 Yuehui Wang
Yuehui Wang Yiran Liu
Yiran Liu Yue Han
Yue Han Yuanyuan Jing
Yuanyuan Jing Fang Deng
Fang Deng- Department of Neurology, The First Hospital of Jilin University, Changchun, China
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is an autosomal dominant disorder characterized by midlife-onset cerebrovascular disease and dementia. It is caused by mutations in the NOTCH3 gene, which affects the amount of cysteine in the extracellular domain (ECD) of the receptor, leading to protein misfolding and receptor aggregation. Emerging evidence indicates that beyond classical missense mutations, other variants including cysteine-sparing missense mutations, homozygous mutations, small deletions, duplications, splice site mutations, a deletion/insertion and loss-of-function mutations may lead to distinct phenotypes with variable severity and disease penetrance. The marked heterogeneity in genotypes and phenotypes poses significant challenges for CADASIL diagnosis and clinical management. The aim of this review is to summarize the mutational spectrum of CADASIL, explore the possible genotype–phenotype correlations and discuss the phenotypic heterogeneity of NOTCH3 mutations. More studies are needed in the future to demonstrate whether CADASIL can be expanded from classical cerebral small vessel disease to a new spectrum of diseases that share the same pathogenesis as mutations in the NOTCH3 gene.
1 Introduction
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is a monogenic inherited small vessel disease, now recognized as the most prevalent genetic cause of stroke and dementia in adults. This autosomal dominant disorder is caused by mutations in the NOTCH3 gene (1). To date, over 220 mutations have been reported in the NOTCH3 gene, of which NOTCH3 missense mutations altering cysteine residue count serve as the primary diagnostic criterion and most common mutation type (2). The accumulation of granular osmiophilic material (GOM), which contains the NOTCH3 extracellular domain (NOTCH3ECD), is detected via skin biopsies and is considered a hallmark pathological feature of CADASIL. Notably, GOM primarily consists of the extracellular structural domain (ECD) of NOTCH3 (3). However, mutations that preserve cysteine and other mutations including homozygous mutations, small deletions, duplications, splice site mutations, a deletion/insertion and loss-of-function mutations in NOTCH3 may also induce structural alterations in NOTCH3, leading to clinical manifestations resembling those of CADASIL. Thus, these diverse genotypes have challenged conventional diagnostic criteria for CADASIL-associated mutations.
CADASIL is a cerebral small-vessel disease in which patients may present with multiple clinical features, including migraine with aura, cerebrovascular events in young to middle-aged individuals, mood disorders, affective apathy, cognitive decline progressing to dementia, and diffuse white matter lesions with subcortical infarcts on neuroimaging findings (4). Elucidation of genotype–phenotype correlations and their mechanisms is essential to explain the clinical heterogeneity of CADASIL. Recent studies have found various manifestations of intracranial large artery anomalies, coronary artery disease, and abnormalities in lipid and glucose metabolism in patients with mutation of NOTCH3 gene. This suggests that NOTCH3 mutations cause more than just classical cerebral small-vessel disease and may involve large intracranial arteries and extracranial diseases. Therefore, further studies are needed to investigate whether the CADASIL phenotype is not limited to the small cerebral vessels. Moreover, the establishment of novel terminology to describe the phenotypic heterogeneity associated with this gene mutation may be necessary.
Therefore, we conducted a comprehensive evaluation of the diversity of mutations in the NOTCH3 gene, the polymorphisms in the clinical phenotype of CADASIL, and the relationship between genotype and phenotype. We first systematically cataloged NOTCH3 mutation types to expand the established CADASIL mutational spectrum. Second, we investigated genotype–phenotype correlations through integrated pathogenesis studies, providing mechanistic insights into clinical heterogeneity. Finally, based on the findings of intracranial large artery anomalies, coronary artery disease and metabolic abnormalities caused by NOTCH3 mutations, we need to revisit the concept of CADASIL disease. Whether this association is part of the NOTCH3 mutation phenotype or whether it affects the clinical course is uncertain, but should be investigated further.
2 Diversity of mutations in the NOTCH3 gene
2.1 Typical NOTCH gene mutations
Classical CADASIL is caused by mutations in the NOTCH3 gene located on chromosome 19p13, which is involved in the differentiation and maturation of vascular smooth muscle cells, vascular development during embryogenesis, and vascular integrity. The NOTCH3 gene contains 33 exons and encodes a transmembrane receptor consisting of three structural domains, including the NOTCH extracellular domain (NECD), the transmembrane domain, and the NOTCH intracellular domain (NICD) (5). Among these, exons 2–24 encode Notch3ECD, which includes 34 epidermal growth factor-like repeats (EGFr), each of which typically includes highly conserved six cysteine residues that form three disulfide bonds. Mutations in the NOTCH3 gene in CADASIL always cause an increase or deletion in the number of cysteine residues within 1 of the EGFr, resulting in an odd number of cysteine residues (usually 5 or 7). That unpaired cysteine residue, which is expected to disrupt normal disulfide bond formation, leads to misfolding of EGFr and increased NOTCH3 multimerization, ultimately causing GOM formation (6). GOM formation has been recognized as a typical pathologic features of CADASIL and has been validated in most patients with classical CADASIL.
Heterozygous missense mutations causing altered cysteine numbers have long been recognized as the typical mutational form of CADASIL, and this pathogenic mutation is located on exons 2–24 encoding the 34 EGFR structural domains. In particular, exon 2–6 is considered a mutational hotspot (7).
2.2 Cysteine-sparing NOTCH3 gene mutations
With advances in gene sequencing and more and more studies, we have found that some patients harboring NOTCH3 missense mutations that preserve cysteine, in particular the p. Arg61Trp, p. Arg75Pro, p. Asp80Gly and p. Arg213Lys mutations, exhibit clinical syndromes consistent with CADASIL, and skin biopsies of these patients have also found GOM deposits in their skin (8). The newly identified p. Gly73Ala, p. Arg75Gln, p. Ser1418Leu and p. Arg1761His mutations are also potentially pathogenic (9). Converging evidence suggests that in the genetic profile of NOTCH3 cysteine-sparing mutations, mutations are more frequently detected in exon 3, with p. Arg75Pro being the most common type.
2.3 Other NOTCH3 mutation types
In addition to this, many other mutations in NOTCH3 have been reported in recent years, causing CADASIL-like clinical phenotypes. The first category is some reports on homozygous mutations. There is a case report of a homozygous missense mutation c. C1759T (p. Arg587Cys) in NOTCH3 gene found in two brothers from China, both patients showed stroke, gait instability, cognitive impairment and psychiatric disorders. Fused white matter hypersignal and multiple lacunar infarcts were demonstrated on MRI, especially by skin biopsy which revealed characteristic GOM deposits in these two patients (10). Consistently, Tuominen et al. also described a patient with a homozygous mutation in NOTCH3 causing a CADASIL-like clinical phenotype, with the mutation located in c. C475T (p. Arg133Cys), and found GOM deposits (11). However, there have also been case reports of earlier onset and more severe disease phenotypes in patients with pure mutations compared with classic CADASIL (12). Overall, the phenotypes of patients with pure synaptic mutations described to date are almost exclusively within the normal CADASIL spectrum, and the reasons for the variation in clinical severity need to be further explored. The second group consists of small NOTCH3 deletions, duplications, splice site mutations and a deletion/insertion leading to a numerical cysteine alteration. These patients exhibit a classic CADASIL-like clinical phenotype and most of them underwent biopsy with clear GOM deposition (13, 14). Consistent with classical NOTCH3 mutations, almost all of these reported mutations involve alterations in the number of cysteine residues in EGFr, and thus they may be lethal. The paucity of reports of such mutations may be due in part to technical limitations of direct sequencing that miss intronic mutations, large deletions, multiplications, or large rearrangements of the NOTCH3 gene. However, a CADASIL phenotype caused by a 12-base pair deletion that does not result in a change in the number of cysteines was reported in one study. This deletion may alter the spacing between the two cysteines in EGFr, resulting in disruption of the normal disulfide bond (15). The last category is NOTCH3 loss-of-function mutations, including nonsense mutations causing premature stop codons and frameshift deletion. Such mutations do not trigger changes in the number of EGFr cysteine residues and GOM deposition, but cause hereditary small vessel disease phenotypes of varying severity or penetrance, similar to CADASIL (16–19). Although some NOTCH3 mutations outside the exons encoding EGFr have also been reported, whether these mutations cause the CADASIL phenotype remains highly controversial, and more patients with such mutations should be found and examined to clarify the conclusions (20–23).
In summary, in the past 30 years since the identification of classical mutations in the CADASIL-causing gene NOTCH3, we have identified an increasing number of novel NOTCH3 variant types that exhibit clinical manifestations similar t1o those of CADASIL, implying a diversification of the NOTCH3 genotypes that cause CADASIL, partly explaining why this disease is much more prevalent than initially assumed, and is even considered to be the most common type of inherited vascular dementia (Table 1). Therefore, we believe that continuous additions to the CADASIL mutational spectrum are essential to clarify the diagnosis of this disease. However, to avoid erroneous mutation interpretations and diagnoses, our analysis of these patients with NOTCH3 mutations should always include comprehensive NOTCH3 molecular screening, as well as comprehensive clinical (re)evaluation, including skin biopsie.
