Abstract
Introduction:
The CACNA1A gene encodes the pore-forming subunit of the Cav2.1 (P/Q type) neuronal calcium channel and pathogenic variants cause a variety of neurological disorders including episodic and congenital ataxia, familial hemiplegic migraine, developmental delay and epilepsy. Multiple types of seizures have been described in affected patients, including status epilepticus as the first manifestation. In mice harboring the homozygous gain-of-function variant p.Ser218Leu, seizures leading to SUDEP triggered by brainstem spreading depolarization with subsequent apnea and cardiac arrest have been reported.
Methods:
Clinical, genetic and functional data are presented.
Results and discussion:
The 9-year-old boy with global developmental delay and congenital ataxia developed recurrent seizures and status epilepticus with prolonged, life-threatening apnea implying a high risk for SUDEP. Genetic testing showed a novel de novo missense variant in CACNA1A (c.5398T>A, p.Phe1800Ile). Functional analysis revealed a gain of channel function as the molecular pathomechanism. Therefore, an increased risk of SUDEP in patients with CACNA1-associated epilepsy seems reasonable and preventive strategies should be discussed with caregivers.
1 Introduction
The CACNA1A gene encodes the pore-forming α1A subunit of the neuronal voltage-gated calcium channel Cav2.1 (P/Q type). The Cav2.1 channel consists of four homologous domains, each comprising six transmembrane helices (S1-S6) connected by intra- and extracellular loops. Segments S1-S4 of each repeat form the voltage sensing domains, segments S5-S6 of all four repeats together form the conduction pore with the selectivity filter and the activation gate. In the central and peripheral nervous system, Cav2.1 plays a key role in initiating neurotransmitter release in excitatory and inhibitory synapses and is involved in intracellular signaling, transcriptional regulation and neuronal viability (1–4). Pathogenic variants of the CACNA1A gene result in allelic disorders with a broad clinical spectrum including episodic ataxia type 2 (OMIM: 108500), familial hemiplegic migraine type 1 (OMIM: 141500), spinocerebellar ataxia type 6 (OMIM: 183086) (5, 6), and overlapping features between these conditions in individual patients. In addition, CACNA1A-related epilepsy (OMIM: 617106) with multiple seizures types and status epilepticus have been described (7–10). Sudden unexpected death in patients with epilepsy (SUDEP) is a rare but significant risk in the cohort of patients with epilepsy of heterogeneous etiology. Brainstem spreading depolarization has been discussed to play an important role, possibly leading to brainstem dysfunction followed by respiratory and cardiac arrest (11–15). Variants in CACNA1A have been found to be one of the susceptibility genes to SUDEP (16).
Here, we report a 9-year-old boy who initially presented with global developmental delay and congenital ataxia caused by a novel de novo missense variant in CACNA1A. Functional analysis of the variant revealed altered gating properties leading to a predominantly gain of function phenotype. Epilepsy began with recurrent status epilepticus after the age of 6 years. The seizures were followed by prolonged and life-threatening central apneas, constituting a high risk of sudden unexpected death.
2 Materials and methods
The patient charts were reviewed for the clinical history, the laboratory (including metabolic and genetic) and radiological investigations. Blood samples from the patient and his parents were obtained after informed consent. Whole exome sequencing was performed after written informed consent according to national regulations on genetic diagnostics.
2.1 Whole exome sequencing and data analysis
DNA samples from whole blood were isolated by standard procedures. Trio whole-exome sequencing (trio-WES) was performed with DNA samples of both healthy parents and the index patient, as described previously (17).
The functional impact of the identified variants was predicted using CADD, REVEL, M-CAP in silico tools (18–21).
2.2 Plasmids
To generate the GFP-CaV2.1 expression plasmid, the cDNA sequence of human CaV2.1 (nt 1–676) was amplified in separate PCR reactions using pβA-CaV2.1 [GenBankTM FJ040507; (22)] as template with a primer introducing a SalI site at the 5’end. The obtained PCR product was then SalI/NotI digested, the remaining cDNA sequence coding for CaV2.1 was isolated from pβA-CaV2.1 by NotI/BamHI digestion and the two fragments were ligated into the corresponding sites of GFP-CaV1.2 (23).
To generate the GFP-CaV2.1-F1800I expression plasmid, the F1802I mutation in GenBank™ FJ040507 (corresponding to F1800I in the CACNA1A variant GenBank™ AAB64179.1) was introduced into GFP-CaV2.1 by splicing by overlap extension (SOE) PCR. For simplicity reasons, henceforth we will be using the name CaV2.1-F1800I. Briefly, the cDNA sequence of human CaV2.1 (nt 5,072–5,980) was amplified in separate PCR reactions using GFP-2.1 as template with overlapping primers mutating the c.5404T>A. The two separate PCR products were then used as templates for a PCR reaction with flanking primers to connect the nucleotide sequences. The resulting fragment was then XhoI/BglII digested and ligated into the corresponding sites of GFP-CaV2.1.
All newly generated sequences were verified by sequencing (Eurofins genomics).
2.3 Electrophysiology
The experiments were conducted on an in-house produced cell line (A2MG), a HEK 293 cell line that stably expresses human β3 and α2δ-1 calcium channel subunits (24, 25). Calcium currents were recorded using the ruptured whole-cell patch-clamp technique in voltage-clamp mode. Patch pipettes (borosilicate glass, Harvard Apparatus, Holliston, MA) had a resistance of 2.5–4.5 MΩ when filled with a solution containing (mM) 135 CsCl, 1 MgCl2, 10 HEPES, 4 ATP-Na2, and 10 EGTA (pH 7.4 with CsOH). 10 mM concentration of EGTA prevents calcium-dependent inactivation (CDI) (26). The extracellular bath solution comprised (mM) 15 CaCl2, 150 choline-chloride, 10 HEPES, and 1 MgCl2 (pH 7.4 with CsOH). All experimental groups were analyzed in transiently transfected cells from three to six independent cell passages. For each cell, the stimulation protocol was only recorded once (no technical replicates). The recordings were acquired with Axopatch 200A amplifier (Axon Instruments, Foster City, CA). Data acquisition and command potentials were controlled by pClamp software (version 10.7, Axon Instruments). Current–voltage (I–V) relationships were obtained by applying a voltage-step square pulse protocol starting from a holding potential (Vhold) of −80 mV followed by the command potential (Vcmd) of 500 ms, ranging from −60 mV to +80 in 10 mV increment. The resulting I–V curves were fitted to the following equation:
Where Gmax is the maximum conductance of the calcium channels, Vrev is the extrapolated reversal potential of the current, V1/2 is the potential for half-maximal conductance, and k is the slope.
