 Yang Li
Yang Li Xiaojie Hu2
Xiaojie Hu2 Yanhong Jiang
Yanhong Jiang Xingchen Wang
Xingchen Wang- 1The Second Clinical Medical College, Shandong University of Traditional Chinese Medicine, Jinan, China
- 2Department of Neurology, The Second Affiliated Hospital of Shandong University of Traditional Chinese Medicine, Jinan, China
- 3Department of Cardiology, Zibo Central Hospital, Zibo, China
Neurocutaneous syndromes are a group of genetic disorders involving the nervous and cutaneous systems, including Tuberous Sclerosis Complex (TSC), neurofibromatosis type 1 (NF1), and Sturge–Weber syndrome (SWS), and others. The incidence of epilepsy, a core clinical manifestation, is significantly higher than in the general population. The purpose of this narrative review is to provide an updated overview of the genetic mechanisms and recent advances in precise treatment for neurocutaneous syndrome-related epilepsy. We conducted a comprehensive search of the PubMed, Scopus, EMBASE, and Web of Science databases using all MeSH terms related to ‘Neurocutaneous Syndromes’, ‘Epilepsy/genetics’, ‘Signal Transduction’, and ‘Precision Medicine’. Selected papers underwent review and risk of bias (RoB) assessment to evaluate core questions. Somatic or germline mutations dysregulate key signaling pathways (e.g., mTOR, Ras-MAPK, PI3K-AKT), inducing malformations of cortical development (MCD) and neuronal-glial dysfunction that collectively form epileptogenic networks. This constitutes the primary pathogenic mechanism underlying neurocutaneous syndrome-related epilepsy. Precise treatment strategies based on molecular mechanisms have achieved breakthroughs: mTOR inhibitors significantly reduce seizure frequency in TSC patients, and cannabidiol (CBD) demonstrates broad-spectrum antiepileptic efficacy in TSC and Dravet syndrome. Advances in surgical techniques, such as multimodal imaging-guided resection, improve outcomes in refractory epilepsy. However, clinical translation faces challenges including technical limitations in detecting mosaic mutations, insufficient specificity of targeted drugs, and interdisciplinary collaboration gaps. Future directions require integrating multi-omics technologies, developing novel gene therapies (e.g., CRISPR-based approaches), and establishing multicenter databases linking genotype–phenotype-treatment responses to advance personalized precision medicine.
1 Introduction
Neurocutaneous syndromes (NCS), also termed phakomatoses, represent a heterogeneous group of multisystem genetic disorders characterized by concomitant neurological and cutaneous manifestations. This category encompasses TSC, NF1, SWS, Epidermal Nevus Syndrome (ENS), and neurocutaneous melanosis (NCM), and others (1). These diseases are mostly caused by somatic mutations or germline mutations, resulting in dysregulation of key signaling pathways, which leads to abnormal neural and vascular development, manifesting as MCD, epilepsy, and various skin manifestations (1).
Epilepsy, a hallmark neurological complication of NCS, exhibits strikingly high prevalence across subtypes. In TSC, seizure incidence reaches 80–90%, with approximately 60% of cases progressing to pharmacoresistant epilepsy (2, 3). Similarly elevated rates are observed in SWS (75–90%) and NF1 (4–7%), significantly exceeding population baselines (4, 5). Emerging evidence highlights the pivotal role of somatic mosaicism in cortical malformation-associated epileptogenesis, providing mechanistic insights for targeted interventions (1). Notably, mTOR inhibitors (e.g., sirolimus, everolimus) demonstrate therapeutic efficacy in TSC by modulating aberrant signaling, achieving seizure frequency reduction in 50% of patients and seizure-free outcomes in select cases (6). Concurrently, CBD shows broad antiepileptic potential across refractory epilepsies including TSC, Dravet syndrome, and Lennox–Gastaut syndrome, underscoring the promise of genotype-driven precision therapeutics (7).
Deciphering the genetic architecture and advancing mechanism-based therapies for NCS-related epilepsy hold dual significance: optimizing clinical management through molecular stratification while revolutionizing epilepsy treatment paradigms. Integrating molecular diagnostics with pathway-specific modulation may substantially improve patient prognoses and catalyze the evolution of precision medicine in neurology.
2 The genetic mechanisms of neurocutaneous syndrome-related epilepsy
The genetic pathogenesis of neurocutaneous syndromes is primarily attributed to somatic mosaic mutations that dysregulate key signaling pathways.
TSC caused by germline or somatic inactivating mutations in tumor suppressor genes TSC1 (9q34) or TSC2 (16p13.3), manifests through disrupted negative regulation of the mTORC1 pathway. This dysregulation participates in epileptogenesis through multi-level mechanisms: At the neuronal level, mTOR hyperactivation promotes protein synthesis while inhibiting autophagy, leading to neuronal hypertrophy, aberrant dendritic arborization, and synaptic plasticity dysregulation – collectively establishing epileptogenic networks (8, 9). Concurrently, glial dysfunction exacerbates neuronal hyperexcitability through decreased glutamate transporter (GLT-1) expression in astrocytes, causing extracellular glutamate accumulation, and metabolic uncoupling that disrupts the neuron-glial lactate-glutamine cycle (10–13). The TSC1/TSC2 protein complex normally maintains cellular homeostasis, but mutation-induced hyperactivation of mTOR signaling promotes multiorgan hamartoma formation, including cerebral tubers and renal angiomyolipomas (4, 14–17). Furthermore, somatic mosaic mutations in genes such as TSC2 or AKT3 drive PI3K-AKT–mTOR pathway overactivation, inducing focal cortical dysplasia or hemimegalencephaly-critical epileptogenic substrates (18, 19).
NF1 arises from mutations in the NF1 gene (17q11.2), whose product neurofibromin functions as a Ras GTPase-activating protein (GAP) that negatively regulates Ras-MAPK signaling (20, 21). Loss of NF1 activity results in constitutive Ras activation, triggering Schwann cell and glial proliferation. This pathological process disrupts prostaglandin E (PGE) metabolism, elevates neuronal excitability, and creates cortical excitation-inhibition imbalance that underlies spontaneous seizure generation. Characteristic phenotypes encompass café-au-lait macules and skeletal abnormalities (16, 20–22).
SWS is principally mediated by the somatic mosaic mutation GNAQ c.548G > A (p. R183Q), which activates the Gαq-PLCβ-PKC axis and Rho-ROCK signaling. This molecular derangement induces pathological vascular endothelial proliferation and leptomeningeal angiomatosis, clinically presenting with the classic triad of facial port-wine stains, glaucoma, and neurological calcifications (23–26). Notably, GNAQ mutations exhibit cross-activation of both Ras-MAPK and PI3K-AKT–mTOR pathways, suggesting therapeutic potential for MEK inhibitors (e.g., selumetinib) and mTOR inhibitors (e.g., everolimus) (27, 28). The Figure 1 illustrates the cellular signaling network initiated by the G protein-coupled receptor α subunit GNAQ and its molecular association with three genetic diseases, ultimately regulating the process of cell proliferation.
