Abstract
Background and aims:
Charcot–Marie-Tooth type 1A (CMT1A) is the most common inherited demyelinating peripheral neuropathy caused by duplication of the peripheral myelin protein 22 (PMP22) gene. Although there is currently no approved treatment for CMT1A, reducing PMP22 expression has emerged as a promising therapeutic approach. The PMP22-C3 mouse model is a widely used CMT1A model that carries human PMP22 (hPMP22) and mouse Pmp22 (mPmp22) genes, complicating the relationship between reduced PMP22 levels and the recovery of phenotypes by drug candidates targeting only human PMP22. To address this, we developed humanized C3 mouse lines lacking the mPmp22 gene. Here, we characterized these models to confirm their utility as novel disease models for CMT1A.
Methods:
Heterozygous (hetero-humanized) and homozygous (homo-humanized) hPMP22 transgenic mice, with an mPmp22 homozygous knockout background, were investigated using biochemical, electrophysiological, histopathological, and behavioral analyses.
Results:
Homo-humanized mice exhibited abnormal mRNA expression of myelin-related genes, slow nerve conduction velocity, reduced compound muscle action potential, demyelinated peripheral nerves, higher levels of plasma neurofilament light chain, muscle weakness and motor/balance disabilities, alterations in electrical impedance myography, and muscle fiber atrophy. In contrast, the hetero-humanized mice did not display any of the previously described impairments.
Interpretation:
Homo-humanized mice reflect various aspects of CMT1A characteristics in an hPMP22 gene dosage-dependent manner. This model will help us better understand the relationship between PMP22 reduction levels and the recovery of CMT1A-related phenotypes, contributing to the translation of preclinical findings into clinically relevant human treatments and dosing strategies.
1 Introduction
Charcot–Marie-Tooth type 1A (CMT1A) is the most common autosomal dominant inherited demyelinating peripheral neuropathy, accounting for more than 50% of all CMT cases (1–3). The primary symptoms include distal muscle weakness/atrophy, balance problems, high-arched feet, tremors, and pain (1, 3, 4). CMT1A is caused by a duplication on chromosome 17p11.2, containing the gene encoding peripheral myelin protein 22 kDa (PMP22), which is a critical component of the myelin sheath (4, 5). Proper expression of PMP22 and other myelin-related proteins, such as sterol-C5-desaturase (SC5D), myelin protein zero (MPZ), and POU Class 3 homeobox 1 (POU3F1), is important for the development and maintenance of myelin structure in peripheral nerves (2, 6–8). In patients with CMT1A, increased levels of PMP22 (1.5–2-fold) and demyelination in peripheral nerves have been demonstrated (9–13). These changes result in slow nerve conduction velocity (NCV) (4, 9, 14, 15) and reduced compound muscle action potential (CMAP) amplitude that reflects axonal dysfunction secondary to demyelination (3, 16). In contrast, heterozygous deletion of PMP22 (approximately 0.5-fold reduction in expression) leads to another demyelinating neuropathy, Hereditary Neuropathy with Liability to Pressure Palsies (HNPP) (4, 10, 17, 18). Therefore, maintaining PMP22 expression within the appropriate range is essential for the treatment of CMT1A.
Since CMT1A is caused by overexpression of PMP22, directly reducing PMP22 expression, for example, by antisense oligonucleotides (ASO), is expected to be a promising therapeutic approach. Treatment with PMP22-targeting ASO or microRNA ameliorated the deficits in CMT1A mouse models, including declines in NCV and CMAP, demyelination in peripheral nerves, and motor disabilities (19, 20); however, an excessive reduction in PMP22 is associated with a risk of HNPP-like symptoms. Thus, a better understanding of the relationship between PMP22 expression and pathological phenotypes in animal models is important for predicting the therapeutic effects of PMP22-targeting drug candidates.
Transgenic mouse models of CMT1A overexpressing the human PMP22 (hPMP22) gene, including PMP22-C3 mice that contain 3–4 copies of hPMP22, have been widely used to elucidate disease mechanisms and facilitate drug discovery (21–23). These models carry the hPMP22 gene and mouse Pmp22 (mPmp22) gene, which makes it difficult to estimate the level of “total” PMP22 reduction by drug candidates targeting only hPMP22. For instance, the complete elimination of hPMP22 means just “normalization” in PMP22-C3 mice due to the remaining intrinsic mPmp22. In other words, PMP22-C3 mice can lead to a misunderstanding of the functional efficacy and adverse effects of human PMP22-targeting drugs on hPMP22-reducing effects. To address this issue, we generated two humanized PMP22-C3 mouse models that completely lack mPmp22 gene: heterozygous or homozygous hPMP22 transgene expression (hetero-humanized with 3–4 copies or homo-humanized with 6–8 copies) in an mPmp22 homozygous knockout (KO) background. We characterized these mouse lines by investigating peripheral nerve deterioration and subsequent muscle impairment to assess their utility as new CMT1A disease models for a more precise preclinical evaluation of drug candidates specifically targeting hPMP22.
2 Materials and methods
2.1 Animals
PMP22-C3 Tg (hetero) mice [B6.Cg-Tg(PMP22)C3Fbas/J] were purchased from Jackson Laboratory (Bar Harbor, ME, USA) with permission for breeding and genetical modifying. They were bred in Axcelead Drug Discovery Partners, Inc. (ADDP, Kanagawa, Japan) and male PMP22-C3 mice and age-matched wild-type (WT) littermates were used. Humanized mice [hPMP22-C3 Tg/mPmp22 KO (ho/ho); homo-humanized, hPMP22-C3 Tg/mPmp22 KO (he/ho); hetero-humanized] were generated by crossing the PMP22-C3 mouse line with mPmp22 KO mouse line that lacks the full length (31 kbp) mPmp22 gene using the CRISPR/Cas9 method and supplied by ADDP. In detail, hPMP22-C3 Tg /mPmp22 KO (he/he) mice were initially generated by crossing hPMP22-C3 Tg (hetero) mice with mPmp22 KO (homo) mice using In Vitro Fertilization method. Then hPMP22-C3 Tg /mPmp22 KO (he/he) mice were crossed with hPMP22-C3 Tg /mPmp22 KO (wt/he) mice to generate hPMP22-C3 Tg /mPmp22 KO (he/ho) mice. Consequently, hPMP22-C3 Tg /mPmp22 KO (he/ho) mice were bred to generate hPMP22-C3 Tg /mPmp22 KO (ho/ho) mice. The ratio of these mouse lines was broadly consistent with Mendel’s Laws. Genotyping analysis using PCR method (50 °C for 2 min, then 95 °C for 20 s, following 40 cycles of 95 °C, 1 s and 60 °C, 20 s) was conducted to identify the genotypes of each mouse. The following primer sets were used: mPmp22 (Forward: 5′-GCGTCCTCTTTCATGAATCAAAA, Reverse: 5′-TCTACCCCTAACCTCTGGTTCCT, Probe: 5′-CATTCACGAATTGCC; Thermo Fisher Scientific, Waltham, MA, USA), hPMP22 (Forward: 5′-GTGCTGCGGCCATCTACAC, Reverse: 5′-GTAGGCGAAACCGTAGGAGTAATC, Probe: 5′-CCGGAGTGGCATCT; Thermo Fisher Scientific). The phenotypic characteristics of these two humanized mice were stable across generations. As the control group for humanized models, age-matched C57BL/6 J Jcl mice (WT), the background mouse line of PMP22-C3 mice and mPmp22 KO mice were purchased from CLEA Japan, Inc. (Tokyo, Japan). The mice were housed in groups with unrestricted access to food and water and maintained under a 12-h light/dark cycle (lights on at 7:00 a.m.) at a temperature of 22 ± 1 °C. To study the natural history of NCV and CMAP, mice were subjected to continuous NCV and CMAP recording at 6, 10, 14, and 18 weeks of age from the same individuals (n = 8–10/group). The mice were then sacrificed, and histopathological analysis and evaluation of mRNA expression were performed. Behavioral assessments and electrical impedance myography (EIM) measurements were performed at 17 and 18 weeks of age, respectively (n = 8–10/group). Another group of 14-week-old humanized and PMP22-C3 mice was used for the PMP22 protein assay (n = 4/group). The overall experimental schedule is shown in Figure 1. All experimental procedures in this study were conducted in accordance with animal care protocols approved by the Institutional Animal Care and Use Committee of Shonan Health Innovation Park (protocol number: AU-00031034 and AU-00031278), in compliance with Act on Welfare and Management of Animals (Act No. 105 of October 1, 1973), harmonized with the American Association for Accreditation of Laboratory Animal Care (AAALAC) International.
Figure 1

Experimental schedule for characterization of humanized mice. NCV and CMAP assessments were conducted at 6, 10, 14, and 18 weeks of age. Plasma NfL levels were measured at 9, 14, and 18 weeks of age. PMP22 protein levels were investigated at 14 weeks. Analyses of mRNA levels of PMP22 and myelin-related genes utilized samples from 18-week-old mice. Behavioral testing and EIM measurements were performed at 17 and 18 weeks of age, respectively. Histopathological examinations of the sciatic nerves and GC muscles were conducted at 18 weeks of age.
