Targeting CDK1 and copper homeostasis in breast cancer via a nanopolymer drug delivery system. Cell Biol Toxicol. (2024) 41:16. doi: 10.1007/s10565-024-09958-2
114.
Shteinfer-Kuzmine A Santhanam M Shoshan-Barmatz V . VDAC1-based peptides as potential modulators of VDAC1 interactions with its partners and as a therapeutic for cancer, NASH, and diabetes. Biomol Ther. (2024) 14:1139. doi: 10.3390/biom14091139
115.
Gao F Zhang Y Hou X Tao Z Ren H Wang G . Dependence of PINK1 accumulation on mitochondrial redox system. Aging Cell. (2020) 19:e13211. doi: 10.1111/acel.13211
116.
Tafuri F Ronchi D Magri F Comi GP Corti S . SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis. Front Cell Neurosci. (2015) 9:336. doi: 10.3389/fncel.2015.00336
117.
Hilton JBW Kysenius K Liddell JR Mercer SW Paul B Beckman JS et al . Evidence for disrupted copper availability in human spinal cord supports Cu(II)(atsm) as a treatment option for sporadic cases of ALS. Sci Rep. (2024) 14:5929. doi: 10.1038/s41598-024-55832-w
118.
Gupta G Cappellini F Farcal L Gornati R Bernardini G Fadeel B . Copper oxide nanoparticles trigger macrophage cell death with misfolding of Cu/Zn superoxide dismutase 1 (SOD1). Part Fibre Toxicol. (2022) 19:33. doi: 10.1186/s12989-022-00467-w
119.
Tokuda E Okawa E Watanabe S Ono S Marklund SL . Dysregulation of intracellular copper homeostasis is common to transgenic mice expressing human mutant superoxide dismutase-1s regardless of their copper-binding abilities. Neurobiol Dis. (2013) 54:308–19. doi: 10.1016/j.nbd.2013.01.001
120.
Farrawell NE Yerbury JJ . Mutant Cu/Zn superoxide dismutase (A4V) turnover is altered in cells containing inclusions. Front Mol Neurosci. (2021) 14:771911. doi: 10.3389/fnmol.2021.771911
121.
Furukawa Y O’Halloran TV . Posttranslational modifications in Cu, Zn-superoxide dismutase and mutations associated with amyotrophic lateral sclerosis. Antioxid Redox Signal. (2006) 8:847–67. doi: 10.1089/ars.2006.8.847
122.
Koehn LM Steele JR Schittenhelm RB Turner BJ Nicolazzo JA . Sex-dependent changes to the intestinal and hepatic abundance of drug transporters and metabolizing enzymes in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Mol Pharm. (2024) 21:1756–67. doi: 10.1021/acs.molpharmaceut.3c01089
123.
Swindell WR Kruse CPS List EO Berryman DE Kopchick JJ . ALS blood expression profiling identifies new biomarkers, patient subgroups, and evidence for neutrophilia and hypoxia. J Transl Med. (2019) 17:170. doi: 10.1186/s12967-019-1909-0
124.
Wang H Morman C Sternke-Hoffmann R Huang CY Prota A Ma P et al . Cu2+ ions modulate the interaction between alpha-synuclein and lipid membranes. J Inorg Biochem. (2022) 236:111945. doi: 10.1016/j.jinorgbio.2022.111945
125.
Cyboran-Mikolajczyk S Jurkiewicz P Hof M Kleszczynska H . The impact of O-glycosylation on cyanidin interaction with POPC membranes: structure-activity relationship. Molecules. (2018) 23:2771. doi: 10.3390/molecules23112771
126.
LeBlanc A Cuperlovic-Culf M Morin PJ Touaibia M . Structurally related Edaravone analogues: synthesis, antiradical, antioxidant, and copper-chelating properties. CNS Neurol Disord Drug Targets. (2019) 18:779–90. doi: 10.2174/1871527318666191114092007
127.
Andreassen OA Dedeoglu A Friedlich A Ferrante KL Hughes D Szabo C et al . Effects of an inhibitor of poly(ADP-ribose) polymerase, desmethylselegiline, trientine, and lipoic acid in transgenic ALS mice. Exp Neurol. (2001) 168:419–24. doi: 10.1006/exnr.2001.7633
128.
Gale J Aizenman E . The physiological and pathophysiological roles of copper in the nervous system. Eur J Neurosci. (2024) 60:3505–43. doi: 10.1111/ejn.16370
129.
