Abstract
Background:
It has been proposed that amyloid-β (Aβ) deposition may trigger neurodegeneration and cognitive decline. The elevated levels of presynaptic growth-associated protein 43 (GAP-43) in cerebrospinal fluid (CSF) were significantly associated with Alzheimer’s disease (AD). To examine whether GAP-43 was associated with faster amyloid-associated neurodegeneration or cognitive decline, it was necessary to further explore whether Aβ deposition affected CSF GAP-43 through inflammation.
Methods:
A total of 671 participants from Alzheimer’s Disease Neuroimaging Initiative (ADNI) were enrolled with available baseline CSF GAP-43, microglia activation [measured by CSF soluble triggering receptor expressed on myeloid cells (sTREM2) and progranulin (PGRN)], neurodegeneration (measured by CSF t-tau), and Aβ pathology (measured by amyloid-PET). To compare CSF GAP-43 levels across different Aβ and clinical stages, the analysis of variance (ANOVA) and Bonferroni post hoc tests were conducted. Multiple linear regression models were used to explore the association of CSF GAP-43 with sTREM2, PGRN, amyloid-PET, p-tau, t-tau and cognitive measures at baseline. Moreover, mediation models with 10,000 bootstrapped iterations were performed to investigate whether CSF GAP-43 was related to accelerated amyloid-associated neurodegeneration, then further contribute to cognitive decline, and how Aβ deposition affected CSF GAP-43 leading to neurodegeneration.
Results:
Compared with the amyloid− (A−) group, CSF GAP-43 was significantly higher at baseline in the amyloid+ (A+) group. When stratified by diagnosis, similar results were observed in A+ cognitively normal (CN) and A+ mild cognitive impairment (MCI), compared with A−CN or A−MCI participants. We found that baseline of CSF GAP-43 was positively related to CSF sTREM2, PGRN, amyloid-PET and t-tau, whereas it was negatively associated with cognition. Besides, CSF GAP-43 mediated the faster progression of amyloid-associated neurodegeneration and cognitive decline. Furthermore, the mediation analysis revealed that CSF sTREM2/PGRN was related to CSF t-tau mediated by CSF GAP-43 in the A+ group.
Conclusion:
Our findings provided evidence that CSF GAP-43 was related to the accelerated amyloid-associated neurodegeneration, and further contributed to cognitive decline. We also demonstrated that Aβ deposition may act as a trigger for synaptic dysfunction by promoting inflammation.
1 Introduction
Alzheimer’s disease (AD) as the usual type of dementia, is characterized by the accumulation of amyloid-β (Aβ) plaques, aggregated hyperphosphorylated tau, neurodegeneration, synaptic degeneration, increased neuroinflammation, and cognitive decline (1, 2). It is widely recognized that increased Aβ may act as the trigger and promote the progression of AD. In particular, Aβ changes initiate AD through the activation of harmful cascade processes with the development and aggregation of hyperphosphorylated tau, ultimately leading to neurodegeneration and dementia (3, 4).
Synaptic damage is generally recognized as a core feature of AD, and is closely related to cognitive impairment (5). Previous studies have demonstrated that presynaptic proteins appeared to be more affected than postsynaptic proteins in AD (6, 7). Growth-associated protein-43 (GAP-43) is a presynaptic membrane protein marker, predominantly expressed in the entorhinal cortex and hippocampus, also involved in synaptogenesis in the adult brain (8, 9). Elevated levels of cerebrospinal fluid (CSF) GAP-43 were observed in AD patients, reflecting the dysfunction of the presynaptic membrane (10, 11). A previous study has reported that CSF GAP-43 is closely associated with tau pathology and neurodegeneration in AD patients (12). Meanwhile, both animal and human studies have also indicated that synaptic loss could precede neurodegeneration (13, 14). And there were no difference in CAP-43 levels between healthy group and A−T− group in former research (15). However, a recent evidence showed that CSF GAP-43 was correlated with Aβ deposition in AD patients (16). Concurrently, a cross-sectional study also found CSF GAP-43 levels were elevated in the A+T− group compared with A−T− group in cognitively normal individuals (17). With the increasing number and size of Aβ plaques, it might induce microglial dysfunction, triggering excessive pro-inflammatory responses that impair synaptic and neuronal function, potentially leading to cognitive decline (18, 19). These findings suggest that Aβ deposition and related inflammation may contribute critically to synaptic injury. Therefore, we hypothesized that Aβ deposition served as a trigger factor, leading to subsequent synaptic dysfunction and neurodegeneration, and eventually causing cognitive impairments.
A study indicates that dysregulation of the innate immune system may play a pivotal role in triggering the initial events of the amyloid cascade (20). And there is also evidence that reactive microglia is frequently colocalized with amyloid plaques in the AD brain, implying an interaction between microglia and this critical pathological hallmark of AD (21). In addition, emerging finding from both animal models and human brains study, indicates the synaptotoxic role of amyloid deposition and microglia-induced neuroinflammation (22, 23). It is well known that AD susceptibility gene—triggering receptor expressed on myeloid cell 2 (TREM2) is associated with microglia activation (24). This gene can undergo ectodomain shedding by ADAM proteases, generating a soluble fragment known as soluble TREM2 (sTREM2) (25). Since sTREM2 is detectable in CSF, it serves as a marker of microglia activation and neuroinflammation. Meanwhile, progranulin (PGRN) is a secreted protein which is predominantly expressed in microglia within the central nervous system. Recently, CSF PGRN has been proposed as a hallmark of microglia-mediated neuroinflammation (26). Herein, we employed CSF sTREM2 and PGRN as biomarkers of microglial activation. Given the critical role of microglial activation in the link between Aβ deposition and synaptic dysfunction, an important question arises as to whether Aβ deposition induces synaptic injury via microglia-mediated inflammation, ultimately leading to neurodegeneration.