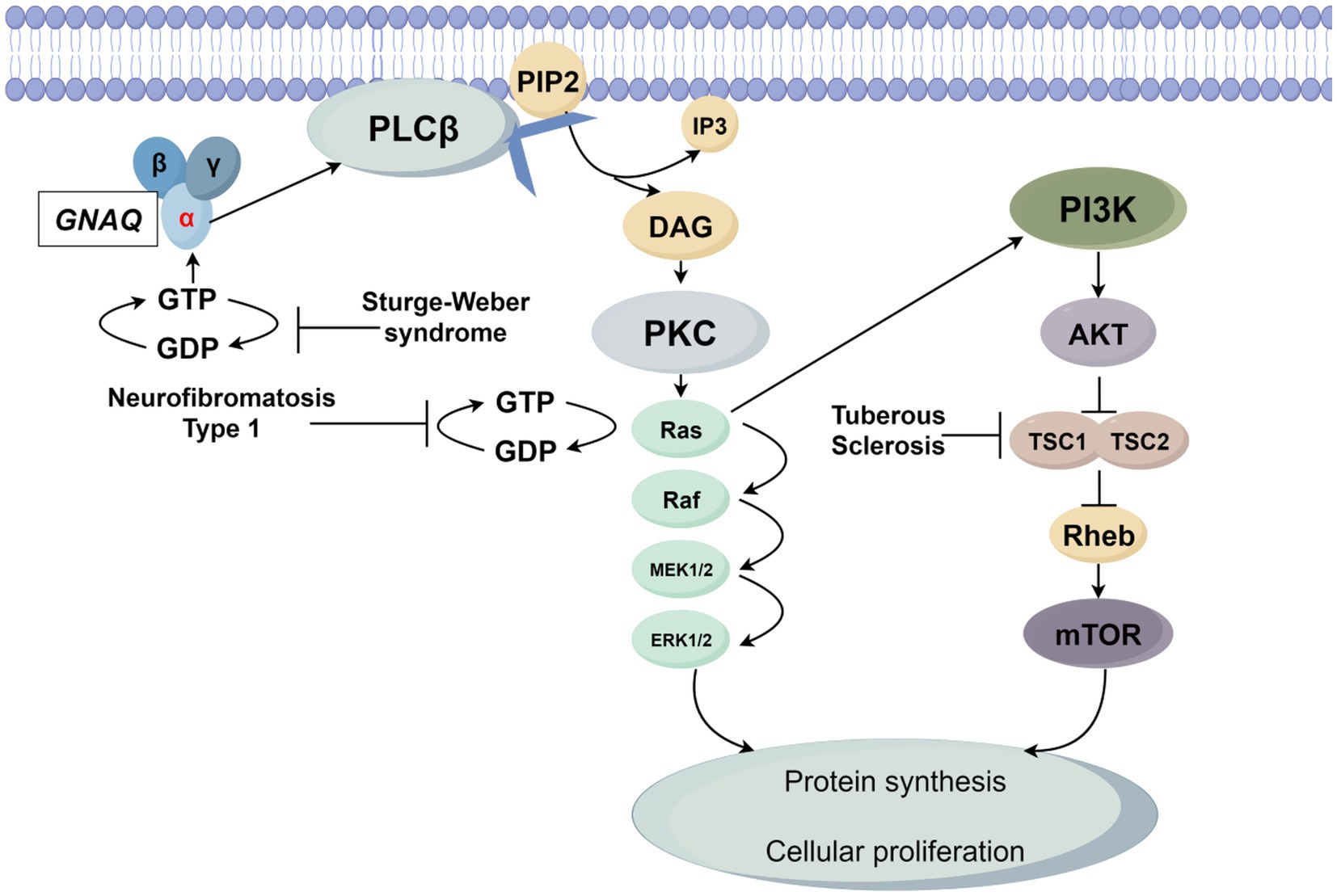
Figure 1. GNAQ is activated upon ligand binding via GDP-GTP exchange, triggering phospholipase Cβ (PLCβ) to hydrolyze membrane-bound phosphatidylinositol 4,5-bisphosphate (PIP2) into diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). DAG activates protein kinase C (PKC), which in turn promotes GTP binding to the small G protein Ras, rendering it active. Active Ras propagates signals through two key pathways: the mitogen-activated protein kinase (MAPK) cascade (Raf → MEK1/2 → ERK1/2) and the phosphoinositide 3-kinase (PI3K)-AKT–mTOR axis. The MAPK cascade drives ERK1/2 activation, while the PI3K-AKT–mTOR pathway involves AKT-mediated inhibition of the TSC1/TSC2 complex (a Rheb negative regulator), leading to mTOR activation. Ultimately, ERK1/2 and mTOR converge to enhance protein synthesis and drive cell proliferation. Three diseases are linked to network dysregulation: SWS arises from GNAQ gain-of-function mutations, causing persistent GTP binding and constitutive PLCβ pathway activation; NF1 results from NF1 loss-of-function mutations, abrogating neurofibromin (a Ras GTPase-activating protein) and leading to Ras hyperactivation due to impaired GTP hydrolysis; TSC is caused by TSC1/TSC2 mutations, disrupting Rheb regulation and allowing mTOR hyperactivation.
Other neurocutaneous syndromes also exhibit distinct genetic patterns. NCM is associated with somatic mutations in NRAS that dysregulate the MAPK pathway, resulting in aberrant melanocyte proliferation and melanin deposition in cutaneous and leptomeningeal tissues (27, 29, 30). ENS arises from activating mutations in PIK3CA or AKT3, which drive hyperactivation of the PI3K-AKT–mTOR signaling axis, clinically manifesting as epidermal nevi, hemimegalencephaly, and drug-resistant epilepsy (19, 30). Additionally, Cerebrofacial Arteriovenous Metameric Syndrome (CAMS), characterized by metameric arteriovenous malformations, requires differential diagnosis from SWS. Emerging evidence suggests its pathogenesis may involve somatic mutations in vascular patterning genes such as EPHB4 or RASA1 (31).
The shared genetic hallmark of neurocutaneous syndromes lies in somatic mutations disrupting core developmental pathways—including mTOR, Ras-MAPK, and Gαq-PLCβ signaling—leading to pluripotent progenitor cell dysregulation, tissue malformations, and tumorigenesis (32). These mechanistic insights have enabled the successful clinical translation of mTOR inhibitors (rapamycin, everolimus) in TSC-associated epilepsy, demonstrating significant seizure frequency reduction (50% responder rate) and cognitive improvement (33, 34). Emerging therapeutic strategies targeting upstream PI3K-AKT–mTOR pathway components (PIK3CA, AKT1) are undergoing clinical evaluation, heralding new precision medicine approaches (35, 36).