Hilton JB Kysenius K White AR Crouch PJ . The accumulation of enzymatically inactive cuproenzymes is a CNS-specific phenomenon of the SOD1(G37R) mouse model of ALS and can be restored by overexpressing the human copper transporter hCTR1. Exp Neurol. (2018) 307:118–28. doi: 10.1016/j.expneurol.2018.06.006
130.
Solovyev N Lucio M Mandrioli J Forcisi S Kanawati B Uhl J et al . Interplay of metallome and metabolome in amyotrophic lateral sclerosis: a study on cerebrospinal fluid of patients carrying disease-related gene mutations. ACS Chem Neurosci. (2023) 14:3035–46. doi: 10.1021/acschemneuro.3c00128
131.
Xiao G Fan Q Wang X Zhou B . Huntington disease arises from a combinatory toxicity of polyglutamine and copper binding. Proc Natl Acad Sci USA. (2013) 110:14995–5000. doi: 10.1073/pnas.1308535110
132.
Bates GP Dorsey R Gusella JF Hayden MR Kay C Leavitt BR et al . Huntington disease. Nat Rev Dis Primers. (2015) 1:15005. doi: 10.1038/nrdp.2015.5
133.
Tabrizi SJ Estevez-Fraga C van Roon-Mom WMC Flower MD Scahill RI Wild EJ et al . Potential disease-modifying therapies for Huntington’s disease: lessons learned and future opportunities. Lancet Neurol. (2022) 21:645–58. doi: 10.1016/S1474-4422(22)00121-1
134.
Lobato AG Ortiz-Vega N Zhu Y Neupane D Meier KK Zhai RG . Copper enhances aggregational toxicity of mutant huntingtin in a Drosophila model of Huntington’s disease. Biochim Biophys Acta Mol basis Dis. (2024) 1870:166928. doi: 10.1016/j.bbadis.2023.166928
135.
Gaetke LM Chow-Johnson HS Chow CK . Copper: toxicological relevance and mechanisms. Arch Toxicol. (2014) 88:1929–38. doi: 10.1007/s00204-014-1355-y
136.
Cordeiro LM Soares MV da Silva AF Dos Santos LV de Souza LI da Silveira TL et al . Toxicity of copper and zinc alone and in combination in Caenorhabditis elegans model of Huntington’s disease and protective effects of rutin. Neurotoxicology. (2023) 97:120–32. doi: 10.1016/j.neuro.2023.06.005
137.
Cordeiro LM Machado ML da Silva AF Obetine Baptista FB da Silveira TL Soares FAA et al . Rutin protects Huntington’s disease through the insulin/IGF1 (IIS) signaling pathway and autophagy activity: study in Caenorhabditis elegans model. Food Chem Toxicol. (2020) 141:111323. doi: 10.1016/j.fct.2020.111323
138.
Cui Z Lockman PR Atwood CS Hsu CH Gupte A Allen DD et al . Novel D-penicillamine carrying nanoparticles for metal chelation therapy in Alzheimer’s and other CNS diseases. Eur J Pharm Biopharm. (2005) 59:263–72. doi: 10.1016/j.ejpb.2004.07.009
139.
Pfalzer AC Yan Y Kang H Totten M Silverman J Bowman AB et al . Alterations in metal homeostasis occur prior to canonical markers in Huntington disease. Sci Rep. (2022) 12:10373. doi: 10.1038/s41598-022-14169-y
140.
Simunkova M Barbierikova Z Jomova K Hudecova L Lauro P Alwasel SH et al . Antioxidant vs. prooxidant properties of the flavonoid, kaempferol, in the presence of Cu(II) ions: a ROS-scavenging activity, fenton reaction and DNA damage study. Int J Mol Sci. (2021) 22:1619. doi: 10.3390/ijms22041619
141.
McGee TP Houston CM Brickley SG . Copper block of extrasynaptic GABAA receptors in the mature cerebellum and striatum. J Neurosci. (2013) 33:13431–5. doi: 10.1523/JNEUROSCI.1908-13.2013
142.
Huang T Li C Sun X Zhu Z Fu Y Liu Y et al . The antitumor mechanism of di-2-pyridylketone 2-pyridine carboxylic acid hydrazone and its copper complex in ROS generation and topoisomerase inhibition, and hydrazone involvement in oxygen-catalytic iron mobilization. Int J Oncol. (2015) 47:1854–62. doi: 10.3892/ijo.2015.3158
143.
Li F Liu H Wu X Liu M Yue Z Liu L et al . Copper modulates mitochondrial oxidative phosphorylation to enhance dermal papilla cells proliferation in rex rabbits. Int J Mol Sci. (2022) 23:6209. doi: 10.3390/ijms23116209
144.