The purpose of this study was to investigate whether the relationship between Aβ deposition and neurodegeneration was mediated by CSF GAP-43, eventually leading to cognitive impairment. We further aimed to explore whether Aβ deposition induced synaptic dysfunction via microglia-induced neuroinflammation. To address this, CSF biomarkers were employed to reflect synaptic dysfunction (GAP-43) and microglia activation (sTREM2, PGRN). Firstly, we examined the associations among CSF GAP43, microglia activation markers (sTREM2, PGRN), amyloid-PET, CSF p-tau, t-tau and cognitive measurements. Then, a mediation model was used to study whether CSF GAP-43 mediated the relationship between amyloid-PET and CSF t-tau. Finally, we studied the role of microglia-induced neuroinflammation in this pathway. Our underlying hypothesis was that CSF GAP-43 was positively related to microglia activation, Aβ deposition, neurodegeneration and cognitive decline. Simultaneously, the relationship between CSF GAP-43 and CSF sTREM2/PGRN in microglia-mediated synaptic dysfunction, may play a crucial role in participants with abnormal Aβ levels, leading to subsequent neurodegeneration.
2 Methods
2.1 Study design
All data for this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database in January 2023. ADNI is a public–private project launched in 2003 and led by principal investigator Michael W. Weiner, MD. The primary aim of ADNI is to examine whether imaging, biochemical biomarker, genetic and clinical assessment can be combined for early determination and tracking of AD. And the inclusion criteria could be available in ADNI website.1 The ADNI study was conducted according to the Declaration of Helsinki. Meanwhile, the institutional review boards of all participating centers approved the procedures of the ADNI study. Furthermore, all participants provided written informed consent. As for up-to-date information, researchers can refer to the ADNI website.
2.2 Participants
A total of 671 participants were enrolled from the ADNI database with available baseline CSF GAP-43, CSF sTREM2, PGRN, t-tau, amyloid-PET, cognitive measurements as well as basic clinical characteristics. The ADNI investigators classified clinical status as cognitively normal CN: (Mini-Mental State Examination) MMSE >24, (Clinical Dementia Rating) CDR = 0, mild cognitive impairment (MCI: MMSE>24, CDR = 0.5) or AD dementia following former criteria (27). Moreover, participants with subjective memory complaints at baseline were included as the CN group in this study. Aβ status was determined based on the previously established cut-off value of 1.11 for Florbetapir (28). Participants were categorized into the A− group (amyloid-PET <1.11), and A+ group (amyloid-PET ≥1.11). And we combined Aβ status with clinical diagnosis to classify the participants. There were six groups were obtained: “A−CN,” “A+CN,” “A+MCI,” “A−MCI,” “A−AD” and “A+AD.”
2.3 CSF biomarkers
CSF samples were obtained via lumbar puncture, following the standardized protocol outlined in the ADNI procedures manual (see text footnote 1). The concentration of CSF p-tau and t-tau were assessed by the team at the University of Pennsylvania. The fully automated Roche Elecsys with Innogenetics (INNO-BIA AlzBio3; Ghent, Belgium) immunoassay kit-based reagents were used to analyze CSF core biomarkers (29). For the CSF sTREM2 and CSF PGRN measurements, a previously described enzyme-linked immunoassay (ELISA) approach was employed (26). The values of CSF sTREM2 were corrected based on the values of 4 internal standards (ISs). And the values of CSF PGRN were corrected based on inter-batch variation. Corrected values of sTREM2 and PGRN can be found under the name “MSD_sTREM2CORRECTED” and “MSD_PGRNCORRECTED” in the ADNI database. CSF GAP-43 was detected by an in-house ELISA at the Sahlgrenska University Hospital. And the details of ELISA procedure have been described previously (10). In brief, the procedure employed a combination of the monoclonal GAP-43 antibody NM4 and the polyclonal GAP-43 antibody ABB-135 to specifically detect GAP-43 with ranging from 312 to 20,000 pg/mL.
2.4 Amyloid-β PET
Following standardized protocols, all ADNI site performed 18F Flortaucipir PET, and amyloid-PET was acquired 50–70 min (4 × 5 min frames) after 18F-florbetapir injection. A more detailed description of amyloid-PET acquisition and analysis has been provided elsewhere (see text footnote 1). A COMPOSITE standardized uptake value ratio (SUVR) was calculated by regional Florbetapir to the mean uptake of the whole cerebellum. A cortical summary SUVR was figured by a composite cortical region, containing frontal, cingulate, parietal, and temporal areas (30). All data were obtained from the UCBERKELEYAV45_08_09_18.csv dataset.
2.5 Cognitive assessment
To reflect memory function and executive function, we applied ADNI composite executive function score (ADNI-EF) and memory function score (ADNI-MEM) measuring by psychometrically optimized approaches with items (31), and the detailed methods document applicable to this dataset was available from the ADNI database (see text footnote 1). The global cognition was reflected by CDR scores and Alzheimer Disease Assessment Scale (ADAS) 11, ADAS 13, ADAS delayed word recall (ADASQ4), the Mini-Mental State Examination (MMSE) (32). Notably, the scores of CDR, ADAS-11, ADAS-13, and ADAS Q4 are inversely related to cognitive performance; that is, higher scores on these scales indicate greater cognitive impairment. And higher scores of ADNI-EF, ADNI-MEM, and MMSE indicate better cognitive performance.