3 Progress in precise treatment of neurocutaneous syndrome related epilepsy
Emerging therapeutic strategies targeting mTOR-associated somatic mutations and glial dysfunction have entered translational phases, encompassing both gene-editing technologies (e.g., CRISPR-Cas systems) and pathway-specific inhibitors (37–39). Clinical validation of mTOR inhibitors (e.g., everolimus) in TSC demonstrates substantial therapeutic efficacy, with phase III trials reporting ≥50% seizure frequency reduction in 50% of patients and seizure-free outcomes in subsets, alongside cognitive improvement (37, 40, 41). Notably, expanding applications in RASopathies-related epilepsy (e.g., NF1) are under investigation, though mechanistic validation remains ongoing (40, 42). For GNAQ-mutated SWS, preclinical studies using mTOR inhibitors show reduced neurovascular inflammation in animal models (43), yet clinical evidence remains limited to anecdotal reports (43, 44). Key unanswered questions include: whether GNAQ mutations exert mTOR activation via PI3K-AKT crosstalk (43, 45), and the potential involvement of downstream effectors like HIF-1α in therapeutic responses (45) Future multicenter trials incorporating biomarker-driven designs (e.g., mTOR activation status via phosphor-S6 immunohistochemistry) and combinatorial anti-angiogenic approaches may address current translational challenges (46). Table 1 shows the genetic molecular targets and treatment evidence for various neurocutaneous syndrome-related epilepsy.
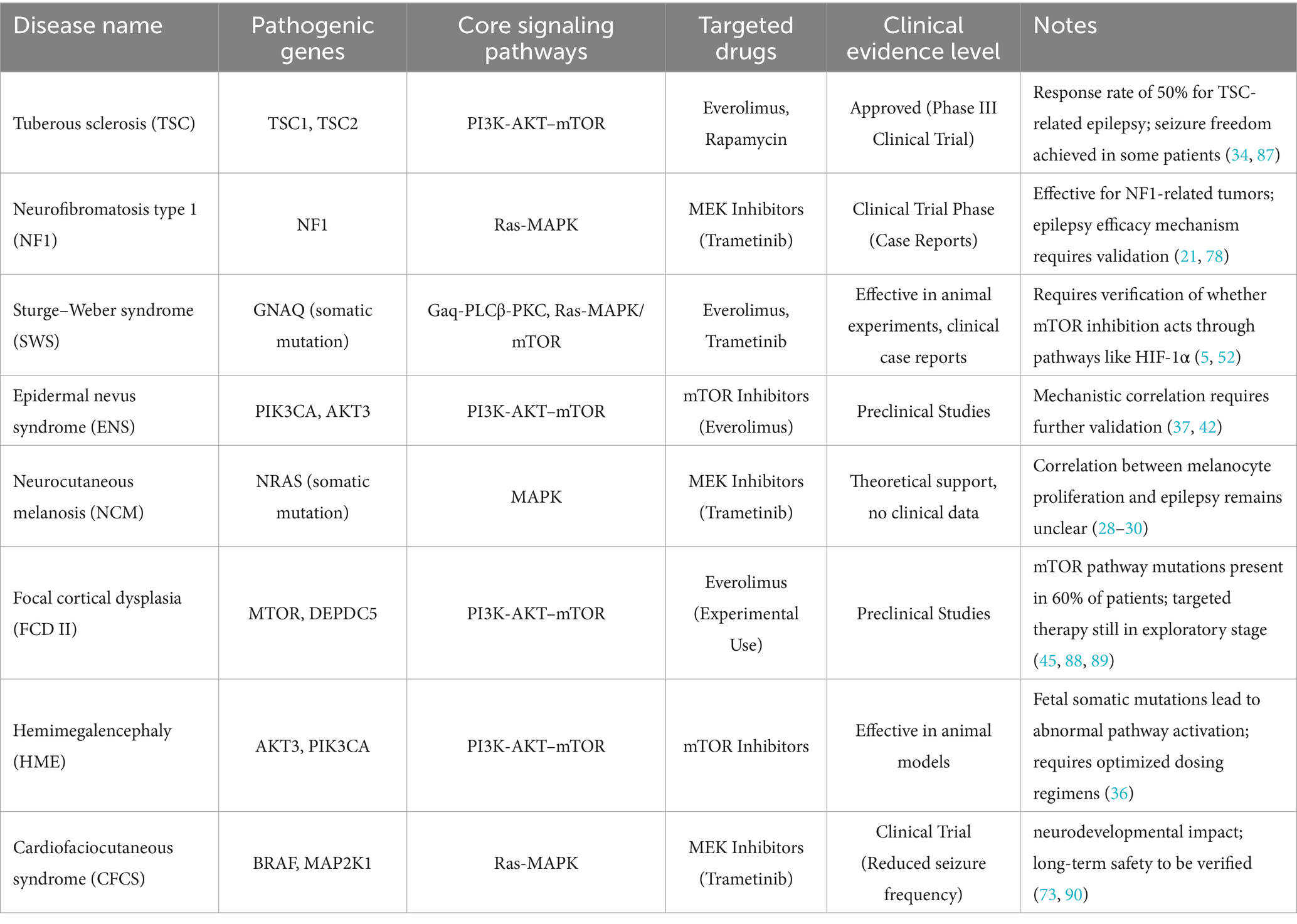
Table 1. Genetic-molecular targets and treatment evidence for neurocutaneous syndrome-related epilepsy.
Cutting-edge methodologies integrating single-cell transcriptomics and spatial proteomics are revolutionizing our understanding of epileptogenic niches. These techniques enable high-resolution mapping of neuron–glia-vascular unit interactions within seizure foci, identifying novel therapeutic targets such as senescent cell populations and dysregulated lactate shuttling (9, 47, 48). This paradigm shift from histomorphological characterization to molecular network analysis provides critical insights for refractory epilepsy management. Ultimately, the convergence of multi-omics profiling, advanced neuroimaging, and clinical phenotyping will catalyze the development of personalized therapeutic frameworks for neurocutaneous syndrome-related epilepsy.
In the domain of novel antiepileptic therapeutics, CBD has emerged as a pharmacological intervention with multi-target mechanisms, including modulation of AMPA, GABA, and GPR55 receptors, demonstrating significant therapeutic potential (41, 49, 50). CBD antagonizes G protein-coupled receptor 55 (GPR55), inhibiting its mediation of intracellular calcium release and mTOR pathway activation. This reduces downstream protein synthesis, regulates autophagy processes, clears abnormal protein accumulation, and alleviates abnormal neural proliferation and seizures (51). Currently approved for Dravet syndrome, Lennox–Gastaut syndrome, and TSC-associated epilepsy, CBD adjunctive therapy achieves ≥50% seizure reduction in 50–60% of patients, with particularly notable efficacy in controlling epileptic spasms among TSC patients (45–50% responder rate) (41, 52, 53). However, hepatotoxicity risk requires vigilant monitoring when co-administered with valproic acid, evidenced by elevated liver enzyme levels (54–56). While short-term safety profiles appear favorable, long-term administration necessitates individualized risk–benefit assessment, particularly regarding cardiovascular parameters and pharmacokinetic interactions. The current evidence base lacks extended longitudinal data beyond 5-year follow-up, underscoring the need for syndrome-specific outcome studies (57). Notably, advancements in precision dosing technologies, including ultra-performance liquid chromatography–tandem mass spectrometry (UPLC-MS/MS), are revolutionizing personalized therapeutic regimens (58).