Scholefield M Unwin RD Cooper GJS . Shared perturbations in the metallome and metabolome of Alzheimer’s, Parkinson’s, Huntington’s, and dementia with Lewy bodies: a systematic review. Ageing Res Rev. (2020) 63:101152. doi: 10.1016/j.arr.2020.101152
145.
Petro A Sexton HG Miranda C Rastogi A Freedman JH Levin ED . Persisting neurobehavioral effects of developmental copper exposure in wildtype and metallothionein 1 and 2 knockout mice. BMC Pharmacol Toxicol. (2016) 17:55. doi: 10.1186/s40360-016-0096-3
146.
Martinez-Lazcano JC Montes S Sanchez-Mendoza MA Rodriguez-Paez L Perez-Neri I Boll MC et al . Sub-chronic copper pretreatment reduces oxidative damage in an experimental Huntington’s disease model. Biol Trace Elem Res. (2014) 162:211–8. doi: 10.1007/s12011-014-0127-0
147.
Zeng Z Cen Y Xiong L Hong G Luo Y Luo X . Dietary copper intake and risk of Parkinson’s disease: a Cross-sectional study. Biol Trace Elem Res. (2024) 202:955–64. doi: 10.1007/s12011-023-03750-9
148.
Zhang Y Zhou Q Lu L Su Y Shi W Zhang H et al . Copper induces cognitive impairment in mice via modulation of cuproptosis and CREB signaling. Nutrients. (2023) 15:972. doi: 10.3390/nu15040972
149.
Bloem BR Okun MS Klein C . Parkinson’s disease. Lancet. (2021) 397:2284–303. doi: 10.1016/S0140-6736(21)00218-X
150.
Tolosa E Garrido A Scholz SW Poewe W . Challenges in the diagnosis of Parkinson’s disease. Lancet Neurol. (2021) 20:385–97. doi: 10.1016/S1474-4422(21)00030-2
151.
Mammen JR Speck RM Stebbins GT Muller M Yang PT Campbell M et al . Relative meaningfulness and impacts of symptoms in people with early-stage Parkinson’s disease. J Parkinsons Dis. (2023) 13:619–32. doi: 10.3233/JPD-225068
152.
Morris HR Spillantini MG Sue CM Williams-Gray CH . The pathogenesis of Parkinson’s disease. Lancet. (2024) 403:293–304. doi: 10.1016/S0140-6736(23)01478-2
153.
Dominguez-Fernandez C Egiguren-Ortiz J Razquin J Gomez-Galan M De Las Heras-Garcia L Paredes-Rodriguez E et al . Review of technological challenges in personalised medicine and early diagnosis of neurodegenerative disorders. Int J Mol Sci. (2023) 24:3321. doi: 10.3390/ijms24043321
154.
Wallert ED van de Giessen E Knol RJJ Beudel M de Bie RMA Booij J . Imaging dopaminergic neurotransmission in neurodegenerative disorders. J Nucl Med. (2022) 63:27S–32S. doi: 10.2967/jnumed.121.263197
155.
Becerra-Calixto A Mukherjee A Ramirez S Sepulveda S Sinha T Al-Lahham R et al . Lewy body-like pathology and loss of dopaminergic neurons in midbrain organoids derived from familial Parkinson’s disease patient. Cells. (2023) 12:625. doi: 10.3390/cells12040625
156.
Burre J Sharma M Sudhof TC . Cell biology and pathophysiology of alpha-synuclein. Cold Spring Harb Perspect Med. (2018) 8:a024091. doi: 10.1101/cshperspect.a024091
157.
Lill CM . Genetics of Parkinson’s disease. Mol Cell Probes. (2016) 30:386–96. doi: 10.1016/j.mcp.2016.11.001
158.
So YJ Lee JU Yang GS Yang G Kim SW Lee JH et al . The potentiality of natural products and herbal medicine as novel medications for Parkinson’s disease: a promising therapeutic approach. Int J Mol Sci. (2024) 25:1071. doi: 10.3390/ijms25021071
159.
Lu X Song N Wang W Liu Y Song H Xu L et al . Generation of integration-free human iPSC line LCPHi001-A from a Parkinson’s disease patient carrying the RecNciI mutation in GBA gene. Stem Cell Res. (2021) 56:102514. doi: 10.1016/j.scr.2021.102514
160.
Hartmann A Michel PP Troadec JD Mouatt-Prigent A Faucheux BA Ruberg M et al . Is Bax a mitochondrial mediator in apoptotic death of dopaminergic neurons in Parkinson’s disease?J Neurochem. (2001) 76:1785–93. doi: 10.1046/j.1471-4159.2001.00160.x
161.
