Abstract
Purpose:
This study aimed to explore the role of additional mitochondrial DNA (mtDNA) variants in the development of Leber hereditary optic neuropathy (LHON) by screening the entire mitochondrial genome in individuals who had previously tested negative for the three common mtDNA variants: m.3460G > A (MT-ND1), m.11778G > A (MT-ND4), and m.14484 T > C (MT-ND6), by conventional Sanger sequencing.
Methods:
Forty-one individuals with a suspected clinical diagnosis of LHON were recruited from the neuro-ophthalmology clinic. Each participant had undergone a comprehensive neuro-ophthalmic examination, including slit lamp examination, indirect ophthalmoscopy, visual field perimetry, optical coherence tomography, and MRI of the brain and orbits. Targeted re-sequencing was conducted using next-generation sequencing (NGS) on the HiSeqX 10 platform (Illumina, San Diego, California) with a 2 × 150 bp paired-end configuration. The sequencing reads were aligned to the human mitochondrial genome sequence (hg19). Variants were filtered with the VariMAT tool (v.2.3.9). Haplogroup analysis was performed using Haplogrep 2 (v2.0). To assess the deleteriousness of nonsynonymous variations, bioinformatics prediction tools such as PolyPhen2, SIFT, CADD, and Mutation Assessor were utilized. In addition, while tools like Consurf, PredictSNP, DynaMut, ENCoM, DUET, SDM, mCSM, were employed to evaluate evolutionary conservation, pathogenicity, structural stability, and functional impact.
Results:
Whole mitochondrial genome sequencing of 41 clinically suspected LHON cases identified a total of 1,518 mtDNA variants. Of these, 822 were located in the coding regions, including 555 synonymous and 273 non-synonymous variants. Two heteroplasmic disease-causing variants (m.11778G > A and m.3460G > A) were identified in one individual each (90.0 and 63.6%, respectively). Additionally, rare mtDNA variants listed in Mitomap were found in five individuals (5/41, 12.1%), namely, MT-ND1 (m.3335 T > C, m.3394 T > C, m.3395A > G), MT-ND4L (m.10680G > A), and MT-ND6 (m.14502 T > C), with variants in MT-ND1 being the most prevalent (3/41, 7.3%).
Conclusion:
Our study of a well-characterized Indian LHON cohort uncovered rare mtDNA variants that should be considered when assessing undiagnosed optic neuropathy cases. Additionally, it underscores the effectiveness of NGS in identifying heteroplasmic mtDNA variants. This indicates that whole mitochondrial genome sequencing via NGS is a more efficient and preferred approach for routine molecular genetic testing.
Introduction
Leber Hereditary Optic Neuropathy (LHON; OMIM: 535000) is an inherited optic neuropathy that predominantly affects young adult men (1, 2). LHON is characterized by bilateral subacute loss of central vision loss, either simultaneous at onset as seen in ~25% of cases or sequential within a few weeks of each other (3). It affects about 1:30,000–1:50,000 people (4, 5) with a peak age of onset in the second and third decades of life, but with reports ranging from 2 to 87 years old (6, 7).
The m.3460G > A (A52T), m.11778G > A (R340H), m.14484 T > C (M64V) mitochondrial DNA (mtDNA) variants occurring in the MT-ND1, MT-ND4, and MT-ND6 genes, respectively, are the three most common primary mutations that are involved in the pathogenesis of LHON (8–10). Cohort-based studies in various populations found varying proportions of these three mtDNA LHON variants, Denmark −98% (11), Mainland China –68% (12), Japan –100% (13), South Korea −73% (14), Finland −67% (15), and the Northern European –86% (16).
The oxidative phosphorylation (OXPHOS) system, which is located in the inner mitochondrial membrane, consists of five protein complexes and each complex is assembled by multimeric subunit proteins of mitochondrial as well as nuclear origin. The pathophysiology of LHON involves dysfunction of the complex I subunit of the mitochondrial respiratory chain, resulting in increased production of reactive oxygen species and decreased production of ATP.
mtDNA contains 37 genes that encode 13 structural proteins involved in the assembly and function of OXPHOS complexes; 22 tRNAs; and 2 rRNAs together involved in mitochondrial protein synthesis (17). LHON is caused by mtDNA variants that disrupt the protein complexes of the OXPHOS systemoften occurring in the genes encoding the subunits of complex I, the NADH: ubiquinone oxidoreductase. In addition to the three common LHON mtDNA variants, 16 other pathogenic variants have been reported1 that account for <5% of LHON cases. Previous studies indicate that specific mitochondrial haplogroups and their subgroups influence the pathogenicity and disease penetrance of the three common LHON mtDNA variants (18). A meta-analysis of 3,613 subjects from 159 white Caucasian LHON pedigrees, showed that the risk of visual failure is greater when the m.11778G > A and m.14484 T > C variants are present in specific subgroups of haplogroup J (J2 for m.11778G > A and J1 for m.14484 T > C), and when the m.3460G > A variants is present in haplogroup K (18). In the Chinese Han population, the M7b1’2 haplogroup increased the risk of visual loss whereas the M8a haplogroup had a protective effect (19–21). In the Indian population, the m.11778G > A and m.14484 T > C variants did not demonstrate significant haplogroup associations (22, 23). The M haplogroup was found to be the most common in both LHON cases and healthy controls in another South Indian study (24).
A reduced frequency of the three common LHON mutations has been identified in the North (27.5%) (25) and South Indian (43.6%) (26) populations. We have previously reported a figure of 29.4% in a combined North and South Indian population (27) emphasizing that the Indian population may exhibit a different mutational LHON profile with the involvement of pathogenic variants other than m.3460G > A, m.11778G > A and m.14484 T > C. In this study, we have further explored these pathogenic variants by analyzing the entire mitochondrial genome in a cohort of individuals with a clinical diagnosis of LHON. Phylogenetic analysis was also carried out to investigate the association of specific mtDNA haplogroups with LHON in the North and South Indian populations, and to predict the group of carriers at higher risk of developing visual loss.
Materials and methods
Clinical examination
The study protocol was approved by the Institutional Review Board (IRB) and ethics committee, and all procedures were performed according to institutional guidelines and the Declaration of Helsinki. Study participants underwent detailed ophthalmic examination by an experienced neuro-ophthalmologist (SA) and her team, including slit lamp examination, indirect ophthalmoscopy, visual field perimetry, optical coherence tomography, and MRI of the brain and orbits. Visual field analysis was performed according to the Humphrey automated visual field analyzer Swedish Interactive Threshold Algorithm (SITA) 30–2 standard. A diagnosis of LHON was based on classical clinical features, namely, a young patient with bilateral subacute progressive central visual loss, color vision defects, and a central or centrocecal scotoma, following exclusion of other acquired causes. These cases were referred for genetic screening.
DNA isolation and primary mutation screening
In patients referred for genetic testing, a detailed pedigree was taken, followed by blood collection for DNA extraction. Genomic DNA was extracted using a Nucleospin blood XL kit (Macherey-Nagel, GmbH, Düren, Germany) according to the manufacturer’s instructions. The three common LHON mtDNA mutations were screened according to the protocol used for our previous study (27).
Next generation sequencing of whole mitochondrial genome
Library preparation and purity were performed according to the manufacturer’s protocol. Briefly PCR products were amplified (Qiagen long-range PCR kit) with MTL-F1 5′-AAA GCA CAT ACC AAG GCC AC-3′ MTL-F2 5′- TAT CCG CCA TCC CAT ACA TT-3′ MTL-R1 5′-TTG GCT CTC CTT GCA AAG TT-3′ MTL-R2 5′-AAT GTT GAG CCG TAG ATG CC-3′ these sets of specific primers and fragments pooled together for NGS library preparation. These two fragments were pooled equally for NGS library preparation. Firstly, the DNA was fragmented (~250 bp), repairing ends, adenylation of 3’ends, followed by adapter ligation. At each step the products were purified using AMPure beads (Beckman Coulter). The adapter sequences were added onto the ends of DNA fragments to generate paired-end libraries. The resulting adaptor-ligated libraries were purified, and index tags were added by amplification, followed by purification. Libraries were assessed to check the quality and quantity by using an (Agilent 2200 Tape station).
Prepared libraries were quantified using the Qubit HS Assay (Invitrogen). The obtained libraries were pooled and diluted to the final optimal loading concentration (2 nm in 10 μL) before cluster amplification on an Illumina flow cell. Once the cluster generation was completed, the cluster flow cell was loaded on the Illumina HiSeq X instrument following the manufacturer’s instructions (Illumina) to generate 2*150 bp paired-end reads at >500X sequencing depth.
Sequence data analysis
The coverage and depth were analyzed after the quality check and alignment to the human mitochondrial genome (hg19). Alignment for all samples was in the range of 94–99%. The average depth for all samples was above 10,000X. GATK version 3.6 (28) was used for calling germline variants and annotated using the in-house VariMAT program (v.2.3.9). It integrates multiple clinical-grade databases, variant class prediction, and variant pathogenicity prediction tools for annotating the variants that rely on the Variant effect predictor [VEP] (29). Some of the annotated information by VariMAT was from population frequency, computational pathogenicity prediction, variant type, and predicted impact of the variant on the protein (e.g., missense and loss of function). A total sequencing coverage of more than 10,000X of the mitochondrial genome was achieved across all the samples. At such coverage, heteroplasmy at 5% levels would be detectable. We determined whether the variations were Novel, Reported, or with any disease association by investigating their presence in the MITOMAP, Human Mitochondrial Genome Database (HmtDB), ClinVar databases, Genome Aggregation Database (GnomAD v3.1.1), as well as Google and PubMed searches for individual studies reporting mtDNA variations. Haplogroup classification of mitochondrial samples was done by using the Haplogrep3 tool (30).