2.6 Statistical analysis
Kolmogorov–Smirnov test was employed to assess the normality distribution of CSF t-tau, amyloid-PET, CSF GAP-43, CSF sTREM2, PGRN and cognitive measurements. Abnormal distributions values were log-transformed. The log10-transforemed values were used for all statistical analyses conducted in this study. And continuous and categorical data were compared between different groups via t-tests and χ2-tests, respectively. To examine levels of CSF GAP-43 across different Aβ and clinical statuses, an analysis of covariance (ANCOVA) was used, followed by Bonferroni post hoc analysis. Subsequently, multiple linear regressions were applied to evaluate the association between CSF GAP-43 with CSF sTREM2, PGRN, amyloid-PET, CSF p-tau, t-tau and cognitive measurements, adjusting age, sex, education and APOE ε4 status. Furthermore, three mediation models were employed in this study. First, we tested whether the associations between amyloid-PET and CSF t-tau were mediated by CSF GAP-43. The second mediation model was used to explore whether the association between amyloid–PET and cognitive measurements was mediated by CSF GAP-43 and CSF t-tau. The third mediation model was employed to explore whether CSF GAP-43 mediated the relationship between CSF sTREM2/PGRN and t-tau in the A+ group. This mediation analysis aimed to examine whether Aβ deposition can lead to synaptic injury through inflammation. All of those mediation analyses were performed with 10,000 bootstraps replications controlling for age, sex, APOE ε4 status and education.
R software version 4.1.0 and IBM SPSS Statistics 23 were utilized for all the data analyses. The “lmer,” “lme4,” “car,” “mediate” and “ggplot2” in R version 4.1.0 software were employed to conduct analyses. For serial multiple mediators, we analyzed through the PROCESS Macro for SPSS developed by Hayes (33). And two-tailed p < 0.05 was set as the statistically significant.
3 Results
3.1 Participant characteristics
The detailed demographic, biomarker and clinical characteristics were displayed in Table 1. A total of 671 participants (310 A− group and 361 A+ group) were included. In this study, the mean age was 72.17 years old, with 45.16% female and 46.35% APOE ε4 carrier. The cognitive decline in the A+ group was significantly higher than that of the A− group (MMSE, p < 0.001). Compared with the A− group, there was higher amyloid-PET (p < 0.001), p-tau (p < 0.001), t-tau (p < 0.001), and GAP-43 (p < 0.001) in the A + group. No significant differences were observed between the two groups in terms of education (p = 0.535), CSF sTREM2 (p = 0.251), or CSF PGRN (p = 0.794).
Table 1
| Characteristics | A− group | A+ group | p |
|---|---|---|---|
| N | 310 | 361 | |
| Age (mean ± SD, years) | 70.81 ± 7.16 | 73.35 ± 7.33 | 0.598a |
| Gender (female/male) | 135/175 | 167/194 | 0.535b |
| Education (mean ± SD, years) | 16.47 ± 2.45 | 15.96 ± 2.69 | 0.364a |
| APOE ε4 (n, %) | 73 (23.55) | 239 (66.20) | <0.001b |
| MMSE score (mean ± SD) | 28.60 ± 1.78 | 26.66 ± 2.85 | <0.001a |
| Amyloid-PET (centiloid) | 1.01 ± 0.05 | 1.40 ± 0.17 | <0.001a |
| CSF p-tau (pg/ml) | 19.13 ± 8.05 | 33.79 ± 15.62 | <0.001a |
| CSF t-tau (pg/ml) | 216.75 ± 85.15 | 339.62 ± 143.31 | <0.001a |
| CSF GAP-43 (pg/ml) | 4504.44 ± 2327.76 | 6038.70 ± 3082.78 | <0.001a |
| CSF sTREM2 (pg/ml) | 3748.44 ± 2116.59 | 4091.40 ± 2188.04 | 0.251a |
| CSF PGRN (pg/ml) | 1582.05 ± 376.64 | 1585.83 ± 447.00 | 0.794a |
Characteristics of participants based on group classification.
Categorical variables are presented as numbers and percentages. Continuous variables are presented as Means ± SDs. APOE, Apolipoprotein E; SD, Standard deviations; MMSE, Mini Mental State Examination; CSF, cerebrospinal fluid; t-tau, total-tau; GAP-43: Growth-associated protein-43; sTREM2: soluble triggering receptor expressed on myeloid cells 2; PGRN: progranulin.
Comparison of subgroups were employed by Student’s t-tests.
Comparison of subgroups were employed by χ2-tests.
3.2 Levels of CSF GAP-43 in different Aβ status and clinical statuses
At baseline, our finding displayed CSF GAP-43 was obviously increased in the A + group compared with the A– group (p < 0.001, Figure 1A). As shown in Figure 1B, after combing with clinical staging, higher levels of CSF GAP-43 were seen in A+MCI compared A-MCI (p < 0.001). The levels of CSF GAP-43 also were observed an increase in A+CN compared with A–CN group (p = 0.909). In addition, there was no statistical differences of CSF GAP-43 in A+AD and A–AD group (p = 1.000).
Figure 1

Scatterplots of CSF GAP-43 levels by Aβ status and clinical diagnosis. (A) Distribution of CSF GAP-43 in different Aβ groups, showing the A+ group with significant higher levels of CSF GAP-43 levels as compared with A− group (p < 0.001). (B) When stratified by clinical diagnosis, CSF GAP-43 levels were increased in A+CN (p = 0.909) and A+MCI (p < 0.001).
3.3 Association of CSF GAP-43 with sTREM2/PGRN, Aβ depotion and neurodegeneration
Linear regression was performed to examine the relationships of CSF GAP-43 with CSF sTREM2/PGRN, Aβ pathology, and neurodegeneration across all participants (Supplementary Table S1). Amyloid-PET was regarded as a marker of Aβ pathology, while CSF t-tau served as a marker of neurodegeneration. We found the increased level of CSF GAP-43 was associated with higher level of CSF sTREM2 (p < 0.001, β = 0.447, Figure 2A), and PGRN (p < 0.001, β = 0.250, Figure 2B). As shown in Figure 3, individuals with higher levels CSF GAP-43 exhibited elevated amyloid-PET (p < 0.001, β = 0.188), as well as higher levels of CSF p-tau (p < 0.001, β = 0.714) and CSF t-tau (p < 0.001, β = 0.774) across the entire cohort.