Surgical innovations have substantially enhanced therapeutic outcomes through multimodal localization strategies. The integration of 18F-fluorodeoxyglucose positron emission tomography (18F-FDG PET; sensitivity 70–80%) with magnetoencephalography (MEG) enables precise epileptogenic zone delineation, particularly for TSC cortical tubers (44). Resective surgery achieves seizure-free outcomes in 60–70% of unifocal TSC cases, while hemispheric disconnection procedures yield 80% seizure freedom rates in SWS, albeit with potential neurological sequelae requiring careful preoperative evaluation (43, 44). Emerging evidence suggests that mTOR pathway activation status, as determined by immunohistochemical markers, may serve as a predictive biomarker for postoperative recurrence (37).
The optimization of individualized therapeutic regimens faces dual challenges in bridging mechanistic research and clinical translation. Current investigations into epileptogenic mechanisms have yet to fully elucidate critical pathway interactions. For instance, the causal relationship between BRAF/MAP2K1 variants and epileptic encephalopathy in CFCS remains ambiguous, significantly impeding targeted drug selection (40, 44, 59). Existing therapeutic strategies remain fragmented, with most antiseizure medications (ASMs) primarily addressing symptomatic management rather than correcting underlying genetic defects (e.g., avoidance of sodium channel blockers in SCN1A mutations), while advanced interventions such as gene replacement therapy remain confined to preclinical development (60, 61). This therapeutic impasse is further compounded by the lack of standardized efficacy evaluation systems, particularly regarding dynamic monitoring of biomarkers such as electroencephalographic (EEG) signatures and molecular imaging parameters (62, 63). Establishing multimodal “genotype–phenotype-treatment response” databases, advancing molecular stratification-based clinical trials (e.g., MEK inhibitors for RASopathies-associated epilepsy), and developing companion diagnostic tools emerge as pivotal strategies to overcome these barriers (40, 64).
The development of novel targeted therapies demonstrates diversification but confronts technical and commercial complexities. RAS-MAPK pathway inhibitors (e.g., selumetinib) exhibit seizure frequency reduction potential in CFCS-related epilepsy, though their long-term neurodevelopmental impacts and safety profiles require rigorous validation (40, 65). As emerging experimental strategies CRISPR-based gene editing technologies hold transformative potential for neurocutaneous syndromes. Precision manipulation of disease-associated genes (e.g., TSC1/TSC2, NF1) enables accurate modeling of patient-specific mutations and elucidation of epileptogenic pathways (66). Preclinical studies confirm that CRISPR-Cas9-mediated correction of TSC1/TSC2 mutations effectively suppresses mTOR pathway hyperactivation and reduces seizure incidence, providing mechanistic validation for gene therapy (57, 67). However, ethical concerns persist across developmental stages, including risks of off-target genomic alterations (e.g., unintended CRISPR/Cas9 activity), immune responses to viral vectors (e.g., AAVs), and unpredictable neurocircuitry remodeling (68–71). Technical hurdles further include achieving stable regulation of vector-mediated gene expression (72) and maintaining excitatory/inhibitory balance during neural network modulation (71). While AAV-based strategies targeting SCN1A and MECP2 mutations face challenges in blood–brain barrier penetration and immunogenicity (73, 74), mTOR inhibitors like everolimus demonstrate dual antiepileptic and antitumor efficacy in tuberous sclerosis, though optimal dosing regimens require refinement (29, 75). Notably, most therapies remain mutation-specific with limited capacity to reverse established neural circuit abnormalities, compounded by recruitment challenges and limited profitability in rare disease drug development (60, 64).
Implementing multidisciplinary care systems is paramount for optimizing therapeutic outcomes. Neurocutaneous syndromes’ multisystem involvement (neurological, dermatological, ocular) demands coordinated expertise, yet current collaboration models exhibit critical deficiencies. Diagnostic delays persist in conditions like Aicardi syndrome due to underrecognized dermal/retinal manifestations (45, 76), while discrepancies between neurologists’ genetic counseling proficiency and geneticists’ epilepsy expertise result in fragmented decision-making (29, 77). Data silos across genomic, imaging, and histopathological platforms further obstruct comprehensive evaluations (62, 78). Addressing these systemic gaps requires establishing specialized neurocutaneous syndrome centers with standardized multidisciplinary workflows (e.g., tumor boards for TSC), integrated clinical databases, and genetics competency training programs to cultivate an integrated diagnostic-therapeutic ecosystem (62, 76).
4 Discussion
Research on genetic mechanisms of neurocutaneous syndrome-related epilepsy has elucidated pathogenic pathways across distinct syndromes. TSC1/TSC2 mutations in TSC activate mTOR signaling to promote epileptogenesis, establishing molecular foundations for targeted therapies (79). Similarly, the CFCS, driven by RAS-MAPK signaling pathway variants (BRAF, KRAS), demonstrates epileptogenic mechanisms involving neuronal hyperexcitability and synaptic plasticity dysregulation (40, 65). The pathogenic mechanisms of different syndromes are both specific (such as over-activation of the mTOR pathway in TSC and cross-activation of multiple pathways by GNAQ mutations in SWS) and common (such as cross-disease effects of glial cell metabolism imbalance and excitotoxicity). Their interactions form a complex network in epileptogenesis. These advances not only delineate genotype–phenotype correlations [e.g., specific mutations associated with Infantile epileptic spasms syndrome (IESS) versus focal epilepsy (80, 81)], but also directly inform therapeutic development. MEK inhibitors like selumetinib targeting the RAS-MAPK pathway exhibit clinically significant seizure reduction in CFCS patients (40). Genetic diagnostics further optimize antiepileptic drug selection, exemplified by avoiding sodium channel blockers in SCN1A mutation carriers while prioritizing valproate (77, 82).
Clinically, synergistic approaches combining CRISPR-based gene editing, single-cell sequencing, and molecularly targeted therapies (mTOR inhibitors, ion channel modulators) show transformative potential (59, 61, 73). Establishing dynamic genotype–phenotype-treatment response databases will refine personalized regimens (80, 83), while prospective trials must validate the long-term efficacy and safety of emerging inter ventions like neuromodulation and gene replacement therapies (83, 84). Future directions should address genetic-psychiatric comorbidities (autism spectrum disorders, cognitive impairment) through integrated treatment paradigms that simultaneously optimize seizure control and neurodevelopmental outcomes (85, 86).
5 Conclusion
This review systematically clarifies the core pathogenic mechanisms and precise treatment strategies of neurocutaneous syndrome-related epilepsy, and reveals a molecular pathological network characterized by abnormal activation of mTOR, Ras-MAPK and other signaling pathways. Somatic/germline mutations drive the formation of epileptogenic networks by regulating neuron-glial dysfunction and cortical development malformations; interventions targeting pathways such as mTOR and MEK have demonstrated clinical potential. Current research faces challenges such as insufficient accuracy in the detection of chimeric mutations, unclear neurodevelopmental effects of targeted drugs, and lack of efficacy evaluation systems, which limit the in-depth implementation of personalized treatment.