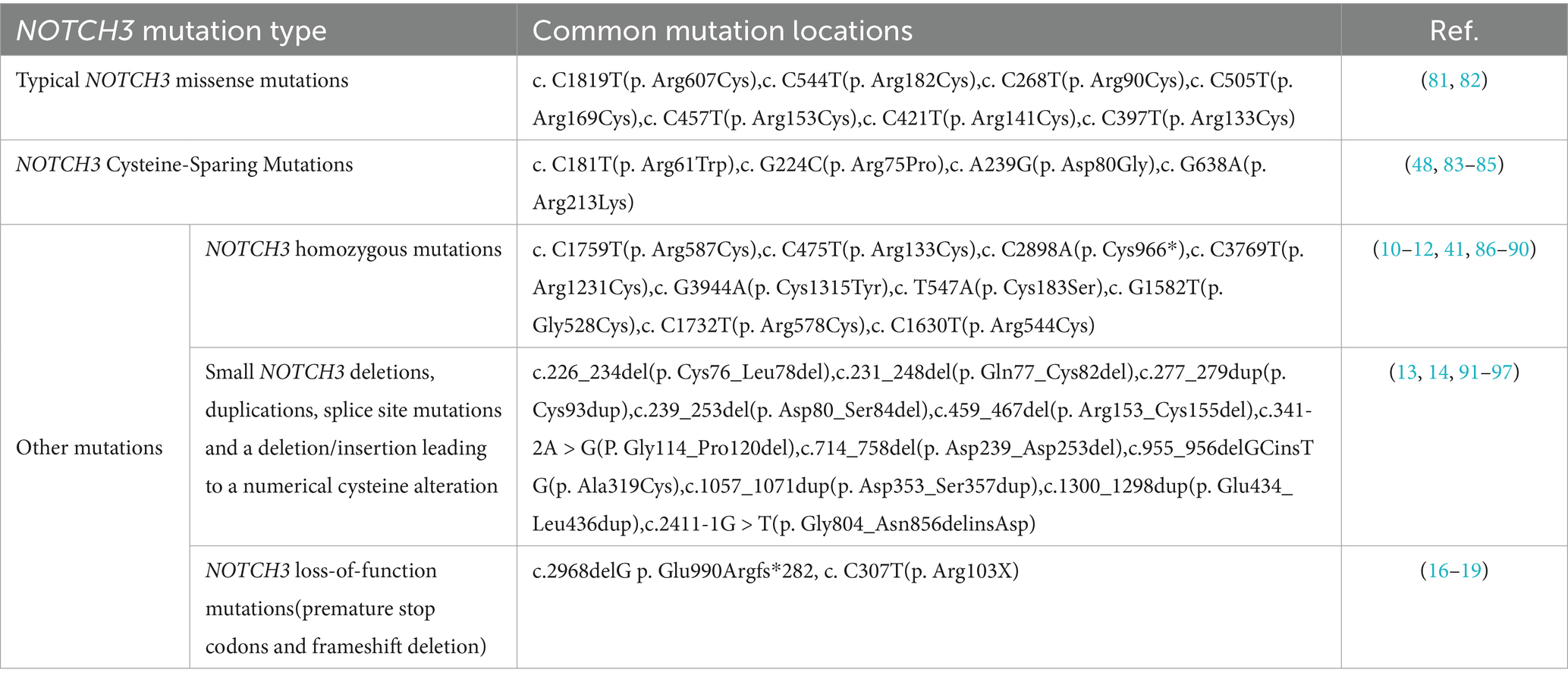
Table 1. Classification of NOTCH3 mutations associated with CADASIL.
3 Pathogenesis of CADASIL
There is no consensus on the pathogenesis of CADASIL. Two key pathophysiological mechanisms are so far thought to contribute to the emergence and progression of CADASIL: aberrant NOTCH3 aggregation and aberrant NOTCH3 signaling (Figure 1). The typical pathologic feature of CADASIL is the presence of GOM deposits within or adjacent to the basement membrane of VSMC or pericytes by electron microscopy. GOM may disrupt VSMC function by inhibiting NOTCH3-regulated smooth muscle transcripts (24). Although the exact composition of GOM remains largely unknown, in vivo and in vitro studies suggest that Notch3ECD is a major component of GOM. Meanwhile, extracellular matrix proteins such as Metalloproteinase inhibitor 3 (TIMP3) and Vitronectin (VTN) co-localized with Notch3ECD, which may jointly contribute to the formation of GOM deposits (25).
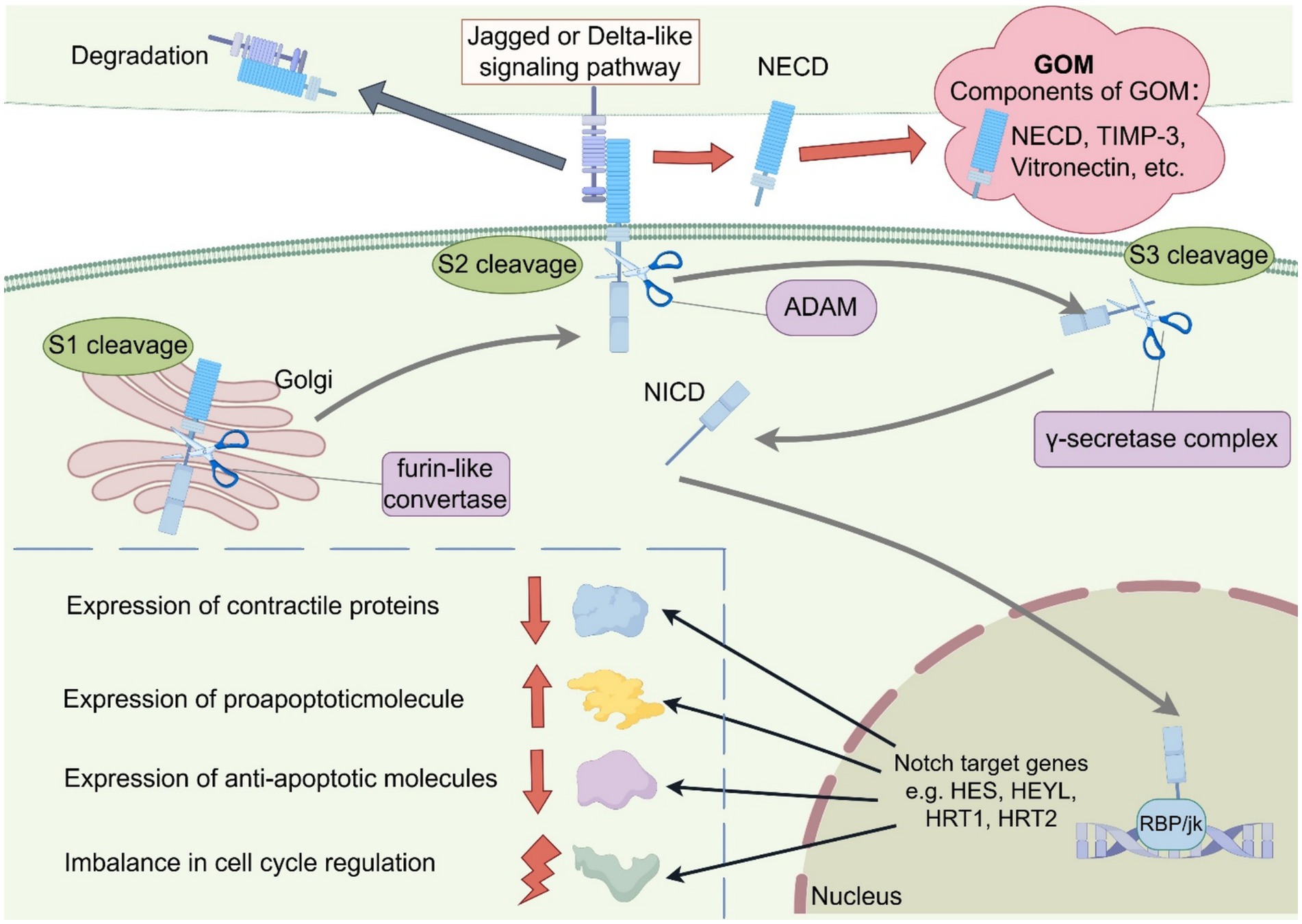
Figure 1. Schematic representation of the two key Pathogenesis of CADASIL. The schematic diagrams were designed by FigDraw (www.figdraw.com). The two main hypotheses being proposed are aberrant NOTCH3 aggregation and aberrant NOTCH3 signaling: (1) Mutations in the NOTCH3 gene in CADASIL can lead to an increase or deletion in the number of 1 cysteine residues in EGFr. This unpaired cysteine residue disrupts normal disulfide bond formation, leading to misfolding of EGFr and increased NOTCH3 multimerization, ultimately leading to the formation of GOM. In addition, proteins such as TIMP-3 and Vitronectin are involved in GOM formation. (2) In the NOTCH signaling pathway, the NOTCH receptor undergoes S1 cleavage by furin-like convertase in the Golgi apparatus and then transfers to the cytoplasmic membrane where it binds to Jagged or Delta-like ligands. When the NOTCH receptor binds to the ligand, it is cleaved by ADAM for S2 cleavage. After S2 cleavage the transmembrane structural domain and NICD enter the cytoplasm to participate in S3 cleavage induced by the γ-secretase complex, generating active NICD. NICD forms a complex with the transcription factor RBP/Jk, leading to the up-regulation of NOTCH target gene expression (HES1, HEYL, HRT1, HRT2, etc.), possibly by influencing the expression of contractile proteins, apoptotic factors and cell cycle proteins to regulate cell growth and apoptosis. GOM, granular osmiophilic material; NECD, NOTCH extracellular domain; NICD, NOTCH intracellular domain; ADAM, a disintegrin and metalloproteinase.