Figure 2

Scatterplots of the association between CSF GAP-43 with CSF sTREM2 and CSF PGRN. The scatterplots showed the relationships between CSF GAP-43 with CSF sTREM2 (A) and CSF PGRN (B) in the whole participants. The correlation analyses were performed using liner regression models. These plots exhibited that standardized beta-estimates (β) and p-values, which were adjusted for the effects of age, sex, APOE ε4, education and diagnosis.
Figure 3

The relationship of between CSF GAP-43 with amyloid PET, CSF p-tau, CSF t-tau and cognitive measurements in the participants. Linear regression analyses were performed to examine the relationships, adjusting for potential confounding variables including age, education, sex, and APOE ε4. ADAS, Alzheimer Disease Assessment Scale; ADASQ4, ADAS delayed word recall; MMSE, Mini-Mental State Examination; ADNI-MEM, memory function score; ADNI-EF, ADNI composite executive function score.
3.4 Association of CSF GAP-43 with cognitive measurements
Next, the associations between CSF GAP-43 and cognitive measurements was studied. For the global cognitive scores, Figure 3 showed the increased levels of CSF GAP-43 were related to higher scores of ADAS11 (p = 0.020, β = 0.090), ADAS13 (p = 0.010, β = 0.090), ADASQ4 (p = 0.006, β = 0.102) and CDR (p = 0.001, β = 0.221) in the whole participants. The CSF GAP-43 level was negatively associated with MMSE (p < 0.001, β = −0.138). Regarding memory function, the CSF GAP-43 level was negatively correlated with ADNI-MEM scores (p < 0.001, β = −0.124). But there was no a significant relationship between CSF GAP-43 and ADNI-EF scores (p = 0.721, β = −0.013). After correction for multiple comparisons using the false discovery rate (FDR), the associations of amyloid PET, CSF p-tau, CSF t-tau and all cognitive measurements still remained statistically significant (PFDR < 0.05), except for ADNI-EF (PFDR = 0.721).
3.5 GAP-43 mediated the association between Aβ deposition and neurodegeneration
Moreover, we investigated whether CSF GAP-43 was associated with accelerated amyloid-related neurodegeneration. To this end, we performed the initial mediation pathway analysis in all participants: Amyloid-PET → CSF GAP-43 → CSF t-tau. Using bootstrapped mediation analyses with 10,000 iterations, we found the indirect effect of amyloid-PET on CSF t-tau via CSF GAP-43 was significant (p < 0.001, β = 0.163, 95%CI: 0.096 to 0.220, Figure 4A).
Figure 4

Mediation analyses with CSF t-tau as outcomes. (A) CSF GAP-43 mediated the relationship between amyloid-PET and CSF t-tau. (B) The relationship between CSF sTREM2 and CSF t-tau, was mediated by CSF GAP-43 in the A+ group. Beta estimates and p values for each path were displayed along the corresponding arrows. IE, indirect effect.
We further tested whether CSF GAP-43 and CSF t-tau contribute to the association between amyloid-PET and cognition. And the second mediation model was used: Amyloid-PET → CSF GAP-43 → CSF t-tau → ADAS11. The significant relationships were seen between amyloid-PET and ADAS11 (Figure 5). And mediation pathway revealed the effect of amyloid-PET with ADAS11 through CSF GAP-43 and CSF t-tau. For the sensitivity analysis, five different cognitive measurements were utilized to conduct mediation analysis, containing ADAS13, ADASQ4, MMSE, CDR, and ADNI-MEM. These obtained results were same as ADAS11 (Supplementary Table S2). Our data suggested that Aβ deposition maybe a key role in leading to synaptic dysfunction, ultimately causing neurodegeneration and cognitive impairment.
Figure 5
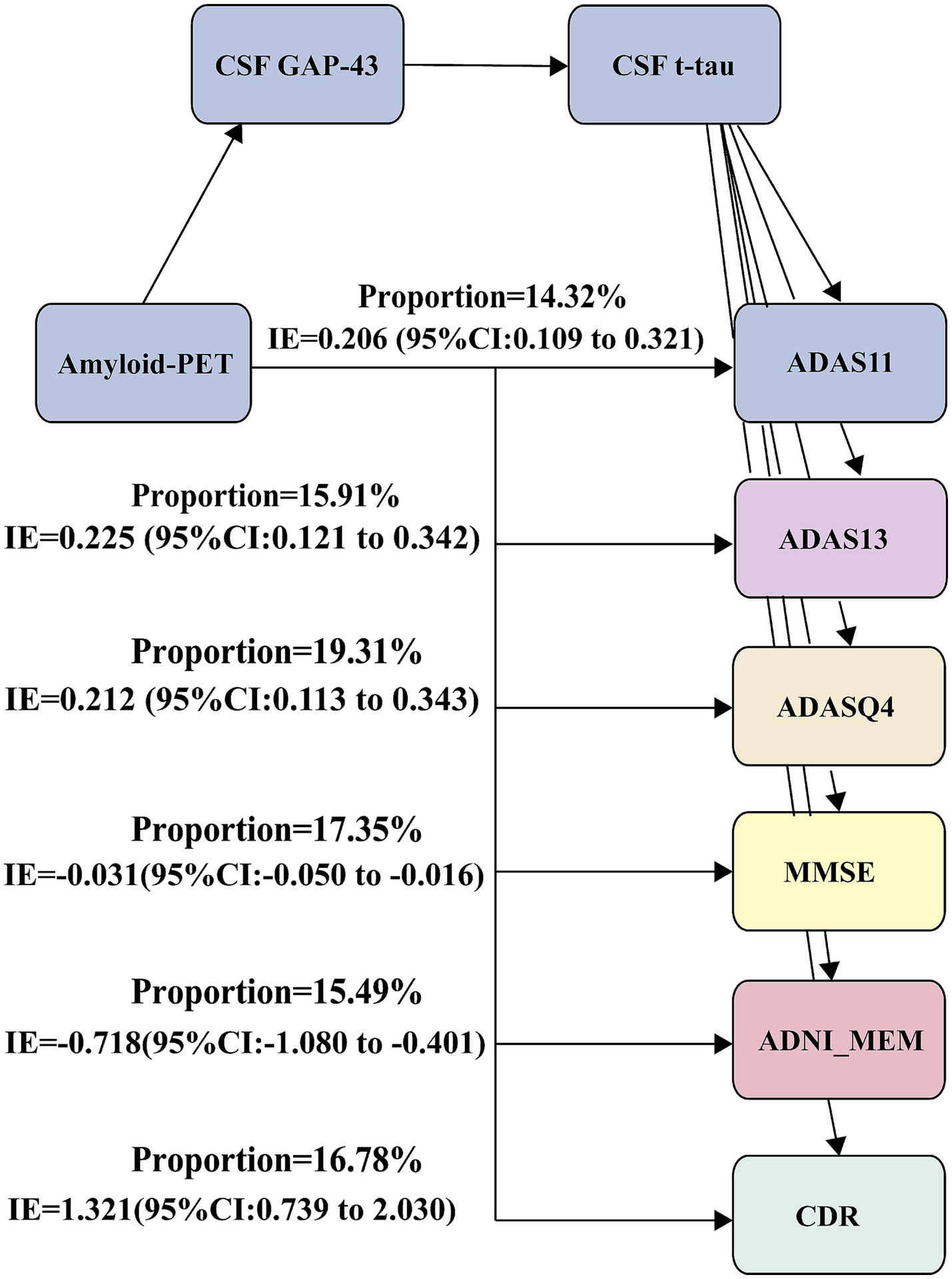
Mediation analyses with cognitive measurements as outcomes. Mediation analysis between amyloid-PET and cognitive measurements: Amyloid-PET → CSF GAP-43 → CSF t-tau → cognitive measurements. ADAS, Alzheimer Disease Assessment Scale; ADASQ4, ADAS delayed word recall; MMSE, Mini-Mental State Examination; ADNI-MEM, memory function score.