Analyze the spatio-temporal specific activation patterns of signal pathways in the epileptogenic microenvironment, identify the core regulatory nodes of abnormal neuron-glial metabolic coupling; develop precise hierarchical treatment plans based on mutation lineages, combining CRISPR gene editing and multimodal imaging technology to achieve etiological intervention; Build a dynamic database of “genotype-treatment response” and verify the long-term safety and neuroprotective effects of new therapies (such as MEK inhibitors and gene replacement therapy) through multi-center collaboration. These studies will promote the transformation of diagnosis and treatment models from symptom control to pathological mechanism targeting, laying an important foundation for the precise treatment practice of neurocutaneous syndrome-related epilepsy.
Author contributions
YL: Formal analysis, Project administration, Writing – review & editing, Methodology, Writing – original draft, Validation, Visualization, Investigation, Data curation. XH: Visualization, Methodology, Investigation, Conceptualization, Formal analysis, Supervision, Writing – review & editing, Data curation. XC: Writing – original draft, Methodology, Software, Formal analysis, Data curation, Project administration. YC: Project administration, Validation, Formal analysis, Methodology, Visualization, Software, Conceptualization, Writing – review & editing, Supervision. YJ: Supervision, Methodology, Writing – review & editing, Software, Conceptualization, Investigation, Validation, Data curation, Project administration. XW: Conceptualization, Writing – review & editing, Supervision, Visualization, Writing – original draft, Software, Funding acquisition, Resources.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by “the National Famous Old Chinese Medicine Experts Inheritance Studio Construction Project, grant number [2022] no. 75” and “the Qilu BianCang Traditional Chinese Medicine Talent Cultivation Project, grant number [2024] no. 78.”
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Iffland, PHN, and Crino, PB. The role of somatic mutational events in the pathogenesis of epilepsy. Curr Opin Neurol. (2019) 32:191–7. doi: 10.1097/WCO.0000000000000667
2. Jansen, FE, van Huffelen, AC, Algra, A, and van Nieuwenhuizen, O. Epilepsy surgery in tuberous sclerosis: a systematic review. Epilepsia. (2007) 48:1477–84. doi: 10.1111/j.1528-1167.2007.01117.x
3. Evans, LT, Morse, R, and Roberts, DW. Epilepsy surgery in tuberous sclerosis: a review. Neurosurg Focus. (2012) 32:E5. doi: 10.3171/2012.1.FOCUS11330
4. Miyata, H, Kuwashige, H, Hori, T, Kubota, Y, Pieper, T, Coras, R, et al. Variable histopathology features of neuronal dyslamination in the cerebral neocortex adjacent to epilepsy-associated vascular malformations suggest complex pathogenesis of focal cortical dysplasia ILAE type IIIc. Brain Pathol. (2022) 32:e13052. doi: 10.1111/bpa.13052
5. Vinters, HV, Kerfoot, C, Catania, M, Emelin, JK, Roper, SN, and DeClue, JE. Tuberous sclerosis-related gene expression in normal and dysplastic brain. Epilepsy Res. (1998) 32:12–23. doi: 10.1016/S0920-1211(98)00036-9
6. Sahin, M, Miller, I, Holmes, GL, and Sheth, RD. Pediatric epileptology. Epilepsy Behav. (2011) 22:32–7. doi: 10.1016/j.yebeh.2011.02.010
7. Talwar, A, Estes, E, Aparasu, R, and Reddy, DS. Clinical efficacy and safety of cannabidiol for pediatric refractory epilepsy indications: a systematic review and meta-analysis. Exp Neurol. (2023) 359:114238. doi: 10.1016/j.expneurol.2022.114238
8. Gelot, A, Draia-Nicolau, TO, Mathieu, R, Gelot, AB, Silvagnoli, L, Watrin, F, et al. Cytomegalic parvalbumin neurons in fetal cases of hemimegalencephaly. Epilepsia. (2025) 66:2099–109. doi: 10.1111/epi.18325
9. Ribierre, T, Bacq, A, Donneger, F, Doladilhe, M, Maletic, M, Roussel, D, et al. Targeting pathological cells with senolytic drugs reduces seizures in neurodevelopmental mTOR-related epilepsy. Nat Neurosci. (2024) 27:1125–36. doi: 10.1038/s41593-024-01634-2
10. Aquiles, A, Fiordelisio, T, Luna-Munguia, H, and Concha, L. Altered functional connectivity and network excitability in a model of cortical dysplasia. Sci Rep. (2023) 13:12335. doi: 10.1038/s41598-023-38717-2
11. Lee, M, Kim, E, Kim, M, Kim, E-J, Kim, M-J, and Yum, M-S. Rapamycin cannot reduce seizure susceptibility in infantile rats with malformations of cortical development lacking mTORC1 activation. Mol Neurobiol. (2022) 59:7439–49. doi: 10.1007/s12035-022-03033-9
12. Soeung, V, Puchalski, RB, and Noebels, JL. The complex molecular epileptogenesis landscape of glioblastoma. Cell Rep Med. (2024) 5:101691. doi: 10.1016/j.xcrm.2024.101691
13. Varella, FJ, Xavier, FAC, Zanirati, G, Gonçalves, JIB, Previato, TTR, Pazzin, DB, et al. Increased activation of the WNT pathway in brain tissue from patients with cortical dysplasia type IIb. Sci Rep. (2025) 15:8049. doi: 10.1038/s41598-025-90045-9
14. Ferrante, L, and Ortman, C. Genetic principles related to neurocutaneous disorders. Semin Pediatr Neurol. (2024) 51:101150. doi: 10.1016/j.spen.2024.101150
15. Pratico, AD, Falsaperla, R, Comella, M, Belfiore, G, Polizzi, A, Ruggieri, M, et al. Case report: a gain-of-function of hamartin may lead to a distinct "inverse TSC1-hamartin" phenotype characterized by reduced cell growth. Front Pediatr. (2023) 11:1101026. doi: 10.3389/fped.2023.1101026
16. Tovani-Palone, MR, Bistagnino, F, and Shah, PA. Multidisciplinary team for patients with neurocutaneous syndromes: the little discussed importance of dentistry. Clinics (Sao Paulo). (2024) 79:100332. doi: 10.1016/j.clinsp.2024.100332
17. Jurca, CM, Kozma, K, Petchesi, CD, Zaha, DC, Magyar, I, Munteanu, M, et al. Tuberous sclerosis, type II diabetes mellitus and the PI3K/AKT/mTOR signaling pathways-case report and literature review. Genes. (2023) 14:433. doi: 10.3390/genes14020433
