Abstract
Objective:
We herein described the clinical characteristics of autoimmune glial fibrillary acidic protein astrocytopathy (GFAP-A) patients with epileptic seizures in the disease course.
Methods:
A single-center retrospective analysis of autoimmune GFAP-A with seizures was conducted.
Results:
There were 14 patients (35.9%, 14/39) with seizures among 39 pediatric GFAP-A patients, nine were boys and five were girls. Nine patients (64.3%, 9/14) manifested focal seizures, four (28.6%, 4/14) showed generalized tonic–clonic seizures, one (7.1%, 1/14) exhibited both forms, and five (35.7%, 5/14) manifested status epilepticus. In addition to seizures, clinical presentations included fever (71.4%), disorders of consciousness (71.4%), dyskinesia (42.9%), psychiatric symptoms (35.7%), headache (28.6%), and involuntary movements (28.6%). Electroencephalograms were all abnormal during the acute phase, principally presenting as focal or diffuse slow waves. During the acute phase, the control rate of epilepsy with immunotherapy was 50%, and seven patients still needed to be treated with antiseizure medication. After 2 years and 6 months to 4 years and 6 months of follow-up, we observed seven patients (50%, 7/14) with recurrence of seizures at 0.5–15 months after discharge, seven patients were treated with one or more antiseizure medications. Epileptic seizures were ultimately controlled in two patients, seizures diminished in one patient, treatment was ineffective in three patients, and one patient died.
Conclusion:
GFAP-A is an important cause of epileptic seizures in children and immunotherapy plays a crucial role. Several patients experienced chronic epileptic seizures after the acute phase and require long-term antiseizure medication, with a few showing refractory characteristics.
1 Introduction
Recent studies have identified immune factor-mediated neuroinflammation as an important etiology of epileptic seizures (1). Autoimmune encephalitis is an immune-mediated inflammatory encephalopathy in which sudden epileptic seizures are common in the acute phase. During the chronic phase, seizures can then develop into persistent autoimmune seizures (2). A meta-analysis of 3,722 antibody-positive patients with autoimmune encephalitis showed that 69.9% of patients experienced seizures during the course of their illness (3). Different types of antineuronal antibodies have been uncovered in patients with epileptic seizures and autoimmune encephalitis over the past decade, including those directed against the N-methyl-D-aspartate receptor (NMDAR), leucine-rich glioma-inactivated 1 (LGI1), and gamma-aminobutyric acid receptor (GABAR) (4), and their pathogenicity of these antineuronal surface-antigen antibodies has been documented (5–7). In recent years anti-glial fibrillary acidic protein (anti-GFAP) antibodies have been associated with autoimmune central nervous system diseases that present with epileptic seizures. The chief clinical presentations of autoimmune GFAP astrocytopathy (GFAP-A) include meningeal, brain parenchymal, or spinal cord inflammation, or a combination of these, with a prevalence rate of 0.6 per 100, 000 (8, 9). The characteristic imaging feature is linear perivascular radial enhancement in the white matter extending radially outward from the ventricles on magnetic resonance imaging (MRI) (10). Detection of GFAP-immunoglobulin G (GFAP-IgG) in cerebrospinal fluid (CSF) through a cell-based assay (CBA) and a tissue-based assay (TBA) is a biomarker of GFAP-A (11). In addition, GFAP-A principally occurs in adults, and only approximately 10% in children (12). Most cases of GFAP-A respond favorably to high-dose corticosteroids. Epileptic seizures constitute a characteristic clinical presentation of GFAP-A, and the incidence of epilepsy in GFAP-A patients is 10–20% (10, 11, 13, 14). The majority of studies only revealed the proportion of epileptic seizures in GFAP-A patients but did not describe the clinical characteristics of seizures in detail. Only two articles provided detailed reports on three patients with GFAP-A-associated epilepsy, including two cases of focal epilepsy and one case of super refractory status epilepticus (15, 16). There are few articles specifically related to the clinical characteristics of GFAP-A patients with seizures. The clinical spectrum, characterization of seizure semiology and data regarding long-term seizure outcomes remain unknown. We conducted a retrospective analysis of pediatric GFAP-A patients with epileptic seizures at our center to further understand the disease’s general, clinical, and imaging characteristics, electroencephalographic changes, and treatment and prognosis.
2 Study participants and methods
2.1 Patient information research methods
Our study participants were 14 patients with epileptic seizures among 39 autoimmune GFAP-A patients treated in the Neurology Department of Hunan Children’s Hospital from January 2015 to April 2024. The inclusion criteria were as follows: ① onset age is less than 18 years; ② patients presenting with meningitis, encephalitis, myelitis, or a combination of the above; ③ positive CSF exhibiting GFAP-IgG; ④ epileptic seizures in the disease course. The exclusion criterion was a definite diagnosis of other diseases. Status epilepticus (SE) is defined as an epileptic seizure continuing beyond a certain time (according to the ILAE 2015 criteria) or recurring within the same timeframe before the patient recovers baseline clinical status (17). This study was approved by the Ethics Committee of the Hunan Children’s Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. All methods were performed in accordance with the relevant guidelines and regulations.
2.2 Research methods
We collected children’s general data (sex, age, prodromal symptoms, initial symptoms, and clinical symptoms), epilepsy-related data (type of seizure, seizure frequency, and changes in disease), auxiliary examinations (laboratory tests, imaging, pathological tests, and neuroelectrophysiologic tests), treatment (drugs used for epilepsy, control results, encephalitis-treatment regimen, and treatment outcomes), and follow-up status. Indirect cellular immunofluorescence was performed to detect central nervous system demyelination antibodies and associated antibodies in the serum and CSF of all enrolled patients.
2.3 Laboratory studies
Testing for GFAP antibodies was conducted by TBA and CBA. For CBA, HEK293 cells were cotransfected with full-length human GFAP and pLV-mCherry-N. After 36 h of transfection in 96 well plate, the cells were fixed with 4% paraformaldehyde for 20 min, washed with PBS and permeabilized with 0.1% Triton X-100 in PBS for 20 min, which was ready for antibody detection. Serum diluted at 1:10 and CSF in PBS-10% goat serum and incubated on cells for 2 h at room temperature. Cells were then washed in PBS-0.1% Tween 20 for three times, incubated for 30 min with goat anti-human IgG (1:500, Thermo Scientific), washed again in PBS-0.1% Tween 20, and evaluated by immunofluorescence microscopy (DMI8, Germany). For TBA, Antibody detection was performed using an indirect immunefluorescence (IF) assay using standard monkey hippocampus and cerebellum tissue. Undiluted CSF sample was allowed to react with tissue sections on glass slides for 3 h at room temperature. Serum was diluted 1:100 before use and reacted with tissue sections on glass slides for 3 h at room temperature. After sample incubation, the slides were rinsed twice with phosphate-buffer saline before being incubated with fluorescein-conjugated goat anti-human IgG for 2 h. Finally, the slides were rinsed with phosphate-buffer and the fluorescence pattern was examined under a microscope. GFAP antibodies were reported as positive if both tests showed concordant results.
2.4 Statistical analysis
We used SPSS 24.0 for all analyses. Normally distributed quantitative data are presented as mean ± standard deviation, while non-normally distributed data are presented as the medians. Qualitative data are presented as the number of patients (percentage).