3.6 GAP-43 mediated the association between CSF sTREM2/PGRN and CSF t-tau at the A+ group
Considering the above findings, Aβ deposition appeared to play a crucial role in synaptic dysfunction. We further studied how Aβ deposition caused synaptic dysfunction, thereby leading to neurodegeneration. The third mediation model was used in the A+ group: CSF sTREM2/PGRN → CSF GAP-43 → CSF t-tau. The results indicated the significant indirect effects (p < 0.001, βsTREM2 = 0.295, 95%CI: 0.224 to 0.370, Figure 4B; p < 0.001, βPGRN = 0.158, 95%CI: 0.083 to 0.240, Supplementary Figure S1). These results suggested that Aβ deposition may be associated with synaptic dysfunction via inflammation.
4 Discussion
Herein, we assessed whether CSF GAP-43 was associated with accelerated amyloid-related neurodegeneration, ultimately leading to cognitive decline. Furthermore, we investigated whether microglia-mediated inflammation played a critical role in Aβ-induced synaptic injury, which in turn led to neurodegeneration. Our data demonstrated that CSF GAP-43 was significantly increased in the A+ group compared to A− group and positively related to CSF t-tau, amyloid-PET, CSF sTREM2/PGRN, and cognitive decline at baseline. Mediation results revealed that CSF GAP-43 mediated the association of amyloid-PET with CSF t-tau, which eventually contributed to cognitive decline. Importantly, the mediation results further exhibited that the relationship between CSF sTREM2/PGRN and CSF t-tau was mediated by CSF GAP-43 in the A+ group. These results supported our hypothesis that the accumulation of Aβ deposition to a certain extent may injure synapses and result in neurodegeneration as well as cognitive impairment through microglia-mediated neuroinflammation in AD.
GAP-43 is essential for axonal growth and early synapse formation, highlighting its role in synaptogenesis. And neurogranin is mainly localized in the postsynaptic density and is linked to synaptic strength and calcium signaling. Moreover, SNAP-25 regulates presynaptic membrane fusion and neurotransmitter release (34). Neurogranin and SNAP-25 are more related to synaptic maturation and function. In this study, we used CSF GAP-43 as a biomarker to reflect early synaptic plasticity and axonal remodeling associated with neurodegenerative processes (35). We found that levels of CSF GAP-43 were increased at baseline in the A+ group compared to A− group. When stratified participants by Aβ status and clinical diagnosis, the same findings were obtained in the A+CN or A+MCI group compared with the A−CN or A+MCI group. Consistent with previous studies, increased levels of CSF GAP-43 were observed in the A+CN group compared with A−CN participants (36), as well as in A+MCI compared with the A–MCI group (10). There was no significant difference in CSF GAP-43 in the A+AD group (110 participants) compared with the A−AD group (13 participants). We suspected that the relatively small size of the A−AD group might have limited the statistical power during our analysis process, potentially affecting the reliability of the estimated differences of CSF GAP-43 levels in this subgroup. In addition, our results also showed CSF GAP-43 was positively related to amyloid-PET, CSF p-tau, and t-tau in the participants. Consistent with previous studies, CSF GAP-43 was related to Aβ deposition and tau pathology, occurring prior to neurodegeneration (10, 14, 16, 37). However, some previous studies reported that there was no obvious relationship between Aβ pathology and CSF GAP-43, which we suspect may be due to their use of CSF Aβ42 (15, 38). In our study, we employed amyloid-PET, which more accurately reflected Aβ deposition. Our data also demonstrated that CSF GAP-43 levels was higher in the A+ group and was associated with Aβ deposition, suggesting that Aβ deposition may trigger synaptic damage. However, the mechanism by which Aβ deposition influences CSF GAP-43 remains still unclear.