18. Kim, SH, Kwon, SS, Park, MR, Lee, HA, Kim, JH, Cha, JH, et al. Detecting low-variant allele frequency mosaic pathogenic variants of NF1, TSC2, and AKT3 genes from blood in patients with neurodevelopmental disorders. J Mol Diagn. (2023) 25:583–91. doi: 10.1016/j.jmoldx.2023.04.003
19. Secco, L, Coubes, C, Meyer, P, Secco, LP, Chenine, L, Roubertie, A, et al. Dermatological and genetic data in tuberous sclerosis: a prospective single-center study of 38 patients. Ann Dermatol Venereol. (2022) 149:241–4. doi: 10.1016/j.annder.2022.02.007
20. Almahmood, H, Al-Sayed, S, and Agab, W. Lambdoid suture defect in a 12-year-old Neurofibromatosis patient. Cureus. (2024) 16:e54567. doi: 10.7759/cureus.54567
21. Xu, L, Jang, H, and Nussinov, R. Allosteric modulation of NF1 GAP: differential distributions of catalytically competent populations in loss-of-function and gain-of-function mutants. Protein Sci. (2025) 34:e70042. doi: 10.1002/pro.70042
22. Chlopek, M, Lasota, J, Thompson, LDR, Szczepaniak, M, Kuźniacka, A, Hińcza, K, et al. Alterations in key signaling pathways in sinonasal tract melanoma. A molecular genetics and immunohistochemical study of 90 cases and comprehensive review of the literature. Mod Pathol. (2022) 35:1609–17. doi: 10.1038/s41379-022-01122-7
23. Galeffi, F, Snellings, DA, Wetzel-Strong, SE, Kastelic, N, Bullock, J, Gallione, CJ, et al. A novel somatic mutation in GNAQ in a capillary malformation provides insight into molecular pathogenesis. Angiogenesis. (2022) 25:493–502. doi: 10.1007/s10456-022-09841-w
24. Nasim, S, Bichsel, C, Dayneka, S, Mannix, R, Holm, A, Vivero, M, et al. MRC1 and LYVE1 expressing macrophages in vascular beds of GNAQ p.R183Q driven capillary malformations in Sturge weber syndrome. Acta Neuropathol Commun. (2024) 12:47. doi: 10.1186/s40478-024-01757-4
25. Nasim, S, Bichsel, C, Pinto, A, Alexandrescu, S, Kozakewich, H, and Bischoff, J. Similarities and differences between brain and skin GNAQ p.R183Q driven capillary malformations. Angiogenesis. (2024) 27:931–41. doi: 10.1007/s10456-024-09950-8
26. Wetzel-Strong, SE, Galeffi, F, Benavides, C, Patrucco, M, Bullock, JL, Gallione, CJ, et al. Developmental expression of the Sturge-weber syndrome-associated genetic mutation in Gnaq: a formal test of Happle's paradominant inheritance hypothesis. Genetics. (2023) 224:iyad077. doi: 10.1093/genetics/iyad077
27. Seront, E, Queisser, A, Boon, LM, and Vikkula, M. Molecular landscape and classification of vascular anomalies. Hematology Am Soc Hematol Educ Program. (2024) 2024:700–8. doi: 10.1182/hematology.2024000598
28. Zecchin, D, Knopfel, N, Gluck, AK, Stevenson, M, Sauvadet, A, Polubothu, S, et al. GNAQ/GNA11 mosaicism causes aberrant calcium signaling susceptible to targeted therapeutics. J Invest Dermatol. (2024) 144:811–819.e4. doi: 10.1016/j.jid.2023.08.028
29. Kioutchoukova, I, Foster, D, Thakkar, R, Ciesla, C, Cabassa, JS, Strouse, J, et al. Neurocutaneous diseases: diagnosis, management, and treatment. J Clin Med. (2024) 13:1648. doi: 10.3390/jcm13061648
30. Rose, AL, and Cathey, SS. Genetic causes of vascular malformations and common signaling pathways involved in their formation. Dermatol Clin. (2022) 40:449–59. doi: 10.1016/j.det.2022.07.002
31. Barros, LL, Lima, PLGD, de Oliveira Junior, PH, Dias, DA, Santos, CF, Braga-Neto, P, et al. Peculiar aetiology for orbital apex syndrome: Wyburn-Mason syndrome as orbital apex lesion. BMJ Neurol Open. (2024) 6:e559. doi: 10.1136/bmjno-2023-000559
32. Payne, JM, Haebich, KM, Mitchell, R, Bozaoglu, K, Giliberto, E, Lockhart, PJ, et al. Brain volumes in genetic syndromes associated with mTOR dysregulation: a systematic review and meta-analysis. Mol Psychiatry. (2025) 30:1676–88. doi: 10.1038/s41380-024-02863-4
33. Pineau, L, Buhler, E, Tarhini, S, Bauer, S, Crepel, V, Watrin, F, et al. Pathogenic MTOR somatic variant causing focal cortical dysplasia drives hyperexcitability via overactivation of neuronal GluN2C N-methyl-D-aspartate receptors. Epilepsia. (2024) 65:2111–26. doi: 10.1111/epi.18000
34. Bonniaud, B, Luu, M, Cormier, C, Racine, C, Espitalier, A, Malbranche, C, et al. Lack of behavioural improvement with sirolimus in a patient with MTOR-related macrocephaly with pigmentary mosaicism: a new case report. Eur J Med Genet. (2025) 75:105012. doi: 10.1016/j.ejmg.2025.105012
35. Fujita, A, Kato, M, Sugano, H, Iimura, Y, Suzuki, H, Tohyama, J, et al. An integrated genetic analysis of epileptogenic brain malformed lesions. Acta Neuropathol Commun. (2023) 11:33. doi: 10.1186/s40478-023-01532-x
36. Itoh, K, Pooh, R, Shimokawa, O, and Fushiki, S. Somatic mosaicism of the PI3K-AKT-MTOR pathway is associated with hemimegalencephaly in fetal brains. Neuropathology. (2023) 43:190–6. doi: 10.1111/neup.12875
37. Knowles, JK, Helbig, I, Metcalf, CS, Lubbers, LS, Isom, LL, Demarest, S, et al. Precision medicine for genetic epilepsy on the horizon: recent advances, present challenges, and suggestions for continued progress. Epilepsia. (2022) 63:2461–75. doi: 10.1111/epi.17332
38. Miziak, B, and Czuczwar, SJ. Approaches for the discovery of drugs that target K Na 1.1 channels in KCNT1-associated epilepsy. Expert Opin Drug Discov. (2022) 17:1313–28. doi: 10.1080/17460441.2023.2150164
39. Zimmern, V, and Korff, C. Updates on the diagnostic evaluation, genotype-phenotype correlation, and treatments of genetic epilepsies. Curr Opin Pediatr. (2022) 34:538–43. doi: 10.1097/MOP.0000000000001170
40. D'Onofrio, G, Delrue, M, Lortie, A, Delrue, M-A, Marquis, C, Striano, P, et al. Treatment of refractory epilepsy with MEK inhibitor in patients with RASopathy. Pediatr Neurol. (2023) 148:148–51. doi: 10.1016/j.pediatrneurol.2023.08.019