However, converging evidence suggests aberrant NOTCH3 signaling as another potential pathogenic mechanism. The NOTCH signaling process is complex. The Notch receptor is first produced in the endoplasmic reticulum, and then undergoes its first cleavage in the Golgi apparatus by furin-like convertase. After cleavage it is transferred to the cytoplasmic membrane to bind to the five corresponding ligands (Jagged 1 and 2 and Delta-like 1, 3, and 4). When the Notch receptor binds to the ligand, specific sites within the negative regulatory region are cleaved by a disintegrin and metalloproteinase domain (ADAM)-type metalloproteinase, which is the second hydrolysis of the notch signaling pathway and a key step in its activation (26). The ECD is removed and taken up and degraded by the ligand-expressing cells, and the remaining transmembrane structural domains and the NICD participate in the third cleavage induced by the γ-secretase complex, generating active NICD. NICD forms a complex with the transcription factor RBP/Jk, leading to the upregulation of NOTCH target gene expression (HES1, HEYL, HRT1, HRT2, etc.), which may promote cell growth and regulate apoptosis (27). NOTCH3 is expressed almost exclusively in vascular smooth muscle cells (VSMC). Aberrant NOTCH3 signaling may affect VSMC differentiation, maturation, growth, and apoptosis by influencing the expression of contractile proteins, the expression of apoptotic factors (including pro- and anti-apoptotic molecules), and the expression of cell cycle proteins (28). A study by Celine et al. concluded that the p. Arg169Cys heterozygous missense mutation that causes CADASIL leads to NOTCH3 hyperactivation (29). There is also evidence that NOTCH3 mutations in EGFr10 to 11 that cause cysteine alterations not only cause GOM deposition, but also lead to defective NOTCH3 receptor function, such as p. Cys455Arg located in EGFr10. A report from Colombia found that individuals with the p. Cys455Arg mutation exhibited ligand-induced NOTCH3 signaling was severely reduced and an early-onset CADASIL phenotype (including early-onset stroke and extensive MRI abnormalities), which may be related to the attenuation of Jagged-1-induced NOTCH3 receptor signaling through RBP-Jk by the p. Cys455Arg mutation (30). These studies suggest that while many mutations do not affect NOTCH3 signaling, certain mutations appear to have varying effects on NOTCH3 signaling activity, causing overactivity or attenuation of the signaling pathway. These diverse findings raise the possibility that different NOTCH3 mutation types and locations can have different effects on NOTCH3 signaling and can promote vascular abnormalities through different mechanisms (eg, loss of mural cells, etc). In models with NOTCH3 mutations, either an increase or decrease in NOTCH3 signaling may be interpreted in the context of a signaling threshold model (or goldilocks) (31). However, of all the current studies on the mechanisms of CADASIL, only a small number have systematically elucidated the effects of NOTCH3 mutations on the NOTCH3 signaling pathway. Therefore, in addition to GOM deposition, we need more studies to elucidate the role of NOTCH3 signaling in pathogenesis.
4 Genotype–phenotype correlations in CADASIL
The core clinical manifestations of CADASIL include early and recurrent cerebral ischemic events (transient ischemic attack and ischemic stroke) and progressive vascular cognitive impairment. Other clinical manifestations include migraine with aura, mood disorders, gait disturbances, intracerebral hemorrhage, motor abnormalities (e.g., Parkinson’s disease), and seizures. Typical Brain magnetic resonance imagin (MRI) abnormalities are progressive symmetrical white matter hyperintensities (WMHs) involving the anterior temporal lobe, the external capsule, and the superior frontal gyrus, lacunar infarcts, cerebral microbleeds (CMBs), and cerebral atrophy (32). The diversity of mutations in the NOTCH3 gene and clinical heterogeneity of the patients have revealed significant and complex genotype–phenotype correlations: mutations in NOTCH3 mutations located in different EGFr regions lead to different severity of clinical phenotypes, the effect of some specific NOTCH3 gene mutation types on clinical phenotypes as well as the fact that cysteine-conserving NOTCH3 mutation types and classically altered cysteine number NOTCH3 mutation types exhibit similar, but not identical, clinical phenotypes.
4.1 Effect of different mutation regions on disease phenotypes
To date, there have been many genotype–phenotype correlations on NOTCH3 mutations caused by typical cysteine alterations. Several studies have been conducted to analyze the genotype–phenotype correlations from several common specific mutant types (Table 2).
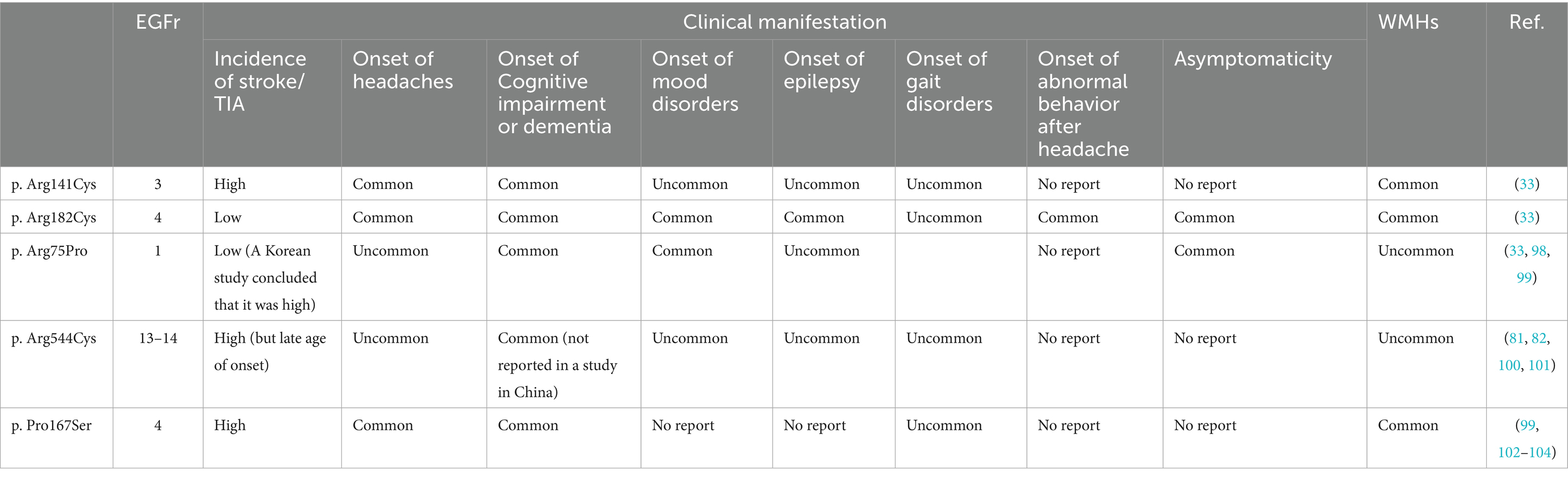
Table 2. Comparison of clinical phenotypes of 5 common NOTCH3 mutations (p. Arg141Cys, p. Arg182Cys, p. Arg75Pro, p. Arg544Cys, p. Pro167Ser).
Rutten et al. found from a large-scale genetic variation database study that the frequency of EGFr cysteine-altered NOTCH3 mutations in the general population was 3. 4/ 1,000, a frequency 100-fold higher than the currently estimated prevalence of CADASIL. Those individuals, in whom the mutation accumulates predominantly in EGFr 7–34, have an increased lifetime risk of developing stroke and vascular dementia, albeit with a later onset than in classic CADASIL patients. In contrast, in patients with CADASIL, typical cysteine-altering mutations are predominantly located in EGFr 1–6 (6). This led us to realize that the spectrum of diseases with NOTCH3 mutations could be very broad, including CADASIL. subsequently, in a further study, Rutten et al. demonstrated that in patients diagnosed with CADASIL, mutations in EGFr 1–6 predispose to the classical, more severe CADASIL phenotype, whereas pathogenic variants in EGFr 7–34 more frequently lead to a milder phenotype. Specifically, patients with mutations in EGFr 1–6 had an earlier age of stroke onset by 12 years, shorter survival, and increased white matter high-signal volume compared with patients with mutations in EGFr 7–34, which is consistent with a previous study in Japan (33). Hack et al. reported a novel three-tiered risk classification for the NOTCH3 variants that included not only EGFr structural domains 1–6 but also EGFr structural domains 8, 11, and 26 in high risk, as they found that patients with variants in EGFr structural domains 8, 11, and 26 had similar imaging burden and clinical presentation as patients with variants in EGFr structural domains 1–6 (34). A recent large cohort study not only confirmed the finding that pathogenic variants in EGFr 7–34 cause a milder clinical phenotype than pathogenic variants in EGFr 1–6, but further found that the severity of stroke was similar in EGFr 1–9 and 18–34, and that patients with variants in EGFr 10–17 had a lower risk of stroke than those with EGFr 3–4 (35). Similar to classical CADASIL, the location of mutations in the NOTCH3 retention cysteine gene likewise influences the severity of the disease: exons 1–6 mutations present a more severe clinical manifestation compared to retention cysteine mutations in exons 7–34. This further explains the fact that the cysteine-sparing mutations that have been diagnostic of CADASIL to date are more frequently detected in exon 3: since these patients have more severe clinical manifestations and disease progression, they are more likely to be detected and diagnosed (8, 9).
The current study allows us to confirm location-dependent genotype–phenotype correlations in CADASIL, and that the location of NOTCH3 pathogenicity variants is a key determinant of disease severity in CADASIL, above cardiovascular risk factors and gender, which is an important guideline for the development of a predictive model for NOTCH3 to help us to identify these patients at risk for early onset, severe disease. We need to further explore the molecular mechanisms associated with the location of pathogenic variants of NOTCH3.