Bioinformatics analysis
Conservation and functionality analysis of variants
Bioinformatics predictions were conducted for missense variants that lack pathogenicity data in ClinVar, as well as for variants classified as Variants of Unknown Significance (VUS) and helper variants. These variants were first subjected to conservation analysis using ConSurf (31) to assess the evolutionary conservation of the affected amino acids and predict their functional significance. Subsequently, sequence-based pathogenicity, structural stability, and functional impact were evaluated using multiple prediction tools, including PredictSNP (32), MAPP Prediction (33), PhD-SNP Prediction (34), PolyPhen-1 (35), PolyPhen-2 (36), and SIFT (37). To further examine the effect of these variants on protein structural stability, in silico tools such as DynaMut (38), ENCoM (39), DUET, SDM (40), and mCSM (41) were employed. In addition, the MitImpact 3D tool (55),2 which exclusively provides insights into the genetic and clinical implications of the non-synonymous substitutions in human mitochondrial protein-coding genes, was also used to determine the conservation and pathogenicity of the variants. Prior to stability analysis, Alphafold-derived protein structures were retrieved and refined. Variants were generated using PyMOL, followed by refinement and preprocessing of both wild-type and variant forms with the Schrodinger suite. Finally, the potential impact of these variants on protein functionality was assessed using the MutPred2 server (42).
Results
Demographics and clinical data
This study included 41 unrelated probands who were negative for the three primary LHON mutations based on sanger sequencing. The median age at disease onset was 20.2 years (standard deviation 10.6 years), with a range from 1 to 45 years. All patients had either diffuse or temporal optic disk pallor on fundus examination. The clinical characteristics and examination findings have been summarized in Supplementary Table 1.
Next generation sequencing of whole mitochondrial genome
A comprehensive analysis of the whole mitochondrial genome was conducted for 41 individuals using NGS, with all samples meeting quality control standards. Our findings revealed a widespread distribution of variants across the mitochondrial genome, particularly in the D-loop region (Figure 1), and with most (98%) being homoplasmic (Figure 2). Detailed case-wise distributions of missense variants observed in suspected LHON cases have been provided in Supplementary Table 2. Among the probands, two individuals who were previously found to be negative for the three primary mutations via Sanger screening were found to be positive for m.11778G > A and m.3460G > A in a heteroplasmic state (90.0 and 63.6%, respectively) through NGS (2/41).
Figure 1

Distribution of variants across the mitochondrial genome.
Figure 2
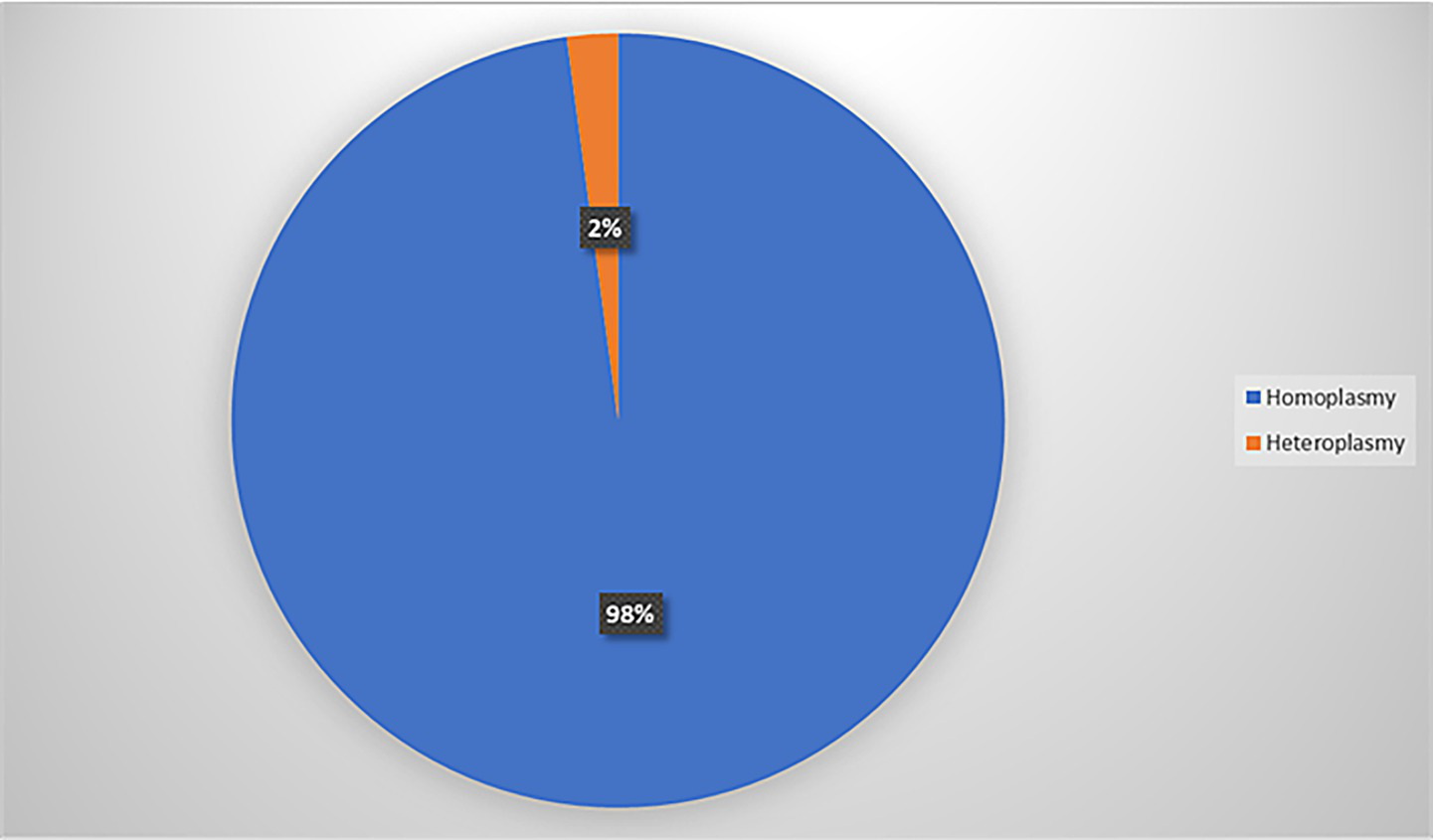
Distribution of homoplasmic and heteroplasmic variants.
Rare pathogenic variants were reported in five probands: MT-ND1 (3/41, 7.3%), MT-ND4L1 (1/41, 2.4%), and MT-ND6 (1/41, 2.4%) are listed in Table 1. They are MT-ND1 (m.3335 T > C, m.3394 T > C, m.3395A > G), MT-ND4L (m.10680G > A), and MT-ND6 (m.14502 T > C). Additionally, two probands shared the same helper variant in MT-CO3 (2/41, 4.8%). The overall mutation detection rate in suspected LHON cases was 17.0% (7/41). Notably, we identified seven cases with variants classified as having conflicting interpretations of pathogenicity in MT-ND1 and MT-ND2 (Table 2). Furthermore, common missense variants in MT-ATP6, MT-ND3, and MT-CYB were observed that are present at high frequencies in GenBank, as referenced in MITOMAP and GnomAD (Table 3).
Table 1
| S. no | Sample no | Age/sex | Age of onset | Levels of visual impairment | Neuroimaging (MRI) brain/orbit | Rare pathogenic variants |
|---|---|---|---|---|---|---|
| 1. | Sample 9 | 29/M | 24 | Moderate | Normal | G10680A |
| 2. | Sample 29 | 14/M | 11 | Moderate | Normal | T3355C |
| 3. | Sample 36 | 27/M | 27 | Moderate | Mild T2 hyperintensities with mild diffusion restriction involving the bilateral optic nerve sheath complexes (R > L) | T3394C |
| 4. | Sample 39 | 28/M | 15 | Severe | Cisternal segment of the left 6th nerve is thinned out | A3395G |
| 5. | Sample 47 | 22/M | 13 | Moderate | Normal | T14502C |
Rare pathogenic variants identified in the study.
Table 2
| Gene | Mutation | REF | ALT | cDNA | Variation | Amino acid | Zygosity | Genbank frequency (GB) (Mitomap) % | GnomAD allele frequency% | Clinvar | Sample nos |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MT-ND1 | m.3316 | G | A | c.10G > A | Missense | p. A4T | Homozygous | R30a1b1 | 0.093 | Benign | 4 |
| MT-ND1 | m.4216 | T | C | c.910 T > C | Missense | p. Y304H | Homozygous | R2d | 10.236 | Benign | 11,13,16 |
| MT-ND2 | m.5460 | G | A | c.991G > A | Missense | p. A331T | Homozygous | M58 | 6.754 | Benign | 36,41,53 |
Conflicting interpretations of pathogenicity.