The conductance was extrapolated using:
The conductance voltage dependence was calculated according to the Boltzmann distribution:
The time constant of activation (τact) was obtained by applying a mono-exponential fit to the rising phase of the current using the equation:
In contrast, the time constant of inactivation (τinact.) was obtained by fitting the decay phase of the current with a mono-exponential function described by:
For both equations, A is the current amplitude, and τ corresponds to the time constant of either activation or inactivation.
The voltage dependence of inactivation was adapted from Gambeta et al. (27) by application of two test pulses to Vmax (at +20 mV for WT and at +10 mV fore F1800I) before and after holding cells at various conditioning test potentials (ranging from −80 to +50 mV) for a duration of 5 s (60 s intersweep interval). Inactivation was calculated as the ratio between the current amplitudes of the test pulses. Steady-state inactivation parameters were obtained by fitting the data to the modified Boltzmann equation, as follows:
Where V1/2,inact. is the half-maximal inactivation voltage, kinact is the inactivation slope factor and Gni is the fraction of non-inactivating current in steady-state.
The kinetics of recovery from inactivation was assessed by application of a 5-s-long pre-pulse followed by a test pulse both at Vmax. The test pulse was recorded at various time points (between 20 ms and 45 s, using a logarithmic increase) after the 5 s pre-pulse (30 s intersweep interval). Per each sweep, the rate of recovery from inactivation was calculated as the ratio between the Imax obtained during the test pulse and the Imax collected during the pre-pulse. The rate of recovery from inactivation was best fit using a double exponential equation.
2.4 Statistical analysis
SigmaPlot (version 12.0; SPSS) was used for statistical analyses and curve fitting; GraphPad Prism (version 8.0.1; Graphpad Softaware LLC) and CorelDRAW2021 (version 23.0.0.363; Corel Corporation) were used to make the figures. All data are presented as mean ± SEM. First, all the data were assessed for the normality of the distribution using a Shapiro–Wilk test with significant criteria alpha = 0.05. Statistical comparison of the fit parameters were obtained by using either Student’s t test or mixed-effects analysis matched with Šídák’s multiple comparisons test, with significance criteria, .
3 Results
Clinical and genetic data are summarized in Table 1.
Table 1
| genetic data | results |
|---|---|
| genomic position | c.5398T>A |
| aminoacid change | p.Phe1800Ile |
| protein domain | domain IV, segment S6 |
| mode of inheritance | de novo |
| general data | |
| ethnicity | caucasian / german |
| gender | male |
| gestational age (weeks) | 40+5 |
| pregnancy, birth | uneventful, spontaneous delivery |
| Apgar score | 9/10/10 |
| birth weight (g) (perc.) | 3340 (P21) |
| birth height (cm) (perc.) | 56 (P91) |
| birth head circumference (cm) (perc.) | 35.5 (P41) |
| age at first examination (months) | 23 |
| age at latest examination (years) | 9 |
| weight at latest examination (kg) (perc.) | 43.9 (P89) |
| height at latest examination (cm) (perc.) | 144.5 (P65) |
| head circumference | macrocephalus after the age of 4 yrs. |
| head circumference at latest examination (cm) (perc.) | 57 (P99) |
| clinical findings | |
| dysmorphism | none |
| first symptoms | muscular hypotonia |
| further neurological symptoms | ataxia, dysarthria |
| development | |
motor development
|
delayed 12 30 |
speech development
|
delayed 17 |
| cognitive development | moderate intellectual deficits |
| school performance | special needs school |
| behavioural disturbances | no |
| epilepsy | |
| age at seizure onset (years) | 6 |
| seizure types | focal, focal to bilateral tonic clonic |
| febrile seizures | yes |
| Status epilepticus | yes |
| complications after status epilepticus | encephalopathy, apnea |
| postictal EEG findings | focal slowing over posterior regions |
| interictal EEG findings (including 24-hour-EEG) | normal |
| current antiepileptic drugs | levetiracetam, lamotrigine, topiramate |
| further antiepileptic drugs (discontinued) | acetacolamide, zonisamide, ethosuximide |
| further diagnostics | |
| MRI (incl. spectroscopy) at 11 months, 4, 6 and 7 years | normal |
| echocardiography | normal |
| audiometry | normal |
| eye examination | normal |
| metabolic work-up including amino acids (blood, CSF), acylcarnitine profile (blood), organic acids (urine), isoelectric transferrin focusing (blood), glycosaminoglycans (urine) | normal |
Clinical and genetic data of our patient.
3.1 Patient data
The 9-year-old boy was born after an uneventful pregnancy as the first child of healthy, unrelated Caucasian parents. At birth, his weight, length and head circumference were within the normal range. A global developmental delay was evident in the boy from infancy onward, and ataxia manifested when he started walking at 27 months of age. His speech was slurred but he learned to speak complete sentences. On neurological examination at the age of 30 months, ataxia and dysarthria were the prominent features. Head circumference became macrocephalic (+2z) after the age of 4 years. At the age of 9 years, the boy shows moderate developmental delay in speech and learning.
The first brain MRI was performed at 11 months of age, and was unremarkable. Subsequent brain MRI including MR spectroscopy at the age of 4, 6, and 7 years were also normal. At the age of 3 years and 9 months, a comprehensive metabolic work-up in blood, urine and cerebrospinal fluid as well as EEG showed no abnormalities, but whole-exome-sequencing revealed the novel de novo missense variant [c.5398T>A, p.(Phe1800Ile)] in the CACNA1A gene which was consistent with the patient’s symptoms. Treatment with acetazolamide was initiated due to the known positive effects of carbonic anhydrase inhibitors in cases of CACNA1A-associated ataxia (28), but was discontinued shortly thereafter because of diarrhea.
After an uneventful period without seizures, headache or hemiplegic episodes, the boy was admitted at the age of 6 years and 2 months with the first generalized, tonic–clonic status epilepticus that was followed by a prolonged period of more than 1 week with somnolence, confusion and inability to speak, sit or walk. Treatment with levetiracetam was initiated despite absence of epileptic discharges on EEG. MRI revealed no abnormalities. The boy recovered over the next few months, slowly regaining all of his former abilities. During the next 17 months, the boy was admitted to the emergency department with multiple focal to bilateral tonic–clonic status epileptici and seizures followed by prolonged central apnea up to 50 min, bradycardia, and hypothermia requiring intubation and mechanical ventilation. The apnea was not on the consequence of benzodiazepine use, as it occurred in part before the administration of a dose of benzodiazepine and was not accompanied by convulsions or increased muscle tone. Postictal EEG showed focal slowing over posterior regions. Several interictal EEGs, including a 24-h EEG, showed normal background activity and no epileptic discharges. After the first status epilepticus, levetiracetam was started, and because of recurrent seizures, the treatment was changed to multiple antiepileptic drugs including carboanhydrase inhibitors, and calcium channel inhibitors, i.e., levetiracetam plus zonisamide, levetiracetam plus ethosuximide, levetiracetam plus ethosuximide plus lamotrigine, and levetiracetam plus lamotrigine plus topiramate, respectively. The latter resulted in a stable situation with only single seizures with mainly spontaneous termination and without postictal apnea. After the first status epilepticus with prolonged and life-threatening postictal apnea, the patient’s risk of dying during seizures seemed extremely high. Therefore, pulse oximetry monitoring during sleep was initiated. At the time of the most recent visit, the boy had been seizure-free for 4 months and had not experienced episodes of severe headache or hemiplegia.