Kim R Kin T Beck WT . Impact of complex apoptotic signaling pathways on cancer cell sensitivity to therapy. Cancers. (2024) 16:984. doi: 10.3390/cancers16050984
162.
Joppe K Nicolas JD Grunewald TA Eckermann M Salditt T Lingor P . Elemental quantification and analysis of structural abnormalities in neurons from Parkinson’s-diseased brains by X-ray fluorescence microscopy and diffraction. Biomed Opt Express. (2020) 11:3423–43. doi: 10.1364/BOE.389408
163.
Beauchamp LC Liu XM Vella LJ Adlard PA Bush AI Finkelstein DI et al . ATH434 rescues pre-motor hyposmia in a mouse model of parkinsonism. Neurotherapeutics. (2022) 19:1966–75. doi: 10.1007/s13311-022-01300-0
164.
Huang L Zeng Y Li Y Zhu Y He Y Liu Y et al . Distribution in rat blood and brain of TDMQ20, a copper chelator designed as a drug-candidate for Alzheimer’s disease. Pharmaceutics. (2022) 14:2691. doi: 10.3390/pharmaceutics14122691
165.
Yu HJ Zhao W Zhou Y Cheng GJ Sun M Wang L et al . Salen-based bifunctional chemosensor for copper (II) ions: inhibition of copper-induced amyloid-beta aggregation. Anal Chim Acta. (2020) 1097:144–52. doi: 10.1016/j.aca.2019.10.072
166.
Li G Peng L Wu M Zhao Y Cheng Z Li G . Appropriate level of cuproptosis may be involved in alleviating pulmonary fibrosis. Front Immunol. (2022) 13:1039510. doi: 10.3389/fimmu.2022.1039510
167.
Villadsen B Thygesen C Grebing M Kempf SJ Sandberg MB Jensen P et al . Ceruloplasmin-deficient mice show changes in PTM profiles of proteins involved in messenger RNA processing and neuronal projections and synaptic processes. J Neurochem. (2023) 165:76–94. doi: 10.1111/jnc.15754
168.
Ilyechova EY Miliukhina IV Karpenko MN Orlov IA Puchkova LV Samsonov SA . Case of early-onset Parkinson’s disease in a heterozygous mutation carrier of the ATP7B gene. J Pers Med. (2019) 9:41. doi: 10.3390/jpm9030041
169.
Zhao S Zhang L Ji W Shi Y Lai G Chi H et al . Machine learning-based characterization of cuprotosis-related biomarkers and immune infiltration in Parkinson’s disease. Front Genet. (2022) 13:1010361. doi: 10.3389/fgene.2022.1010361
170.
Wu J Qin C Cai Y Zhou J Xu D Lei Y et al . Machine learning screening for Parkinson’s disease-related cuproptosis-related typing development and validation and exploration of personalized drugs for cuproptosis genes. Ann Transl Med. (2023) 11:11. doi: 10.21037/atm-22-5756
171.
Li L Cai Q Wu Z Li X Zhou W Lu L et al . Bioinformatics construction and experimental validation of a cuproptosis-related lncRNA prognostic model in lung adenocarcinoma for immunotherapy response prediction. Sci Rep. (2023) 13:2455. doi: 10.1038/s41598-023-29684-9
172.
Dudzik CG Walter ED Millhauser GL . Coordination features and affinity of the Cu2+ site in the alpha-synuclein protein of Parkinson’s disease. Biochemistry. (2011) 50:1771–7. doi: 10.1021/bi101912q
173.
Synhaivska O Bhattacharya S Campioni S Thompson D Nirmalraj PN . Single-particle resolution of copper-associated annular alpha-synuclein oligomers reveals potential therapeutic targets of neurodegeneration. ACS Chem Neurosci. (2022) 13:1410–21. doi: 10.1021/acschemneuro.2c00021
174.
Choi TS Lee J Han JY Jung BC Wongkongkathep P Loo JA et al . Supramolecular modulation of structural polymorphism in pathogenic alpha-synuclein fibrils using copper(II) coordination. Angew Chem Int Ed Engl. (2018) 57:3099–103. doi: 10.1002/anie.201712286
175.
Li Y Yang C Wang S Yang D Zhang Y Xu L et al . Copper and iron ions accelerate the prion-like propagation of alpha-synuclein: a vicious cycle in Parkinson’s disease. Int J Biol Macromol. (2020) 163:562–73. doi: 10.1016/j.ijbiomac.2020.06.274
176.