2.2 Messenger RNA extraction and real-time quantitative polymerase chain reaction (RT-qPCR)
Mice were anesthetized via intraperitoneal injection of a combination of anesthetics (medetomidine, 0.3 mg/kg; midazolam, 4 mg/kg; and butorphanol, 5 mg/kg) and then euthanized by exsanguination. The left sciatic nerve was removed and homogenized using a FastPrep-24 5G homogenizer (MP Biochemicals LLC, Irvine, CA, USA) in Lysing Matrix I tubes (6918–100, MP Biochemicals LLC) filled with QIAzol Lysis Reagent (79,306, Qiagen, Hilden, Germany). Total RNA was extracted using the RNeasy Plus Universal Mini Kit (73,404, Qiagen) according to the manufacturer’s instructions. Total RNA (0.1 μg) was used for complementary DNA synthesis using SuperScript IV VILO Master Mix (11,756,500, Thermo Fisher Scientific). RT-qPCR was conducted using QuantStudio (Thermo Fisher Scientific) with TaqPath qPCR Master Mix, CG (A15297; Thermo Fisher Scientific). Relative copy numbers of mPmp22 and hPMP22 were calculated. The mRNA expression levels of myelin-related genes were normalized to those of Gldn, a Schwann cell-specific gene, and compared with each WT group. The following primer sets were used: mPmp22 (Forward: 5′-CTTCCAAATCCTTGCTGGTCTG, Reverse: 5′-GGATGTAGGCGAAGCCATAGG, Probe: 5′-ATGCCACTCACTGTGCCTCACTGTGTAGAT; Sigma-Aldrich, Saint Louis, USA), hPMP22 (Forward: 5′-CTTGCTGGTCTGTGCGTGAT, Reverse: 5′-ACCGTAGGAGTAATCCGAGTTGAG, Probe: 5′-CATCTACACGGTGAGGCACCCGG; Sigma-Aldrich), Sc5d (Mm. PT.58.33540072; IDT, San Jose, CA, USA), Mpz (Mm. PT.58.5771188; IDT), Pou3f1 (Mm. PT.58.33607006. g; IDT), Gldn (Mm. PT.58.43925599; IDT).
2.3 Quantitative evaluation of mPmp22 and hPMP22 protein expression
After euthanasia by exsanguination, the left sciatic nerve was removed. 2.5% homogenate of sciatic nerve was homogenized by Precellys Evoluation (Beritin, France) with lysis buffer including 50 mM Tris pH 7.5, 4% sodium dodecyl sulfate, 150 mM NaCl, Complete (04693132001, Roche, Switzerland), and PhosSTOP (04906837001, Roche). After incubation at room temperature (20–25 °C) for 30 min, the samples were subjected to ultrasonication at 40 kHz for 10 min and then incubated at 95 °C for 10 min using an Eppendorf ThermoMixer C (Eppendorf, Germany). After cooling to room temperature, the samples were centrifuged at 21,600 × g for 10 min at room temperature. The protein concentration in the supernatant was measured using a bicinchoninic acid assay. Sciatic nerve (7.5 μg) was mixed with 1 M phosphoric acid buffer (10 μL), 5 M NaCl (10 μL), and cold acetone (0.5 mL) at −30 °C for 2 h. After centrifugation (21,600 × g, 4 °C, 20 min), the supernatant was completely removed and the precipitant was kept at room temperature for 15 min. The precipitant was dissolved in 8 M urea (50 μL), 0.5 M Tris(2-carboxyethyl) phosphine (5 μL), and 0.5 M iodoacetamide (5 μL) and incubated for 15 min at room temperature. This was followed by sonication for 10 min and a subsequent incubation for 10 min at 37 °C. Next, 800 μL of 0.1 M Tris–HCl (pH 8.0) was added, and proteolytic digestion was performed using 12.5 μg of chymotrypsin (Promega, WI, USA) overnight at 25 °C. The resulting sample was mixed with 4% phosphoric acid (200 μL) and 100 mg/mL of internal standard peptides (10 μL), then purified via solid-phase extraction using Oasis MCX cartridges (Waters, MA, USA). The final eluted solution was dried by nitrogen gas. The residue was reconstituted in 200 μL of a formic acid/acetonitrile/purified water (1,40,960, v/v/v), and an aliquot of 0.2 μL was injected into an EASY-nLC 1,200 system/Q Exactive HF-X Orbitrap mass spectrometer (Thermo Fisher Scientific). The signature peptides were monitored in parallel reaction monitoring mode. The detailed analytical conditions are listed in Supplementary Tables S1, S2.
2.4 NCV and CMAP measurements
NCV and CMAP were recorded from the right sciatic nerve of WT and humanized mice using a Natus UltraPro S100 (Natus Medical Inc., Middleton, WI, USA). Disposable monopolar needle electrodes (TECA® Elite, 902-DMF25-TP (28G), Natus Medical Inc.) were used for stimulating and recording. Mice were anesthetized with 1.5–2.0% isoflurane on a heating pad to maintain body temperature. To record the NCV and CMAP, a sensing recording electrode was inserted into the tibialis anterior muscle, and the reference electrode was inserted into the ipsilateral plantar muscle. For stimulation, the stimulating cathodes were subcutaneously placed at the sciatic nerve, 4 mm (distal) and 16 mm (proximal) from the sensing recording electrode. The anode was inserted into the contralateral plantar muscle. Square-wave pulses of 0.1 ms duration were delivered to the distal and proximal sites at a 0.5 Hz frequency, and each CMAP was measured at supramaximal stimulation. The NCV was calculated from the difference in peak latencies between the distal and proximal CMAP responses and the distance (12 mm) between the stimulating electrodes. The CMAP evoked by proximal stimulation was used to analyze the CMAP amplitude.
2.5 Sciatic nerve histopathology
The right sciatic nerve was immersed in 4% paraformaldehyde phosphate buffer solution (163–20,145, FUJIFILM Wako Pure Chemical Corp., Osaka, Japan) for 5–7 days at 4 °C. The fixative was replaced with 0.01 mol/L phosphate-buffered saline and stored at 4 °C until the histopathological analysis. For morphometric assessment of axons and myelin, the sciatic nerve was embedded in resin and sectioned transversely (0.5 μm thick), and stained with toluidine blue (104,172, Merck, Darmstadt, Germany). Digital images were obtained using a digital slide scanner (NanoZoomer S60; Hamamatsu Photonics, Shizuoka, Japan), and the inner and outer areas/diameters of the axon and myelin were subsequently analyzed using HALO image analysis software (Indica Labs, Albuquerque, NM, USA). The g-ratio of each axon was calculated as a ratio between axon outer diameter and inner diameter.
2.6 Measurement of plasma NfL levels
Blood samples were collected from the jugular vein of the mouse, which was gently restrained by hand, at 9 and 14 weeks of age, and from the abdominal vena cava of the mouse anesthetized with the combination anesthetic described above at 18 weeks of age. Approximately 200 μL of whole blood was collected using a 1 mL syringe with a 27-guage needle coated with 0.1 mol/L EDTA-2Na (344–09255, Dojindo Laboratories, Kumamoto, Japan) and kept on ice. After the centrifugation at 17,400 × g at 4 °C for 1 min, 40 μL of plasma was collected into a Protein LoBind® Tube (0030108434, Eppendorf), then chilled on dry ice and stored at −80 °C until analysis. NfL levels in plasma were measured using an HD-X analyzer (Quanterix Corporation, Massachusetts, USA) with a Simoa NF-Light™ V2 Advantage Kit (104,073, Quanterix) according to the manufacturer’s instructions.
2.7 Behavioral assessments
2.7.1 Grip strength test
The grip strengths of the forelimbs and hindlimbs were measured using a grip strength meter (MK-380CM; Muromachi Kikai Co., Ltd., Tokyo, Japan). The apparatus consisted of a triangular metal bar or a T-shaped metal bar connected to a strength transducer to measure the grip strength of the forelimbs or hindlimbs, respectively. Each mouse was held by its tail and forced to grasp the metal bar with its forelimbs or hindlimbs. The mouse was then gently pulled backward with a consistent force until the grip was lost. This step was repeated five times, and the data were averaged after excluding the maximal and minimal values.
2.7.2 Beam walking test
The beam walking test was performed to evaluate gait and balance. A square metal bar (width 1.2 cm, length 85 cm) with a dark box at the arrival point was placed 60 cm above the ground. Each mouse was habituated to the box for 5 min and then trained to walk to the box five times. The test session was performed once, at least 1 h later. The mice were placed at the departure point and allowed to walk to the box. The transit time and the number of slips were measured. Trials in which the mouse fell off, paused (over 5 s), or ran backward were excluded, and the mouse was retested.
2.7.3 Rotarod test
To evaluate motor coordination, the mice were tested on a rotarod apparatus (MK-670; Muromachi Kikai Co., Ltd., Tokyo, Japan). Each mouse was trained on a rotating rod (three 5-min sessions at 4, 8 and 15 rpm). During the test trials (at least 1 h later), each mouse was placed on the rotarod at increasing speeds from 4 to 40 rpm for 300 s. The test trials were performed three times, at least 15 min apart. The latency to fall off the rotarod during Trials 2 and 3 was recorded, and the mean value was used in the analysis.