Table 3
| Gene | Mutation | REF | ALT | cDNA | Variation | Amino acid | Zygosity | Genbank frequency (GB) (Mitomap) % | GnomAD allele frequency% | Clinvar | Sample nos |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MT-ATP6 | m.8701 | A | G | c.175A > G | MISSENSE | p. T59A | Homozygous | 31.5 | 30 | Benign | 3,6,9,12,16.19,21,23,24,26,27,34,35,37,38,39,40,41,42,46,48,50 |
| MT-ATP6 | m.8860 | A | G | c.334A > G | MISSENSE | p. T112A | Homozygous | 98.4 | 99 | Benign | 3,6,8,9,11,12,13,16,18,19,21,22,23,24,25,26,27,28,29,30,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,50,51,52,53 |
| MT-ND3 | m.10398 | A | G | c.340A > G | MISSENSE | p. T114A | Homozygous | 42.5 | 41 | Benign | 3,6,9,12,16,19,21,23,24,26,27,34,35,37,38,39,40,41,42,46,48,50,51,52,53 |
| MT-CYB | m.14766 | C | T | c.20C > T | MISSENSE | p. T7I | Homozygous | 75.6 | 70 | Benign | 3,6,8,9,11,12,13,16,18,19,21,22,23,24,25,26,27,28,29,30,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,50,51,52,53 |
| MT-CYB | m.15326 | A | G | c.580A > G | MISSENSE | p. T194A | Homozygous | 98.5 | 99 | Benign | 3,6,8,9,11,12,13,16,18,19,21,22,23,24,25,26,27,28,29,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,50,51,52,53 |
Frequently observed Benign missense variants with high GB and GnomAD frequency.
Conservation and functionality analysis of variants
Evolutionary conservation analysis was conducted using ConSurf (Table 4; Figures 3, 4). For MT-ND1 (Figure 3A), residues m.3316G > A (p. A4T). m.4099C > T (p. L265F), show the least conservation and are exposed, with m.3392G > C (p. G29A) first substitution, and m.3460G > A (p. A52T) having average conservation. For MT-ND2 (Figure 3b), residues m.4638A > G (p. I57V) and m.5279C > A (p. F270L) are highly conserved, but only m.4638A > G (p. F57V) has functional implications. For MT-ND3 (Figure 3c), m.10327C > T (p. S90L) shows the least conservation. For MT-ATP6 (Figure 3d), residue m.8594 T > C (p. I23T) is highly conserved and predicted to have functional impact, while residues m.9059C > T (p. T178I), and m.9106A > G (p. T194A) are least conserved and buried, suggesting that variants at these tyrosine residues will not significantly affect the protein’s structure or function. For MT-ATP8 (Figure 3e), residue m.8420A > G (p. T19A) is highly conserved. For MT-CO2 and MT-CO3, residue m.7685A > G (p. I34V) in MT-CO2 exhibits greater conservation compared to the least conserved residues in MT-CO3 (Figures 4a,b).
Table 4
| S. no | Sample ID | Gene | Variant | Amino acid | Wild residue | Position | Target residue | Conservation score | S/F |
|---|---|---|---|---|---|---|---|---|---|
| 1. | Sample 50 | MT-ND1 | m.3392G > C | G29A | G | 29 | A | 6 | B |
| 2. | Sample 50 | MT-ND1 | m.3460G > A | A52T | A | 52 | T | 7 | E |
| 3. | Sample 27,49 | MT-ND1 | m.4099C > T | L265F | L | 265 | F | 1 | E |
| 4. | Sample 40 | MT-ND2 | m.4638A > G | I57V | I | 57 | V | 8 | F/E |
| 5. | Sample 34 | MT-ND2 | m.5444C > A | F325L | F | 325 | L | 8 | E |
| 6. | Sample 3 | MT-ND2 | m.5279C > A | F270L | F | 270 | L | 7 | B |
| 7. | Sample 50 | MT-ND3 | m.10327C > T | S90L | S | 90 | L | 3 | E |
| 8. | Sample 35 | MT-ATP8 | m.8420A > G | T19A | T | 19 | A | 8 | F/E |
| 9. | Sample18, 44, 47 | MT-ATP6 | m.8594 T > C | I23T | I | 23 | T | 8 | F/E |
| 10. | Sample 12 | MT-ATP6 | m.9059C > T | T178I | T | 178 | I | 4 | B |
| 11. | Sample 49 | MT-ATP6 | m.9106A > G | T194A | T | 194 | A | 3 | E |
| 12. | Sample 27,49 | MT-CO3 | m.9966G > A | V254I | V | 254 | I | 4 | E |
| 13. | Sample 28 | MT-CO2 | m.7685A > G | I34V | I | 34 | V | 9 | F/E |
Conservation analysis of all the variants using Consurf.
Figure 3
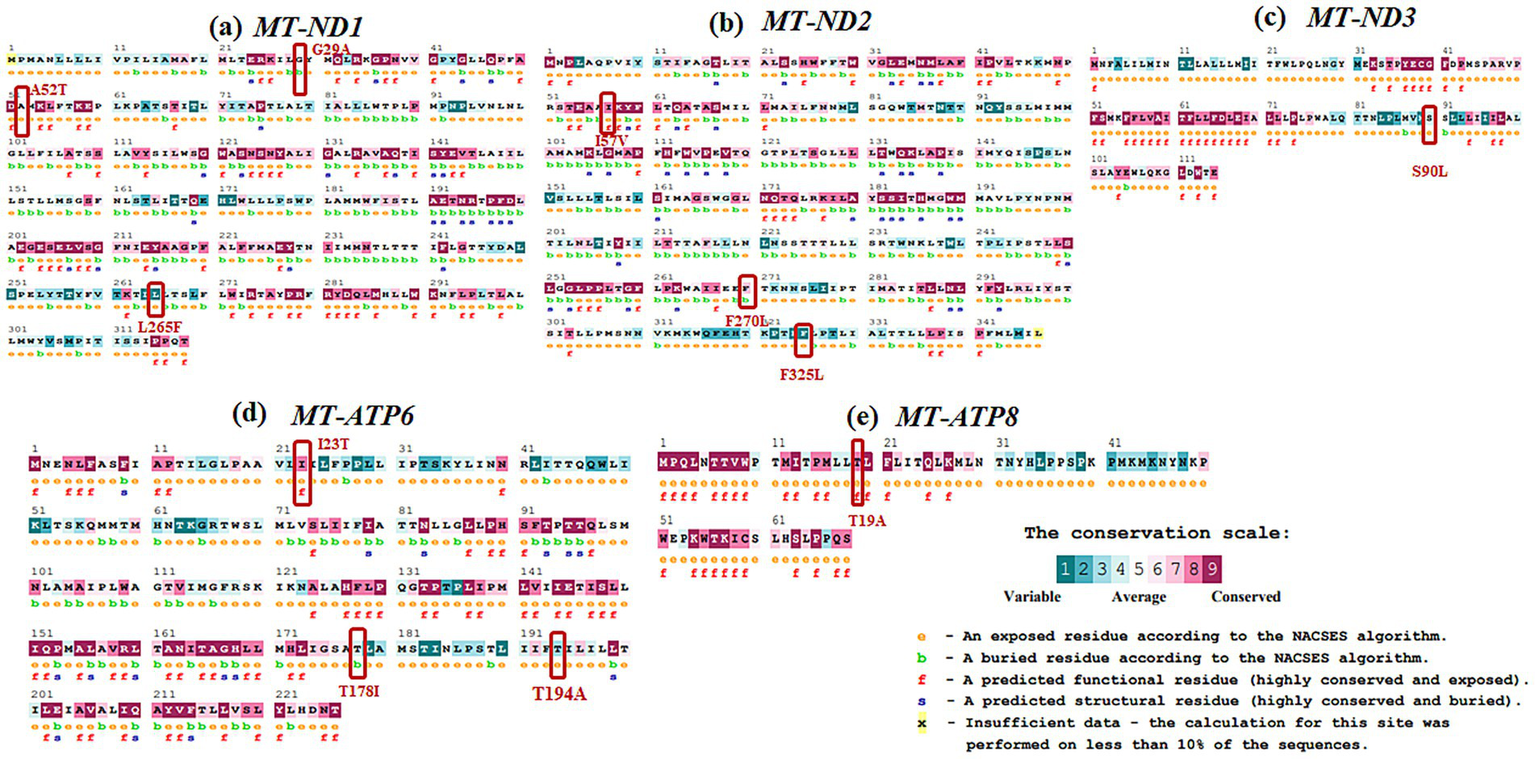
Evolutionary conservation analysis of (a)MT-ND1, (b)MT-ND2, (c)MT-ND3, (d)MT-ATP6, (e)MT-ATP8 with the positions of the variants highlighted in boxes.