3 Results
3.1 Clinical presentation
3.1.1 General data
Of the 39 patients with autoimmune GFAP-A, 14 (35.9%, 14/39) exhibited epileptic seizures during the disease course. The mean patient age was 6.43 ± 3.34 years (range: 1 year 4 months to 11 years), comprising nine boys and five girls. Five patients had a history of prodromal upper respiratory tract infection history (the specific pathogen was not determined, one had EB virus infection, and one experienced an intestinal EV-RNA virus infection). One patient was misdiagnosed with tuberculous meningitis during the early phase. This patient was negative for GFAP antibody within 1 week of disease onset, but blood and cerebrospinal fluid GFAP antibodies were positive at reexamination after 1 month and diagnosis was confirmed (Patient 1 in Table 1). Of the 14 patients, nine had meningoencephalitis and four had encephalomyelitis; one patient only had epileptic seizures as the presentation but was diagnosed due to head MRI abnormalities and blood and cerebrospinal fluid positivity for GFAP antibodies (Patient 2 in Table 1).
Table 1
| Patient no. | Sex | Age at onset | Clinical symptoms besides epilepsy | CSF white blood cell count,/L; protein, g/L | Serum antibody titer | CSF antibody titer | MRI findings | Immunomodulatory therapy | ICU admission | mRS at admission/discharge | Response to therapy |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Female | 7y5 m | Fever, headache, disorders of consciousness, bowel or micturition disorder, dyskinesia | wbc:240; p:1.56 | GFAP-IgG (0 → 1:100) | GFAP-IgG (0 → 1:32) | Brain: T2-hyperintense lesions in frontal, temporal, parietal, occipital, basal ganglia, thalamus, periventricular, hippocampus, corpus callosum, brain stem Spine: lesions in T1-L1 (Lesion disappeared after 6 months) |
IVIG, IVMP | Yes | 5/3 | Symptoms improved |
| 2 | Male | 7y1 m | Only epilepsy | wbc:6; p:0.19 | GFAP-IgG (1:50) | GFAP-IgG (1:1) | Brain: T2-hyperintense lesions in right cerebellum Spine: normal |
IVMP | No | 1/0 | Symptoms disappeared |
| 3 | Male | 11y | Memory disorder | wbc:28; p:0.21 | NMDAR-IgG (1:10) MOG-IgG (1:100) |
GFAP-IgG(1:32) NMDAR-IgG(1:1) MOG-IgG(1:100) |
Brain: T2-hyperintense lesions in frontal, temporal, parietal, occipital, optic nerve Spine: normal (Lesion disappeared after 2 months) |
IVIG, IVMP | No | 3/0 | Symptoms disappeared |
| 4 | Male | 1y4 m | Fever, disorders of consciousness, dyskinesia | wbc:50; p:0.6 | Antibody (−) | GFAP-IgG (1:32) | Brain: T2-hyperintense lesions in frontal, temporal, parietal, occipital, cerebellum, corpus callosum, brain stem Spine: NA |
IVMP | Yes | 5/5 | Dead |
| 5 | Female | 5y8m | Fever, headache, psychiatric symptoms | wbc:0; p:0.191 | GFAP-IgG (1:320) | GFAP-IgG (1:32) | Brain: T2-hyperintense lesions in right frontal, left periventricular Spine: normal |
IVIG, IVMP | Yes | 4/1 | Symptoms improved |
| 6 | Male | 3y9m | Fever, disorders of consciousness, peripheral facial palsy, impaired hearing, dyskinesia | wbc:400; p:0.63 | GFAP-IgG (1:32) | GFAP-IgG (1:100) | Brain: T2-hyperintense lesions in frontal, temporal, parietal, occipital, basal ganglia, thalamus, periventricular, corpus callosum Spine: normal |
IVMP | Yes | 5/3 | Symptoms improved |
| 7 | Female | 1y10m | Fever, disorders of consciousness | wbc:233; p:0.46 | GFAP-IgG (1:32) | GFAP-IgG (1:32) | Brain: T2-hyperintense lesions in thalamus, brain stem, cerebellum, left frontal Spine: lesions in C3–C6 (lesion disappeared after 2 months) |
IVIG, IVMP | No | 3/0 | Symptoms disappeared |
| 8 | Male | 6y6m | Fever | wbc:26; p:0.189 | GFAP-IgG (1:32) | GFAP-IgG (1:32) | Brain: normal Spine: normal |
IVMP | No | 3/0 | Symptoms disappeared |
| 9 | Male | 10y9m | Fever, headache, disorders of consciousness, bowel or micturition disorder, dyskinesia, automatic nervous disorder, peripheral motor or sensory nerve damage | wbc:382; p:0.84 | Antibody (−) | GFAP-IgG (1:32) | Brain: T2-hyperintense lesions in basal ganglia, caudate nucleus, thalamus, brain stem, cerebellum Spine: extensive lesions |
IVIG, IVMP, PLEX | Yes | 5/4 | Symptoms improved |
| 10 | Female | 9y3m | disorders of consciousness, psychiatric symptoms, sleep disorder, involuntary movements | wbc:120; p:0.21 | GFAP-IgG (1:32) NMDAR-IgG (1:10) |
GFAP-IgG (1:32) NMDAR-IgG (1:30) |
Brain: normal Spine: NA |
IVIG, IVMP | No | 5/4 | Symptoms improved |
| 11 | Male | 10y6m | Fever, headache, disorders of consciousness, psychiatric symptoms, involuntary movements | wbc:8; p:0.19 | GFAP-IgG (1:100) | GFAP-IgG (1:32) | Brain: T2-hyperintense lesions in parietal, occipital, corpus callosum Spine: NA (lesion disappeared after 2 years and 3 months) |
IVIG, IVMP | Yes | 5/2 | Symptoms disappeared |
| 12 | Male | 1y8m | Fever, disorders of consciousness, dyskinesia, peripheral motor or sensory nerve damage | wbc:80; p:0.53 | GFAP-IgG (1:10) | GFAP-IgG(1:1) | Brain: T2-hyperintense lesions in frontal, temporal, parietal, occipital, thalamus Spine: normal |
IVIG, IVMP | Yes | 5/4 | Symptoms improved |
| 13 | Male | 7y6m | Fever, disorders of consciousness, dyskinesia, psychiatric symptoms, involuntary movements | wbc:2; p:0.05 | GFAP-IgG (1:32) | GFAP-IgG(1:32) | Brain: T2-hyperintense lesions in frontal, temporal, parietal, occipital, periventricular Spine: normal |
IVMP | Yes | 5/5 | Symptoms improved |
| 14 | Female | 5y9m | Fever, disorders of consciousness, psychiatric symptoms, involuntary movements, bulbar palsy | wbc:14; p:0.1 | GFAP-IgG (1:320) | GFAP-IgG (1:1) | Brain: T2-hyperintense lesions in frontal, parietal, occipital Spine: NA |
IVIG, IVMP | Yes | 5/3 | Symptoms improved |
Clinical features, auxiliary examinations, treatment strategies, prognosis in GFAP astrocytopathy patients with seizures.