2.8 EIM measurement
EIM measurements were performed as previously described (24). Briefly, mice were anesthetized with 1.5–2.0% isoflurane and placed on a heating pad to maintain their body temperature. The left hindlimb was shaved and pretreated with skin preparation gel (Nuprep®; Weaver and Company, Aurora, CO, USA) to reduce impedance, and positioned at a 45° angle. After application of an electrode cream (SignaCreme®; Parker Labs, Fairfield, NJ, USA), a four-electrode array with a weight of 20 g was placed over the gastrocnemius (GC) muscle in the longitudinal direction and then, a weak electrical current with multi-frequencies was applied. The EIM parameters (reactance, resistance, and phase) were obtained using an impedance spectroscopy system (mScan-V™; Myolex Inc., Boston, MA, USA). Measurements were performed three times and averaged within the 1–10,000 kHz range. Values from 10 to 1,000 kHz were used for the analysis to avoid artifacts.
2.9 Skeletal muscle histopathology
The hindlimbs were removed from each mouse and immersed in 4% paraformaldehyde phosphate buffer solution (163–20,145; FUJIFILM Wako Pure Chemical Corp., Osaka, Japan). After 24 h, the fixative was replaced with 0.01 mol/L phosphate-buffered saline and stored at 4 °C. To assess muscle pathology, GC muscle tissues were assessed using laminin immunohistochemical staining as previously described (24). Digital images were obtained using a digital slide scanner (NanoZoomer S60; Hamamatsu Photonics, Shizuoka, Japan), and the cross-sectional area (CSA) and muscle fiber count were subsequently analyzed using HALO image analysis software (Indica Labs).
2.10 Data analysis
Statistical analyses were performed using the SAS software (version 9.4; SAS Institute Japan, Tokyo, Japan). For multiple comparison for PMP22 mRNA and protein levels, one-way ANOVA with a post hoc Bonferroni’s test was utilized. To compare two groups genetic differences between humanized or PMP22-C3 and WT mice, data were assessed using Student’s t-test for homogeneous data or Mann–Whitney U test for nonhomogeneous data. For the analysis of repeated measures data of NAV, CMAP, and plasma NfL, two-way Repeated Measures ANOVA with a post hoc Bonferroni’s test was performed. Statistical significance was set at p < 0.05. Data were reported as the mean ± standard error of the mean (SEM).
3 Results
3.1 Expression of mPmp22 or hPMP22 mRNA and protein in sciatic nerves of humanized mice and PMP22-C3 mice
To confirm the genetic characteristics of humanized mice, we investigated the mRNA expression of mPmp22 and hPMP22 in the sciatic nerves of WT, humanized, and PMP22-C3 mice at 18 weeks of age using RT-qPCR. mPmp22 was not detected in hetero-humanized or homo-humanized mice, whereas WT and PMP22-C3 mice showed similar levels of intrinsic mPmp22 expression (Figure 2A). The expression level of hPMP22 in hetero-humanized mice was similar to that in PMP22-C3 mice. In contrast, homo-humanized mice showed higher levels of hPMP22 expression than PMP22-C3 or hetero-humanized mice (2.5-fold increase compared to PMP22-C3 mice). Consistent with the mRNA expression of mPmp22 and hPMP22, hetero- and homo-humanized mice showed no expression of mPmp22 protein, and homo-humanized mice exhibited a higher level of hPMP22 protein than PMP22-C3 mice or hetero-humanized mice (2.3-fold increase compared to PMP22-C3 mice) (Figure 2B). These data confirmed that humanized mice had no mPmp22 gene or protein and that gene dosage-dependent hPMP22 mRNA and protein expression was observed in the sciatic nerve.
Figure 2

Expression of mouse Pmp22 and human PMP22 mRNA and protein in the sciatic nerve of WT, humanized, and PMP22-C3 mice. (A) Relative copy numbers of mouse Pmp22 and human PMP22 mRNA or (B) relative amounts of mouse Pmp22 and human PMP22 protein were analyzed in the sciatic nerve of WT (C57BL/6 J), homo-humanized, hetero-humanized, and PMP22-C3 mice (n = 4/group) at 18 or 14 weeks of age, respectively. Data are shown as the ratio to PMP22-C3 mice and as mean ± SEM. Statistical comparisons were performed using one-way ANOVA with a post hoc Bonferroni’s test (**p < 0.01, ***p < 0.001 vs. PMP22-C3 mice). WT, wild-type; SEM, standard error of the mean.
3.2 Alterations in transcript levels of myelin-related genes in humanized mice
Transcriptional changes in several myelin-related genes, such as Sc5d, Mpz, and Pou3f1, were investigated in the sciatic nerves of humanized and PMP22-C3 mice because changes in the transcript levels of these genes have been reported in the CMT1A rodent model (19). The mRNA expression of these genes in the sciatic nerve was assessed using RT-qPCR at 18 weeks of age. In homo-humanized mice, the transcript levels of Sc5d and Mpz were decreased, and Pou3f1 was increased relative to those in WT mice (Sc5d: 0.46-fold reduction, Mpz: 0.58-fold reduction, Pou3f1: 3.2-fold increase; p < 0.005) (Figure 3A), consistent with those in PMP22-C3 mice (Figure 3C) and a previous report (19). In contrast, hetero-humanized mice showed transcript levels of these genes comparable to those in WT mice (Figure 3B). These data suggest that the mRNA expression levels of myelin-related genes changed depending on PMP22 levels in the sciatic nerve of humanized mice.
Figure 3

Quantitative analysis of myelin-related genes in the sciatic nerve of humanized mice and PMP22-C3 mice. Transcript levels of myelin-related genes (Sc5d, Mpz, and Pou3f1) were analyzed in the sciatic nerves of WT, (A) homo-humanized, (B) hetero-humanized, and (C) PMP22-C3 mice at 18 weeks of age using RT-qPCR. Data are shown as the ratio to the WT group and as mean ± SEM (n = 4/group). Statistical comparisons were performed using Mann–Whitney U test (*p < 0.05 vs. WT mice). RT-qPCR, real-time quantitative polymerase chain reaction; WT, wild-type; SEM, standard error of the mean.
3.3 Longitudinal deficits in NCV and CMAP in humanized mice
Patients with CMT1A and animal models of this disease exhibit electrophysiological impairment of peripheral nerves due to demyelination (9, 15, 21, 22). To investigate functional changes in the peripheral nerves, NCV and CMAP were repeatedly measured in humanized and PMP22-C3 mice at 6, 10, 14, and 18 weeks of age. Individual values of NCV and CMAP are shown in Supplementary Table S3. Robustly slower NCV (6.8, 7.1, 8.5, and 9.7% of WT mice at 6, 10, 14, and 18 weeks of age, respectively; p < 0.01 or 0.001) and reduced CMAP amplitude (21.7, 24.0, 20.4, and 32.3% of WT mice at 6, 10, 14, and 18 weeks of age, respectively; p < 0.001) were observed in homo-humanized mice compared to WT mice (Figures 4A,D), reflecting demyelination in the sciatic nerve and indicating an abnormality in the lower motor unit of this model. These results were comparable to those observed in the PMP22-C3 mice (Figures 4C,F). In contrast, hetero-humanized mice showed no differences from WT mice in NCV (102.2, 107.4, 105.5, and 99.7% of WT mice at 6, 10, 14, and 18 weeks of age, respectively) or CMAP (103.0, 99.6, 105.2, and 106.0% of WT mice at 6, 10, 14, and 18 weeks of age, respectively) at all ages (Figures 4B,E). These findings suggest that homo-humanized mice show peripheral nerve dysfunction similar to that in PMP22-C3 mice, which depends on the total amount of PMP22 expression.
Figure 4

Longitudinal data of NCV and CMAP in humanized mice and PMP22-C3 mice. NCV and CMAP were measured repeatedly at 6, 10, 14, and 18 weeks of age in the WT, (A,D) homo-humanized, (B,E) hetero-humanized, and (C,F) PMP22-C3 mice. Data are represented as mean ± SEM (n = 8–10/group). Statistical comparisons were performed using two-way Repeated Measures ANOVA with a post hoc Bonferroni’s test (**p < 0.01, ***p < 0.001 vs. WT mice). NCV, nerve conduction velocity; CMAP, compound muscle action potential; WT, wild-type; SEM, standard error of the mean.
3.4 Morphological changes in the sciatic nerve of humanized mice
As homo-humanized and PMP22-C3 mice showed changes in myelin-related genes and demyelination-related nerve dysfunction in the sciatic nerve, histopathological analyses of the sciatic nerve were performed using these mice at 18 weeks of age. In homo-humanized mice, many demyelinated axons were observed (Figure 5A, middle panel, arrowhead), and quantitative analysis revealed a smaller myelin area (58.3% of WT mice, p < 0.001) than in WT mice (Figure 5B). To further investigate the characteristics of demyelination in homo-humanized mice, the g-ratio of each axon was calculated and analyzed by axon size (Figure 5C). Homo-humanized mice exhibited higher g-ratio, especially in larger axons, consistent with previous reports for PMP22-C3 mice (25, 26). In addition, the axonal inner (54.4% of WT mice, p < 0.001) and outer areas (56.8% of WT mice, p < 0.001) were significantly smaller in homo-humanized mice than in WT mice (Figure 5B), and the distribution of axon size in homo-humanized mice shifted toward smaller compared to that in WT mice (Figure 5D), indicating axonal atrophy in the sciatic nerve of this model. These morphological changes were comparable to those observed in the PMP22-C3 mice (Figure 5A, right panel, E–G).