3.2 Genetic data
Using WES, we identified the novel de novo missense variant c.5398T>A, [p.(Phe1800Ile)] in CACNA1A (RefSeq accession number NM_001127221.2). The detected alteration had not been reported in the dbSNP1 or GnomAD2 databases and was computationally predicted to be functionally relevant with the following scores—CADD: 27.7, REVEL: 0.911 and M-CAP: 0.629. The affected Phe1800 is highly conserved across species and is located within the transmembrane segment S6 of the domain IV, which is part of the channel conduction pore and near a hot-spot for disease-associated variants in calcium channels (29). According to the scoring criteria of American College of Medical Genetics and Genomics (ACMG) this variant was evaluated as pathogenic [applied criteria: PS2, PM2, PP3 and PS3 (see below)] (30). No other candidate genes or known disease genes have been identified that could contribute to the patient’s phenotype.
3.3 Functional data
To examine the effects of the Phe1800Ile substitution on the properties of the CaV2.1 calcium channel we introduced the corresponding mutation in the human CaV2.1 variant (GeneBank™ FJ040507). The domain model in Figure 1B shows the position of the mutation in the S6 helix of domain IV, which forms part of the activation gate. The expression plasmids coding for the wildtype and mutant channel N-terminally tagged with green fluorescent protein (GFP-CaV2.1, GFP-CaV2.1-F1800I) were heterologously expressed in HEK 293 cells. Using the patch-clamp technique, whole-cell calcium currents in response to 500 ms voltage steps from a holding potential of −80 mV to varying test potentials were recorded (Figures 1A,C–E). While peak amplitudes of the calcium currents were not significantly different in wildtype and mutant constructs (Figures 1F,H), the voltage-dependence of activation of CaV2.1-F1800I was shifted by 10 mV to less depolarized potentials (Figures 1F,G,I; Table 2). Consequently, cells expressing the disease-associated variant CaV2.1-F1800I experienced calcium influx at 0 mV, a membrane potential at which wildtype CaV2.1 channels barely opened (Figure 1C). Comparing normalized currents indicated slowed activation kinetics of the CaV2.1-F1800I variant (Figure 1J). Indeed, the time-to-peak measured in the maximal current traces (Vmax) was significantly slower in CaV2.1-F1800I compared to wildtype control (Figure 1K). However, the time constants of current activation measured at the test potentials between 0 and +60 mV revealed no statistical significant difference (Figure 1L).
Figure 1

Functional analysis of the CaV2.1-F1800I variant heterologously expressed in HEK-293 cells—Current activation. (B) Domain structure of the CaV2.1 α1 subunit with the approximate location of the F1800I mutation in the IVS6 gate-forming helix. (A) Voltage-clamp protocol. (C–E) Calcium currents (mean ± SEM) of wildtype CaV2.1 (WT, blue) and mutant CaV2.1-F1800I (F1800I, orange) in response to 500-ms voltage steps to 0 mV (C), +10 mV (D) and +20 mV (E) demonstrates that currents are elicited at lower potentials in the disease variant. (F,G) I/V curves and fractional activation curves showing a 9.3 mV left-shift of the V1/2 of activation for F1800I relative to its control. (H,I) Scatter plots of current density and the voltage of half-maximal activation (H: p = 0.3; I: p < 0.0001). (J) Normalized calcium currents at Vmax (+20 or +30 mV for WT; +10 or +20 mV for F1800I) show the relative slowing of activation kinetics of the mutant (F1800I, orange) compared to the wild type (WT, blue). (K) Scatter plot of the time to peak obtained at Vmax displaying a significantly slowed activation kinetics of F1800I compared to its control (p = 0.005). (L) Time constants of current activation calculated at voltage steps (τact) between 0 and +60 mV indicate no significant difference (p = 0.1). Mean ± SEM; mutant and WT controls compared by t-tests or mixed-effects analysis matched with Šídák’s multiple comparisons test, using significance criteria, .
Table 2
| Ca V 2.1 WT | F1800I | p-value | |
|---|---|---|---|
| Activation | |||
| n | 8 | 6 | |
| ICa (pA/pF) | -152.7 ± 45.5 | -96.4 ± 22.2 | 0.34 |
| V0.5 (mV) | 12.2 ± 1.2 | 2.9 ± 0.4 | < 0.0001 |
| TTP (ms) | 7.3 ± 0.9 | 13.7 ± 1.8 | 0.005 |
| % inact. at 100 ms | 47.5 ± 4.4 | 26.2 ± 4.4 | 0.006 |
| % inact. at 250 ms | 77.5 ± 3.0 | 60.3 ± 5.5 | 0.01 |
| % inact. at 500 ms | 88.7 ± 1.9 | 76.3 ± 4.8 | 0.02 |
| Steady state inactivation | |||
| n | 9 | 7 | |
| V0.5 (mV) | -13.7 ± 1.9 | -24.7 ± 3.0 | 0.006 |
| Time constants of activation (τact/ms) | |||
| n | 8 | 6 | |
| at 0 mV | 2.4 ±1.3 | 3.4 ± 0.5 | 0.9947 |
| at 10 mV | 2.1 ± 0.2 | 3.1 ± 0.5 | 0.6277 |
| at 20 mV | 1.6 ± 0.2 | 1.2 ± 0.1 | 0.4283 |
| at 30 mV | 0.9 ± 0.1 | 0.7 ± 0.1 | 0.9162 |
| at 40 mV | 0.7 ± 0.1 | 0.6 ± 0.1 | 0.9976 |
| at 50 mV | 0.4 ± 0.1 | 0.4 ± 0.1 | >0.9999 |
| Time constants of activation (τinact/ms) | |||
| n | 8 | 6 | |
| at 0 mV | 584.1 ±174.8 | 572.9 ± 222.0 | >0.9999 |
| at 10 mV | 260.8 ± 40.9 | 267.9 ± 52.5 | >0.9999 |
| at 20 mV | 149.1 ± 25.5 | 194.8 ± 18.9 | 0.7439 |
| at 30 mV | 123.1 ± 10.1 | 235.9 ± 36.5 | 0.1682 |
| at 40 mV | 140.4 ± 9.5 | 233.1 ± 30.7 | 0.1811 |
| at 50 mV | 166.1 ± 15.9 | 347.9 ± 53.1 | 0.1558 |
| at 60 mV | 190.4 ± 14.5 | 317.9 ± 63.8 | 0.6455 |
Current properties of WT and F1800I mutant CaV2.1.