Ramis R Ortega-Castro J Vilanova B Adrover M Frau J . A systematic DFT study of some plausible Zn(II) and Al(III) interaction sites in N-terminally acetylated alpha-synuclein. J Phys Chem A. (2018) 122:690–9. doi: 10.1021/acs.jpca.7b10744
177.
Liu W Lim KL Tan EK . Intestine-derived alpha-synuclein initiates and aggravates pathogenesis of Parkinson's disease in Drosophila. Transl Neurodegener. (2022) 11:44. doi: 10.1186/s40035-022-00318-w
178.
Tripathi T . A master regulator of alpha-synuclein aggregation. ACS Chem Neurosci. (2020) 11:1376–8. doi: 10.1021/acschemneuro.0c00216
179.
Doumi I Lang L Vileno B Deponte M Faller P . Glutathione protects other cellular thiols against oxidation by Cu(II)-Dp44mT. Chemistry. (2024) 30:e202304212. doi: 10.1002/chem.202304212
180.
Ramis R Ortega-Castro J Vilanova B Adrover M Frau J . Cu2+, Ca2+, and methionine oxidation expose the hydrophobic alpha-synuclein NAC domain. Int J Biol Macromol. (2021) 169:251–63. doi: 10.1016/j.ijbiomac.2020.12.018
181.
Bacchella C Camponeschi F Kolkowska P Kola A Tessari I Baratto MC et al . Copper binding and redox activity of alpha-synuclein in membrane-like environment. Biomol Ther. (2023) 13:287. doi: 10.3390/biom13020287
182.
Savva L Platts JA . How Cu(II) binding affects structure and dynamics of alpha-synuclein revealed by molecular dynamics simulations. J Inorg Biochem. (2023) 239:112068. doi: 10.1016/j.jinorgbio.2022.112068
183.
Meena R Sahoo SS Sunil A Manna D . Cuproptosis: a copper-mediated programmed cell death. Chem Asian J. (2025) 20:e202400934. doi: 10.1002/asia.202400934
184.
Deng J Zhuang H Shao S Zeng X Xue P Bai T et al . Mitochondrial-targeted copper delivery for cuproptosis-based synergistic cancer therapy. Adv Healthc Mater. (2024) 13:e2304522. doi: 10.1002/adhm.202304522
185.
Lin CH Chin Y Zhou M Sobol RW Hung MC Tan M . Protein lipoylation: mitochondria, cuproptosis, and beyond. Trends Biochem Sci. (2024) 49:729–44. doi: 10.1016/j.tibs.2024.04.002
186.
Dolgova NV Yu C Cvitkovic JP Hodak M Nienaber KH Summers KL et al . Binding of copper and cisplatin to Atox1 is mediated by glutathione through the formation of metal-sulfur clusters. Biochemistry. (2017) 56:3129–41. doi: 10.1021/acs.biochem.7b00293
187.
Zhang F Chen F Shen R Chen YX Zhao Z Zhang B et al . Naphthalimide fluorescent skeleton for facile and accurate quantification of glutathione. Anal Chem. (2023) 95:4301–9. doi: 10.1021/acs.analchem.2c04098
188.
Rebai A Chbili C Ben Amor S Hassine A Ben Ammou S Saguem S . Effects of glutathione S-transferase M1 and T1 deletions on Parkinson's disease risk among a North African population. Rev Neurol. (2021) 177:290–5. doi: 10.1016/j.neurol.2020.03.013
189.
Gonzalez-Alcocer A Gopar-Cuevas Y Soto-Dominguez A Castillo-Velazquez U de Jesus Loera-Arias M Saucedo-Cardenas O et al . Combined chronic copper exposure and aging lead to neurotoxicity in vivo. Neurotoxicology. (2023) 95:181–92. doi: 10.1016/j.neuro.2023.02.002
190.
Huo S Wang Q Shi W Peng L Jiang Y Zhu M et al . ATF3/SPI1/SLC31A1 signaling promotes cuproptosis induced by advanced glycosylation end products in diabetic myocardial injury. Int J Mol Sci. (2023) 24:1667. doi: 10.3390/ijms24021667
191.
Pienkowska N Bartosz G Sadowska-Bartosz I . Effect of 6-hydroxydopamine increase the glutathione level in SH-SY5Y human neuroblastoma cells. Acta Biochim Pol. (2023) 70:457–64. doi: 10.18388/abp.2020_6847
192.
Matsumura N Kinoshita C Bhadhprasit W Nakaki T Aoyama K . A purine derivative, paraxanthine, promotes cysteine uptake for glutathione synthesis. J Pharmacol Sci. (2023) 151:37–45. doi: 10.1016/j.jphs.2022.11.001
193.