Figure 4
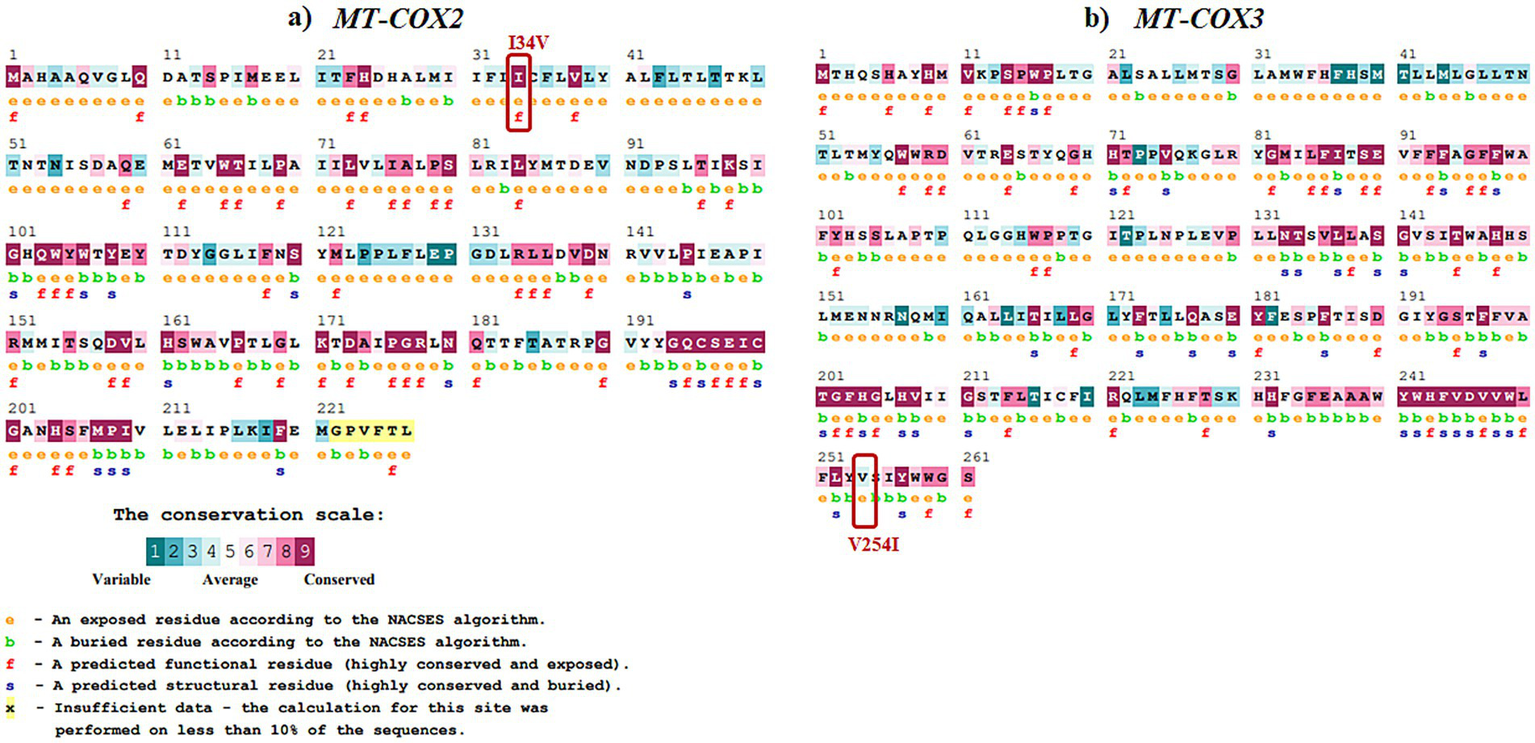
Evolutionary conservation analysis of (a)MT-COX2 and (b)MT-COX3 with the positions of the variants highlighted in boxes.
From the conservation prediction using MitImpact 3D, of the 13 variants, only five variants occurring at amino acid positions have been observed to be highly conserved. In specific, the MT-ND1 variants, m.3392G > C and m.3460G > A, and other gene variants namely, MT-ND2 (m.4638A > G), MT-ATP6 (m.8594 T > C), MT-CO2 (m.7685A > G) were observed to be highly conserved in all the predictors except for PhastCons 470way predictor (Supplementary Table 3).
Overall, from the consensus analysis of all the predictors, it was observed that the amino acid positions of the following variants, namely, m.4099C > T (MT-ND1), m.5444C > A (MT-ND2), m.10327C > T (MT-ND3), m.9059C > T and m.9106A > G (MT-ATP6), m.9966G > A (MT-CO3) are non-conserved.
Pathogenicity analysis and structural stability analysis
The m.8594 T > C variant in MT-ATP6 was identified as highly deleterious by all bioinformatics tools (Table 5). In terms of the meta-predictors, it was observed that the MT-ND1 variants (m.3392G > C and m.3460G > A) were observed to be damaging and deleterious among the five meta-predictors. In case of APOGEE1-based prediction, only ND1 variants (m.3392G > C and m.3460G > A) and MT-CO2 variant m.7685A > G are pathogenic (Supplementary Table 4).
Table 5
| S. no | Sample ID | Gene | Variant | Amino acid | PredictSNP prediction | PredictSNP expected accuracy | MAPP prediction | MAPP expected accuracy | PhD-SNP prediction | PhD-SNP expected accuracy | PolyPhen-1 prediction | PolyPhen-1 expected accuracy | PolyPhen-2 prediction | PolyPhen-2 expected accuracy | SIFT prediction | SIFT expected accuracy |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1. | Sample 18, 44, 47 | MT-ATP6 | m.8594 T > C | I23T | DELETERIOUS | 0.719 | DELETERIOUS | 0.427 | DELETERIOUS | 0.773 | DELETERIOUS | 0.594 | DELETERIOUS | 0.551 | DELETERIOUS | 0.793 |
| 2. | Sample 12 | MT-ATP6 | m.9059C > T | T178I | NEUTRAL | 0.603 | NEUTRAL | 0.705 | NEUTRAL | 0.719 | DELETERIOUS | 0.594 | DELETERIOUS | 0.503 | DELETERIOUS | 0.793 |
| 3. | Sample 49 | MT-ATP6 | m.9106A > G | T194A | NEUTRAL | 0.753 | NEUTRAL | 0.717 | NEUTRAL | 0.832 | NEUTRAL | 0.669 | NEUTRAL | 0.703 | DELETERIOUS | 0.793 |
| 5. | Sample 35 | MT-ATP8 | m.8420A > G | T19A | NEUTRAL | 0.748 | DELETERIOUS | 0.877 | NEUTRAL | 0.892 | NEUTRAL | 0.669 | NEUTRAL | 0.644 | NEUTRAL | 0.7 |
| 6. | Sample 28 | MT-CO2 | m.7685A > G | I34V | NEUTRAL | 0.603 | DELETERIOUS | 0.718 | NEUTRAL | 0.892 | NEUTRAL | 0.669 | NEUTRAL | 0.644 | DELETERIOUS | 0.793 |
| 7. | Sample 27,49 | MT-CO3 | m.9966G > A | V254I | NEUTRAL | 0.826 | NEUTRAL | 0.851 | NEUTRAL | 0.832 | NEUTRAL | 0.669 | NEUTRAL | 0.763 | NEUTRAL | 0.671 |
| 8. | Sample 50 | MT-ND1 | m.3392G > C | G29A | DELETERIOUS | 0.607 | DELETERIOUS | 0.771 | NEUTRAL | 0.508 | DELETERIOUS | 0.594 | DELETERIOUS | 0.65 | DELETERIOUS | 0.793 |
| 9. | Sample 27,49 | MT-ND1 | m.4099C > T | L265F | NEUTRAL | 0.653 | DELETERIOUS | 0.409 | NEUTRAL | 0.832 | NEUTRAL | 0.669 | NEUTRAL | 0.723 | DELETERIOUS | 0.793 |
| 10. | Sample 50 | MT-ND1 | m.3460G > A | A52T | DELETERIOUS | 0.756 | DELETERIOUS | 0.842 | DELETERIOUS | 0.589 | DELETERIOUS | 0.745 | DELETERIOUS | 0.811 | DELETERIOUS | 0.528 |
| 12. | Sample 40 | MT-ND2 | m.4638A > G | I57V | NEUTRAL | 0.632 | DELETERIOUS | 0.484 | NEUTRAL | 0.719 | NEUTRAL | 0.669 | NEUTRAL | 0.725 | DELETERIOUS | 0.793 |
| 13. | Sample 3 | MT-ND2 | m.5279C > A | F270L | NEUTRAL | 0.738 | NEUTRAL | 0.796 | NEUTRAL | 0.832 | NEUTRAL | 0.669 | NEUTRAL | 0.763 | DELETERIOUS | 0.459 |
| 14. | Sample 34 | MT-ND2 | m.5444C > A | F325L | NEUTRAL | 0.826 | NEUTRAL | 0.796 | NEUTRAL | 0.783 | NEUTRAL | 0.669 | NEUTRAL | 0.873 | NEUTRAL | 0.682 |
| 15. | Sample 50 | MT-ND3 | m.10327C > T | S90L | NEUTRAL | 0.737 | NEUTRAL | 0.65 | NEUTRAL | 0.661 | DELETERIOUS | 0.594 | NEUTRAL | 0.789 | NEUTRAL | 0.775 |
Pathogenicity analysis of the mitochondrial DNA variants.
Structural stability analysis of the refined structures (Supplementary Figures 1–5) indicated that the variants in MT-ND1, MT-ND2, MT-ATP6, and MT-CO3 led to structural destabilization (Table 6). Specifically, the variants m.8594 T > C (MT-ATP6) and m.5279C > A (MT-ND2), m.7685A > G (MT-CO2), were found to negatively impact their respective protein structures.