Mean current densities (ICa), activation kinetics (time to peak, TTP) and percentage of inactivation at 100, 250, and 500 ms were calculated at Vmax (WT + 20 or +30 mV; F1800I + 10 or +20 mV, respectively). The voltage-dependence of activation (V0.5), tau of activation and inactivation (τact, τinact.) were measured from voltage-step protocols (Figure 1E). The voltage-dependence of inactivation was determined using a steady-state inactivation protocol (Figure 2A). Mean ± SEM; mutant and WT controls compared by t-tests or mixed-effects analysis matched with Šídák’s multiple comparisons test, using significance criteria,
The normalized current traces at Vmax also indicate a slowed inactivation of the CaV2.1-F1800I currents (Figure 2A). Accordingly, the fractional inactivation measured after 100, 250, and 500 ms depolarization was significantly reduced in CaV2.1-F1800I compared to wildtype controls (Figures 2B–D). For example, in test pulses to +10 mV this resulted in a substantially increased calcium current during the declining phase of the current (Figure 1D). However, analyzing the time constants of inactivation at voltages between 0 mV and +60 mV showed no statistically significant differences (Figure 2E). Furthermore, the analysis of steady-state inactivation demonstrated a reduced availability of CaV2.1-F1800I mutant channels after prolonged depolarizations (Figure 2F). The voltage-dependence of inactivation was shifted by 10 mV to more hyperpolarized potentials, in parallel to the left-shift of the voltage-dependence of activation (Figures 2G,H). Recovery from inactivation after a 5 s depolarization reached about 90% within 45 s, and the time course of recovery was very similar in wildtype and mutant channels (Figures 2I,J). Together, the functional analyses demonstrates that the F1800I substitution in CaV2.1 results in altered channel gating properties with a left-shifted voltage-dependence of activation and inactivation, and somewhat reduced activation and inactivation kinetics (Table 2).
Figure 2
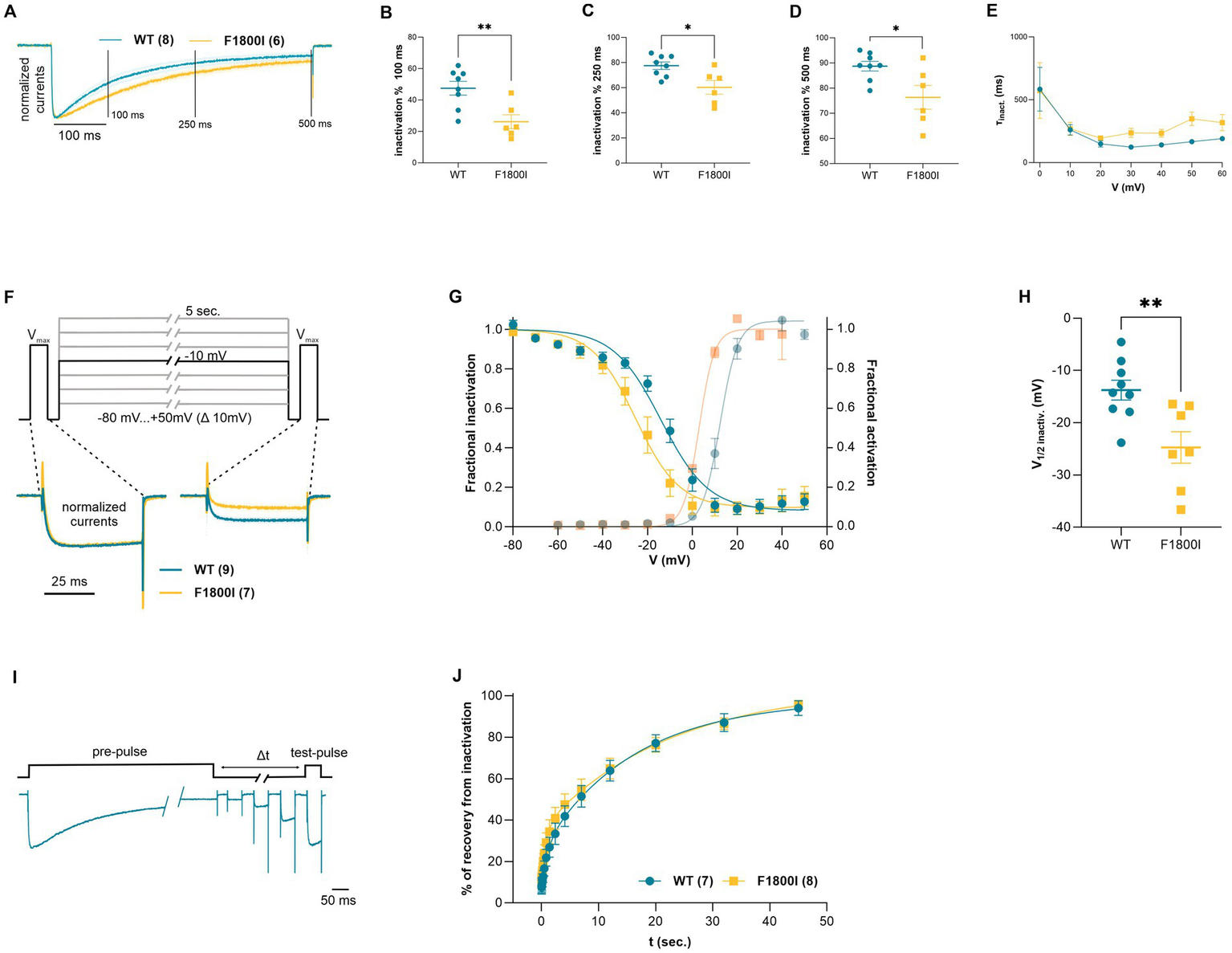
Functional analysis of current inactivation of the CaV2.1-F1800I variant. (A) Normalized current traces at Vmax (blue, wild type, orange, CaV2.1-F1800I; mean ± SEM) showing different time courses of inactivation. The three vertical lines (100, 250 and 500 ms) indicate the specific time points at which fractional inactivation was calculated. (B–D) Percent reduction of peak currents at Vmax after 100, 250 or 500 ms of depolarization is significantly less for F1800I compared with WT (B: p = 0.006; C: p = 0.012; D: p = 0.02). (E) Time constant of current inactivation calculated at voltage steps (τinact.) between 0 and +60 mV reveal no statistically significant effect of the mutation on inactivation kinetics (p = 0.7). (F) Steady state inactivation protocol and typical example traces at −10 mV sweep for Cav2.1 WT (cyan) and Cav2.1-F1800I (yellow). (G,H) Fractional inactivation curves and scatter plot of the V1/2 of inactivation showing a 10.9 mV shift to more hyperpolarized potential of F1800I compared to WT. Mean ± SEM (C: p = 0.006). (I) Pulse protocol and representative current traces for analyzing the recovery of inactivation (∆t 20 ms to 45 s) after a 5 s. pre-pulse to Vmax (+20 mV for WT; +10 mV for F1800I). (J) The time course of recovery from inactivation does not reveal any significant difference between WT and variant (p = 0.58). Mean ± SEM; mutant and WT controls compared by t-tests or mixed-effects analysis matched with Šídák’s multiple comparisons test, using significance criteria, .