Figure 5
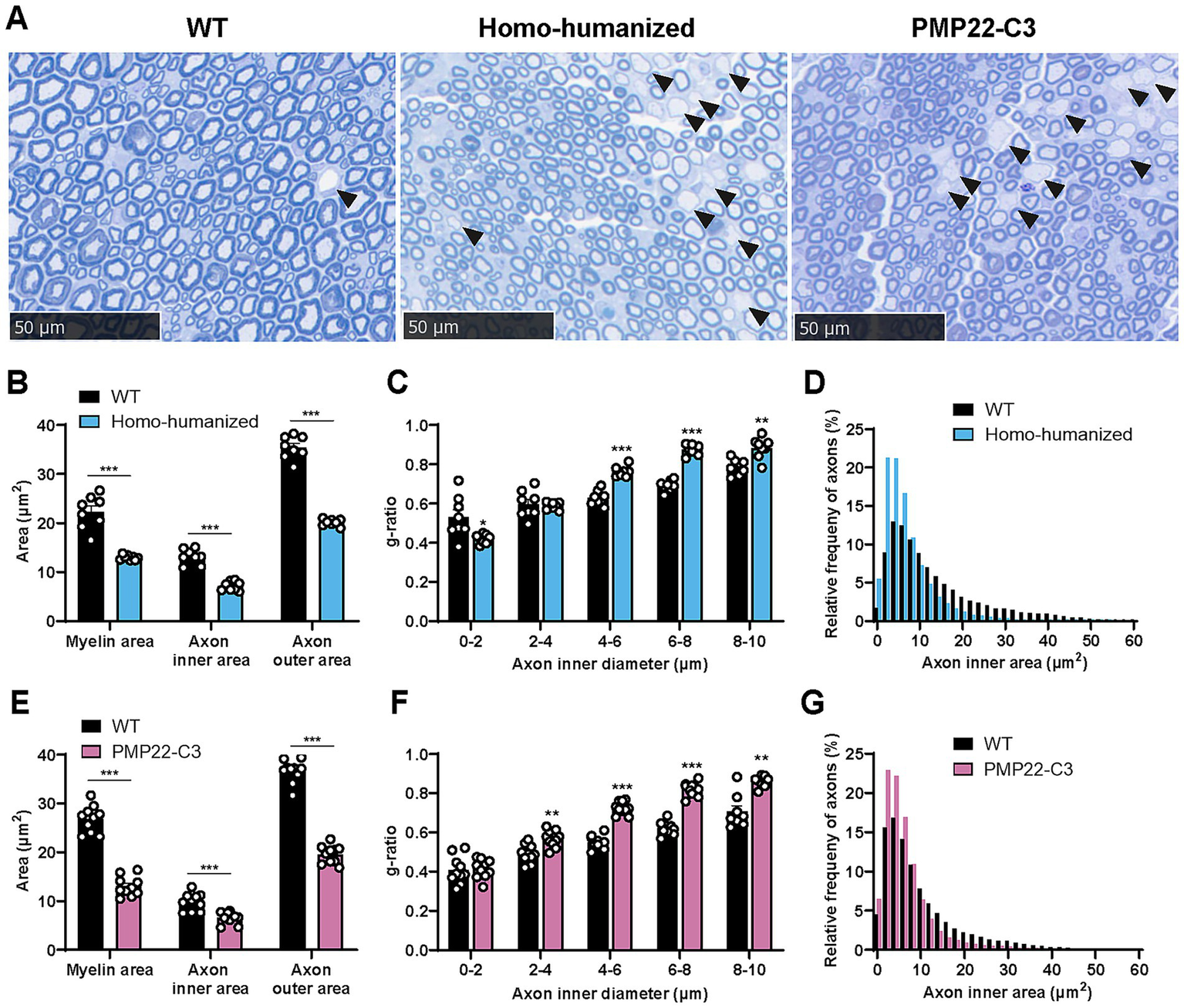
Histopathological analysis in the sciatic nerve of homo-humanized mice and PMP22-C3 mice. (A) Representative images of toluidine blue-stained sciatic nerve in WT, homo-humanized, and PMP22-C3 mice at 18 weeks of age. Demyelinated axons are pointed with black arrowheads. (B,E) Myelin area, axon inner area, and outer area in WT and (B) homo-humanized mice or (E) PMP22-C3 mice. (C,F) The g-ratio of each axon in WT and (C) homo-humanized mice or (F) PMP22-C3 mice was analyzed by axon size. (D,G) Comparison of histograms for axon inner area in the sciatic nerve of WT and (D) homo-humanized mice or (G) PMP22-C3 mice. Analyzed axon frequencies from all animals are plotted for each given axon inner diameter with a 2 μm2-bin. Data are represented as mean ± SEM (n = 8–10/group). Statistical comparisons were performed using Mann–Whitney U test (*p < 0.05, **p < 0.01, ***p < 0.001 vs. WT mice). WT, wild-type; SEM, standard error of the mean.
3.5 Changes in plasma NfL levels in humanized mice
NfL is considered to reflect axonal damage (27), and patients with CMT1A show increased levels of NfL in the blood (28). As axonal atrophy was observed in the sciatic nerves of homo-humanized and PMP22-C3 mice at 18 weeks of age, we investigated plasma NfL levels in these mice at 18 weeks of age and two other earlier points (9 and 14 weeks of age). Individual values of plasma NfL are shown in Supplementary Table S4. Homo-humanized mice showed significantly higher plasma NfL levels than those in WT mice at all ages (8.3-, 5.3-, and 4.2-fold increases at 9, 14, and 18 weeks of age, respectively; p < 0.001) (Figure 6A), as well as PMP22-C3 mice (Figure 6C), implying that axonal atrophy occurs earlier than 18 weeks of age. In contrast, hetero-humanized mice showed no statistically significant difference from WT mice (Figure 6B).
Figure 6
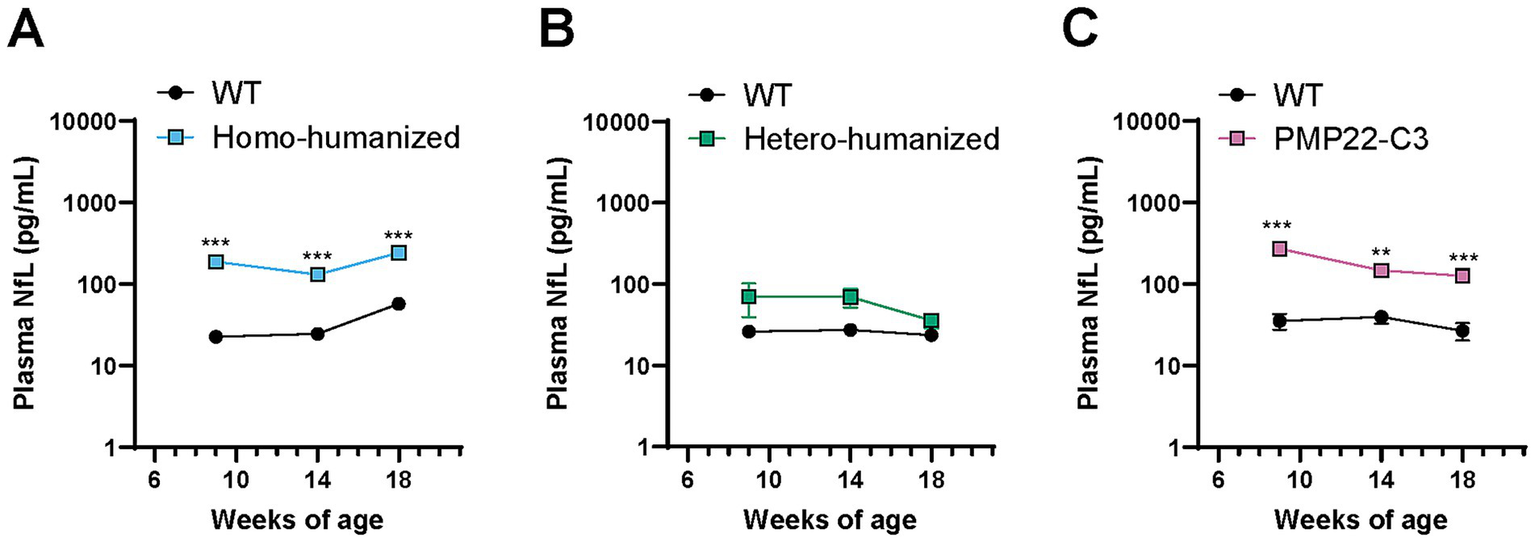
Quantitative analysis of plasma NfL levels in humanized mice and PMP22-C3 mice. Plasma NfL levels were measured using the SiMoA immunoassay in WT, (A) homo-humanized, (B) hetero-humanized, and (C) PMP22-C3 mice at 9, 14, and 18 weeks of age. Data are represented as mean ± SEM (n = 8–10/group). Statistical comparisons were performed using two-way Repeated Measures ANOVA with a post hoc Bonferroni’s test (**p < 0.01, ***p < 0.001 vs. WT mice). WT, wild-type; NfL, neurofilament light chain; SiMoA, single molecule array; SEM, standard error of the mean.
3.6 Motor disabilities in humanized mice
Motor disabilities such as muscle weakness and impaired balance are common features of patients with CMT1A (29, 30) and mouse models (19, 23, 31, 32). To evaluate whether peripheral nerve dysfunction affects motor ability in the humanized mouse model, we performed three behavioral assessments (grip strength, beam walking, and rotarod tests) at 17 weeks of age.
