 Rebeca Pérez-Alfayate1*†
Rebeca Pérez-Alfayate1*† Vanesa García-Barberán2
Vanesa García-Barberán2 Isabel Casado-Fariñas3Desiré Hernández-Martínez3María E. Gómez del Pulgar2Juan Pablo Castaño-Montoya1
Isabel Casado-Fariñas3Desiré Hernández-Martínez3María E. Gómez del Pulgar2Juan Pablo Castaño-Montoya1 Pedro Pérez-Segura4
Pedro Pérez-Segura4 Santiago Cabezas-Camarero4†
Santiago Cabezas-Camarero4†- 1Neurosurgical Department, Hospital Universitario Clínico San Carlos, IdISCC, Madrid, Spain
- 2Medical Oncology Lab, Hospital Universitario Clínico San Carlos, IdISCC, Madrid, Spain
- 3Pathology Department, Hospital Universitario Clínico San Carlos, Madrid., Spain
- 4Medical Oncology Department, Hospital Universitario Clínico San Carlos, IdISCC, Madrid, Spain
Background: Sporadic brain arteriovenous malformations (bAVMs) are rare vascular anomalies characterized by abnormal angiogenesis and direct arteriovenous shunting. While the VEGF pathway is well studied, the genetic landscape contributing to angiogenic dysregulation remains poorly defined. We aimed to characterize the mutational profile of resected bAVMs using a pan-cancer next-generation sequencing panel, with particular focus on angiogenesis-associated pathways and RNA Polymerase II activity.
Methods: A descriptive analysis of clinical and molecular characteristics was conducted In formalin-fixed, paraffin-embedded tissue from the bAVM nidus. DNA was extracted and sequenced using the Oncomine Tumor Mutational Load Assay, covering 409 cancer-related genes. Variants were filtered for pathogenicity, allele frequency, and functional relevance.
Results: Thirteen sporadic bAVMs were retrospectively analyzed. Twelve pathogenic variants were detected in 7/13 (54%) patients, with variant allele frequencies ranging from 3.61 to 50.61%. Most mutations clustered within angiogenesis-related pathways (PI3K/AKT/mTOR, RAS/MAPK), DNA repair mechanisms, and transcriptional regulators of RNA Polymerase II. Notably, six mutations involved genes with known functional links to RNA Pol II activity. These findings suggest a converging role for transcriptional dysregulation and vascular remodeling in bAVM pathogenesis.
Conclusion: This study proposes a novel hypothesis implicating RNA Polymerase II-mediated transcription in the aberrant angiogenesis of bAVMs. While KRAS mutations were detected at low frequency and allele burden, other genetic alterations in DNA repair and transcriptional machinery may drive or sustain vascular instability. Further functional validation is warranted to clarify their pathogenic role and therapeutic potential.
Introduction
Brain arteriovenous malformations (bAVMs) are vascular anomalies characterized by tortuous, morphologically abnormal channels that create direct connections between arteries and veins, bypassing the capillary network. This anatomical defect results in high-pressure arterial blood being shunted directly into the venous drainage system. Affecting approximately 15 per 100,000 individuals, bAVMs represent a major cause of hemorrhagic stroke, particularly in young adults (1–2).
Currently, four treatment options are available for unruptured brain arteriovenous malformations (bAVMs): microsurgical resection, radiosurgery, embolization, and conservative management. The management of unruptured bAVMs remains controversial, and treatment decisions should be guided by the patient’s clinical condition, the natural history of the disease, and the radiological characteristics of each case. Given these factors, existing treatment modalities are not sufficiently safe (1). Therefore, a deeper understanding of the pathogenesis of bAVMs, the identification of potential therapeutic targets, and the development of more personalized treatments are crucial to improve patient outcomes (2).
While the precise etiology of sporadic bAVMs remains unknown, similar vascular lesions have been observed in rare genetic syndromes (3, 4). Sporadic brain arteriovenous malformations (bAVMs) may arise from aberrant molecular signaling pathways, leading to abnormal angiogenesis. While the vascular endothelial growth factor (VEGF) pathway is the most extensively studied (5, 6), recent research suggests that high-flow bAVMs may be due to somatic mutations, affecting mainly the RAS-MAPK pathway, and especially affecting KRAS and BRAF (7–9). It has also been suggested that epigenetic changes such as methylation or hypermethylation may contribute to bAVM pathogenesis (10). On the other hand, some polymorphisms can increase the risk of bAVM rupture by elevating the expression of certain inflammatory cytokines (11).
This study aimed to characterize the mutational profile (MP) of a series of resected bAVMs to identify potentially actionable alterations.
Materials and methods
Patients
A retrospective series of 13 consecutively resected sporadic brain arteriovenous malformations (bAVMs) was analyzed following approval by the Institutional Review Board (IRB), in accordance with the principles outlined in the World Medical Association Declaration of Helsinki (IRB code: 23/332-E). Written informed consent was obtained from all patients prior to study participation. Clinical records were reviewed for patient demographics, presenting symptoms, and medical history, with a focus on intracranial or extracranial vascular lesions (Table 1). Imaging studies were also analyzed to define bAVM Spetzler-Martin and Lawton-Young scores. Family history was assessed for bAVMs, vascular lesions, or stroke.
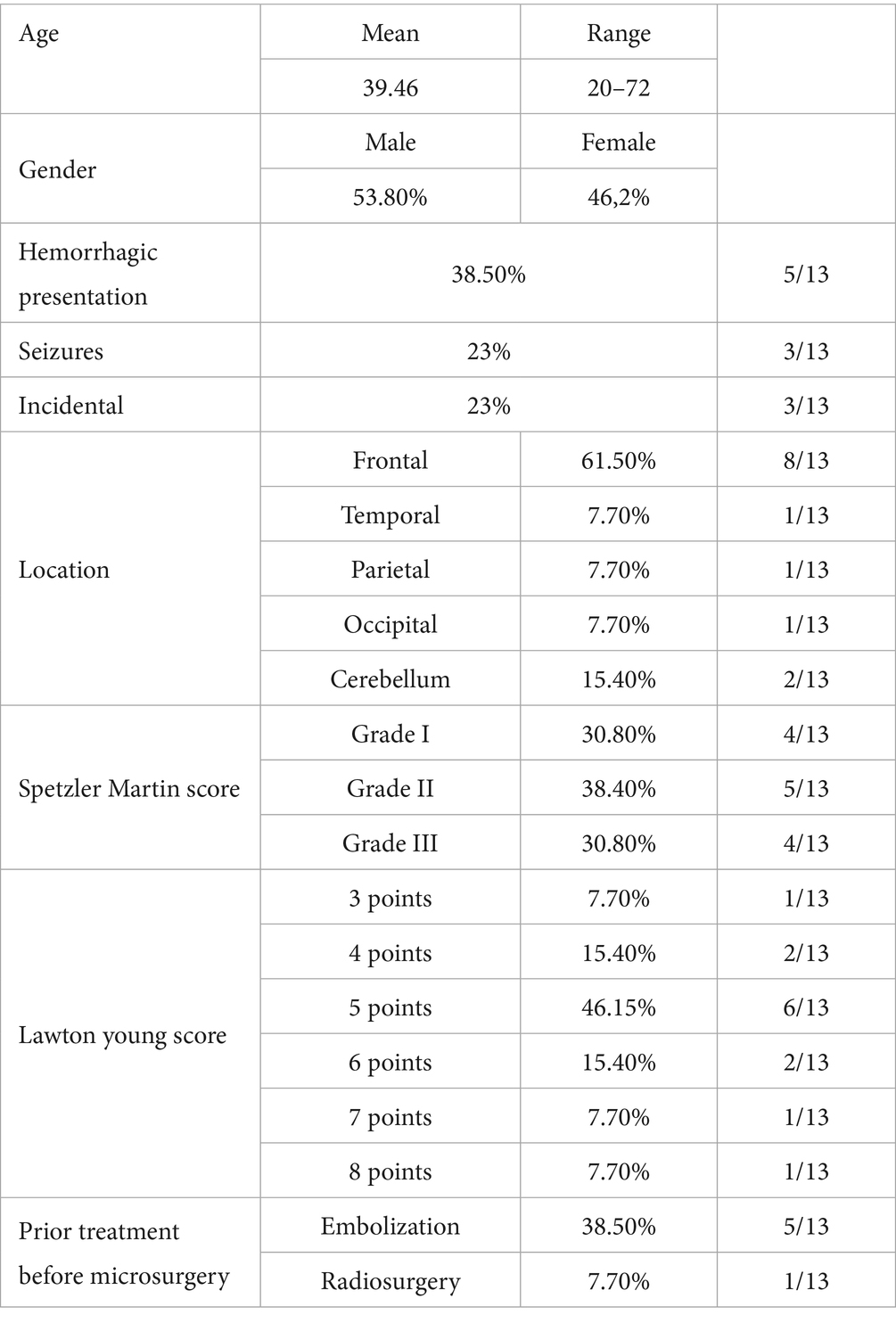
Table 1. Clinical characteristics of patients in this study.