Lucot KL Stevens MY Bonham TA Azevedo EC Chaney AM Webber ED et al . Tracking innate immune activation in a mouse model of Parkinson’s disease using TREM1 and TSPO PET tracers. J Nucl Med. (2022) 63:1570–8. doi: 10.2967/jnumed.121.263039
194.
Campbell BCV Khatri P . Stroke. Lancet. (2020) 396:129–42. doi: 10.1016/S0140-6736(20)31179-X
195.
Alsbrook DL Di Napoli M Bhatia K Biller J Andalib S Hinduja A et al . Neuroinflammation in acute ischemic and hemorrhagic stroke. Curr Neurol Neurosci Rep. (2023) 23:407–31. doi: 10.1007/s11910-023-01282-2
196.
An Y Li S Huang X Chen X Shan H Zhang M . The role of copper homeostasis in brain disease. Int J Mol Sci. (2022) 23:13850. doi: 10.3390/ijms232213850
197.
Nakahara H Nomura A Tokuda S Okamura M Fujisawa K Koitaya T et al . Superoxide dismutase-like activity of zeolitic imidazolate framework nanoparticles comprising biomimetic imidazolato-bridged CuZn units. Chemistry. (2023) 29:e202300881. doi: 10.1002/chem.202300881
198.
Rodrigo R Fernandez-Gajardo R Gutierrez R Matamala JM Carrasco R Miranda-Merchak A et al . Oxidative stress and pathophysiology of ischemic stroke: novel therapeutic opportunities. CNS Neurol Disord Drug Targets. (2013) 12:698–714. doi: 10.2174/1871527311312050015
199.
Przykaza L Kozniewska E . Ligands of the neuropeptide Y Y2 receptors as a potential multitarget therapeutic approach for the protection of the neurovascular unit against acute ischemia/reperfusion: view from the perspective of the laboratory bench. Transl Stroke Res. (2022) 13:12–24. doi: 10.1007/s12975-021-00930-4
200.
Tuo QZ Zhang ST Lei P . Mechanisms of neuronal cell death in ischemic stroke and their therapeutic implications. Med Res Rev. (2022) 42:259–305. doi: 10.1002/med.21817
201.
Ruan Y Luo H Tang J Ji M Yu D Yu Q et al . Curcumin inhibits oxidative stress and autophagy in C17.2 neural stem cell through ERK1/2 signaling pathways. Aging Med. (2024) 7:559–70. doi: 10.1002/agm2.12361
202.
Sakata H Niizuma K Wakai T Narasimhan P Maier CM Chan PH . Neural stem cells genetically modified to overexpress Cu/Zn-superoxide dismutase enhance amelioration of ischemic stroke in mice. Stroke. (2012) 43:2423–9. doi: 10.1161/STROKEAHA.112.656900
203.
Hernandez PP Allende ML . Zebrafish (Danio rerio) as a model for studying the genetic basis of copper toxicity, deficiency, and metabolism. Am J Clin Nutr. (2008) 88:835S–9S. doi: 10.1093/ajcn/88.3.835S
204.
Ma C Li LF Zhang BX . Metallothionein I and II gene knock-out mice exhibit reduced tolerance to 24-h sodium lauryl sulphate patch testing. Clin Exp Dermatol. (2007) 32:417–22. doi: 10.1111/j.1365-2230.2007.02399.x
205.
Gudekar N Shanbhag V Wang Y Ralle M Weisman GA Petris MJ . Metallothioneins regulate ATP7A trafficking and control cell viability during copper deficiency and excess. Sci Rep. (2020) 10:7856. doi: 10.1038/s41598-020-64521-3
206.
Gou X Ying J Yue Y Qiu X Hu P Qu Y et al . The roles of high mobility group box 1 in cerebral ischemic injury. Front Cell Neurosci. (2020) 14:600280. doi: 10.3389/fncel.2020.600280
207.
He H Xiao S Xu G Wang B Zou Z Qin X et al . The NADPH oxidase 4 protects vascular endothelial cells from copper oxide nanoparticles-induced oxidative stress and cell death. Life Sci. (2020) 252:117571. doi: 10.1016/j.lfs.2020.117571
208.
Bak DW Weerapana E . Proteomic strategies to interrogate the Fe–S proteome. Biochim Biophys Acta Mol Cell Res. (2024) 1871:119791. doi: 10.1016/j.bbamcr.2024.119791
209.