Our results also revealed that CSF GAP-43 was positively related to CSF sTREM2/PGRN in the participants. The microglia-synapse pathway has been identified as a key factor in AD progression, yet its underlying mechanism in AD remains unclear (19). Hence, several potential mechanisms may be as follows. One possible explanation was that the synapse was localized by classical complement proteins C1q and C3, which then mediated synapse loss by microglia (39). Another explanation was that microglia may release soluble synaptotoxic factors, ultimately promoting synaptic damage (40). Previous animal experiments had repeatedly shown that microglia activation was related to synaptic injury, which can eventually lead to neurodegeneration and cognitive decline (19). However, to our knowledge, there is lack of studies exploring the associations between microglia activation and synaptic dysfunction in human studies.
A study has suggested that Aβ deposition may be a mechanism to regulate synaptic activity (41). As Aβ pathology progresses, plaques began to deposit and increase, resulting in reduced glutamatergic transmission and synaptic damage in animal experiments (42, 43). Finally, this process might cause the detachment of tau from microtubules and the formation of tau tangles, which in turn contributed to neurodegeneration (44, 45). Some researches and reviews have showed that Aβ deposition initially leads to synaptic injury, which is subsequently followed by tau-related axonal degeneration (17, 46). Based on these research results, we found that CSF GAP-43 mediated the relationship between Aβ deposition and neurodegeneration, which eventually leading to cognitive impairment. And previous studies mainly focused on tau pathology in individuals (12). Our data provided evidence that GAP-43 was associated with faster amyloid-associated neurodegeneration and cognitive decline. Thus, Aβ deposition may serve as a trigger for damage synapses, ultimately leading to neurodegeneration and cognitive impairment.
A widely accepted theory proposed that Aβ deposition damaged synapse and neuronal loss, for which the underlying mechanisms were still controversial (47). We further explored how Aβ deposition caused the synaptic dysfunction and the events that followed. The mediation analysis revealed that in the A+ group, CSF GAP-43 mediated the association between CSF sTREM2/PGRN and CSF t-tau. The underlying mechanism linking Aβ deposition to synaptic dysfunction may involve microglia-induced neuroinflammation, with Aβ deposition acting upstream of microglia activation (48). Recent studies provided solid evidence that microglial activation increased in response to Aβ deposition. And lots of vitro studies have showed that activated microglia released soluble factors, such as TNF-, IL-6, and NO to induce synapse dysfunction (40, 49). To our knowledge, study on how Aβ deposition damages synapses in human remains limited. Herein, our findings provide evidence in human that Aβ deposition may serve as a critical initiating factor for synaptic dysfunction, and that its aggregation could damage microglia function and enhance inflammatory responses, thereby promoting synaptic injury and ultimately leading to neurodegeneration.
This study had significant advantages. As far as we know, it was the first study to reveal the impact of sTREM2/PGRN on the relationship between CSF GAP-43 and AD pathology based on population data. Moreover, we proposed a potential underlying pathway linking Aβ pathology to neurodegeneration. Nevertheless, there were also several limitations in our study. Firstly, our study lacked other synapse-related biomarkers due to sample sizes being limited in the ADNI cohort, including the presynaptic protein SNAP-25 and postsynaptic protein neurogranin. Further studies should obtain other synapse-related biomarkers to explore. Secondly, our findings focused on the cross-sectional analysis, and future studies should include more longitudinal data with additional visits and longer follow-up periods. Finally, animal experiments should be conducted to investigate the underlying mechanisms.
5 Conclusion
In summary, these study findings suggested that CSF GAP-43 was related to accelerated amyloid-associated neurodegeneration and cognitive decline in AD. Our data further indicated that Aβ deposition may promote microglia-mediated inflammation, leading to synaptic dysfunction. Study results provided evidence that Aβ deposition and microglia may sever as targets for future AD treatment to reduce synaptic dysfunction.
Statements
Data availability statement
Publicly available datasets were analyzed in this study. This data can be found at: www.adni-info.org.
Ethics statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent from the patients/participants or patients/participants’ legal guardian/next of kin was not required to participate in this study in accordance with the national legislation and the institutional requirements.
Author contributions
YL: Investigation, Methodology, Writing – original draft, Writing – review & editing. XX: Investigation, Writing – original draft, Writing – review & editing. LT: Formal analysis, Supervision, Writing – original draft, Writing – review & editing.
Group members of Alzheimer’s Disease Neuroimaging Initiative
The data used in preparation for this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in the analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd. and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2025.1629389/full#supplementary-material
Footnotes
References
1.
Scheltens P Blennow K Breteler MMB de Strooper B Frisoni GB Salloway S et al . Alzheimer's disease. Lancet. (2016) 388:505–17. doi: 10.1016/S0140-6736(15)01124-1
2.
Rubio-Perez JM Morillas-Ruiz JM . A review: inflammatory process in Alzheimer's disease, role of cytokines. ScientificWorldJournal. (2012) 2012:756357. doi: 10.1100/2012/756357
3.
Jack CR Jr Knopman DS Jagust WJ Petersen RC Weiner MW Aisen PS et al . Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. (2013) 12:207–16. doi: 10.1016/S1474-4422(12)70291-0
4.
Karran E Mercken M De Strooper B . The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. (2011) 10:698–712. doi: 10.1038/nrd3505
5.
Forner S Baglietto-Vargas D Martini AC Trujillo-Estrada L LaFerla FM . Synaptic impairment in Alzheimer’s disease: a dysregulated symphony. Trends Neurosci. (2017) 40:347–57. doi: 10.1016/j.tins.2017.04.002
6.
Love S Siew LK Dawbarn D Wilcock GK Ben-Shlomo Y Allen SJ . Premorbid effects of APOE on synaptic proteins in human temporal neocortex. Neurobiol Aging. (2006) 27:797–803. doi: 10.1016/j.neurobiolaging.2005.04.008
7.
de Wilde MC Overk CR Sijben JW Masliah E . Meta-analysis of synaptic pathology in Alzheimer’s disease reveals selective molecular vesicular machinery vulnerability. Alzheimers Dement. (2016) 12:633–44. doi: 10.1016/j.jalz.2015.12.005
8.