3.1.2 Epileptic seizure characteristics in initial course of disease
All patients developed epileptic seizures during the initial disease course, of whom seven (50%, 7/14) manifested epileptic seizures as the initial symptom. During hospitalization, nine (64.3%, 9/14) patients exhibited focal motor seizures. Of these, impaired awareness was present in five, awareness was unimpaired in two. Other seizure semiologies included generalized tonic–clonic seizures in four patients (28.6%, 4/14), and one (7.1%, 1/14) manifested both generalized tonic–clonic seizures and focal motor seizures with impaired awareness. During the initial disease course, epileptic seizures occurred only once in two (14.3%, 2/14) patients, twice in one (7.1%, 1/14) patient, and recurrently in the remaining 11 (78.6%, 11/14) patients. The duration of epileptic seizures varied: 11 (78.6%, 11/14) patients experienced epileptic seizures lasting for less than 5 min and three (21.4%, 3/14) patients had epileptic seizures lasting for 5–10 min. Among these patients, five (35.7%, 5/14) exhibited status epilepticus, with the longest lasting 2 h (Table 2).
Table 2
| During admission | Follow up after discharge after discharge | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient no. | Days from symptom onset to first seizure | Type of seizure (seizure frequency) | SE | Interictal EEG | Seizures captured on EEG | ASMs | Total follow-up duration | Time from discharge to recurrent seizures | Type of seizure (seizure frequency) | SE | ASMs | Response to therapy |
| 1 | 1w | Focal (1 episode) | No | Slow waves in the right brain region +spike wave | No | No | 3y8m | 15 m | Focal (1–2 episodes/m) | No | LCS, PER | Failure |
| 2 | 1d | Focal (1episode/2-7d) | No | Spike wave | No | OXC | 2y1 m | 0.5 m | Focal (4–5 episodes/ m) |
No | OXC | Failure |
| 3 | 1d | Focal (1–4 episodes/d) | No | Slow waves in the right parietal and temporal regions | No | No | 3y1 m | No seizure | / | / | No | / |
| 4 | 2d | GTCS (several episodes/4 d) | Yes | Severe and widespread low voltage | No | LEV | 4y1 m | 1 m | Focal (several episodes/w) | No | LEV | Dead |
| 5 | 1d | Focal (1–4 episodes/ d) | No | Slow waves in the left central, parietal, and occipital regions | Three focal seizures | LEV, LCS | 1y8m | 4 m | Focal (9 episodes/4 m) | No | LCS, LEV | Seizure-free |
| 6 | 2 W | Focal (2 episodes) | No | Diffuse slow waves | No | No | 3y4 m | No seizure | / | / | No | / |
| 7 | 1d | Focal (1 episode) | No | Diffuse slow waves | No | No | 3y | No seizure | / | / | No | / |
| 8 | 1d | GTCS+Focal (1–3 episodes/ d) | Yes | Slow waves in the right temporal region | One BIRDs, one electrical seizures | OXC | 1y4 m | 2 m | Focal (3 episodes/3d) | No | OXC + IVMP | Seizure-free |
| 9 | 1 W | GTCS (several episodes/ w) | Yes | Diffuse slow waves | No | No | 2y6m | No seizure | / | / | No | / |
| 10 | 1d | Focal (2–4 episodes/ d) | Yes | Slow waves in the left brain region | No | No | 4y | no seizure | / | / | No | / |
| 11 | 4d | Focal (1–4 episodes/1–2 d) | No | Diffuse slow wave | Five electrical seizures | LEV, CZP | 3y11 m | 0.5 m | Focal (several episodes/m) | No | LEV+OXC + VPA + IVMP | Failure |
| 12 | 5d | GTCS(3 episodes) | No | Diffuse slow wave | No | No | 4y6m | No seizure | / | / | No | / |
| 13 | 1d | GTCS (1–7 episodes/d) | Yes | Diffuse slow wave+spike wave | No | LEV | 3y4 m | No seizure | / | / | LEV | / |
| 14 | 4d | Focal (1–3 episodes/d) | No | Diffuse slow waves+sharp waves in the posterior brain region | One NCSE | LEV, VPA | 2y | 5 m | Focal (1 episode/ several months) | No | VPA + PER | Reduction |
Seizures characteristics, EEG findings, antiseizure medications strategies, prognosis in GFAP astrocytopathy patients with seizures.
CSF, cerebrospinal fluid; EEG, electroencephalography; MRI, magnetic resonance imaging; mRS, modified Rankin Scale; IVIG, intravenous immunoglobulin; IVMP, intravenous methylprednisolone; PLEX, Plasma exchange; MOG, myelin oligodendrocyte glycoprotein; NMDAR, N-methyl-d-aspartate receptor; NA, no available; ASMs, antiseizure medications; SE, status epilepticus; GTCS, generalized tonic–clonic seizure; BIRD, brief potentially ictal rhythmic discharge; NCSE, non-convulsive status epilepticus; VPA, valproate; LEV, levetiracetam; OXC, oxcarbazepine; LCS, lacosamide; CZP, clonazepam; y, year; m, month; d, day; WBC, white blood cell; P, protein.
3.1.3 Clinical presentations other than epileptic seizures
In addition to epileptic seizures, other clinical symptoms during the course of the disease included fever (n = 10), disorders of consciousness (n = 10), dyskinesia (n = 6), psychiatric symptoms (n = 5), headache (n = 4), involuntary movements (n = 4), peripheral motor or sensory nerve damage (n = 2), bowel or micturition disorder (n = 2), bulbar palsy (n = 1), memory disorder (n = 1), sleep disorder (n = 1), peripheral facial palsy (n = 1), automatic nervous disorder (n = 1), and impaired hearing (n = 1). Nine of these children were admitted to the ICU due to severe condition and five received ventilator support due to respiratory failure. Twelve patients developed complications, including seven patients with pneumonia (two with severe pneumonia), five with electrolyte disturbance, four with impaired hepatic function, two with intracranial hypertension, two with severe sepsis, two with myocardial injury, one with cerebral hemorrhage, and one with urinary tract infection. Ten (71.4%) patients had a modified Rankin scale (mRS) score of 4–5 points and four patients (28.6%) had an mRS score of 1–3 points at disease peak; the median mRS score was 5 (3, 5) points (Table 3).