4.2 Effect of NOTCH3 cysteine-sparing mutations on disease phenotypes
There are many reports of NOTCH3 cysteine-sparing mutations with CADASIL-like clinical manifestations. Although the clinical presentation and imaging of patients with NOTCH3 cysteine-sparing mutations are similar to those of classical CADASIL, they are not identical. Cysteine-sparing mutations have a later onset of symptoms than cysteine-associated mutations: the mean age of patients with CADASIL presenting with neurological signs or symptoms is 45–51. 3 year, particularly in men at approximately 50 years and in women at approximately 53 years (36), whereas carriers of NOTCH3 cysteine-sparing mutations have a mean age of 53. 64 years. The most common clinical manifestations in patients with NOTCH3 cysteine-sparing mutations are gait disturbance, cognitive impairment and stroke. The most common cerebral radiologic manifestations were lacunar cerebral infarction and cerebral microhemorrhage. Compared with patients with classical CADASIL, patients with NOTCH3-preserved cysteine mutations had a significantly higher frequency of cognitive impairment and cerebral microhemorrhages, whereas anterior temporal pole and external capsule white matter high signal was rarely observed. The relatively milder white matter involvement in two specific locations, the anterior temporal pole and the external capsule, may be an imaging feature of NOTCH3-preserved galactorrhea mutations (9). We speculate that this may be because the unique convoluted structure and vascularization of the temporal pole is particularly susceptible to myelin depletion or edema, which may result from aggregation of NOTCH3ECD and microvascular changes (37). In patients with preserved cysteine mutations, these mutations are less involved in GOM deposition and have less effect on interstitial fluid drainage and white matter thinning. This may partly explain the different effects observed for different mutation types in the CADASIL phenotype. However, we need to standardize MRI protocols to ensure consistent detection and characterization of white matter lesions and vascular abnormalities across different patient populations. p. Leu1518Met is a variant located outside of the EGFr region that does not involve a change in the number of cysteines, and Park et al. have reported this variant in a GOM-positive patient with CADASIL (38). Schmidt et al. have linked the p. Leu1518Met variant to be associated with severe white matter lesions (39). p. Leu1518Met was detected in a 36-year-old female patient with severe neurological symptoms and a 47-year-old male patient, both of whom showed extensive white matter involvement on neuroimaging, suggesting that the p. Leu1518Met variant is associated with severe white matter lesions (40). In addition to this, patients with a homozygous NOTCH3 mutation also seem to exhibit a clinical phenotype belonging to the classical clinical spectrum of CADASIL, whereas some reports have emphasized an increased disease severity in homozygous carriers, the exact mechanism of which is still unclear (41).
In summary, the location and type of mutation are major determinants of the clinical phenotype, and further studies are needed to clarify the relationship between CADASIL genotype and phenotype and the underlying mechanisms. However, the manifestation and severity of the disease cannot be explained solely by genotype–phenotype, but may also vary among different races and family lines with the same mutation (42), or even among identical twins (43). This may be related to modifier genes, environmental influences, or other concomitant risk factors that together determine the clinical heterogeneity of the disease.
5 Clinical polymorphisms in CADASIL
With the development of diagnostic and experimental techniques, we have identified NOTCH3 mutations in more clinical studies, including anomalies of the large intracranial arteries, coronary artery disease and abnormalities in lipid and glucose metabolism. These clinical alterations may be related to the molecular mechanisms of NOTCH3 signaling. Notch signaling is evolutionarily conserved, and the NOTCH3 gene encodes the single-channel transmembrane receptor NOTCH3. Clinical manifestations of large intracranial arteries and extracranial have led to a reexamination of the CADASIL perspective and there is an urgent need for us to use new terminology to summarize this clinical polymorphism of NOTCH3.
5.1 Relationship between NOTCH3 mutations and intracranial large arteries
In the past, it was thought that NOTCH3 mutations caused pure cerebral small-vessel disease-classical CADASIL. However, several studies have shown that NOTCH3 mutations may be present in a variety of intracranial large-artery anomalies causing infarcts associated with large-artery disease, with most of the patients having altered cysteine (44). Eun J (45) and Hyun (44) both confirmed in their clinical studies that intracranial large artery stenosis may not be an incidental phenomenon unrelated to mutations in the NOTCH3 gene. In a study from China, 28 of 37 CADASIL patients (75. 7%) had intracranial large artery anomalies. Usually WMH distribution is symmetrical in CADASIL, but this study suggested that intracranial large artery disease in CADASIL patients may cause an asymmetrical WMH pattern, which may be due to large artery anomalies altering hemodynamics and exacerbating the WMH in the corresponding region. Therefore, this characteristic imaging manifestation could be an important marker of intracranial large artery stenosis in patients with NOTCH3 (46). Hyun et al. suggested that intracranial large artery stenosis in patients with CADASIL may preferentially involve relatively small intracranial arteries because in these patients, the arterial stenosis is usually located in the anterior cerebral artery, the anterior inferior cerebellar artery, or the middle cerebral artery distal to the smaller vessels M1 and M2 (44). This clinical evidence suggests that NOTCH3 mutations cause more than cerebral small-vessel disease and have an important association with intracranial large-artery stenosis, although most cases have been described only in East Asia. One autopsy showed GOM deposition in the aorta, carotid and renal arteries of a patient with CADASIL (47). An autopsy of a patient with CADASIL in Japan showed arterial stenosis in the basilar, internal carotid, anterior cerebral, middle cerebral, and posterior cerebral arteries, which exhibited atherosclerotic changes in the absence of any risk factors (48). Therefore, we hypothesized that stenosis of large intracranial arteries may be related to atherosclerosis, and that endothelial cell damage associated with GOM deposition accelerates the progression of atherosclerosis. In addition to stenosis of the great arteries, congenital anomalies of the intracranial great arteries in CADASIL patients have also been reported, including basilar artery fenestration, vertebral artery (VA) hypoplasia or agenesis, anterior cerebral artery (ACA) shares common trunk, and fetal posterior cerebral artery (FPCA). However, whether these congenital abnormalities of the cerebral vasculature share a common pathogenesis with the stenosis that develops later in life is unclear, and whether these changes are part of the NOTCH3 mutant phenotype has not been further investigated (46).
The mechanism by which NOTCH3 mutations cause atherosclerosis in large arteries has been less well studied. Atherosclerosis is a chronic inflammatory disease caused by the interaction of lipids, macrophages/foam cells and arterial wall cells (49). Chronic inflammation is central to the pathology of atherosclerotic vascular disease and metabolic disorders. In particular, macrophages are involved in the expanded cascade of reactions and play a key role in maintaining the inflammatory response in atherosclerotic plaques, promoting their structural instability, and thrombus formation (50). There are multiple NOTCH3 receptors and ligands expressed in macrophages, among which DII4 and NOTCH3 are co-localized in macrophages within atherosclerotic plaques (51). Fung et al. revealed the role of the DII4-NOTCH pathway in the inflammatory response characterized by macrophage activation: stimulation of pro-inflammatory factors such as LPS and IL-1β increased the expression of DII4 in macrophage expression, triggered hydrolysis and activation of NOTCH proteins, and induced pro-inflammatory molecules and pathways, such as inducible nitric oxide synthase (iNOS), pentraxin 3 (PTX3), mitogen-activated protein kinase (MAPK), Akt, and NF-κB. Notably, DII4-triggered NOTCH signaling increased the expression of DII4 itself, which further demonstrates that the DII4-NOTCH signaling pathway mediates the inflammatory response by accelerating is a positive feedback loop of cell activation leading to atherosclerotic plaque loading, progression and thrombus formation (52). Consistently, blockade of Dll4-Notch signaling using a neutralizing anti-Dll4 antibody attenuated the development of atherosclerosis in LDL receptor-deficient mice (53). VSMC influence atherosclerotic plaque formation in several ways and may determine susceptibility to plaque rupture (54, 55). Among these, trans-differentiation of VSMCs, or their transition from a contractile/quiescent state to a secretory/inflammatory/migratory state plays an important role during atherosclerosis (56). Although the relationship between VSMC and NOTCH signaling during atherosclerosis is unclear, it has been found that atherosclerotic plaques in human carotid and femoral arteries usually have a lot of NOTCH3 proteins, and it is possible that Jagged1 stimulates the NOTCH signaling pathway in VSMCs, which causes pathological changes (57). Adenylate cyclase type 8 (AC8) is known to be involved in trans-differentiation of VSMC. A study by Zela et al. indicated that Jagged1-triggered inhibition of the NOTCH3-HRT3 and/or HRT1 pathways in the context of pathological vascular remodeling enhanced IL1β-mediated up-regulation of AC8 in trans-differentiated VSMC and amplified its deleterious effect on trans-differentiated VSMC differentiation (58). A recent study by Dave et al. linked elastin deficiency to NOTCH signaling (59): although elastin deficiency does not directly contribute to atherosclerosis, degradation of elastin may allow lipid infiltration into atherosclerotic plaques; elastin deficiency in VSMC resulted in reduced levels of DNA methyltransferase 1 (DNMT1) and DNA methylation (an epigenetic marker that drives gene silencing), which induces the expression of key NOTCH pathway genes Jagged1, γ-secretase catalytic subunits PSEN1 and PSEN2 and triggers NOTCH3 activation. That is, upregulation of the NOTCH3 pathway causes accumulation of pathological VSMC during elastin deficiency (59). Therefore, we suggest that lesions of large intracranial arteries may be one of the clinical phenotypes of NOTCH3 mutant disease. The study of the molecular biological mechanism of NOTCH3 mutation-induced lesions in large vessels led us to hypothesize that NOTCH3 mutation is a general vascular lesion affecting all types of intracranial vessels but manifesting predominantly in small vessels rather than a lesion unique to small vessels. We need to further investigate the mechanism and validate it pathologically in stenotic vessels.
5.2 Association of NOTCH3 mutations with coronary artery disease
To the best of our knowledge, although the vascular lesions in classical CADASIL predominantly involve the intracranium, vascular changes in CADASIL occur along the arterial tree, including the heart (47). Embryological effects of the NOTCH3 receptor pathway responsible for the development of the cardiovascular system support this theory (60). Consistent with the findings of CADASIL intracranial vasculature, we confirmed abnormalities of the coronary microvascular system in myocardial histopathologic examination and found typical CADASIL changes such as GOM deposition, cellular degeneration, and basement membrane thickening (61). In recent years an increasing number of patients with CADASIL present with ischemic coronary artery disease (CAD), and there have been many reports of an association between CADASIL and CAD, albeit with limited evidence. Argirò et al. included 17 patients with CADASIL confirming a blunted coronary flow reserve due to dysfunction of the coronary microcirculation with diffuse hypoperfusion areas (62). In a case report of a patient with CADASIL presenting with angina pectoris without obstructive coronary artery disease, coronary CT angiography demonstrated diffuse vascular irregularities, vascular thickening, and mixed noncalcified/calcified coronary lesions with moderate stenosis (63). Consistently, Rubin et al. performed coronary angiography in patients with CADASIL and found diffuse and irregular stenosis of the involved coronary arteries (64). Thus, these clinical findings suggest that NOTCH3 mutations may have caused coronary microvascular dysfunction and diffuse coronary stenosis rather than coronary atherosclerosis. Also, the more aggressive and accelerated CAD in CADASIL patients may be related to microvascular dysfunction, allowing for a younger age of onset and a broader disease burden (65). NOTCH3 mutations impair the damaged heart through coronary microvascular dysfunction, loss of pericytes, and impaired microvascular maturation recovery, and the main causes of coronary microvascular dysfunction are fibrovascular wall thickening, impaired vascular reactivity, and increased myogenic tone (66).