Table 6
| S. no | Sample ID | Gene | Variant | Amino acid | ΔΔG DynaMut (kcal/mol) | ΔΔG ENCoM (kcal/mol) | ΔΔG mcsm (kcal/mol) | ΔΔG SDM (kcal/mol) | ΔΔG DUET (kcal/mol) |
|---|---|---|---|---|---|---|---|---|---|
| 1. | Sample 12 | MT-ATP6 | m.9059C > T | T178I | 1.178 (Stabilizing) | 0.367 (Destabilizing) | −0.546 (Destabilizing) | 1.200 (Stabilizing) | 0.084 (Stabilizing) |
| 2. | Sample 49 | MT-ATP6 | m.9106A > G | T194A | 0.438 (Stabilizing) | −0.015 (Destabilizing) | −0.686 (Destabilizing) | 1.980 (Stabilizing) | 0.136 (Stabilizing) |
| 3. | Sample 18, 44, 47 | MT-ATP6 | m.8594 T > C | I23T | −1.240 (Destabilizing) | −0.185 (Destabilizing) | −1.854 (Destabilizing) | −1.930 (Destabilizing) | −1.919 (Destabilizing) |
| 4. | Sample 35 | MT-ATP8 | m.8420A > G | T19A | 0.117 (Stabilizing) | −0.218 (Destabilizing) | −0.571 (Destabilizing) | 1.980 (Stabilizing) | 0.249 (Stabilizing) |
| 5. | Sample 28 | MT-CO2 | m.7685A > G | I34V | −0.686 (Destabilizing) | −0.269 (Destabilizing) | −0.751 (Destabilizing) | −0.180 (Destabilizing) | −0.418 (Destabilizing) |
| 6. | Sample 27,49 | MT-CO3 | m.9966G > A | V254I | 0.408 (Stabilizing) | 0.099 (Destabilizing) | −0.024 (Destabilizing) | 0.270 (Stabilizing) | 0.456 (Stabilizing) |
| 7. | Sample 27,49 | MT-ND1 | m.4099C > T | L265F | −0.105 (Destabilizing) | 0.005 (Stabilizing) | −0.67 (Destabilizing) | −0.57 (Destabilizing) | −0.711 (Destabilizing) |
| 8. | Sample 50 | MT-ND1 | m.3392G > C | G29A | 0.616 (Stabilizing) | 0.084 (Stabilizing) | −0.887 (Destabilizing) | 0.65 (Stabilizing) | −0.355 (Destabilizing) |
| 9. | Samplse 50 | MT-ND1 | m.3460G > A | A52T | 0.396 (Stabilizing) | 0.136 (Destabilizing) | −0.989 (Destabilizing) | −1.330 (Destabilizing) | −0.870 (Destabilizing) |
| 10. | Sample 3 | MT-ND2 | m.5279C > A | F270L | −1.182 (Destabilizing) | −0.362 (Destabilizing) | −1.748 (Destabilizing) | −1.53 (Destabilizing) | −1.871 (Destabilizing) |
| 11. | Sample 40 | MT-ND2 | m.4638A > G | I57V | 0.009 (Stabilizing) | −0.056 (Destabilizing) | −0.787 (Destabilizing) | −0.18 (Destabilizing) | −0.457 (Destabilizing) |
| 12. | Sample 34 | MT-ND2 | m.5444C > A | F325L | −0.065 (Destabilizing) | 0.027 (Stabilizing) | −0.396 (Destabilizing) | 0.39 (Stabilizing) | −0.196 (Destabilizing) |
| 13. | Sample 50 | MT-ND3 | m.10327C > T | S90L | 0.375 (Stabilizing) | 0.162 (Stabilizing) | −0.124 (Destabilizing) | 1.310 (Stabilizing) | 0.532 (Stabilizing) |
Structural stability analysis of the identified mitochondrial DNA variants.
Interatomic interaction analysis
In the interatomic interaction analysis (Supplementary Figures 1–5), except for the m.8594 T > C variant, the two other MT-ATP6 variants, m.9059C > T and m.9106A > G, exhibited stabilization through additional non-bonded interactions. No significant interatomic variations or alterations were detected for MT-ATP8, MT-CO2 and MT-CO3 variants.
Haplogroup distribution
The phylogenetic analysis of mtDNA haplogroups in the LHON patient cohort revealed five distinct haplogroups (M, R, U, D, and W). The M haplogroup was the most prevalent (26/41, 63.4%), followed by R (9/41, 22.0%) and U (6/41, 14.6%). Haplogroups W and D each contributed one case.
Discussion
In the current study, we analyzed the whole mitochondrial genome of 41 individuals with a clinical diagnosis of LHON who were negative for the three common mtDNA mutations based on conventional Sanger sequencing. Of these cases, NGS identified the m.11778G > A and m.3460G > A variant in one patient each, present in the heteroplasmic state (90.0 and 63.6%, respectively). A possible explanation for these mtDNA variants being missed could be the lower sensitivity of Sanger sequencing, which analyses a single DNA fragment, limiting its ability to detect heteroplasmic variants.
In our previous study (27), 82 out of 278 suspected LHON cases (29.4%) were found to be positive for one of the three primary mutations. This diagnostic rate is significantly lower than the figure of ~ 90% reported in other studies (6, 43), but it is comparable to findings from other studies of Indian populations (24, 26, 27). In this study, we explored the possible pathogenicity of other mtDNA variants in those patients who had tested negative for m.3460G > A, m.11778G > A and m.14484 T > C. We identified five previously reported LHON variants: three located in MT-ND1 (m.3335 T > C, m.3394 T > C, m.3395A > G), one in MT-ND4L (m.10680G > A), and one in MT-ND6 (m.14502 T > C).
MT-ND1 variants
The MT-ND1 variants m.3335 T > C and m.3394 T > C were reported in a Chinese study involving 1,281 subjects. The m.3335 T > C variant was identified in four individuals with mild visual impairment. In contrast, the m.3394 T > C variant was found in 38 probands with varying degrees of visual impairment, accounting for an incidence rate of 2.97% (44). Another variant, m.3395A > G, was reported in a single suspected LHON (Leber’s Hereditary Optic Neuropathy) case from Argentina and has recently been reclassified from Likely Benign to Likely Pathogenic based on ACMG/AMP guidelines for mitochondrial DNA variant interpretation (45). The m.3394 T > C variant has also been reported in the Tibetan population, where it is associated with reduced complex I activity, influenced by mitochondrial haplogroup background and environment (46). It is notably enriched in the M9 haplogroup in Tibet. However, in our study, this variant was identified in the W3 haplogroup, which is relatively rare. The identification of this variant within W3, a lineage not typically associated with LHON, suggests that its pathogenic potential may vary by haplogroup. This highlights the influence of genetic background on disease expression. Importantly, no other rare variants were observed in the family, supporting a potential pathogenic role of this variant in the given haplogroup context. The MT-ND1 polypeptide includes transmembrane, outer membrane, and intermembrane domains; notably, all three variants identified in this study are located within the transmembrane domain, which plays a critical role in mitochondrial complex I function (44).
MT-ND4L and MT-ND6 variants
The MT-ND4L variant m.10680G > A occurs at a highly conserved position, resulting in the amino acid change from alanine to threonine (A71T), with conservation rates of 86% in eukaryotes and 97% in mammals, and within an invariant stretch of 16 amino acids in primates (47). Studies of cybrid cell lines harboring this mtDNA variant demonstrated a respiration defect comparable to that observed in LHON cells (47). The variant has been identified in 14 haplogroups and has been previously reported as the sole pathogenic change in three LHON families, occurring as independent mutational events in haplogroups B4a1e, M13a1b, and D6a1 (48, 49). Additionally, this mutation has been found in association with the m.14484 T > C/MT-ND6 mutation in another LHON family with a B4d1 haplogroup background (50). A separate family-based study also revealed the co-occurrence of three missense variants: m.10680G > A/MT-ND4L, m.12033A > G/MT-ND4, and m.14258G > A/MT-ND6, present in the proband, mother, and sister (47). The MT-ND6 variant m.14502 T > C has been reported in three unrelated subjects from a Chinese LHON cohort, leading to the substitution of isoleucine for valine (I58V), a conserved amino acid across multiple species (51). Additionally, Zhang et al. (52) described four Chinese families in which individuals co-harboring both m.14502 T > C and m.11778G > A exhibited increased penetrance of visual loss, suggesting a potential synergistic effect. However, m.14502 T > C has also been identified at a relatively high frequency in population databases, including in healthy individuals, which challenges its role as a primary pathogenic LHON mutation. Given this, it is more plausible that m.14502 T > C serves as a secondary modifier or risk factor that may influence disease expression in the presence of other LHON-associated variants. Further functional and population-based studies are needed to clarify its exact role.
MT-CO3 variant
The missense variant m.9966G > A in MT-CO3 is considered a helper variant, as it has been found to co-occur with LHON mtDNA variants (47) However, one Indian study identified this variant solely in two LHON families without any secondary variations (53). In the current study, we detected this variant in two families, Sample 27 and Sample 49, both belonging to the same haplogroup M38a. Interestingly, both families also carried the MT-ND1 variant m.4099C > T, which was called as being deleterious, albeit only by CADD scoring. Additionally, Sample 49 carried a Variant of Unknown Significance (VUS) located in MT-ATP6, m.9106A > G, with very low frequencies in Mitomap (0.005%) and gnomAD (0.009%). Further evidence is needed to clarify the pathogenic role of the m.9966G > A variant in individuals diagnosed with LHON.