Table 3
| Items | Incidence |
|---|---|
| Feature | |
| Males:females | 9:5 |
| Mean age (y) | 6.43 ± 3.34 |
| Intensive care unit | 9/14 (64.3%) |
| Main symptoms | |
| Fever | 10/14 (71.4%) |
| Disorders of consciousness | 10/14 (71.4%) |
| Dyskinesia | 6/14 (42.9%) |
| Psychiatric symptoms | 5/14 (35.7%) |
| Headache | 4/14 (28.6%) |
| Involuntary movements | 4/14 (28.6%) |
| Peripheral motor or sensory nerve damage | 2/14 (14.3%) |
| Bowel or micturition disorder | 2/14 (14.3%) |
| Seizure characteristics during the acute phase | |
| Focal seizures | 9/14 (64.3%) |
| GTCS | 4/14 (28.6%) |
| Both GTCS and focal seizures | 1/14 (7.1%) |
| SE | 5/14 (35.7%) |
| Epileptic seizures as the initial symptom | 7/14 (50.0%) |
| EEG findings | |
| Abnormal | 14/14 (100.0%) |
| Severe low voltage | 1/14 (7.1%) |
| Focal slow waves | 5/14 (35.7%) |
| Diffuse slow wave | 7/14 (50.0%) |
| Epileptiform discharge | 4/14 (28.6%) |
| Neuroimaging | |
| Brain | 12/14 (85.7%) |
| Spinal cord | 3/14 (21.4%) |
| Optic nerve | 1/14 (7.1%) |
| Enhancement | 8/14 (57.1%) |
| CSF abnormality | |
| Elevated protein | 5/14 (35.7%) |
| Elevated leukocyte | 9/14 (64.3%) |
| Multiple antibodies positive | 2/14 (14.3%) |
| Immunotherapy during the acute phase | |
| IVMP | 5/14 (35.7%) |
| IVMP+IVIG | 8/14 (57.1%) |
| IVMP+IVIG+PLEX | 1/14 (7.1%) |
| Recurrent seizure characteristics after discharge | |
| Patients of recurrent seizures | 7/14 (50.0%) |
| Time from discharge to recurrent seizures | 0.5–15 m |
| focal seizures | 7/7 (100.0%) |
| Seizure freedom achieved | 2/7 (28.6%) |
| seizure reduction | 1/7 (14.3%) |
| Seizure failure | 3/7 (42.9%) |
| Dead | 1/7 (14.3%) |
Summary of clinical presentation and associated conditions in GFAP astrocytopathy patients with seizures.
3.2 Laboratory tests
Routine cerebrospinal fluid biochemical tests were completed in the acute phase in all 14 pediatric patients, with nine showing elevated cerebrospinal fluid white blood cell count (normal values, 0–20 × 106/L; range, 26–400 × 106/L). Among the patients, three, one, and five had white blood cell count of 20–50 × 106/L, 50–100 × 106/L, and >100 × 106/L, respectively. Five patients exhibited elevated cerebrospinal fluid protein levels, ranging from 0.53 to 1.56 g/L. Cytologic test results were principally monocyte and lymphocyte elevations, and neutrophils and activated monocytes were observed in several patients. Eleven of the 14 patients tested positive for both blood and cerebrospinal fluid GFAP antibodies, while three patients were only positive for antibodies in the cerebrospinal fluid (Figure 1). One child’s fluid reflected overlapping NMDA antibody and one showed both overlapping NMDA antibody and myelin oligodendrocyte glycoprotein (MOG) antibody.
Figure 1

Detection of anti-GFAP antibody by CBA (A1–F1) and TBA (A2–D2) in patient 14. (A1–F1) GFAP-IgG were tested by a CBA using HEK293 cells transiently cotransfected with full-length human GFAP and pLV-mCherry-N. The patient’s IgG bound to GFAP-transfected cells and showed green fluorescence as a positive control. (A1–C1) GFAP-IgG in serum were tested. (D1–F1) GFAP-IgG in CSF were tested. (A2–D2) TBA was performed with an indirect immunofluorescence assay using standard monkey hippocampus and cerebellum tissue. (A2–B2) Serum specific IgG binds to the white matter astrocytes in monkey cerebellar tissue, consistent with GFAP distribution. (C2–D2) CSF specific IgG binds to the white matter astrocytes in monkey cerebellar tissue, consistent with GFAP distribution.
3.3 Electroencephalogram
All patients underwent at least one video electroencephalography during the initial disease onset, and the monitoring duration was 4–15 h. All patients had abnormal electroencephalographic results: one with severe low voltage, one with epileptiform discharge but no slowing, five with focal slow waves involving one to multiple brain regions and with epileptiform discharge simultaneously detected in one patient, and seven with diffuse slow waves, of which two showed epileptiform discharge. All study subjects had epileptic seizures, but epileptiform discharge was only detected in four patients. Epileptiform discharge involved multiple brain regions, of which the temporal region (n = 4), center (n = 2), parietal region (n = 2), and frontal region (n = 1) were common. We detected three focal seizures, six electrical seizures, one brief potentially ictal rhythmic discharge (BIRD), and one episode of non-convulsive status epilepticus (NCSE) (Figure 2) using video electroencephalography of four patients. Focal or electrical seizures primarily originate in the central, parietal, temporal, and occipital regions. The initial electroencephalographic pattern was mainly spike waves or sharp waves, and seizures lasted for 40–115 s. Four patients demonstrated suspected epileptiform activity, which was ruled out using synchronized video electroencephalography. Abnormal activity primarily presented as positive bilateral eye rolling, frequent bilateral upward gazing, paroxysmal bilateral dazed eyes, right lower limb convulsion, upper limb lifting, forceful limb movements, strabismus, or blinking, waveforms were not detected in the seizure phase.
Figure 2
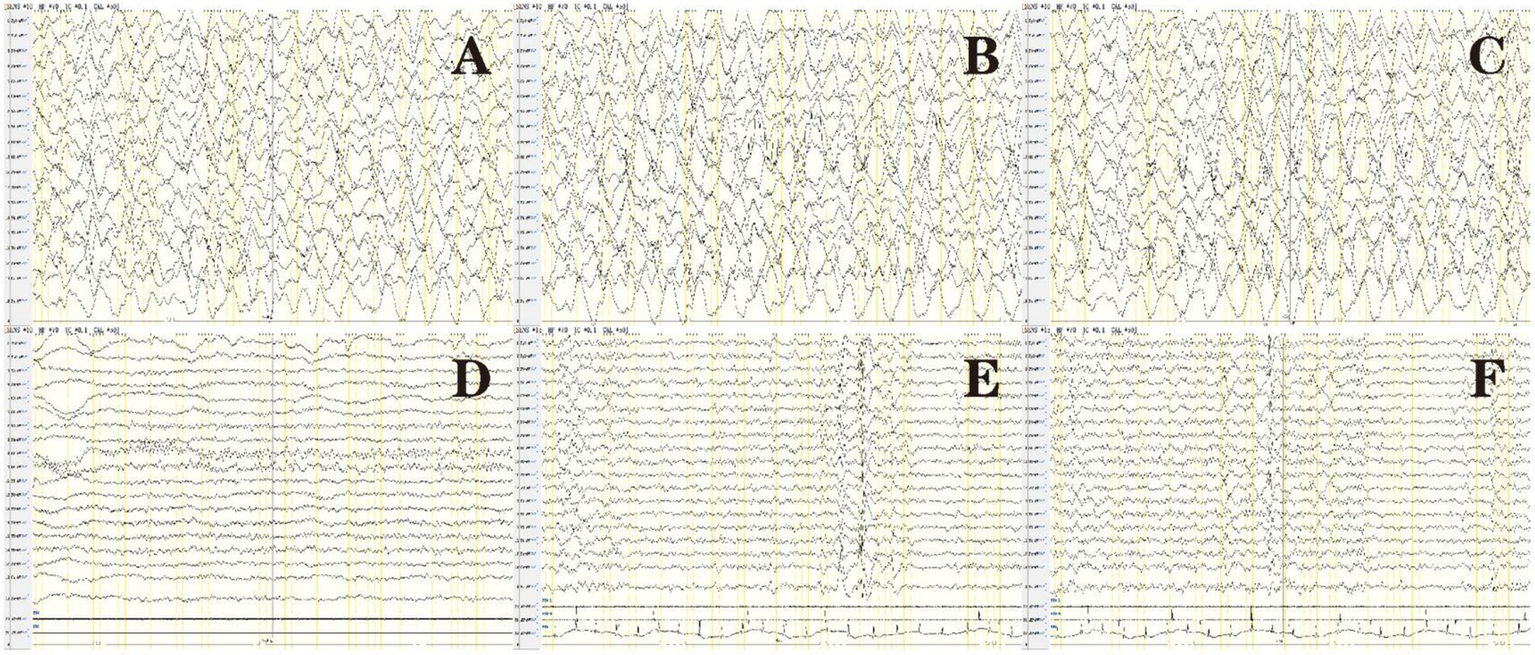
Electroencephalogram of patient 14 in Table 1: before treatment (A–C), after treatment (D), and after seizure recurrence (E,F). (A) Background diffuse slow waves. (B) Spike waves were primarily present in the bilateral occipital and posterior temporal regions. (C) Eye opening in a patient, poor response to external stimuli, and absence of convulsions. Synchronized electroencephalography showed diffuse high-extremely high wave amplitude in various brain regions and the absence of intermittent discharge at the 1–1.5 Hz δ area, indicating NCSE. (D) Normal electroencephalography after treatment. (E,F) After epileptic seizures recurred, electroencephalograms showed right frontal pole, frontal, and anterior temporal spike waves, and several discharges of spike-slow waves were observed during sleep.