Several studies have been conducted on the molecular mechanisms of how NOTCH3 mutations cause CAD. Mutations in components of the NOTCH pathway lead to structural abnormalities and dysfunction in the heart. In particular, activation of NOTCH exerts a protective effect against myocardial infarction, cardiac hypertrophy, and alcoholic cardiomyopathy. Binding of NOTCH3 receptors on human coronary smooth muscle cells and Jagged1 ligands on human coronary endothelial cells contributes to increased expression of smooth muscle-α-actinin and calmodulin (67). A recent study demonstrated that the NOTCH3-RBPJk signaling pathway maintains the integrity of the coronary vascular system by regulating the vascular endothelial growth factor A (VEGFA)/vascular endothelial growth factor receptor 2 (VEGFR2)/DII4 pathway. Thus, the absence of the NOTCH3-RBPJk signaling pathway in VSMC leads to heart failure during pressure overload, and alterations in the cardiac vasculature are a direct causative factor (68). Oncostatin M (OSM) is known to be an inflammatory cytokine, and inhibition of OSM signaling inhibits cardiomyocyte remodeling, leading to deterioration of cardiac function after myocardial infarction. A study showed that the mechanism of OSM on cardiac ischemia/reperfusion injury is partially mediated by the NOTCH3/Akt pathway. Thus, a novel role of NOTCH3/Akt signaling contributes to OSM-induced protection against cardiac ischemia/reperfusion injury (69).
5.3 Association of NOTCH3 mutations with metabolic abnormalities such as lipids and blood glucose
NOTCH signaling is considered a key player in metabolism (70), and activation of the NOTCH3 gene may lead to hyperlipidemia. Gong et al. reported 1 family with CADASIL with elevated lipoprotein levels that caused a higher risk of disease (71). Apolipoprotein D (APOD) is known to be a secreted glycoprotein that regulates smooth muscle cells (72, 73). It is associated with lipid metabolism, particularly in plasma mainly associated with High-density lipoprotein HDL, and is abundantly present in atherosclerotic lesions (74). Several studies have demonstrated a correlation between APOD expression and healthy lipid profiles (75). It was shown that cell contact-dependent Notch signaling contributes to the regulation of APOD in mural cells (76). Further experiments revealed that NOTCH3 contributes to the down-regulation of APOD, possibly through the activation of the downstream NOTCH signaling pathway by JAGGED-1 ligand on endothelial cells, which increases the regulation of APOD in wall cells by endothelial cells (77). Therefore, we suggest that mutations in the NOTCH3 gene may cause impairment of the corresponding signaling pathway and increased APOD expression, leading to severe dyslipidemia. Wimmer et al. identified the Dll4-NOTCH3 pathway as a key driver of diabetic vasculopathy in the human vasculature. They constructed vascular organoid models and placed them in a high-glucose environment and observed by electron microscopy a marked increase in collagen IV, as well as fibronectin, laminin, perlecan, and the pro-inflammatory cytokines TNFα and IL6. Mainly pericytes, but also endothelial and MSC-like cells, upregulated ECM synthesis. And we also observed substantial thickening and splitting of the basement membrane layer without changes in vessel diameter. Basement membrane thickening was reduced after blocking NOTCH3 protein and its ligand DLL4, suggesting that the DLL4-NOTCH3 pathway plays an important role in diabetic vasculopathy (78). We speculate that NOTCH3 gene mutations may interfere with the Dll4-NOTCH3 pathway thereby causing blood glucose abnormalities, which suggests that we investigate the association between the CADASIL clinical phenotype and diabetes through more clinical series. However, there are limited studies on the relationship between NOTCH3 mutations and metabolisms such as lipids and glucose, and we need further studies on pathogenesis to clarify the relationship.
In addition to this, NOTCH3 has many associations with human diseases through mutations, altered expression, or dysregulation of its activity or turnover. Although the main function of NOTCH3 is to regulate angiogenesis, dysregulation of NOTCH3 activity may play an important role in tumorigenesis and tumor maintenance. The apparent alteration (and in many cases overactivation) of NOTCH3 signaling activity causes a number of changes, including controlling the tumor-initiating cell phenotype, regulating known upstream or downstream tumor-associated signaling factors, facilitating angiogenesis or tumor invasion, regulating the cell cycle, etc. Recent studies have found that NOTCH3 expression is found in most T-cell acute lymphoblastic leukemias, possibly through the activation of Notch3 signaling by the expression of Dll in endothelial cells thereby promoting the proliferation of T-cell acute lymphoblastic leukemia cells (79). Furthermore, it has been demonstrated that aging is associated with a significant progressive decline in NOTCH3 signaling. This age-related lack of NOTCH3 triggers a decline in vascular function, which subsequently affects lymphatic flow and ultimately leads to degenerative changes (80). Thus, the complex and highly variable functions of NOTCH3 factors have led to diverse associations in human development and disease, and the pathogenesis of these NOTCH3-related diseases almost always involves alterations in the NOTCH3 signaling pathway. However, whether these diseases share the same mechanisms as CADASIL has not been systematically elucidated. Perhaps, further studies of the mechanisms and a more complete understanding of NOTCH signaling will help to generalize the clinical heterogeneity of NOTCH mutations. At the same time, this will underpin the development of means to more specifically target its aberrant activity in different disease contexts.
To date, there have been no case reports of CADASIL patients with concomitant intracranial large artery and coronary artery stenosis. However, previous studies have found typical CADASIL pathologic manifestations such as deposition of GOM in both intracranial large arteries and coronary arteries. Meanwhile, activation or inhibition of the NOTCH signaling pathway caused by NOTCH3 mutations also leads to pathological changes such as VSMCs. In conclusion, NOTCH3 mutations may lead to intracranial aortic anomalies, coronary artery disease, and abnormalities in lipid and glucose metabolism through different mechanisms. NOTCH3 mutations may no longer be simply a disease of the small blood vessels of the brain-typical of CADASIL-but a new spectrum of diseases that share the same pathogenesis as NOTCH3 mutations. However, there is evidence that traditional risk factors such as hypertension, diabetes mellitus, or smoking also contribute to the more severe clinical phenotype of CADASIL. It is possible that the observed cases of intracranial large artery stenosis or coronary artery disease reflect traditional vascular risk factors and diseases (e.g., higher prevalence of intracranial atheromatous plaques in Southeast Asia) rather than representing a true characterization of the pathology associated with NOTCH3 mutations. Therefore, more robust clinical studies and mechanistic investigations are needed to demonstrate that clinical polymorphisms do indeed characterize genetically determined diseases.
6 Conclusion
In conclusion, this study synthesizes the CADASIL mutational landscape and systematically delineates genotype–phenotype correlations with their underlying pathophysiological mechanisms. The correlation between CADASIL genotypes and phenotypes depends mainly on the NOTCH3 mutation location and mutation type. Environmental factors, epigenetic regulation and multigene interactions also influence the correlation to some extent. Despite great progress, the exact mechanisms have not been clarified. Continued future research on CADASIL genotype–phenotype correlation is essential to reveal its heterogeneity, identify therapeutic targets, achieve early diagnosis and mitigate disease progression. Moreover, we further demonstrated that NOTCH3 mutations cause anomalies of the large intracranial arteries, coronary artery disease, and metabolic abnormalities through a pathogenesis similar to that of classical CADASIL. Unfortunately, this association has not been confirmed by large clinical series and potential mechanistic studies. Still, these findings suggest a possible need to revisit the concept of CADASIL (characterized by cerebral small-vessel disease caused by NOTCH3 mutations) as a disease. We attempt the establishment of a new term- NOTCH3 mutaion spectrum diseases -to explain this novel disease entity that differs from traditional CADASIL. It refers to NOTCH3 mutation-associated diseases that contain multiple heterogeneous presentations with similar pathogenesis. The application of this terminology is expected to provide a more comprehensive overview of the complexity and heterogeneity of NOTCH3 mutation diseases and support the development of precision medicine approaches. Future studies will explore the association of NOTCH3 mutations with intracranial large arteries, coronary artery disease, and metabolic abnormalities and their associated pathogenesis based on a larger sample.
Author contributions
YW: Conceptualization, Visualization, Writing – original draft. YL: Visualization, Writing – original draft. HM: Writing – original draft. YH: Writing – original draft. YJ: Writing – original draft. FD: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Natural Science Foundation of Jilin Province (Grant no. YDZJ202401446ZYTS).