Homo-humanized mice showed a weaker grip strength in both forelimb (WT: 163.6 ± 3.4 g, homo-humanized: 130.6 ± 3.2 g, p < 0.001) and hindlimb (WT: 58.8 ± 2.4 g, homo-humanized: 14.7 ± 2.4 g, p < 0.001) than those in WT mice (Figure 7A), while hetero-humanized mice showed a similar level of grip strength as WT mice in both forelimb (WT: 164.3 ± 4.9 g, hetero-humanized: 175.0 ± 3.3 g) and hindlimb (WT: 64.4 ± 2.5 g, hetero-humanized: 70.2 ± 2.3 g) (Figure 7B).
Figure 7
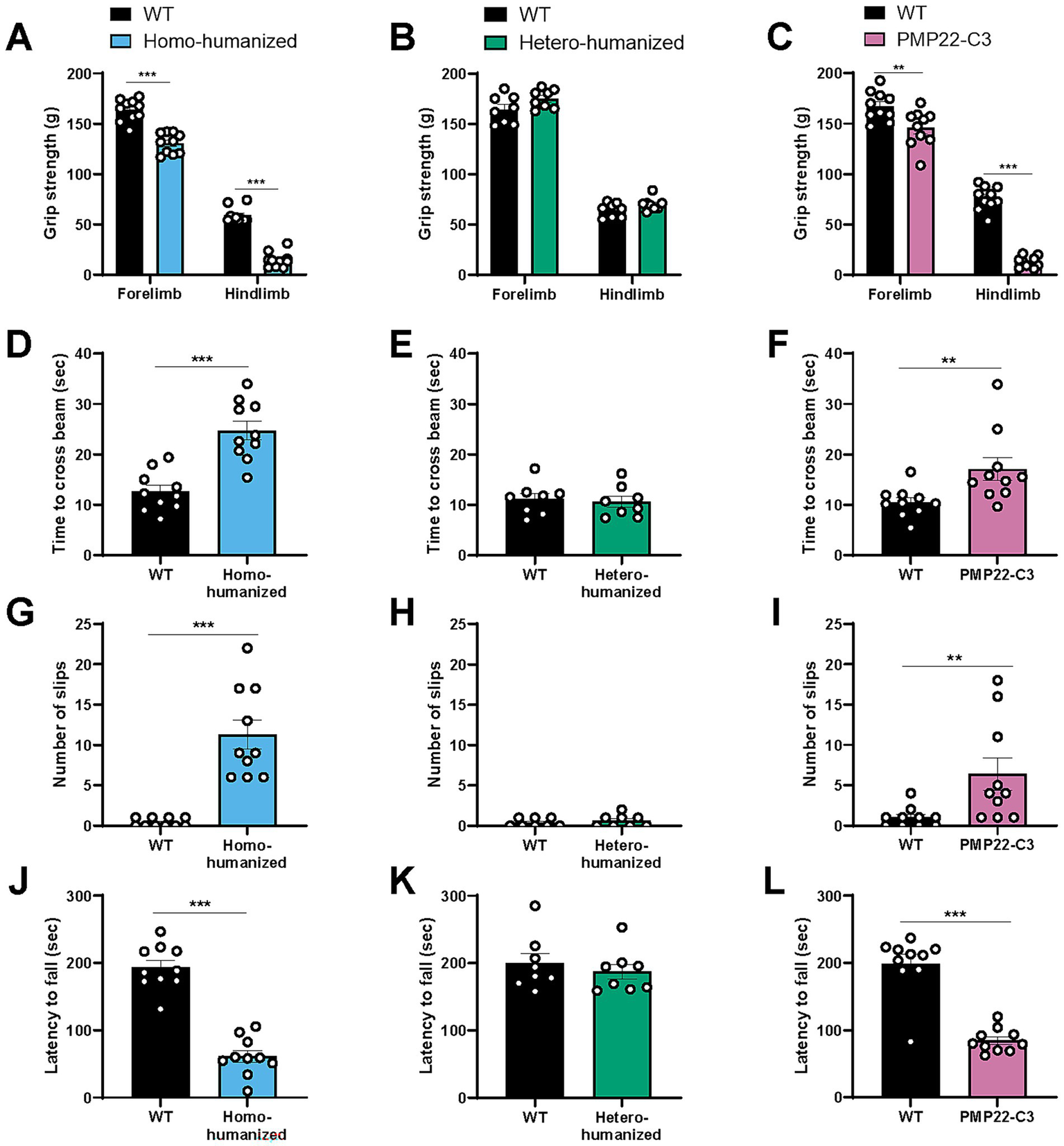
Behavioral characteristics in humanized mice and PMP22-C3 mice. Behavioral assessments were performed at 17 weeks of age. (A-C) Grip strength of forelimbs and hindlimbs in WT, (A) homo-humanized, (B) hetero-humanized, and (C) PMP22-C3 mice. Individual data, excluding the maximal and minimal values from five measurements, were averaged. (D–I) Time to cross the beam and slip counts of WT, (D,G) homo-humanized, (E,H) hetero-humanized, and (F, I) PMP22-C3 mice in the beam walking test. (J–L) Latency to fall off the rotating rod in WT, (J) homo-humanized, (K) hetero-humanized, and (L) PMP22-C3 mice in the rotarod test. Each mouse was tested three times, and the data from Trials 2 and 3 were averaged. Data are represented as mean ± SEM (n = 8–10/group). Statistical comparisons were performed using Mann–Whitney U test (**p < 0.01, ***p < 0.001 vs. WT mice). WT, wild-type; SEM, standard error of the mean.
In the beam walking test that assesses the motor balance and gait, WT mice typically crossed the beam in approximately 12 s with less than one slip, while homo-humanized mice took longer to cross the beam (WT: 12.6 ± 1.2 s, homo-humanized: 24.7 ± 1.9 s, p < 0.001) (Figure 7D) with many slips (WT: 0.40 ± 0.16, homo-humanized: 11.3 ± 1.8, p < 0.001) (Figure 7G), indicating the abnormal motor balance and gait. In contrast, no difference was observed in crossing time (WT: 11.2 ± 1.1 s, hetero-humanized: 10.6 ± 1.1 s) (Figure 7E) or slip counts (WT: 0.38 ± 0.18, hetero-humanized: 0.63 ± 0.26) (Figure 7H) between WT mice and hetero-humanized mice.
The motor coordination was tested using a rotarod. Homo-humanized mice dropped off the rotating rod much earlier than WT mice (WT: 193.3 ± 10.5 s, homo-humanized: 61.1 ± 9.0 s, p < 0.001) (Figure 7J). In contrast, hetero-humanized mice showed a latency comparable to that of WT mice (WT: 199.8 ± 14.3 s, hetero-humanized: 187.1 ± 11.2 s) (Figure 7K).
Similar to homo-humanized mice, PMP22-C3 mice showed weaker grip strength in both forelimbs and hindlimbs, longer crossing time and slipping on the beam, and shorter latency to fall off the rod (Figures 7C,F,I,L). These behavioral findings suggested that homo-humanized mice exhibited motor disabilities similar to those of PMP22-C3 mice, whereas hetero-humanized mice maintained normal motor function. This is consistent with the morphological and functional peripheral nerve characteristics observed in all three mouse lines.
3.7 Alteration in EIM parameters in the GC muscle of humanized mice
We recently reported that PMP22-C3 mice exhibited muscle weakness and alterations in EIM parameters in the GC muscle innervated by tibial nerve branched off from the sciatic nerve (24). To further confirm that muscle weakness can be detected as changes in muscle composition, EIM data were obtained from the GC muscles of WT, homo-humanized, and hetero-humanized mice at 18 weeks of age. Individual values of EIM parameters are shown in Supplementary Table S5. Homo-humanized mice showed obvious alterations in EIM parameters (Figure 8A), which were consistent with EIM changes in PMP22-C3 mice (24). Statistical comparison using EIM parameters at 50 kHz revealed significant decreases in reactance (79.1% of WT mice, p < 0.05) and phase (69.0% of WT mice, p < 0.001) and an increase in resistance (119.0% of WT mice, p < 0.001) in homo-humanized mice than those in WT mice (Figure 8B). In contrast, hetero-humanized mice showed similar values for all parameters as WT mice (Figure 8C), and no statistical difference in the 50 kHz parameters was detected (Figure 8D). As decreased EIM reactance and phase or increased EIM resistance indicate a reduction in muscle fiber size or muscle mass, respectively (33–35), homo-humanized mice have skeletal muscular deterioration, whereas hetero-humanized mice maintain a healthy muscle state.
Figure 8

EIM parameters in the GC muscle of humanized mice. Multi-frequency data (10–1,000 kHz) for the EIM parameters were obtained from the GC muscles of WT, (A) homo-humanized, and (C) hetero-humanized mice at 18 weeks of age. Comparison of EIM parameters at 50 kHz between WT and (B) homo-humanized or (D) hetero-humanized mice. The EIM data are shown as three parameters: reactance (left), resistance (middle), and phase (right). Data are represented as mean ± SEM (n = 8/group). Statistical comparisons were performed using Mann–Whitney U test (**p < 0.05, **p < 0.001 vs. WT mice). EIM, electrical impedance myography; WT, wild-type; SEM, standard error of the mean.