41. Kuhne, F, Becker, L, Bast, T, Bertsche, A, Borggraefe, I, Boßelmann, CM, et al. Real-world data on cannabidiol treatment of various epilepsy subtypes: a retrospective, multicenter study. Epilepsia Open. (2023) 8:360–70. doi: 10.1002/epi4.12699
42. Qu, X, Lai, X, He, M, Zhang, J, Xiang, B, Liu, C, et al. Investigation of epilepsy-related genes in a Drosophila model. Neural Regen Res. (2026) 21:195–211. doi: 10.4103/NRR.NRR-D-24-00877
43. Ranji, S, Akbari, B, Javani, M, Taghilou, A, Tafakhori, A, Salehizadeh, S, et al. Triple pathology in a patient with uncontrolled epilepsy: a case report. J Med Case Rep. (2025) 19:141. doi: 10.1186/s13256-025-05137-x
44. Saini, L, Mukherjee, S, Gunasekaran, PK, Saini, AG, Ahuja, C, Sharawat, IK, et al. The profile of epilepsy and its characteristics in children with neurocutaneous syndromes. J Neurosci Rural Pract. (2024) 15:233–7. doi: 10.25259/JNRP_510_2023
45. Izzedine, H, Begum, F, Kashfi, S, Rouprêt, M, Bridges, A, and Jhaveri, KD. Renal involvement in genetic neurocutaneous syndromes. Clin Nephrol. (2024) 102:351–69. doi: 10.5414/CN111425
46. Arenas-Cabrera, C, Cabezudo-Garcia, P, Calvo-Medina, R, Galeano-Bilbao, B, Martínez-Agredano, P, Ruiz-Giménez, J, et al. Advances and guidance in the treatment of drug-resistant epilepsy: a review by the Andalusian epilepsy society of the new drugs cenobamate, fenfluramine and cannabidiol. Rev Neurol. (2024) 79:161–73. doi: 10.33588/rn.7906.2024086
47. Specchio, N, Trivisano, M, Aronica, E, Balestrini, S, Arzimanoglou, A, Colasante, G, et al. The expanding field of genetic developmental and epileptic encephalopathies: current understanding and future perspectives. Lancet Child Adolesc Health. (2024) 8:821–34. doi: 10.1016/S2352-4642(24)00196-2
48. Sran, S, and Bedrosian, TA. RAS pathway: the new frontier of brain mosaicism in epilepsy. Neurobiol Dis. (2023) 180:106074. doi: 10.1016/j.nbd.2023.106074
49. O'Sullivan, SE, Jensen, SS, Nikolajsen, GN, Bruun, HZ, Bhuller, R, and Hoeng, J. The therapeutic potential of purified cannabidiol. J Cannabis Res. (2023) 5:21. doi: 10.1186/s42238-023-00186-9
50. Riva, A, D'Onofrio, G, Pisati, A, Roberti, R, Amadori, E, Bosch, F, et al. Cannabidiol add-on in glycosylphosphatidylinositol-related drug-resistant epilepsy. Cannabis Cannabinoid Res. (2024) 9:990–5. doi: 10.1089/can.2022.0255
51. Davis, BH, Beasley, TM, Amaral, M, Szaflarski, JP, Gaston, T, Perry Grayson, L, et al. Pharmacogenetic predictors of Cannabidiol response and tolerability in treatment-resistant epilepsy. Clin Pharmacol Ther. (2021) 110:1368–80. doi: 10.1002/cpt.2408
52. Borowicz-Reutt, K, Czernia, J, and Krawczyk, M. CBD in the treatment of epilepsy. Molecules. (2024) 29:1981. doi: 10.3390/molecules29091981
53. Perriguey, M, Succar, ME, Clement, A, Lagarde, S, Ribes, O, Dode, X, et al. High-purified cannabidiol efficacy and safety in a cohort of adult patients with various types of drug-resistant epilepsies. Rev Neurol (Paris). (2024) 180:147–53. doi: 10.1016/j.neurol.2023.07.012
54. Beers, JL, Zhou, Z, and Jackson, KD. Advances and challenges in modeling Cannabidiol pharmacokinetics and hepatotoxicity. Drug Metab Dispos. (2024) 52:508–15. doi: 10.1124/dmd.123.001435
55. Lakhani, VV, Generaux, G, Howell, BA, Longo, DM, and Watkins, PB. Assessing liver effects of Cannabidiol and valproate alone and in combination using quantitative systems toxicology. Clin Pharmacol Ther. (2023) 114:1006–14. doi: 10.1002/cpt.3004
56. Silvinato, A, Floriano, I, and Bernardo, WM. Use of cannabidiol in the treatment of epilepsy: Lennox-Gastaut syndrome, Dravet syndrome, and tuberous sclerosis complex. Rev Assoc Med Bras (1992). (2022) 68:1345–57. doi: 10.1590/1806-9282.2022D689
57. Klein, P, Kaminski, RM, Koepp, M, and Löscher, W. New epilepsy therapies in development. Nat Rev Drug Discov. (2024) 23:682–708. doi: 10.1038/s41573-024-00981-w
58. Franco, V, Palmisani, M, Marchiselli, R, Crema, F, Fattore, C, de Giorgis, V, et al. On-line solid phase extraction high performance liquid chromatography method coupled with tandem mass spectrometry for the therapeutic monitoring of Cannabidiol and 7-Hydroxy-cannabidiol in human serum and saliva. Front Pharmacol. (2022) 13:915004. doi: 10.3389/fphar.2022.915004
59. Zimmern, V, Minassian, B, and Korff, C. A review of targeted therapies for monogenic epilepsy syndromes. Front Neurol. (2022) 13:829116. doi: 10.3389/fneur.2022.829116
60. Striano, P, and Minassian, BA. From genetic testing to precision medicine in epilepsy. Neurotherapeutics. (2020) 17:609–15. doi: 10.1007/s13311-020-00835-4
61. Specchio, N, Pietrafusa, N, Perucca, E, and Cross, JH. New paradigms for the treatment of pediatric monogenic epilepsies: progressing toward precision medicine. Epilepsy Behav. (2022) 131:107961. doi: 10.1016/j.yebeh.2021.107961
62. Byrne, S, Enright, N, and Delanty, N. Precision therapy in the genetic epilepsies of childhood. Dev Med Child Neurol. (2021) 63:1276–82. doi: 10.1111/dmcn.14929
63. Taha, M, Nordli, DR, Park, C, and Nordli, DR Jr. Innovative epilepsy management: a combined figure of EEG categorization and medication mechanisms. Front Neurol. (2025) 16:1534913. doi: 10.3389/fneur.2025.1534913
64. Goodspeed, K, Bailey, RM, Prasad, S, Sadhu, C, Cardenas, JA, Holmay, M, et al. Gene therapy: novel approaches to targeting monogenic epilepsies. Front Neurol. (2022) 13:805007. doi: 10.3389/fneur.2022.805007
65. Kenney-Jung, DL, Rogers, DJ, Kroening, SJ, Zatkalik, AL, Whitmarsh, AE, Roberts, AE, et al. Infantile epileptic spasms syndrome in children with cardiofaciocutanous syndrome: clinical presentation and associations with genotype. Am J Med Genet C Semin Med Genet. (2022) 190:501–9. doi: 10.1002/ajmg.c.32022
66. Jamiolkowski, RM, Nguyen, Q, Farrell, JS, Nguyen, Q-A, McGinn, RJ, Hartmann, DA, et al. The fasciola cinereum of the hippocampal tail as an interventional target in epilepsy. Nat Med. (2024) 30:1292–9. doi: 10.1038/s41591-024-02924-9