Samples and preparation
Formalin-fixed, paraffin-embedded (FFPE) tissue sections, selected by a pathologist from the bAVM nidus, were used for DNA extraction and quantification. Slides were assessed to determine tissue adequacy and viability for molecular testing. Cases were excluded if the tissue quantity was insufficient or if extensive artifact-related damage compromised sample integrity. These samples were retrospectively selected from an institutional biobank, ensuring they met quality criteria such as tissue integrity and absence of significant contamination. Prior to extraction, FFPE tissue sections were deparaffinized manually.
DNA extraction
DNA extraction was performed using the QIAamp DNA FFPE Tissue Kit (QIAGEN, Germantown, MD, USA), specifically designed for FFPE samples where DNA may be fragmented and cross-linked due to formalin fixation. This kit employs silica-based column technology that allows selective binding of DNA to a membrane under chaotropic conditions, followed by washes to remove inhibitors such as proteins, salts, and formalin residues. The protocol involved: (1) tissue lysis with proteinase K to digest proteins and release DNA; (2) incubation at elevated temperatures (approximately 56–90 °C) to reverse formalin-induced cross-links; (3) column-based purification with specific buffers (AW1 and AW2 for washes, and AE for elution). This yields high-purity DNA suitable for downstream applications like sequencing. Multiple aliquots per sample were processed to ensure reproducibility, and over-extraction was avoided to minimize degradation.
DNA quantification
DNA quantification was carried out using the QUBIT 3.0 fluorometer (Thermo Fisher Scientific, Waltham, MA, USA), which uses dsDNA-specific fluorescent dyes. QUBIT provides a selective and sensitive measurement (detection range of 0.2–100 ng/μL). The protocol involves mixing 1–20 μL of sample with the QUBIT dsDNA HS (high sensitivity) or BR (broad range) reagent, brief incubation, and fluorescence measurement excited at ~502 nm with emission at ~523 nm. A minimum of 20 ng of DNA per sample was required to proceed with library preparation, with adjustments to elution volume if necessary to concentrate the DNA.
Next-Generation sequencing and mutational profiling
The mutational profile and tumor mutational burden (TMB, defined as the number of somatic mutations per megabase of coding DNA) were assessed using next-generation sequencing with the Oncomine Tumor Mutation Load Assay (Thermo Fisher Scientific, Waltham, MA, USA). This targeted panel covers 1.65 Mb of exonic and intronic regions across 409 genes frequently altered in cancer (including oncogenes such as KRAS, BRAF, PIK3CA, and tumor suppressors like TP53), optimized for detecting low-frequency somatic variants in FFPE samples with limited DNA. The assay uses AmpliSeq technology, which amplifies target regions via ultra-deep multiplex PCR, enabling uniform coverage (>95% at 500x average depth) and detection of variants with allelic frequencies (VAF) as low as 5–10%.
Library construction was automated using Chef-Ready Kits with 20 ng of input DNA, minimizing bias from manual handling. This step involved: (1) multiplex amplification of target amplicons (typically 12–24 PCR cycles to avoid artifacts); (2) partial primer digestion with FuPa reagent; (3) ligation of Ion Torrent adapters with barcodes for sample multiplexing; and (4) purification with magnetic beads (AMPure XP) to select fragments of optimal size (~200–300 bp). Libraries were loaded onto an Ion 540 chip using the Ion Chef Instrument, which performs automated emulsification and enrichment of sequencing particles (Ion Sphere Particles, ISPs) loaded with DNA. Sequencing was performed on the Ion GeneStudio S5 System (Thermo Fisher Scientific, Waltham, MA, USA), based on semiconductor sequencing technology (Ion Torrent). This method detects pH changes caused by proton release during nucleotide incorporation, eliminating the need for laser optics and enabling rapid runs (~2–4 h per chip). It was configured for single-end reads with an average length of 200 bp, achieving an average coverage depth of 500-1000x for optimal TMB sensitivity.
Bioinformatic analysis
Raw data (BAM/FASTQ files) were analyzed using Ion Reporter version 5.12 (Thermo Fisher Scientific, Waltham, MA, USA), a cloud-based platform for automated processing of Ion Torrent data. The Coverage Analysis plugin was used to assess coverage uniformity, read quality (Phred score >20), and metrics such as the percentage of on-target bases (>90% expected). The specific workflow “Oncomine Tumor Mutation Load-w3.4-LOD0.1” was applied for variant calling, incorporating alignment to the hg19/GRCh37 reference genome, filtering of artifacts (e.g., homopolymers common in Ion Torrent), and TMB calculation. The limit of detection (LOD) of 0.1 indicates sensitivity for variants with allelic frequency ≥10%, adjusted for background noise in FFPE samples.
Variant allele frequency (VAF) was calculated as the proportion of reads supporting the variant allele divided by the total reads covering that genomic position, expressed as a percentage. A reporting threshold of ≥5% VAF was applied in line with the validated sensitivity limits of the Oncomine assay.
Variants were annotated using “Oncomine Tumor Mutation Load Assay Annotations v1.5,” which integrates databases like COSMIC, dbSNP, and 1,000 Genomes for functional context (e.g., synonymous, nonsynonymous, frameshifts). The “Oncomine Variants (5.20)” filter was applied to prioritize cancer-relevant variants, excluding common polymorphisms (MAF > 1%) and technical artifacts. Each gene variant was classified manually or semi-automatically using the ClinVar database,1 a NIH-curated repository providing evidence-based clinical interpretations. Variants were categorized as pathogenic if classified as “pathogenic” or “likely pathogenic.” Non-pathogenic variants included: (1) “likely benign” or “benign,” based on lack of functional impact; (2) variants of uncertain significance (VUS), where evidence is insufficient; and (3) those not documented in ClinVar, considered benign by default unless additional functional analyses (e.g., in silico with SIFT/PolyPhen) suggested otherwise. Cross-validation with tools like Variant Effect Predictor (VEP) was performed if needed to resolve ambiguities.
Results
Patients
Among the 13 patients included in the study, six were female and seven were male. The mean age was 39.5 years (range: 20–72 years). The most common clinical presentation was intracranial hemorrhage (5/13, 38.5%), followed by seizures (3/13, 23.1%) and incidental findings (3/13, 23.1%); less frequent presentations included headache and cerebellar ataxia. No patient had relevant comorbidities. Data related to the angioarchitectonic characteristics of the bAVMs are summarized in Table 1. All patients had a surgical indication for bAVM. Preoperative embolization was required in five cases due to the presence of flow-related aneurysms or acute bleeding. One patient had previously undergone stereotactic radiosurgery (SRS), which failed to achieve complete bAVM closure. The lowest TMB was observed in those cases that had undergone prior embolization (Table 2).