Acknowledgments
We sincerely appreciated all participants.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Choi, JC. Genetics of cerebral small vessel disease. J Stroke. (2015) 17:7–16. doi: 10.5853/jos.2015.17.1.7
2. Di Donato, I, Bianchi, S, De Stefano, N, Dichgans, M, Dotti, MT, Duering, M, et al. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (cadasil) as a model of small vessel disease: update on clinical, diagnostic, and management aspects. BMC Med. (2017) 15:41. doi: 10.1186/s12916-017-0778-8
3. Yuan, L, Chen, X, Jankovic, J, and Deng, H. Cadasil: a notch3-associated cerebral small vessel disease. J Adv Res. (2024) 66:223–35. doi: 10.1016/j.jare.2024.01.001
4. Viswanathan, A, Gschwendtner, A, Guichard, JP, Buffon, F, Cumurciuc, R, O'Sullivan, M, et al. Lacunar lesions are independently associated with disability and cognitive impairment in CADASIL. Neurology. (2007) 69:172–9. doi: 10.1212/01.wnl.0000265221.05610.70
5. Joutel, A, Andreux, F, Gaulis, S, Domenga, V, Cecillon, M, Battail, N, et al. The ectodomain of the notch3 receptor accumulates within the cerebrovasculature of cadasil patients. J Clin Invest. (2000) 105:597–605. doi: 10.1172/jci8047
6. Rutten, JW, Dauwerse, HG, Gravesteijn, G, van Belzen, MJ, van der Grond, J, Polke, JM, et al. Archetypal notch3 mutations frequent in public exome: implications for cadasil. Ann Clin Transl Neurol. (2016) 3:844–53. doi: 10.1002/acn3.344
7. Heidari, P, Taghizadeh, M, and Vakili, O. Signaling pathways and molecular mechanisms involved in the onset and progression of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (cadasil); a focus on notch3 signaling. J Headache Pain. (2025) 26:96. doi: 10.1186/s10194-025-02025-z
8. Muiño, E, Gallego-Fabrega, C, Cullell, N, Carrera, C, Torres, N, Krupinski, J, et al. Systematic review of cysteine-sparing notch3 missense mutations in patients with clinical suspicion of cadasil. Int J Mol Sci. (2017) 18:18. doi: 10.3390/ijms18091964
9. Cao, Y, Zhang, DD, Han, F, Jiang, N, Yao, M, and Zhu, YC. Phenotypes associated with notch3 cysteine-sparing mutations in patients with clinical suspicion of cadasil: a systematic review. Int J Mol Sci. (2024) 25:25. doi: 10.3390/ijms25168796
10. He, R, Li, H, Sun, Y, Chen, M, Wang, L, Zhu, Y, et al. Homozygous notch3 p.R587c mutation in Chinese patients with cadasil: a case report. BMC Neurol. (2020) 20:72. doi: 10.1186/s12883-020-01660-0
11. Tuominen, S, Juvonen, V, Amberla, K, Jolma, T, Rinne, JO, Tuisku, S, et al. Phenotype of a homozygous cadasil patient in comparison to 9 age-matched heterozygous patients with the same r133c notch3 mutation. Stroke. (2001) 32:1767–74. doi: 10.1161/01.str.32.8.1767
12. Vinciguerra, C, Rufa, A, Bianchi, S, Sperduto, A, De Santis, M, Malandrini, A, et al. Homozygosity and severity of phenotypic presentation in a cadasil family. Neurol Sci. (2014) 35:91–3. doi: 10.1007/s10072-013-1580-9
13. Dichgans, M, Herzog, J, and Gasser, T. Notch3 mutation involving three cysteine residues in a family with typical cadasil. Neurology. (2001) 57:1714–7. doi: 10.1212/wnl.57.9.1714
14. Saiki, S, Sakai, K, Saiki, M, Kitagawa, Y, Umemori, T, Murata, K, et al. Varicose veins associated with cadasil result from a novel mutation in the notch3 gene. Neurology. (2006) 67:337–9. doi: 10.1212/01.wnl.0000224758.52970.19
15. Mazzei, R, Conforti, FL, Lanza, PL, Sprovieri, T, Lupo, MR, Gallo, O, et al. A novel notch3 gene mutation not involving a cysteine residue in an italian family with cadasil. Neurology. (2004) 63:561–4. doi: 10.1212/01.wnl.0000133399.37716.84
16. Yoon, CW, Kim, YE, Seo, SW, Ki, CS, Choi, SH, Kim, JW, et al. Notch3 variants in patients with subcortical vascular cognitive impairment: a comparison with typical cadasil patients. Neurobiol Aging. (2015) 36:2443.e2441–7. doi: 10.1016/j.neurobiolaging.2015.04.009
17. Dotti, MT, De Stefano, N, Bianchi, S, Malandrini, A, Battisti, C, Cardaioli, E, et al. A novel notch3 frameshift deletion and mitochondrial abnormalities in a patient with cadasil. Arch Neurol. (2004) 61:942–5. doi: 10.1001/archneur.61.6.942
18. Moccia, M, Mosca, L, Erro, R, Cervasio, M, Allocca, R, Vitale, C, et al. Hypomorphic notch3 mutation in an Italian family with cadasil features. Neurobiol Aging. (2015) 36:547.e545–11. doi: 10.1016/j.neurobiolaging.2014.08.021
19. Erro, R, Moccia, M, Cervasio, M, Penco, S, De Caro, MB, and Barone, P. Are granular osmiophilic material deposits an epiphenomenon in cadasil? Folia Neuropathol. (2015) 53:168–71. doi: 10.5114/fn.2015.52414
20. Fouillade, C, Chabriat, H, Riant, F, Mine, M, Arnoud, M, Magy, L, et al. Activating notch3 mutation in a patient with small-vessel-disease of the brain. Hum Mutat. (2008) 29:452. doi: 10.1002/humu.9527
21. Joutel, A, Corpechot, C, Ducros, A, Vahedi, K, Chabriat, H, Mouton, P, et al. Notch3 mutations in cadasil, a hereditary adult-onset condition causing stroke and dementia. Nature. (1996) 383:707–10. doi: 10.1038/383707a0
22. Martignetti, JA, Tian, L, Li, D, Ramirez, MC, Camacho-Vanegas, O, et al. Mutations in pdgfrb cause autosomal-dominant infantile myofibromatosis. Am J Hum Genet. (2013) 92:1001–7. doi: 10.1016/j.ajhg.2013.04.024
23. Bersano, A, Ranieri, M, Ciammola, A, Cinnante, C, Lanfranconi, S, Dotti, MT, et al. Considerations on a mutation in the notch3 gene sparing a cysteine residue: a rare polymorphism rather than a cadasil variant. Funct Neurol. (2012) 27:247–52.
24. Meng, H, Zhang, X, Yu, G, Lee, SJ, Chen, YE, Prudovsky, I, et al. Biochemical characterization and cellular effects of cadasil mutants of notch3. PLoS One. (2012) 7:e44964. doi: 10.1371/journal.pone.0044964
25. Nagatoshi, A, Ueda, M, Ueda, A, Tasaki, M, Inoue, Y, Ma, Y, et al. Serum amyloid p component: a novel potential player in vessel degeneration in cadasil. J Neurol Sci. (2017) 379:69–76. doi: 10.1016/j.jns.2017.05.033
26. Zhao, Y, Lu, Y, Wang, F, Wang, Y, Li, Y, Sun, R, et al. Mechanistic advances in factors influencing phenotypic variability in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy: a review. Front Neurol. (2025) 16:1573052. doi: 10.3389/fneur.2025.1573052
27. Martinez Arias, A, Zecchini, V, and Brennan, K. Csl-independent notch signalling: a checkpoint in cell fate decisions during development? Curr Opin Genet Dev. (2002) 12:524–33. doi: 10.1016/s0959-437x(02)00336-2
28. Morris, HE, Neves, KB, Montezano, AC, Mac Lean, MR, and Touyz, RM. Notch3 signalling and vascular remodelling in pulmonary arterial hypertension. Clin Sci (Lond). (2019) 133:2481–98. doi: 10.1042/cs20190835
29. Baron-Menguy, C, Domenga-Denier, V, Ghezali, L, Faraci, FM, and Joutel, A. Increased notch3 activity mediates pathological changes in structure of cerebral arteries. Hypertension. (2017) 69:60–70. doi: 10.1161/hypertensionaha.116.08015
30. Peters, N, Opherk, C, Zacherle, S, Capell, A, Gempel, P, and Dichgans, M. Cadasil-associated notch3 mutations have differential effects both on ligand binding and ligand-induced notch3 receptor signaling through rbp-jk. Exp Cell Res. (2004) 299:454–64. doi: 10.1016/j.yexcr.2004.06.004
31. Coupland, K, Lendahl, U, and Karlström, H. Role of notch3 mutations in the cerebral small vessel disease cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Stroke. (2018) 49:2793–800. doi: 10.1161/strokeaha.118.021560
32. Mizuno, T, Mizuta, I, Watanabe-Hosomi, A, Mukai, M, and Koizumi, T. Clinical and genetic aspects of cadasil. Front Aging Neurosci. (2020) 12:91. doi: 10.3389/fnagi.2020.00091
33. Mukai, M, Mizuta, I, Watanabe-Hosomi, A, Koizumi, T, Matsuura, J, Hamano, A, et al. Genotype-phenotype correlations and effect of mutation location in japanese cadasil patients. J Hum Genet. (2020) 65:637–46. doi: 10.1038/s10038-020-0751-9
34. Hack, RJ, Gravesteijn, G, Cerfontaine, MN, Santcroos, MA, Gatti, L, Kopczak, A, et al. Three-tiered EGFR domain risk stratification for individualized notch3-small vessel disease prediction. Brain. (2023) 146:2913–27. doi: 10.1093/brain/awac486
35. Cho, BPH, Jolly, AA, Nannoni, S, Tozer, D, Bell, S, and Markus, HS. Association of notch3 variant position with stroke onset and other clinical features among patients with cadasil. Neurology. (2022) 99:e430–9. doi: 10.1212/wnl.0000000000200744