4 Discussion
CACNA1A-related disorders include a spectrum of distinct clinical phenotypes such as episodic ataxia, familial hemiplegic migraine, and epilepsy, but also overlapping phenotypes with additional symptoms such as developmental delay and cognitive disability. Here, we describe a case of CACNA1A-associated disease in a boy with developmental delay and congenital ataxia who developed recurrent status epilepticus and life-threatening postictal apnea after the age of 6 years. A broad spectrum of seizure types has been reported in CACNA1A-related epilepsy with status epilepticus often being the initial manifestation. Both gain-of-function and loss-of-function variants have been found in patients with epilepsy (10, 31). However, in patients with status epilepticus mainly gain-of-function variants, located in the transmembrane regions, particularly in segments 4–6, have been reported (8, 9). Accordingly, the novel missense variant p.Phe1800Ile, found in our patient, is located in the transmembrane segment 6 of domain IV (Table 3).
Table 3
| Ca V 2.1 WT | F1800I | p-value | |
|---|---|---|---|
| % of Recovery from inactivation (∆t / ms) | |||
| n | 7 | 8 | |
| at 20 ms | 8.3 ± 3.7 | 11.4 ± 2.8 | >0.9999 |
| at 35 ms | 7.6 ± 3.4 | 11.5 ± 2.9 | 0.9996 |
| at 60 ms | 8.4 ± 3.1 | 13 ± 3.1 | 0.9965 |
| at 100 ms | 9.2 ± 3.1 | 13.3 ± 3.3 | 0.9992 |
| at 170 ms | 10.9 ± 3.2 | 15.9 ± 3.3 | 0.9945 |
| at 290 ms | 12.8 ± 2.9 | 20.2 ± 3.9 | 0.9002 |
| at 490 ms | 16.6 ± 3.6 | 23.9 ± 4.5 | 0.9783 |
| at 830 ms | 21.8 ± 4.1 | 29.1 ± 5 | 0.9913 |
| at 1400 ms | 26.9 ± 4.8 | 34.4 ± 6.1 | 0.9985 |
| at 2400 ms | 33.4 ± 5.2 | 40.9 ± 4.6 | 0.9982 |
| at 4100 ms | 41.8 ± 5.0 | 47.5 ± 5.6 | >0.9999 |
| at 7000 ms | 51.4 ± 5.2 | 54.7 ± 5.5 | >0.9999 |
| at 12000 ms | 63.8 ± 5.0 | 64.7 ± 5.3 | >0.9999 |
| at 20000 ms | 77.1 ± 4.1 | 76.3 ± 3.9 | >0.9999 |
| at 32000 ms | 87.1 ± 4.3 | 86.5 ± 2.8 | >0.9999 |
| at 45000 ms | 94.1 ± 3.5 | 95.9 ± 1.5 | >0.9999 |
Rate of recovery from inactivation was measured from a double step protocol using different ∆t (Figure 2I).
Mean ± SEM; mutant and WT controls compared by mixed-effects analysis matched with Šídák’s multiple comparisons test, using significance criteria,
Functional analysis of the variant in our patient showed that Cav2.1-F1800I channels opened at lower voltages, but also inactivated at lower voltages. Specifically, the left-shifted voltage-dependence of activation and delayed inactivation correspond to a gain of channel function resulting in increased calcium influx during brief depolarizations. Both effects on channel gating of the F1800I mutation in the S6 helix of the fourth repeat are very similar to the electrophysiological effects reported for a deletion variant of the corresponding phenylalanine in the S6 helix of the third repeat of CaV2.1 (ΔF1502) found in patients with congenital ataxia and hemiplegic migraine (32), thus, supporting the causative role of mutations of this highly conserved amino acid in the pore domain of CaV2.1. As CaV2.1 is the major pre-synaptic channel in the central and peripheral nervous system, such altered channel gating would translate in increased neurotransmitter release and synaptic transmission. Whereas CaV2.1 controls neurotransmitter release in both, excitatory and inhibitory synapses, previous studies of gain-of-function CaV2.1 variants revealed enhanced excitatory neurotransmission at glutamatergic pyramidal cell synapses without affecting GABA-ergic neurotransmission at interneuron synapses (33–35). Such a propensity for enhancing excitatory neurotransmission might result in hyperexcitability and thereby explain the seizures in the patients.
The further observed left-shifted voltage-dependence of steady-state inactivation results in a decreased availability of CaV2.1 channels in neurons persistently depolarized to potentials above −30 mV, thus representing a loss-of-function effect of this disease variant. However, under physiological conditions, such persistent depolarizations of neurons are not to be expected and therefore reduced channel availability due to left-shifted voltage-dependence of steady-state inactivation probably is of lesser importance for the pathogenicity of the CaV2.1-F1800I variant. In contrast, the observed gain of channel function could lead to an increased neurotransmitter release probability as well as to synaptic remodeling due to increased calcium influx (36). Although epilepsy has been reported in patients with both gain-of-function variants and loss-of-function CACNA1A variants, status epilepticus was more frequently associated with gain-of-function variants (8). Apneic seizures also occurred in a boy with epileptic encephalopathy and the p.Val1808Leu variant, which is located next to the variant found in our patient. However, no functional data are available (37).