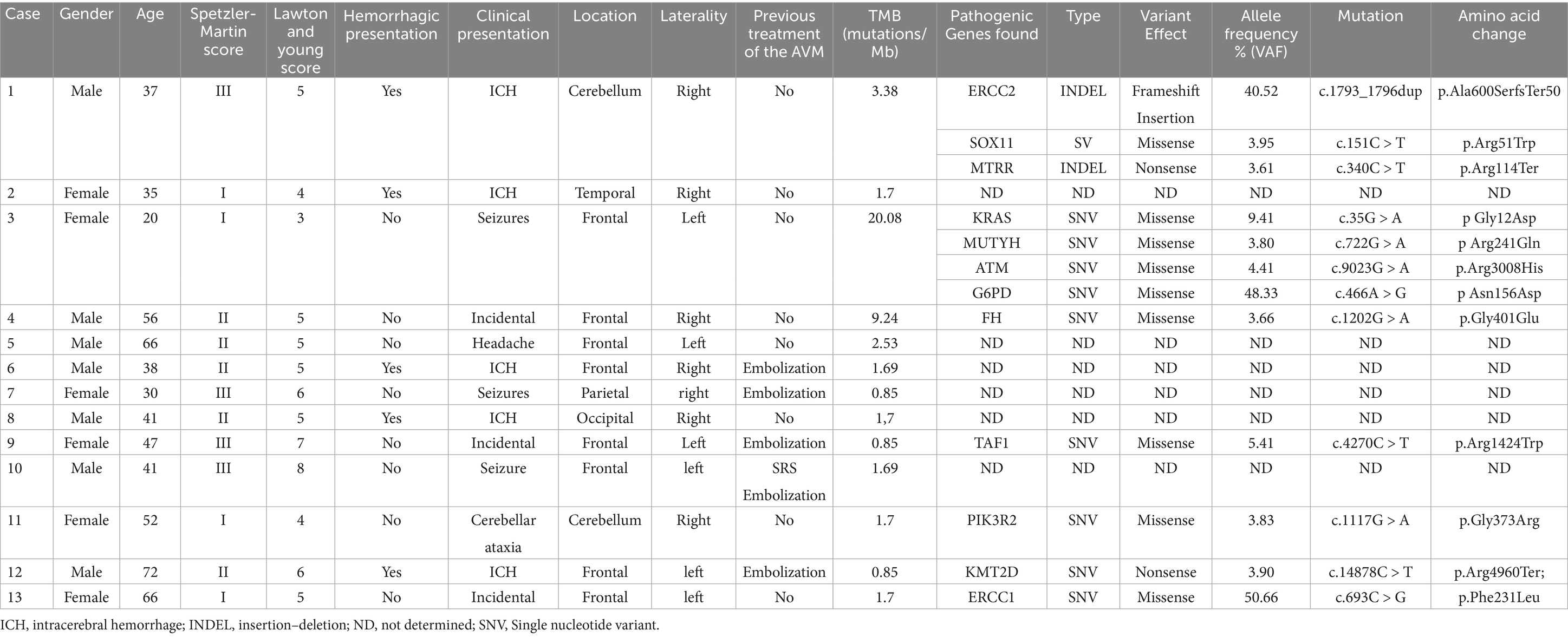
Table 2. Summary of the cases and the pathogenic mutations found in the study.
Mutational analysis
Following next-generation sequencing analysis. Within the panel of 409 analyzed genes, 224 mutations were identified. Among these, 12 genes harbored pathogenic variants (Figure 1).
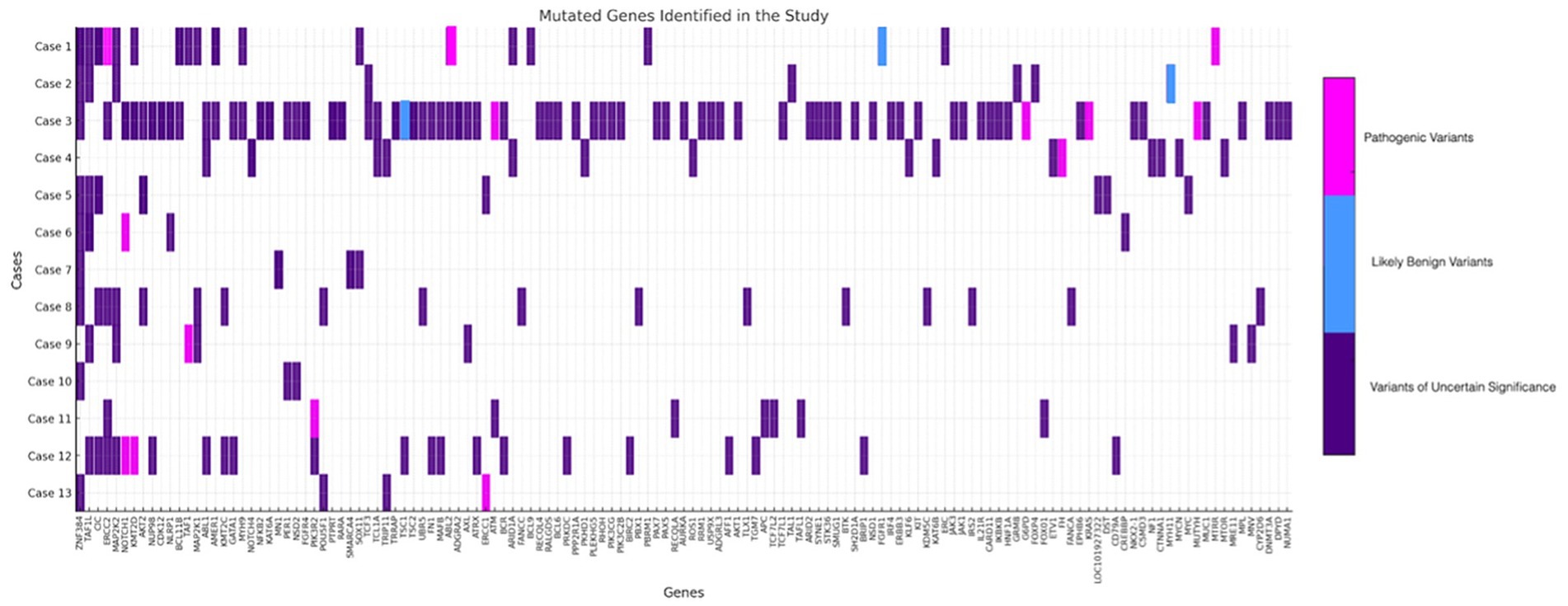
Figure 1. Schematic representation of all variants with an allele frequency of approximately 5% or higher. Variants of uncertain significance are depicted in purple, likely benign variants in blue, and pathogenic variants in pink.
The TMB ranged from 0.85 to 20.08 mutations per megabase. The analysis of mutation frequency within the sample revealed a heterogeneous distribution of genetic alterations across multiple genes.
The 12 pathogenic variants were identified in seven out of the 13 patients. Allele frequencies (VAF) ranged from 3.61 to 50.61%, suggesting a somatic origin. In case 1, pathogenic variants were detected in ERCC2 (c.1793_1796dup; VAF: 40.52%), SOX11 (c.151C > T; VAF: 3.95%), and MTRR (c.340C > T; VAF: 3.61%). Case 3 exhibited mutations in KRAS (c.35G > A; VAF: 9.41%), MUTYH (c.722G > A; VAF: 3.80%), ATM (c.9023G > A; VAF: 4.41%), and G6PD (c.466A > G; VAF: 48.33%). In case 4, a pathogenic variant was identified in FH (c.1202G > A; VAF: 3.66%). Case 9 presented a mutation in TAF1 (c.4270C > T; VAF: 5.4%), while case 11 exhibited a pathogenic variant in PIK3R2 (c.1117G > A; VAF: 3.83%). Additionally, case 12 carried a mutation in KMT2D (c.14878C > T; VAF: 3.90%), and case 13 harboured a pathogenic variant in ERCC1 (c.693C > G; VAF: 50.66%; Table 2).