36. Juhosová, M, Chandoga, J, Cisárik, F, Dallemule, S, Ďurina, P, Jarásková, D, et al. Influence of different spectra of notch3 variants on the clinical phenotype of cadasil - experience from Slovakia. Neurogenetics. (2023) 24:1–16. doi: 10.1007/s10048-022-00704-6
37. Yamamoto, Y, Ihara, M, Tham, C, Low, RW, Slade, JY, Moss, T, et al. Neuropathological correlates of temporal pole white matter hyperintensities in cadasil. Stroke. (2009) 40:2004–11. doi: 10.1161/strokeaha.108.528299
38. Park, DG, Min, JH, Sohn, SH, Sohn, YB, and Yoon, JH. Ataxia associated with CADASIL: a pathology-confirmed case report and literature review. Cerebellum. (2020) 19:907–10. doi: 10.1007/s12311-020-01173-z
39. Schmidt, H, Zeginigg, M, Wiltgen, M, Freudenberger, P, Petrovic, K, Cavalieri, M, et al. Genetic variants of the notch3 gene in the elderly and magnetic resonance imaging correlates of age-related cerebral small vessel disease. Brain. (2011) 134:3384–97. doi: 10.1093/brain/awr252
40. Gorukmez, O, Gorukmez, O, Topak, A, Seferoglu, M, Sivaci, AO, Ali, A, et al. Notch3 variants in patients with suspected cadasil. Ann Indian Acad Neurol. (2023) 26:484–90. doi: 10.4103/aian.aian_989_22
41. Abou Al-Shaar, H, Qadi, N, Al-Hamed, MH, Meyer, BF, and Bohlega, S. Phenotypic comparison of individuals with homozygous or heterozygous mutation of notch3 in a large cadasil family. J Neurol Sci. (2016) 367:239–43. doi: 10.1016/j.jns.2016.05.061
42. Liu, X, Zuo, Y, Sun, W, Zhang, W, Lv, H, Huang, Y, et al. The genetic spectrum and the evaluation of cadasil screening scale in chinese patients with notch3 mutations. J Neurol Sci. (2015) 354:63–9. doi: 10.1016/j.jns.2015.04.047
43. Mykkänen, K, Junna, M, Amberla, K, Bronge, L, Kääriäinen, H, Pöyhönen, M, et al. Different clinical phenotypes in monozygotic cadasil twins with a novel notch3 mutation. Stroke. (2009) 40:2215–8. doi: 10.1161/strokeaha.108.528661
44. Kang, HG, and Kim, JS. Intracranial arterial disease in cadasil patients. J Neurol Sci. (2015) 359:347–50. doi: 10.1016/j.jns.2015.11.029
45. Choi, EJ, Choi, CG, and Kim, JS. Large cerebral artery involvement in cadasil. Neurology. (2005) 65:1322–4. doi: 10.1212/01.wnl.0000180965.79209.50
46. Zhang, C, Li, W, Li, S, Niu, S, Wang, X, Yu, X, et al. Intracranial large artery abnormalities and association with cerebral small vessel disease in cadasil. Front Neurol. (2020) 11:726. doi: 10.3389/fneur.2020.00726
47. Ruchoux, MM, Guerouaou, D, Vandenhaute, B, Pruvo, JP, Vermersch, P, and Leys, D. Systemic vascular smooth muscle cell impairment in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Acta Neuropathol. (1995) 89:500–12. doi: 10.1007/bf00571504
48. Santa, Y, Uyama, E, Chui, DH, Arima, M, Kotorii, S, Takahashi, K, et al. Genetic, clinical and pathological studies of cadasil in Japan: a partial contribution of notch3 mutations and implications of smooth muscle cell degeneration for the pathogenesis. J Neurol Sci. (2003) 212:79–84. doi: 10.1016/s0022-510x(03)00109-6
49. Libby, P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. (2012) 32:2045–51. doi: 10.1161/atvbaha.108.179705
50. Deguchi, JO, Aikawa, E, Libby, P, Vachon, JR, Inada, M, Krane, SM, et al. Matrix metalloproteinase-13/collagenase-3 deletion promotes collagen accumulation and organization in mouse atherosclerotic plaques. Circulation. (2005) 112:2708–15. doi: 10.1161/circulationaha.105.562041
51. Fukuda, D, and Aikawa, M. Expanding role of delta-like 4 mediated notch signaling in cardiovascular and metabolic diseases. Circ J. (2013) 77:2462–8. doi: 10.1253/circj.cj-13-0873
52. Fung, E, Tang, SM, Canner, JP, Morishige, K, Arboleda-Velasquez, JF, Cardoso, AA, et al. Delta-like 4 induces notch signaling in macrophages: implications for inflammation. Circulation. (2007) 115:2948–56. doi: 10.1161/circulationaha.106.675462
53. Fukuda, D, Aikawa, E, Swirski, FK, Novobrantseva, TI, Kotelianski, V, Gorgun, CZ, et al. Notch ligand delta-like 4 blockade attenuates atherosclerosis and metabolic disorders. Proc Natl Acad Sci USA. (2012) 109:E1868–77. doi: 10.1073/pnas.1116889109
54. Gomez, D, and Owens, GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res. (2012) 95:156–64. doi: 10.1093/cvr/cvs115
55. Shankman, LS, Gomez, D, Cherepanova, OA, Salmon, M, Alencar, GF, Haskins, RM, et al. Klf 4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. (2015) 21:628–37. doi: 10.1038/nm.3866
56. Jarad, S, Gill, G, Amadi, P, Gu, HM, and Zhang, DW. Vsmcs in atherosclerosis: implications on the role of inflammation and extracellular matrix remodelling. Pharmacol Res. (2025) 218:107833. doi: 10.1016/j.phrs.2025.107833
57. Davis-Knowlton, J, Turner, JE, Turner, A, Damian-Loring, S, Hagler, N, Henderson, T, et al. Characterization of smooth muscle cells from human atherosclerotic lesions and their responses to notch signaling. Lab Investig. (2019) 99:290–304. doi: 10.1038/s41374-018-0072-1
58. Keuylian, Z, de Baaij, JH, Gueguen, M, Glorian, M, Rouxel, C, Merlet, E, et al. The notch pathway attenuates interleukin 1β (il 1β)-mediated induction of adenylyl cyclase 8 (ac 8) expression during vascular smooth muscle cell (vsmc) trans-differentiation. J Biol Chem. (2012) 287:24978–89. doi: 10.1074/jbc.M111.292516
59. Dave, JM, Chakraborty, R, Ntokou, A, Saito, J, Saddouk, FZ, Feng, Z, et al. Jagged 1/notch3 activation promotes aortic hypermuscularization and stenosis in elastin deficiency. J Clin Invest. (2022) 132:338. doi: 10.1172/JCI142338
60. Luxán, G, D'Amato, G, Mac Grogan, D, and de la Pompa, JL. Endocardial notch signaling in cardiac development and disease. Circ Res. (2016) 118:e1–e18. doi: 10.1161/circresaha.115.305350
61. Lesnik Oberstein, SA, Jukema, JW, Van Duinen, SG, Macfarlane, PW, van Houwelingen, HC, Breuning, MH, et al. Myocardial infarction in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (cadasil). Medicine (Baltimore). (2003) 82:251–6. doi: 10.1097/01.md.0000085054.63483.40
62. Argirò, A, Sciagrà, R, Marchi, A, Beltrami, M, Spinelli, E, Salvadori, E, et al. Coronary microvascular function is impaired in patients with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Eur J Neurol. (2021) 28:3809–13. doi: 10.1111/ene.14678
63. Langer, C, Adukauskaite, A, Plank, F, Feuchtner, G, and Cartes-Zumelzu, F. Cerebral autosomal dominant arteriopathy (cadasil) with cardiac involvement (anoca) and subcortical leukencephalopathy. J Cardiovasc Comput Tomogr. (2020) 14:e1–6. doi: 10.1016/j.jcct.2018.08.005
64. Rubin, CB, Hahn, V, Kobayashi, T, and Litwack, A. A report of accelerated coronary artery disease associated with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Case Rep Cardiol. (2015) 2015:167513. doi: 10.1155/2015/167513
65. Servito, M, Gill, I, Durbin, J, Ghasemlou, N, Popov, AF, Stephen, CD, et al. Management of coronary artery disease in cadasil patients: review of current literature. Medicina (Kaunas). (2023) 59:586. doi: 10.3390/medicina59030586
66. Miao, Q, Paloneva, T, Tuominen, S, Pöyhönen, M, Tuisku, S, Viitanen, M, et al. Fibrosis and stenosis of the long penetrating cerebral arteries: the cause of the white matter pathology in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Brain Pathol. (2004) 14:358–64. doi: 10.1111/j.1750-3639.2004.tb00078.x
67. Bhattacharyya, A, Lin, S, Sandig, M, and Mequanint, K. Regulation of vascular smooth muscle cell phenotype in three-dimensional coculture system by jagged 1-selective notch3 signaling. Tissue Eng Part A. (2014) 20:1175–87. doi: 10.1089/ten.TEA.2013.0268
68. Ragot, H, Monfort, A, Baudet, M, Azibani, F, Fazal, L, Merval, R, et al. Loss of notch3 signaling in vascular smooth muscle cells promotes severe heart failure upon hypertension. Hypertension. (2016) 68:392–400. doi: 10.1161/hypertensionaha.116.07694
69. Zhang, M, Wang, C, Hu, J, Lin, J, Zhao, Z, Shen, M, et al. Notch3/akt signaling contributes to osm-induced protection against cardiac ischemia/reperfusion injury. Apoptosis. (2015) 20:1150–63. doi: 10.1007/s10495-015-1148-7
70. Bi, P, and Kuang, S. Notch signaling as a novel regulator of metabolism. Trends Endocrinol Metab. (2015) 26:248–55. doi: 10.1016/j.tem.2015.02.006
71. Gong, M, Rueschendorf, F, Marx, P, Schulz, H, Kraft, HG, Huebner, N, et al. Clinical and genetic features in a family with cadasil and high lipoprotein (a) values. J Neurol. (2010) 257:1240–5. doi: 10.1007/s00415-010-5496-5