Our patient had recurrent status epilepticus occurring during sleep with prolonged central apnea postictally requiring intensive care and implicating a high risk of death without appropriate intervention. In a case review of 130 patients, 6 deaths were reported in children aged 3 months to 5 years with CACNA1A-related disease (8). Causes of death were listed as “fatal cerebral edema” and “epileptic encephalopathy” (not referred to as SUDEP) (38, 39). The pathophysiology of SUDEP is assumed to be heterogeneous and not fully understood. Several mechanisms have been discussed, including the spread of cortical depolarization to the brainstem during a seizure and the resulting suppression of cardiorespiratory control (14). SUDEP has a frequency of 1,2/1,000 years of epilepsy patients and higher in patients with generalized tonic–clonic seizures. Several genes, especially channelopathies, have been identified in patients who died of SUDEP including gene variants associated with seizures (e.g., SCN1A, SCN8A, SCN2A) or long QT-Syndrome (SCN5A, KCNH2, KCNQ1) (40–42). Conversely, however, only a few genes have been shown to be associated with an increased SUDEP risk, and it is often unclear whether this is due to a high seizure frequency in these patients or to additional pathophysiological factors caused by the gene variant. Therefore, SUDEP is thought to have a multifactorial origin with a genetic predisposition. In addition to the genes listed above, CACNA1A was identified as a potential candidate gene in a cohort of 14 patients who died of SUDEP (43). The CACNA1A missense variant p.Ser218Leu is associated with a gain of channel function and in the homozygous CACNA1A218 mouse, in contrast to the wildtype, seizures led to SUDEP triggered by brainstem spreading depolarization with subsequent apnea and cardiac arrest (44, 45). The shift in the voltage dependence of activation observed here in the CaV2.1-F1800I variant is similar in direction and extent as reported in both neurons of the CACNA1A218 mouse and human recombinant S218L mutant channels (35, 46). Further, Cain et al. (47) demonstrated that the superior colliculi play an important role in the propagation of seizures to the brainstem in CACNA1A218 mice, leading to fatal seizures. Hyperexcitability of superior colliculus neurons as a result of gain of channel function with lower voltage threshold for calcium influx and prolonged channel opening was speculated by the authors as the underlying mechanism. These data from the CACNA1A mouse model closely fit the combination of the gain of channel function observed in our patient, as determined by the functional analysis of our patient’s variant, and the prolonged seizure-related but nonconvulsive apneas leading to multiple life-threatening situations.
In our patient, the combination of levetiracetam plus topiramate plus lamotrigine resulted in the cessation of status epilepticus. Since no selective Cav2.1 channel inhibitors are available, we aimed to modulate channel activity and neuronal calcium homeostasis. To address the gain-of-function in channel activity, we chose the aforementioned unselective calcium channel inhibitors. Different therapeutic approaches in patients with CACNA1-related disorder have been discussed, mainly targeting channel activity and its cellular function, such as the use of carbonic anhydrase inhibitors, for example acetazolamide, in both gain- and loss-of-function CACNA1A mutations (48–50) as well as non-selective calcium channel blockers or openers to modulate channel activity (51–53). The best benefit of lamotrigine, which acts on the P/Q-type calcium channel, was observed in a patient with epileptic encephalopathy and the CACNA1A missense variant p.Ser1373Leu, but functional data on the variant were not reported (54). In the case series of 18 patients, Le Roux et al. (10) found the best efficacy in seizure reduction for topiramate, levetiracetam, lamotrigine, and valproate, which is consistent with the observation in our patient.
In conclusion, patients with CACNA1A-related epilepsy are prone to develop status epilepticus. Life-threatening, seizure-related apnea in these patients increases the risk of sudden death in epilepsy and prevention strategies such as pulse oximetry monitoring should be discussed with the families.
Statements
Data availability statement
All electrophysiological data presented in the study are included in Tables 2 and 3, further inquiries can be directed to the corresponding author.
Ethics statement
Ethical approval was not required for the study involving human samples in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
SP: Investigation, Methodology, Writing – review & editing. MC: Investigation, Methodology, Writing – review & editing. YE: Investigation, Methodology, Writing – review & editing. TB: Investigation, Methodology, Writing – review & editing. MH: Investigation, Methodology, Writing – review & editing. JD: Conceptualization, Formal analysis, Writing – review & editing. BF: Conceptualization, Formal analysis, Funding acquisition, Methodology, Supervision, Writing – original draft. JJ: Conceptualization, Formal analysis, Investigation, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded in part by the Austrian Science Fund (FWF) Grant-DOI 10.55776/P35618 to BF. For open access purposes, the authors have applied a CC BY public copyright license to any author accepted manuscript version arising from this submission.
Acknowledgments
We thank the child and its family for participation in this report. We thank Nicole Kranebitter, Sandra Demetz, and Enikö Török for technical help.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
Eilers J Plant T Konnerth A . Localized calcium signalling and neuronal integration in cerebellar Purkinje neurones. Cell Calcium. (1996) 20:215–26. doi: 10.1016/S0143-4160(96)90108-6
2.
Fletcher CF Tottene A Lennon VA Wilson SM Dubel SJ Paylor R et al . Dystonia and cerebellar atrophy in Cacna1a null mice lacking P/Q calcium channel activity. FASEB J. (2001) 15:1288–90. doi: 10.1096/fj.00-0562fje
3.
Pietrobon D . CaV2.1 channelopathies. Pflugers Arch. (2010) 460:375–93. doi: 10.1007/s00424-010-0802-8
4.
Catterall WA . Voltage-gated calcium channels. Cold Spring Harb Perspect Biol. (2011) 3:a003947. doi: 10.1101/cshperspect.a003947
5.
Ophoff RA Terwindt GM Vergouwe MN van Eijk R Oefner PJ Hoffman SMG et al . Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. (1996) 87:543–52. doi: 10.1016/S0092-8674(00)81373-2
6.
Zhuchenko O Bailey J Bonnen P Ashizawa T Stockton DW Amos C et al . Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the α1A-voltage-dependent calcium channel. Nat Genet. (1997) 15:62–9. doi: 10.1038/ng0197-62
7.
Epi4K Consortium, Epilepsy Phenome/Genome Project Allen AS Berkovic SF Cossette P Delanty N . De novo mutations in epileptic encephalopathies. Nature. (2013) 501:217–21. doi: 10.1038/nature12439
8.
Kessi M Chen B Pang N Yang L Peng J He F et al . The genotype–phenotype correlations of the CACNA1A-related neurodevelopmental disorders: a small case series and literature reviews. Front Mol Neurosci. (2023) 16:321. doi: 10.3389/fnmol.2023.1222321
9.
Niu X Yang Y Chen Y Cheng M Liu M Ding C et al . Genotype–phenotype correlation of CACNA1A variants in children with epilepsy. Dev Med Child Neurol. (2022) 64:105–11. doi: 10.1111/dmcn.14985
10.