3.8 Morphological changes in the GC muscle of homo-humanized mice
The histopathology of the GC muscle of homo-humanized mice was investigated at 18 weeks of age to identify morphological changes in muscle fibers underlying EIM alterations. A higher number of thin muscle fibers were observed in homo-humanized mice than in WT mice (Figure 9A). In WT mice, the CSA histogram of individual muscle fibers showed a wider distribution pattern with a peak at approximately 1,300 μm2, while homo-humanized mice shifted the distribution to a right-skewed pattern with a peak at the smallest muscle fiber group in the GC muscle (Figure 9B). This distribution change in muscle fibers led to a significant decrease in the average CSA of the GC muscle of homo-humanized mice compared to that in WT mice (65.0% of WT mice, p < 0.001) (Figure 9C). These data indicate muscle fiber atrophy in the GC muscles of homo-humanized mice, resulting in alterations in EIM parameters.
Figure 9
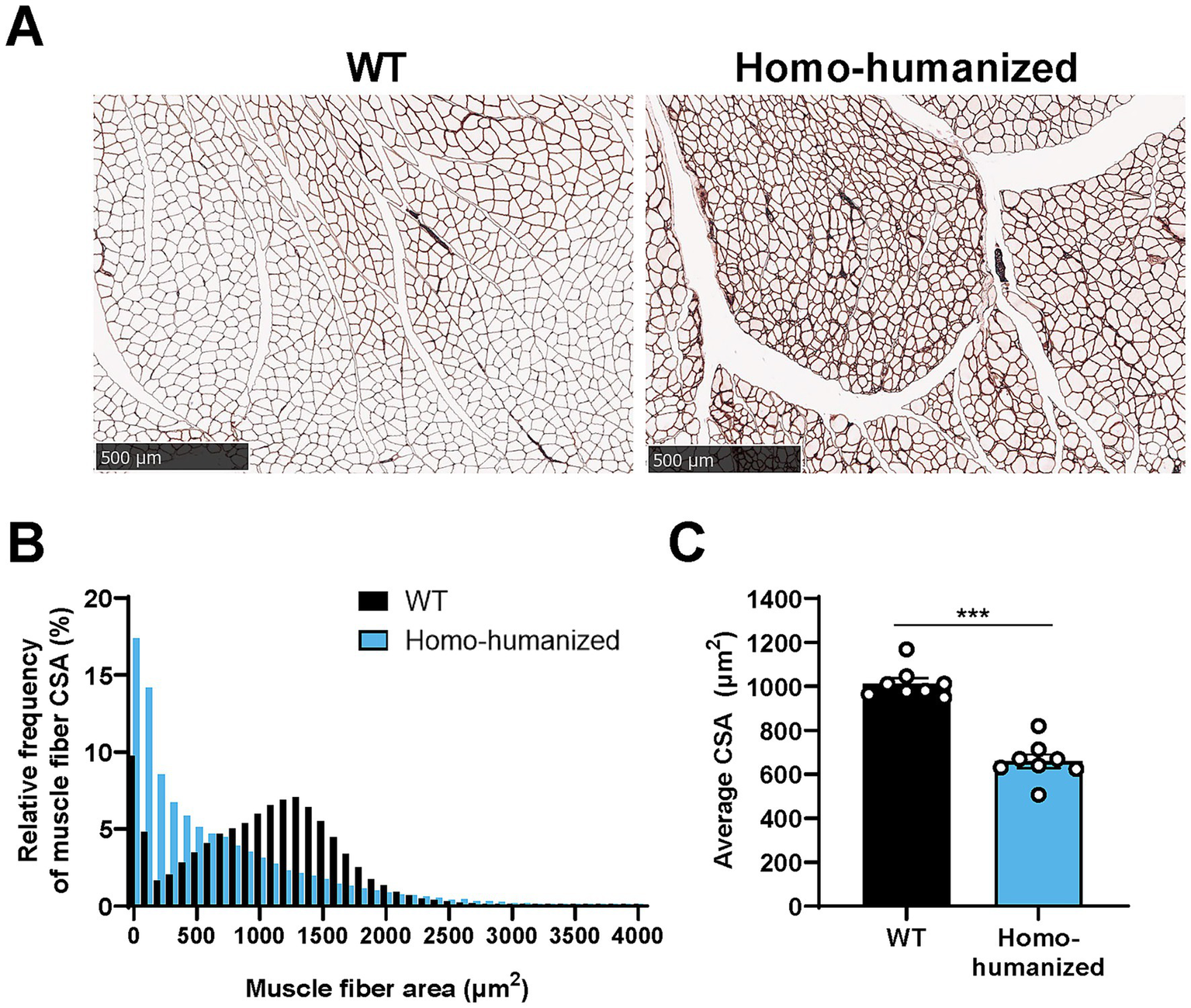
Histopathological analysis of the GC muscle in homo-humanized mice. (A) Representative images of laminin-stained cross sections of the GC muscle in WT and homo-humanized mice at 18 weeks of age. (B) Comparison of histograms for muscle fiber CSA in the GC muscle of WT and homo-humanized mice. Analyzed muscle fiber frequencies from all animals are plotted for each given muscle fiber CSA with a 100 μm2-bin. (C) Average muscle fiber CSA in the GC muscle calculated from individual animal values. Data are represented as mean ± SEM (n = 8/group). Statistical comparisons were performed using Student’s t-test (***p < 0.001 vs. WT mice). GC, gastrocnemius; WT, wild-type; CSA, cross-sectional area; SEM, standard error of the mean.
4 Discussion
In this study, we characterized two humanized PMP22-C3 mouse lines as novel animal models of CMT1A and investigated their utility. Our biochemical, electrophysiological, histopathological, and behavioral studies demonstrated that homo-humanized mice exhibited various aspects of CMT1A pathology.
Quantitative assessments of PMP22 mRNA and/or PMP22 protein expression levels, compared to the gold-standard PMP22-C3 mouse model, are important for validating the reliability of our newly generated humanized mouse lines. Although PMP22 protein expression can be detected using immunoassays such as western blotting (20), there is room for improvement in its quantitative accuracy. In addition, because PMP22-C3 mice express hPMP22 and mPmp22, the selective detection of each protein is required. In this study, we established a highly sensitive and selective quantitative method using nanoflow liquid chromatography-high-resolution mass spectrometry (nLC-HRMS) for measuring PMP22 protein levels. The chromatogram in Supplementary Figure S1 shows that nLC-HRMS specifically detects hPMP22 and mPmp22. The quantified levels of PMP22 protein correlated with its mRNA levels across mouse models with different genotypes, both hetero- and homo-transgenic, in a gene dosage-dependent manner (Figure 2). Considering that knockdown by PMP22 mRNA-targeting treatment correlates with phenotypic improvement in CMT1A animal models (19), quantitative measurements are important for estimating the disease state and its change by therapeutic intervention in CMT1A. The mRNA turnover rate is higher than that of protein (36). Therefore, quantifying the PMP22 protein is useful for understanding the time lag between mRNA and protein reduction caused by PMP22-targeting drug candidates. In addition to mRNA detection in the shorter term, the quantitative method for PMP22 protein expression levels will benefit detailed investigations in preclinical models, including dose dependency and time course.
A gene dosage-dependent manifestation of peripheral neuropathy caused by hPMP22 overexpression was observed in transgenic mouse models of CMT1A (37). The two humanized mouse lines exhibited gene dosage-dependent phenotypes. Homo-humanized mice showed abnormalities in the peripheral nerves and muscles, while hetero-humanized mice, which expressed approximately half the amount of hPMP22 mRNA/protein compared to homo-humanized mice, did not exhibit any abnormalities. Therefore, the PMP22 levels in hetero-humanized mice were within the normal range. This indicates that the extrinsic hPMP22 protein functions properly at the proper place in mice and that its reduction by 50% or less can normalize the phenotypes of homo-humanized mice. This relationship between PMP22 expression levels and phenotypes would be closer to that of patients with CMT1A (approximately 40% for normalization) than that of PMP22-C3 mice when evaluating drug candidates targeting only hPMP22. Both homo-humanized and PMP22-C3 mice showed similar degrees of peripheral nerve and motor dysfunctions. These findings suggest that intrinsic mPmp22 contributes to the severity of disease phenotypes in PMP22-C3 mice, potentially complicating the efficacy and adverse effects of drug candidates targeting only hPMP22. Regarding the severity of phenotypes, other CMT1A models carrying more hPMP22 copies, such as C22 mice (7 copies) (22) and TgN248 mice (16 copies) (38), generally have more severe neuropathy phenotypes than mild models including PMP22-C3 mice (3–4 copies), C61 mice (4 copies) (31), and JP18/JY13 mice (2 copies) (39), which may be more relevant to severely affected patients in the CMT1A. In contrast, homo-humanized mice, which exhibit a phenotype comparable to that of PMP22-C3 mice, better mimic the mild myelin and axonal pathology observed in most human patients with CMT1A than a severe model (22). Therefore, with an advantage of overcoming the mPmp22 issue, homo-humanized mice may serve as a more reliable animal model of CMT1A for drug discovery, focusing on a direct approach to hPMP22.