It is worth mentioning Case 3 which involved a 20-year-old woman with no relevant personal or family medical history, diagnosed with a Spetzler-Martin I, Lawton-Young 3 bAVM. The patient initially presented with a seizure, prompting further investigation. The case is illustrated in Figure 2.
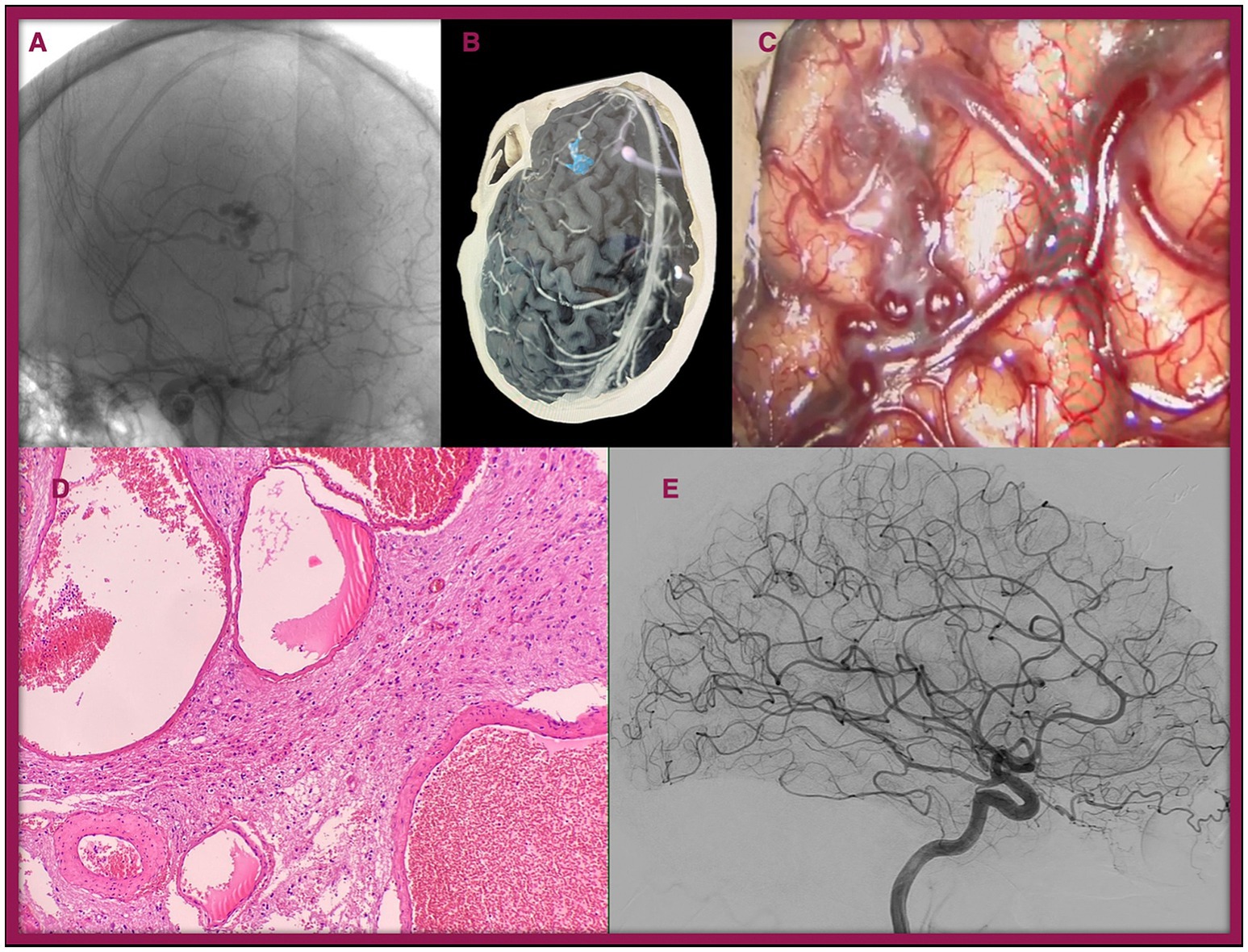
Figure 2. Illustration of Case 3. A 20-year-old female presented with seizures. During the workup, a bAVM (Spetzler-Martin grade I, Lawton-Young 3) was identified in the left frontal lobe. (A) Preoperative conventional angiogram. (B) 3D reconstruction of the lesion. (C) Intraoperative image of the lesion. (D) Histopathological view with Hematoxylin–Eosin staining (×40): Cluster of arterial and venous vessels with dilated lumens lined by mature endothelium, lacking an intervening capillary bed, and associated with brain parenchyma showing reactive gliosis. (E) Postoperative angiogram showing complete resection of the bAVM.
Discussion
This study reveals a potentially novel convergence of pathogenic mutations affecting angiogenesis, DNA repair, and transcriptional regulation via RNA Polymerase II (Pol II) in sporadic bAVMs. While previous research has identified somatic mutations in KRAS and BRAF as potential drivers of vascular malformations (7–9). Our results suggest that the mutational landscape of bAVMs is broader and functionally interconnected. The detection of mutations in genes related to transcriptional machinery and genome integrity introduces a more complex model of disease pathogenesis that extends beyond canonical angiogenic pathways.
Molecular heterogeneity and the role of KRAS in bAVM pathogenesis
Our cohort included mostly low-grade (Spetzler-Martin I–II) bAVMs, which reflects the surgical selection bias common in most tissue-based studies (9). Haemorrhagic presentation was present in 38.5% of cases, consistent with natural history data (12). The lack of high-grade lesions limits the generalizability of our findings, as these bAVMs may exhibit a different molecular signature. This limitation is shared by previous studies, such as that by Tao-Hong et al. (9) which included only one Spetzler-Martin IV case. Alternative tissue-sampling techniques, such as liquid biopsy, have been proposed but remain limited in sensitivity. Nikolaev et al. (7) for instance, failed to detect KRAS mutations in paired plasma samples from patients with KRAS-positive nidus tissue. Endoluminal biopsy, recently demonstrated by Winkler et al. (13) in four bAVM cases, may offer a minimally invasive way to sample tissue from high-grade or unresectable lesions in vivo.
We detected a KRAS mutation in only 1 of 13 patients (7.7%), a much lower rate than previously reported by Nikolaev et al. (7) (62.5%) and Tao-Hong et al. (9) (up to 87.1% including BRAF). These discrepancies likely reflect differences in sequencing technology and sensitivity. Our study used a pan-cancer amplicon-based panel optimized for tumor mutational burden (TMB), with a ~ 5% variant allele frequency (VAF) detection limit. In contrast, Nikolaev et al. used whole-exome sequencing with ~100 × −200 × coverage (7), while Tao-Hong et al. combined panel Next Generation Sequencing with ddPCR validation and ultra-deep sequencing (>1,000×), enabling detection of subclonal mutations with lower VAF (9).