Neve RL Finch EA Bird ED Benowitz LI . Growth-associated protein GAP-43 is expressed selectively in associative regions of the adult human brain. Proc Natl Acad Sci USA. (1988) 85:3638–42. doi: 10.1073/pnas.85.10.3638
9.
Benowitz LI Perrone-Bizzozero NI Finklestein SP Bird ED . Localization of the growth-associated phosphoprotein GAP-43 (B-50, Fl) in the human cerebral cortex. J Neurosci. (1989) 9:990–5. doi: 10.1523/JNEUROSCI.09-03-00990.1989
10.
Sandelius A Portelius E Källén Å Zetterberg H Rot U Olsson B et al . Elevated CSF GAP-43 is Alzheimer’s disease specific and associated with tau and amyloid pathology. Alzheimers Dement. (2019) 15:55–64. doi: 10.1016/j.jalz.2018.08.006
11.
Mila-Aloma M Brinkmalm A Ashton NJ Kvartsberg H Shekari M Operto G et al . CSF synaptic biomarkers in the preclinical stage of Alzheimer disease and their association with MRI and PET: a cross-sectional study. Neurology. (2021) 97:e2065–78. doi: 10.1212/WNL.0000000000012853
12.
Lan G Cai Y Li A Liu Z Ma S Guo T et al . Association of presynaptic loss with Alzheimer’s disease and cognitive decline. Ann Neurol. (2022) 92:1001–15. doi: 10.1002/ana.26492
13.
Yoshiyama Y Higuchi M Zhang B Huang SM Iwata N Saido TC et al . Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. (2007) 53:337–51. doi: 10.1016/j.neuron.2007.01.010
14.
Lan G Li A Liu Z Ma S Guo T for the Alzheimer's Disease Neuroimaging Initiative . Presynaptic membrane protein dysfunction occurs prior to neurodegeneration and predicts faster cognitive decline. Alzheimers Dement. (2022) 19:2408–19. doi: 10.1002/alz.12890
15.
Bergström S Remnestål J Yousef J Olofsson J Markaki I Carvalho S et al . Multi-cohort profiling reveals elevated CSF levels of brain-enriched proteins in Alzheimer's disease. Ann Clin Transl Neurol. (2021) 8:1456–70. doi: 10.1002/acn3.51402
16.
Zhang H Lyu D Jia J for the Alzheimer’s Disease Neuroimaging Initiative . The trajectory of cerebrospinal fluid growth-associated protein 43 in the Alzheimer’s disease continuum: a longitudinal study. J Alzheimer's Dis. (2022) 85:1441–52. doi: 10.3233/JAD-215456
17.
Pereira JB Janelidze S Ossenkoppele R Kvartsberg H Brinkmalm A Mattsson-Carlgren N et al . Untangling the association of amyloid-beta and tau with synaptic and axonal loss in Alzheimer’s disease. Brain. (2021) 144:310–24. doi: 10.1093/brain/awaa395
18.
Piccioni G Mango D Saidi A Corbo M Nisticò R . Targeting microglia–synapse interactions in Alzheimer's disease. Int J Mol Sci. (2021) 22:2342. doi: 10.3390/ijms22052342
19.
Xie J Wang H Lin T Bi B . Microglia-synapse pathways: promising therapeutic strategy for Alzheimer’s disease. Biomed Res Int. (2017) 2017:2986460. doi: 10.1155/2017/2986460
20.
Pascoal TA Benedet AL Ashton NJ Kang MS Therriault J Chamoun M et al . Microglial activation and tau propagate jointly across Braak stages. Nat Med. (2021) 27:1592–9. doi: 10.1038/s41591-021-01456-w
21.
Tooyama I Kimura H Akiyama H McGeer PL . Reactive microglia express class I and class II major histocompatibility complex antigens in Alzheimer’s disease. Brain Res. (1990) 523:273–80. doi: 10.1016/0006-8993(90)91496-4
22.
Palop JJ Mucke L . Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat Neurosci. (2010) 13:812–8. doi: 10.1038/nn.2583
23.
Hong S Beja-Glasser VF Nfonoyim BM Frouin A Li S Ramakrishnan S et al . Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. (2016) 352:712–6. doi: 10.1126/science.aad8373
24.
Wang Y Cella M Mallinson K Ulrich JD Young KL Robinette ML et al . TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell. (2015) 160:1061–71. doi: 10.1016/j.cell.2015.01.049
25.
Schlepckow K Kleinberger G Fukumori A Feederle R Lichtenthaler SF Steiner H et al . An Alzheimer-associated TREM2 variant occurs at the ADAM cleavage site and affects shedding and phagocytic function. EMBO Mol Med. (2017) 9:1356–65. doi: 10.15252/emmm.201707672
26.
Suárez‐Calvet M Capell A Araque Caballero MÁ Morenas‐Rodríguez E Fellerer K Franzmeier N et al . CSF progranulin increases in the course of Alzheimer's disease and is associated with sTREM2, neurodegeneration and cognitive decline. EMBO Mol Med. (2018) 10:e9712. doi: 10.15252/emmm.201809712
27.
Aisen PS Petersen RC Donohue MC Gamst A Raman R Thomas RG et al . Clinical core of the Alzheimer's disease neuroimaging initiative: progress and plans. Alzheimers Dement. (2010) 6:239–46. doi: 10.1016/j.jalz.2010.03.006
28.
Landau SM Mintun MA Joshi AD Koeppe RA Petersen RC Aisen PS et al . Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann Neurol. (2012) 72:578–86. doi: 10.1002/ana.23650
29.
Shaw LM Vanderstichele H Knapik-Czajka M Clark CM Aisen PS Petersen RC et al . Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol. (2009) 65:403–13. doi: 10.1002/ana.21610
30.