3.4 Imaging
A head MRI scan was completed in 14 patients and a spinal cord MRI scan was completed in 10 patients. Eight of these scans showed abnormalities on head MRI, three reflected head and spinal cord MRI abnormalities, and one manifested head and optic nerve MRI abnormalities. Eight patients also exhibited enhanced shadows. The heads of the children chiefly showed cortical and subcortical white matter involvement, with 1–10 sites of involvement. Bilateral lesions were the most common (n = 8) and all were asymmetric, numbering twice that of the unilateral lesions (n = 4). The involved sites included the frontal lobe (n = 9), parietal lobe (n = 8), occipital lobe (n = 8), temporal lobe (n = 6), thalamus (n = 5), periventricular white matter (n = 4), cerebellum (n = 4), brain stem (n = 3), basal ganglia (n = 3), corpus callosum (n = 3), caudate nucleus (n = 1) and hippocampus (n = 1). Only three patients presented with spinal cord lesions: one comprised thoracic-lumbar spinal cord T1-L1, one involved the cervical spinal cord C3-C6, and one encompassed extensive spinal cord lesions. Lesions diminished or disappeared after treatment in the 11 patients with imaging abnormalities, and four children had complete disappearance of the lesion on MRI reexamination, occurring 2 months to 2 years and 3 months after disease onset (Figures 3, 4).
Figure 3

Brain MRI of patient 14 in Table 1: upon admission (A–D) and 1 year later (E–H). (A) T2 image showed widening of the cerebral sulcus. (B) T2-hyperintense lesions in the white matter of the frontal lobe (red arrow) and parietal lobe (blue arrow). (C) T2-hyperintense lesions in the white matter of the occipital lobe (yellow arrow). (D) Enhancement of T2 flair showed small strip like high signal shadows in the sulci of the brain. (E–H) Follow-up images of improved T2 lesions and enhancement.
Figure 4
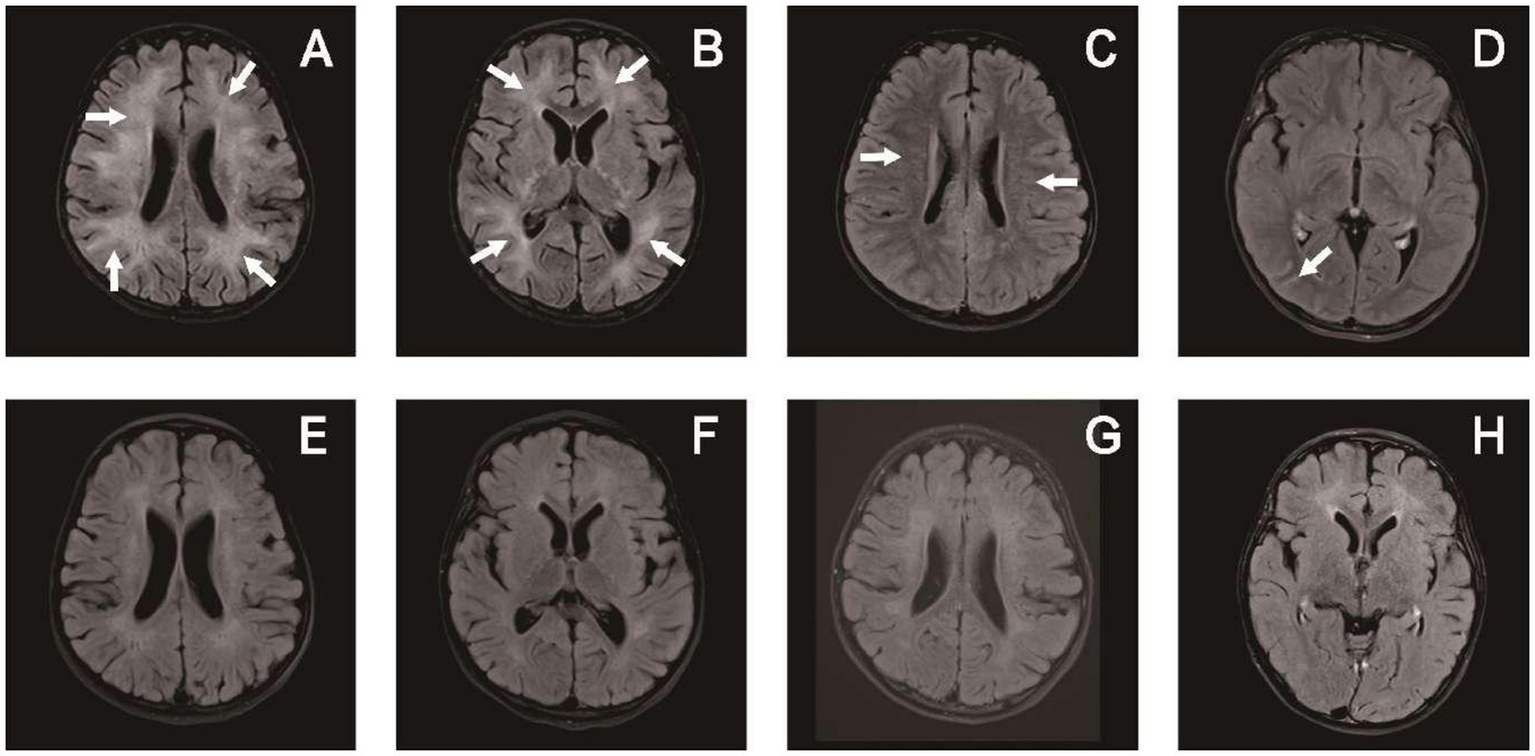
Brain MRI of patient 6 in Table 1: upon admission (A–D) and 3 months later (E–H). (A,B) T2-hyperintense lesions in the white matter of bilateral cerebral hemispheres. (C) Periventricular radial linear enhancement. (D) Enhanced shadow of occipital lobe sulci. (E–H) Follow-up images of improved T2 lesions and enhancement.