Variants with conflicting interpretations
We identified two variants in MT-ND1: m.3316G > A in one family and m.4216 T > C in three different families. The pathogenicity of these variants remains unclear, with the reported incidences of these variants in a Chinese cohort being 4.0 and 1.09%, respectively (12). The m.3316G > A variant has been noted to co-occur with m.11778G > A mutations, and it has been suggested to increase the penetrance of LHON (12). We did not find any primary LHON mutations in the family associated with this variant in our cohort. The m.4216 T > C variant has been observed in both patient and control populations, and its pathogenic significance remains uncertain (12).
Intermolecular interaction analysis
The sequence-based pathogenicity analysis (Table 5) revealed that confirmed pathogenic LHON variants were found to be deleterious with high accuracy, primarily through MAPP prediction. All predictors identified the m.3460G > A variant in MT-ND1 and the m.8594 T > C variant in MT-ATP6 as highly deleterious. The presence of multiple variants, such as m.3460G > A and m.3392G > C could increase the pathogenicity score. Structural stability analysis of refined structures (Figure 5) showed that only the variants in MT-ATP6, MT-ND2, MT-CO3, and MT-ND1 were associated with structural destabilization (Table 5). Specifically, the m.8594 T > C variant in MT-ATP6, m.5279C > A in MT-ND2, and m.7685A > G in MT-CO2 negatively impacted their respective protein structures.
Figure 5
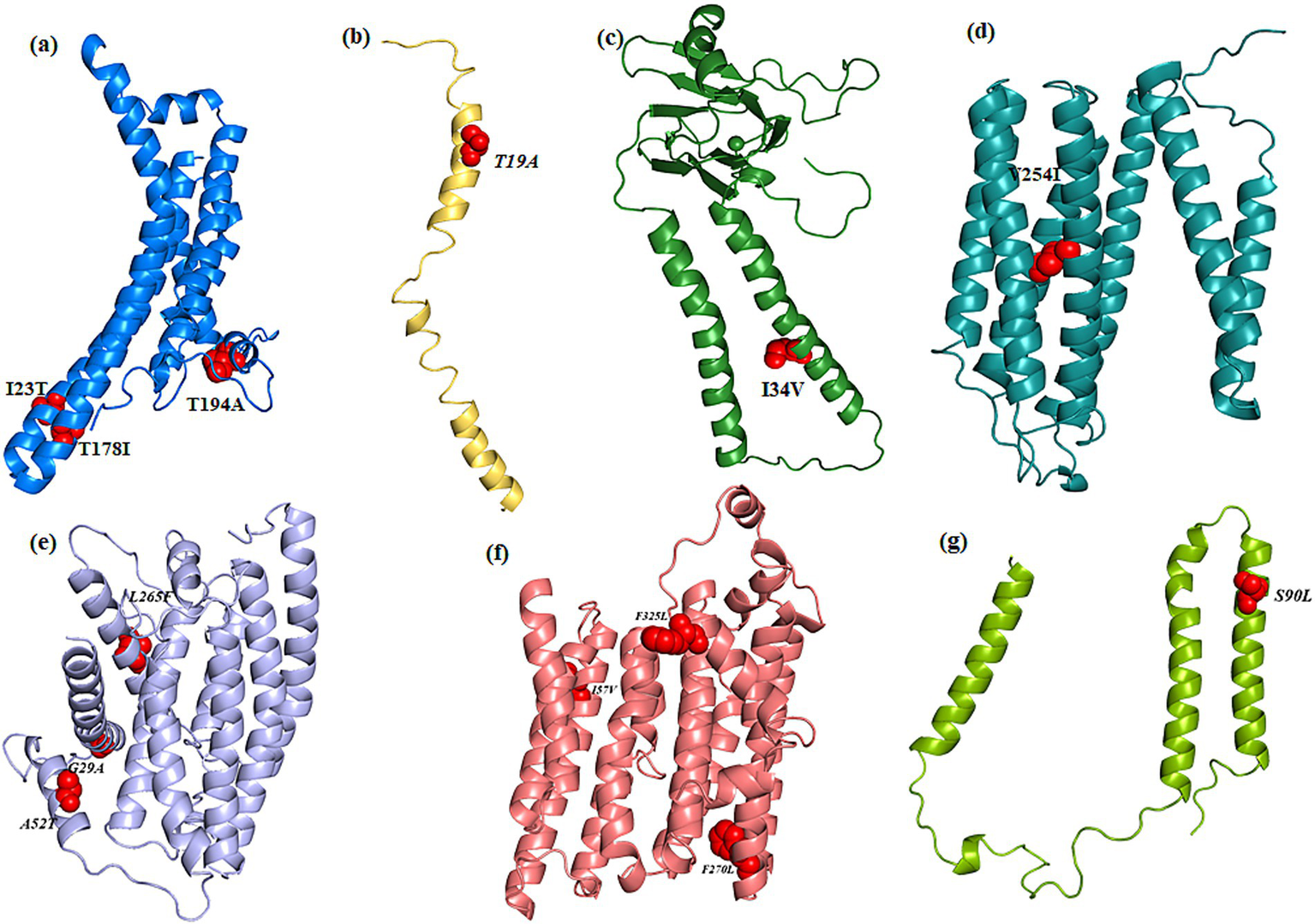
Cartoon representation of the modeled and refined structure of (a) MT-ATP6, (b)MT-ATP8, (c)MT-COX2, (d)MT-COX3, (e)MT-ND1, (f)MT-ND2, (g)MT-ND3 wherein the variants are showed as spheres (red color).
All the MT-ATP6 variants maintained their interactions with the wild-type variant, except for m.8594 T > C (p. I23T), where T23 has lost its hydrophobic interaction with P12. The m.9059C > T (p. T178I) variant, I178, gained additional hydrophobic interactions with I193 and T189. Despite losing an H-bond with I191, the m.9106A > G (p. T194A) variant, which encodes A194, stabilizes MT-ATP6 through strong van der Waals interactions with I192. In MT-ATP8, although the m.8420A > G (p. T19A) variant, A19, resulted in the loss of H-bonds with I23 and M16, all other interactions were preserved compared to the wild-type variant. For MT-CO2, the m.7685A > G (p. I34V) variant, V34 maintained H-bond interactions with I30 and V38 and gained an additional van der Waals interaction with I31. In MT-CO3, the m.9966G > A (p. V254I) variant, I254, did not induce interatomic changes, maintaining H-bond interactions with L250 and T259, and van der Waals interactions with L250, similar to the wild-type variant.
All the m.3392G > C, m.3460G > A, and m.4099C > T in MT-ND1 maintained the interatomic interactions found with the wild-type variant. In MT-ND2, m.4638A > G (p. I57V), V57 formed van der Waals interactions with E54, differing from the wild type. Notably, variant 5279C > A (p. F270L), L270 lost hydrophobic interactions with Y196 and ionic interactions with M282.
For MT-ND3, the H-bond interactions were similar for the wild-type and the 10327C > T (p. S90L) variants. Functional impact analysis of all variants (Table 6) indicated that only the MT-ND1 variant m.3392G > C (p. G29A) had a significant impact, such as alterations in disordered interfaces, ordered interfaces (Pr = 0.26 | p = 0.01), DNA binding, and transmembrane protein interactions (Table 7).
Table 7
| S. no | Sample ID | Gene | Variant | Amino acid | Functional/structural loss |
|---|---|---|---|---|---|
| 1. | Sample 12 | MT-ATP6 | m.9059C > T | T178I | – |
| 2. | Sample 49 | MT-ATP6 | m.9106A > G | T194A | – |
| 3. | Sample 18, 44, 47 | MT-ATP6 | m.8594 T > C | I23T | – |
| 4. | Sample 27, 49 | MT-ND1 | m.4099C > T | L265F | – |
| 5. | Sample 50 | MT-ND1 | m.3392G > C | G29A | Altered Disordered interface (Pr = 0.28 | p = 0.04); Altered Ordered interface (Pr = 0.26 | P = 0.01); Altered DNA binding (Pr = 0.24 | P = 0.01); Altered Transmembrane protein (Pr = 0.20 | p = 5.2e-03) |
| 6. | Sample 50 | MT-ND1 | m.3460G > A | A52T | – |
| 7. | Sample 3 | MT-ND2 | m.5279C > A | F270L | – |
| 8. | Sample 40 | MT-ND2 | m.4638A > G | I57V | – |
| 9. | Sample 34 | MT-ND2 | m.5444C > A | F325L | – |
| 10. | Sample 50 | MT-ND3 | m.10327C > T | S90L | – |
| 11. | Sample 35 | MT-ATP8 | m.8420A > G | T19A | – |
| 12. | Sample 28 | MT-CO2 | m.7685A > G | I34V | – |
| 13. | Sample 27, 49 | MT-CO3 | m.9966G > A | V254I | – |
Functional impact analysis of the identified mitochondrial DNA variants.
The bold highlights the significant functional impact attained by the variant.
The analysis of missense mtDNA variants revealed a high proportion of variants that could be deleterious by disrupting protein structure and stability. Functional impact and conservation analyses further support the pathogenicity of several variants, particularly those in MT-ATP6, MT-ND1, and MT-ND2. Additionally, the co-occurrence of variants, such as those in MT-ND1, MT-ATP6, and MT-CO3, suggests potential synergistic effects on disease expression and severity, further complicating the interpretation of mitochondrial genetics in LHON. It has previously been reported that combinations of individually non-pathogenic missense mtDNA variants could cause low penetrance LHON (47).