Huang CY Kuo WW Ho TJ Chiang SF Pai PY Lin JY et al . Rab9-dependent autophagy is required for the IGF-IIR triggering mitophagy to eliminate damaged mitochondria. J Cell Physiol. (2018) 233:7080–91. doi: 10.1002/jcp.26346
210.
Zangiabadi S Chamoun KP Nguyen K Tang Y Sweeney G Abdul-Sater AA . Copper infused fabric attenuates inflammation in macrophages. PLoS One. (2023) 18:e0287741. doi: 10.1371/journal.pone.0287741
211.
Ambi A Stanisavljevic A Victor TW Lowery AW Davis J Van Nostrand WE et al . Evaluation of copper chelation therapy in a transgenic rat model of cerebral amyloid angiopathy. ACS Chem Neurosci. (2023) 14:378–88. doi: 10.1021/acschemneuro.2c00483
212.
Wang R Hou L Lu H Zhang Y Guo T Zhou B et al . Unveiling the interplay of MAPK/NF-kappaB/MLKL axis in brain health: omega-3 as a promising candidates against copper neurotoxicity. J Environ Manag. (2024) 370:122791. doi: 10.1016/j.jenvman.2024.122791
213.
Unzeta M Hernandez-Guillamon M Sun P Sole M . SSAO/VAP-1 in cerebrovascular disorders: a potential therapeutic target for stroke and Alzheimer’s disease. Int J Mol Sci. (2021) 22:3365. doi: 10.3390/ijms22073365
214.
Xiao Y Nie X Han P Fu H James Kang Y . Decreased copper concentrations but increased lysyl oxidase activity in ischemic hearts of rhesus monkeys. Metallomics. (2016) 8:973–80. doi: 10.1039/C6MT00037A
215.
Gromadzka G Tarnacka B Flaga A Adamczyk A . Copper dyshomeostasis in neurodegenerative diseases-therapeutic implications. Int J Mol Sci. (2020) 21:9259. doi: 10.3390/ijms21239259
216.
Huang S Black SA Huang J Stys PK Zamponi GW . Mutation of copper binding sites on cellular prion protein abolishes its inhibitory action on NMDA receptors in mouse hippocampal neurons. Mol Brain. (2021) 14:117. doi: 10.1186/s13041-021-00828-0
217.
Schlief ML West T Craig AM Holtzman DM Gitlin JD . Role of the Menkes copper-transporting ATPase in NMDA receptor-mediated neuronal toxicity. Proc Natl Acad Sci USA. (2006) 103:14919–24. doi: 10.1073/pnas.0605390103
218.
Hodgkinson VL Zhu S Wang Y Ladomersky E Nickelson K Weisman GA et al . Autonomous requirements of the Menkes disease protein in the nervous system. Am J Physiol Cell Physiol. (2015) 309:C660–8. doi: 10.1152/ajpcell.00130.2015
219.
Jian Z Guo H Liu H Cui H Fang J Zuo Z et al . Oxidative stress, apoptosis and inflammatory responses involved in copper-induced pulmonary toxicity in mice. Aging. (2020) 12:16867–86. doi: 10.18632/aging.103585
220.
Li LY Zhu XQ Tao WW Yang WM Chen HZ Wang Y . Acute onset neurological symptoms in Wilson disease after traumatic, surgical or emotional events: a cross-sectional study. Medicine. (2019) 98:e15917. doi: 10.1097/MD.0000000000015917
221.
Liu WQ Lin WR Yan L Xu WH Yang J . Copper homeostasis and cuproptosis in cancer immunity and therapy. Immunol Rev. (2024) 321:211–27. doi: 10.1111/imr.13276
222.
Peng G Huang Y Xie G Tang J . Exploring Copper’s role in stroke: progress and treatment approaches. Front Pharmacol. (2024) 15:1409317. doi: 10.3389/fphar.2024.1409317
223.
Cukierman DS Lazaro DF Sacco P Ferreira PR Diniz R Fernandez CO et al . X1INH, an improved next-generation affinity-optimized hydrazonic ligand, attenuates abnormal copper(I)/copper(II)-alpha-syn interactions and affects protein aggregation in a cellular model of synucleinopathy. Dalton Trans. (2020) 49:16252–67. doi: 10.1039/D0DT01138J
224.
Cox C Teknos TN Barrios M Brewer GJ Dick RD Merajver SD . The role of copper suppression as an antiangiogenic strategy in head and neck squamous cell carcinoma. Laryngoscope. (2001) 111:696–701. doi: 10.1097/00005537-200104000-00024
225.