Landau SM Fero A Baker SL Koeppe R Mintun M Chen K et al . Measurement of longitudinal beta-amyloid change with 18F-florbetapir PET and standardized uptake value ratios. J Nucl Med. (2015) 56:567–74. doi: 10.2967/jnumed.114.148981
31.
Crane PK Carle A Gibbons LE Insel P Mackin RS Gross A et al . Development and assessment of a composite score for memory in the Alzheimer’s disease neuroimaging initiative (ADNI). Brain Imaging Behav. (2012) 6:502–16. doi: 10.1007/s11682-012-9186-z
32.
Folstein MF Folstein SE McHugh PR . "Mini-mental state". A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. (1975) 12:189–98. doi: 10.1016/0022-3956(75)90026-6
33.
Hayes A.F. , Introduction to mediation, moderation, and conditional process Analysis_ a regression-based approach. Alberta, Canada. (2013).
34.
Zhang C Xie S Malek M . SNAP-25: a biomarker of synaptic loss in neurodegeneration. Clin Chim Acta. (2025) 571:120236. doi: 10.1016/j.cca.2025.120236
35.
Rohden F Ferreira PCL Bellaver B Ferrari-Souza JP Aguzzoli CS Soares C et al . Glial reactivity correlates with synaptic dysfunction across aging and Alzheimer’s disease. Nat Commun. (2025) 16:5653. doi: 10.1038/s41467-025-60806-1
36.
Öhrfelt A Benedet AL Ashton NJ Kvartsberg H Vandijck M Weiner MW et al . Association of CSF GAP-43 with the rate of cognitive decline and progression to dementia in amyloid-positive individuals. Neurology. (2023) 100:e275–85. doi: 10.1212/WNL.0000000000201417
37.
Casaletto KB Zetterberg H Blennow K Brinkmalm A Honer W Schneider JA et al . Tripartite relationship among synaptic, amyloid, and tau proteins: an in vivo and postmortem study. Neurology. (2021) 97:e284–97. doi: 10.1212/WNL.0000000000012145
38.
Qiang Q Skudder-Hill L Toyota T Wei W Adachi H . CSF GAP-43 as a biomarker of synaptic dysfunction is associated with tau pathology in Alzheimer’s disease. Sci Rep. (2022) 12:17392. doi: 10.1038/s41598-022-20324-2
39.
Schafer DP Lehrman EK Kautzman AG Koyama R Mardinly AR Yamasaki R et al . Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. (2012) 74:691–705. doi: 10.1016/j.neuron.2012.03.026
40.
Wang WY Tan MS Yu JT Tan L . Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann Transl Med. (2015) 3:136. doi: 10.3978/j.issn.2305-5839.2015.03.49
41.
Fagiani F Lanni C Racchi M Pascale A Govoni S . Amyloid-beta and Synaptic vesicle dynamics: a cacophonic orchestra. J Alzheimer's Dis. (2019) 72:1–14. doi: 10.3233/JAD-190771
42.
Wu HY Hudry E Hashimoto T Kuchibhotla K Rozkalne A Fan Z et al . Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J Neurosci. (2010) 30:2636–49. doi: 10.1523/JNEUROSCI.4456-09.2010
43.
Burgold S Bittner T Dorostkar MM Kieser D Fuhrmann M Mitteregger G et al . In vivo multiphoton imaging reveals gradual growth of newborn amyloid plaques over weeks. Acta Neuropathol. (2011) 121:327–35. doi: 10.1007/s00401-010-0787-6
44.
Howlett DR Bowler K Soden PE Riddell D Davis JB Richardson JC et al . Abeta deposition and related pathology in an APP x PS1 transgenic mouse model of Alzheimer's disease. Histol Histopathol. (2008) 23:67–76. doi: 10.14670/HH-23.67
45.
Jack CR Jr Bennett DA Blennow K Carrillo MC Dunn B Haeberlein SB et al . NIA-AA research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. (2018) 14:535–62. doi: 10.1016/j.jalz.2018.02.018
46.
Edwards FA . A unifying hypothesis for Alzheimer's disease: from plaques to neurodegeneration. Trends Neurosci. (2019) 42:310–22. doi: 10.1016/j.tins.2019.03.003
47.
Masters CL Cappai R Barnham KJ Villemagne VL . Molecular mechanisms for Alzheimer's disease: implications for neuroimaging and therapeutics. J Neurochem. (2006) 97:1700–25. doi: 10.1111/j.1471-4159.2006.03989.x
48.
Selkoe DJ Hardy J . The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. (2016) 8:595–608. doi: 10.15252/emmm.201606210
49.
Azevedo EP Ledo JH Barbosa G Sobrinho M Diniz L Fonseca ACC et al . Activated microglia mediate synapse loss and short-term memory deficits in a mouse model of transthyretin-related oculoleptomeningeal amyloidosis. Cell Death Dis. (2013) 4:e789. doi: 10.1038/cddis.2013.325
Summary
Keywords
Alzheimer’s disease, growth-associated protein 43 (GAP-43), neurodegeneration, cognition, Alzheimer’s Disease Neuroimaging Initiative (ADNI)
Citation
Li Y, Xu X, Tang L and on behalf of the Alzheimer’s Disease Neuroimaging Initiative (2025) GAP-43 is associated with faster amyloid-associated neurodegeneration and cognitive decline in Alzheimer’s disease. Front. Neurol. 16:1629389. doi: 10.3389/fneur.2025.1629389
Received
15 May 2025
Accepted
15 September 2025
Published
26 September 2025
Volume
16 - 2025
Edited by
Qiang Qiang, Fudan University, China
Reviewed by
Wenchuan Zhou, Chongqing Medical University, China
Yi-Ming Huang, Capital Medical University, China
Updates
Copyright
© 2025 Li, Xu and Tang.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yaxin Li, 15605573756@163.com
†These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.