Our findings underscore the complexity of LHON genetics, revealing not only known pathogenic mtDNA variants, but previously unreported variants that could play a role in disease pathogenesis. Future work is needed to determine whether a proportion of our undiagnosed LHON cohort carries nuclear genetic defects that are increasingly being recognized as an important cause of recessive LHON (2, 54).
Overlapping clinical features suggest broader genetic etiology in LHON
All patients who presented with clinical features suggestive of Leber Hereditary Optic Neuropathy (LHON) but remained genetically unsolved (i.e., tested negative for known mitochondrial mutations) demonstrated a clinical profile similar to that of genetically confirmed (solved) LHON cases. The majority of patients in the unsolved group were male, with only one female patient identified. Most exhibited moderate vision loss without progression to severe visual impairment. Universally, patients in both cohorts showed temporal optic disk pallor at presentation, along with cecocentral visual field defects. No systemic manifestations were observed in either group.
MRI brain imaging revealed optic nerve signal changes in two patients from the solved group and one patient from the unsolved group. However, these findings were non-specific and not characteristic of LHON. Importantly, no distinct clinical or radiological differences were identified between the solved and unsolved cases. This clinical overlap underscores the need for further investigation, particularly nuclear gene analysis, with the help of Whole Exome Sequencing (WES) or Whole Genome Sequencing (WGS) to uncover the underlying genetic etiology in the unsolved cohort.
Conclusion
A comprehensive analysis of the whole mitochondrial genome, preferably with NGS, is recommended in individuals with a clinical diagnosis of LHON in whom conventional Sanger sequencing has failed to uncover one of the three common mtDNA mutations. Additional research involving larger cohorts of Indian patients and complementary functional studies will be essential to validate our results and further clarify the full range of genetic contributors to LHON in this particular population.
Statements
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Ethics statement
The studies involving human participants were reviewed and approved by the Institutional Review Board (Ethics Committee) of the Medical Research Foundation (Approval number: PR_0000023). Written informed consent to participate in this study was provided by the patient/participants or patient/participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
SS: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Validation, Writing – original draft. SA: Project administration, Supervision, Validation, Writing – review & editing. NH: Data curation, Formal analysis, Software, Writing – review & editing. DV: Investigation, Methodology, Writing – review & editing. PY-W-M: Funding acquisition, Project administration, Resources, Supervision, Visualization, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was funded by the Roche Collaborative Research Fellowships from ARVO Foundation and we thank Indian Council of Medical Research (ICMR), Govt. of India for the award Long term ICMR-DHR International Fellowship—No. INDO/FRC/452/Y-06/2022-23-IH&HRD to SS. PY-W-M was supported by an Advanced Fellowship Award (NIHR301696) from the UK National Institute of Health Research (NIHR). PY-W-M also received funding from the Rosetrees Trust (PGL23/100048), Fight for Sight (UK), the Isaac Newton Trust (UK), Moorfields Eye Charity (GR001376), the Addenbrooke’s Charitable Trust, the National Eye Research Centre (UK), the International Foundation for Optic Nerve Disease (IFOND), the NIHR as part of the Rare Diseases Translational Research Collaboration, the NIHR Cambridge Biomedical Research Centre (NIHR203312), and the NIHR Biomedical Research Centre based at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology (NIHR203322). This research was supported by the LifeArc under grant no. 10748. LifeArc is a charity registered in England and Wales under no. 1015243 and in Scotland under no. SC037861.
Acknowledgments
We thank the patients and their family for their kind co-operation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer J-WT declared a shared affiliation with the author PY-W-M to the handling editor at the time of review.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2025.1584748/full#supplementary-material
References
1.
Yu-Wai-Man P Turnbull DM Chinnery PF . Leber hereditary optic neuropathy. J Med Genet. (2002) 39:162–9. doi: 10.1136/jmg.39.3.162
2.
Newman NJ Yu-Wai-Man P Biousse V Carelli V . Understanding the molecular basis and pathogenesis of hereditary optic neuropathies: towards improved diagnosis and management. Lancet Neurol. (2023) 22:172–88. doi: 10.1016/S1474-4422(22)00174-0
3.
Mashima Y Kimura I Yamamoto Y Ohde H Ohtake Y Tanino T et al . Optic disc excavation in the atrophic stage of Leber's hereditary optic neuropathy: comparison with normal tension glaucoma. Graefes Arch Clin Exp Ophthalmol. (2003) 241:75–80. doi: 10.1007/s00417-002-0598-0
4.
Puomila A Hämäläinen P Kivioja S Savontaus M-L Koivumäki S Huoponen K et al . Epidemiology and penetrance of Leber hereditary optic neuropathy in Finland. Eur J Hum Genet. (2007) 15:1079–89. doi: 10.1038/sj.ejhg.5201828
5.
Spruijt L Kolbach DN de Coo RF Plomp AS Bauer NJ Smeets HJ et al . Influence of mutation type on clinical expression of Leber hereditary optic neuropathy. Am J Ophthalmol. (2006) 141:676–676.e8. doi: 10.1016/j.ajo.2005.11.007
6.
Newman NJ . Hereditary optic neuropathies: from the mitochondria to the optic nerve. Am J Ophthalmol. (2005) 140:517.e1–9. doi: 10.1016/j.ajo.2005.03.017
7.
Giraudet S Lamirel C Amati-Bonneau P Reynier P Bonneau D Miléa D et al . Never too old to harbour a young man's disease?Br J Ophthalmol. (2011) 95:887. doi: 10.1136/bjo.2009.161539
8.
Huoponen K Vilkki J Aula P Nikoskelainen EK Savontaus ML . A new mtDNA mutation associated with Leber hereditary optic neuroretinopathy. Am J Hum Genet. (1991) 48:1147–53.
9.
Wallace DC Singh G Lott MT Hodge JA Schurr TG Lezza AMS et al . Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science. (1988) 242:1427–30. doi: 10.1126/science.3201231
10.
Johns DR Neufeld MJ Park RD . An ND-6 mitochondrial DNA mutation associated with leber hereditary optic neuropathy. Biochem Biophys Res Commun. (1992) 187:1551–7. doi: 10.1016/0006-291X(92)90479-5
11.
Rosenberg T Nørby S Schwartz M Saillard J Magalhães PJ Leroy D et al . Prevalence and genetics of Leber hereditary optic neuropathy in the Danish population. Invest Ophthalmol Vis Sci. (2016) 57:1370–5. doi: 10.1167/iovs.15-18306
12.
Dai Y Wang C Nie Z Han J Chen T Zhao X et al . Mutation analysis of Leber’s hereditary optic neuropathy using a multi-gene panel. Biomed Rep. (2018) 8:51–8. doi: 10.3892/br.2017.1014
13.
Yamada K Oguchi Y Hotta Y Nakamura M Isashiki Y Mashima Y . Multicenter study on the frequency of three primary mutations of mitochondrial DNA in Japanese pedigrees with Leber's hereditary optic neuropathy: comparison with American and British counterparts. Neuro-Ophthalmology. (1999) 22:187–93. doi: 10.1076/noph.22.3.187.3724
14.
Kim JY Hwang JM Chang BL Park SS . Spectrum of the mitochondrial DNA mutations of Leber's hereditary optic neuropathy in Koreans. J Neurol. (2003) 250:278–81. doi: 10.1007/s00415-003-0985-4
15.
Jiang P Liang M Zhang J Gao Y He Z Yu H et al . Prevalence of mitochondrial ND4 mutations in 1281 Han Chinese subjects with Leber’s hereditary optic neuropathy. Invest Ophthalmol Vis Sci. (2015) 56:4778–88. doi: 10.1167/iovs.14-16158
16.
Mascialino B Leinonen M Meier T . Meta-analysis of the prevalence of Leber hereditary optic neuropathy mtDNA mutations in Europe. Eur J Ophthalmol. (2012) 22:461–5. doi: 10.5301/ejo.5000055
17.
Taanman JW . The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta. (1999) 1410:103–23. doi: 10.1016/S0005-2728(98)00161-3
18.
Hudson G Carelli V Spruijt L Gerards M Mowbray C Achilli A et al . Clinical expression of Leber hereditary optic neuropathy is affected by the mitochondrial DNA-haplogroup background. Am J Hum Genet. (2007) 81:228–33. doi: 10.1086/519394
19.
Ji Y Zhang AM Jia X Zhang YP Xiao X Li S et al . Mitochondrial DNA Haplogroups M7b1′2 and M8a affect clinical expression of Leber hereditary optic neuropathy in Chinese families with the m.11778G→a mutation. Am J Hum Genet. (2008) 83:760–8. doi: 10.1016/j.ajhg.2008.11.002
20.
Ji Y Jia X Zhang Q Yao YG . mtDNA haplogroup distribution in Chinese patients with Leber's hereditary optic neuropathy and G11778A mutation. Biochem Biophys Res Commun. (2007) 364:238–42. doi: 10.1016/j.bbrc.2007.09.111
21.
Zhang AM Jia X Bi R Salas A Li S Xiao X et al . Mitochondrial DNA haplogroup background affects LHON, but not suspected LHON, in Chinese patients. PLoS One. (2011) 6:e27750. doi: 10.1371/journal.pone.0027750
22.
Khan NA Govindaraj P Soumittra N Sharma S Srilekha S Ambika S et al . Leber's hereditary optic neuropathy-specific mutation m.11778G>a exists on diverse mitochondrial Haplogroups in India. Invest Ophthalmol Vis Sci. (2017) 58:3923–30. doi: 10.1167/iovs.16-20695
23.