Yang L Chen X Cheng H Zhang L . Dietary copper intake and risk of stroke in adults: a case-control study based on National Health and Nutrition Examination Survey 2013–2018. Nutrients. (2022) 14:409. doi: 10.3390/nu14030409
226.
Song X Meng J Li J Shen B Li J Xu M et al . Association of plasma metals with resting-state functional connectivity in ischemic stroke. Neurotoxicology. (2024) 104:56–65. doi: 10.1016/j.neuro.2024.07.011
227.
Xiao Y Yuan Y Liu Y Yu Y Jia N Zhou L et al . Circulating multiple metals and incident stroke in Chinese adults. Stroke. (2019) 50:1661–8. doi: 10.1161/STROKEAHA.119.025060
228.
Zhang M Li W Wang Y Wang T Ma M Tian C . Association between the change of serum copper and ischemic stroke: a systematic review and meta-analysis. J Mol Neurosci. (2020) 70:475–80. doi: 10.1007/s12031-019-01441-6
229.
Jiang Y Wang LP Dong XH Cai J Jiang GJ Zhang C et al . Trace amounts of copper in drinking water aggravate cerebral ischemic injury via impairing endothelial progenitor cells in mice. CNS Neurosci Ther. (2015) 21:677–80. doi: 10.1111/cns.12427
230.
Yang N Tian H Zhan E Zhai L Jiao P Yao S et al . Reverse-D-4F improves endothelial progenitor cell function and attenuates LPS-induced acute lung injury. Respir Res. (2019) 20:131. doi: 10.1186/s12931-019-1099-6
231.
Gaya-Bover A Hernandez-Lopez R Alorda-Clara M Ibarra de la Rosa JM Falco E Fernandez T et al . Antioxidant enzymes change in different non-metastatic stages in tumoral and peritumoral tissues of colorectal cancer. Int J Biochem Cell Biol. (2020) 120:105698. doi: 10.1016/j.biocel.2020.105698
232.
Jahan J Monte de Oca I Meissner B Joshi S Maghrabi A Quiroz-Olvera J et al . Transforming growth factor-beta1/thrombospondin-1/CD47 axis mediates dysfunction in CD34+ cells derived from diabetic older adults. Eur J Pharmacol. (2022) 920:174842. doi: 10.1016/j.ejphar.2022.174842
233.
Yang Z von Ballmoos MW Faessler D Voelzmann J Ortmann J Diehm N et al . Paracrine factors secreted by endothelial progenitor cells prevent oxidative stress-induced apoptosis of mature endothelial cells. Atherosclerosis. (2010) 211:103–9. doi: 10.1016/j.atherosclerosis.2010.02.022
234.
Groleau J Dussault S Haddad P Turgeon J Menard C Chan JS et al . Essential role of copper-zinc superoxide dismutase for ischemia-induced neovascularization via modulation of bone marrow-derived endothelial progenitor cells. Arterioscler Thromb Vasc Biol. (2010) 30:2173–81. doi: 10.1161/ATVBAHA.110.212530
235.
Xue Q Kang R Klionsky DJ Tang D Liu J Chen X . Copper metabolism in cell death and autophagy. Autophagy. (2023) 19:2175–95. doi: 10.1080/15548627.2023.2200554
236.
Jiang S Li H Zhang L Mu W Zhang Y Chen T et al . Generic diagramming platform (GDP): a comprehensive database of high-quality biomedical graphics. Nucleic Acids Res. (2025) 53:D1670–6. doi: 10.1093/nar/gkae973
Summary
Keywords
copper homeostasis, cuproptosis, neurodegenerative diseases, immune response, cell death mechanisms
Citation
Li L, Lv L, Wang Z, Liu X, Wang Q, Zhu H, Jiang B, Han Y, Pan X, Zhou X, Ren L and Chang Z (2025) From copper homeostasis to cuproptosis: a new perspective on CNS immune regulation and neurodegenerative diseases. Front. Neurol. 16:1581045. doi: 10.3389/fneur.2025.1581045
Received
21 February 2025
Accepted
28 April 2025
Published
29 May 2025
Corrected
29 July 2025
Volume
16 - 2025
Edited by
Gabriela Cristina De Paula, Institute for Research in Biomedicine (IRB), Switzerland
Reviewed by
Agostinho Almiro Almeida, University of Porto, Portugal
Mini Jose Deepak, St. Jude Children’s Research Hospital, United States
Arun Kumar Yadawa, Texas A&M University, United States
Updates
Copyright
© 2025 Li, Lv, Wang, Liu, Wang, Zhu, Jiang, Han, Pan, Zhou, Ren and Chang.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhuo Chang, 1650843841@qq.com
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.