This raises the question of whether low-VAF KRAS mutations are merely passenger mutations or true drivers of vascular dysregulation. Although our results support a broader mutational landscape, the biological relevance of KRAS cannot be discounted. As a dominant oncogene, even subclonal KRAS mutations may exert strong downstream effects on MAPK signaling and angiogenesis. In cancer and other vascular malformations, low-frequency oncogenic mutations have been shown to act as early drivers that expand under selective conditions (14). Tao-Hong et al. (9) found an inverse correlation between VAF and nidus size, further suggesting a possible growth-promoting role for early KRAS/BRAF events. Conversely, Al-Olabi et al. (15) demonstrated in a zebrafish model that expression of BRAFV600E alone caused vascular dysplasia in only 10–20% of cases, supporting a two-hit model in which an initial mutation sets the stage for further disruption. Our identification of multiple co-occurring mutations in angiogenic, DNA repair, and metabolic genes—particularly in Case 3—suggests that KRAS may act in concert with other lesions to promote lesion development and progression. In addition, the overall mutational profile in our cohort was highly heterogeneous, with most variants occurring in single cases. The fact that only one patient harbored a KRAS mutation, in contrast to prior reports of recurrent KRAS alterations, underscores the exploratory nature of our findings and highlights the need for cautious interpretation.
Importantly, this interpretation is reinforced by recent endothelial models demonstrating that somatic activation of KRAS or BRAF in vascular endothelium is sufficient to induce AVM formation, with MEK/ERK identified as the critical downstream effector pathway (16). These preclinical findings strengthen the biological plausibility of our observations and highlight the translational potential of pathway-targeted therapies.
Beyond angiogenesis: DNA repair, transcriptional dysregulation, and pol II pathways
In addition to KRAS, we identified 12 pathogenic variants across genes involved in angiogenesis (e.g., PIK3R2, SOX11, KRAS) (17–19). DNA repair (ERCC2, ERCC1, ATM, MUTYH, G6PD, FH) (20–25). DNA transcription (TAF1) (26, 27). and epigenetic modulation (KMT2D, MTRR) (28–30). Notably, several of these genes intersect with RNA Polymerase II (Pol II) function (ERCC2, ATM, KRAS, G6PD, TAF1, KMT2D), a transcriptional hub that mediates angiogenic signaling downstream of VEGF, KRAS-MAPK, and HIF-1α (26, 27). While Pol II is not typically viewed as an angiogenic regulator per se, its disruption could impair endothelial gene expression programs and promote abnormal vessel formation. To our knowledge, this connection between Pol II dysfunction and bAVMs has not been previously described. However, this proposed link remains hypothetical, as our study did not include functional assays to confirm pathway activation. Therefore, the role of Pol II dysfunction in AVM pathogenesis should be interpreted as exploratory and will require validation in future cellular and animal models.
Further supporting a developmental transcriptional dysregulation model, recent single-cell RNA-sequencing of human brain vasculature demonstrated reactivation of embryonic gene programs in bAVM endothelial cells (31). Our findings align with this notion, suggesting that genetic lesions affecting chromatin remodelers (KMT2D), DNA repair factors (ATM, MUTYH), and Pol II regulators (TAF1) may collectively produce a vascular phenotype that retains fetal-like characteristics and abnormal angiogenic responsiveness.
Taken together, these observations raise the hypothesis that alterations in DNA repair, transcriptional regulation, and angiogenic pathways could converge to create a permissive environment for AVM development. Defective DNA repair may facilitate genomic instability, while dysregulated transcriptional programs could amplify abnormal endothelial responses to angiogenic cues. These combined alterations may not act in isolation, but rather interact to promote aberrant vascular remodeling. Such a model suggests that bAVMs may arise from the interplay of multiple disrupted pathways, extending beyond canonical angiogenesis alone.
Toward a network model of vascular instability
The interplay of DNA repair, oxidative stress, and angiogenesis becomes especially evident in Case 3, which carried mutations in KRAS, ATM, MUTYH, and G6PD. These genes converge functionally on the cellular response to oxidative stress and genomic instability (17, 18, 20–24, 32). MUTYH is critical in base-excision repair of oxidative lesions (33), ATM regulates DNA damage checkpoints (21), and G6PD controls the redox balance through NADPH generation (23). Disruption in these pathways may promote secondary oncogenic events, such as KRAS activation, and create a permissive environment for clonal expansion. Such cases support a network model of pathogenesis, in which no single mutation is sufficient, but together they impair vascular stability and remodelling.
Notably, Case 3 was also the youngest patient in our series (20 years old), raising the hypothesis that higher mutational burden could be linked to earlier clinical onset. This is consistent with prior observations that pediatric and young-adult AVMs often exhibit distinct clinical behavior, including higher recurrence rates after treatment. Hak et al. (34) conducted a meta-analysis showing an overall recurrence rate of 10.9% in pediatric patients, with recurrence risk decreasing significantly with each additional year of age at diagnosis (RR 0.97, 95% CI 0.93–0.99; p = 0.046).
This concept has therapeutic implications. Bevacizumab, an anti-VEGF agent, showed modest clinical effects in a small pilot study of two bAVM patients conducted by Muster et al. (35). Our findings suggest that targeting VEGF alone may not be sufficient, as the dysregulation extends beyond classic angiogenic signaling. Intervening in transcriptional regulation, DNA repair, or redox homeostasis may be needed to fully correct the molecular imbalance. As summarized in Table 3, several of the pathogenic variants identified in our cohort affect genes that are already known targets—or are mechanistically linked to targets, of approved or investigational drugs, including inhibitors of KRAS [e.g., adagrasib (36), sotorasib (36, 37)], PI3K [e.g., alpelisib (29), duvelisib (28)], ATM (e.g., imatinib), and epigenetic modulators (e.g., entacapone). This highlights the translational relevance of our mutational profiling and warrants further validation in preclinical models and single-cell profiling studies.
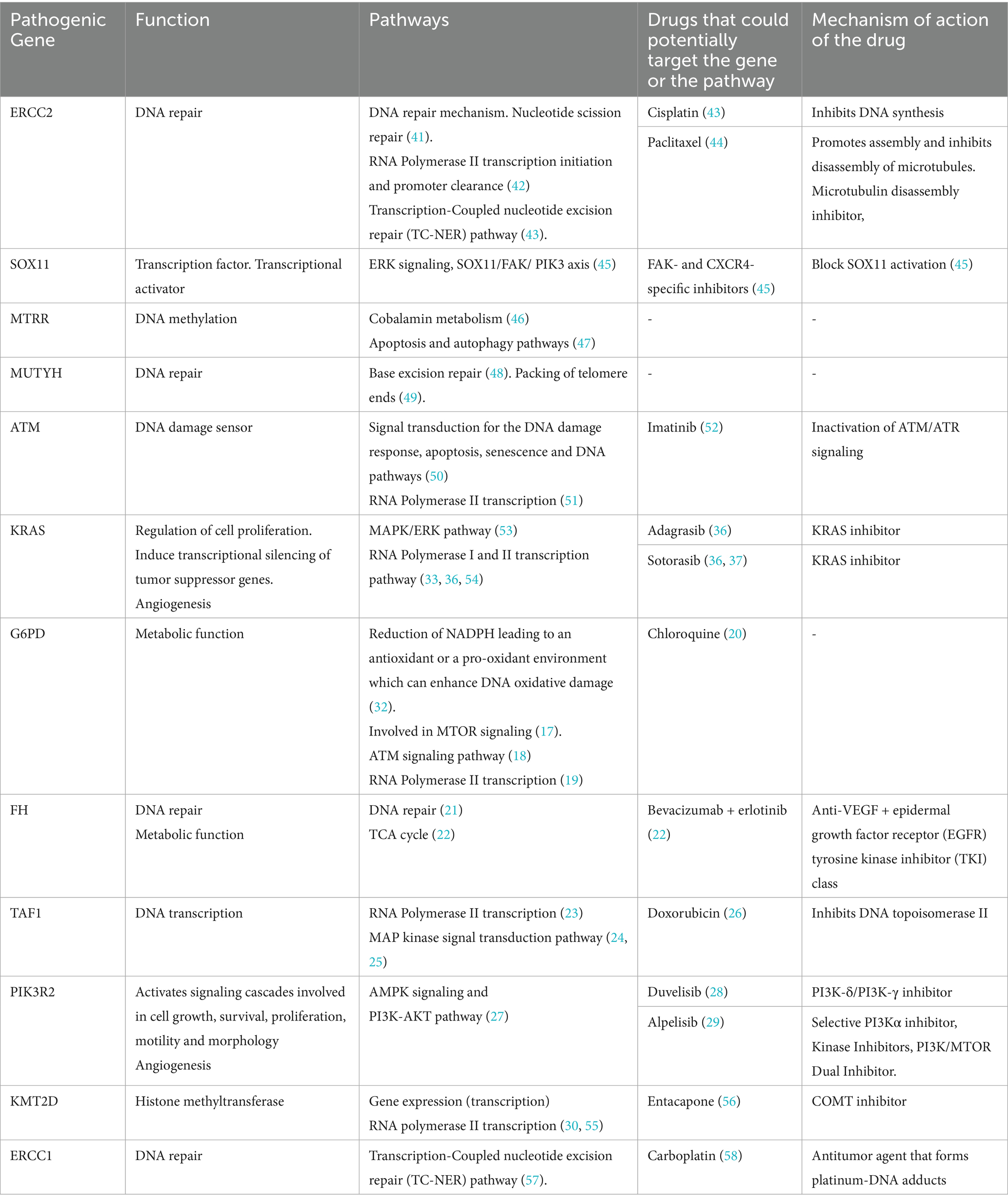
Table 3. Altered pathways and potential targeted therapies for each of the pathogenic gene variants detected.