Le Roux M Barth M Gueden S Desbordes de Cepoy P Aeby A Vilain C et al . CACNA1A-associated epilepsy: Electroclinical findings and treatment response on seizures in 18 patients. Eur J Paediatr Neurol. (2021) 33:75–85. doi: 10.1016/j.ejpn.2021.05.010
11.
Aiba I Noebels JL . Spreading depolarization in the brainstem mediates sudden cardiorespiratory arrest in mouse SUDEP models. Sci Transl Med. (2015) 7:282ra46. doi: 10.1126/scitranslmed.aaa4050
12.
Lhatoo S Noebels J Whittemore V N.C.f.S. Research . Sudden unexpected death in epilepsy: identifying risk and preventing mortality. Epilepsia. (2015) 56:1700–6. doi: 10.1111/epi.13134
13.
Richter F Bauer R Lehmenkühler A Schaible H-G . The relationship between sudden severe hypoxia and ischemia-associated spreading depolarization in adult rat brainstem in vivo. Exp Neurol. (2010) 224:146–54. doi: 10.1016/j.expneurol.2010.03.004
14.
Katayama PL . Cardiorespiratory dysfunction induced by brainstem spreading depolarization: A potential mechanism for SUDEP. J Neurosci. (2020) 40:2387–9. doi: 10.1523/JNEUROSCI.3053-19.2020
15.
Jansen NA Schenke M Voskuyl RA Thijs RD van den Maagdenberg AMJM Tolner EA . Apnea associated with brainstem seizures in Cacna1aS218L mice is caused by medullary spreading depolarization. J Neurosci. (2019) 39:9633–44. doi: 10.1523/JNEUROSCI.1713-19.2019
16.
Coll M Oliva A Grassi S Brugada R Campuzano O . Update on the genetic basis of sudden unexpected death in epilepsy. Int J Mol Sci. (2019) 20:1979. doi: 10.3390/ijms20081979
17.
Hempel M Cremer K Ockeloen CW Lichtenbelt KD Herkert JC Denecke J et al . De novo mutations in CHAMP1 cause intellectual disability with severe speech impairment. Am J Hum Genet. (2015) 97:493–500. doi: 10.1016/j.ajhg.2015.08.003
18.
Adzhubei IA Schmidt S Peshkin L Ramensky VE Gerasimova A Bork P et al . A method and server for predicting damaging missense mutations. Nat Methods. (2010) 7:248–9. doi: 10.1038/nmeth0410-248
19.
Jagadeesh KA Wenger AM Berger MJ Guturu H Stenson PD Cooper DN et al . M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet. (2016) 48:1581–6. doi: 10.1038/ng.3703
20.
Kircher M Witten DM Jain P O'Roak BJ Cooper GM Shendure J . A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. (2014) 46:310–5. doi: 10.1038/ng.2892
21.
Ioannidis NM Rothstein JH Pejaver V Middha S McDonnell SK Baheti S et al . REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. (2016) 99:877–85. doi: 10.1016/j.ajhg.2016.08.016
22.
Watschinger K Horak SB Schulze K Obermair GJ Wild C Koschak A et al . Functional properties and modulation of extracellular epitope-tagged Cav2. 1 voltage-gated calcium channels. Channels. (2008) 2:461–73. doi: 10.4161/chan.2.6.6793
23.
Grabner M Dirksen RT Beam KG . Tagging with green fluorescent protein reveals a distinct subcellular distribution of L-type and non-L-type Ca2+ channels expressed in dysgenic myotubes. Proc Natl Acad Sci. (1998) 95:1903–8. doi: 10.1073/pnas.95.4.1903
24.
Ortner NJ Bock G Dougalis A Kharitonova M Duda J Hess S et al . Lower affinity of isradipine for L-type Ca2+ channels during substantia nigra dopamine neuron-like activity: implications for neuroprotection in Parkinson's disease. J Neurosci. (2017) 37:6761–77. doi: 10.1523/JNEUROSCI.2946-16.2017
25.
El Ghaleb Y Ortner NJ Posch W Fernández-Quintero ML Tuinte WE Monteleone S et al . Calcium current modulation by the γ1 subunit depends on alternative splicing of CaV1. 1. Biophys J. (2022) 121:88a. doi: 10.1016/j.bpj.2021.11.2257
26.
Lee A Scheuer T Catterall WA . Ca2+/calmodulin-dependent facilitation and inactivation of P/Q-type Ca2+ channels. J Neurosci. (2000) 20:6830–8. doi: 10.1523/JNEUROSCI.20-18-06830.2000
27.
Gambeta E Gandini MA Souza IA Ferron L Zamponi GW . A CACNA1A variant associated with trigeminal neuralgia alters the gating of Cav2. 1 channels. Mol Brain. (2021) 14:1–6. doi: 10.1186/s13041-020-00725-y
28.
Battistini S Stenirri S Piatti M Gelfi C Righetti PG Rocchi R et al . A new CACNA1A gene mutation in acetazolamide-responsive familial hemiplegic migraine and ataxia. Neurology. (1999) 53:38–8. doi: 10.1212/WNL.53.1.38
29.
El Ghaleb Y Flucher BE . Channelopathies. Handb Exp Pharmacol. (2023) 279:263–288. doi: 10.1007/164_2022_631
30.
Richards S Aziz N Bale S Bick D das S Gastier-Foster J et al . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
31.
Jiang X Raju PK D'Avanzo N Lachance M Pepin J Dubeau F et al . Both gain-of-function and loss-of-function de novo CACNA 1A mutations cause severe developmental epileptic encephalopathies in the spectrum of Lennox-Gastaut syndrome. Epilepsia. (2019) 60:1881–94. doi: 10.1111/epi.16316
32.
Isabel Bahamonde M Serra SA Drechsel O Rahman R Marcé-Grau A Prieto M et al . A Single Amino Acid Deletion (ΔF1502) in the S6 Segment of CaV2.1 Domain III Associated with Congenital Ataxia Increases Channel Activity and Promotes Ca2+ Influx. PLoS One. (2015) 10:35. doi: 10.1371/journal.pone.0146035
33.
Tottene A Conti R Fabbro A Vecchia D Shapovalova M Santello M et al . Enhanced excitatory transmission at cortical synapses as the basis for facilitated spreading depression in CaV2. 1 knockin migraine mice. Neuron. (2009) 61:762–73. doi: 10.1016/j.neuron.2009.01.027
34.