3.5 Treatment protocol
3.5.1 Immunotherapy
Five patients underwent methylprednisolone pulse therapy (10–20 mg/kg, 3–5 days), and nine patients received methylprednisolone pulse therapy and intravenous immunoglobulin (IVIG), one of whom underwent plasmapheresis before IVIG. All patients received oral prednisone for maintenance therapy.
3.5.2 Antiseizure medication
During their first hospitalization, 14 patients were treated with immunotherapy, and seven of them had acute seizure control. The control rate of epilepsy with immunotherapy was 50%, and seven patients still needed to be treated with antiseizure medication, four of whom received monotherapy and three received combination therapy. Monotherapy comprised oxcarbazepine (n = 2) and levetiracetam (LEV, n = 2), and combination therapy comprised LEV + lacosamide (LCM, n = 1), LEV + clonazepam (CZP, n = 1), and LEV + sodium valproate (VPA, n = 1). Five patients received diazepam and midazolam as temporary antispasmodic treatments due to frequent convulsions or status epilepticus.
3.6 Determination of response and follow-up
After discharge, six of the 14 patients achieved favorable outcomes, while eight (57.1%) had poor outcomes, including residual cognitive disability, movement disorder, and convulsions. Post-discharge outpatient or telephone follow-up was conducted and the mean follow-up duration was 3.04 ± 0.98 years. After discharge, seven patients did not develop epileptic seizures after 6 months to 4 years and 6 months of follow-up, and the mean follow-up duration was 2.68 ± 1.16 years. The remaining seven patients (50.0%) developed epileptic seizures that occurred 0.5–15 months after discharge, with a median duration of 2 (0.5, 5) months. While these seven patients all showed focal seizures after discharge, they did not develop status epilepticus. All pediatric patients who developed convulsions after discharge received antiseizure medications: two patients received monotherapy and five received combination therapy, of whom two underwent combined pulse steroid therapy. Of the seven patients, epileptic seizures were controlled in two patients (including one patient receiving repeated immunotherapy) and seizures did not recur after more than 1 year, one patient experienced a reduction in seizures, three patients (including one patient receiving repeated immunotherapy) showed low antiseizure medication effectiveness and still had several seizures each month, and the remaining patient died owing to recurrent convulsions and a secondary lung infection after discharge.
4 Discussion
Autoimmune GFAP-A is a relatively new autoimmune disease of the central nervous system that was first named by Fang et al. (18) from the Mayo Clinic, USA, in 2016; the authors reported that an IgG that specifically targeted GFAP was present in experimental animals and patients with this disease. The chief clinical presentation of autoimmune GFAP astrocytopathy (GFAP-A) includes meningeal, brain parenchymal, or spinal cord inflammation, or a combination of these. Although studies have shown that epileptic seizure are a clinical presentation of GFAP-A (19), only two small-scale case series (a total of three cases) have provided detailed descriptions of epileptic seizures in GFAP-A (15, 16). Most studies have only revealed the proportion of patients with GFAP-A who experienced seizures, without detailing the clinical features of seizures. Therefore, little is known about the characteristics of epileptic seizures in GFAP-A, especially in pediatric patients. In our study, we reported 14 patients (35.9%) who presented with seizures among 39 pediatric patients with GFAP-A. To the best of our knowledge, this is the largest clinical study of GFAP-A patients with seizures.
In the present study, 35.9% of GFAP-A patients developed epileptic seizures during the disease course (with significantly more males than females), and our proportion of patients was significantly higher than that of pediatric and adult GFAP-A patients with epileptic seizures (10–20%), as reported in the literature (10, 11, 13, 14). With the exception of one patient with a clinical presentation of epileptic seizures alone, the other patients presented with encephalopathy, primarily meningoencephalitis and encephalomyelitis. GFAP-A exhibits diverse types of epileptic seizures. During the acute phase, it mainly presents as focal seizures, followed by generalized tonic–clonic seizures; however, after the acute phase, 50.0% of patients redevelop epileptic seizures that present as focal seizures. Most studies have revealed that epileptic seizures are present in patients with GFAP-A, but these did not describe the type of seizure in detail. These investigators ascertained that five patients (35.7%, 5/14) developed status epilepticus in the early phase of the disease, which was generally consistent with the proportion (6–40%) of NMDAR encephalitis patients with status epilepticus (20). Table 2 shows that among five patients with status epilepticus during the early phase of the disease, only two (40%, 2/5) experienced epileptic seizures during the post-discharge follow-up, and five of nine patients without status epilepticus (55.6%, 5/9) developed epileptic seizures again during follow-up. This finding supports the concept that status epilepticus in the early phase of the disease cannot be used as an indicator of subsequent autoimmune encephalitis-related epilepsy. In addition to epileptic seizures, the other clinical presentations in our 14 patients were fever, disorders of consciousness, dyskinesia, psychiatric symptoms, headache, and involuntary movements, which are generally consistent with previous reports (21). During the peak of disease, 10 of our patients (71.4%) had an mRS score of 4–5 points, nine patients (64.3%) were admitted to the ICU due to a severe condition, and five (35.7%) patients were provided ventilator support due to respiratory failure; these rates were all higher than the mean levels for GFAP-A patients (21, 22). This indicates that epileptic seizures in patients with GFAP-A may be associated with initial disease severity.
In our study, 12 of 14 patients showed imaging abnormalities of the head and two had normal imaging presentations. During follow-up, one of these two patients with a normal head MRI developed epileptic seizures again, which were ultimately controlled after antiseizure medication. Of the remaining 12 patients with head MRI abnormalities, head lesions on MRI completely disappeared 2 months to 2 years and 3 months after the disease onset. Of these four patients, two had epileptic seizures again during follow-up and antiseizure medication effectiveness was low in these patients. This shows that epileptic seizures caused by imaging abnormalities of the head are associated with poor outcome. We asked, ‘What is the pathogenesis of epilepsy in patients with a normal imaging presentation?’ Studies have shown that astrocyte activation increases the risk of epilepsy and astrocyte proliferation and that increased astrocyte GFAP expression is associated with the severity of epilepsy (23, 24). These microscopic astrocyte changes may explain why epilepsy occurs in GFAP-A with a normal imaging presentation.
In the present analysis, 100% of pediatric patients showed abnormal electroencephalograms, which was higher than the head MRI abnormality rate and mainly presented as diffuse or focal slow waves. Epileptiform discharge was detected in several patients, and this was for the most part consistent with the majority of immune encephalitis cases (25, 26). Herein, we noted only one patient with extensive severe low voltage during the early phase of the disease. Although electroencephalography findings improved after treatment, the patient died due to recurrent convulsions and secondary lung infection after discharge. Thus, a low voltage in electroencephalograms may be associated with a poor prognosis. Involuntary movements are also present in patients with GFAP-A, and it is sometimes difficult to distinguish these from epileptic seizures based on clinical presentation alone. In the current study, we determined the clinical and subclinical seizures in four patients. Thus, of the 14 patients, four had suspected epileptiform activity that was finally ruled out through synchronized video electroencephalography. We recognize that electroencephalography plays an important role in distinguishing epileptic events from non-epileptic events in patients with GFAP-A, and that subclinical electrical seizures can be detected in an effort to guide treatment.