Limitations
This study has several limitations. First, the small cohort and the fact that all cases were Spetzler–Martin grade I–III surgically resected bAVMs limit the generalizability of our findings to higher-grade lesions (38–40). In addition, no pediatric patients were included in this series, which may limit extrapolation of our findings to younger populations, as pediatric AVMs have been associated with distinct clinical behavior and higher recurrence rates after treatment.
Second, although the use of a pan-cancer sequencing panel could be perceived as a limitation due to its design focus on oncogenic mutations, this approach is, in fact, strategically justified in the context of bAVMs. Currently, there are no Next generation sequencing panels specifically optimized for the genetic study of sporadic brain arteriovenous malformations. Therefore, using a broad, oncology-based panel offers the advantage of covering many of the genes already implicated in bAVM pathogenesis. Notably, somatic mutations in KRAS, BRAF, and PIK3R2, all well-established oncogenes, have been repeatedly reported in sporadic bAVMs (7, 9, 36). These genes play central roles in angiogenesis-related signaling pathways, including RAS-MAPK and PI3K-AKT, which are essential to both tumor biology and vascular development. In this sense, the pan-cancer panel serves not only as a pragmatic solution in the absence of a bAVM-specific tool, but also as a biologically relevant platform to explore the somatic landscape of these lesions. Nevertheless, we acknowledge that the pathogenic relevance of the detected variants remains uncertain, and our results should be interpreted as exploratory and hypothesis generating rather than definitive.
Third, the panel’s 5% VAF threshold likely missed subclonal variants detectable only through ultra-deep or ddPCR-based approaches (8). Fourth, lack of functional validation (e.g., protein expression, pathway activation) precludes mechanistic conclusions.
Finally, although functional validation (e.g., protein expression, pathway activation) was not performed in this study, we view this not solely as a limitation but as a critical avenue for future research. Functional studies in cellular and animal models will be essential to confirm the mechanistic contribution of these mutations and to assess their potential as therapeutic targets. In addition, the restricted gene coverage of the panel and the absence of recurrently mutated genes across patients further limit the strength of our conclusions, underscoring that these findings should be considered exploratory and hypothesis-generating. Moreover, patient heterogeneity in treatment history (embolization, radiosurgery) could introduce confounding.
Conclusion
Our findings support the presence of a complex mutational profile in sporadic brain AVMs, with convergence on angiogenesis, DNA repair, and RNA Polymerase II-mediated transcription pathways. The identification of multiple mutations associated with Pol II function suggests a novel mechanism of vascular dysregulation, potentially linking genetic and epigenetic signals to aberrant vessel formation.
Although KRAS mutations were infrequent and low in allele frequency, other functionally relevant alterations may contribute to a broader molecular network underlying bAVM pathogenesis. These insights provide a framework for future studies exploring transcriptional regulation in AVMs and open the door for potential therapeutic interventions targeting these pathways.
Data availability statement
The datasets generated and analyzed for this study are contained within the article. Additional anonymized data underlying the findings of this study are available from the corresponding author upon reasonable request, in accordance with institutional and ethical guidelines.
Ethics statement
The studies involving humans were approved by the Ethics Committee of Hospital Clínico San Carlos (Approval number: 23/332-E). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
RP: Data curation, Methodology, Validation, Conceptualization, Writing – original draft, Supervision, Writing – review & editing, Investigation. VG-B: Formal analysis, Writing – review & editing, Validation, Methodology, Data curation. IC-F: Data curation, Writing – review & editing, Formal analysis. DH-M: Writing – review & editing, Formal analysis, Data curation. MG: Formal analysis, Writing – review & editing, Data curation. JC-M: Writing – review & editing, Validation. PP-S: Validation, Writing – review & editing. SC-C: Writing – original draft, Conceptualization, Validation, Data curation, Writing – review & editing, Methodology, Supervision.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
The authors acknowledge the contributions of all team members, each of whom played a role in the development and execution of this study. We also extend our sincere gratitude to the patients who provided their informed consent, allowing us to conduct this research.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
References
1. Pérez-Alfayate, R, Grasso, G, Pérez, CF, Arias-Díaz, J, and Sallabanda-Díaz, K. Does endovascular treatment with curative intention have benefits for treating high-grade arteriovenous malformation versus radiosurgery? Efficacy, safety, and cost-effectiveness analysis. World Neurosurg. (2021) 149:e178–87. doi: 10.1016/j.wneu.2021.02.053
2. Pérez-Alfayate, R, and Grasso, G. State of the art and future direction in diagnosis, molecular biology, genetics, and treatment of brain arteriovenous malformations. World Neurosurg. (2022) 159:362–72. doi: 10.1016/j.wneu.2021.08.111
3. Gallione, CJ, Repetto, GM, Legius, E, Rustgi, AK, Schelley, SL, Tejpar, S, et al. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet. (2004) 363:852–9. doi: 10.1016/S0140-6736(04)15732-2
4. Amyere, M, Revencu, N, Helaers, R, Pairet, E, Baselga, E, Cordisco, M, et al. Germline loss-of-function mutations in EPHB4 cause a second form of capillary malformation-arteriovenous malformation (CM-AVM2) deregulating RAS-MAPK signaling. Circulation. (2017) 136:1037–48. doi: 10.1161/CIRCULATIONAHA.116.026886
5. Hao, Q, Wang, H, Lu, JL, Ma, L, Chen, XL, Ye, X, et al. Activin receptor-like kinase 1 combined with VEGF-A affects migration and proliferation of endothelial cells from sporadic human cerebral AVMs. Front Cell Neurosci. (2019) 12:12. doi: 10.3389/FNCEL.2018.00525
6. Wang, K, Zhao, S, Liu, B, Zhang, Q, Li, Y, Liu, J, et al. Perturbations of BMP/TGF-β and VEGF/VEGFR signalling pathways in non-syndromic sporadic brain arteriovenous malformations (BAVM). J Med Genet. (2018) 55:675–84. doi: 10.1136/jmedgenet-2017-105224
7. Morita, H, and Komuro, I. Somatic activating KRAS mutations in arteriovenous malformations of the brain. N Engl J Med. (2018) 378:1561–2. doi: 10.1056/NEJMc1802190
8. Goss, JA, Huang, AY, Smith, E, Konczyk, DJ, Smits, PJ, Sudduth, CL, et al. Somatic mutations in intracranial arteriovenous malformations. PLoS One. (2019) 14:e0226852. doi: 10.1371/journal.pone.0226852