Both homo-humanized and PMP22-C3 mice, but not hetero-humanized mice with lower levels of PMP22 than the homo-genotype, showed alterations in the transcript levels of myelin-related genes, such as Sc5d, Mpz, and Pou3f1 in the peripheral nerve, similar to the C22 mouse model (19). This is reasonable because transcriptional changes in C22 mice were ameliorated by treatment with PMP22-targeting ASO (19). The transcriptional changes in myelin-related genes could be associated with the demyelination observed in homo-humanized and PMP22-C3 mice, and smaller myelin areas and increased g-ratio, resulting in slow nerve conduction. In addition, these mouse lines show axonal atrophy, represented by a smaller inner axon area, which could increase plasma NfL levels. The relatively milder increase in plasma NfL compared to other neurodegenerative disease models with robust increases, such as amyotrophic lateral sclerosis (ALS) (40), corresponds to clinical observations that patients with CMT1A show moderate NfL level elevation (28), whereas patients with ALS show a robust increase (41). This could be due to neuropathy confined to the peripheral nerves in PMP22-C3 mice, unlike ALS, which affects both the central and peripheral nervous systems.
Distal muscle weakness/atrophy and balance problems reminiscent of CMT1A (29, 30) were observed in homo-humanized and PMP22-C3 mice but not in hetero-humanized mice (Figure 7). Like PMP22-C3 mice (24), muscular impairments, such as EIM parameter changes and histopathological muscle fiber atrophy, were observed in the hindlimbs of homo-humanized, but not hetero-humanized mice (Figures 8A,B, 9). Considering the selective expression of the extrinsic hPMP22 gene in peripheral nerves, similar to intrinsic mPmp22 in PMP22-C3 mice (Supplementary Figure S2), muscle deterioration in homo-humanized mice may stem from peripheral nerve abnormalities caused by PMP22 overexpression. Since PMP22-C3 mice exhibit progressive muscular deficits similar to those observed in patients (24), the time-dependent changes in muscle deterioration in homo-humanized mice remain to be elucidated in future studies. Collectively, the electrophysiological and histological characterizations indicated that homo-humanized mice mimic the features of neurogenic muscle atrophy and motor disabilities observed in patients with CMT1A.
Given that treatments that excessively reduce PMP22 have the potential to cause HNPP-like symptoms, an accurate estimation of total PMP22 expression is crucial for predicting the therapeutic window of PMP22-targeting drugs between efficacy and adverse effects. As mentioned above, the presence of intrinsic mPmp22 in existing CMT1A animal models hampers the estimation with drugs targeting hPMP22 but not mPmp22. In contrast, the homo-humanized mice that we developed can simplify the discussion of the relationship between hPMP22 reduction levels and functional efficacy due to the absence of the mPmp22 gene. Additionally, since mPmp22 KO mice, the background mouse line of our humanized mice, show nerve conduction deficits and demyelination, including prominent hypomyelination and tomacula formation (42, 43), homo-humanized mice have the potential to assess the therapeutic effect of a proper reduction of hPMP22 and the adverse effects of overreduction of hPMP22. Further studies investigating the effects of hPMP22-targeting drug candidates are required to clarify the utility of this homo-humanized mouse model. Moreover, it will be important to investigate how the duration of hPMP22 knockdown, as well as the degree of knockdown, impact outcomes in this model.
In conclusion, the newly developed homo-humanized mice exhibited biochemical, electrophysiological, histopathological, and behavioral abnormalities that closely mimicked the features of CMT1A pathology. This model could serve as a promising tool for optimizing the balance between efficacy and potential risks associated with excessive PMP22 reduction and contributing to the translation of preclinical findings into clinically relevant human treatments and dosing strategies.
Statements
Data availability statement
All data generated or analyzed in this study will be made available by the authors, without undue reservation.
Ethics statement
The animal study was approved by the Institutional Animal Care and Use Committee of Shonan Health Innovation Park. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
AT: Validation, Conceptualization, Investigation, Methodology, Writing – review & editing, Visualization, Writing – original draft, Formal analysis. SM: Writing – review & editing, Validation, Investigation. YM: Writing – review & editing, Investigation, Validation. TH: Writing – review & editing, Supervision, Methodology, Conceptualization. AH: Resources, Writing – review & editing, Project administration, Methodology. YK: Supervision, Writing – review & editing, Methodology. MN: Writing – review & editing, Visualization, Writing – original draft, Project administration, Conceptualization, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the Takeda Pharmaceutical Company Limited. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
Acknowledgments
The authors thank Sayuri Nakamura, Ryotaro Hori, and Kotaro Yokoyama in ADDP for their contribution to the histopathological analysis. We are grateful to Dr. Naohiro Nrita and Hiroaki Shida for contributing to the selection of signature peptides in the PMP22 protein assay; Dr. Kazumi Ohuchi for providing technical support to measure NfL; Dr. Tomoya Sano and Dr. Chie Matsuda for analytical suggestions for histopathology; and Dr. Hiroki Iwashita for reviewing this manuscript at Takeda Pharmaceutical Company.
Conflict of interest
AT, SM, YM, TH, AH, YK, MN were employed by the Takeda Pharmaceutical Company Limited.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2025.1658204/full#supplementary-material
- ALS
Amyotrophic lateral sclerosis
- ASO
Antisense oligonucleotide
- CMT1A
Charcot–Marie-Tooth type 1A
- CMAP
Compound muscle action potential
- CSA
Cross-sectional area
- EIM
Electrical impedance myography
- GC
Gastrocnemius
- hPMP22
Human PMP22
- KO
Knockout
- nLC-HRMS
Nanoflow liquid chromatography-high-resolution mass spectrometry
- mPmp22
Mouse Pmp22
- MPZ
Myelin protein zero
- NCV
Nerve conduction velocity
- NfL
Neurofilament light chain
- PMP22
Peripheral myelin protein 22
- POU3F1
POU Class 3 Homeobox 1
- RT-qPCR
Real-time quantitative polymerase chain reaction
- SC5D
Sterol-C5-desaturase
- SEM
Standard error of the mean
- SiMoA
Single molecule array
- WT
Wild-type
Glossary
References
1.
Morena J Gupta A Hoyle JC . Charcot-Marie-Tooth: from molecules to therapy. Int J Mol Sci. (2019) 20:3419. doi: 10.3390/ijms20143419
2.
Fridman V Saporta MA . Mechanisms and treatments in demyelinating CMT. Neurotherapeutics. (2021) 18:2236–68. doi: 10.1007/s13311-021-01145-z
3.
Yiu EM Burns J Ryan MM Ouvrier RA . Neurophysiologic abnormalities in children with Charcot-Marie-Tooth disease type 1A. J Peripher Nerv Syst. (2008) 13:236–41. doi: 10.1111/j.1529-8027.2008.00182.x
4.
van Paassen BW van der Kooi AJ van Spaendonck-Zwarts KY Verhamme C Baas F de Visser M . PMP22 related neuropathies: Charcot-Marie-Tooth disease type 1A and hereditary neuropathy with liability to pressure palsies. Orphanet J Rare Dis. (2014) 9:38. doi: 10.1186/1750-1172-9-38
5.
Raeymaekers P Timmerman V Nelis E Jongheb P Hoogendukc JE Baas F et al . Duplication in chromosome 17p11.2 in Charcot-Marie-Tooth neuropathy type 1a (CMT 1a). The HMSN collaborative research group. Neuromuscul Disord. (1991) 1:93–7. doi: 10.1016/0960-8966(91)90055-w
6.
Snipes GJ Suter U Welcher AA Shooter EM . Characterization of a novel peripheral nervous system myelin protein (PMP-22/SR13). J Cell Biol. (1992) 117:225–38. doi: 10.1083/jcb.117.1.225
7.
Ryu EJ Wang JY Le N Wang JYT Baloh RH Gustin JA et al . Misexpression of Pou 3f1 results in peripheral nerve hypomyelination and axonal loss. J Neurosci. (2007) 27:11552–9. doi: 10.1523/jneurosci.5497-06.2007
8.
Li J Parker B Martyn C Natarajan C Guo J . The PMP22 gene and its related diseases. Mol Neurobiol. (2013) 47:673–98. doi: 10.1007/s12035-012-8370-x
9.
Thomas PK Marques W Davis MB Jr Gugenheim M Tardieu S Maisonobe T et al . The phenotypic manifestations of chromosome 17p11.2 duplication. Brain. (1997) 120:465–78. doi: 10.1093/brain/120.3.465
10.
Katona I Wu X Feely SM Feely SME Sottile S Siskind CE et al . PMP22 expression in dermal nerve myelin from patients with CMT1A. Brain. (2009) 132:1734–40. doi: 10.1093/brain/awp113
11.
Svaren J Moran JJ Wu X Zuccarino R Bacon C Bai Y et al . Schwann cell transcript biomarkers for hereditary neuropathy skin biopsies. Ann Neurol. (2019) 85:887–98. doi: 10.1002/ana.25480
12.
Vallat JM Sindou P Preux PM Vallat J‐M Preux P‐M Tabaraud F et al . Ultrastructural PMP22 expression in inherited demyelinating neuropathies. Ann Neurol. (1996) 39:813–7. doi: 10.1002/ana.410390621
13.
Yoshikawa H Nishimura T Nakatsuji Y Fujimura H Himoro M Hayasaka K et al . Elevated expression of messenger RNA for peripheral myelin protein 22 in biopsied peripheral nerves of patients with Charcot-Marie-Tooth disease type 1A. Ann Neurol. (1994) 35:445–50. doi: 10.1002/ana.410350412
14.
Birouk N Gouider R Le Guern E et al . Charcot-Marie-Tooth disease type 1A with 17p11.2 duplication. Clinical and electrophysiological phenotype study and factors influencing disease severity in 119 cases. Brain. (1997) 120:813–23. doi: 10.1093/brain/120.5.813
15.