Two of our patients exhibited overlap syndrome, one manifesting overlapping NMDA antibodies and one with both overlapping NMDA and MOG antibodies. A recent meta-analysis revealed that the probability of acute epileptic seizures in patients with MOG antibody disease is generally 20.5% (27). Another analysis showed that approximately 70% of anti-NMDAR encephalitis patients developed epileptic seizures (28, 29). We then asked, ‘What epileptogenic mechanisms are present in GFAP overlap-syndrome patients, and which antibodies reflect a dominant role?’ However, the specific mechanisms underlying these effects remain unclear. One study showed that the simultaneous presence of GFAP antibody and other well-characterized antibodies (such as NMDAR and MOG) in autoimmune overlap syndrome was due to elevated astrocyte activation or destruction, exposing the GFAP antigen and increasing GFAP immune responses. Thus, the production of GFAP antibodies may be critical for the pathogenicity of GFAP-A (30, 31). Another study speculated that immune reconstitution during the tapering of immunosuppressive drugs might activate production of new autoantibodies; therefore, special attention should be paid to slow tapering of steroids (32). Overlapping autoantibodies are common in GFAP astrocytopathy, involving MOG-IgG, NMDAR-IgG, or other neuronal antibodies. The exact difference between GFAP-A patients with overlapping and non overlapping syndromes is still unclear. The system screening patients with GFAP-A for other antibodies is helpful in understanding patient’s condition.
In this study, all patients received pulse steroid therapy: nine patients experienced combined gamma globulin therapy, one patient underwent additional plasmapheresis, and several patients received antiseizure medication. There are disparities in therapeutic strategies for GFAP-A. Currently, there is no treatment standard or consensus, and it is still unclear whether long-term antiseizure medication is required for epileptic seizures. Some investigators have indicated good responses to corticosteroids, and the majority of their patients showed clinical improvement after immunotherapy, including decreased acute epileptic seizures (30, 33). Therefore, acute long-term antiseizure medication is not recommended. In the present study, seven patients developed epileptic seizures again during the post-discharge follow-up, and these patients received long-term antiseizure medication after epileptic seizures recurred. Only two patients (patients 8 and 11) underwent repeated combined pulse steroid therapy. Patient 8 received steroids combined with antiseizure medication after convulsions, with no convulsions occurring 1 year after treatment. However, patient 11 still manifested recurrent convulsions after administration of steroids combined with antiseizure medication. The limitations of the current study include the difficulty in determining the prophylactic effects of immunosuppressants combined with antiseizure medication in patients who develop epileptic seizures after their condition has stabilized. Sriram et al. (16) reported a female patient diagnosed with GFAP-A-associated super-refractory status epilepticus, which was initiated on intravenous methylprednisolone (IVMP) but showed no improvement. She subsequently underwent plasma exchange (PLEX) and showed a reduction in number. She was administered intravenous immunoglobulin (IVIG), bortezomib, rituximab, and tocilizumab. She showed gradual yet significant improvement and was ultimately seizure-free. Therefore, it is crucial to initiate early and aggressive immunotherapy with multiple agents that target various parts of the autoimmune pathway to successfully manage the disease. The sample size of this study was small, and the follow-up duration of some patients was short. Therefore, a cohort study with a larger sample size and longer duration is required to explore this area further.
In summary, epileptic seizures were common in pediatric patients with GFAP-A, with most male patients, and the most common type of seizure was focal in our study. Head MRI chiefly showed cortical and subcortical white matter involvement, and electroencephalograms primarily exhibited focal and diffuse slow waves. Acute glucocorticoid and/or immunoglobulin treatment can also be used to control the disease. Several of our patients demonstrated secondary epileptic seizures after the acute phase and required long-term antiseizure medication or immunotherapy, while several patients had refractory disease. We posit that electroencephalography and imaging are helpful for patients with early acute epileptic seizures or isolated epileptic seizures without any cause and recommend that GFAP antibody testing be carried out as soon as possible to determine the diagnosis.
Statements
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by the Ethics Committee of Hunan Children’s Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
HF: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Writing – original draft, Writing – review & editing. WH: Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – review & editing. ZJ: Data curation, Formal analysis, Methodology, Project administration, Software, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by grants from the Natural Science Foundation of Hunan Province (No. 2022JJ70087).
Acknowledgments
We appreciate the patients’ participation in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
Scheffer IE Berkovic S Capovilla G Connolly MB French J Guilhoto L et al . ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. (2017) 58:512–21. doi: 10.1111/epi.13709
2.
Steriade C Britton J Dale RC Gadoth A Irani SR Linnoila J et al . Acute symptomatic seizures secondary to autoimmune encephalitis and autoimmune-associated epilepsy: conceptual definitions. Epilepsia. (2020) 61:1341–51. doi: 10.1111/epi.16571
3.
Yeshokumar AK Coughlin A Fastman J Psaila K Harmon M Randell T et al . Seizures in autoimmune encephalitis-a systematic review and quantitative synthesis. Epilepsia. (2021) 62:397–407. doi: 10.1111/epi.16807
4.
Spatola M Dalmau J . Seizures and risk of epilepsy in autoimmune and other infammatory encephalitis. Curr Opin Neurol. (2017) 30:345–53. doi: 10.1097/WCO.0000000000000449
5.
Vanli-Yavuz EN Erdag E Tuzun E Ekizoglu E Baysal-Kirac L Ulusoy C et al . Neuronal autoantibodies in mesial temporal lobe epilepsy with hippocampal sclerosis. J Neurol Neurosurg Psychiatry. (2016) 87:684–92. doi: 10.1136/jnnp-2016-313146
6.
Baysal-Kirac L Tuzun E Erdag E Ulusoy C Vanli-Yavuz EN Ekizoglu E et al . Neuronal autoantibodies in epilepsy patients with peri-ictal autonomic findings. J Neurol. (2016) 263:455–66. doi: 10.1007/s00415-015-8002-2
7.
Masdeu JC Dalmau J Berman KF . NMDA receptor internalization by autoantibodies: a reversible mechanism underlying psychosis?Trends Neurosci. (2016) 39:300–10. doi: 10.1016/j.tins.2016.02.006
8.
Kunchok A Zekeridou A McKeon A . Autoimmune glial fbrillary acidic protein astrocytopathy. Curr Opin Neurol. (2019) 32:452–8. doi: 10.1097/WCO.0000000000000676
9.
Long Y Liang J Xu H Huang Q Yang J Gao C et al . Autoimmune glial fibrillary acidic protein astrocytopathy in Chinese patients: a retrospective study. Eur J Neurol. (2018) 25:477–83. doi: 10.1111/ene.13531
10.
Ke G Jian S Yang T Zhao X . Clinical characteristics and MRI features of autoimmune glial fibrillary acidic protein astrocytopathy: a case series of 34 patients. Front Neurol. (2024) 15:1375971. doi: 10.3389/fneur.2024.1375971
11.
Gklinos P Athanasopoulos F Giatrakou V Arkoudis N-A Pournara D Giagkou E et al . Unveiling GFAP astrocytopathy: insights from case studies and a comprehensive review of the literature. Antibodies (Basel). (2024) 13:79. doi: 10.3390/antib13040079
12.
Cheng W He L Luo H Jiang Y Tan C Fan X . Magnetic resonance imaging characteristics of autoimmune glial fibrillary acidic protein (GFAP) astrocytopathy: a pediatric series in Southwest China. Neuropsychiatr Dis Treat. (2023) 19:1685–93. doi: 10.2147/NDT.S417492
13.