9. Hong, T, Yan, Y, Li, J, Radovanovic, I, Ma, X, Shao, YW, et al. High prevalence of KRAS/BRAF somatic mutations in brain and spinal cord arteriovenous malformations. Brain. (2019) 142:23–34. doi: 10.1093/BRAIN/AWY307
10. Thomas, JM, Sasankan, D, Abraham, M, Surendran, S, Kartha, CC, and Rajavelu, A. DNA methylation signatures on vascular differentiation genes are aberrant in vessels of human cerebral arteriovenous malformation nidus. Clin Epigenetics. (2022) 14:1–10. doi: 10.1186/S13148-022-01346-Z
11. Germans, MR, Sun, W, Sebök, M, Keller, A, and Regli, L. Molecular signature of brain arteriovenous malformation hemorrhage: a systematic review. World Neurosurg. (2022) 157:143–51. doi: 10.1016/J.WNEU.2021.10.114
12. Laakso, A, Dashti, R, Juvela, S, Niemelä, M, and Hernesniemi, J. Natural history of arteriovenous malformations: presentation, risk of hemorrhage and mortality. Acta Neurochir Suppl. (2010) 107:65–9. doi: 10.1007/978-3-211-99373-6_10
13. Winkler, EA, Kim, CN, Ross, JM, Garcia, JH, Gil, E, Oh, I, et al. A single-cell atlas of the normal and malformed human brain vasculature. Science. (2022) 375:eabi7377. doi: 10.1126/SCIENCE.ABI7377
14. Greaves, M, and Maley, CC. Clonal evolution in cancer. Underw Nat. (2012) 481:306–13. doi: 10.1038/nature10762
15. Al-Olabi, L, Polubothu, S, Dowsett, K, Andrews, KA, Stadnik, P, Joseph, AP, et al. Mosaic RAS/MAPK variants cause sporadic vascular malformations which respond to targeted therapy. J Clin Invest. (2018) 128:5185. doi: 10.1172/JCI124649
16. Tu, T, Yu, J, Jiang, C, Zhang, S, Li, J, Ren, J, et al. Somatic BrafV600E mutation in the cerebral endothelium induces brain arteriovenous malformations. Angiogenesis. (2024) 27:441–60. doi: 10.1007/s10456-024-09918-8
17. Deng, H, Chen, Y, Wang, L, Zhang, Y, Hang, Q, Li, P, et al. PI3K/mTOR inhibitors promote G6PD autophagic degradation and exacerbate oxidative stress damage to radiosensitize small cell lung cancer. Cell Death Dis. (2023) 14:652–17. doi: 10.1038/s41419-023-06171-7
18. Zhang, Y, Lee, JH, Paull, TT, Gehrke, S, D’Alessandro, A, Dou, Q, et al. Mitochondrial redox sensing by the kinase ATM maintains cellular antioxidant capacity. Sci Signal. (2018) 11:538. doi: 10.1126/scisignal.aaq0702
19. Luzzatto, L, Ally, M, and Notaro, R. Glucose-6-phosphate dehydrogenase deficiency. Blood. (2020) 136:1225–40. doi: 10.1182/blood.2019000944
20. Kane, M. (2023). Chloroquine therapy and G6PD genotype. Med Genet Summ. Available online at: http://europepmc.org/books/NBK591833 (Accessed February 22, 2025).
21. Zyla, RE, and Hodgson, A. Gene of the month: FH. J Clin Pathol. (2021) 74:615–9. doi: 10.1136/jclinpath-2021-207830
22. Bai, X, Xiang, D, Huang, M, and Chen, Y. Case report: successful response to bevacizumab combined with erlotinib for a novel FH gene mutation hereditary leiomyoma and renal cell carcinoma. Front Pharmacol. (2024) 15:1373020. doi: 10.3389/fphar.2024.1373020
23. Malik, S, and Roeder, RG. Regulation of the RNA polymerase II pre-initiation complex by its associated coactivators. Nat Rev Genet. (2023) 24:767–82. doi: 10.1038/s41576-023-00630-9
24. Tsai, P-F. UC, and Riverside, UC (2009). Riverside electronic theses and dissertations title TAF1 regulation of gene expression: Genome-wide localization and transcription profiling. Available online at: https://escholarship.org/uc/item/1810n0pv (Accessed February 22, 2025).
25. Katzenberger, RJ, Marengo, MS, and Wassarman, DA. ATM and ATR pathways signal alternative splicing of Drosophila TAF1 pre-mRNA in response to DNA damage. Mol Cell Biol. (2006) 26:9256–67. doi: 10.1128/MCB.01125-06
26. Harati, K, Daigeler, A, Hirsch, T, Lehnhardt, M, Steinstraesser, L, Langer, S, et al. Tumor-associated fibroblasts promote the proliferation and decrease the doxorubicin sensitivity of liposarcoma cells. Int J Mol Med. (2016) 37:1535–41. doi: 10.3892/IJMM.2016.2556/HTML
27. Shorning, BY, Dass, MS, Smalley, MJ, and Pearson, HB. The PI3K-AKT-mTOR pathway and prostate Cancer: at the crossroads of AR, MAPK, and WNT signaling. Int J Mol Sci. (2020) 21:4507. doi: 10.3390/ijms21124507
28. Yang, J, Nie, J, Ma, X, Wei, Y, Peng, Y, and Wei, X. Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol Cancer. (2019) 18:26–8. doi: 10.1186/S12943-019-0954-X
29. LoRusso, PM. Inhibition of the PI3K/AKT/mTOR pathway in solid tumors. J Clin Oncol. (2016) 34:3803–15. doi: 10.1200/JCO.2014.59.0018
30. Ladopoulos, V, Hofemeister, H, Hoogenkamp, M, Riggs, AD, Stewart, AF, and Bonifer, C. The histone methyltransferase KMT2B is required for RNA polymerase II association and protection from DNA methylation at the MagohB CpG Island promoter. Mol Cell Biol. (2013) 33:1383–93. doi: 10.1128/MCB.01721-12
31. Wälchli, T, Ghobrial, M, Schwab, M, Takada, S, Zhong, H, Suntharalingham, S, et al. Single-cell atlas of the human brain vasculature across development, adulthood and disease. Underw Nat. (2024) 632:603–13. doi: 10.1038/s41586-024-07493-y
32. Yang, HC, Stern, A, and Chiu, DTY. G6PD: a hub for metabolic reprogramming and redox signaling in cancer. Biom J. (2021) 44:285–92. doi: 10.1016/j.bj.2020.08.001
33. Zhang, Z, Li, H, Deng, Y, Schuck, K, Raulefs, S, Maeritz, N, et al. AGR2-dependent nuclear import of RNA polymerase II constitutes a specific target of pancreatic ductal adenocarcinoma in the context of wild-type p53. Gastroenterology. (2021) 161:1601–1614.e23. doi: 10.1053/j.gastro.2021.07.030
34. Hak, J-F, Boulouis, G, Kerleroux, B, Benichi, S, Stricker, S, Gariel, F, et al. Pediatric brain arteriovenous malformation recurrence: a cohort study, systematic review and meta-analysis. J Neurointerv Surg. (2022) 14:611–7. doi: 10.1136/neurintsurg-2021-017777
35. Muster, R, Ko, N, Smith, W, Su, H, Dickey, MA, Nelson, J, et al. Proof-of-concept single-arm trial of bevacizumab therapy for brain arteriovenous malformation. BMJ Neurol Open. (2021) 3:e000114. doi: 10.1136/bmjno-2020-000114
36. Hussain, MS, Moglad, E, Afzal, M, Bansal, P, Kaur, H, Deorari, M, et al. Circular RNAs in the KRAS pathway: emerging players in cancer progression. Pathol Res Pract. (2024) 256:155259. doi: 10.1016/J.PRP.2024.155259
37. Hong, DS, Fakih, MG, Strickler, JH, Desai, J, Durm, GA, Shapiro, GI, et al. KRAS G12C inhibition with Sotorasib in advanced solid tumors. N Engl J Med. (2020) 383:1207–17. doi: 10.1056/NEJMoa1917239
38. Stefani, MA, Porter, PJ, terBrugge, KG, Montanera, W, Willinsky, RA, and Wallace, MC. Large and deep brain arteriovenous malformations are associated with risk of future hemorrhage. Stroke. (2002) 33:1220–4. doi: 10.1161/01.STR.0000013738.53113.33
39. Fleetwood, IG, and Steinberg, GK. Arteriovenous malformations. Lancet. (2002) 359:863–73. doi: 10.1016/S0140-6736(02)07946-1
40. Kader, A, Young, WL, Pile-Spellman, J, Mast, H, Sciacca, RR, Mohr, JP, et al. The influence of hemodynamic and anatomic factors on hemorrhage from cerebral arteriovenous malformations. Neurosurgery. (1994) 34:801–7.