72. Rassart, E, Bedirian, A, Do Carmo, S, Guinard, O, Sirois, J, Terrisse, L, et al. Apolipoprotein D. Biochim Biophys Acta. (2000) 1482:185–98. doi: 10.1016/s0167-4838(00)00162-x
73. Weech, PK, Provost, P, Tremblay, NM, Camato, RN, Milne, RW, Marcel, YL, et al. Apolipoprotein d--an atypical apolipoprotein. Prog Lipid Res. (1991) 30:259–66. doi: 10.1016/0163-7827(91)90023-x
74. Perdomo, G, and Henry Dong, H. Apolipoprotein d in lipid metabolism and its functional implication in atherosclerosis and aging. Aging (Albany NY). (2009) 1:17–27. doi: 10.18632/aging.100004
75. Vijayaraghavan, S, Hitman, GA, and Kopelman, PG. Apolipoprotein-d polymorphism: a genetic marker for obesity and hyperinsulinemia. J Clin Endocrinol Metab. (1994) 79:568–70. doi: 10.1210/jcem.79.2.7913935
76. Liu, H, Kennard, S, and Lilly, B. Notch3 expression is induced in mural cells through an autoregulatory loop that requires endothelial-expressed jagged 1. Circ Res. (2009) 104:466–75. doi: 10.1161/circresaha.108.184846
77. Pajaniappan, M, Glober, NK, Kennard, S, Liu, H, Zhao, N, and Lilly, B. Endothelial cells downregulate apolipoprotein d expression in mural cells through paracrine secretion and notch signaling. Am J Physiol Heart Circ Physiol. (2011) 301:H784–93. doi: 10.1152/ajpheart.00116.2011
78. Wimmer, RA, Leopoldi, A, Aichinger, M, Wick, N, Hantusch, B, Novatchkova, M, et al. Human blood vessel organoids as a model of diabetic vasculopathy. Nature. (2019) 565:505–10. doi: 10.1038/s41586-018-0858-8
79. Zhou, B, Lin, W, Long, Y, Yang, Y, Zhang, H, Wu, K, et al. Notch signaling pathway: architecture, disease, and therapeutics. Signal Transduct Target Ther. (2022) 7:95. doi: 10.1038/s41392-022-00934-y
80. Romay, MC, Knutsen, RH, Ma, F, Mompeón, A, Hernandez, GE, Salvador, J, et al. Age-related loss of notch3 underlies brain vascular contractility deficiencies, glymphatic dysfunction, and neurodegeneration in mice. J Clin Invest. (2024) 134:e166134. doi: 10.1172/JCI166134
81. Liao, YC, Hsiao, CT, Fuh, JL, Chern, CM, Lee, WJ, Guo, YC, et al. Characterization of cadasil among the han chinese in Taiwan: distinct genotypic and phenotypic profiles. PLoS One. (2015) 10:e0136501. doi: 10.1371/journal.pone.0136501
82. Chen, S, Ni, W, Yin, XZ, Liu, HQ, Lu, C, Zheng, QJ, et al. Clinical features and mutation spectrum in chinese patients with cadasil: a multicenter retrospective study. CNS Neurosci Ther. (2017) 23:707–16. doi: 10.1111/cns.12719
83. Wollenweber, FA, Hanecker, P, Bayer-Karpinska, A, Malik, R, Bäzner, H, Moreton, F, et al. Cysteine-sparing cadasil mutations in notch3 show proaggregatory properties in vitro. Stroke. (2015) 46:786–92. doi: 10.1161/strokeaha.114.007472
84. Mizuno, T, Muranishi, M, Torugun, T, Tango, H, Nagakane, Y, Kudeken, T, et al. Two japanese cadasil families exhibiting notch3 mutation r75p not involving cysteine residue. Intern Med. (2008) 47:2067–72. doi: 10.2169/internalmedicine.47.1391
85. Brass, SD, Smith, EE, Arboleda-Velasquez, JF, Copen, WA, and Frosch, MP. Case records of the Massachusetts General Hospital. Case 12-2009. A 46-year-old man with migraine, aphasia, and hemiparesis and similarly affected family members. N Engl J Med. (2009) 360:1656–65. doi: 10.1056/NEJMcpc0810839
86. Pippucci, T, Maresca, A, Magini, P, Cenacchi, G, Donadio, V, Palombo, F, et al. Homozygous notch3 null mutation and impaired notch3 signaling in recessive early-onset arteriopathy and cavitating leukoencephalopathy. EMBO Mol Med. (2015) 7:848–58. doi: 10.15252/emmm.201404399
87. Ragno, M, Pianese, L, Tiberi, S, Cacchiò, G, Paci, C, and Trojano, L. First report of a homozygous mutation on exon 24 of the notch3 gene in a paucisymptomatic cadasil elderly patient. Neurol Sci. (2022) 43:1457–8. doi: 10.1007/s10072-021-05706-0
88. Monet-Leprêtre, M, Haddad, I, Baron-Menguy, C, Fouillot-Panchal, M, Riani, M, Domenga-Denier, V, et al. Abnormal recruitment of extracellular matrix proteins by excess notch3 ecd: a new pathomechanism in cadasil. Brain. (2013) 136:1830–45. doi: 10.1093/brain/awt092
89. Liem, MK, Lesnik Oberstein, SA, Vollebregt, MJ, Middelkoop, HA, van der Grond, J, and Helderman-van den Enden, AT. Homozygosity for a notch3 mutation in a 65-year-old cadasil patient with mild symptoms: a family report. J Neurol. (2008) 255:1978–80. doi: 10.1007/s00415-009-0036-x
90. Soong, BW, Liao, YC, Tu, PH, Tsai, PC, Lee, IH, Chung, CP, et al. A homozygous notch3 mutation p.R544c and a heterozygous trex1 variant p.C99mfsx3 in a family with hereditary small vessel disease of the brain. J Chin Med Assoc. (2013) 76:319–24. doi: 10.1016/j.jcma.2013.03.002
91. Dichgans, M, Ludwig, H, Müller-Höcker, J, Messerschmidt, A, and Gasser, T. Small in-frame deletions and missense mutations in cadasil: 3d models predict misfolding of notch3 egf-like repeat domains. Eur J Hum Genet. (2000) 8:280–5. doi: 10.1038/sj.ejhg.5200460
92. Opherk, C, Peters, N, Herzog, J, Luedtke, R, and Dichgans, M. Long-term prognosis and causes of death in cadasil: a retrospective study in 411 patients. Brain. (2004) 127:2533–9. doi: 10.1093/brain/awh282
93. Wang, Z, Yuan, Y, Zhang, W, Lv, H, Hong, D, Chen, B, et al. Notch3 mutations and clinical features in 33 mainland chinese families with cadasil. J Neurol Neurosurg Psychiatry. (2011) 82:534–9. doi: 10.1136/jnnp.2010.209247
94. Tikka, S, Mykkänen, K, Ruchoux, MM, Bergholm, R, Junna, M, Pöyhönen, M, et al. Congruence between notch3 mutations and gom in 131 cadasil patients. Brain. (2009) 132:933–9. doi: 10.1093/brain/awn364
95. Mazzei, R, Guidetti, D, Ungaro, C, Conforti, FL, Muglia, M, Cenacchi, G, et al. First evidence of a pathogenic insertion in the notch3 gene causing cadasil. J Neurol Neurosurg Psychiatry. (2008) 79:108–10. doi: 10.1136/jnnp.2007.128009
96. Lackovic, V, Bajcetic, M, Lackovic, M, Novakovic, I, Labudović Borović, M, Pavlovic, A, et al. Skin and sural nerve biopsies: ultrastructural findings in the first genetically confirmed cases of cadasil in Serbia. Ultrastruct Pathol. (2012) 36:325–35. doi: 10.3109/01913123.2012.679352
97. Skehan, SJ, Hutchinson, M, and Mac Erlaine, DP. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy: Mr findings. AJNR Am J Neuroradiol. (1995) 16:2115–9.
98. Richards, S, Aziz, N, Bale, S, Bick, D, Das, S, Gastier-Foster, J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
99. Min, JY, Park, SJ, Kang, EJ, Hwang, SY, and Han, SH. Mutation spectrum and genotype-phenotype correlations in 157 korean cadasil patients: a multicenter study. Neurogenetics. (2022) 23:45–58. doi: 10.1007/s10048-021-00674-1
100. Lee, JS, Ko, K, Oh, JH, Park, JH, and Lee, HK. Phenotypic features of cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy subjects with r544c mutation. Dement Neurocogn Disord. (2016) 15:15–9. doi: 10.12779/dnd.2016.15.1.15
101. Choi, JC, Song, SK, Lee, JS, Kang, SY, and Kang, JH. Headache among cadasil patients with r544c mutation: prevalence, characteristics, and associations. Cephalalgia. (2014) 34:22–8. doi: 10.1177/0333102413497598
102. Choi, BW, Park, S, and Kim, HJ. Possible role of a missense mutation of p.P167s on notch3 gene associated with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Dement Neurocogn Disord. (2016) 15:52–4. doi: 10.12779/dnd.2016.15.2.52
103. Qin, W, Ren, Z, Xia, M, Yang, M, Shi, Y, Huang, Y, et al. Clinical features of 4 novel notch3 mutations of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy in China. Med Sci Monit Basic Res. (2019) 25:199–209. doi: 10.12659/msmbr.918830
Keywords: CADASIL, NOTCH3 , NOTCH3 mutaion spectrum diseases, hereditary cerebral small vessel disease, the phenotypic heterogeneity
Citation: Wang Y, Liu Y, Mo H, Han Y, Jing Y and Deng F (2025) CADASIL or NOTCH3 mutaion spectrum diseases? Interpretation of NOTCH3 mutations and clinical heterogeneity in CADASIL. Front. Neurol. 16:1662012. doi: 10.3389/fneur.2025.1662012
Edited by:
Li Wang, First Hospital of Tsinghua University, ChinaReviewed by:
Orhan Görükmez, Şevket Yılmaz Hospital, Bursa, TürkiyeWan Wang, Sun Yat-sen University, China
Sánchez-Lanzas Raúl, Queen Mary University of London, United Kingdom
Copyright © 2025 Wang, Liu, Mo, Han, Jing and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fang Deng, ZGVuZ19mYW5nQGpsdS5lZHUuY24=