Dubey D Hinson SR Jolliffe EA Zekeridou A Flanagan EP Pittock SJ et al . Autoimmune GFAP astrocytopathy: prospective evaluation of 90 patients in 1 year. J Neuroimmunol. (2018) 321:157–63. doi: 10.1016/j.jneuroim.2018.04.016
14.
Hagbohm C Ouellette R Flanagan EP Jonsson DI Piehl F Banwell B et al . Clinical and neuroimaging phenotypes of autoimmune glial fibrillary acidic protein astrocytopathy: a systematic review and meta-analysis. Eur J Neurol. (2024) 31:e16284. doi: 10.1111/ene.16284
15.
Savaş M Tzartos J Küçükali Cİ Dursun E Karagiorgou K Gezen-Ak D et al . Glial fbrillary acidic protein (GFAP)-antibody in children with focal seizures of undetermined cause. Acta Neurol Belg. (2021) 121:1275–80. doi: 10.1007/s13760-020-01361-y
16.
Sriram M Shivarthi T Narayanan S Rohan P Nikhilesh M Kannoth S et al . Super refractory status epilepticus secondary to autoimmune glial fibrillary acidic protein astrocytopathy. Epileptic Disord. (2025) 1–4. doi: 10.1002/epd2.70001
17.
Trinka E Cock H Hesdorffer D Rossetti AO Scheffer IE Shinnar S et al . A definition and classification of status epilepticus-report of the ILAE task force on classification of status epilepticus. Epilepsia. (2015) 56:1515–23. doi: 10.1111/epi.13121
18.
Fang B McKeon A Hinson SR Kryzer TJ Pittock SJ Aksamit AJ et al . Autoimmune glial fibrillary acidic protein astrocytopathy: a novel meningoencephalomyelitis. JAMA Neurol. (2016) 73:1297–307. doi: 10.1001/jamaneurol.2016.2549
19.
Iorio R Damato V Evoli A Gessi M Gaudino S Di Lazzaro V et al . Clinical and immunological characteristics of the spectrum of GFAP autoimmunity: a case series of 22 patients. J Neurol Neurosurg Psychiatry. (2018) 89:138–46. doi: 10.1136/jnnp-2017-316583
20.
Vogrig A Joubert B André-Obadia N Gigli GL Rheims S Honnorat J . Seizure specificities in patients with antibody-mediated autoimmune encephalitis. Epilepsia. (2019) 60:1508–25. doi: 10.1111/epi.16282
21.
Zhang W Xie Y Wang Y Liu F Wang L Lian Y et al . Clinical characteristics and prognostic factors for shortterm outcomes of autoimmune glial fibrillary acidic protein astrocytopathy: a retrospective analysis of 33 patients. Front Immunol. (2023) 14:1136955. doi: 10.3389/fimmu.2023.1136955
22.
Du J Cao S Xia L Li Q Tian Y . Plasma exchange for two patients with autoimmune GFAP astrocytopathy with rapid progression to respiratory failure: a case report. Front Immunol. (2023) 14:1265609. doi: 10.3389/fimmu.2023.1265609
23.
Li D Li P He Z Cen D Meng Z Liang L et al . Human intravenous immunoglobulins suppress seizure activities and inhibit the activation of GFAP-positive astrocytes in the hippocampus of picrotoxin-kindled rats. Int J Neurosci. (2012) 122:200–8. doi: 10.3109/00207454.2011.639470
24.
Simani L Elmi M Asadollahi M . Serum GFAP level: a novel adjunctive diagnostic test in diferentiate epileptic seizures from psychogenic attacks. Seizure. (2018) 61:41–4. doi: 10.1016/j.seizure.2018.07.010
25.
Zhang Y Liu G Jiang MD Li LP Su YY . Analysis of electroencephalogram characteristics of anti-NMDA receptor encephalitis patients in China. Clin Neurophysiol. (2017) 128:1227–33. doi: 10.1016/j.clinph.2017.04.015
26.
Chusak L Denlertchaikul C Saraya AW Jirasakuldej S . Predictive values and specificity of electroencephalographic findings in autoimmune encephalitis diagnosis. Epilepsy Behav. (2018) 84:29–36. doi: 10.1016/j.yebeh.2018.04.007
27.
Shen C-H Zheng Y Cai M-T Yang F Fang W Zhang Y-X et al . Seizure occurrence in myelin oligodendrocyte glycoprotein antibody-associated disease: a systematic review and meta-analysis. Mult Scler Relat Disord. (2020) 42:102057. doi: 10.1016/j.msard.2020.102057
28.
Gaspard N Foreman BP Alvarez V Cabrera Kang C Probasco JC Jongeling AC et al . New-onset refractory status epilepticus: etiology, clinical features, and outcome. Neurology. (2015) 85:1604–13. doi: 10.1212/WNL.0000000000001940
29.
Xu L Yan B Wang R Li C Chen C Zhou D et al . Seizure outcomes in patients with anti-NMDAR encephalitis: a follow-up study. Epilepsia. (2017) 58:2104–11. doi: 10.1111/epi.13929
30.
Yang X Xu H Ding M Huang Q Chen B Yang H et al . Overlapping autoimmune syndromes in patients with glial fibrillary acidic protein antibodies. Front Neurol. (2018) 9:251. doi: 10.3389/fneur.2018.00251
31.
Papantoniou M Panagou G Kanavouras K . Clinical, lab, and radiological evolution of an adult patient with unilateral cortical lesion in anti-myelin oligodendrocyte glycoprotein (MOG)-associated encephalitis with seizures and anti-glial fibrillary acidic protein (GFAP) positive antibodies. Cureus. (2024) 16:e70546. doi: 10.7759/cureus.70546
32.
Ding J Ren K Wu J Li H Sun T Yan Y et al . Overlapping syndrome of MOG-IgG-associated disease and autoimmune GFAP astrocytopathy. J Neurol. (2020) 267:2589–93. doi: 10.1007/s00415-020-09869-2
33.
Liu Q Yang X Bao JZ Ma B Niu X Wang X et al . Clinical characteristics of patient with GFAP-IgG: a review of 31 patients from two tertiary referral centers in China. Int J Neurosci. (2023) 134:1383–94. doi: 10.1080/00207454.2023.2277664
Summary
Keywords
autoimmune glial fibrillary acidic protein astrocytopathy, GFAP-IgG antibodies, seizure, children, GFAP-A
Citation
Fang H, Hu W and Jiang Z (2025) A single-center retrospective analysis of autoimmune glial fibrillary acidic protein astrocytopathy with seizures in children. Front. Neurol. 16:1591835. doi: 10.3389/fneur.2025.1591835
Received
13 March 2025
Accepted
19 May 2025
Published
05 June 2025
Volume
16 - 2025
Edited by
Jinming Han, Capital Medical University, China
Reviewed by
Dominica Hudasch, Hannover Medical School, Germany
Michail Papantoniou, General Hospital of Athens G. Genimatas, Greece
Tuğçe Aksu Uzunhan, İstanbul Atlas University, Türkiye
Updates
Copyright
© 2025 Fang, Hu and Jiang.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongjun Fang, fangcaozeng@163.com
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.