Tottene A Favero M Pietrobon D . Enhanced thalamocortical synaptic transmission and dysregulation of the excitatory–inhibitory balance at the thalamocortical feedforward inhibitory microcircuit in a genetic mouse model of migraine. J Neurosci. (2019) 39:9841–51. doi: 10.1523/JNEUROSCI.1840-19.2019
35.
Vecchia D Tottene A van den Maagdenberg AM Pietrobon D . Abnormal cortical synaptic transmission in CaV2.1 knockin mice with the S218L missense mutation which causes a severe familial hemiplegic migraine syndrome in humans. Front Cell Neurosci. (2015) 9:8. doi: 10.3389/fncel.2015.00008
36.
Gambardella A Labate A . The role of calcium channel mutations in human epilepsy. Prog Brain Res. (2014) 213:87–96. doi: 10.1016/B978-0-444-63326-2.00004-1
37.
Hayashida T Saito Y Ishii A Yamada H Itakura A Minato T et al . CACNA1A-related early-onset encephalopathy with myoclonic epilepsy: a case report. Brain Dev. (2018) 40:130–3. doi: 10.1016/j.braindev.2017.08.006
38.
Gauquelin L Hawkins C Tam EWY Miller SP Yoon G . Pearls & Oy-sters: fatal brain edema is a rare complication of severe CACNA1A-related disorder. Neurology. (2020) 94:631–4. doi: 10.1212/WNL.0000000000009223
39.
Reinson K Õiglane-Shlik E Talvik I Vaher U Õunapuu A Ennok M et al . Biallelic CACNA1A mutations cause early onset epileptic encephalopathy with progressive cerebral, cerebellar, and optic nerve atrophy. Am J Med Genet A. (2016) 170:2173–6. doi: 10.1002/ajmg.a.37678
40.
Mantegazza M Curia G Biagini G Ragsdale DS Avoli M . Voltage-gated sodium channels as therapeutic targets in epilepsy and other neurological disorders. Lancet Neurol. (2010) 9:413–24. doi: 10.1016/S1474-4422(10)70059-4
41.
Tu E Bagnall RD Duflou J Semsarian C . Post-mortem review and genetic analysis of sudden unexpected death in epilepsy (SUDEP) cases. Brain Pathol. (2011) 21:201–8. doi: 10.1111/j.1750-3639.2010.00438.x
42.
Whitney R Sharma S Jones KC RamachandranNair R . Genetics and SUDEP: challenges and future directions. Seizure. (2023) 110:2. doi: 10.1016/j.seizure.2023.07.002
43.
Coll M Allegue C Partemi S Mates J del Olmo B Campuzano O et al . Genetic investigation of sudden unexpected death in epilepsy cohort by panel target resequencing. Int J Legal Med. (2016) 130:331–9. doi: 10.1007/s00414-015-1269-0
44.
Loonen IC Loonen ICM Jansen NA Cain SM Schenke M Voskuyl RA et al . Brainstem spreading depolarization and cortical dynamics during fatal seizures in Cacna1a S218L mice. Brain. (2019) 142:412–25. doi: 10.1093/brain/awy325
45.
van den Maagdenberg AM Pietrobon D Pizzorusso T Kaja S Broos LA Cesetti T et al . A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron. (2004) 41:701–10. doi: 10.1016/s0896-6273(04)00085-6
46.
Tottene A Pivotto F Fellin T Cesetti T van den Maagdenberg AMJM Pietrobon D . Specific kinetic alterations of human CaV2. 1 calcium channels produced by mutation S218L causing familial hemiplegic migraine and delayed cerebral edema and coma after minor head trauma. J Biol Chem. (2005) 280:17678–86. doi: 10.1074/jbc.M501110200
47.
Cain SM Bernier LP Zhang Y Yung AC Kass J Bohnet B et al . Hyperexcitable superior colliculus and fatal brainstem spreading depolarization in a model of sudden unexpected death in epilepsy. Brain Commun. (2022) 4:fcac006. doi: 10.1093/braincomms/fcac006
48.
Kotagal V . Acetazolamide-responsive ataxia. Semin Neurol. (2012) 32:533–7. doi: 10.1055/s-0033-1334475
49.
Omata T Takanashi JI Wada T Arai H Tanabe Y . Genetic diagnosis and acetazolamide treatment of familial hemiplegic migraine. Brain Dev. (2011) 33:332–4. doi: 10.1016/j.braindev.2010.05.006
50.
Rajakulendran S Kaski D Hanna MG . Neuronal P/Q-type calcium channel dysfunction in inherited disorders of the CNS. Nat Rev Neurol. (2012) 8:86–96. doi: 10.1038/nrneurol.2011.228
51.
Nimmrich V Gross G . P/Q-type calcium channel modulators. Br J Pharmacol. (2012) 167:741–59. doi: 10.1111/j.1476-5381.2012.02069.x
52.
Rajakulendran S Graves TD Labrum RW Kotzadimitriou D Eunson L Davis MB et al . Genetic and functional characterisation of the P/Q calcium channel in episodic ataxia with epilepsy. J Physiol. (2010) 588:1905–13. doi: 10.1113/jphysiol.2009.186437
53.
Pisani A Bonsi P Martella G de Persis C Costa C Pisani F et al . Intracellular calcium increase in epileptiform activity: modulation by levetiracetam and lamotrigine. Epilepsia. (2004) 45:719–28. doi: 10.1111/j.0013-9580.2004.02204.x
54.
Byers HM Beatty CW Hahn SH Gospe SM Jr . Dramatic response after lamotrigine in a patient with epileptic encephalopathy and a de novo CACNA1A variant. Pediatr Neurol. (2016) 60:79–82. doi: 10.1016/j.pediatrneurol.2016.03.012
Summary
Keywords
P/Q-type calcium channels, CaV2.1, status epilepticus, brainstem spreading depolarization, preventive strategies
Citation
Pelizzari S, Campiglio M, El Ghaleb Y, Bierhals T, Hempel M, Denecke J, Flucher BE and Johannsen J (2025) Prolonged apnea in a boy with epilepsy and a novel gain-of-function missense CACNA1A variant indicating SUDEP risk. Front. Neurol. 16:1582548. doi: 10.3389/fneur.2025.1582548
Received
24 February 2025
Accepted
10 June 2025
Published
09 July 2025
Volume
16 - 2025
Edited by
Hua-Jun Feng, Massachusetts General Hospital and Harvard Medical School, United States
Reviewed by
Daniela Pietrobon, University of Padua, Italy
Roope Mannikko, University College London, United Kingdom
Updates
Copyright
© 2025 Pelizzari, Campiglio, El Ghaleb, Bierhals, Hempel, Denecke, Flucher and Johannsen.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jessika Johannsen, j.johannsen@uke.de
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.