67. Barber, HM, Pater, AA, Gagnon, KT, Damha, MJ, and O’Reilly, D. Chemical engineering of CRISPR-Cas systems for therapeutic application. Nat Rev Drug Discov. (2025) 24:209–30. doi: 10.1038/s41573-024-01086-0
68. Bettegazzi, B, Cattaneo, S, Simonato, M, Zucchini, S, and Soukupova, M. Viral vector-based gene therapy for epilepsy: what does the future hold? Mol Diagn Ther. (2024) 28:5–13. doi: 10.1007/s40291-023-00687-6
69. Cai, A, Gao, K, Zhang, F, Cai, A-J, and Jiang, Y-W. Recent advances and current status of gene therapy for epilepsy. World J Pediatr. (2024) 20:1115–37. doi: 10.1007/s12519-024-00843-w
70. Majumder, S, Moriarty, KL, Lee, Y, and Crombleholme, TM. Placental gene therapy for fetal growth restriction and preeclampsia: preclinical studies and prospects for clinical application. J Clin Med. (2024) 13:5647. doi: 10.3390/jcm13185647
71. Michetti, C, and Benfenati, F. Homeostatic regulation of brain activity: from endogenous mechanisms to homeostatic nanomachines. Am J Physiol Cell Physiol. (2024) 327:C1384–99. doi: 10.1152/ajpcell.00470.2024
72. Sullivan, KA, Vitko, I, Blair, K, Gaykema, RP, Failor, MJ, San Pietro, JM, et al. Drug-inducible gene therapy effectively reduces spontaneous seizures in kindled rats but creates off-target side effects in inhibitory neurons. Int J Mol Sci. (2023) 24:1347. doi: 10.3390/ijms241411347
73. Brock, DC, Demarest, S, and Benke, TA. Clinical trial Design for Disease-Modifying Therapies for genetic epilepsies. Neurotherapeutics. (2021) 18:1445–57. doi: 10.1007/s13311-021-01123-5
74. Muller, P, and Lerche, H. Gene therapy for epilepsy: clinical studies are on the road. Fortschr Neurol Psychiatr. (2023) 91:135–40. doi: 10.1055/a-1995-5405
75. Carvill, GL, Matheny, T, Hesselberth, J, and Demarest, S. Haploinsufficiency, dominant negative, and gain-of-function mechanisms in epilepsy: matching therapeutic approach to the pathophysiology. Neurotherapeutics. (2021) 18:1500–14. doi: 10.1007/s13311-021-01137-z
76. Engelhard, SB, Kiss, S, and Gupta, MP. Retinal manifestations of the neurocutaneous disorders. Curr Opin Ophthalmol. (2020) 31:549–62. doi: 10.1097/ICU.0000000000000712
77. Steinbart, D, Gaus, V, Kowski, AB, and Holtkamp, M. Valproic acid use in fertile women with genetic generalized epilepsies. Acta Neurol Scand. (2021) 144:288–95. doi: 10.1111/ane.13446
78. Wang, G, Wu, W, Xu, Y, Yang, Z, Xiao, B, and Long, L. Imaging genetics in epilepsy: current knowledge and new perspectives. Front Mol Neurosci. (2022) 15:891621. doi: 10.3389/fnmol.2022.891621
79. Toscano-Prat, C, Garcia-Sanchez, C, Ros-Castello, V, Barguilla-Arribas, A, Saladich, IG, Rodríguez-Clifford, K, et al. Cognitive and neuro-psychiatric profile in adult patients with epilepsy secondary to tuberous sclerosis complex. Epilepsy Behav. (2025) 166:110380. doi: 10.1016/j.yebeh.2025.110380
80. Kovacevic, M, Jankovic, M, Brankovic, M, Milićević, O, Novaković, I, Sokić, D, et al. Novel GATOR1 variants in focal epilepsy. Epilepsy Behav. (2023) 141:109139. doi: 10.1016/j.yebeh.2023.109139
81. Murthy, MC, Banerjee, B, Shetty, M, Mariappan, M, and Sekhsaria, A. A retrospective study of the yield of next-generation sequencing in the diagnosis of developmental and epileptic encephalopathies and epileptic encephalopathies in 0-12 years aged children at a single tertiary care hospital in South India. Epileptic Disord. (2024) 26:609–25. doi: 10.1002/epd2.20254
82. Dwivedi, R, Kaushik, M, Tripathi, M, Dada, R, and Tiwari, P. Unraveling the genetic basis of epilepsy: recent advances and implications for diagnosis and treatment. Brain Res. (2024) 1843:149120. doi: 10.1016/j.brainres.2024.149120
83. Hoffman, CE, Parker, WE, Rapoport, BI, Zhao, M, Ma, H, and Schwartz, TH. Innovations in the neurosurgical Management of Epilepsy. World Neurosurg. (2020) 139:775–88. doi: 10.1016/j.wneu.2020.03.031
84. Goldberg, EM. Rational small molecule treatment for genetic epilepsies. Neurotherapeutics. (2021) 18:1490–9. doi: 10.1007/s13311-021-01110-w
85. Specchio, N, Di Micco, V, Aronica, E, Auvin, S, Balestrini, S, Brunklaus, A, et al. The epilepsy-autism phenotype associated with developmental and epileptic encephalopathies: new mechanism-based therapeutic options. Epilepsia. (2025) 66:970–87. doi: 10.1111/epi.18209
86. Watkins, LV, O'Dwyer, M, and Shankar, R. A review of the pharmacotherapeutic considerations for managing epilepsy in people with autism. Expert Opin Pharmacother. (2022) 23:841–51. doi: 10.1080/14656566.2022.2055461
87. Auvin, S, and Baulac, S. mTOR-therapy and targeted treatment opportunities in mTOR-related epilepsies associated with cortical malformations. Rev Neurol (Paris). (2023) 179:337–44. doi: 10.1016/j.neurol.2022.12.007
88. Elziny, S, Sran, S, Yoon, H, Corrigan, RR, Page, J, Ringland, A, et al. Loss of Slc35a2 alters development of the mouse cerebral cortex. Neurosci Lett. (2024) 836:137881. doi: 10.1016/j.neulet.2024.137881
89. Chong, D, Jones, NC, Schittenhelm, RB, Anderson, A, and Casillas-Espinosa, PM. Multi-omics integration and epilepsy: towards a better understanding of biological mechanisms. Prog Neurobiol. (2023) 227:102480. doi: 10.1016/j.pneurobio.2023.102480
Keywords: neurocutaneous syndromes, epilepsy, genetic mechanisms, precise treatment, mTOR pathway
Citation: Li Y, Hu X, Chen X, Cheng Y, Jiang Y and Wang X (2025) Progress in genetic mechanisms and precise treatment of neurocutaneous syndrome-related epilepsy. Front. Neurol. 16:1642299. doi: 10.3389/fneur.2025.1642299
Edited by:
Xiaoxu Yang, The University of Utah, United StatesReviewed by:
Maria Jones-Muhammad, University of Alabama at Birmingham, United StatesLip Yuen Teng, Tuanku Ja'afar Hospital, Malaysia
Copyright © 2025 Li, Hu, Chen, Cheng, Jiang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xingchen Wang, c2RsY3d4YzE5NjRAMTYzLmNvbQ==