41. Walter, RB, and Morizot, DC. Conservation of genome and gene structure from fishes to mammals. Adv Struct Biol. (1996) 4:1–24. doi: 10.1016/S1064-6000(96)80003-2
42. Chalut, C, Moncollin, V, and Egly, JM. Transcription by RNA polymerase II: a process linked to DNA repair. BioEssays. (1994) 16:651–5. doi: 10.1002/BIES.950160910
43. Li, Q, Damish, AW, Frazier, Z, et al. ERCC2 helicase domain mutations confer nucleotide excision repair deficiency and drive cisplatin sensitivity in muscle-invasive bladder cancer. Clin Cancer Res. (2019) 25:977–88. doi: 10.1158/1078-0432.CCR-18-1001/87550/AM/ERCC2-HELICASE-DOMAIN-MUTATIONS-CONFER-NUCLEOTIDE
44. Moisan, F, Laroche-Clary, A, Auzanneau, C, Ricard, N, Pourquier, P, Robert, J, et al. Deciphering the role of the ERCC2 gene polymorphism on anticancer drug sensitivity. Carcinogenesis. (2012) 33:962–8. doi: 10.1093/carcin/bgs107
45. Balsas, P, Palomero, J, Eguileor, Á, Rodríguez, ML, Vegliante, MC, Planas-Rigol, E, et al. SOX11 promotes tumor protective microenvironment interactions through CXCR4 and FAK regulation in mantle cell lymphoma. Blood. (2017) 130:501–13. doi: 10.1182/BLOOD-2017-04-776740
46. McCorvie, TJ, Ferreira, D, Yue, WW, and Froese, DS. The complex machinery of human cobalamin metabolism. J Inherit Metab Dis. (2023) 46:406–20. doi: 10.1002/JIMD.12593
47. Chen, J, Wang, Q, Zhang, W, and Li, L. Effect of MTRR gene on apoptosis and autophagy pathways in multiresistant epithelial ovarian cancer. Zhonghua Fu Chan Ke Za Zhi. (2016) 51:285–92. doi: 10.3760/CMA.J.ISSN.0529-567X.2016.04.008
48. Kairupan, C, and Scott, RJ. Base excision repair and the role of MUTYH. Hered Cancer Clin Pract. (2007) 5:1–11. doi: 10.1186/1897-4287-5-4-199
49. De Rosa, M, Barnes, RP, Detwiler, AC, Nyalapatla, PR, Wipf, P, and Opresko, PL. OGG1 and MUTYH repair activities promote telomeric 8-oxoguanine induced senescence in human fibroblasts. Nat Commun. (2025) 161:18. doi: 10.1038/s41467-024-55638-4
50. Stracker, TH, Roig, I, Knobel, PA, and Marjanović, M. The ATM signaling network in development and disease. Front Genet. (2013) 4:4(MAR). doi: 10.3389/FGENE.2013.00037
51. Shanbhag, NM, Rafalska-Metcalf, IU, Balane-Bolivar, C, Janicki, SM, and Greenberg, RA. Atm-dependent chromatin changes silence transcription in cis to dna double-strand breaks. Cell. (2010) 141:970–81. doi: 10.1016/j.cell.2010.04.038
52. Morii, M, Fukumoto, Y, Kubota, S, Yamaguchi, N, Nakayama, Y, and Yamaguchi, N. Imatinib inhibits inactivation of the ATM/ATR signaling pathway and recovery from adriamycin/doxorubicin-induced DNA damage checkpoint arrest. Cell Biol Int. (2015) 39:923–32. doi: 10.1002/CBIN.10460
53. Guo, Y, Pan, W, Liu, S, Shen, Z, Xu, Y, and Hu, L. ERK/MAPK signalling pathway and tumorigenesis (review). Exp Ther Med. (2020) 19:1997–2007. doi: 10.3892/ETM.2020.8454
54. Cinque, G, Ferino, A, Pedersen, EB, and Xodo, LE. Role of poly [ADP-ribose] polymerase 1 in activating the Kirsten ras (KRAS) gene in response to oxidative stress. Int J Mol Sci. (2020) 21:6237. doi: 10.3390/ijms21176237
55. Hu, S, Song, A, Peng, L, Tang, N, Qiao, Z, Wang, Z, et al. H3K4me2/3 modulate the stability of RNA polymerase II pausing. Cell Res. (2023) 33:403–6. doi: 10.1038/s41422-023-00794-3
56. Gao, W, Liu, JL, Lu, X, and Yang, Q. Epigenetic regulation of energy metabolism in obesity. J Mol Cell Biol. (2021) 13:480–99. doi: 10.1093/JMCB/MJAB043
57. Sijbers, AM, Van der Spek, PJ, Odijk, H, et al. Mutational analysis of the human nucleotide excision repair gene ERCC1. Nucleic Acids Res. (1996) 24:3370–80. doi: 10.1093/nar/24.17.3370
Keywords: brain arteriovenous malformation, angiogenesis, somatic mutation, RNA polymerase II, PI3K pathway, DNA repair, next-generation sequencing, intracranial arteriovenous malformations
Citation: Pérez-Alfayate R, García-Barberán V, Casado-Fariñas I, Hernández-Martínez D, Gómez del Pulgar ME, Castaño-Montoya JP, Pérez-Segura P and Cabezas-Camarero S (2025) Somatic mutations in angiogenesis-related pathways and RNA polymerase II activity in sporadic brain arteriovenous malformations. Front. Neurol. 16:1660604. doi: 10.3389/fneur.2025.1660604
Edited by:
Yasushi Takagi, Tokushima University, JapanReviewed by:
Hitoshi Fukuda, Kōchi University, JapanTomohito Hishikawa, Kawasaki Medical School, Japan
Hideo Chihara, Graduate School of Science Kyoto University, Japan
Copyright © 2025 Pérez-Alfayate, García-Barberán, Casado-Fariñas, Hernández-Martínez, Gómez del Pulgar, Castaño-Montoya, Pérez-Segura and Cabezas-Camarero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rebeca Pérez-Alfayate, cmViZWNhcC5hbGZheWF0ZUBnbWFpbC5jb20=; cmVicGVyMDFAdWNtLmVz
†ORCID: Rebeca Pérez-Alfayate, orcid.org/0000-0002-2594-1988
Santiago Cabezas-Camarero, orcid.org/0000-0003-4756-7031