Pareyson D Scaioli V Laurà M . Clinical and electrophysiological aspects of Charcot-Marie-Tooth disease. NeuroMolecular Med. (2006) 8:3–22. doi: 10.1385/nmm:8:1-2:3
16.
Krajewski KM Lewis RA Fuerst DR Turansky C Hinderer SR Garbern J et al . Neurological dysfunction and axonal degeneration in Charcot-Marie-Tooth disease type 1A. Brain. (2000) 123:1516–27. doi: 10.1093/brain/123.7.1516
17.
Gabriel JM Erne B Pareyson D Sghirlanzoni A Taroni F Steck AJ . Gene dosage effects in hereditary peripheral neuropathy. Expression of peripheral myelin protein 22 in Charcot-Marie-Tooth disease type 1A and hereditary neuropathy with liability to pressure palsies nerve biopsies. Neurology. (1997) 49:1635–40. doi: 10.1212/wnl.49.6.1635
18.
Schenone A Nobbio L Mandich P Bellone E Abbruzzese M Aymar F et al . Underexpression of messenger RNA for peripheral myelin protein 22 in hereditary neuropathy with liability to pressure palsies. Neurology. (1997) 48:445–9. doi: 10.1212/wnl.48.2.445
19.
Zhao HT Damle S Ikeda-Lee K Kuntz S Li J Mohan A et al . PMP22 antisense oligonucleotides reverse Charcot-Marie-Tooth disease type 1A features in rodent models. J Clin Invest. (2018) 128:359–68. doi: 10.1172/JCI96499
20.
Stavrou M Kagiava A Choudury SG Jennings MJ Wallace LM Fowler AM et al . A translatable RNAi-driven gene therapy silences PMP22/PMP 22 genes and improves neuropathy in CMT1A mice. J Clin Invest. (2022) 132:e159814. doi: 10.1172/JCI159814
21.
Sereda MW Nave KA . Animal models of Charcot-Marie-Tooth disease type 1A. NeuroMolecular Med. (2006) 8:205–16. doi: 10.1385/nmm:8:1-2:205
22.
Verhamme C King RH ten Asbroek AL Muddle JR Nourallah M Wolterman R et al . Myelin and axon pathology in a long-term study of PMP22-overexpressing mice. J Neuropathol Exp Neurol. (2011) 70:386–98. doi: 10.1097/NEN.0b013e318217eba0
23.
Moss KR Johnson AE Bopp TS Yu AT Perry K Chung T et al . SARM1 knockout does not rescue neuromuscular phenotypes in a Charcot-Marie-Tooth disease type 1A mouse model. J Peripher Nerv Syst. (2022) 27:58–66. doi: 10.1111/jns.12483
24.
Taruta A Hiyoshi T Harada A Nakashima M . Electrical impedance myography detects progressive pathological alterations in the hindlimb muscle of the PMP22-C3 mice, an animal model of CMT1A. Exp Neurol. (2025) 385:115111. doi: 10.1016/j.expneurol.2024.115111
25.
Prior R Verschoren S Vints K Jaspers T Rossaert E Klingl YE et al . HDAC3 inhibition stimulates myelination in a CMT1A mouse model. Mol Neurobiol. (2022) 59:3414–30. doi: 10.1007/s12035-022-02782-x
26.
Bai Y Treins C Volpi VG Scapin C Ferri C Mastrangelo R et al . Treatment with IFB-088 improves neuropathy in CMT1A and CMT1B mice. Mol Neurobiol. (2022) 59:4159–78. doi: 10.1007/s12035-022-02838-y
27.
Perrot R Berges R Bocquet A Eyer J . Review of the multiple aspects of neurofilament functions, and their possible contribution to neurodegeneration. Mol Neurobiol. (2008) 38:27–65. doi: 10.1007/s12035-008-8033-0
28.
Sandelius Å Zetterberg H Blennow K Adiutori R Malaspina A Laura M et al . Plasma neurofilament light chain concentration in the inherited peripheral neuropathies. Neurology. (2018) 90:e518–24. doi: 10.1212/wnl.0000000000004932
29.
Tozza S Bruzzese D Severi D Spina E Iodice R Ruggiero L et al . The impact of symptoms on daily life as perceived by patients with Charcot-Marie-Tooth type 1A disease. Neurol Sci. (2022) 43:559–63. doi: 10.1007/s10072-021-05254-7
30.
Johnson NE Heatwole CR Dilek N Sowden J Kirk CA Shereff D et al . Quality-of-life in Charcot-Marie-Tooth disease: the patient's perspective. Neuromuscul Disord. (2014) 24:1018–23. doi: 10.1016/j.nmd.2014.06.433
31.
Norreel JC Jamon M Riviere G Passage E Fontes M Clarac F . Behavioural profiling of a murine Charcot-arie-tooth disease type 1A model. Eur J Neurosci. (2001) 13:1625–34. doi: 10.1046/j.0953-816x.2001.01535.x
32.
Ha N Choi YI Jung N Song JY Bae DK Kim MC et al . A novel histone deacetylase 6 inhibitor improves myelination of Schwann cells in a model of Charcot-Marie-Tooth disease type 1A. Br J Pharmacol. (2020) 177:5096–113. doi: 10.1111/bph.15231
33.
Pandeya SR Nagy JA Riveros D Semple C Taylor RS Mortreux M et al . Estimating myofiber cross-sectional area and connective tissue deposition with electrical impedance myography: a study in D2-mdx mice. Muscle Nerve. (2021) 63:941–50. doi: 10.1002/mus.27240
34.
Clark BC Rutkove S Lupton EC Padilla CJ Arnold WD . Potential utility of electrical impedance Myography in evaluating age-related skeletal muscle function deficits. Front Physiol. (2021) 12:666964. doi: 10.3389/fphys.2021.666964
35.
Kapur K Taylor RS Qi K Nagy JA Li J Sanchez B et al . Predicting myofiber size with electrical impedance myography: a study in immature mice. Muscle Nerve. (2018) 58:106–13. doi: 10.1002/mus.26111
36.
Schwanhäusser B Busse D Li N Dittmar G Schuchhardt J Wolf J et al . Global quantification of mammalian gene expression control. Nature. (2011) 473:337–42. doi: 10.1038/nature10098
37.
Huxley C Passage E Robertson AM Youl B Huston S Manson A et al . Correlation between varying levels of PMP22 expression and the degree of demyelination and reduction in nerve conduction velocity in transgenic mice. Hum Mol Genet. (1998) 7:449–58. doi: 10.1093/hmg/7.3.449
38.
Magyar JP Martini R Ruelicke T Aguzzi A Adlkofer K Dembic Z et al . Impaired differentiation of Schwann cells in transgenic mice with increased PMP22 gene dosage. J Neurosci. (1996) 16:5351–60. doi: 10.1523/jneurosci.16-17-05351.1996
39.
Boutary S Caillaud M El Madani M Vallat JM Loisel-Duwattez J Rouyer A et al . Squalenoyl si RNA PMP22 nanoparticles are effective in treating mouse models of Charcot-Marie-Tooth disease type 1 a. Commun Biol. (2021) 4:317. doi: 10.1038/s42003-021-01839-2
40.
Loeffler T Schilcher I Flunkert S Hutter-Paier B . Neurofilament-light chain as biomarker of neurodegenerative and rare diseases with high translational value. Front Neurosci. (2020) 14:579. doi: 10.3389/fnins.2020.00579
41.
Behzadi A Pujol-Calderón F Tjust AE Wuolikainen A Höglund K Forsberg K et al . Neurofilaments can differentiate ALS subgroups and ALS from common diagnostic mimics. Sci Rep. (2021) 11:22128. doi: 10.1038/s41598-021-01499-6
42.
Bai Y Zhang X Katona I Saporta MA Shy ME O'Malley HA et al . Conduction block in PMP22 deficiency. J Neurosci. (2010) 30:600–8. doi: 10.1523/jneurosci.4264-09.2010
43.
Adlkofer K Martini R Aguzzi A Zielasek J Toyka KV Suter U . Hypermyelination and demyelinating peripheral neuropathy in Pmp 22-deficient mice. Nat Genet. (1995) 11:274–80. doi: 10.1038/ng1195-274
Summary
Keywords
Charcot–Marie-Tooth type 1A, PMP22 , humanized model, demyelination, electrophysiology, muscle deterioration
Citation
Taruta A, Matsumoto S-i, Masuda Y, Hiyoshi T, Harada A, Kosugi Y and Nakashima M (2025) Characterization of a humanized mouse model of Charcot-Marie-Tooth type 1A for the discovery of human PMP22-targeting drugs. Front. Neurol. 16:1658204. doi: 10.3389/fneur.2025.1658204
Received
02 July 2025
Accepted
06 October 2025
Published
29 October 2025
Volume
16 - 2025
Edited by
Stefano Carlo Previtali, San Raffaele Scientific Institute (IRCCS), Italy
Reviewed by
Devesh Pant, Emory University, United States
Marina Stavrou, The Cyprus Institute of Neurology and Genetics, Cyprus
Updates
Copyright
© 2025 Taruta, Matsumoto, Masuda, Hiyoshi, Harada, Kosugi and Nakashima.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Masato Nakashima, masato.nakashima@takeda.com
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.