Khan NA Govindaraj P Soumittra N Srilekha S Ambika S Vanniarajan A et al . Haplogroup heterogeneity of LHON patients carrying the m.14484T>C mutation in India. Invest Ophthalmol Vis Sci. (2013) 54:3999–4005. doi: 10.1167/iovs.13-11925
24.
Saikia BB Dubey SK Shanmugam MK Sundaresan P . Whole mitochondrial genome analysis in south Indian patients with Leber's hereditary optic neuropathy. Mitochondrion. (2017) 36:21–8. doi: 10.1016/j.mito.2016.10.006
25.
Mishra A Devi S Saxena R Gupta N Kabra M Chowdhury M . Frequency of primary mutations of Leber's hereditary optic neuropathy patients in north Indian population. Indian J Ophthalmol. (2017) 65:1156–60. doi: 10.4103/ijo.IJO_380_17
26.
Gowri P Kumar SM Vanniarajan A Bharanidharan D Sundaresan P . A hospital-based five-year prospective study on the prevalence of Leber's hereditary optic neuropathy with genetic confirmation. Mol Vis. (2020) 26:789–96.
27.
Sundaramurthy S Selvakumar A Dharani V Soumittra N Mani J Thirumalai K et al . Prevalence of primary mutations in Leber hereditary optic neuropathy: a six-year report from a tertiary eye care center in India. Mol Vis. (2021) 27:718–24.
28.
Van der Auwera GA O'Connor BD . Genomics in the cloud: using Docker, GATK, and WDL in Terra. 1st ed. Sebastopol, CA: O'Reilly Media (2020).
29.
McLaren W Gil L Hunt SE Riat HS Ritchie GRS Thormann A et al . The Ensembl variant effect predictor. Genome Biol. (2016) 17:122. doi: 10.1186/s13059-016-0974-4
30.
Schönherr S Weissensteiner H Kronenberg F Forer L . Haplogrep 3 - an interactive haplogroup classification and analysis platform. Nucleic Acids Res. (2023) 51:W263–8. doi: 10.1093/nar/gkad284
31.
Ben Chorin A Masrati G Kessel A Narunsky A Sprinzak J Lahav S et al . ConSurf-DB: an accessible repository for the evolutionary conservation patterns of the majority of PDB proteins. Protein Sci. (2020) 29:258–67. doi: 10.1002/pro.3779
32.
Bendl J Stourac J Salanda O Pavelka A Wieben ED Zendulka J et al . PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput Biol. (2014) 10:e1003440. doi: 10.1371/journal.pcbi.1003440
33.
Stone EA Sidow A . Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome Res. (2005) 15:978–86. doi: 10.1101/gr.3804205
34.
Capriotti E Fariselli P . PhD-SNPg: a webserver and lightweight tool for scoring single nucleotide variants. Nucleic Acids Res. (2017) 45:W247–52. doi: 10.1093/nar/gkx369
35.
Adzhubei IA Schmidt S Peshkin L Ramensky VE Gerasimova A Bork P et al . A method and server for predicting damaging missense mutations. Nat Methods. (2010) 7:248–9. doi: 10.1038/nmeth0410-248
36.
Adzhubei I Jordan DM Sunyaev SR . Predicting functional effect of human missense mutations using polyphen-2. Curr Protoc Hum Genet. (2013) Chapter 7:Unit7.20-Unit7. doi: 10.1002/0471142905.hg0720s76
37.
Sim NL Kumar P Hu J Henikoff S Schneider G Ng PC . SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. (2012) 40:W452–7. doi: 10.1093/nar/gks539
38.
Rodrigues CH Pires DE Ascher DB . DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. (2018) 46:W350–5. doi: 10.1093/nar/gky300
39.
Kang J Tang Q He J Li L Yang N Yu S et al . RNAInter v4.0: RNA interactome repository with redefined confidence scoring system and improved accessibility. Nucleic Acids Res. (2022) 50:D326–32. doi: 10.1093/nar/gkab997
40.
Anderson BA Freestone GC Low A De-Hoyos CL Iii WJD Ostergaard ME et al . Towards next generation antisense oligonucleotides: mesylphosphoramidate modification improves therapeutic index and duration of effect of gapmer antisense oligonucleotides. Nucleic Acids Res. (2021) 49:9026–41. doi: 10.1093/nar/gkab718
41.
Pires DE Ascher DB Blundell TL . Mcsm: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics. (2014) 30:335–42. doi: 10.1093/bioinformatics/btt691
42.
Pejaver V Urresti J Lugo-Martinez J Pagel KA Lin GN Nam HJ et al . Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat Commun. (2020) 11:5918. doi: 10.1038/s41467-020-19669-x
43.
Shemesh A Sood G Blair K Margolin E . Leber Hereditary Optic Neuropathy (LHON). Treasure Island, FL: StatPearls (2025).
44.
Ji Y Liang M Zhang J Zhu L Zhang Z Fu R et al . Mitochondrial ND1 variants in 1281 Chinese subjects with Leber's hereditary optic neuropathy. Invest Ophthalmol Vis Sci. (2016) 57:2377–89. doi: 10.1167/iovs.16-19243
45.
Buonfiglio PI Menazzi S Francipane L Lotersztein V Ferreiro V Elgoyhen AB et al . Mitochondrial DNA variants in a cohort from Argentina with suspected Leber's hereditary optic neuropathy (LHON). PLoS One. (2023) 18:e0275703. doi: 10.1371/journal.pone.0275703
46.
Ji F Sharpley MS Derbeneva O Alves LS Qian P Wang Y et al . Mitochondrial DNA variant associated with Leber hereditary optic neuropathy and high-altitude Tibetans. Proc Natl Acad Sci USA. (2012) 109:7391–6. doi: 10.1073/pnas.1202484109
47.
Caporali L Iommarini L La Morgia C Olivieri A Achilli A Maresca A et al . Peculiar combinations of individually non-pathogenic missense mitochondrial DNA variants cause low penetrance Leber's hereditary optic neuropathy. PLoS Genet. (2018) 14:e1007210. doi: 10.1371/journal.pgen.1007210
48.
Zou Y Jia X Zhang AM Wang WZ Li S Guo X et al . The MT-ND1 and MT-ND5 genes are mutational hotspots for Chinese families with clinical features of LHON but lacking the three primary mutations. Biochem Biophys Res Commun. (2010) 399:179–85. doi: 10.1016/j.bbrc.2010.07.051
49.
Zhang AM Jia X Guo X Zhang Q . Yao YG mitochondrial DNA mutation m.10680G > a is associated with Leber hereditary optic neuropathy in Chinese patients. J Transl Med. (2012) 10:43. doi: 10.1186/1479-5876-10-43
50.
Yang J Zhu Y Tong Y Zhang Z Chen L Chen S et al . The novel G10680A mutation is associated with complete penetrance of the LHON/T14484C family. Mitochondrion. (2009) 9:273–8. doi: 10.1016/j.mito.2009.04.003
51.
Qian Y Zhou X Hu Y Tong Y Li R Lu F et al . Clinical evaluation and mitochondrial DNA sequence analysis in three Chinese families with Leber's hereditary optic neuropathy. Biochem Biophys Res Commun. (2005) 332:614–21. doi: 10.1016/j.bbrc.2005.05.003
52.
Zhang S Wang L Hao Y Wang P Hao P Yin K et al . T14484C and T14502C in the mitochondrial ND6 gene are associated with Leber's hereditary optic neuropathy in a Chinese family. Mitochondrion. (2008) 8:205–10. doi: 10.1016/j.mito.2008.02.003
53.
Jha RK Dawar C Hasan Q Pujar A Gupta G Vishnu VY et al . Mitochondrial genetic heterogeneity in Leber's hereditary optic neuropathy: original study with Meta-analysis. Genes. (2021) 12:1300. doi: 10.3390/genes12091300
54.
Moore AT Yu-Wai-Man P . Mitochondrial disorders and the eye: a new era for diagnosis. Ophthalmology. (2021) 128:632–3. doi: 10.1016/j.ophtha.2020.12.032
55.
Castellana S Rónai J Mazza T . MitImpact: an exhaustive collection of pre-computed pathogenicity predictions of human mitochondrial non-synonymous variants. Hum Mutat. (2015) 36:E2413-22. doi: 10.1002/humu.22720
Summary
Keywords
next generation sequencing, homoplasmy, rare variants, bioinformatics analysis, haplogroup analysis
Citation
Srilekha S, Ambika S, Hemavathy N, Vidhya D and Yu-Wai-Man P (2025) Whole mitochondrial genome sequencing in individuals with Leber hereditary optic neuropathy negative for the common pathogenic mitochondrial DNA variants. Front. Neurol. 16:1584748. doi: 10.3389/fneur.2025.1584748
Received
27 February 2025
Accepted
22 July 2025
Published
01 September 2025
Volume
16 - 2025
Edited by
Christos Proukakis, University College London, United Kingdom
Reviewed by
Claudio Fiorini, IRCCS Institute of Neurological Sciences of Bologna (ISNB), Italy
Jan-Willem Taanman, University College London, United Kingdom
Updates
Copyright
© 2025 Srilekha, Ambika, Hemavathy, Vidhya and Yu-Wai-Man.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sundaramurthy Srilekha, srilekhasundar@gmail.com; drsrilekha@snmail.org
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.